

Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains remarkably lethal with a 5-year survival rate of 8%. This is mainly attributed to the late stage of presentation, as well as widespread resistance to conventional therapy. Additionally, PDAC tumors are largely non-immunogenic, and most patients have displayed incomplete responses to cancer immunotherapies. Our group has previously identified Transforming Growth Factor Beta (TGFβ) as a crucial repressor of anti-tumor immune function in PDAC, particularly with respect to cytotoxic T lymphocytes. However, pharmacologic inhibition of TGFβ signaling has had limited efficacy in clinical trials, failing to promote a significant anti-tumor immune response. Hence, in this work we extend our analysis to identify and circumvent the mechanisms of resistance to TGFβ signal inhibition in PDAC. Consistent with our previous observations, adoptive transfer of TGFβ-insensitive CD8+ T-cells led to the near compete regression of neoplastic disease in vivo. However, we demonstrate that this cannot be recapitulated via global reduction in TGFβ signaling, either through genetic ablation or pharmacologic inhibition of TGFBR1. In fact, tumors with TGFβ signal inhibition displayed increased PD-L1 expression and had no observable change in anti-tumor immunity. Using genetic models of advanced PDAC, we then determined that concomitant inhibition of both TGFβ and PD-L1 receptors led to a reduction in the neoplastic phenotype, improving survival and reducing disease-associated morbidity in vivo. Combined, these data strongly suggest that TGFβ and PD-L1 pathway inhibitors may synergize in PDAC, and this approach warrants clinical consideration.
INTRODUCTION
Transforming Growth Factor Beta (TGFβ) is a well-established modifier of the pancreatic tumor microenvironment (TME), though its overall contributions to carcinogenesis are poorly understood. Previous in vivo studies have found that, upon epithelial loss of TGFβ signaling, the P48-Cre/LSL-Kras (KC) mouse model of PanIN disease developed advanced pancreatic cancer with increased incidence and severity of developing adenocarcinoma from neoplastic lesions (1–3). In contrast, systemic deficiency of Tgfbr1 in the Elastase-KrasG12D model of cystic papillary neoplastic (CPN) disease (4) led to a reduction in the neoplastic phenotype (5). Combined, these data underscore the need for increased understanding of the multiple roles of TGFβ in PDAC pathobiology. This is especially important prior to the introduction of TGFβ inhibitors to the management of PDAC patients.
In our previous work, we determined that TGFβ represses the activation of cytotoxic T lymphocytes (CTLs) in early PDAC development. Indeed, adoptive transfer of Tgfbr1 haplo-insufficient (Tgfbr1+/−) T-cells into KC mice lead to a cytotoxic response towards developing lesions and a significant reduction in tumor burden (6). Clinically, TGFβ expression was inversely associated with that of GranzymeB, a surrogate marker of cytotoxicity (6). These findings appear to support therapeutic targeting of TGFBRs in pancreatic cancers to promote T-cell mediated cytotoxicity. In this work, we extend our analysis to determine the potential efficacy of systemic TGFβ signal inhibition, particularly with respect to anti-tumor immunity. We found that, while selective TGFβ inhibition in CD8+ T-cells leads to regression of neoplastic disease, systemic blockade of TGFβ signaling fails to promote cytotoxicity due to compensatory upregulation of PD-L1 on the cancer epithelium. By targeting both TGFβ and PD-L1 receptors, we were able to promote the T-cell mediated clearance of PDAC, and this approach warrants further exploration in the management of pancreatic cancer patients.
MATERIALS AND METHODS
Chemicals and Reagents
Galunisertib (LY2157299 (7), Figure S1A) was purchased from Selleck Chem (Houston, Tx), reconstituted per the manufacturer instructions, and used at 75 mg/kg in vivo. The anti-PD-1 antibody RMP1–14 was purchased from BioXCell (West Lebanon, NH) and used at fixed dose of 200 μg per injection.
Flow Cytometry
Mesenteric lymph nodes or spleens were isolated, ruptured, washed in PBS, and contents filtered. Two million cells were seeded into a round-bottom 96-well plate, washed in PBS, incubated with a golgi plug/protein transport inhibitor (BD biosciences, San Jose, CA) and stained with anti-CD4-APC-Cy7, CD25-PE (BD biosciences), CD69-APC, CD8-PerCP-Cy5.5 (Biolegend, San Diego, CA), and an Alive/Dead kit (Invitrogen, Grand Island, NY) at 1:200–1:1500 in PBS at room temperature for 20 minutes. Cells were then fixed with 1% PFA in PBS for 10 minutes at room temperature, and stained with either anti-FoxP3-FITC (eBioSci, San Diego, CA) or anti-Perforin-FITC (Biolegend) at 1:150 in perm/stain buffer (BD biosciences) for 30 minutes over ice and washed three times with perm/wash buffer (BD biosciences). Cells were analyzed with a BD Fortessa Cytometer. All flow plots correspond to live, single cells and are representative of 100,000 events unless otherwise stated.
CD8 Isolation and Adoptive Transfer
Cells were isolated form the spleen, and CD8+ cells isolated by a dynabead CD8 positive isolation kit used per manufacturer specification (Life Technologies, Grand Island, NY). After isolation, 2 million CD8 cells were re-suspended in PBS and injected into the anesthetized recipient animals via the retro-orbital vein. Recipient animals were euthanized after one month and tissues subject to pathological analysis.
Mice
P48-Cre x LSL-KRASG12D (KC), Tgfbr1+/−, KC/ Tgfbr1+/− (KCT), and Pdx1-Cre x LSL-KRASG12D x LSL-TP53R172H (KPC), mice were generated as described. KC mice were administered adoptive transfer of either naïve or Tgfbr1+/− CD8 T cells and euthanized at 3 months. Similarly, 6 week KPC mice were administered intraperitoneal injection of either a PBS vehicle, Galunisertib, anti-PD-1, or Galunisertib/anti-PD-1 once every other day. This cohort was euthanized either after 2.5 months or when moribund. For euthanasia, animals were anesthetized with xylazine/ketamine until unresponsive to toe tap and/or agonal breathing. Thoracotomy served as the primary method of euthanasia and exsanguination the secondary method.
Histology, Immunohistochemistry, Immunofluorescence, and Slide Scoring
Age-matched mice were euthanized and the pancreas, colon, small bowel, liver, and spleen were subjected to pathologic examination. Tissues were fixed in 10% formalin, paraffin-embedded, and sections at 4 mm interval were cut from each tissue, and stained with hematoxylin and eosin (H&E), trichrome (Sigma Aldrich), or via immunohistochemistry (IHC) or immunofluorescence (IF).
For IHC, slides were deparaffinized by xylenes and rehydrated by ethanol gradient, then heated in a pressure cooker using DAKO retrieval buffer. Endogenous peroxidases were quenched in 3% hydrogen peroxide in methanol for 30 minutes. Tissues were blocked with 0.5% BSA in PBS for 30 minutes and incubated with primary antibodies against pSMAD2 or PD-L1 (Cell Signaling Technology) at 1:50–1:200 overnight at 4°C. Slides were developed using either Streptavidin or secondary antibodies, followed by DAB substrate/buffer (DAKO).
For IF, slides were heated via pressure cooker in DAKO retrieval buffer and tissues blocked with 0.5% BSA in PBS for 1 hour at room temperature. Sections were exposed to primary antibodies against CK19 (University of Iowa Hybridoma Bank), PCNA, CD3, (Santa Cruz Biotechnology), or Granzyme B (abcam, Cambridge, MA) at 1:50–1:200 overnight at 4#C. Slides were developed using AlexaFluor 488- or 594–conjugated secondary antibodies (1:200–1:1,000, Abcam), mounted in DAPI-containing media (Santa Cruz Biotechnology), exposed to DAPI, FITC, and Texas Red filters, and images superimposed. All counts and scores were performed by two blinded investigators. For any contradicting scores, a third investigator was consulted.
Authentication of Key Resources
KC, Tgfbr1+/−, KCT, and KPC mice were bred in house and genotypes verified by PCR for all relevant transgenes (Pdx1-Cre, P48-Cre, Tgfbr1+/−, LSL-KRASG12D, LSL-TP53R172H). Galunisertib is routinely verified in our lab by the ability to neutralize TGFβ1-induced pSMAD2 phosphorylation in vitro by western blot as seen in our previous publication (6). The effectiveness of Galunisertib in vivo is similarly validated by the absence or reduction of pSMAD2 by immunohistochemistry. Additionally, the anti-PD-1 antibody RMP1–14 is verified by the manufacturer for the ability to recognize purified mouse PD-1 by western blot and has been successfully used to neutralize PD-1 signaling in murine pancreatic ductal adenocarcinoma (8).
Statistical Analysis
Data were analyzed by two-way ANOVA and fit to a general linear model in Minitab16, the validity of which was tested by adherence to the normality assumption and the fitted plot of the residuals. Results were arranged by the Tukey method, and were considered significant at p < 0.05. Results are presented as mean ± S.E.M. unless otherwise noted.
Study Approval
All experiments involving the use of mice were performed following protocols approved by the IACUC at the University of Illinois at Chicago. Slides were obtained from patients who provided written informed consent. Tissues were de-identified and provided by the Northwestern University Pathcore following local IRB approval.
RESULTS
TGFBR1-Deficient CTLs Reduce Tumor Burden, but Systemic Reduction of TGFβ Signals Fails to Promote Cytotoxicity
To determine the clinical utility of strategies targeting the TGFβ pathway, we first generated KC mice and conducted adoptive transfer experiments as described in our previous study (6), but this time comparing them to animals with systemic ablation of TGFβ signaling akin to that seen using a TGFβ pathway inhibitor. In brief, KC mice were allowed to reach 2.5 months of age, at which time they develop significant neoplastic disease. We then administered retro-orbital injection of either 2×106 wild type (WT) CD8+ T cells, or CD8+ T cells from Tgfbr1+/− mice, and euthanized the mice after 30 days. Consistent with our previous findings (6), WT CD8+ T-cells failed to significantly alter lesion development, yet Tgfbr1+/− CD8+ T-cells led to rapid clearance of the neoplastic phenotype and an increase in both GranzymeB producing T-cells (Figure S1B,C), as well as GranzymeB deposition in the remaining neoplastic tissues (Figure 1A,B, N=3/group).
Figure 1. TGFBR1-Deficient CTLs Reduce Tumor Burden, but Systemic Reduction of TGFβ Signals Fails to Promote Cytotoxicity.
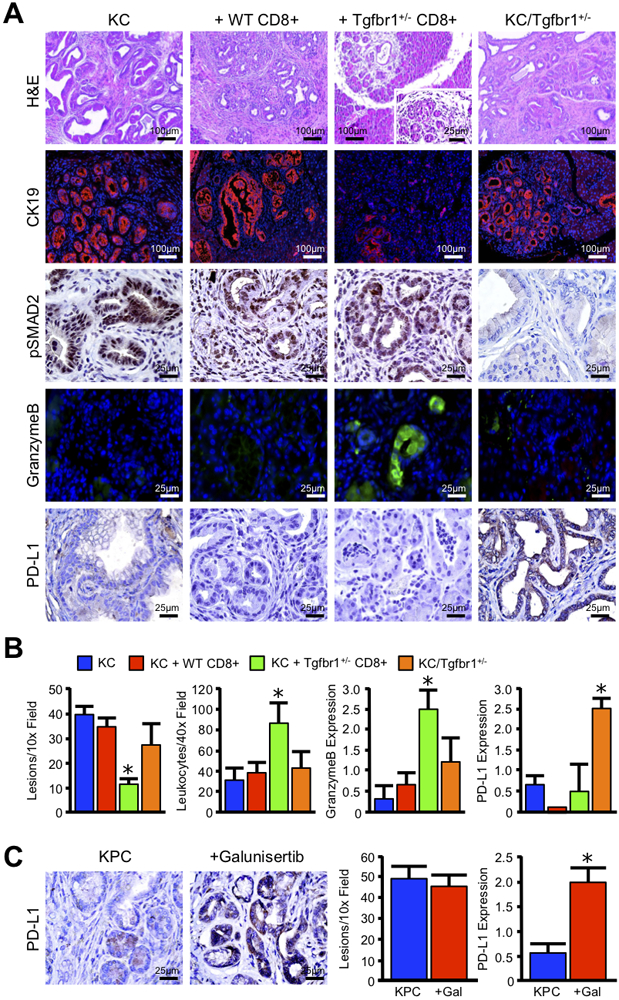
(A) P48-Cre x LSL-KRASG12D (KC) mice were bred to generate a model of conditional expression of oncogenic KRASG12D. Mice were then administered retro-orbital injection of either 2×106 wild type CD8+ T-cells (WT CD8+), or T-cells from Tgfbr1 haplo-insufficient mice (Tgfbr1+/− CD8+). Tissues were collected 30 days after adoptive transfer. Additionally, KC mice were crossed to the Tgfbr1 haplo-insufficient animals, from which the protective Tgfbr1+/− T-cells were derived, to generate KC/Tgfbr1+/−. Tissues were then collected and stained with H&E, or via immunohistochemistry for the ductal marker CK19, TGFβ effector pSMAD2, the cytotoxic surrogate GranzymeB, or for the immune checkpoint PD-L1. (B) Tissue sections were quantified by two independent investigators and averages displayed ± S.E.M (N=3–6/group). (C) Pdx1-Cre x LSL-KRASG12D x LSL-TP53R172H (KPC) mice were used to concomitantly target expression of oncogenic KRASG12D and P53R172H mutations to the pancreas to generate a model of aggressive PDAC. At 6 weeks of age, KPC mice were treated every other day with intraperitoneal injection of 75mg/kg of the TGFBR1 inhibitor Galunisertib, and tissues were collected after 2.5 months. Tissue sections were then stained by H&E or for PD-L1 by immunohistochemistry, quantified by two independent investigators, and averages displayed ± S.E.M (*p < 0.05, N=4/group).
While these findings indicate that TGFβ signal depletion enhances the cytotoxic immune response, they do not necessarily predict responsiveness to broad spectrum or globally administered TGFβ inhibitors. Therefore, we crossed KC mice into the Tgfbr1+/− background to generate KC/Tgfbr1+/−. Despite KC/Tgfbr1+/− mice harboring the protective Tgfbr1+/− transgene in T-cells, these animals had no significant alteration in disease course, nor any observable increase in cell mediated cytotoxicity or GranzymeB deposition (Figure 1A,B, N=3–6/group).
In addition to TGFβ, immune checkpoints such as PD-L1/PD-1 have been identified as a primary mediator of immune tolerance in the pancreas during islet cell transplantation (9). While TGFβ signal inhibition has enhanced immune checkpoint inhibition in immunogenic cancer types (10), this combination has yet to be explored in the management of PDAC. We therefore first assessed mice for expression of PD-L1, which is often upregulated in the pancreatic TME (Figure S2). Interestingly, only KC/Tgfbr1+/− mice displayed an increase in PD-L1 expression on the surface of the neoplastic epithelium (Figure 1A,B, N=3–6/group). To determine whether pharmacologic inhibition of TGFβ signals would similarly cause the upregulation of PD-L1, we next used the Pdx1-Cre x LSL-KRASG12D x LSL-TP53R172H (KPC) model of advanced PDAC and administered the most widely used TGFβ pathway inhibitor Galunisertib (LY2157299). In accordance with our previous results, when 6-week-old KPC mice were treated with 75 mg/kg of the TGFBR1 inhibitor Galunisertib, mice also displayed increased PD-L1 expression and had no significant alteration in lesion development (Figure 1C, N=4/group).
Concomitant TGFBR1 and PD-1 inhibition significantly reduces tumor burden in vivo
In order to determine the physiologic importance of PD-L1 upregulation in response to Galunisertib, we generated KPC mice and after allowing PDAC to develop, administered an IP injection every other day of either a saline vehicle (N=9), 75mg/kg Galunisertib (N=4), a fixed 200μg dose of the PD-1 neutralizing antibody RMP1–14 (anti-PD-1, N=4), or a combination of Galunisertib and anti-PD-1 (Figure 2A, N=7). Both Galunisertib and PD-1 neutralizing antibodies (e.g. Nivolumab or Pembrolizumab) have been used in clinical trials for PDAC (11,12). However, as human and murine PD-1 proteins differ significantly in the extracellular domain (13), it was necessary to utilize a mouse-specific PD-1 neutralizing antibody for this study.
Figure 2. Concomitant TGFBR1 and PD1 inhibition significantly reduces tumor burden in vivo.
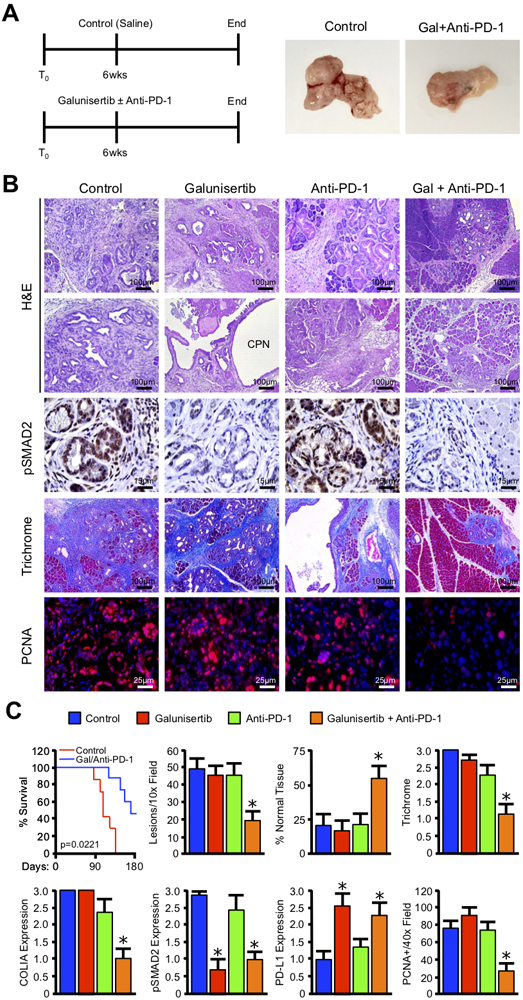
(A) Pdx1-Cre x LSL-KRASG12D x LSL-TP53R172H (KPC) mice were regenerated as a model of aggressive PDAC. At 6 weeks of age, mice were administered intraperitoneal injection every other day either a PBS vehicle (Control), 75 mg/kg of the TGFBR1 inhibitor Galunisertib, 200 μg of an anti-PD-1 neutralizing antibody (Anti-PD-1), or a combination of Galunisertib and Anti-PD-1. The pancreas was then collected either when the animals were moribund or at the conclusion of the study (180 days post enrollment), and gross changes in pancreas gland structure evaluated. (B) The pancreas from control and drug treated mice was then stained with either H&E or Masson’s Trichrome. Sections were also stained via immunohistochemistry for the TGFβ effector pSMAD2 or the proliferation surrogate PCNA. (C) Kaplan-Meier curve indicating survival for Control and Galunisertib/Anti-PD-1 treated mice measured in days after treatment began (N=7–9/group). Additionally, tissue sections were quantified by two investigators and averages displayed ± S.E.M. (*p < 0.05, N=3–4/group).
While Galunisertib was highly effective at neutralizing SMAD2 phosphorylation (Figure 2B,C), once again there was no significant effect on tumor development apart from the development of regions of cystic papillary neoplasms (CPN). Similarly, anti-PD-1 failed to significantly alter disease course as a monotherapy as previously reported (8). However, when the two agents were combined the KPC mice displayed a significant reduction in the neoplastic phenotype despite the increased PD-L1 expression (Figure 2A-C and S3-5). This was also associated with a modest survival benefit in dual-treated mice compared to untreated mice, which was not seen in single agent controls (Figure 2C and S6, P=0.0221). This was also accompanied by improvement in weight gain (Figure S3B), as well as reduction in both fibrosis and proliferation in remaining areas of disease (Figure 2B,C). Further, while there was no observable change in pancreas length (Figure S4B), mice treated with both Galunisertib and anti-PD-1 had reductions in pancreas weight consistent with a reduction in the tumor stroma (Figure S4C). Despite the reduced tumor burden, there were no obvious signs of autoimmunity or drug toxicity in mice treated with the combination regimen (Figure S5).
Galunisertib Cooperates with PD-1 inhibition to promote T-cell mediated regression of Advanced PDAC
Consistent with the observed reduction in tumor burden, on histology mice treated with the combination regimen displayed an overwhelming lymphocyte infiltrate, which localized strongly to remaining areas of disease (Figure 3A,B). These infiltrating cells were uniformly positive for CD3, affirming their T-cell lineage. Additionally, the T-cell infiltrate of Galuinisertib and anti-PD-1 dual-treated mice had robust expression of GranzymeB, which also localized to any remaining areas of neoplastic disease (Figure 3A,B and S7, N=3/group). These local changes in immune function were further verified by flow cytometry of the mesenteric lymph nodes, which revealed similar increase in T-helper activation (CD4+CD69+) cells exclusively in mice treated with the combination regimen (Figure 3C,G and S8). As macrophages are a primary target of Th1 cells and may have an important role in the response to immune checkpoint inhibitors (14), we also determined the presence of CD11b/GR1+ granulocytes. While these cells were largely absent from the lymph nodes, we found a significant increase in CD11b/GR1+ cells in the spleen. Further, these CD11b/GR1+ cells were largely positive for the mouse macrophage marker F4/80 (Figure 3D,G and S8). Most importantly, mice treated with the combination regimen also had increased CTL activation in the mesenteric lymph nodes (CD8+CD69+ cells), as well as enhanced Interferon-γ (IFNγ), GranzymeB (GrzB), and Perforin production (Figure 3D-G and S8), further affirming an increase in cell-mediated cytotoxicity.
Figure 3. Galunisertib Cooperates with PD-1 inhibition to promote T-cell mediated regression of Advanced PDAC.
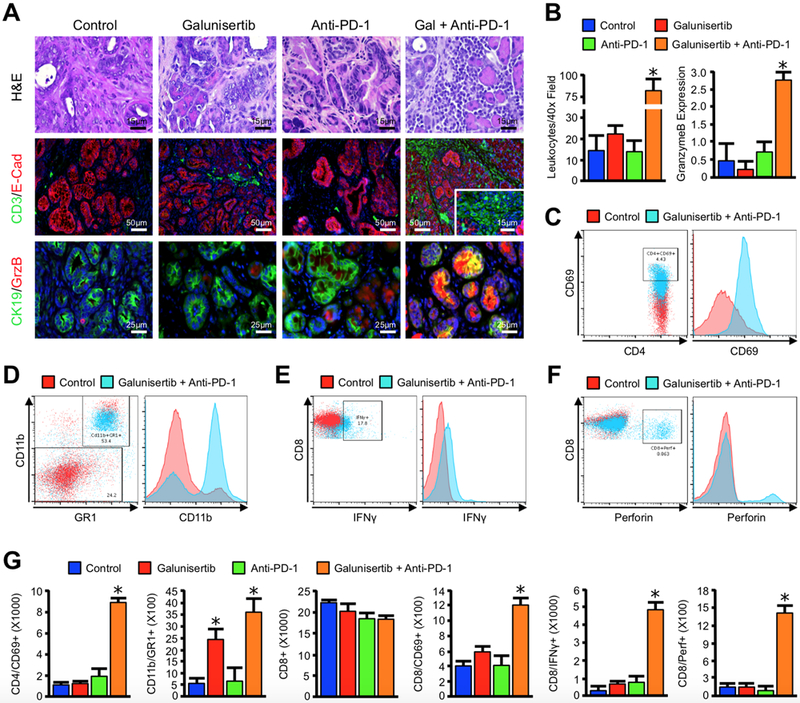
(A) Pdx1-Cre x LSL-KRASG12D x LSL-TP53R172H (KPC) mice were used as a model of advanced PDAC, and animals were treated with either a PBS vehicle (Control), 75 mg/kg of the TGFBR1 inhibitor Galunisertib, 200 μg of an anti-PD-1 neutralizing antibody (Anti-PD-1), or a combination of Galunisertib and Anti-PD-1. The pancreas from control and drug-treated mice were stained with H&E and evaluated under high power, or dual-stained for the T-cell marker CD3 and the epithelial surrogate E-Cadherin (E-Cad), or for the duct cell marker CK19 and the cytotoxic surrogate GranzymeB (GrzB). (B) Tissue sections were subsequently quantified and averages displayed ± S.E.M. (*p < 0.05, N=3–6/group). Lymphoid tissues were harvested and evaluated by flow cytometry for (C) CD4+CD69+ dual positive populations in the mesenteric lymph nodes (D) CD11b+Gr1+ granulocytes in the spleen (E,F) CD8+IFNγ+ or CD8+Perforin+ dual positive cells in the mesenteric lymph nodes. (G) Populations identified by cytometry across all four groups were quantified and averages displayed ± S.E.M. (*p < 0.05).
DISCUSSION
To date there are no curative strategies in the management of advanced pancreatic cancer patients. As PDAC often presents in late stages, most efforts focus on extending life when possible and providing supportive/palliative care. Immune checkpoint inhibition has been a major therapeutic breakthrough in many cancers. The PD-1 neutralizing antibody Pembrolizumab is now approved for non-small cell lung cancers, melanoma, some squamous cell carcinomas, and Hodgkin’s lymphoma. Similarly, the anti-PD-1 antibody Nivolumab is first line in metastatic melanoma, and has been approved for progressive squamous cell carcinomas of the head and neck, renal cell carcinoma, and non-small cell lung cancer (15).
As a result, investigators have been eager to determine whether similar strategies targeting PD-1/PD-L1 are applicable in the management of PDAC. Importantly, PD-L1 is often overexpressed in PDAC and is associated with poor outcomes (16). Though ineffective as a monotherapy, the combination of Nivolumab with Abraxane is showing early promise, as is the combination of Nivolumab with GVAX-based immunotherapy (17). Though encouraging, these trials appear to only modestly extend survival, and overall mortality remains high.
The mechanisms underlying the reduced efficacy of immune checkpoint inhibition in pancreatic cancer remains unclear. Therefore, it is essential to dissect alternate means through which tumor cells promote peripheral immune tolerance in order to combine with immune checkpoint inhibitors to improve their efficacy and further extend survival. As discussed and demonstrated, TGFβ is an important immunomodulator that plays a role both in physiologic and pathologic tolerance. In the absence of TGFβ signaling, T-lymphocytes rapidly acquire high levels of FasL and GranzymeB, two main functional components for cytotoxicity. Similarly, T-cells expressing a constitutively active form of TGFBR1 also remain refractory to full activation (18).
However, much like anti-PD-1 antibodies, drugs targeting the TGFβ pathway have also only shown partial efficacy in the clinic. In an ongoing Phase II clinical trial, Galunisertib was added to the first line agent Gemcitabine, which extended overall survival from 7.2 to 10.9 months, and substantially reduced serum levels of TGFβ1 (19). However, should therapies targeting the TGFβ pathway induce PD-L1 upregulation, this may substantially limit their therapeutic efficacy. Similarly, as TGFβ directly suppresses effector T-cell function independent of PD-L1, its near ubiquitous overexpression in pancreatic cancer patients may also explain the limited efficacy of PD-1/PD-L1 inhibitors in pancreatic cancer patients (Figure 4).
Figure 4. Model for mechanism of action for combined TGFβ and PD-1 pathway inhibition.
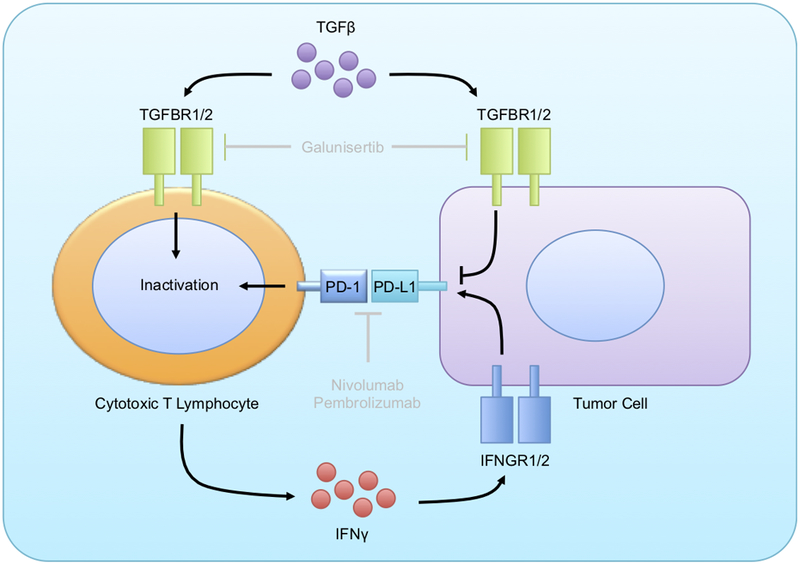
Both TGFβ and PD-L1 are known to repress the function of CD8+ cytotoxic T-cells in pancreatic ductal adenocarcinoma. However, our data appears to suggest that TGFβ negatively regulates the expression of PD-L1 on the neoplastic epithelium, either directly (as shown) or indirectly, potentially through IFNγ as shown. Hence, pharmacologic ablation of TGFβ signals via the drug Galunisertib leads to the accumulation of PD-L1 in the cancer epithelium, failing to promote a substantial anti-tumor immune response. Hence, combination therapy with an anti-PD-1 antibody and a TGFβ pathway inhibitor is necessary to have therapeutic efficacy, and such a combination regimen warrants clinical consideration.
The link between TGFβ signaling and immune checkpoint inhibition is now becoming clear, and their combination is showing early efficacy in other cancers types (10). For instance, in metastatic urothelial cancer patients, a poor response to the anti-PD-L1 antibody Atezolizumab was associated with a TGFβ signaling signature in fibroblasts. Similarly, the combination of TGFβ and PD-L1 inhibition greatly enhanced anti-tumor immune function (20). Additionally, concomitant TGFβ/PD-L1 pathway inhibition greatly enhanced T-cell responses in esophageal squamous cell carcinoma (21), and blockade of greatly sensitized genetically reconstituted models of colon cancer metastasis (22). Further, TGFβ pathway inhibitors enhanced responsiveness to PD-1/PD-L1 neutralizing antibodies in mice bearing poorly immunogenic 4T1-LP breast tumors (23).
Despite this increasing body of evidence, this combination has not been fully explored in PDAC. As it is well established that TGFβ is almost ubiquitously overexpressed in the pancreatic TME and is itself a primary means for immune evasion, it is essential to further study the ability of TGFβ inhibitors to promote immune cell clearance of pancreatic tumors, as well as overcome the innate resistance of pancreatic cancers to immune checkpoint inhibitors and improve patient outcomes.
Supplementary Material
ACKNOWLEDGEMENTS
This work is dedicated to the memory of our friends Geraldine Leonardo and Rodney Hughes, both of whom recently lost their lives to pancreatic cancer. The authors also would like to thank our summer students Matt Nabutis and Shravya Chanamolu for all their hard work and dedication. This work was supported by University of Illinois College of Medicine Hazel I. Craig Fellowships to D.R. Principe and A. Park, NIH R01CA186885 and Veterans Affairs Merit Award I01BX002922 to H.G Munshi, and by Veterans Affairs Merit Awards BX002703 and BX002355 to A. Rana. Additionally, D.R. Principe is supported by NIH 1F30CA236031.
Financial Information: This work was supported by University of Illinois College of Medicine Hazel I. Craig Fellowships to D.R. Principe and A. Park, NIH R01CA186885 and Veterans Affairs Merit Award I01BX002922 to H.G. Munshi, and by Veterans Affairs Merit Awards BX002703 and BX002355 to A. Rana. D.R. Principe is supported by NIH 1F30CA236031.
Abbreviations:
- PDAC
Pancreatic Ductal Adenocarcinoma
- PanIN
Pancreatic Intrapithelial Neoplasm
- TGFβ
Transforming Growth Factor β
- KC
P48-Cre/LSL-Kras
- KPC
Pdx1-Cre x LSL-KRASG12D x LSL-TP53R163H
Footnotes
Conflict of Interest Disclosure: The authors have no conflicts to disclose.
REFERENCES
- 1.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20(22):3147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007;11(3):229–43. [DOI] [PubMed] [Google Scholar]
- 3.Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer research 2007;67(17):8121–30. [DOI] [PubMed] [Google Scholar]
- 4.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer research 2003;63(9):2016–9. [PubMed] [Google Scholar]
- 5.Adrian K, Strouch MJ, Zeng Q, Barron MR, Cheon EC, Honasoge A, et al. Tgfbr1 haploinsufficiency inhibits the development of murine mutant Kras-induced pancreatic precancer. Cancer Res 2009;69(24):9169–74. [DOI] [PubMed] [Google Scholar]
- 6.Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res 2016;76(9):2525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Center for Biotechnology Information. PubChem Compound Database; CID=10090485, https://pubchem.ncbi.nlm.nih.gov/compound/10090485 (accessed Nov. 7, 2018).
- 8.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 2015;3(4):399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baas M, Besancon A, Goncalves T, Valette F, Yagita H, Sawitzki B, et al. TGFbeta-dependent expression of PD-1 and PD-L1 controls CD8(+) T cell anergy in transplant tolerance. Elife 2016;5:e08133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanpouille-Box C, Formenti SC. Dual Transforming Growth Factor-beta and Programmed Death-1 Blockade: A Strategy for Immune-Excluded Tumors? Trends Immunol 2018;39(6):435–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 2015;9:4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune checkpoint therapy for pancreatic cancer. World J Gastroenterol 2016;22(43):9457–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li D, Xu J, Wang Z, Gong Z, Liu J, Zheng Y, et al. Epitope mapping reveals the binding mechanism of a functional antibody cross-reactive to both human and murine programmed death 1. MAbs 2017;9(4):628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suda K Tumor-associated macrophages-additional effectors at anti-PD-1/PD-L1 therapy? J Thorac Dis 2017;9(11):4197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, Turrini O, et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget 2016;7(44):71198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett 2017;407:57–65. [DOI] [PubMed] [Google Scholar]
- 18.Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst 2014;106(2):djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. A phase II, double-blind study of galunisertib+gemcitabine (GG) vs gemcitabine+placebo (GP) in patients (pts) with unresectable pancreatic cancer (PC). Journal of Clinical Oncology 2016;34(15_suppl):4019–19. [Google Scholar]
- 20.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Wang L, Li P, Song M, Qin G, Gao Q, et al. Dual TGF-beta and PD-1 blockade synergistically enhances MAGE-A3-specific CD8(+) T cell response in esophageal squamous cell carcinoma. Int J Cancer 2018. [DOI] [PubMed] [Google Scholar]
- 22.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554(7693):538–43. [DOI] [PubMed] [Google Scholar]
- 23.Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer 2018;6(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.