

Abstract
Even after a century of investigations, our knowledge of how enzymes work remains far from complete. In particular, several factors that enable enzymes to achieve the high catalytic efficiency still remain only poorly understood. A number of theories have been developed, which propose or reaffirm that enzymes work as structural scaffolds, serving to bringing together and properly orienting the participants so that the reaction can proceed; therefore, only viewing enzymes as passive participants in the catalyzed reaction. Increasing evidence shows that enzymes are not rigid structures but they are constantly undergoing a wide range of internal motions and conformational fluctuations. In this perspective, based on studies from our group, we discuss the emerging biophysical model for enzyme catalysis that provides detailed understanding of the inter-connection between internal protein motions, conformational sub-states, enzyme mechanisms and catalytic efficiency of enzymes. For a number of enzymes, networks of conserved residues have been discovered that span from the surface of the enzyme all the way to the active-site. These networks are hypothesized to serve as pathways of energy transfer that enables thermodynamical coupling of surrounding solvent with enzyme catalysis, and play a role in promoting the enzyme function. Additionally, the role of enzyme structure and electrostatic effects is already well known for quite some time. Collectively, the recent knowledge gained about enzyme mechanisms suggest that the conventional paradigm of enzyme structure encodes function is incomplete and needs to be extended to structure encodes dynamics and the catalytic rate-acceleration, and together these enzyme features encode function.
Keywords: Enzyme dynamics, conformational sub-states, energy flow, solvent conditions, enzyme engineering
Graphical abstract
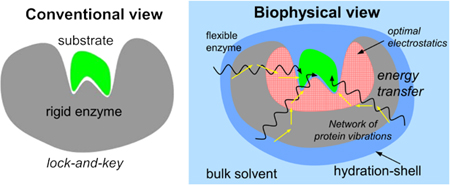
Introduction
Enzymes are known to accelerate reactions by more than 1017 folds.1 Understanding of the detailed mechanism of enzyme catalysis and the factors that enable enzymes to achieve the remarkable catalytic efficiency has now been sought for more than a century.2 A wealth of information about enzymes has been obtained through experimental and computational investigations, particularly revealing the important role of enzyme structure in catalysis. In the original lock-and-key hypothesis and in the revised forms including the induced-fit and the transition-state stabilization hypotheses, enzymes are proposed to serve as structural scaffolds that selectively recognize and bind their substrates.2–4 After binding and proper positioning of the participants, enzymes are proposed to create a reaction environment with properties that differ significantly from the properties in the bulk solvent, and favor the reaction to proceed faster than possible in solution.5 Enzyme residues that make direct contacts with the substrate (and cofactor) have been shown to have critical roles in holding the reaction participants in fixed positions;6, 7 and mutations of these residues are known to alter the speed and/or outcome of the catalyzed reaction.7 It is widely discussed that the shapes of active-sites are complementary to the transition-state of the catalyzed reaction, and the electrostatic environment of the active-sites have been shown to be different from that in the bulk solvent, such that they favor the progress of reactions.8 Therefore, enzyme function has long been understood in terms of structural scaffolds where enzyme active-sites serve to provide a complementary structural and electrostatic environment to preferentially bind and/or stabilize the transition-state.
Even after many decades of investigations, a number of important aspects of enzyme catalysis still remain unexplained. Enzymes have large complex structures, what roles do the enzyme regions beyond the active-site play in catalysis? It is well-known that residues located far away from the active-site when altered through mutations can have drastic consequences for the enzyme catalyzed reaction;9 the role of these residues in the catalytic mechanism is poorly understood.10 Evidence suggest that the bulk and hydration-shell solvent can have drastic impact on enzyme dynamics and activity.11, 12 However, the understanding of the linkage between solvent, and enzyme structure and function remains rather limited. Moreover, bio-organic and even protein molecules that are specially designed to mimic the active-site, with complementary environment to the transition-state of catalyzed reaction, have much lower catalytic efficiencies than their naturally occurring counterparts.13, 14
The search for a fundamental understanding of factors that control enzyme catalytic efficiency has led investigators to look beyond viewing enzymes only as rigid structural scaffolds. In pioneering work Arieh Warshel has described the critical role of electrostatic effects in mechanisms of many enzymes.8 More recently, other factors including the internal motions of proteins have attracted considerable interest for their role in enzyme function.15–36 Like all objects, enzyme molecules also undergo a range of internal motions driven by the surrounding environment and temperature. Emerging evidence indicates that enzymes, as suggested by the conventional paradigm, are not rigid structure but are intrinsically flexible molecules and the internal motions play an active role during the enzyme catalyzed reactions. Recent investigations have shown that a number of enzymes contain network of conserved residues that connect distal region of the enzyme to the active-site.37–40 These networks show the presence of protein motions coupled to the enzyme reaction that play a promoting role in catalysis.
Based on investigations of enzymes performed in our group, we present a biophysical model of how enzymes function (see Figure 1), with the following hypothesized aspects:
Figure 1: The inter-connection between protein structure, dynamics and function has significance for improving catalytic efficiency of enzymes.
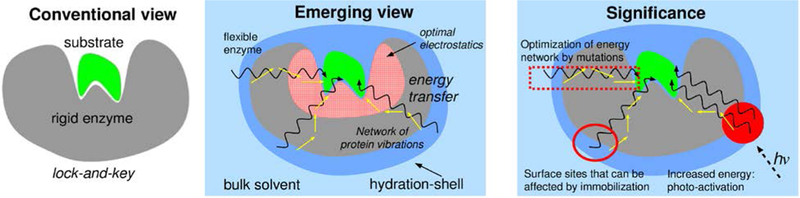
The active-site region provides environment with optimal electrostatics to promote the catalyzed reaction. Several enzymes contain conserved networks of residues that connect surface regions to the active-site. These networks provide thermo-dynamical coupling between the hydration-shell and bulk solvent, and the catalyzed reaction. The discovery of these enzyme energy networks allows new strategies for increasing the catalytic efficiency through enhancing the energy flow through these networks and optimization through mutations.
Internal motions, particularly in the conserved network of residues, play important roles in enzyme function;
A hierarchy of conformational fluctuations enable energetic coupling between the external solvent and the enzyme’s active-site;
The thermodynamical energy selectively directed into the active-site from solvent allows access to functionally relevant conformational sub-states and allows overcoming the activation energy barrier;
The functionally important dynamical parts of the enzyme (particularly the ones that exhibit slow conformational fluctuations) are conserved over evolution; and
Using this new understanding of enzymes, it is possible to identify and modulate reaction promoting conformational fluctuations to develop hyper-catalytic enzymes.41
These aspects of this hypothesized model are discussed in this perspective. This review describes a perspective about how enzymes work based on studies performed in our group. Note, this perspective is not meant to claim that we completely understand all aspects of enzyme catalysis; and it is also noted that investigations from many other research groups also continue to provide new and vital insights into enzyme function as well.15–25 Moreover, various enzyme structural elements and electro-static environment, enabled by the optimal positioning of the active-site and distal residues, play critical roles during enzyme catalyzed reactions (Figure 1), and no biophysical models would be complete without inclusion of their significant contributions to enzyme mechanisms.5, 8
I. Protein motions and conserved networks in enzyme catalysis
Internal motions of proteins span at least 12 orders of magnitude, ranging from femtoseconds (10−15 s) to microseconds and higher (>10−6 s).16, 39 The process of enzyme function involves internal movements of participating molecules during the binding and positioning of substrate (and cofactor) and the removal of product(s). Inherently these steps involve motions of enzyme residues, opening and closing of large regions due to movements at hinge regions or even entire domains in facilitating these events to occur. These types of concerted motions of protein parts and their role in enzyme function are well documented.42, 43 The more interesting question is regarding the direct role of enzyme motions in accelerating the rate limiting step, which determines the overall efficiency of enzyme catalysis. The rate limiting step could be the binding step, the chemical (substrate turnover) step or removal step. The identification of enzyme motions or conformational fluctuations that actively participate and promote the rate limiting step will have important implications in understanding the catalytic efficiency.12
The role of internal motions in enzyme function a highly debated topic with conflicting views being reported.6, 26, 40, 44–46 The confusion, in part, comes from different definitions of protein motions.26 The term protein dynamics has been used broadly to describe a wide variety of motions often leading to confusing and conflicting results. We distinguish between the following types of protein motions (Figure 2):
Figure 2: Various types of internal protein motions.
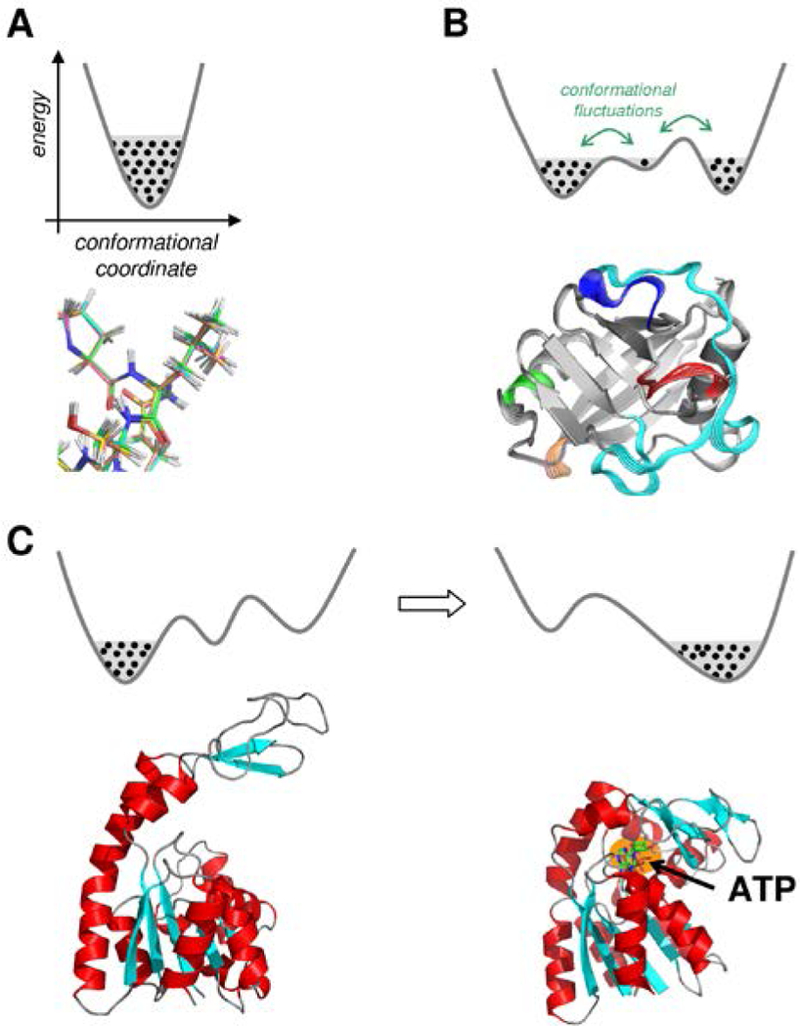
(A) Fast motions. Each black dot indicates a unique conformation. (B) The conformational fluctuations correspond to large scale movements within protein domains or entire protein. These types of motions allow sampling large areas of conformational landscape that could span over multiple minima. (C) Structural changes are induced by events such as binding (or release) of other molecules. Shown here is the enzyme adenylate kinase in the apo form (left) and adenosine triphosphate (ATP) bound (right).
The motions at femtoseconds to sub picoseconds time-scales involving a small number of atoms or a few residues, which repeatedly sample the same area of the conformational landscape and over time average around the same mean structure, and more importantly can be described by harmonic potential wells are considered as fast motions or dynamical motions (see Figure 2A). These types of motions are random in nature and are driven by thermodynamical fluctuations associated with temperature.
Conformational fluctuations are movements that occur at intermediate (nanoseconds) to slow time-scales (microseconds and higher) and enable sampling of distant areas of protein landscape. In particular, these types of motions involve movements of large protein domains (loops, secondary structure elements) and allow overcoming barriers between various conformational wells. These types of motions require more energy than what is available from temperature related thermodynamical fluctuations. Most likely these conformational fluctuations are driven by a group of faster motions, through an enslaving type process.47 As shown in Figure 2B, a single conformational fluctuation can allow sampling of conformational wells located along an anharmonic landscape.
A movement that results in temporary (or even permanent) change in protein structure is considered as a structural change. This type of movement differs from the previous two types of motions, as it entails a shift in the thermodynamic equilibrium. For example, as shown in Figure 2C binding of a small molecule changes the average structure that is sampled by enzyme (such as the binding of adenosine triphosphate to enzyme adenylate kinase).48 Such changes mostly commonly occur on binding and/or release of reactants and products, leading to a change in average structure. Most likely they are a result of changes in interaction between the protein and binding molecule(s). Structural changes also results in change of types of motions that are sampled, but structural change is fundamentally different from sampling of equilibrium motions. Binding or removal of substrate/cofactor or ligands causes changes in the energetic landscape and, therefore, corresponds to major shifts in conformational sub-states and the related sampling of motions (see section III below).
A number of recent studies and reviews have described theoretical approaches for quantifying the contributions of various types of dynamical motions in enzyme kinetics.6, 49–52 Experimental investigations including nuclear magnetic resonance (NMR) experiments,33, 44, 53 single molecule techniques,54 hydrogen/deuterium exchange,55 neutron scattering56 and computational techniques15, 33 have been used to identify motions that are closely coupled to the reaction mechanism of enzyme reactions.31, 44 These techniques have also led to the discovery that enzyme residues that show functionally relevant motions are arranged in networks that connect surface loop regions of the enzymes to the active-sites,37, 38, 40, 41, 57 such as shown in Figure 3. As an illustration we briefly discuss enzyme cyclophilin A (CypA), which has been investigated extensively for the link between protein motions in enzyme catalysis.
Figure 3: Identified network of promoting motions/vibrations in three different enzyme folds,40.
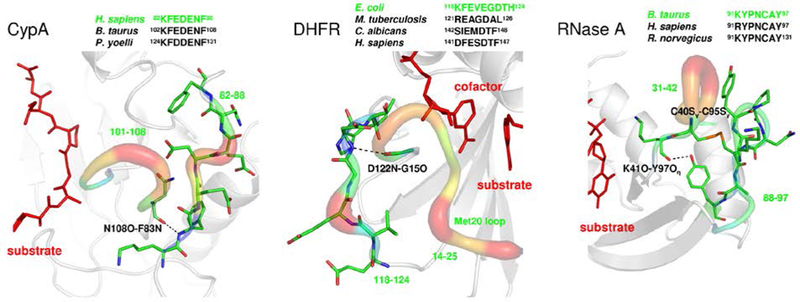
cyclophilin A (CypA), dihydrofolate reductase (DHFR) and ribonuclease A (RNaseA).The flexible surface loop regions with high flexibility show the presence of residues with long side-chains and are interconnected to the active-site through preserved hydrogen bonds. Experimental studies have confirmed presence of these networks.33, 61–67 Reproduced from Ramanathan A et al. (2011),PLoS Biol 9(11): e1001193.
Cyclophilin A:
The enzyme human CypA catalyzes the cis/trans isomerization of peptidyl-prolyl amide bonds in peptides and proteins substrates.33, 58 NMR spin relaxation studies by Kern and coworkers provided important insights into the movement of the protein residues in presence and absence of the substrate which coincides with the catalytic cycle.59, 60 The data indicated that the intrinsic dynamics of the protein in this enzyme coincides with the time-scale (about 9000/second) of the substrate turnover step (the rate limiting step) and promotes the reaction mechanism.
Computational studies by Agarwal and coworkers identified a hierarchy of motions coupled to the reaction.38 Analysis of fast motions, slow conformational fluctuations and cross-correlation of the motions over the course of isomerization reaction led to the discovery of a network of protein vibrations in CypA coupled to its reaction mechanism (Figure 3). This network extends from surface loop residues of the enzyme all the way to the active-site, through a series of hydrogen bonds and hydrophobic interactions.38, 57 The protein motions in this network occur on the time-scales ranging from picosecond to microsecond-millisecond (time scale of the reaction). Genomic and structural analyses indicated that the residues and hydrogen-bonds forming crucial points in the network are conserved in several cyclophilin structures of species ranging from bacteria to human. NMR relaxation studies have observed motions in the network residues coupled to the substrate turnover step.59
The biophysical impact of discovered network on the CypA reaction mechanism was revealed by monitoring changes in the active-site environment that are introduced by the conformational fluctuations of the network residues.38 Computational analysis of these changes showed that the progress of reaction is correlated with the fluctuations in the substrate-enzyme interactions, which in turn are controlled by the motions and conformational fluctuations in the CypA network.12 The relative orientations and the motions of these network residues have shown to be conserved aspect of isomerase fold.40 These observations are consistent with the reaction mechanism from previous X-ray crystallographic studies that indicate that the interaction between target proline residue of the substrate and active-site hydrophilic and hydrophobic residues of enzyme play a crucial role during the reaction.68 An interesting observation is that the maximum enzyme stabilization of the substrate occurs close to transition-state (consistent with the transition-state stabilization theory). The role of these conformational fluctuations could, therefore, be also interpreted as concerted internal protein movements, which facilitate in stabilization of the transition-state. Hence, these are conformational fluctuations are termed as reaction promoting as they: (1) bring together the enzyme active-site in an organized state such that the all the structural elements are correctly positioned and (2) allow more reaction trajectories to become productive. Note, other investigators have discovered that active-site electrostatic environment also plays an important role in CypA mechanism.69
A number of other enzymes have also shown the presence of networks and the role of internal motions on enzyme catalysis including dihydrofolate reductase (DHFR), ribonuclease A, liver alcohol dehydrogenase, lipase B and others.7, 37, 40, 41, 66, 70–75 In some enzyme systems, the thermodynamical motions or fast motions have been shown to aid in enzyme catalysis.22, 76 However, CypA and DHFR specifically show the presence of motions and conformational fluctuations that are intrinsic motions associated with the topology of the enzyme. Therefore, these motions are not random in nature but are designed part of the enzyme structure as they have been observed to be present even in apo enzyme and enzyme complexes, and the rate of these repeated motions can be measured reproducibly using different techniques.44, 59, 77
It should be noted that distal regions in all proteins can be connected through a serious of interactions, however, experimental data (with controls) is required to before a series of interactions can be considered as a network impacting enzyme catalysis. In particular the effect of elimination or substitution of the network interactions (through mutations) on enzyme activity should be clearly demonstrated. For non-enzyme systems the existence of such networks is difficult to characterize and validate. The conversion of substrate(s) to product(s) by enzyme systems offer development of ways to monitor change in protein function associated with changes introduced in protein structure by techniques such as mutations. PDZ domain is an example of non-enzyme systems where the existence of such networks has been demonstrated using statistical coupling approach and tested with the use of mutational studies.78, 79
II. Long-distance effects and energetic coupling
A puzzling aspect of enzymes is the presence of long-range effects and distal control of activity.27, 63, 78, 80 It is widely known that regions of enzymes located far away from the active-site, with no obvious connection between the distal regions and the functional site, can have drastic impact on protein and enzyme function. The allosteric and cooperative effects refer to the control of activity by binding of ligands or biological molecules at a site other than the primary site of enzyme function (the active-site).81, 82 Allosteric modulation is commonly used mechanism for feed-positive and feed-negative regulation of biochemical processes.83 The understanding of biophysical mechanism of allosteric/cooperative effects in enzymes still remains another unsolved mystery. Investigations initially suggested that electrostatics and correlated structural interactions enabled allosteric modulation.84 More recently, the long-range effects in enzyme function have been understood in terms of energy connectivity in the enzyme structure.10, 63, 80, 85
It may initially appear un-related to long range effects but there is another important question about enzymes that still remains unanswered. Most enzyme catalyzed reactions have an activation energy barrier that needs to be surmounted for reaction to be productive. Note that even though enzymes lower the effective transition-state barrier (by raising the ground state and/or lowering the transition-state through stabilization), but still there are barriers typically of 5–10 kcal/mol that need to be overcome by the enzyme assisted reaction.86 How do enzymes assist in overcoming these barriers? The random thermal fluctuations associated with enzyme motions (about 1 kcal/mol at ambient temperature) are not enough to provide enough energy. It has been hypothesized that the thermodynamical energy of the solvent on the surface of the protein provides the required energy to overcome these barriers.12, 39, 40 As described above, conserved networks which span from the surface regions of the enzyme to the active-site have been widely reported for a number of enzymes. It has also been hypothesized that these network pathways serve as channels of long-range connectivity between the solvent and the active-site. The role of these energetic pathways is to couple the surrounding solvent to the enzyme reaction.11 Put another way, a hierarchy of protein motions allows the collection of energy from fast motions on the surface of the protein to intermediate motions associated with loop regions and ultimately this energy is transferred into the large-scale global conformations of the enzyme which are conserved part of the protein fold.12, 40, 41 Once the correct reaction promoting conformational fluctuations are activated they provide the energy for overcoming the activation energy barrier (Figure 4).38 Such a model also provides a biophysical mechanism for long range effects including allostery. Supporting evidence for such a model of enzyme networks acting as energy channels is already available. The investigations of PDZ domain by Ranganathan and coworkers discovered that enzyme residues have co-evolved for long range connectivity.79 Informatic theoretic approaches have also confirmed the presence of pathways of long-distance communication in protein structure.87, 88 The question arises if these pathways have a role in the designated function of the enzymes.
Figure 4: A hypothesized model for the conformational and energetic coupling between enzyme and solvent.
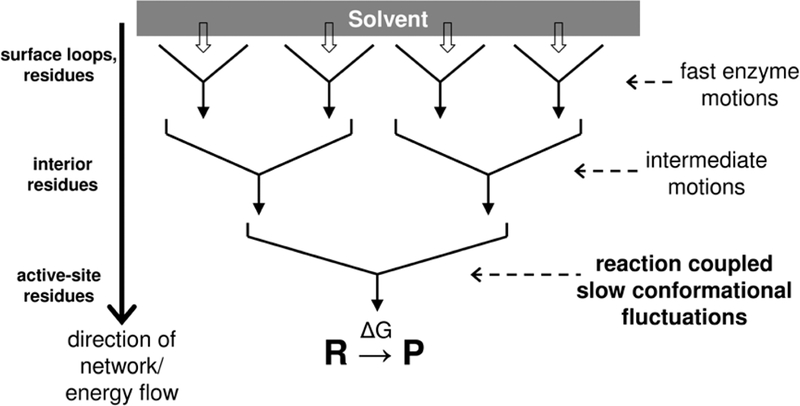
Random motions of solvent molecules slave the motions of surface residues through collisions. These fast motions drive the conformational fluctuations of the interior regions of the protein. These intermediate motions eventually drive the slow conformational fluctuations, some of which promote the catalytic reaction. The hierarchy of protein motions enables the transfer of thermo-dynamical energy from the surface regions to the active-site.
Conformational fluctuations as pathways for energy flow within proteins:
In the hypothesized model of enzymes (Figure 1 and Figure 4) the energy is collected by fast motions of the surface protein residues through thermo-dynamical coupling with the solvent.31, 89–91 This energy is transferred to the intermediate motions and eventually fed to the slow protein motions, which are coupled to the reaction.12 Note that this process is not dissipative (that is energy is not lost by random collisions) but rather leads to directed flow of energy through discovered networks of coupled motions.85, 92 Previous studies have already shown that vibrational energy from a specific normal mode is not lost by dissipation but instead transferred to a few other resonant modes, with a mode coupling term that is correlated with the geometric overlap between the modes.92 Similar studies have also been carried out for studying vibrational energy transfer between α-helices.85, 93 Additional evidence in support of the proposed model comes from another study showing that the slower global motions in proteins are enslaved by faster but local motions.47
Thermo-dynamical energy from solvent enables overcoming the activation energy barrier:
Computational work performed by our group has shown that the discovered energy networks or pathways are closely connected to the overall functioning of the enzyme mechanism particularly the rate limiting step where an energy barrier needs to be overcome.12 The discovered pathways in enzyme structure function by channeling energy from different areas of the protein into the active-site. Terahertz spectroscopy has revealed protein-hydration shell coupling in human lysozyme at fast time-scales (picoseconds).94 Neutron scattering and computational studies have already shown that the motions of solvent slave the motions of the protein motions therefore indicating thermo-dynamical coupling between the solvent and the protein motions.11 The collected energy is channeled into the active-site through a detailed mechanism involving motions of the network residues.
The motions and energy in the networks alters the active-site environment by subtle changes in enzyme-substrate interactions so that more reactive trajectories become productive. For CypA, it has been shown that presence of extra kinetic energy in the reaction promoting conformational fluctuations improves the success rate of reactive trajectories, which corresponds to improving rate kinetics for the enzyme. It is only the presence of kinetic energy in the correct protein motions that shows improvement in reaction rate; which consists of only a small fraction of the total motions of a protein.12 The majority of motions do not have any effect on the reaction. Supporting evidence for this energy flow model also comes from studies where microwave activation95 has been used to improve the rate catalysis as well as photo-activation for improved catalysis through channeling energy into the active-site.41
Significance of these pathways and energy flow in enzyme catalysis could possibly be very wide. These networks provide a biophysical explanation of the presence of allosteric and long-range effects in protein structure including enzymes. It has been widely described that allosteric effects are possibly present in the enzyme molecules to provide nature a mechanism for regulation. However, the model presented here provides an explanation for the wide presence of these pathways and long-range effects in a variety of protein molecules, as they serve a mechanism for to enable the energy transfer for designated function of the protein, and the control through regulation is use of these pathways to modulate the activity of enzyme molecules.
III. Conformational sub-states in enzyme catalysis
An enzyme goes through a catalytic cycle involving a series of intermediate states that includes enzyme in unbound form, enzyme with substrate and cofactor bound (ground state), and enzyme in activated complex form with substrate and cofactor (activated state). In these states the enzyme exhibits different conformations, with functionally important residues and secondary structural elements arranged in different orientations. The probability of sampling these different conformations along the catalytic cycle is not equally distributed; instead depends on enzyme’s energetic landscape. As illustrated in Figure 5, the fast time-scale motions of the protein allow sampling of the conformations in the near vicinity (within the wells) while the conformational fluctuations at long time-scales allow inter-conversion between different groups of populations or conformational sub-states (between conformational wells). These fluctuations that allow inter- conversion between different sub-states are known as conformational transitions and have been investigated in proteins. Austin and coworkers have reported a connection between slow anharmonic energy relaxation rate at nanosecond time-scale and the rate of conformational transitions in proteins, which require multilevel vibrational excitations.96
Figure 5: Internal protein motions and conformational fluctuations allow sampling of conformational sub-states.
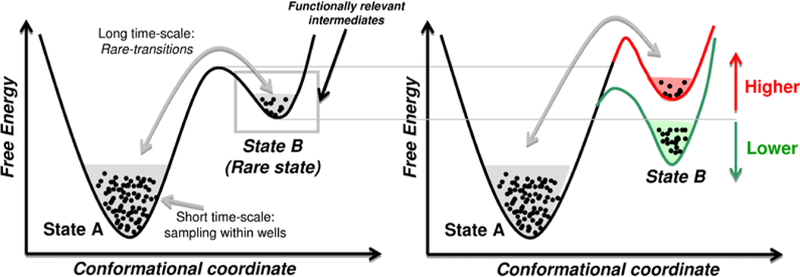
Protein motions at fast time-scales allow sampling within a sub-state, while fluctuations at long time-scales allow access to different sub-states. Some of these higher energy states may contain function promoting structure and dynamical features. Any factors that change the sampling of the higher energy conformational will change access to these functionally important states and therefore change the rate of the reaction.
Recent studies indicate that certain conformational sub-states with small populations contain the structural and dynamical features that aid in promoting catalysis;97 and these sub-states are known as functionally relevant conformational sub-states. A number of recent studies have shown that the intrinsic dynamics of the enzyme enables it to sample higher energy conformational sub-states (the activated complex), which contain geometric and dynamical features of residues that favor the reaction to proceed to the product state,35 therefore, promoting the reaction. In other words, these conformational sub-states have the active-site residues correctly oriented for stabilizing the transition-state, providing the appropriate electrostatic environment, and allowing enzyme-substrate interactions to sample the right motions at the fast time-scales that enable successful completion of reactive trajectories.36, 98
Characterizing the populations of enzyme conformations, particularly in relation to the designated function, has been extremely difficult. Experimental techniques such as X-ray crystallography and NMR provide some information about enzyme conformations associated with ground state, some intermediate states as well as the functioning ensemble of the activated complex.66, 99 However, obtaining quantitative information about the sub-states and conformational populations has been very difficult due to the transient nature of these short lived sub-states and the wide range of time-scales involved. Nonetheless, emerging evidence for a number of systems including ubiquitin, lysozyme, cyclophilin A have shown the connection between populations that contain the right structural and dynamical features facilitate in the reaction mechanism.98
Most commonly projection of conformations on principal component vectors is used to obtain information about conformational sub-states. The underlying argument is that the vectors obtained from the principal component analysis identify the relevant conformational fluctuations, and projection of sampled protein conformations on lowest frequency modes would identify sub-states with meaningful populations. However, this approach suffers from a serious limitation that it provides only provides populations with mixed properties.98 It is widely acknowledged that the conformations located in the same sub-state share similar properties including internal energy, geometrical and dynamical properties.100 The principal component techniques have limited ability to identify conformational sub-states with homogeneous populations that correspond to the ones observed from experimental techniques. The break-through in conformational sub-state analysis came by realization that protein motions at long time-scales are anharmonic,89, 101, 102 and needs to be quantified using higher-order statistics.98 Using this rationale a number of groups have developed techniques for identifying functionally important conformational sub-states, including a new computational technique known as quasi-anharmonic analysis (QAA) developed by Chennubhotla, Agarwal and coworkers.98
This new technique, QAA, allowed the quantitative characterization of conformation sub-states associated with the reaction catalyzed by enzyme CypA, particularly identifying the ones associated with the transition-state.98 Further, it also allowed identification of the long time-scales conformational fluctuations (conformational transitions) that are required to access these transition-state enabling sub-states. These long time-scales conformational fluctuations identified by computational method QAA provide the same information corresponding to the NMR spin relaxation studies.59, 100 Results indicated that the enzyme regions which show large dynamical motions in these long-range and long time-scales fluctuations include the surface loop areas as well as loop in the proximity of active-site. Overall, QAA offers the unique opportunity for the identification and quantitative characterization of rare enzyme conformations that have the geometrical features that facilitate the attainment of transition-state. Note that this is related to the short-lived hidden conformational sub-states that have been discovered using X-ray and NMR techniques, and influence enzyme catalysis in CypA.77
Enzymes in cell operate in different conditions than those of laboratory scale experiments. In laboratory, the enzyme studies are typically conducted in dilute aqueous conditions while in the cells the enzymes are surrounded by a wide variety of biomolecules. Recently, our group used miscible organic solvents to explore the linkage between protein dynamics, functionally relevant conformational sub-states and enzyme catalysis. Use of binary mixture of isopropanol and water indicated that the hydride transfer by E. coli DHFR under rate limiting conditions (measure by steady state kinetics) shows 2.2-fold decrease, at 20% isopropanol concentration. X-ray crystallography showed no change in enzyme structure, however, quasi-elastic neutron scattering shows that enzyme motions are suppressed with increasing concentration of isopropanol.103 Computational modeling indicated that there is no change in active-site structural or electrostatic environment but the access to conformational sub-states altered is significantly. In particular, the rate of transitions into functionally relevant conformational sub-states was about 3-fold smaller, close to the experimentally observed decreased in hydride transfer rate. Collectively, this study using experimental techniques (including steady state enzyme kinetics, X-ray crystallography, neutron scattering and isothermal calorimetry) and computational methods indicates that solvent conditions altered the energetic landscape such that enzyme is unable to sample the natural motions required for accessing the functionally relevant conformational sub-states. These results have important implication for investigating enzyme mechanisms in vivo as the cellular conditions could impact dynamics and therefore catalysis.
IV. Conservation of functionally important dynamics
Evidence collected from an increasing number of enzyme systems where conformational fluctuations and internal motions play a promoting role in enzyme catalysis, leads to several questions – how common is this phenomenon and if protein dynamics makes significant contribution to the catalytic efficiency (rate enhancement) of enzymes? It is difficult to answer these questions conclusively for all enzymes as obtaining quantitative estimates is challenging. Evidence suggests that different enzymes may show different contributions of dynamics along with other factors.6, 46 However, a number of investigations have attempted to quantitatively assess the contribution of protein dynamics in enzyme catalysis.19, 104 In particular, enzyme super-families show preservation of functionally important dynamics.104
A test for the proposed role of dynamics in catalysis has come from investigating the conservation of reaction promoting conformational fluctuations as a part of the enzyme fold.40 The familiar paradigm for role of structure in enzyme catalysis suggests that if it is important for function then the structural element is conserved over evolution. The same question could be asked for reaction promoting motions, if the motions and conformational fluctuations are important for function, then are they also conserved as a part of overall fold? One of our studies identified the reaction promoting dynamics in three enzyme systems catalyzing diverse chemical reactions: cis/trans isomerization catalyzed by CypA, hydride transfer catalyzed by DHFR and single-stranded RNA hydrolysis by ribonuclease A (RNase A).40 The study showed that even with low sequence similarity (as low as 30%) these enzymes that share the same overall fold show the conservation of reaction promoting dynamics as a part of the enzyme fold (Figure 3). The reaction promoting dynamics show movements in distal regions of the protein with large displacements and these regions are located in the same area of the fold, even though the sequences can be significantly different. These regions include surface loops showing high flexibility that are connected to the active-site through the conserved network of residues. The common features of these networks are that they include surface residues with long side-chains, possibly to increase thermodynamical coupling with the solvent. (Terahertz spectroscopy has shown coupling of protein-hydration shell solvent at picosecond time-scale.94) It is interesting to note that the exact type of surface residue is not conserved as long as the replacement residue has the long side chain needed for the enzyme-solvent coupling. Additionally the networks show the presence of preserved hydrogen bonds and hydrophobic interactions all the way to the active-site (Figure 3). It is further interesting to note that these residues may themselves not be conserved as long as the interactions forming the networks are conserved as a part of the overall fold. It has been seen that mutation in these network residues, causing disruption of the network, can have drastic impact on enzyme catalysis.65, 105
The enzymes with DHFR activity also present an interesting case for investigating the role of dynamics and catalysis. There are two structural forms of DHFR: the chromosomally encoded DHFR, present in bacteria to humans, with the Rossmann fold (including the E. coli DHFR). The other type of DHFR is encoded by a plasmid and is known as type II R67DHFR.106 These two enzymes catalyze same reaction however share no structural or sequence homology. Detailed studies have shown that in addition for these enzymes in addition to bringing together the cofactor NADPH and substrate DHF through interactions in the active-site of the enzyme folds also contain designed features in the enzyme fold that promote the reaction through dynamical motions. As mentioned above, a network of coupled promoting motions in chromosomal encoded DHFR promotes the reaction.37 Enzyme’s assistance in the movement of donor carbon and the acceptor carbon towards each other are also part of the reaction mechanism in R67DHFR. However, in R67DHFR these movements are introduced by a residue position behind the nicotinamide ring and the flexibility of DHF tail which is present in a solvent exposed hole.107 Therefore, function promoting dynamics is present in the two non-homologous enzymes that catalyze the same chemistry.
Pancreatic-type ribonucleases (RNases) are an interesting superfamily of enzymes which catalyze the cleavage of phosphodiester bonds in ribonucleic acid (RNA) substrates. The rate-limiting event in the catalytic cycle for the archetypical member of this family, namely the bovine ribonuclease A (bRNaseA), is the product removal. The catalytic efficiency of phosphodiester bond cleavage among the 8 human RNases differs about 106 fold, which is intriguingly similar to range of the observed rates of conformational exchange which differs by 5 orders of magnitude. A joint computational-NMR study of 23 members of the RNases superfamily revealed some very interesting insights about linkage between conformational dynamics and function.108 The superfamily members share the common chemical function of ribonucleolytic cleavage or RNA substrates, but their biological functions are diverse including antiviral, antibacterial and angiogenesis activities. Phylogenetic classification revealed the grouping of RNases into subfamilies with distinct biological functions, with members within a subfamily sharing similar biological functions. More interestingly, members within subfamilies that share similar biological functions also display conservation of dynamical properties, while significant variation in dynamics was observed between members from different subfamilies. The subfamily with the highest catalytic efficiency for the ribonucleolytic cleavage showed protein regions with the largest flexibility, on the other hand members of angiogenin subfamily that shows a million fold less catalytic activity shows most rigid structures in the entire RNase superfamily. Furthermore, swapping a loop region that was previously shown to be important for optimal catalysis, from bRNaseA with that from a different subfamily switched the dynamical profile of the chimeric protein, further highlighting the conservation of dynamical properties within a subfamily. The evidence from this study further indicates that functionally important dynamics is conserved over evolution as a part of the enzyme structural fold to optimize for function.
V. Designing hyper-catalytic enzymes through conformational modulation
In this article, we have described a biophysical model for enzyme catalysis. This model emphasizes enzymes not as rigid bodies that passively enhance catalysis but as internally flexible molecules that work closely in association with the surrounding environment to promote catalysis. In particular, examples from a number of soluble proteins show a hierarchy of motions with the surface motions enslaved by the surrounding solvent. The thermo-dynamical coupling between the solvent and the surface enzyme residues provide energy (through directed energy flow into the active-site) for the slow conformational fluctuations that have important role in promoting the enzyme catalyzed reaction. Moreover, it is also hypothesized that these set of reaction promoting conformational fluctuations are conserved part of the enzyme fold. Nevertheless, the role of protein motions or protein dynamics is a widely debated topic. Therefore, the biophysical model of enzyme catalysis described here needs to be tested thoroughly.
As an important first test of this model, it should be possible to identify and modulate the conformational fluctuations that are involved in promoting the reaction. In other words it should be possible to excite the functionally relevant conformations through addition of extra kinetic energy into the channels of the network pathways that are present in the enzyme (Figure 1), and if this model is correct then it should increase the efficiency of enzymes through conformational modulation of the enzyme. We used this concept to design a strategy for enzyme engineering for the development of hyper-catalytic enzymes.41 A challenging aspect of this design is to develop a strategy that allows selective modulation of the correct conformations that promote the reaction. The engineering strategy was made possible by recent developments that show peptide/protein conformations can be modulated through attachment of a photo-activatable linker (containing azo-benzene group) and providing an external stimulus such as exposure to light. Under exposure of the appropriate wavelength of light azo-benzene molecule undergoes change from the low-energy trans form to the high-energy cis form, which induces conformational modulation of the protein through mechanical strain.109 A number of investigators have attached linkers containing azo-benzene on peptides and proteins to change the conformation at fast time-scales enabled by exposure to light.109, 110
Using the enzyme lipase B from Candida antarctica (CALB) as a model system, we were successfully able to design the hyper-catalytic enzyme.41 The reaction coupled (and reaction promoting) conformational fluctuations were identified using computations, and a photo-activatable linker containing the azo-benzene was conjugated to enable modulation of the enzyme conformations. Once the photo activatable bridge is correctly conjugated on the enzyme, external stimulus through exposure of two wavelength of light allows the conformational modulation of the function promoting conformations. To estimate the increase in catalytic efficiency of the engineered enzyme, it is important to quantify the amount of enzyme that has conjugated bridge in the correct position. Estimates show that in our preliminary and un-optimized chemical strategy, the yield is expected to be 0.48%−3.35%. Using this yield, we estimated that the engineered enzyme is able to achieve 8 to 52 fold (or ~3000% average) improvement in enzyme activity over the naturally occurring form of the enzyme. It should be noted that a number of control experiments, including the exposure of wild type enzyme to light and attaching photo-activatable group to non-specific locations and exposing them to light, did not show any increase in catalytic activity.
The successful design of this hyper-catalytic enzyme provides support for the various aspects of the biophysical model of enzyme catalysis. As we hypothesized that conformational fluctuations are important for function, the ability to identify the reaction promoting conformations and then modulating them through an external stimulus for better catalytic efficiency provides vital support for this model. Further preliminary model also provides evidence that the mechanical energy from the azo-benzene could also be transferred into the active-site (through the network of residues) allowing the enzyme to overcome the reaction at much faster rate. Moreover, the lack of increased enzyme activity during the control experiments also support the observations that not all motions are able to promote the reaction; only specific regions and specific motions have reaction promoting capability. Other groups have also used modulation of enzyme dynamics to develop enzyme engineering strategies.34
VI. Summary
In this article, we have presented a biophysical model for how enzymes work. It has been acknowledged for a number of decades now that the enzymes function as a structural scaffold that bind and correctly orient the reaction participants and provide a chemical environment different from bulk solvent to facilitate catalysis. Emerging evidence, from a wide variety of experimental and computational techniques, has also revealed that enzymes play a more active role in catalyzing the reaction through internal protein motions and conformational fluctuations. These conformational fluctuations occur over a wide range time-scales, including the time-scale of reaction, and allow the enzyme to access the region of conformational landscape that contains dynamical and structural features for promoting the reaction that is the functionally relevant transient state sub-states. It has also been suggested that the hierarchy of protein motions enable the coupling of the bulk and hydration-shell solvent with the catalyzed reaction. In our lab, this hypothesized model has already been used to design a hyper catalytic enzyme through conformational modulation through photo-activation, and preliminary results show ~30 fold increase in catalytic activity over the naturally occurring enzyme. Overall, the evidence discussed here indicated that the paradigm that structure encodes function is incomplete and it needs to be extended to enzyme structure encodes dynamics, and structure-dynamics together encode function.
A number of studies have questioned the role of protein motions, or dynamics, in catalysis. Part of the confusion comes from the lack of proper description/definition of protein dynamics. Much like all other objects, enzymes undergo a range of motions; however, this does not mean that any or all protein motions would promote the function. The issue is complicated by a number of computational (as well as some experimental) studies, which only provide explanations for already known observations but unfortunately fail independent validation and/or cannot make clear and testable predictions. Further, increasing number of literature reports have proposed existence of networks without providing clear evidence showing the effect of these networks on enzyme activity. Therefore, efforts dedicated to developing models that provide mechanistic insights and allow making predictions that can be independently validated and experimentally tested would help in resolving this debate. As an example, the study of enzyme in miscible organic solvents have already provided some insights that not all types of enzyme dynamics affects catalysis, however, it is a particular sub-set of motions (or conformational transitions into the functionally relevant sub-states) that alter enzyme rates.103
Note, the aim of this article is not to claim that dynamics is the only contributor to enzyme catalysis. In addition to the well-known role of direct structural interactions, a number of other factors also contribute to catalysis through long range effects. These include electrostatics effects and solvent molecules (both external and internal). Suggestions have also been made that the energy released from binding of the substrate and/or factor could also provide energy required to overcome the activation energy barrier. These are all interesting mechanisms and possibly do make contributions. The exact contribution each factor makes would depend on the enzyme system and the type of chemistry being catalyzed. High quality models that allow quantitative estimates of contributions for each of these factors would also be extremely valuable.
Acknowledgements:
This work was supported in part by a multi-PI grant from NIH to PKA (GM105978). The author would like thank Nicolas Doucet, Elizabeth Howell, Khushboo Bafna and Chitra Narayanan for interesting discussions.
References
- [1].Richard JP (2013) Enzymatic Rate Enhancements: A Review and Perspective, Biochemistry-Us 52, 2009–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fischer E (1894) Einfluss der Configuration auf die Wirkung den Enzyme, Ber. Dtsch. Chem. Ges. 27, 2985–2993. [Google Scholar]
- [3].Koshland DE (1958) Application of a Theory of Enzyme Specificity to Protein Synthesis, P Natl Acad Sci USA 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robertus JD, Alden RA, Birktoft JJ, and Kraut J (1972) Subtilisin - Stereochemical Mechanism Involving Transition-State Stabilization, Biochemistry-Us 11, 4293–4303. [DOI] [PubMed] [Google Scholar]
- [5].Bruice TC, and Benkovic SJ (2000) Chemical basis for enzyme catalysis, Biochemistry-Us 39, 6267–6274. [DOI] [PubMed] [Google Scholar]
- [6].Nagel ZD, and Klinman JP (2009) A 21(st) century revisionist’s view at a turning point in enzymology, Nat Chem Biol 5, 543–550. [DOI] [PubMed] [Google Scholar]
- [7].Nagel ZD, Cun SJ, and Klinman JP (2013) Identification of a Long-range Protein Network That Modulates Active Site Dynamics in Extremophilic Alcohol Dehydrogenases, J Biol Chem 288, 14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Warshel A, Sharma PK, Kato M, Xiang Y, Liu HB, and Olsson MHM (2006) Electrostatic basis for enzyme catalysis, Chem Rev 106, 3210–3235. [DOI] [PubMed] [Google Scholar]
- [9].Meyer MP, Tomchick DR, and Klinman JP (2008) Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase-1 (vol 105, pg 1146, 2008), P Natl Acad Sci USA 105, 19562–19562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reynolds KA, McLaughlin RN, and Ranganathan R (2011) Hot Spots for Allosteric Regulation on Protein Surfaces, Cell 147, 1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fenimore PW, Frauenfelder H, McMahon BH, and Young RD (2004) Bulk-solvent and hydration-shell fluctuations, similar to alpha- and beta-fluctuations in glasses, control protein motions and functions, P Natl Acad Sci USA 101, 14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Agarwal PK (2005) Role of protein dynamics in reaction rate enhancement by enzymes, J Am Chem Soc 127, 15248–15256. [DOI] [PubMed] [Google Scholar]
- [13].Benkovic SJ (1992) Catalytic Antibodies, Annual Review of Biochemistry 61, 29–54. [DOI] [PubMed] [Google Scholar]
- [14].Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, Clair JLS, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, and Baker D (2010) Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction, Science 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Garcia-Viloca M, Gao J, Karplus M, and Truhlar DG (2004) How enzymes work: Analysis by modern rate theory and computer simulations, Science 303, 186–195. [DOI] [PubMed] [Google Scholar]
- [16].Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, and Kern D (2007) A hierarchy of timescales in protein dynamics is linked to enzyme catalysis, Nature 450, 913–916. [DOI] [PubMed] [Google Scholar]
- [17].Quaytman SL, and Schwartz SD (2007) Reaction coordinate of an enzymatic reaction revealed by transition path sampling, P Natl Acad Sci USA 104, 12253–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schramm AM, Mehra-Chaudhary R, Furdui CM, and Beamer LJ (2008) Backbone flexibility, conformational change, and catalysis in a phosphohexomutase from Pseudomonas aeruginosa, Biochemistry-Us 47, 9154–9162. [DOI] [PubMed] [Google Scholar]
- [19].Arora K, and Brooks CL (2009) Functionally Important Conformations of the Met20 Loop in Dihydrofolate Reductase are Populated by Rapid Thermal Fluctuations, J Am Chem Soc 131, 5642–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hirschi JS, Arora K, Brooks CL, and Schramm VL (2010) Conformational Dynamics in Human Purine Nucleoside Phosphorylase with Reactants and Transition-State Analogues, J Phys Chem B 114, 16263–16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Machleder SQ, Pineda JRET, and Schwartz SD (2010) On the origin of the chemical barrier and tunneling in enzymes, Journal of Physical Organic Chemistry 23, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Antoniou D, and Schwartz SD (2011) Protein Dynamics and Enzymatic Chemical Barrier Passage, J Phys Chem B 115, 15147–15158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clouthier CM, Morin S, Gobeil SMC, Doucet N, Blanchet J, Nguyen E, Gagne SM, and Pelletier JN (2012) Chimeric beta-Lactamases: Global Conservation of Parental Function and Fast Time-Scale Dynamics with Increased Slow Motions, Plos One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Klinman JP (2013) Importance of Protein Dynamics during Enzymatic C-H Bond Cleavage Catalysis, Biochemistry-Us 52, 2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Micheletti C (2013) Comparing proteins by their internal dynamics: Exploring structure-function relationships beyond static structural alignments, Physics of Life Reviews 10, 1–26. [DOI] [PubMed] [Google Scholar]
- [26].Kohen A (2015) Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies, Accounts Chem Res 48, 466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lisi GP, and Loria JP (2016) Solution NMR Spectroscopy for the Study of Enzyme Allostery, Chem Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Onions S, Ito K, Charron C, Brown R, Colucci M, Frickel F, Hardy G, Joly K, King-Underwood J, Kizawa Y, Knowles I, Murray J, Novak A, Rani A, Rapeport G, Smith A, Strong P, Taddei D, and Williams J (2016) The Discovery of Narrow Spectrum Kinase Inhibitors: New Therapeutic Agents for the Treatment of COPD and Steroid-Resistant Asthma, J Med Chem. [DOI] [PubMed] [Google Scholar]
- [29].Kondo T, Pinnola A, Chen WJ, Dall’Osto L, Bassi R, and Schlau-Cohen GS (2017) Single-molecule spectroscopy of LHCSR1 protein dynamics identifies two distinct states responsible for multi-timescale photosynthetic photoprotection, Nat Chem 9, 772–778. [DOI] [PubMed] [Google Scholar]
- [30].Kim TH, Mehrabi P, Ren Z, Sljoka A, Ing C, Bezginov A, Ye L, Pomes R, Prosser RS, and Pai EF (2017) The role of dimer asymmetry and protomer dynamics in enzyme catalysis, Science 355. [DOI] [PubMed] [Google Scholar]
- [31].Klinman JP, Offenbacher AR, and Hu S (2017) Origins of Enzyme Catalysis: Experimental Findings for C-H Activation, New Models, and Their Relevance to Prevailing Theoretical Constructs, J Am Chem Soc 139, 18409–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Choy MS, Li Y, Machado L, Kunze MBA, Connors CR, Wei X, Lindorff-Larsen K, Page R, and Peti W (2017) Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery, Mol Cell 65, 644–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Holliday MJ, Camilloni C, Armstrong GS, Vendruscolo M, and Eisenmesser EZ (2017) Networks of Dynamic Allostery Regulate Enzyme Function, Structure 25, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Boehr DD, D’Amico RN, and O’Rourke KF (2018) Engineered control of enzyme structural dynamics and function, Protein Sci 27, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Aviram HY, Pirchi M, Mazal H, Barak Y, Riven I, and Haran G (2018) Direct observation of ultrafast large-scale dynamics of an enzyme under turnover conditions, Proc Natl Acad Sci U S A 115, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Otten R, Liu L, Kenner LR, Clarkson MW, Mavor D, Tawfik DS, Kern D, and Fraser JS (2018) Rescue of conformational dynamics in enzyme catalysis by directed evolution, Nat Commun 9, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Agarwal PK, Billeter SR, Rajagopalan PTR, Benkovic SJ, and Hammes-Schiffer S (2002) Network of coupled promoting motions in enzyme catalysis, P Natl Acad Sci USA 99, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Agarwal PK, Geist A, and Gorin A (2004) Protein dynamics and enzymatic catalysis: Investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A, Biochemistry-Us 43, 10605–10618. [DOI] [PubMed] [Google Scholar]
- [39].Agarwal PK (2006) Enzymes: An integrated view of structure, dynamics and function, Microbial Cell Factories 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ramanathan A, and Agarwal PK (2011) Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis, PLoS Biology 9, e1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Agarwal PK, Schultz C, Kalivretenos A, Ghosh B, and Broedel SE (2012) Engineering a Hyper-catalytic Enzyme by Photoactivated Conformation Modulation, Journal of Physical Chemistry Letters 3, 1142–1146. [Google Scholar]
- [42].Sawaya MR, and Kraut J (1997) Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence, Biochemistry-Us 36, 586–603. [DOI] [PubMed] [Google Scholar]
- [43].Berendsen HJC, and Hayward S (2000) Collective protein dynamics in relation to function, Curr Opin Struc Biol 10, 165–169. [DOI] [PubMed] [Google Scholar]
- [44].Boehr DD, Dyson HJ, and Wright PE (2006) An NMR perspective on enzyme dynamics, Chem Rev 106, 3055–3079. [DOI] [PubMed] [Google Scholar]
- [45].Benkovic SJ, Hammes GG, and Hammes-Schiffer S (2008) Free-energy landscape of enzyme catalysis, Biochemistry-Us 47, 3317–3321. [DOI] [PubMed] [Google Scholar]
- [46].Kamerlin SCL, and Warshel A (2010) At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis?, Proteins 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hawkins RJ, and McLeish TCB (2006) Coupling of global and local vibrational modes in dynamic allostery of proteins, Biophysical Journal 91, 2055–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ramanathan A, Savol AJ, Agarwal PK, and Chennubhotla CS (2012) Event detection and sub-state discovery from biomolecular simulations using higher-order statistics: Application to enzyme adenylate kinase, Proteins 80, 2536–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hay S, and Scrutton NS (2012) Good vibrations in enzyme-catalysed reactions, Nat Chem 4, 161–168. [DOI] [PubMed] [Google Scholar]
- [50].Klinman JP, and Kohen A (2013) Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis, Annu Rev Biochem 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Min W, Xie XS, and Bagchi B (2008) Two-dimensional reaction free energy surfaces of catalytic reaction: Effects of protein conformational dynamics on enzyme catalysis, J Phys Chem B 112, 454–466. [DOI] [PubMed] [Google Scholar]
- [52].Min W, Xie XS, and Bagchi B (2009) Role of conformational dynamics in kinetics of an enzymatic cycle in a nonequilibrium steady state, J Chem Phys 131. [DOI] [PubMed] [Google Scholar]
- [53].Palmer AG 3rd. (2015) Enzyme dynamics from NMR spectroscopy, Acc Chem Res 48, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang ZQ, Rajagopalan PTR, Selzer T, Benkovic SJ, and Hammes GG (2004) Single-molecule and transient kinetics investigation of the interaction of dihydrofolate reductase with NADPH and dihydrofolate, P Natl Acad Sci USA 101, 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zavodszky P, Kardos J, Svingor A, and Petsko GA (1998) Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins, P Natl Acad Sci USA 95, 7406–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tehei M, Franzetti B, Wood K, Gabel F, Fabiani E, Jasnin M, Zamponi M, Oesterhelt D, Zaccai G, Ginzburg M, and Ginzburg BZ (2007) Neutron scattering reveals extremely slow cell water in a Dead Sea organism, P Natl Acad Sci USA 104, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Agarwal PK (2004) Cis/trans isomerization in HIV-1 capsid protein catalyzed by cyclophilin A: Insights from computational and theoretical studies, Proteins 56, 449–463. [DOI] [PubMed] [Google Scholar]
- [58].Fischer G, Wittmannliebold B, Lang K, Kiefhaber T, and Schmid FX (1989) Cyclophilin and Peptidyl-Prolyl Cis-Trans Isomerase Are Probably Identical Proteins, Nature 337, 476–478. [DOI] [PubMed] [Google Scholar]
- [59].Eisenmesser EZ, Bosco DA, Akke M, and Kern D (2002) Enzyme dynamics during catalysis, Science 295, 1520–1523. [DOI] [PubMed] [Google Scholar]
- [60].Henzler-Wildman K, and Kern D (2007) Dynamic personalities of proteins, Nature 450, 964–972. [DOI] [PubMed] [Google Scholar]
- [61].Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, and Kern D (2005) Intrinsic dynamics of an enzyme underlies catalysis, Nature 438, 117–121. [DOI] [PubMed] [Google Scholar]
- [62].Chi CN, Vogeli B, Bibow S, Strotz D, Orts J, Guntert P, and Riek R (2015) A Structural Ensemble for the Enzyme Cyclophilin Reveals an Orchestrated Mode of Action at Atomic Resolution, Angew Chem Int Ed Engl 54, 11657–11661. [DOI] [PubMed] [Google Scholar]
- [63].Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, and Ranganathan R (2008) Surface sites for engineering allosteric control in proteins, Science 322, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Boehr DD, Schnell JR, McElheny D, Bae SH, Duggan BM, Benkovic SJ, Dyson HJ, and Wright PE (2013) A Distal Mutation Perturbs Dynamic Amino Acid Networks in Dihydrofolate Reductase, Biochemistry-Us 52, 4605–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang L, Goodey NM, Benkovic SJ, and Kohen A (2006) Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase, P Natl Acad Sci USA 103, 15753–15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gagne D, Charest LA, Morin S, Kovrigin EL, and Doucet N (2012) Conservation of Flexible Residue Clusters among Structural and Functional Enzyme Homologues, J Biol Chem 287, 44289–44300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gagne D, Narayanan C, and Doucet N (2015) Network of long-range concerted chemical shift displacements upon ligand binding to human angiogenin, Protein Sci 24, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Howard BR, Vajdos FF, Li S, Sundquist WI, and Hill CP (2003) Structural insights into the catalytic mechanism of cyclophilin A, Nature Structural Biology 10, 475–481. [DOI] [PubMed] [Google Scholar]
- [69].Camilloni C, Sahakyan AB, Holliday MJ, Isern NG, Zhang FL, Eisenmesser EZ, and Vendruscolo M (2014) Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism, P Natl Acad Sci USA 111, 10203–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Agarwal PK, Billeter SR, and Hammes-Schiffer S (2002) Nuclear quantum effects and enzyme dynamics in dihydrofolate reductase catalysis, J Phys Chem B 106, 3283–3293. [Google Scholar]
- [71].Agarwal PK, Webb SP, and Hammes-Schiffer S (2000) Computational studies of the mechanism for proton and hydride transfer in liver alcohol dehydrogenase, J Am Chem Soc 122, 4803–4812. [DOI] [PubMed] [Google Scholar]
- [72].Billeter SR, Webb SP, Agarwal PK, Iordanov T, and Hammes-Schiffer S (2001) Hydride transfer in liver alcohol dehydrogenase: Quantum dynamics, kinetic isotope effects, and role of enzyme motion, J Am Chem Soc 123, 11262–11272. [DOI] [PubMed] [Google Scholar]
- [73].Bandaria JN, Dutta S, Hill SE, Kohen A, and Cheatum CM (2008) Fast enzyme dynamics at the active site of formate dehydrogenase, J Am Chem Soc 130, 22–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Veeraraghavan N, Bevilacqua PC, and Hammes-Schiffer S (2010) Long-Distance Communication in the HDV Ribozyme: Insights from Molecular Dynamics and Experiments, J Mol Biol 402, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Layfield JP, and Hammes-Schiffer S (2013) Calculation of Vibrational Shifts of Nitrile Probes in the Active Site of Ketosteroid Isomerase upon Ligand Binding, J Am Chem Soc 135, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mincer JS, and Schwartz SD (2004) Rate-promoting vibrations and coupled hydrogen-electron transfer reactions in the condensed phase: A model for enzymatic catalysis, J Chem Phys 120, 7755–7760. [DOI] [PubMed] [Google Scholar]
- [77].Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, and Alber T (2009) Hidden alternative structures of proline isomerase essential for catalysis, Nature 462, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Suel GM, Lockless SW, Wall MA, and Ranganathan R (2003) Evolutionarily conserved networks of residues mediate allosteric communication in proteins (vol 10, pg 59, 2003), Nature Structural Biology 10, 232–232. [DOI] [PubMed] [Google Scholar]
- [79].Lockless SW, and Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families, Science 286, 295–299. [DOI] [PubMed] [Google Scholar]
- [80].Frauenfelder H, McMahon BH, Austin RH, Chu K, and Groves JT (2001) The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin, P Natl Acad Sci USA 98, 2370–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cook RA, and Koshland DE (1970) Positive and Negative Cooperativity in Yeast Glyceraldehyde 3-Phosphate Dehydrogenase, Biochemistry-Us 9, 3337–&. [DOI] [PubMed] [Google Scholar]
- [82].Levitzki A, and Koshland DE (1969) Negative Cooperativity in Regulatory Enzymes, P Natl Acad Sci USA 62, 1121–&. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Monod J, Changeux JP, and Jacob F (1963) Allosteric Proteins and Cellular Control Systems, J Mol Biol 6, 306–&. [DOI] [PubMed] [Google Scholar]
- [84].Baldwin J, and Chothia C (1979) Hemoglobin - Structural-Changes Related to Ligand-Binding and Its Allosteric Mechanism, J Mol Biol 129, 175–200. [DOI] [PubMed] [Google Scholar]
- [85].Leitner DM (2008) Energy flow in proteins, Annual Review of Physical Chemistry 59, 233–259. [DOI] [PubMed] [Google Scholar]
- [86].The contribution by enzymes could be described as a combination of biochemical and biophysical effects. The biochemical contribution includes the relative lowering of barrier when compared to the reaction in solution, which is achieved by structural and electrostatic complementary environment of the active-site and critical enzyme-substrate interactions. The biophysical contribution includes providing a mechanism (and energy) to overcome the (lowered) activation energy barrier. .
- [87].Chennubhotla C, and Bahar I (2007) Signal propagation in proteins and relation to equilibrium fluctuations, Plos Comput Biol 3, 1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McClendon CL, Friedland G, Mobley DL, Amirkhani H, and Jacobson MP (2009) Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles, J Chem Theory Comput 5, 2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Borreguero JM, He JH, Meilleur F, Weiss KL, Brown CM, Myles DA, Herwig KW, and Agarwal PK (2011) Redox-Promoting Protein Motions in Rubredoxin, J Phys Chem B 115, 8925–8936. [DOI] [PubMed] [Google Scholar]
- [90].Ramanathan A, and Agarwal PK (2009) Computational Identification of Slow Conformational Fluctuations in Proteins, J Phys Chem B 113, 16669–16680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bellissent-Funel MC, Hassanali A, Havenith M, Henchman R, Pohl P, Sterpone F, van der Spoel D, Xu Y, and Garcia AE (2016) Water Determines the Structure and Dynamics of Proteins, Chem Rev 116, 7673–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Moritsugu K, and Kidera A (2004) Protein motions represented in moving normal mode coordinates, J Phys Chem B 108, 3890–3898. [Google Scholar]
- [93].Yu X, and Leitner DM (2005) Heat flow in proteins: Computation of thermal transport coefficients, J Chem Phys 122. [DOI] [PubMed] [Google Scholar]
- [94].Novelli F, Ostovar Pour S, Tollerud J, Roozbeh A, Appadoo DRT, Blanch EW, and Davis JA (2017) Time-Domain THz Spectroscopy Reveals Coupled Protein-Hydration Dielectric Response in Solutions of Native and Fibrils of Human Lysozyme, J Phys Chem B 121, 4810–4816. [DOI] [PubMed] [Google Scholar]
- [95].Young DD, Nichols J, Kelly RM, and Deiters A (2008) Microwave activation of enzymatic catalysis, J Am Chem Soc 130, 10048–10049. [DOI] [PubMed] [Google Scholar]
- [96].Xie AH, van der Meer AFG, and Austin RH (2002) Excited-state lifetimes of far-infrared collective modes in proteins, Phys Rev Lett 88. [DOI] [PubMed] [Google Scholar]
- [97].Ramanathan A, Savol A, Burger V, Chennubhotla C, and Agarwal PK (2013) Protein Conformational Populations and Functionally Relevant Sub-states, Accounts Chem Res, [DOI] [PubMed] [Google Scholar]
- [98].Ramanathan A, Savol AJ, Langmead CJ, Agarwal PK, and Chennubhotla CS (2011) Discovering Conformational Sub-States Relevant to Protein Function, PLoS ONE 6, e15827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Boehr DD, McElheny D, Dyson HJ, and Wright PE (2006) The dynamic energy landscape of dihydrofolate reductase catalysis, Science 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- [100].Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, and de Groot BL (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution, Science 320, 1471–1475. [DOI] [PubMed] [Google Scholar]
- [101].Ichiye T, and Karplus M (1988) Anisotropy and Anharmonicity of Atomic Fluctuations in Proteins - Implications for X-Ray-Analysis, Biochemistry-Us 27, 3487–3497. [DOI] [PubMed] [Google Scholar]
- [102].Pontiggia F, Colombo G, Micheletti C, and Orland H (2007) Anharmonicity and self-similarity of the free energy landscape of protein G, Phys Rev Lett 98. [DOI] [PubMed] [Google Scholar]
- [103].Duff MR Jr., Borreguero JM, Cuneo MJ, Ramanathan A, He J, Kamath G, Chennubhotla SC, Meilleur F, Howell EE, Herwig KW, Myles DAA, and Agarwal PK (2018) Modulating Enzyme Activity by Altering Protein Dynamics with Solvent, Biochemistry-Us 57, 4263–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zen A, Carnevale V, Lesk AM, and Micheletti C (2008) Correspondences between low-energy modes in enzymes: Dynamics-based alignment of enzymatic functional families, Protein Science 17, 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wang L, Tharp S, Selzer T, Benkovic SJ, and Kohen A (2006) Effects of a distal mutation on active site chemistry, Biochemistry-Us 45, 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Feng J, Goswami S, and Howell EE (2008) R67, the other dihydrofolate reductase: Rational design of an alternate active site configuration, Biochemistry-Us 47, 555–565. [DOI] [PubMed] [Google Scholar]
- [107].Kamath G, Howell EE, and Agarwal PK (2010) The Tail Wagging the Dog: Insights into Catalysis in R67 Dihydrofolate Reductase, Biochemistry-Us 49, 9078–9088. [DOI] [PubMed] [Google Scholar]
- [108].Narayanan C, Bernard DN, Bafna K, Gagne D, Chennubhotla CS, Doucet N, and Agarwal PK (2018) Conservation of Dynamics Associated with Biological Function in an Enzyme Superfamily, Structure 26, 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Beharry AA, and Woolley GA (2011) Azobenzene photoswitches for biomolecules, Chemical Society Reviews 40, 4422–4437. [DOI] [PubMed] [Google Scholar]
- [110].Bredenbeck J, Helbing J, Sieg A, Schrader T, Zinth W, Renner C, Behrendt R, Moroder L, Wachtveitl J, and Hamm P (2003) Picosecond conformational transition and equilibration of a cyclic peptide (vol 100, pg 6452, 2003), P Natl Acad Sci USA 100, 10580–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]