

Abstract
Eicosanoids and specialized proresolving mediators (SPM) regulate leukocyte function and inflammation. They are ideally positioned at the interphase of innate and adaptive immune responses when lymphocytes interact with leukocytes. Receptors for LTB4, PGE2 and SPM are expressed in lymphocytes. Evidence points toward an essential role of these lipid mediators for directly regulating lymphocyte functions. SPMs, which include lipoxins, demonstrate comprehensive protective actions with lymphocytes. LTB4 and PGE2 regulation of lymphocyte is diverse and depends on the interaction of lymphocytes with other cells. Importantly, both LTB4 and PGE2 are essential regulators of T cell anti-tumor activity. These lipid mediators are attractive therapeutic targets to control dysregulated innate and adaptive immune responses, promote lymphocyte antitumor activity and prevent tumor immune evasion.
Keywords: adaptive immune response, lipoxins, leukotrienes, prostglandins, resolvins, lymphocytes, cancer, Treg, helper cells, T cell, B cell, tumor immune evasion, NK cell
Intrinsic Lipid Mediator Signaling in the Immune System
The immune system consists of an intricate network of continuous signaling and communication; where cells regulate one another in complementary and interloping manners for stratified defense and response. Acute inflammation elicits a tissue-specific immune program through the activation of tissue resident cells and innate immune cells, which secrete signaling molecules to enlist and attune adaptive immune cells equipped with an enhanced and specific response to previously encountered antigens. Lipoxygenase- and cyclooxygenase-derived lipid mediators (LMs), eicosanoids and specialized proresolving mediators (SPMs), are early response signaling molecules generated as an essential response to inflammatory triggers and regulators of leukocyte-mediated inflammation. Eicosanoids and SPMs are tissue-specific autocrine and paracrine signals that regulate the activation, amplitude, and resolution of acute inflammation [1, 2]. The production of eicosanoids and SPMs is temporally defined, and in acute inflammation they often have opposing and counter-regulatory actions. Every human cell type and tissue express one or more cyclooxygenase and lipoxygenase enzyme(s) and G-protein coupled receptors for prostaglandins, leukotrienes and/or SPMs. A large body of work has established the integral roles of these LMs in innate immune cell function that drives inflammation and its resolution [1–4]. However, current understanding of the direct actions of eicosanoids and SPMs on lymphocytes is still limited.
The interaction between innate immune cells and antigen-specific lymphocytes is critical to initiate or prevent adaptive immune responses. Eicosanoids and SPMs are important regulators for demarcating healthy inflammation and immune disease. Interests in eicosanoid and SPM regulation of lymphoid cells have intensified, as dysregulated lymphocyte function is a primary cause of various debilitating diseases such as allergies, asthma, autoimmunity and cancer. Tissue-specific and temporal profiles of eicosanoids and SPMs determine whether acute inflammation is a balanced, healthy response and actively resolves, or morphs into chronic inflammation. Dysregulated inflammation can trigger amplified effector lymphocyte responses often resulting in sustained tissue damage, increased risk of autoantigen recognition or tumor immune evasion. Eicosanoids and SPMs are important in both innate and adaptive immunity, and are promising therapeutic targets due to their potent bioactions with leukocytes. However, mechanisms of LM regulation of lymphocyte functions such as cytokine secretion, trafficking and differentiation remain ambiguous. This review will focus on the current understanding of SPMs, leukotriene B4 (LTB4), and prostaglandin E2 (PGE2) regulation of lymphoid-derived cells in adaptive immunity and cancer.
LXA4 Regulation of Lymphoid-derived Cells
The eicosanoid lipoxin A4 (LXA4) was discovered as the first SPM in 1984, which was followed in the 2000s by the discoveries of a large super family of DHA-, EPA- and DPA- derived ω-3 SPMs. The formation and actions of SPMs are active areas of research, and their therapeutic potential is of considerable interest that has propelled the burgeoning resolution pharmacology field [5, 6]. Despite an impressive body of work, the direct action of SPMs on lymphocytes is not well characterized. Hallmark bioactions of LXA4 that define the large SPM family are the abilities to inhibit vascular neutrophil migration, enhance macrophage efferocytosis and downregulate pro-inflammatory cytokines, chemokines and cell adhesion molecules, all of which reduce the amplitude of inflammation and drive active resolution [7]. Initially, SPMs bioactions on lymphocyte function in inflammatory disease models were considered secondary, since ALX/FPR2 was originally discovered in myeloid cells [8] as the first non-prostanoid eicosanoid receptor. In addition, SPMs demonstrated potent bioaction in controlling innate immune cell function and antigen presenting cell (APC) activation. This paradigm shifted with the discovery of the LXA4 receptor/formyl peptide receptor 2 (ALX/FPR2) on T cells and the ability of LXA4 stable mimetics to inhibit TNF-α secretion in activated human T cells [9]. Additional SPM receptors have recently been discovered in conjunction with SPM proresolving functions, which includes a second LXA4 receptor GPR32 in humans [10].
Recent studies have uncovered the important roles of endogenous and tissue resident LXA4 circuits on T cell function. The immune regulatory function of LXA4 on cells was investigated in vivo in an immune-driven dry eye model, where neutrophil-derived LXA4 was a critical resident signal to control pathogenic T helper cell type 1 (Th1) and T helper cell type 17 (Th17) effector cells and increased the number of T regulatory cells (Tregs) in the eye draining lymph nodes [11, 12]. More importantly, sex-specific regulation of the LXA4 circuit in resident lymph nodes was identified as a key factor that drives female-specific immune-driven dry eye disease. The amplified adaptive immune response in females to routine ocular surface stress can be rescued by treatment with LXA4.
An in vitro study showed that LXA4 promotes the differentiation of naïve T cells into T follicular cells, which in turn induces B cells to form germinal centers [13], demonstrating that LXA4 mediates cellular signaling among lymphocytes. Direct LXA4 regulation of B cells has also been established. In vitro LXA4 treatment reduces IgG and IgM production from B cells and decreases memory B cell proliferation in an ALX/FPR2 receptor-dependent mechanism [14]; and in vivo, LXA4 treatment protects against LPS-induced sepsis by promoting generation and migration of splenic B cells [15].
ALX/FPR2 receptor expression has been identified in human natural killer (NK) cells [16] and LXA4 can induce protective functions of K cells during airway inflammation. NK cells from asthma patients treated with LXA4 ex vivo maintain functional killing responses [17], alleviate airway inflammation by increasing NK cell-mediated eosinophil apoptosis, and reduce interleukin-13 (IL-13) release by group 2 innate lymphoid cells (ILC2) [18].
Recent reports have demonstrated that lipoxins are not only formed during inflammation and the resolution phase of inflammation, but that they are also part of normal signaling in healthy tissues and actively regulate homeostasis and the threshold for activation of immune responses in the cornea, lymph nodes, lacrimal glands and retina [11, 12, 19, 20]. Regulation and therapeutic amplification of this homeostatic SPM circuit in health and diseases is the focus of several NIH-funded projects.
EPA- and DHA- derived SPM Regulation of Lymphoid-derived Cells
The field of SPMs emerged from the discovery of distinct EPA- and DHA- derived mediators that shared some of the basic pro-resolving and protective actions of lipoxins and displayed potent bioactions in several inflammatory disease models. Distinct SPM receptors that were originally identified in innate leukocytes are also expressed in lymphocytes [10, 21]: FPR2/ALX for LXA4, resolvin D1 (RvD1); G protein-coupled receptor 32 (GPR32) for LXA4, RvD1; Gprotein- coupled receptor 18 (GPR18) for RvD2; chemokine-like receptor 1 (ChemR23) for resolvin E1 (RvE1) [22].
Identification of SPM receptors on lymphocytes [23] spurred efforts to investigate direct lymphocyte regulation by SPMs. In vitro, RvD1 and RvE1 downregulate Th1 and Th17 differentiation, cytokine production and expression of T cell lineage transcription factors T-bet, GATA3 and RORc, as well as induce de novo iTreg generation [24]. This may suggest a role of SPMs in T cell lineage commitment. Several reports have also demonstrated in vivo lymphocyte regulation by RvD1 in inflammation and infection models. RvD1 treatment in LPS-induced uveitis reduces infiltration of CD4+ cells, CD8+ T cells, B cells and CD11b+ cells in the eye [25,26]. Consistent with its protective function in inflammation, RvD1 increases local Treg cell counts in the inflamed tissue in experimental autoimmune neuritis [27]. It is important to note that the DHA- derived RvD1 is a structural homolog of LXA4 and mediates its action via the same two receptors (FPR2/ALX and GPR32) as LXA4. Hence, it is expected that LXA4 and RvD1 have similar direct actions on lymphocytes.
As a treatment, the RvD1 epimer 17R-RvD1 can quell infection by reducing the number of Th1 and Th17 cells and inhibiting the production of proinflammatory cytokines in stromal keratitis [28]. RvD1, like LXA4, also has direct actions on human B cells by suppressing IgE production and differentiation of naïve B cells [29]. In a follow up study, RvD1 reduces IgE production by B cells in asthma patients treated with low dose steroids [30]. Other members of the SPM family such as maresin-1 (MaR1) also demonstrated its therapeutic and protective effects in vivo by restraining IL-13 cytokine production from ILCs and increasing de novo generation of induced Tregs (iTregs) to resolve lung inflammation [31]. A receptor for MaR1 has yet to be identified, therefore it is unclear if these are direct or indirect actions on lymphocytes.
Consistent with their broad protective actions in acute inflammation, SPMs downregulate effector T cell and B cell function. Hence, they are attractive therapeutic targets for controlling dysregulated innate and adaptive immune responses. A hot area of cancer research is the development of biological therapy, which is aimed at amplifying the adoptive T cell response to cancer cells. Hence, how SPM downregulation of adaptive immune responses potentially impacts immune evasion in the tumor environment needs to be investigated.
LTB4 Regulation of Lymphoid-derived Cells
Expression of the leukotriene B4 receptor 1 (BLT1) was identified on cells in 2003 [32, 33], and the initial findings on LTB4-mediated T cell response were investigated using allergic lung inflammation models [34]. These experiments established LTB4-mediated T cell recruitment, and implicated CD8+ T cells as the main pathogenic cell type driving allergic airway inflammation. BLT1 expression was higher on cells of human asthma patients than healthy individuals, which corresponded to disease severity and confirmed a role of the BLT1-LTB4 axis in pathogenic cell recruitment in asthma [35].
In vitro, LTB4 has dichotomous effects on T lymphocytes. In T cell differentiation assays, LTB4 inhibits de novo iTreg generation and increases interleukin-17 (IL-17) cytokine production [36], whereas LTB4-activated T cells inhibit proliferation of Epstein-barr virus-infected B cells [37], demonstrating that LTB4, like LXA4, can mediate cellular interactions among T cells and B cells.
Recent work has provided evidence of LTB4 regulating migration of various lymphoid-derived cell types. In an experimental autoimmune encephalomyelitis model, LTB4 guides the migration of Th17 cells into the central nervous system and induces pathology [38]. In contact dermatitis, inhibition of the LTB4-BLT1 axis ameliorates disease by preventing neutrophil and CD8+ T cell recruitment [39]. Although eicosanoid and SPM generation by the different lymphoid cell types is not well defined, it has been shown that virus-infected human CD4+ T cells can secrete LTB4 to further recruit T cells and propagate virus infection, and inhibition of LTB4 synthesis reduces viral load [40]. The LTB4-BLT1 axis also directs γδ T cell migration in murine pleural cavities in an LPS inflammation model [41], and induces NK cell chemotaxis in vitro in a BLT1 receptor-specific manner [42].
Most recently, BLT1 and the cysteinyl leukotriene receptor 1 (CysLT1R) were identified on group 2 innate lymphoid cells (ILC2) [43, 44], and LTB4 was shown to activate ILC2 and the downstream helper cell type 2 cytokine production in a NFAT-dependent manner during lung inflammation [44]. Thus LTB4 may indirectly regulate cell proliferation and differentiation through NFAT-mediated IL-2 production.
Current state of the field indicates that LTB4 has pleiotropic actions on lymphocytes to dynamically regulate the immune response in a cell type- and context-dependent manner. The underlying mechanisms warrant further investigation, especially since many drugs that target the LTB4 pathways and BLT1 are in clinical trials or FDA-approved.
PGE2 Regulation of Lymphoid-derived Cells
PGE2 exercises complex and multidimensional actions due to having four distinct receptors EP1, EP2, EP3 and EP4, expressed in many cell types. PGE2 regulates normal physiology but is also a key mediator of acute inflammation and autoimmune disorders. APC serve as prominent cellular sources of PGE2 to suppress T cell activation and regulate innate immune cells via paracrine or autocrine signaling [45, 46].
The immunomodulatory actions of PGE2 on T lymphocytes have been studied extensively. PGE2 exerts many physiological actions on T cells, including thymic T cell development, T helper cell differentiation, migration, and cytokine production [47, 48]. The main prostaglandin receptor expressed on effector cells are EP2, involved in Th17 cytokine production, and EP4 that regulates IFN-γ and interleukin-10 (IL-10) production [49].
The contrasting effects of PGE2 on T cell activation and cytokine production are in part due to different expression of costimulation molecules and the state of activation of T cells [50]. More importantly, the diverse and often contrasting actions PGE2 also stem from the heterogeneous cellular sources of PGE2 and cell type-specific interaction with T effector cells. Macrophage-produced PGE2 enhances IFN-γ and IL-17A production by CD4+ T cells [51], thereby augmenting T effector functions. In contrast, multipotent adult progenitor cell-produced PGE2 upregulates suppressor of cytokine signaling-2 (SOCS2) and growth arrest and DNA-damage-inducible protein alpha (GADD45A) expression in T cells, thereby preventing effector cell expansion [52]. However, PGE2 appears to downregulate effector cell function in vivo [53]. The intrinsic role of PGE2 in regulating lymphocyte function is underscored by the discovery that activated human CD4+ T cells generate PGE2, which in turn serves as an autocrine signal to further upregulate EP2 and EP4 expression in T cells [54]. More importantly, T cell-secreted PGE2 is a determinant for T cell cytokine response and polarization to Treg, Th1 or Th17 phenotypes [55].
PGE2 has conflicting roles in T cell differentiation. Keratinocyte-produced PGE2 inhibits T helper cell proliferation in a psoriasis model. In vitro, PGE2 can upregulate Foxp3, a lineage specification marker expressed by Treg [56], consistent with cancer models where PGE2 produced in the tumor microenvironment can polarize T cells toward the iTreg phenotype to suppress anti-tumor responses [57, 58]. However, in another in vitro study, PGE2 was shown inhibit iTreg differentiation via EP2 the receptor [59], emphasizing differential roles that depend on the tissue environment.
Direct bioactions of PGE2 on NK cells and the regulation of cognate EP receptors have been elucidated mostly in vitro. Tissue-secreted PGE2 suppresses NK cell activation [60, 61], suggesting protection against cytotoxic cell damage. All four EP receptors are functionally expressed on NK cells but PGE2 primarily mediates its suppressive action via EP2 and EP4. PGE2 regulation of NK cells leads to loss of function by blocking cell migration, inhibiting NK cell-mediated cytotoxicity and IFN-γ and TNF-α cytokine production[62–64]
Protective functions of PGE2 also include direct regulation of innate lymphoid cells (ILCs), which express the EP4 receptor. In a systemic inflammation mouse model, PGE2 maintains gut barrier homeostasis by triggering interleukin-22 released by type 3 innate lymphoid cells (ILC3s) [65]. PGE2 can also directly inhibit ILC2 function in allergic airway inflammation by reducing eosinophilia, interleukin-5 and cytokine production [66].
PGE2 is a complex signaling molecule with disparate functions that are contingent on the cell type, tissue, EP receptor signaling and immune response. It is clear that PGE2 and receptors are an integral part of lymphocyte responses; however, PGE2 as a therapeutic target so far has largely been overlooked by the immunology field, likely due to its complex and diverse actions in and health and disease.
Eicosanoid and SPM Regulation of T cell Function in Cancer
In view of advancements in cancer immunotherapy that aims to eradicate cancer in a targeted and personalized approach, recent clinical trials also have shone light on the drawbacks of altered T cell function. To amplify endogenous immune responses, checkpoint inhibitor treatments prevent the “deactivation” of cells to continuously combat tumor cells. However, such approach sometimes lead to over-reactive immune responses that drive the immune system toward autoimmunity [67], thus the fine balance of administering checkpoint inhibitors to enhance anti-tumor responses of T cells without eliciting autoimmune responses becomes a challenging feat. Conversely, autoimmunity (and infection, not covered in the scope of this review) sustained by chronic inflammation could increase the risk of developing cancer, as long-term exposure to inflammation causes DNA damage and/or mutation, and the damaged cells in turn proliferate in an inflammatory environment that potentiates neoplasia [68, 69]. The duality of autoimmunity and cancer thus poses an immunological conundrum, in which the interconnected relationship between LMs and inflammation could be considered as a target of interest to reverse inflammation-induced immunological aberrations underlying cancer and autoimmunity.
The tumorigenic role of cyclooxygenase-2 (COX-2) is well characterized and amplified formation of PGE2 by COX-2 has been established as a cause of tumor progression and metastasis [70]. As cancer therapeutics, COX-2 inhibitors are effective in preventing tumor growth and cancer metastasis of human breast, lung, colorectal and prostate cancers [71]. Despite extensive studies, the mechanism of COX-2 tumorigenicity is still not well defined. Studies have demonstrated COX-2 expression in tumors and tumor-secreted PGE2 induce Foxp3 expression and Treg activity within the tumor microenvironment to allow tumor immune evasion [72, 73], while administration of COX-2 inhibitors promotes local anti-tumor effect by reducing locally converted Tregs in a renal cell carcinoma model [74]. In vitro, tumor necrosis induces COX-2 expression, which amplifies PGE2 release that leads to impaired cytotoxic T cell (CTL) function and enables tumor growth [75]. In lung carcinoma models, activation of endothelial cells by tumor-derived vascular endothelial growth factor (VEGF) leads to PGE2 production and subsequent suppression of anti-tumor T cell functions [76]. PGE2 inhibition of T effector responses and induction of Tregs support the notion that impaired anti-tumor T cell activity and subsequent tumor growth are caused in part by dysregulated COX-2 expression and PGE2 levels.
The consistent suppressive action of PGE2 on lymphoid cells is also observed in γδ T cells in vitro simulating the tumor microenvironment. PGE2 inhibits T cell receptor (TCR) -mediated cytotoxicity of γδ cells [64], downregulate γδ T cell cytokine production and proliferation via signaling through EP2 and EP4 [77]. Therefore, PGE2 secreted by tumor cells can potentially thwart antitumor activities by γδ T cells.
As a potent chemoattractant for T cells and neutrophils, the LTB4-BLT1 axis can be a significant regulator of both innate and adaptive immune cells in the inflammatory tumor microenvironment. Therefore, the role of LTB4 in cancer becomes incongruous depending on the cell type and context of inflammation. LTB4 can either attract CTLs to help kill tumor cells, or promote tumor growth by inducing neutrophilic inflammation [78, 79]. Established as a pro-inflammatory mediator, LTB4 recruitment of T cells in cancer can render the inflammatory event beneficial as CTL recruitment is a critical step for effective tumor cell killing. Deletion of 5-lipoxygenase (5-LOX), the rate-limiting enzyme of LTB4 production, increases primary tumor volume and liver metastases in mice. Tumors in 5-LOX deficient mice have decreased numbers of CD4+ T cells and CTL, indicating impaired cell recruitment and killing, which correlates with increased tumor growth when CTLs are depleted [80]. In a cervical cancer model, expression of BLT1 on CTLs and NK cells was shown to be essential for effector anti-tumor response. The indispensable role of LTB4-BLT1 axis for reducing tumor growth and survival was established with adaptive transfer of CTLs from tumor-bearing BLT1+/+ or BLT1−/− mice [81]. In another similar experiment, BLT1−/− mice bred onto a spontaneous tumor model showed increase tumor development and mortality [82]. Hence, multiple lines of evidence have established an essential role of LTB4-BLT1 axis in preventing tumor progression.
The infiltration of Tregs into tumors is an indicator of poor disease outcome as regulatory immune cells can help cancer cells bypass immune surveillance. In a murine breast cancer model, LTB4 can induce generation of B regulatory cells (Bregs) by upregulating proliferator-activated receptor alpha (PPARα) expression, increased Bregs in turn facilitate cancer metastasis [83]. Whether LTB4 can also induce generation of Treg cells is of interest, since the role of LTB4 depends on the balanced recruitment of CTLs and Tregs, which would dictate tumor survival or elimination.
Since SPM regulation of T cells is an emerging field of research, there are no published reports of direct SPM regulation of cells in cancer to date. Research efforts have primarily focused on SPMs’ ability to inhibit inflammation, promote efferocytosis and inhibit angiogenesis in cancer models and their direct interactions with cancer cells. LXA4 can attenuate cancer cell invasion and metastasis, RvD2 inhibits oral cancer growth in vivo and in vitro by reducing cancer-derived cytokines and chemokines, resolvins D1, D2 and E1 inhibit cancer progression by inducing macrophage efferocytosis and reducing inflammatory cytokine production, and treating gastric cancer cells with LXB4 and RvD1 reduces angiogenesis [84].
Research efforts are underway to define SPM regulation of distinct populations of lymphoid cells, which could complement their established efferocytosis function, as well as anti-tumor, anti-inflammatory and anti-angiogenic activity.
Compelling evidence has established PGE2 and LTB4 as important regulators of T cell anti-tumor activity. Enzyme inhibitors, receptor-specific agonists and antagonists are well developed and/or in clinical use for LTB4, PGE2 and SPMs. Eicosanoids and SPMs should be explored as complementary approaches with cellular immune therapies to ablate the inflammatory tumor microenvironment, promote CTL infiltration into tumors, stop tumor growth and prevent tumor immune evasion.
Concluding Remarks
Inflammation triggers dynamic and diverse arrays of tissue-specific eicosanoids and SPMs. These paracrine and autocrine early response mediators regulate activation, function, and migration of leukocytes and are established therapeutic targets. Reports covered in this review have identified direct regulation of lymphoid cells by PGE2, LTB4, and SPMs. These LMs are ideally positioned at the interphase of innate and adaptive immunity and are determinants for healthy or dysregulated lymphocyte responses in inflammation and cancer. However, the mechanism of eicosanoids and SPMs regulation of lymphoid cells in health and diseases is still not well defined.
SPMs, which include the eicosanoids LXA4 and LXB4, demonstrate comprehensive protective actions with lymphocytes by downregulating effector T cell cytokine release, inhibiting B cell antibody formation and enhancing NK cell protective action to prevent lymphocyte driven disease. LTB4 and PGE2 regulation of lymphocyte function is complex and depends on the inflammatory scenario and the interaction of lymphocytes with other cells. Moreover, LTB4 and PGE2 can be generated by lymphocytes, to either promote or inhibit effector T cell function, migration and differentiation. Importantly, both LTB4 and PGE2 are essential regulators of T cell anti-tumor activity.
There is a striking gap of knowledge in our understanding of the endogenous roles of SPMs, LTB4 and PGE2 in lymph nodes, their function and/or formation in distinct lymphoid cell types and their cellular sources and regulation in the tumor microenvironment. The diverse cellular source of these LMs, autocrine and paracrine actions and their dynamic and prevalent receptor expression present significant experimental hurdles. However, tools for these well-developed therapeutic targets are in place, such as recent advances in CRISPR-Cas9, conditional and/or cell specific knockout or knockin mouse lines can be genetically engineered to clearly define their role in lymphocyte regulation.
Figure 1.
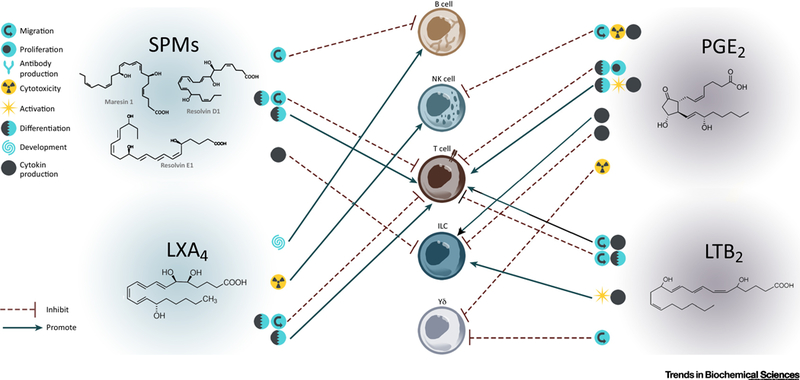
Direct immune regulation of lymphoid cells by SPMs and eicosanoids during inflammation. Lipid mediators and inflammation are conceptually and functionally closely intertwined. Upon cellular activation, lipid mediators are synthesized de novo locally and rapidly metabolized at the site of inflammation to modulate the immune response. SPMs and eicosanoids can directly inhibit or promote lymphocytes functions as depicted by symbols representing migration, proliferation, antibody production, cytotoxicity, activation, differentiation, development and cytokine production. Eicosanoids and SPMs mediate intracellular communication in a cell -specific approach.
Figure 2.
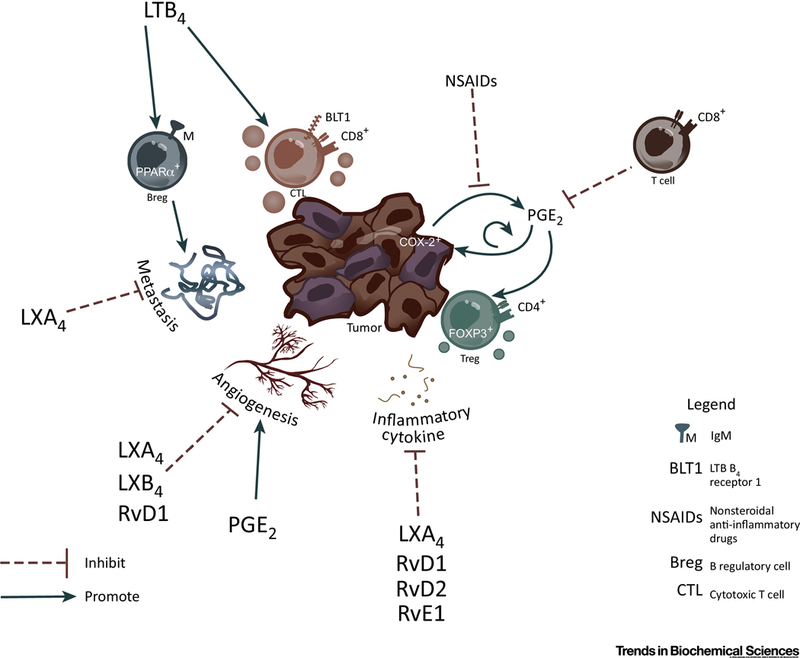
Direct immune regulation of lymphoid cells by SPMs and eicosanoids in cancer. Tumor COX-2 expression induces PGE2 production and tumor growth in a self-feedback loop manner. PGE2 increases angiogenesis that facilitates tumor growth and metastasis, as well as promote the differentiation of Treg to help tumors evade immune surveillance. Actions of COX-2 and PGE2 are inhibited by NSAIDs. Lipoxins and SPMs have broad actions that result in inhibition metastasis, angiogenesis and/or inflammatory cytokine production. LTB4 can have dichotomous functions on lymphocytes in the tumor environment promoting tumor metastasis via upregulating the expression of PPAR□ in Bregs, and on the other hand directly recruiting CTLs to tumors to enhance tumor killing.
Figure 3.
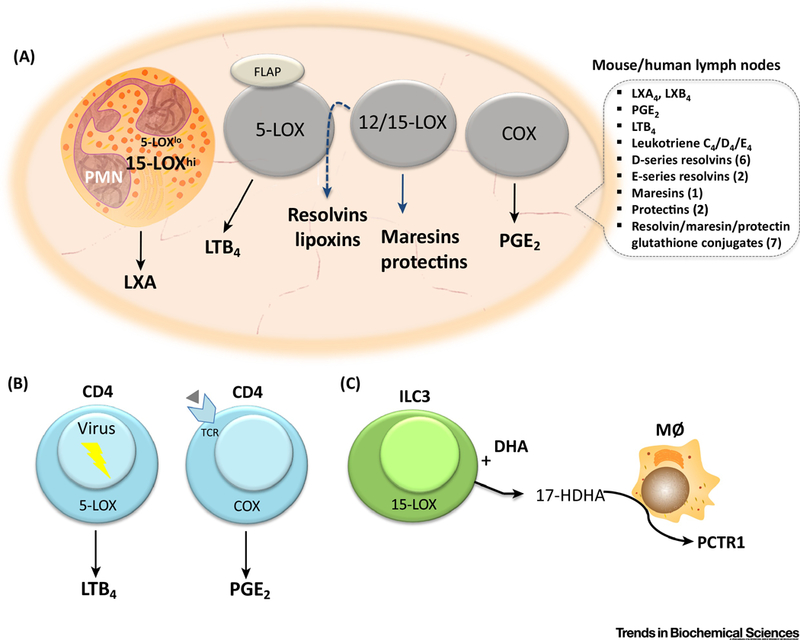
Eicosanoid and SPM biosynthetic pathways in lymphocytes and lymph nodes. a) Eicosanoids and SPMs formed in mouse and human lymph nodes. Indicated are the number of structurally distinct and bioactive resolvins, protectins and resolvin/protectin/maresin glutathione conjugates (PCTR, MCTR, RCTR) that have been documented in human lymph nodes [85, 86]. Gray-colored cells indicating unidentified cellular source of eicosanoids. PMN= neutrophil; 5-LOX= 5-lipoxygenase; FLAP= 5-lipoxygenase activating protein; 12/15-LOX= 12/15-lipoxygenase; COX= cyclooxygenase. b) Direct LTB4 formation by virus infected CD4+ T cells, and direct PGE2 formation by CD4+ T cells upon T cell receptor stimulation. c) Transcellular generation of a protectin glutathione-conjugate, protectin conjugates in tissue regeneration (PCTR) by innate lymphoid cell type 3 (ILC3). ILC3 in presence of DHA forms 17-HDHA, which then is converted by macrophages (MØ) to CTR.
Table 1.
General functions of lymphoid-derived cells.
| Cell Type | Antigen Specificity |
SPM/ Eicosanoid Receptor Expression |
Function |
|---|---|---|---|
| T helper Cell Type 1 |
⌷ | ALX/FPR2, BLT1, ChemR23, EP2, EP4, GPR32 |
CD4+ effector cell against intracellular pathogens, autoimmunity, inflammation |
| T helper Cell Type 17 |
⌷ | ALX/FPR2, BLT1, ChemR23, EP2, EP4, GPR32 |
CD4+ effector cell against extracellular pathogens, autoimmunity, inflammation |
| Cytotoxic T Lymphocyte |
⌷ | BLT1 | CD8+ effector cell that kill cells infected with intracellular pathogens and cancer cells |
| T regulatory Cell |
⌷ | ALX/FPR2, EP2 | CD4+ FOXP3+ cell that suppresses effector cell responses, immune tolerance |
| B Cell | ⌷ | ALX/FPR2, GPR32(?) |
Antibody production against pathogens, humoral immunity |
| B regulatory Cell |
⌷ | BLT1/2 | Suppresses expansion of pathogenic lymphocytes, promote Treg generation |
| γδ T cell | ⌷ ⌷ | BLT1, EP2, EP4 | Immune regulation, inflammation, antigen presentation |
| Innate Lymphoid Cell Type 1/ Natural Killer Cell |
⌷ | ALX/FPR2, BLT1, EP1–4 |
Immunity against intracellular pathogens, cytotoxicity |
| Innate Lymphoid Cell Type 2 |
⌷ | ALX/FPR2, BLT1, CysLT1R, EP4 |
Immunity against parasites, allergies and asthma responses |
| Innate Lymphoid Cell Type 3 |
⌷ | EP4 | Immunity against extracellular bacteria, maintains gut homeostasis and mucosal barrier function |
Highlights.
Eicosanoids and SPMs are ideally positioned at the interphase of innate and adaptive immunity. Their regulation of acute inflammation and innate immune cell function is well established and an important therapeutic target for inflammatory diseases.
Eicosanoid and SPMs are formed in lymph nodes in health and disease
Expression of PGE2, LTB4 and SPM receptors has been identified on lymphoid cells and established direct regulation of lymphocyte function by these lipid mediators.
SPM treatment controls effector T cell function and reduces diseases pathogenesis. LTB4 and PGE2 have tissue- and cell type-specific action that are critical determinants for effector cell function, migration and differentiation.
Eicosanoid and SPM therapeutics have the potential to reduce inflammation in the tumor microenvironment, promote lymphocyte anti-tumor activity, stop tumor growth and prevent tumor immune evasion.
Outstanding Questions.
Lymph nodes initiate, amplify and suppress lymphocyte functional responses. PGE2 and LXA4 are generated in lymph nodes in health and disease. What is their cellular source, role and mechanism of action in lymph nodes?
The diverse lymphocyte populations have been classified in detail in terms of cytokine production, receptors and protein markers. What functional eicosanoid and/or SPM receptors do these distinct lymphocyte cell type express? Is it possible to define the function of different immune cells according to their temporal expression of LM enzymes and receptors and does this knowledge enable us to develop therapeutic that target specific lymphoid cell functions?
The tumor environment can contain a large number of myeloid derived suppressor cells, namely tumor-associated macrophages and neutrophils, which inhibit the anti-tumor activity of lymphocytes. A key feature of some tissue resident macrophages and neutrophils is their high capacity to generate GE2 and LXA4, respectively. Do myeloid derived suppressor cells in the tumor microenvironment generate eicosanoids or SPMs?
Acknowledgement
The authors would like to thank Elizabeth (Liz) Lawler for creating figures in Adobe Illustrator. Cited work from the authors was supported in part by grants (to KG) from the National Institutes of Health (EY022208, EY026082) and Sjögren’s Syndrome Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bennett M and Gilroy DW (2016) Lipid Mediators in Inflammation. Microbiol Spectr 4 (6). [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN and Levy BD (2018) Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 128 (7), 2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dennis EA and Norris PC (2015) Eicosanoid storm in infection and inflammation. Nat Rev Immunol 15 (8), 511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvarez Y et al. (2010) Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm 2010. [DOI] [PMC free article] [PubMed]
- 5.Crean D and Godson C (2015) Specialised lipid mediators and their targets. Semin Immunol 27 (3), 169–76. [DOI] [PubMed] [Google Scholar]
- 6.Basil MC and Levy BD (2016) Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 16 (1), 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godson C et al. (2000) Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 164 (4), 1663–7. [DOI] [PubMed] [Google Scholar]
- 8.Fiore S et al. (1994) Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med 180 (1), 253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariel A et al. (2003) Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol 170 (12), 6266–72. [DOI] [PubMed] [Google Scholar]
- 10.Dalli J and Serhan CN (2018) Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br J Pharmacol [DOI] [PMC free article] [PubMed]
- 11.Gao Y et al. (2015) Female-Specific Downregulation of Tissue Polymorphonuclear Neutrophils Drives Impaired Regulatory T Cell and Amplified Effector T Cell Responses in Autoimmune Dry Eye Disease. J Immunol 195 (7), 3086–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y et al. (2018) Dietary DHA amplifies LXA4 circuits in tissues and lymph node PMN and is protective in immune-driven dry eye disease. Mucosal Immunol [DOI] [PMC free article] [PubMed]
- 13.Nagaya T et al. (2017) Lipid mediators foster the differentiation of T follicular helper cells. Immunol Lett 181, 51–57. [DOI] [PubMed] [Google Scholar]
- 14.Ramon S et al. (2014) Lipoxin A(4) modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur J Immunol 44 (2), 357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Q et al. (2016) Lipoxin A4 protects against lipopolysaccharide-induced sepsis by promoting innate response activator B cells generation. Int Immunopharmacol 39, 229–235. [DOI] [PubMed] [Google Scholar]
- 16.Kim SD et al. (2009) Functional expression of formyl peptide receptor family in human NK cells. J Immunol 183 (9), 5511–7. [DOI] [PubMed] [Google Scholar]
- 17.Duvall MG et al. (2017) Natural killer cell-mediated inflammation resolution is disabled in severe asthma. Sci Immunol 2 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnig C et al. (2013) Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med 5 (174), 174ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livne-Bar I et al. (2017) Astrocyte-derived lipoxins A4 and B4 promote neuroprotection from acute and chronic injury. J Clin Invest 127 (12), 4403–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei J and Gronert K (2017) The role of pro-resolving lipid mediators in ocular diseases. Mol Aspects Med 58, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510 (7503), 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshimura T and Oppenheim JJ (2011) Chemokine-like receptor 1 (CMKLR1) and chemokine (C-motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp Cell Res 317 (5), 674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serhan CN et al. (2011) Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr Top Med Chem 11 (6), 629–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiurchiu V et al. (2016) Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med 8 (353), 353ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Settimio R et al. (2012) Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediators Inflamm 2012, 318621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi S et al. (2015) Protection from endotoxic uveitis by intravitreal Resolvin D1: involvement of lymphocytes, miRNAs, ubiquitin-proteasome, and M1/M2 macrophages. Mediators Inflamm 2015, 149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo B et al. (2016) Resolvin D1 Programs Inflammation Resolution by Increasing TGF-beta Expression Induced by Dying Cell Clearance in Experimental utoimmune Neuritis. J Neurosci 36 (37), 9590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajasagi NK et al. (2017) Frontline Science: Aspirin-triggered resolvin D1 controls herpes simplex virus-induced corneal immunopathology. J Leukoc Biol 102 (5), 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim N et al. (2016) Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur J Immunol 46 (1), 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim N et al. (2017) Corticosteroids inhibit anti-IgE activities of specialized proresolving mediators on B cells from asthma patients. JCI Insight 2 (3), e88588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnamoorthy N et al. (2015) Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J Immunol 194 (3), 863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodarzi K et al. (2003) Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol 4 (10), 965–73. [DOI] [PubMed] [Google Scholar]
- 33.Tager AM et al. (2003) Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol 4 (10), 982–90. [DOI] [PubMed] [Google Scholar]
- 34.Gelfand EW (2017) Importance of the leukotriene B4-BLT1 and LTB4-BLT2 pathways in asthma. Semin Immunol 33, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung EH et al. (2014) Leukotriene B4 receptor 1 is differentially expressed on peripheral T cells of steroid-sensitive and -resistant asthmatics. Ann Allergy Asthma Immunol 112 (3), 211–216.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H et al. (2009) Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+ T regulatory cells and Th17 cells. Prostaglandins Leukot Essent Fatty Acids 80 (4), 195–200. [DOI] [PubMed] [Google Scholar]
- 37.Liu A et al. (2008) Leukotriene B4 activates T cells that inhibit B-cell proliferation in EBV-infected cord blood-derived mononuclear cell cultures. Blood 111 (5), 2693–703. [DOI] [PubMed] [Google Scholar]
- 38.Lee W et al. (2015) Leukotrienes induce the migration of Th17 cells. Immunol Cell Biol 93 (5), 472–9. [DOI] [PubMed] [Google Scholar]
- 39.Lv J et al. (2015) Leukotriene B(4)-leukotriene B(4) receptor axis promotes oxazolone-induced contact dermatitis by directing skin homing of neutrophils and CD8(+) T cells. Immunology 146 (1), 50–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Percher F et al. (2017)HTLV-1-induced leukotriene B4 secretion by cells promotes T cell recruitment and virus propagation. Nat Commun 8, 15890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costa MF et al. (2010) Leukotriene B4 mediates gammadelta T lymphocyte migration in response to diverse stimuli. J Leukoc Biol 87 (2), 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M et al. (2015) Differential Contribution of BLT1 and BLT2 to Leukotriene B4-Induced Human NK Cell Cytotoxicity and Migration. Mediators Inflamm 2015, 389849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doherty TA et al. (2013) Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol 132 (1), 205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Moltke J et al. (2017) Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med 214 (1), 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H et al. (2013) Regulation of T helper cell subsets by cyclooxygenases and their metabolites. Prostaglandins Other Lipid Mediat 104–105, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirata T and Narumiya S (2012) Prostanoids as regulators of innate and adaptive immunity. Adv Immunol 116, 143–74. [DOI] [PubMed] [Google Scholar]
- 47.Kawahara K et al. (2015) Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim Biophys Acta 1851 (4), 414–21. [DOI] [PubMed] [Google Scholar]
- 48.Nicolaou A et al. (2014) Polyunsaturated Fatty Acid-derived lipid mediators and T cell function. Front Immunol 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boniface K et al. (2009) Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med 206 (3), 535–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kickler K et al. (2012) Prostaglandin E2 affects T cell responses through modulation of CD46 expression. J Immunol 188 (11), 5303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato Y et al. (2014) IL-27 affects helper T cell responses via regulation of PGE2 production by macrophages. Biochem Biophys Res Commun 451 (2), 215–21. [DOI] [PubMed] [Google Scholar]
- 52.Reading JL et al. (2015) Suppression of IL-7-dependent Effector T-cell Expansion by Multipotent Adult Progenitor Cells and PGE2. Mol Ther 23 (11), 1783–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zaslona Z et al. (2014) Prostaglandin E(2) suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allergy Clin Immunol 133 (2), 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sreeramkumar V et al. (2016) Efficient T-cell priming and activation requires signaling through prostaglandin E2 (EP) receptors. Immunol Cell Biol 94 (1), 39–51. [DOI] [PubMed] [Google Scholar]
- 55.Maseda D et al. (2018) mPGES1-Dependent Prostaglandin E2 (PGE2) Controls Antigen-Specific Th17 and Th1 Responses by Regulating T Autocrine and Paracrine PGE2 Production. J Immunol 200 (2), 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kopfnagel V et al. (2011) Resting but not CpG stimulated keratinocytes suppress autologous T-helper cell proliferation--importance of PGE2 and T regulatory function. Exp Dermatol 20 (5), 394–400. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu K et al. (2017) Urinary levels of prostaglandin E2 are positively correlated with intratumoral infiltration of Foxp3(+) regulatory T cells in non-small cell lung cancer. Oncol Lett 14 (2), 1615–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whiteside TL (2014) Regulatory cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother 63 (1), 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H et al. (2017) Prostaglandin E2 restrains human Treg cell differentiation via E prostanoid receptor 2-protein kinase A signaling. Immunol Lett 191, 63–72. [DOI] [PubMed] [Google Scholar]
- 60.Noone C et al. (2013) IFN-gamma stimulated human umbilical-tissue-derived cells potently suppress NK activation and resist NK-mediated cytotoxicity in vitro. Stem Cells Dev 22 (22), 3003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang M et al. (2014) Liver myofibroblasts from hepatitis B related liver failure patients may regulate natural killer cell function via PGE2. J Transl Med 12, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dupuy S et al. (2012) Human Herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and Kaposi sarcoma. PLoS Pathog 8 (1), e1002486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holt M et al. (2012) Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J Immunother 35 (2), 179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinet L et al. (2010) PGE2 inhibits natural killer and gamma delta T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol 80 (6), 838–45. [DOI] [PubMed] [Google Scholar]
- 65.Duffin R et al. (2016) Prostaglandin E(2) constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 351 (6279), 1333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y et al. (2018) Prostaglandin E2 Inhibits Group 2 Innate Lymphoid Cell Activation and Allergic Airway Inflammation Through E-Prostanoid 4-Cyclic Adenosine Monophosphate Signaling. Front Immunol 9, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.June CH et al. (2017) Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 23 (5), 540–547. [DOI] [PubMed] [Google Scholar]
- 68.Coussens LM and Werb Z (2002) Inflammation and cancer. Nature 420 (6917), 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shacter E and Weitzman SA (2002) Chronic inflammation and cancer. Oncology (Williston Park) 16 (2), 217–26,229;discussion230–2. [PubMed] [Google Scholar]
- 70.Greene ER et al. (2011) Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat 96 (1–4), 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao X et al. (2017) NSAIDs Use and Reduced Metastasis in Cancer Patients: results from a meta-analysis. Sci Rep 7 (1), 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma S et al. (2005) Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 65 (12), 5211–20. [DOI] [PubMed] [Google Scholar]
- 73.Liu B et al. (2015) Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int 15, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J et al. (2010) Renal cell carcinoma may evade the immune system by converting CD4+Foxp3- T cells into CD4+CD25+Foxp3+ regulatory T cells: Role of tumor COX-2-derived PGE2. Mol Med Rep 3 (6), 959–63. [DOI] [PubMed] [Google Scholar]
- 75.Sha W et al. (2013) Necrosis in DU145 prostate cancer spheroids induces COX-2/mPGES-1-derived PGE2 to promote tumor growth and to inhibit cell activation. Int J Cancer 133 (7), 1578–88. [DOI] [PubMed] [Google Scholar]
- 76.Mulligan JK et al. (2010) Tumor secretion of VEGF induces endothelial cells to suppress T cell functions through the production of PGE2. J Immunother 33 (2), 126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinet L et al. (2009) A regulatory cross-talk between Vgamma9Vdelta2 T lymphocytes and mesenchymal stem cells. Eur J Immunol 39 (3), 752–62. [DOI] [PubMed] [Google Scholar]
- 78.Jala VR et al. (2017) The yin and yang of leukotriene B4 mediated inflammation in cancer. Semin Immunol 33, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Satpathy SR et al. (2015) Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat Commun 6, 7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poczobutt JM et al. (2016) Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating ell Recruitment. J Immunol 196 (2), 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sharma RK et al. (2013) Expression of leukotriene B(4) receptor-1 on CD8(+) T cells is required for their migration into tumors to elicit effective antitumor immunity. J Immunol 191 (6), 3462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jala VR et al. (2017) Leukotriene B4-receptor-1 mediated host response shapes gut microbiota and controls colon tumor progression. Oncoimmunology 6 (12), e1361593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wejksza K et al. (2013) Cancer-produced metabolites of 5-lipoxygenase induce tumor-evoked regulatory B cells via peroxisome proliferator-activated receptor alpha. J Immunol 190 (6), 2575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sulciner ML et al. (2018) Targeting lipid mediators in cancer biology. Cancer Metastasis Rev [DOI] [PubMed]
- 85.Colas RA et al. (2014) Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol 307 (1), C39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de la Rosa X et al. (2018) Identification and Complete Stereochemical Assignments of the New Resolvin Conjugates in Tissue Regeneration in Human Tissues that Stimulate Proresolving Phagocyte Functions and Tissue Regeneration. Am J Pathol 188 (4), 950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]