

Abstract
New classes of alkylated hetarylpropylguanidines with different functionality and variation in spacer length were synthesized to determine their behavior at the four histamine receptor (H1R, H2R, H3R, H4R) subtypes. Alkylated guanidines with different terminal functional groups and varied basicity, like amine, guanidine and urea were developed, based on the lead structure SK&F 91486 (2). Furthermore, heteroatomic exchange at the guanidine structure of 2 led to simple analogues of the lead compound. Radioassays at all histamine receptor subtypes were accomplished, as well as organ bath studies at the guinea pig (gp) ileum (gpH1R) and right atrium (gpH2R). Ligands with terminal functionalization led to, partially, highly affine and potent structures (two digit nanomolar), which showed up a bad selectivity profile within the histamine receptor family. While the benzoylurea derivative 144 demonstrated a preference towards the human (h) H3R, S‐methylisothiourea analogue 143 obtained high affinity at the hH4R (pKi=8.14) with moderate selectivity. The molecular basis of the latter finding was supported by computational studies.
Keywords: histamine H1-4 receptor, ligand design, receptor subtype selectivity, organ pharmacology, computational chemistry
Selectivity first! Structure‐activity relationship of 27 new ligands from the hetarylpropylguanidine‐type was investigated at the four histamine receptor subtypes with detailed pharmacological characterization. Methylisothiourea analogue 143 turned out to have high affinity at the hH4R with perceptible selectivity. Computational studies for 143 at both hH4R and hH3R were accomplished in order to gain more insight into the respective binding modes.
Introduction
The biogenic amine histamine (1, Figure 1) is known to be the endogenous key modulator for histamine receptors in the human body.1 There it regulates a variety of effects via the four histamine receptor (HR) subtypes H1, H2, H3 and H4, each belonging to the superfamily of G‐protein coupled receptors (GPCRs).2, 3, 4, 5, 6 The H1R is expressed in several tissues (e. g., brain, blood vessels, gastrointestinal tract) and couples to a Gq/11‐protein.7,8 For decades, H1‐antihistamines have been successfully used for the treatment of allergic diseases as sedatives and antiemetics.9 The H2R is mainly expressed in gastric parietal cells, in the heart, as well as in the brain and couples to a Gαs‐protein, which activates the adenylyl cyclase (AC).10,11 Prior to proton‐pump inhibitors, such as omeprazole and pantoprazole,12 overstocking the market in the 1990s, H2R antagonists like cimetidine have been one of the first blockbuster drugs for the treatment of gastroesophageal reflux disease (GERD) and peptic ulcer.13 The H3R and H4R are both coupled to Gαi/o‐proteins, but differentiate in their localization in the human body.14, 15, 16 While H3Rs are widely expressed in the central nervous system,17 the H4R is mainly found in immune and mast cells.5,15,18, 19, 20 Due to its function as an auto‐ and heteroreceptor in the brain, the H3R is a promising potential target for various cognitive disorders, like Alzheimer's disease, Parkinson or Tourette syndrome.21, 22, 23, 24, 25 Even if the biological functions of the H4R are not completely apparent, intensive research proved the involvement in allergic and inflammatory processes.26 For this reason, targeting the H4R is expected to be crucial for the treatment of allergic rhinitis, rheumatoid arthritis or pruritus.27, 28, 29, 30
Figure 1.
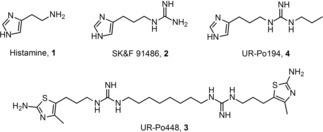
Structures of histamine and selected histamine receptor ligands (2–4).
While the first antihistamines (H1R), like mepyramine and diphenhydramine, as well as their functional behavior on guinea‐pig organs were published in the 1930s, 1940s and 1950s,31, 32, 33 a large number of highly potent H2R agonists like impromidine and arpromidine were released in the 1970s and 1980s.34, 35, 36 Deriving from the lead structure SK&F 91486 (2, Figure 1)37, a long‐known ligand addressing histamine receptors, several classes of newly synthesized monomers were characterized in this study. A couple of previous projects, focusing on the development of potent H2R agonists, observed an overlap of H3R‐ and H4R‐related effects of imidazole‐containing compounds.38 Heterocyclic replacement by amino(methyl)thiazole, following amthamine,39 led to highly selective dimeric H2R agonists, like UR−Po448 (3, Figure 1)40 and associated molecules.41, 42, 43 In addition, a switch away from imidazole‐bearing compounds is recommended as these structures show poor pharmacokinetic properties due to interactions with cytochrome P450.44 Structural modifications around the guanidine group gave cyano‐, carbamoyl‐, acylguanidines and related structures, which showed up selectivity towards the H3R or H4R, respectively.45,46 In this project, we aimed to attaining insights into structure‐activity relationships of novel ligands from the hetarylpropylguanidine‐type. We wanted to close the gap between the monomeric lead structure 2, the highly affine hH4R ligand UR−Po194 (4, Figure 1)40 and the dimeric ligands described in the literature, e. g. 3.40 Therefore, we created alkylated guanidines with various terminal functional groups of different basicity, like amine, guanidine, urea, including variable spacer length. Moreover, we synthesized a class of molecules focusing on the heteroatomic exchange at the guanidine moiety of 2 only to attain (thio)ureas and S‐methylisothioreas. The final compounds were pharmacologically characterized with radioligand binding assays and the GTPγS binding assay to get binding as well as functional data. In addition, we analyzed all compounds by organ pharmacological studies on the guinea pig ileum and right atrium in order to receive information about their functional behavior under physiological conditions (gpH1R (ileum), gpH2R (right atrium)).
Results and Discussion
Chemistry
Syntheses of the amines 5–7 (Figure 2), which were used for the development of the final compounds were carried out according to the literature.39,41,47,48 The required precursors 17–27 for the terminal amines and guanidines were prepared according to previously reported procedures (Scheme 1) and adaptions.41,49,50 The isothiourea 10 proved to be a suitable guanidinylation reagent for the preparation of 23–27 and was obtained in a two‐step synthesis by S‐methylation of 8 and di‐Boc‐protection of 9 with two equivalents of Boc2O (Scheme 1).41,51 Mono‐Boc‐protection of the respective diamines 11–16 was also carried out with Boc2O to get 17–22 (Scheme 1). Due to the possibility of a di‐protection a molar ratio of at least 1 : 5 (Boc2O : diamine) was required in order to achieve yields >90 %.49 The aforementioned di‐Boc‐protected guanidines 23–27 were prepared by dropping the guanidinylation reagent 10 into a solution of the appropriate diamine (12–16, 3 equiv) in DCM (Scheme 1).50
Figure 2.
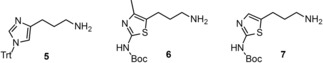
Structures of the amines 5–7, which were used for the preparation of the final compounds 115–136 and 141–145 (cf. Scheme 1–3).
Scheme 1.
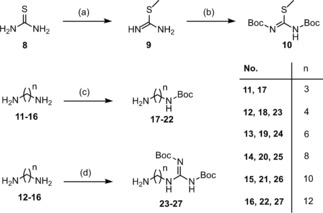
Synthesis of the precursors 17–27. Reagents and conditions: (a) CH3I (1.1 equiv), MeCN, 1 h, reflux; (b) NEt3 (1 equiv), Boc2O (2 equiv), overnight, room temperature (rt); (c) diamine (5 equiv), Boc2O (1 equiv), DCM, 2 h, 0 °C→rt; (d) diamine (3 equiv), 10 (1 equiv), DCM, overnight, rt.
The synthetic route for the preparation of 115–136 and 141–145 was adapted as previously described in the literature (Scheme 2 and 3).40,49,50,52, 53, 54, 55, 56 In a first step the relevant amine 11–27 attacks benzoyl isothiocyanate (28) via nucleophilic substitution to give benzoylthioureas 29–44 (Scheme 2).40,52 After alkaline hydrolysis yielding the corresponding thioureas 45–60, the intermediates were treated with methyl iodide to receive 61–76 (Scheme 2).40,52 Prior to guanidinylation, the S‐methylisothioureas were Boc‐protected obtaining 77–92 (Scheme 2).40 Aminolysis of the guanidinylation reagents 77–92 with some of the amines 5–7 in presence of HgCl2 and triethylamine gave 93–114 (Scheme 2).40,57 For the synthesis of the Boc‐protected amines (93–100) and guanidines (101–109) one equivalent of HgCl2 was used, while four equivalents of HgCl2 were used for the preparation of the Boc‐protected carbodiimides (110–114). The carbodiimides, which were converted into ureas in the next step, were unscheduled. It was planned to create the relative dimers, which were published by Pockes et al.40 The original synthetic description for one‐site coupling was described with two equivalents of mercury chloride.41 This excess should be maintained for this two‐site coupling. Contrary to our expectations the excess of HgCl2 (4 equiv) – which facilitates the elimination of the S‐methyl group by coordination to sulfur – led to mono‐Boc‐carbodiimides, where just one aminolysis was successful. This fact could be proven by NMR spectroscopy and mass spectrometry and a similar issue was already reported by Kim et al. in 1993.57 Afterwards, the use of HgCl2 for one‐site coupling was adjusted as described in 4.2.9, as well as for two‐site coupling (cf. Pockes et al.)40. In a last step the precursors were Boc‐deprotected using trifluoroacetic acid (TFA) to get 115–136 as final compounds (Scheme 2).40
Scheme 2.
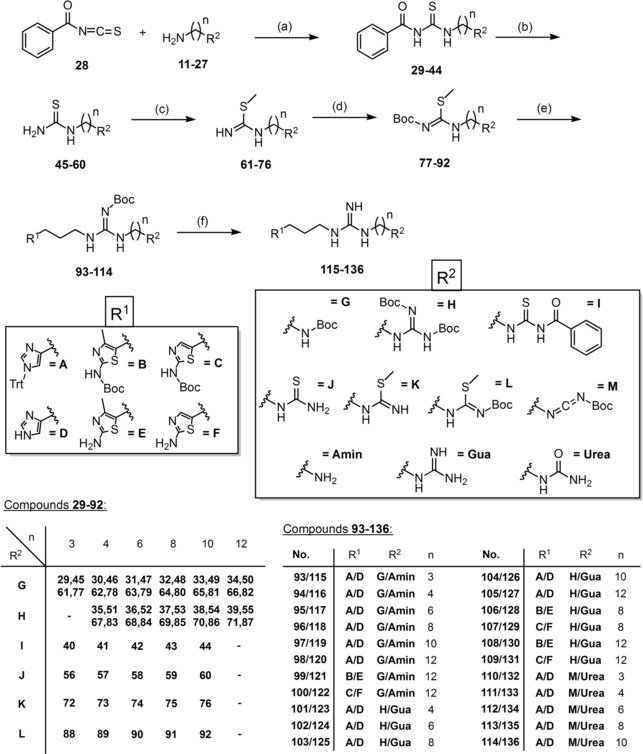
Synthesis of the HR ligands 115–136. Reagents and conditions: (a) amine/diamine (1 equiv), 28 (1 equiv/2 equiv), DCM, 2 h/overnight, 0 °C→rt; (b) K2CO3 (2.1 equiv/4.1 equiv), MeOH/H2O (7/3, v/v), 3–5 h, rt; (c) CH3I (1.1 equiv/2.1 equiv), MeCN, 1 h, reflux; (d) NEt3 (1 equiv/2 equiv), Boc2O (1 equiv/2 equiv), overnight, rt; (e) 5, 6 or 7 (1 equiv/2 equiv), HgCl2 (1 equiv/4 equiv), NEt3 (3 equiv/6 equiv), DCM, overnight, rt; (f) 20 % TFA, DCM, overnight, reflux.
Scheme 3.
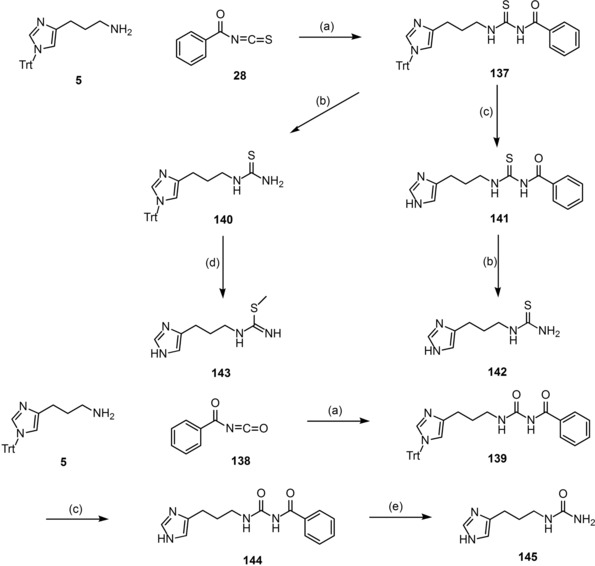
Synthesis of the SK&F 91486 analogues 141–145. Reagents and conditions: (a) 5 (1 equiv), 28 or 138 (1 equiv), DCM, overnight, 0 °C→rt; (b) K2CO3 (2.1 equiv), MeOH/H2O (7/3, v/v), 3–5 h, rt; (c) 20 % TFA, DCM, overnight, reflux; (d) i) 66 % HI, EtOH, rt; ii) CH3I (1.1 equiv), MeOH 1 h, reflux; (e) NaOH (1 M solution), 1 h, reflux.
The synthetic strategy for the final compounds 141–145 is depicted in Scheme 3. The same pattern was used for the nucleophilic addition to get 137 or 139 using 28 or benzoyl isocyanate (138) together with the amine 5 (Scheme 3).40,52 137 was further processed in two different ways, getting 140 by alkaline hydrolysis and 141 by deprotection under acidic conditions (Scheme 3).40,52 Moreover, the benzoyl isothiocyanate 141 was hydrolysed with potassium carbonate to give the thiourea 142 (Scheme 3).40,52 To complete the second route, 140 was first deprotected with hydrogen iodide (66 %) and directly handled with methyl iodide yielding 143 (Scheme 3).53,54 To create the urea analogues, the trityl group of 139 was first cleaved with TFA to get the final compound 144,40 followed by alkaline hydrolysis with sodium hydroxide solution (1 M) under reflux obtaining 145 (Scheme 3).56 Usual basic hydrolysis with potassium carbonate was not successful in this case, not even after several hours of reflux. Compounds 141[ 53], 142[ 53] and 143[ 54,35] were already decribed in the literature. In this study we resynthesized these structures for further pharmacological investigations.
Pharmacology
The ligands 115–136 and 141–145 were pharmacologically characterized using radioligand binding assays (hH1,2,3,4R), the guinea pig ileum assay (gpH1R) as well as the guinea pig right atrium assay (gpH2R). The most interesting compounds were further investigated in the [35S]GTPγS binding assay (hH2,3,4R). The radioassays were performed using membranes of Sf9 cells expressing the respective histamine receptor described in Table 1 and 2.
Table 1.
Binding data (pKi values) of compounds DPH, 1–4, 115–136 and 141–145 determined at human HxRs (x=1–4).[a]
|
compound |
hH1R[b] |
hH2R[c] |
hH3R[d] |
hH4R[e] |
||||
|---|---|---|---|---|---|---|---|---|
|
pKi |
N |
pKi |
N |
pKi |
N |
pKi |
N |
|
|
DPH |
7.62±0.01 |
4 |
n.d. |
– |
n.d. |
– |
n.d. |
‐ |
|
1 |
5.62±0.03 |
3 |
6.58±0.04 |
48 |
7.59±0.01 |
42 |
7.60±0.01 |
45 |
|
2 |
<4 |
3 |
5.39±0.04[f] |
3 |
7.42±0.04 |
3 |
8.13±0.08 |
3 |
|
3 |
<5.5 |
2 |
7.33±0.05 |
3 |
5.25±0.05 |
3 |
5.00±0.05 |
3 |
|
4 |
<4.5 |
2 |
5.52±0.05 |
3 |
7.21±0.02 |
3 |
8.04±0.05 |
3 |
|
115 |
<4 |
2 |
6.63±0.06 |
3 |
5.59±0.03 |
3 |
7.03±0.07 |
3 |
|
116 |
<5 |
2 |
6.07±0.04 |
3 |
5.82±0.03 |
3 |
6.47±0.02 |
3 |
|
117 |
<5 |
2 |
6.12±0.04 |
3 |
6.19±0.01 |
3 |
6.36±0.03 |
3 |
|
118 |
5.91±0.03 |
2 |
6.44±0.02 |
3 |
6.65±0.04 |
3 |
6.33±0.03 |
3 |
|
119 |
6.06±0.03 |
2 |
6.80±0.02 |
3 |
7.14±0.01 |
3 |
6.78±0.01 |
3 |
|
120 |
6.71±0.02 |
2 |
7.28±0.04 |
3 |
7.45±0.02 |
3 |
7.16±0.04 |
3 |
|
121 |
6.30±0.01 |
2 |
6.50±0.09 |
3 |
5.38±0.01 |
3 |
5.33±0.03 |
3 |
|
122 |
6.52±0.01 |
2 |
6.73±0.05 |
3 |
5.29±0.06 |
3 |
4.65±0.06 |
3 |
|
123 |
<4.5 |
2 |
6.15±0.05 |
3 |
6.41±0.01 |
3 |
7.51±0.04 |
3 |
|
124 |
<5 |
2 |
6.24±0.02 |
3 |
6.97±0.03 |
3 |
6.62±0.02 |
3 |
|
125 |
<5.5 |
2 |
6.80±0.10 |
3 |
7.20±0.03 |
3 |
6.84±0.02 |
3 |
|
126 |
6.09±0.01 |
2 |
6.85±0.06 |
3 |
7.43±0.02 |
3 |
7.50±0.02 |
3 |
|
127 |
6.59±0.01 |
2 |
7.06±0.04 |
3 |
7.48±0.05 |
3 |
7.55±0.02 |
3 |
|
128 |
5.75±0.01 |
2 |
6.67±0.01 |
3 |
6.53±0.03 |
3 |
6.40±0.01 |
3 |
|
129 |
6.58±0.01 |
2 |
7.03±0.09 |
3 |
6.25±0.04 |
3 |
5.62±0.05 |
3 |
|
130 |
6.25±0.01 |
2 |
6.93±0.02 |
3 |
7.07±0.01 |
3 |
6.23±0.09 |
3 |
|
131 |
6.28±0.01 |
2 |
6.84±0.05 |
3 |
6.43±0.17 |
3 |
6.47±0.07 |
3 |
|
132 |
<4.5 |
2 |
5.75±0.10 |
3 |
6.80±0.01 |
3 |
6.84±0.01 |
3 |
|
133 |
<4.5 |
2 |
6.57±0.03 |
3 |
6.27±0.05 |
3 |
6.59±0.02 |
3 |
|
134 |
<5 |
2 |
6.55±0.07 |
3 |
6.69±0.02 |
3 |
6.18±0.03 |
3 |
|
135 |
<5.5 |
2 |
6.58±0.11 |
3 |
6.84±0.02 |
3 |
6.56±0.01 |
3 |
|
136 |
<5.5 |
2 |
6.95±0.02 |
3 |
7.00±0.01 |
3 |
6.70±0.01 |
3 |
|
141 |
<4 |
2 |
4.26±0.09 |
3 |
6.78±0.05 |
3 |
6.82±0.04 |
3 |
|
142 |
<4 |
2 |
<4 |
3 |
6.07±0.03 |
3 |
5.77±0.02 |
3 |
|
143 |
<4 |
2 |
4.98±0.09 |
3 |
6.58±0.08 |
3 |
8.14±0.01 |
3 |
|
144 |
<4 |
2 |
<4 |
3 |
6.09±0.01 |
3 |
5.28±0.06 |
3 |
|
145 |
<5 |
2 |
6.17±0.08 |
3 |
6.91±0.03 |
3 |
6.83±0.02 |
3 |
[a]Data represent mean values ± SEM from at least two independent experiments (N), each performed in triplicate. Radioligand competition binding experiments performed with [b][3H]mepyramine (hH1R, K d 4.5 nM, c=5 nM), [c][3H]tiotidine (hH2R, K d 19.7 nM, c=10 nM), [d][3H]Nα‐methylhistamine (hH3R, K d 8.6 nM, c=3 nM) or [e][3H]histamine (hH4R, K d 16.0 nM, c=15 nM) at membranes of Sf9 cells expressing [b]the hH1R plus RGS4, [c]the hH2R plus Gsαs, [d]the hH3R plus Gαi2 plus Gβ1γ2, [e]the hH4R plus Gαi2 plus Gβ1γ2. [f]Displacement of [3H]UR‐DE257 (hH2R, K d 31.3 nM, c=20 nM) instead of [3H]tiotidine; n.d.=not determined.
Table 2.
Agonistic (pEC50) and antagonistic (pKB) activities of 1–3, 120, 121, 127, 130 and 136 at the hH2,3,4R determined in the [35S]GTPγS binding assay.[a]
|
Compound |
hH2R[b] |
hH3R[c] |
hH4R[d] |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
pEC50 [e] |
E max [f] |
N |
pEC50 [e] (pKB)[g] |
E max [f] |
N |
pEC50 [e] (pKB)[g] |
E max [f] |
N |
|
|
1 |
6.01±0.07 |
1.00 |
7 |
8.52±0.10 |
1.00 |
6 |
8.20±0.08 |
1.00 |
3 |
|
2 |
5.59±0.01[h],[46] |
0.66±0.02[h],[46] |
3 |
8.12±0.10[h],[46] |
0.69±0.04[h],[46] |
3 |
8.09±0.04[h],[46] |
0.83±0.01[h],[46] |
3 |
|
3 |
6.61±0.03 |
0.33±0.03 |
5 |
(4.53±0.05) |
0 |
3 |
(3.83±0.03) |
0 |
3 |
|
120 |
6.95±0.04 |
0.66±0.01 |
3 |
(6.72±0.03) |
0 |
3 |
(3.43±0.01) |
0 |
3 |
|
121 |
7.11±0.10 |
0.22±0.01 |
3 |
5.88±0.03 |
−0.69±0.03 |
3 |
(3.75±0.01) |
0 |
3 |
|
127 |
6.86±0.03 |
0.45±0.03 |
3 |
(7.18±0.04) |
0 |
3 |
(3.38±0.01) |
0 |
3 |
|
130 |
7.28±0.10 |
0.22±0.01 |
3 |
4.46±0.02 |
−1.21±0.10 |
3 |
4.52±0.02 |
‐0.98±0.01 |
3 |
|
136 |
6.72±0.06 |
0.45±0.03 |
3 |
4.57±0.03 |
−1.20±0.15 |
3 |
4.44±0.01 |
‐0.96±0.02 |
3 |
[a]Data represent mean values ± SEM from at least three independent experiments (N), each performed in triplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration‐response curves (CRCs). [35S]GTPγS binding assay at membranes of Sf9 cells expressing [b]the hH2R plus Gsαs, [c]the hH3R plus Gαi2 plus Gβ1γ2, [d]the hH4R plus Gαi2 plus Gβ1γ2; [e]pEC50: −logEC50; [f] E max: maximal response relative to histamine (E max=1.00); [g]For determination of antagonism, reaction mixtures contained histamine (1) (100 nM) and ligands were at concentrations from 10 nM and 1 mM; pKB=−logKB. [h]Determined in a steady‐state [32P]GTPase assay on Sf9 cells expressing the related receptors.
Introduction of a third basic moiety was the main focus by developing new ligands as shown in Scheme 2. Therefore, we created terminal amines 115–122, guanidines 123–131 and ureas 132–136 with different spacer lengths. Furthermore, heterocyclic exchange of imidazole by amino(methyl)thiazole should give more insight in the selectivity profile of the ligands. The compounds depicted in Scheme 3 (141–145) were mainly altered by heteroatomic exchange at the guanidine group of 2. The following influence on basicity should give important information about the variability of this partial structure, with respect to histamine receptor affinity and potency.
Radioligand Binding Data
A correlation was found between binding affinities of the amines 115–122 at the hH1R and the respective lipophilicity (Table 1). From C3‐ (115) to C12‐spacer (120–122) there is an upward shift of approximately 3 log units. The tendency at the hH2R is the same, but to a lesser extent The highest affinity value was measured for 120 (pKi=7.28, Table 1). In comparison with compounds bearing small alkylic side chains like UR−Po194 (4), showing high affinity at the hH4R,40 introduction of a terminal amine with similar spacer length (115, 116) led to a remarkable loss in affinity of at least 1 log unit (Table 1). Heterocyclic replacement by amino(methyl)thiazole resulted in the already known affinity decrease at the hH3,4Rs. Related to the hH2R 121 and 122 reveal moderate selectivity towards the hH3,4Rs, but not towards the hH1R (Table 1).
Data for the guanidines 123–131 at the hH1R and the hH2R were similar to those of the amines 115–122. Increasing spacer length led to higher affinity values culminating in 127 (pKi (hH1R)=6.59; pKi (hH2R)=7.06; cf. Table 1). Affinity values at the hH3,4Rs were all in a submicromolar range and this time a switch from imidazole to amino(methyl)thiazole resulted in a moderate affinity loss from maximally 1 log unit, which is noticeably low. A slight selectivity vis‐à‐vis hH2R and hH1,3,4Rs is just seen for 129 (Table 1). All further compounds revealed similar but partially high affinities, especially for the hH2,3,4Rs.
Introducing a terminal urea in the side chain (132–136) comes along with a massive decrease in basicity, but not with a massive loss in affinity. Overall they were in a range with the terminal amines (115–122) and guanidines (123–131), which means slight or no affinity at the hH1R and submicromolar affinities at the hH2,3,4Rs (Table 1). This functionality has no remarkable impact or change with respect to selectivity or affinity at the histamine receptors.
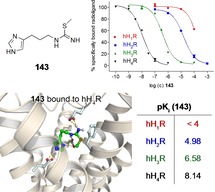
Compounds 141–145 with its nonlipophilic structures presented – as expected – no affinity for the hH1R (Table 1). Values at the hH2R were similar, instead of 145, where the urea analogue of 2 surprisingly demonstrated higher affinity (pKi (145)=6.17; pKi (2)=5.39; cf. Table 1). Scoping at the hH3,4Rs the benzoyl derivatives 141 and 144 illustrated moderate binding values. Even if the affinities of 141 at the hH3,4Rs were higher, 144 tends to be selective towards the hH3R (Table 1). Compound 143 gives an even more pronounced selectivity towards the hH4R (Figure 3). Besides the weak binding data at the hH1,2Rs and a moderate result at the hH3R (pKi (hH3R)=6.58), a pKi of 8.14 showed up a remarkably high tendency for the hH4R (Table 1). In comparison with the binding data of 2, the selectivity profile of the S‐methylated analogue 143 has improved.
Figure 3.
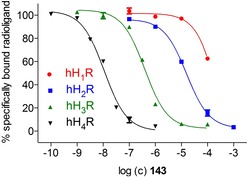
Selectivity profile of 143 with radioligand displacement curves from radioligand binding assays. Experiments were performed with compound 143 and [3H]mepyramine (hH1R, K d 4.5 nM, c=5 nM), [3H]tiotidine (hH2R, K d 19.7 nM, c=10 nM), [3H]Nα‐methylhistamine (hH3R, K d 8.6 nM, c=3 nM) or [3H]histamine (hH4R, K d 16.0 nM, c=15 nM) at membranes of Sf9 cells expressing the respective hHR. Data represent mean values ± SEM from at least two independent experiments, each performed in triplicate.
[35S]GTPγS Binding Data
The functional data of the [35S]GTPγS assay characterize 120 as a partial agonist at the hH2R (pEC50=6.95) and an intrinsic activity of 66 %, relative to histamine (Table 2). At the hH3R an antagonistic activity (pKB=6.72) of 120 could be measured, as well as a negligible weak antagonistic effect at the hH4R (Table 2). The values for 121 were quite similar. The partial agonism at the hH2R is equipotent, but less effective (Emax=0.22) and functionality at the hH3R turns into a partial inverse agonism at one digit micromolar range (Table 2). 120 and 121 are just different in its heterocyclic group (imidazole vs. aminomethylthiazole).
The same structural decision was made for further characterization of terminal guanidines. 127 and 130 both bear a C12‐chain with different hetarylic cycles. Compared to the amines, functional data were similar and in accordance with binding data (Table 2). 130 is a partial agonist at the hH2R (pEC50=7.28, Emax=0.22) and a weak but full inverse agonist at the hH3,4Rs, while 127 acts as a silent antagonist at these receptors (Table 2). The switch from antagonism to inverse agonism at the hH3,4Rs could be assigned to the aminomethylthiazole structure.
The functional experiments of the urea analogue 136 were in line with these data. Partial agonism (pEC50=6.72, Emax=0.45) at the hH2R, as well as a (partial) inverse agonism at the hH3,4Rs could be measured (Table 2). 136 was the only imidazole‐containing compound, that shows up an (partial) inverse agonism at these receptors.
Organ Pharmacological Data
Data from organ bath studies at the guinea pig ileum (gpH1R) and right atrium (gpH2R) provided functional values under physiological conditions. The class of terminal amines (115–122) showed a steady increase in their antagonistic activity at the gpH1R by elongation of the alkyl side chain (pA2 (115–120)=4.78–6.95; cf. Table 3). A further significant increase could be observed by exchange of the heterocycle by aminothiazole (pA2 (122)=8.03; cf. Table 3). Agonistic data at the gpH2R gave potencies in a submicromolar range with high intrinsic activities culminated in 120 as a full agonist (pEC50=6.86, Emax=1.00; cf. Table 3). Heterocyclic exchange with amino(methyl)thiazoles led to a slight decrease in potency and efficacy.
Table 3.
Agonistic (pEC50) and antagonistic (pA2) activities of 1–4, 115–136 and 141–145 determined by organ bath studies at the gpH1R (ileum) and the gpH2R (atrium).[a]
|
Compound |
gpH1R |
gpH2R |
|||
|---|---|---|---|---|---|
|
pA2 [b] (pEC50) |
N |
pEC50 [c],[d] |
E max [e] |
N |
|
|
1 |
(6.68±0.03) |
255 |
6.16±0.01 |
1.00 |
225 |
|
2 |
n.a.f |
24 |
5.16±0.04 |
0.75±0.03 |
5 |
|
3 |
5.88±0.03 |
9 |
8.38±0.05 |
0.78±0.01 |
3 |
|
4 |
4.83±0.05 |
8 |
5.40±0.11 |
0.83±0.03 |
3 |
|
115 |
4.78±0.02 |
8 |
5.33±0.11 |
0.88±0.05 |
3 |
|
116 |
5.42±0.05 |
9 |
5.13±0.05 |
0.68±0.02 |
3 |
|
117 |
5.20±0.07 |
9 |
6.42±0.08 |
0.79±0.01 |
3 |
|
118 |
5.89±0.05 |
9 |
6.51±0.05 |
0.75±0.10 |
3 |
|
119 |
6.17±0.04 |
9 |
6.41±0.05 |
0.78±0.01 |
3 |
|
120 |
6.95±0.06 |
9 |
6.86±0.06 |
1.00±0.06 |
3 |
|
121 |
7.15±0.06 |
8 |
6.49±0.09 |
0.71±0.03 |
3 |
|
122 |
8.03±0.03 |
15 |
6.63±0.07 |
0.90±0.05 |
3 |
|
123 |
5.58±0.02 |
6 |
7.30±0.05 |
1.03±0.03 |
3 |
|
124 |
6.03±0.04 |
6 |
7.67±0.03 |
0.94±0.02 |
3 |
|
125 |
5.79±0.03 |
6 |
7.69±0.02 |
0.83±0.07 |
3 |
|
126 |
6.60±0.04 |
6 |
7.56±0.04 |
0.80±0.02 |
3 |
|
127 |
7.22±0.05 |
6 |
7.25±0.11 |
0.77±0.07 |
3 |
|
128 |
6.67±0.04 |
6 |
6.87±0.04 |
0.72±0.02 |
3 |
|
129 |
7.30±0.05 |
6 |
7.41±0.09 |
1.00±0.01 |
3 |
|
130 |
6.76±0.05 |
6 |
7.02±0.07 |
0.90±0.04 |
3 |
|
131 |
8.06±0.05 |
14 |
6.93±0.09 |
0.85±0.05 |
3 |
|
132 |
5.04±0.04 |
6 |
5.61±0.08 |
0.89±0.03 |
3 |
|
133 |
5.22±0.08 |
6 |
6.54±0.08 |
1.03±0.03 |
3 |
|
134 |
5.13±0.04 |
4 |
6.17±0.09 |
0.96±0.03 |
3 |
|
135 |
5.85±0.04 |
6 |
6.74±0.09 |
0.94±0.05 |
3 |
|
136 |
6.00±0.03 |
6 |
6.53±0.01 |
0.96±0.01 |
3 |
|
141 |
n.a.f |
12 |
4.73±0.05 |
0.43±0.01 |
3 |
|
142 |
n.a.f |
9 |
4.96±0.08 |
0.27±0.02 |
3 |
|
143 |
5.41±0.06 |
6 |
5.13±0.03 |
0.45±0.05 |
3 |
|
144 |
5.02±0.06 |
6 |
4.62±0.05 |
0.15±0.03 |
3 |
|
145 |
<4.5 |
6 |
3.57±0.02 |
0.58±0.02 |
3 |
[a]Data represent mean values ± SEM from at least three independent experiments (N). Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration‐response curves. [b]pA2: −log c(Ant)+log (r–1); r=10ΔpEC50; ΔpEC50 was calculated from pEC50 of histamine and pEC50 of histamine in presence of the respective antagonist; [c]pEC50: −logEC50; [d]pEC50 was calculated from the mean corrected shift ΔpEC50 of the agonist curve relative to the histamine reference curve by equation pEC50=6.16+ΔpEC50; [e] E max: maximal response relative to the maximal increase in heart rate induced by histamine (E max=1.00). [f]n.d.=not determined.
The raise of basicity, with respect to the terminal guanidines (123–131), resulted in slightly higher antagonistic values at the gpH1R compared to the respective amines (e. g. pA2 (127)=7.22; cf. Table 3). In analogy to 122, substitution of imidazole by aminothiazole led to a highly active antagonist at the gpH1R (pA2 (131)=8.06; cf. Table 3). Terminal guanidines (123–131) were developed to more potent and highly efficient agonists at the gpH2R, in comparison to their amine and alkyl analogues (the latter were published by Pockes et al.40). However, it is striking that the alkylic spacer length seems to have no significant influence on the agonistic potency (Table 3). The most potent guanidine 125 (pEC50=7.69, Emax=0.83) and the most efficient guanidine 123 (pEC50=7.30, Emax=1.03) bear a C8‐ and a C4‐spacer, respectively (Table 3).
Organ pharmacological data for the ureas 132–136 were in comparison with the respective amines 115–122 (Table 3). Therefore, 135 and 136 with its lipophilic C8‐ and C10‐spacer, respectively, pointed up highest antagonistic activity at the gpH1R (pA2 (135)=5.85; pA2 (136)=6.00; cf. Table 3). All compounds, instead of 132, exhibited values in a submicromolar range at the gpH2R with remarkable high agonistic efficacy (Emax=0.89‐1.03; cf. Table 3).
Compounds 141–145 boasted only slight or no antagonistic activity at the gpH1R (Table 3). Compared to 2, which reveals a guanidine structure, the less basic thiourea (141, 142) and urea derivatives (144, 145) presented weak partial agonism at the guinea pig right atrium (gpH2R). Isothiourea 143 is equipotent (pEC50=5.13) but less effective (Emax=0.45) referred to SK&F 91486 (2). According to the literature 143 demonstrated a potency comparable to histamine (rel. potency=0.1) at the guinea‐pig right atrium, while the maximum response was higher (0.45 vs. 0.23).35
The maximum responses of the tested compounds (115–136 and 141–145) at the right atrium were completely antagonized after addition of the H2R antagonist cimetidine (pA2=6.1058,59) (30 μM). For compounds 120, 125 (Figure S35, Supporting Information (SI)) and 135 full concentration‐response curves (CRCs) in the presence of cimetidine (30 μM, 30 min preincubation) were determined. The presence of an antagonist resulted in rightward shifted curves. The calculated values via Schild equation (Table S2, SI) were in accordance with the experimental data. This outcome confirms that the increment of the heart frequency in the guinea‐pig right atrium assay was conveyed via the H2R. The most interesting results at the gpH2R were displayed in Figure 4, where CRCs of selected compounds of each group (colored) were depicted together with references (black).
Figure 4.
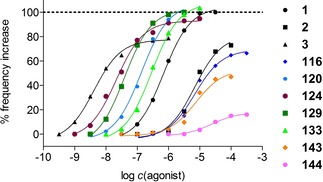
Concentration‐response curves of 1, 2 and 3 (black), as well as 116, 120, 124, 129, 133, 143 and 144 (colored) at the gpH2R (atrium). Histamine (1) was used as a reference (pEC50=6.16, Emax=1.00). Displayed curves are calculated by endpoint determination (N=1).
Computational Studies
143 was “flexibly” docked into the orthosteric binding pocket of both the hH4R and hH3R (cf. Figure 5), two closely related histamine receptor subtypes sharing a high sequence identity.15,60 Of the investigated protonation and/or tautomerization states of the imidazole ring (τ‐H and π‐H, τ‐H, π‐H), docking of 143 resulted in the most reasonable binding poses and in the lowest MM‐GBSA values in case of the protonated (τ‐H and π‐H) form of the imidazole ring. At first, ligand‐receptor interactions of these lowest free energy (MM‐GBSA) binding poses seemed to be highly comparable between both histamine receptor subtypes (cf. Figure 5): The isothiourea moiety and the protonated imidazole ring of 143 formed salt bridges with D943.32, E163ECL2.49 and E1825.46 (hH4R) or D1143.32, E185ECL2.47 and E2065.46 (hH3R). In addition, cation‐π‐interactions were detected between the isothiourea moiety of 143 and F3447.39 (hH4R) or F3987.39 (hH3R). However, by taking a closer look at the differences between binding of 143 to either hH4R or hH3R, it becomes obvious that the location of a certain GLU in the extracellular loop 2 (hH4R: E163ECL2.49, hH3R: E185ECL2.47) is shifted by two amino acids. Therefore, the orientation of this GLU residue seems to slightly differ between both receptor subtypes: Whereas it seems to be still capable of properly forming a salt bridge with the isothiourea moiety of 143 in case of the hH4R, the interactions may be weakened in the case of hH3R. Furthermore, this salt bridge appeared in four of five docking poses in case of the hH4R compared to only one of five in case of the hH3R. Consequently, these molecular differences may, at least in parts, reflect the discrepancies in pKi values of more than one order of magnitude between hH4R and hH3R (hH4R: pKi=8.14, hH3R: pKi=6.58, cf. Table 1) and thus provide a possible molecular explanation.
Figure 5.
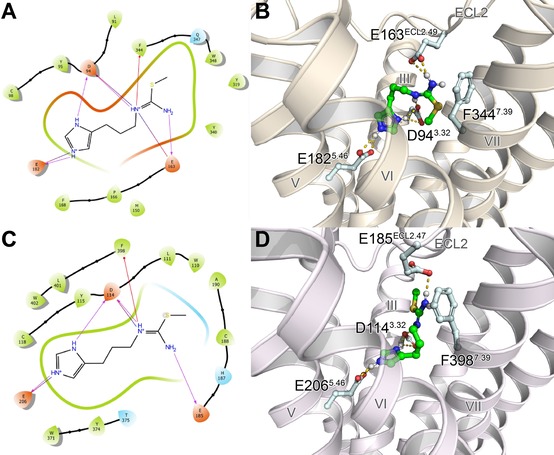
Lowest free energy (MM‐GBSA) docking poses of 143 at both the hH4R (A, B) and hH3R (C, D) showing key ligand‐receptor interactions in the form of ligand interaction diagrams (A, C) or three‐dimensional illustrations (B, D). Hydrogen bonds and salt bridges are colored in magenta (A, C) or yellow (B, D), and cation‐π interactions in red (A, C).
Conclusions
Novel series of alkylated hetarylpropylguanidines with functionalized side chains or new functionality at the guanidine structure were investigated in this project. By introduction of three different functional groups (amine, guanidine, urea) in a terminal position of an alkylic side chain various shades of basicity could be displayed. The respective ligands 115—136 were obtained in a six‐ to nine‐step synthesis in excellent yield, just as for compounds 141–145 (two to three steps). Elongation of the spacer length and, associated therewith, the increase of lipophilicity led to higher affinities and potencies at all four histamine receptors. The most affine and potent derivatives (two digit nanomolar range) could be assigned to guanidines in the terminal position (123–131), in comparison with the appropriate amines (115–122) and ureas (132–136). None of these classes pointed up a distinct selectivity towards any of the four histamine receptors. Although bioisosteric replacement of imidazole by amino(methyl)thiazole led to selectivity towards the H2R, improvement of the selectivity profile could not be determined, in comparison with already described H2‐selective compounds. Heteroatomic exchange at the guanidine group of SK&F 91486 (2) led to benzoylurea derivative 144, with a preference towards the hH3R, and isothiourea 143, with considerable improvement of the selectivity profile towards the hH4R. Thereby, computational studies provided molecular insights into the binding modes of 143 at both hH4R and hH3R and supported the proposal of a possible mechanism of the enhanced selectivity profile. Furthermore, both structures,143 and 144, could be an interesting starting point for future projects facing H3 and H4 receptor selectivity. This is of special interest as to date there are still no drugs available for both receptors (apart from Pitolisant61), although a wide field of applications are reported for the H3R (e. g. several neurodegenerative diseases)21, 22, 23, 24, 25 and the H4R (e. g. inflammation, allergic diseases).26, 27, 28, 29, 30
Experimental Section
General Conditions
Commercially chemicals (8, 11–16, 28 and 138), reagents and solvents were purchased from Acros Organics (Geel, Belgium), Alfa Aesar GmbH & Co. KG (Karlsruhe, Germany), Iris Biotech GmbH (Marktredwitz, Germany), Merck KGaA (Darmstadt, Germany), Sigma‐Aldrich Chemie GmbH (München, Germany) or TCI Europe (Zwijndrecht, Belgium) and were used as received. Deuterated solvents for nuclear magnetic resonance (1H NMR and 13C NMR) spectra were purchased from Deutero GmbH (Kastellaun, Germany). All reactions including dry solvents were carried out in dry flasks under nitrogen or argon atmosphere. For the preparation of buffers, HPLC eluents and stock solutions millipore water was used. Column chromatography was accomplished using Merck silica gel Geduran 60 (0.063–0.200 mm) or Merck silica gel 60 (0.040–0.063 mm) (flash column chromatography). Reactions were monitored by TLC on aluminium sheets with silica gel 60 F254 from Merck. Spots were detected under UV light at 254 nm, by iodine vapor, ninhydrin or fast blue B staining. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were measured on a Bruker (Karlsruhe, Germany) Avance 300 (1H: 300 MHz, 13C: 75 MHz) or Avance 400 (1H: 400 MHz, 13C: 101 MHz) spectrometer using perdeuterated solvents. Chemical shifts (δ) are given in parts per million (ppm). Multiplicities were stated using the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), m (multiplet) and bs (broad signal) and combinations thereof. 13C NMR‐Peaks were measured by DEPT 135 and DEPT 90 (distortionless enhancement by polarization transfer): “+” primary and tertiary carbon atom (positive DEPT 135 signal), “−“ secondary carbon atom (negative DEPT 135 signal), “quat” quaternary carbon atom. NMR spectra were processed with MestReNova 11.0 (Mestrelab Research, Compostela, Spain). High resolution mass spectrometry (HRMS) was performed on an Agilent 6540 UHD Accurate‐Mass Q‐TOF LC/MS system (Agilent Technologies, Santa Clara, CA) using an ESI source. Elemental analyses (EA) were executed on a Heraeus Elementar Vario EL III and are within ±0.4 % unless otherwise noted. Melting points (mp) were detected on a Büchi (Essen, Germany) B‐545 apparatus using an open capillary and are uncorrected. Preparative HPLC was handled with a system from Knauer (Berlin, Germany) consisting of two K‐1800 pumps and a K‐2001 detector. A Eurospher‐100 C18 (250×32 mm, 5 μm) (Knauer, Berlin, Germany) or a Kinetex XB−C18 (250 x 21.2 mm, 5 μm) (Phenomenex, Aschaffenburg, Germany) served as stationary phase. As mobile phase, 0.1 % TFA in millipore water and acetonitrile (MeCN) were used. The temperature was 25 °C, the flow rate 15 mL/min and UV detection was performed at 220 nm. Analytical HPLC was implemented on an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) using a binary pump, autosampler, and DAD detector. Stationary phase was a Kinetex XB−C18 (250 x 4.6 mm, 5 μm) (Phenomenex, Aschaffenburg, Germany). As mobile phase, mixtures of MeCN and aqueous TFA were used (linear gradient: MeCN/TFA (0.1 %) (v/v) 0 min: 5 : 95, 25 min: 50 : 50, 26–35 min: 95 : 5 (method A); flow rate=1.0 mL/min, t0=2.57 min). Capacity factors were calculated pursuant to k=(tR−t0)/t0. Detection was measured at 220 nm. All compounds were examined using method A. Filtration of the stock solutions with PTFE filters (25 mm, 0.2 μm, Phenomenex Ltd., Aschaffenburg, Germany) was carried out before testing. Compound purities determined by HPLC were calculated as the peak area of the analyzed compound in % relative to the total peak area (UV detection at 220 nm). The HPLC purities (see analytical data and Supporting Information) of the final compounds were≥95 %. For all purity runs (see SI) the blank run was subtracted to avoid TFA‐dependent baseline drift. All the tested compounds were screened for PAINS and aggregation by publicly available filters (http://zinc15.docking.org/patterns/home, http://advisor.docking.org).62,63 None of the screened molecules have been previously reported as PAINS or an aggregator. Since Devine et al. described 2‐aminothiazoles as a promiscuous frequent hitting scaffold at different enzymes,64 full dose response curves for all experiments and compounds – not only for the 2‐aminothiazoles – were performed. None of the curves displayed abnormalities, e. g. high Hill slopes, what could be an indication for PAINS.63
Chemical Synthesis and Analytical Data
General Procedure for the Preparation of the Mono‐Boc‐Protected Diamines 17–22
A 0.5 M solution of Boc2O (1 equiv) in DCM was added dropwise over a 2 h period to a 0.25 M solution of diamine 11–16 (5 equiv) in DCM cooled with an ice‐bath. The reaction mixture was stirred over night at room temperature (rt) and filtered. The filtrate was concentrated under vacuum and the resulting oil dissolved in EtOAc was washed with half‐saturated brine (3×150 mL), dried (Na2SO4) and concentrated under vacuum. The crude product was purified by column chromatography (DCM/MeOH/7 M NH3 in MeOH 80/18/2 – 50/48/2 v/v/v).
N‐(tert‐Butoxycarbonyl)‐1,3‐propanediamine (17)49
The reaction was carried out with propane‐1,3‐diamine (11, 3.83 mL, 45.87 mmol), Boc2O (2.0 g, 9.16 mmol) and DCM. The product was obtained as a colorless oil (1.55 g, 97 %): R f=0.40 (DCM/MeOH/NH3 80 : 20:0.1); 1H NMR (300 MHz, CDCl3) δ 5.06 (t, J=5.3 Hz), 3.20 (q, J=6.2 Hz, 2H), 2.86 (bs, 2H), 2.79 (t, J=6.6 Hz, 2H), 1.65 (p, J=6.6 Hz, 2H), 1.43 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 156.23, 79.15, 39.17, 38.19, 32.55, 28.42. HRMS (ESI‐MS): m/z [M+H+] calculated for C8H19N2O2 +: 175.1441, found 175.1445; C8H18N2O2 (174.24).
General Procedure for the Preparation of the N‐Aminoalkyl‐N’,N’’‐di‐Boc‐Protected Guanidines 23–27
A solution of 10 (1 equiv) in DCM (50 mL) was added dropwise to a solution of the respective diamine (12‐16, 3 equiv) in DCM (50 mL) at rt. The resulting mixture was stirred over night and washed with H2O (3x25 mL) and brine (30 mL). The organic solvent was dried over Na2SO4 and the crude product was purified with column chromatography (DCM/MeOH/7 M NH3 in MeOH 95/3/2 – 90/8/2 v/v/v).
1‐(4‐Aminobutyl)‐2,3‐(di‐tert‐butoxycarbonyl)guanidine (23)50
The synthesis was accomplished with 12 (1.37 g, 15.51 mmol) and 10 (1.50 g, 5.17 mmol) according to the general procedure. Column chromatography gave 23 as a yellow oil (1.18 g, 69 %): R f=0.18 (DCM/MeOH/NH3 95 : 5:0.1); 1H NMR (300 MHz, CDCl3) δ 11.47 (bs, 1H), 8.33 (bs, 1H), 3.39 (q, J=7.3 Hz, 2H), 2.71 (t, J=6.8 Hz, 2H), 1.86 (bs, 2H), 1.66 – 1.50 (m, 4H), 1.48 (s, 9H), 1.47 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 163.55, 156.16, 153.30, 83.13, 79.32, 41.60, 40.63, 30.56, 28.29, 28.07, 26.36. HRMS (ESI‐MS): m/z [M+H+] calculated for C15H31N4O4 +: 331.2340, found 331.2346; C15H30N4O4 (330.43).
General Procedure for the Preparation of the Benzoylthioureas 29–39
To an ice‐cold solution of the pertinent amine (11–27, 1 equiv) in DCM, benzoyl isothiocyanate (28, 1 equiv) was added dropwise. The reaction was allowed to stir at room temperature (rt) for 2 h. The reaction mixture was washed three times with H2O and saturated solution of NaCl (each 30 mL). The organic layer was dried over Na2SO4 and the crude product was purified with column chromatography (DCM/MeOH 100/0 – 98/2 v/v).
tert‐Butyl [3‐(3‐benzoylthioureido)propyl]carbamate (29)55
The product was developed using 17 (1.55 g, 8.90 mmol) and 28 (1.20 mL, 8.90 mmol) in DCM (30 mL) and the desired compound was isolated as a yellow oil (2.91 g, 97 %): R f=0.15 (DCM); 1H NMR (300 MHz, CDCl3) δ 10.83 (bs, 1H), 9.12 (bs, 1H), 7.91–7.74 (m, 2H), 7.56–7.32 (m, 3H), 5.05 (bs, 1H), 3.77 (q, J=6.5 Hz, 2H), 3.21 (q, J=7.4 Hz, 2H), 1.86 (p, J=6.6 Hz, 2H), 1.42 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 180.25, 166.91, 156.20, 133.56, 131.74, 129.10, 127.51, 79.50, 42.95, 37.06, 30.20, 28.41. HRMS (ESI‐MS): m/z [M+Na+] calculated for C16H23N3NaO3S+: 360.1352, found 360.1355; C16H23N3O3S (337.44).
The synthesis of 40–44 is described in the literature (cf. 17–21)40 and was carried out with the appropriate diamine 11–15 (1 equiv) and 28 (2 equiv).
General Procedure for the Preparation of the Thioureas 45–55
The general procedure for the synthesis of the thioureas is described in the literature (cf. 4.2.9.)40. The NMR peak splitting due to thione‐thiol tautomerism – described in the reference – also appears for the compounds 45–55.
tert‐Butyl (3‐thioureidopropyl)carbamate (45)65
45 was made out of 29 (2.90 g, 8.59 mmol) and K2CO3 (2.49 g, 18.04 mmol) in 50 ml MeOH/H2O (7/3 v/v) yielding a yellow oil (1.90 g, 95 %): R f=0.29 (DCM/MeOH 95 : 5); 1H NMR (300 MHz, CDCl3) δ 7.42 (bs, 1H), 6.39 (bs, 1H), 5.24 (bs, 1H), 3.56+3.25 (2 bs, 1.4H+0.6H (thione‐thiol tautomerism)), 3.13 (q, J=6.4 Hz, 2H), 1.80 – 1.61 (m, 2H), 1.39 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 183.31, 156.80, 79.72, 41.87, 37.38, 29.73, 28.43. HRMS (ESI‐MS): m/z [M+H+] calculated for C9H20N3O2S+: 234.1271, found 234.1271; C9H19N3O2S (233.33).
The synthesis of 56–60 is described in the literature (cf. 23–27)40 and was carried out with the appropriate dibenzoylthiourea 40–44 (1 equiv) and K2CO3 (4.1 equiv).
General Procedure for the Preparation of the S‐methylisothioureas 61–71
The general procedure for the synthesis of the S‐methylisothioureas is described in the literature (cf. 4.2.10.).40
tert‐Butyl {3‐[(imino(methylthio)methyl)amino]propyl}carbamate (61)66
Compound 45 (1.80 g, 7.71 mmol) was dissolved in MeCN (30 mL) and treated with methyl iodide (0.53 mL, 8.49 mmol) resulting a yellow oil (61 x HI, 2.80 g, 97 %): R f=0.14 (DCM/MeOH 95 : 5); 1H NMR (300 MHz, CDCl3, hydrogen iodide) δ 3.76–3.32 (m, 2H), 3.21 (q, J=6.3 Hz, 2H), 2.76 (s, 3H), 1.88 (p, J=6.6 Hz, 2H), 1.39 (s, 9H). 13C NMR (75 MHz, CDCl3, hydrogen iodide) δ 170.17, 154.28, 78.25, 40.17, 35.13, 26.66, 26.37, 13.41. HRMS (ESI‐MS): m/z [M+H+] calculated for C10H22N3O2S+: 248.1427, found 248.1429; C10H21N3O2S x HI (375.27).
The synthesis of 72–76 is described in the literature (cf. 29–33)40 and was carried out with the appropriate bisthiourea 56–60 (1 equiv) and methyl iodide (2.1 equiv).
General Procedure for the Preparation of the N’‐Boc‐S‐methylisothioureas 77–87
The general procedure for the synthesis of the N’‐Boc‐S‐methylisothioureas is described in the literature (cf. 4.2.11.)40.
tert‐Butyl {3‐[(((tert‐butoxycarbonyl)imino)(methylthio)methyl)amino]propyl}carbamate (77)
The reaction was realized with 61 (2.70 g, 7.19 mmol), NEt3 (1.00 mL, 7.19 mmol) and Boc2O (1.57 g, 7.19 mmol). After column chromatography a colorless oil (2.30 g, 92 %) was obtained: R f=0.41 (DCM/MeOH 98 : 2); 1H NMR (300 MHz, CDCl3) δ 9.52 (bs, 1H), 4.61 (bs, 1H), 3.31 (t, J=7.1 Hz, 2H), 3.12 (q, J=7.2 Hz, 2H), 2.39 (s, 3H), 1.73 (p, J=6.7 Hz), 1.43 (s, 9H), 1.37 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 173.55, 162.07, 156.11, 79.34, 79.20, 41.13, 37.75, 29.96, 28.37, 28.21, 13.65. HRMS (ESI‐MS): m/z [M+H+] calculated for C15H30N3O4S+: 348.1952, found 348.1952; C15H29N3O4S (347.47).
The synthesis of 88–92 is described in the literature (cf. 35–39)40 and was carried out with the appropriate isothiourea 72–76 (1 equiv), NEt3 (2 equiv) and Boc2O (2 equiv).
General Procedure for the Guanidinylation reaction of 93–109
To a suspension of the pertinent amine 5, 6, or 7 (1 equiv), the pertinent N’‐Boc‐S‐methylisothiourea 77–87 (1 equiv) and HgCl2 (1 equiv) in DCM, NEt3 (3 equiv) was added. The mixture was stirred overnight at rt. A possible excess of HgCl2 was quenched with 7 N NH3 in MeOH (3‐5 mL). The resulting suspension was filtered over Celite and the crude product was purified with column chromatography (DCM/MeOH/7N NH3 in MeOH 98/1/1 – 95/3/2 v/v/v).
2‐tert‐Butoxycarbonyl‐1‐(N‐tert‐butoxycarbonylaminopropanyl)‐3‐[3‐(1‐trityl‐1H‐imidazol‐4‐yl)propyl]guanidine (93)
Compound 93 was prepared from 5 (500 mg, 1.36 mmol), 77 (473 mg, 1.36 mmol), HgCl2 (369 mg, 1.36 mmol) and NEt3 (0.57 mL, 4.08 mmol) in DCM (20 mL) conforming to the general procedure yielding a yellow foamlike solid (420 mg, 46 %): R f=0.30 (DCM/MeOH/NH3 98 : 2:0.1); 1H NMR (300 MHz, CDCl3) δ 9.05 (bs, 1H) 7.42–7.27 (m, 10H), 7.15–7.07 (m, 6H), 6.56 (d, J=0.8 Hz, 1H), 5.91 (bs, 1H), 3.55–3.16 (m, 4H), 3.09 (q, J=5.6 Hz, 2H), 2.56 (t, J=6.3 Hz, 2H), 1.87 (p, J=6.7 Hz, 2H), 1.67–1.48 (m, 2H), 1.44 (s, 9H), 1.41 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 164.20, 160.89, 156.55, 142.34, 140.46, 138.02, 129.72, 128.12, 128.10, 118.31, 78.74, 75.26, 75.24, 40.23, 40.20, 36.93, 33.15, 30.58, 29.16, 28.57, 28.51. HRMS (ESI‐MS): m/z [M+H+] calculated for C39H51N6O4 +: 667.3966, found 667.3970; C39H50N6O4 (666.87).
General Procedure for the Guanidinylation Reaction of 110–114
To a suspension of the amine 5 (2 equiv), the pertinent N’‐Boc‐S‐methylisothiourea 88–92 (1 equiv) and HgCl2 (4 equiv) in DCM, NEt3 (6 equiv) was added. The mixture was stirred overnight at rt. A possible excess of HgCl2 was quenched with 7 N NH3 in MeOH (3–5 mL). The resulting suspension was filtered over Celite and the crude product was purified with column chromatography (DCM/MeOH/7 N NH3 in MeOH 98/1/1 – 95/3/2 v/v/v).
2‐tert‐Butoxycarbonyl‐1‐(N′‐tert‐butoxycarbonylcarbodiimidopropyl)‐3‐[3‐(1‐trityl‐1H‐imidazol‐4‐yl)propyl]guanidine (110)
Compound 110 was prepared from 5 (1.0 g, 2.72 mmol), 88 (572 mg, 1.36 mmol), HgCl2 (1.48 g, 5.44 mmol) and NEt3 (1.13 mL, 8.16 mmol) in DCM (20 mL) conforming to the general procedure yielding a yellow oil (430 mg, 46 %): R f=0.44 (DCM/MeOH/NH3 98 : 2 : 0.1); 1H NMR (300 MHz, CDCl3) δ 9.75 (bs, 1H), 7.34–7.21 (m, 10H), 7.14–7.03 (m, 6H), 6.51 (s, 1H), 3.52–3.08 (m, 6H), 2.68–2.46 (m, 2H), 1.99–1.71 (m, 4H), 1.41 (s, 9H), 1.39 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 164.45, 160.71, 158.01, 156.00, 142.52, 140.58, 138.41, 129.73, 128.00, 127.94, 117.91, 85.50, 78.63, 75.02, 53.52, 45.56, 43.58, 28.54, 28.26, 27.93, 25.78, 21.06. MS (LC–MS, ESI): m/z 692.39 [M+H+]; C40H49N7O4 (691.88).
General Procedure for the Preparation of the Title Compounds 115–136
The general procedure for the synthesis of 115–136 is described in the literature (cf. 4.2.7.).40 All compounds were obtained as tri‐trifluoroacetates.
1‐(3‐Aminopropyl)‐3‐[3‐(1H‐imidazol‐4‐yl)propyl]guanidine (115)
The title compound was prepared from 93 (420 mg, 0.63 mmol), TFA (4 mL) and DCM (16 mL) according to the general procedure, yielding a yellow oil (300 mg, 84 %): RP‐HPLC: 100 %, (tR=5.94, k=1.31). 1H NMR (300 MHz, CD3OD, tri‐trifluoroacetate) δ 8.81 (d, J=1.3 Hz, 1H), 7.35 (s, 1H), 3.30–3.23 (m, 4H), 3.01 (t, J=7.4 Hz, 2H), 2.81 (t, J=7.3 Hz, 2H), 1.96 (m, 4H). 13C NMR (75 MHz, CD3OD, tri‐trifluoroacetate) δ 157.66, 134.92, 134.54, 116.99, 41.68, 39.61, 38.13, 28.75, 28.02, 22.55. HRMS (ESI‐MS): m/z [M+H+] calculated for C10H21N6 +: 225.1822, found 225.1822; C10H20N6 x 3 TFA. (566.38).
Synthesis of the SK&F 91486 analogues 141–145
N‐{[3‐(1‐Trityl‐1H‐imidazol‐4‐yl)propyl]thiocarbamoyl}benzamide (137)
Compound 137 was prepared according to the general procedure described in 4.2.5. using 5 (4.80 g, 13.06 mmol) and 28 (1.76 mL, 13.06 mmol) in 100 mL DCM. After column chromatography (EtOAc/PE 1/2 – 1/1 v/v) the product was obtained as a yellow solid (4.60 g, 66 %): R f=0.55 (EtOAc/Hex 1 : 1); mp 139.4 °C. 1H NMR (300 MHz, CDCl3) δ 10.76 (bs, 1H), 9.09 (bs, 1H), 7.85–7.74 (m, 2H), 7.63–7.54 (m, 1H), 7.52–7.42 (m, 2H), 7.37 (d, J=1.3 Hz, 1H), 7.39–7.25 (m, 9H), 7.20–7.05 (m, 6H), 6.60 (s, 1H), 3.72 (q, J=6.9 Hz, 2H), 2.66 (t, J=7.3 Hz, 2H), 2.05 (p, J=7.3 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 179.69, 166.76, 142.53, 140.22, 138.62, 133.46, 131.89, 129.80, 129.08, 128.05, 127.99, 127.49, 118.26, 75.14, 45.35, 27.72, 25.73. HRMS (ESI‐MS): m/z [M+H+] calculated for C33H31N4OS+: 531.2213, found 531.2218; C33H30N4OS (530.69).
1‐[3‐(1‐Trityl‐1H‐imidazol‐4‐yl)propyl]thiourea (140)
Compound 140 was prepared according to the general procedure described in 4.2.6. using 137 (1.50 g, 2.83 mmol) and K2CO3 (781 mg, 5.65 mmol) in 30 mL MeOH/H2O (7/3 v/v). The product was obtained as a beige solid (920 mg, 76 %): R f=0.20 (DCM/MeOH 95 : 5); mp 196.1 °C. 1H NMR (400 MHz, CD3OD) δ 7.45–7.28 (m, 10H), 7.22–7.08 (m, 6H), 6.71 (s, 1H), 3.48+3.13 (2 bs, 1.2H+0.8H, (thione‐thiol tautomerism)), 2.56 (t, J=7.3 Hz, 2H), 1.84 (p, J=7.3 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 179.77, 143.76, 141.40, 139.35, 130.88, 129.28, 129.24, 119.92, 76.77, 45.09, 29.93, 26.03. HRMS (ESI‐MS): m/z [M+H+] calculated for C26H27N4S+: 427.1951, found 427.1955; C26H26N4S (426.58).
N‐{[3‐(1H‐Imidazol‐4‐yl)propyl]thiocarbamoyl}benzamide (141)
The title compound was prepared from 137 (1.0 g, 1.88 mmol), TFA (4 mL) and DCM (16 mL) according to the general procedure (cf. 4.2.11). The crude product was purified by column chromatography (DCM/MeOH/7 M NH3 in MeOH 95/3/2 v/v/v) yielding 141 as free base and yellow solid (350 mg, 64 %): R f=0.12 (DCM/MeOH 95 : 5); mp 139.8 °C. 1H NMR (300 MHz, CD3OD) δ 8.01–7.84 (m, 2H), 7.71–7.62 (m, 1H), 7.61 (s, 1H), 7.57–7.46 (m, 2H), 6.88 (s, 1H), 3.72 (t, J=7.0 Hz, 2H), 2.70 (t, J=7.5 Hz, 2H), 2.04 (p, J=7.3 Hz, 2H). 13C NMR (75 MHz, CD3OD) δ 182.06, 169.56, 137.51, 135.97, 134.24, 133.95, 129.88, 129.19, 117.78, 45.70, 29.01, 25.07. HRMS (ESI‐MS): m/z [M+H+] calculated for C14H17N4OS+: 289.1118, found 289.1120; C14H16N4OS (288.37); Anal. calculated for C14H16N4OS: C 58.31, H 5.59, N 19.43, found: C 58.32, H 5.66, N 19.16.
N‐[3‐(1H‐Imidazol‐4‐yl)propyl]‐S‐methylisothiourea (143)
To an ice‐cold suspension of 140 (500 mg, 1.17 mmol) in EtOH (20 mL) an aqueous solution of HI (66 %, 5 mL) was added dropwise. The resulted yellow precipitate (142 x HI) was filtrated and washed with Et2O. Subsequently, 142 x HI was dissolved in MeOH (5 mL), treated with methyl iodide (0.08 mL, 1.29 mmol) and refluxed for 1 h. After evaporation the title compound was obtained by recrystallization in isopropanol/Et2O to give 143×2 HI as a yellow‐brown solid (300 mg, 56 %): R f=0.10 (DCM/MeOH/NH3 90 : 10:0.1); mp 104.9 °C (2 HI). 1H NMR (300 MHz, CD3OD, di‐hydrogen iodide) δ 8.85 (d, J=1.4 Hz, 1H), 7.43 (d, J=1.4 Hz, 1H), 3.46 (t, J=7.2 Hz, 2H), 2.84 (t, J=7.7 Hz, 2H), 2.66 (s, 3H), 2.02 (p, J=7.5 Hz, 2H). 13C NMR (75 MHz, CD3OD, di‐hydrogen iodide) δ 170.42, 135.07, 134.21, 117.59, 44.74, 27.96, 22.99, 15.40. HRMS (ESI‐MS): m/z [M+H+] calculated for C8H15N4S+: 199.1012, found 199.1012; C8H14N4S x 2 HI (454.11); Anal. calculated for C7H12N4S×2 HI: C 21.16, H 3.55, N 12.34, found: C 21.23, H 3.87, N 12.08.
1‐[3‐(1H‐Imidazol‐4‐yl)propyl]urea (145)
A suspension of 144 (120 mg, 0.44 mmol) in an aqueous solution of NaOH (1 M, 10 mL) was refluxed for 1 h. After cooling of the clear solution (!) a colorless solid precipitated. After filtration the title compound was washed with Et2O to give 145 (50 mg, 67 %): R f=0.12 (DCM/MeOH/NH3 90 : 10); mp 128.0 °C. 1H NMR (400 MHz, DMSO‐d6) δ 7.61 (s, 1H), 6.77 (s, 1H), 6.00 (t, J=5.9 Hz, 1H), 5.40 (s, 2H), 2.97 (q, J=6.5 Hz, 2H), 2.47 (t, J=7.8 Hz, 2H), 1.63 (p, J=7.2 Hz, 2H). 13C NMR (101 MHz, DMSO‐d6) δ 159.25, 135.91, 134.79, 117.58, 39.19, 30.38, 23.94. HRMS (ESI‐MS): m/z [M+H+] calculated for C7H13N4O+: 169.1084, found 169.1088; C7H12N4O (168.20); Anal. calculated for C7H12N4O2 x 0.24 DMSO (sample was a recovery of a NMR sample solved in DMSO): C 48.06, H 7.25, N 29.97, found: C 48.00, H 7.11, N 30.11.
Pharmacological Methods and Materials
Materials
Histamine dihydrochloride was acquired from Alfa Aesar GmbH & Co. KG (Karlsruhe, Germany). [3H]mepyramine (specific activity: 20.0 Ci/mmol), [3H]tiotidine (specific activity: 78.4 Ci/mmol), [3H]Nα‐methylhistamine (specific activity: 85.3 Ci/mmol) and [3H]histamine (specific activity: 25.0 Ci/mmol) were purchased from Hartmann analytic (Braunschweig, Germany). GTPγS was from Roche (Mannheim, Germany), and [35S]GTPγS was bought from PerkinElmer Life Science (Boston, USA) or Hartmann Analytic (Braunschweig, Germany). [3H]UR‐DE257 was synthesized in our laboratories. All stock solutions were dissolved in millipore water or in a mixture of Millipore water/DMSO. In all assays, the final DMSO content included less than 0.5 %.
Methods
All the pharmacological methods used in this study (Membrane Preparation of Sf9 Cells, Radioligand Binding Assay, [35S]GTPγS Binding Assay, Histamine H1 Receptor Assay on Isolated Guinea Pig Ileum, Histamine H2 Receptor Assay on the Isolated Guinea Pig Right Atrium) were already described in the literature.34
Computational Methods
Homology modelling of both hH4R and hH3R, based on the crystal structure of the inactive state hH1R (PDB ID: 3RZE)67 is described by Pockes et al.40 Protein and ligand preparation as well as the assignment of protonation states (Schrödinger LLC, Portland, OR USA) were essentially performed as described in Pegoli et al.68 Disulfide bonds were maintained between C873.25 and C164ECL2 (hH4R) and C1073.25 and C188ECL2 (hH3R). While the isothiourea moiety of 143 was protonated, the imidazole ring was considered in both deprotonated (τ‐H or π‐H) and protonated (τ‐H and π‐H) form, resulting in a net charge of +1 or +2, respectively. „Flexible“ docking of 143 to both the hH4R and hH3R was performed using the induced fit docking module (Schrödinger LLC). 143 was docked within a box of 46×46×46 Å3 around the center of mass of the residues D943.32, E1825.46, Q3477.42 (hH4R) or D1143.32, E2065.46 and L4017.42 (hH3R). Redocking was performed in the extended precision mode. Furthermore, the resulting poses were scored using MM‐GBSA (Schrödinger LLC). Among the reasonable ligand binding poses, the pose corresponding to the lowest MM‐GBSA value, was selected as the most probable pose. Figures showing molecular structures of the hH4R or hH3R in complex with 143 were generated with PyMOL Molecular Graphics system, version 2.2.0 (Schrödinger LLC), and the corresponding ligand interaction diagrams were prepared with Maestro (Schrödinger LLC).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Christine Braun, Kerstin Röhrl and Maria Beer‐Krön for expert technical assistance, as well as the Leibnitz Rechenzentrum (LRZ) in Munich for providing software (Schrödinger Suite) and computing resources.
S. Pockes, D. Wifling, A. Buschauer, S. Elz, ChemistryOpen 2019, 8, 285.
This Paper is dedicated to the memory of Prof. Dr. Armin Buschauer (died on July 18, 2017).
References
- 1. Windaus A., Vogt W., Ber. Dtsch. Chem. Ges. 1907, 40, 3685–3691. [Google Scholar]
- 2. Ash A. S. F., Schild H. O., Br. J. Pharmacol. Chemother. 1966, 27, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Black J. W., Duncan W. A. M., Durant C. J., Ganellin C. R., Parsons E. M., Nature 1972, 236, 385–390. [DOI] [PubMed] [Google Scholar]
- 4. Arrang J.-M., Garbarg M., Schwartz J.-C., Nature 1983, 302, 832–837. [DOI] [PubMed] [Google Scholar]
- 5. Oda T., Morikawa N., Saito Y., Masuho Y., Matsumoto S., J. Biol. Chem. 2000, 275, 36781–36786. [DOI] [PubMed] [Google Scholar]
- 6. Lagerström M. C., Schiöth H. B., Nat. Rev. Drug Discovery 2008, 7, 339–357. [DOI] [PubMed] [Google Scholar]
- 7. Hill S. J., Pharmacol. Rev. 1990, 42, 45–83. [PubMed] [Google Scholar]
- 8. Leurs R., Smit M. J., Timmerman H., Pharmacol. Ther. 1995, 66, 413–463. [DOI] [PubMed] [Google Scholar]
- 9. Hill S. J., Ganellin C. R., Timmerman H., Schwartz J. C., Shankley N. P., Young J. M., Schunack W., Levi R., Haas H. L., Pharmacol. Rev. 1997, 49, 253–278. [PubMed] [Google Scholar]
- 10. Soll A. H., Wollin A., Am. J. Physiol. Gastrointest. Liver Physiol. 1979, 237, G444–G450. [Google Scholar]
- 11. Johnson C. L., Weinstein H., Green J. P., Mol. Pharmacol. 1979, 16, 417–428. [PubMed] [Google Scholar]
- 12. Fischer J., Ganellin C. R., Ganesan A., Proudfoot J., Analogue-Based Drug Discovery, Wiley-VCH, 2010. [Google Scholar]
- 13. Goot H. Van Der, Timmerman H., Eur. J. Med. Chem. 2000, 35, 5–20. [DOI] [PubMed] [Google Scholar]
- 14. Rouleau A., Ligneau X., Tardivel-Lacombe J., Morisset S., Gbahou F., Schwartz J.-C., Arrang J.-M., Br. J. Pharmacol. 2002, 135, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leurs R., Chazot P. L., Shenton F. C., Lim H. D., Esch I. J. De, Br. J. Pharmacol. 2009, 157, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morse K. L., Behan J., Laz T. M., West R. E., Greenfeder S. A., Anthes J. C., Umland S., Wan Y., Hipkin R. W., Gonsiorek W., J. Pharmacol. Exp. Ther. 2001, 296, 1058–1066. [PubMed] [Google Scholar]
- 17. Lovenberg T. W., Roland B. L., Wilson S. J., Jiang X., Pyati J., Huvar A., Jackson M. R., Erlander M. G., Mol. Pharmacol. 1999, 55, 1101–1107. [PubMed] [Google Scholar]
- 18. Zhu Y., Michalovich D., Wu H.-L., Tan K. B., Dytko G. M., Mannan I. J., Boyce R., Alston J., Tierney L. A., Li X., Mol. Pharmacol. 2001, 59, 434–441. [DOI] [PubMed] [Google Scholar]
- 19. Liu C., Ma X.-J., Jiang X., Wilson S. J., Hofstra C. L., Blevitt J., Pyati J., Li X., Chai W., Carruthers N., Mol. Pharmacol. 2001, 59, 420–426. [DOI] [PubMed] [Google Scholar]
- 20. Hofstra C. L., Desai P. J., Thurmond R. L., Fung-Leung W.-P., J. Pharmacol. Exp. Ther. 2003, 305, 1212–1221. [DOI] [PubMed] [Google Scholar]
- 21. Gemkow M. J., Davenport A. J., Harich S., Ellenbroek B. A., Cesura A., Hallett D., Drug Discovery Today 2009, 14, 509–515. [DOI] [PubMed] [Google Scholar]
- 22. Sadek B., Khan N., Darras F. H., Pockes S., Decker M., Physiol. Behav. 2016, 165, 383–391. [DOI] [PubMed] [Google Scholar]
- 23. Sadek B., Saad A., Sadeq A., Jalal F., Stark H., Behav. Brain Res. 2016, 312, 415–430. [DOI] [PubMed] [Google Scholar]
- 24. Rapanelli M., Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 73, 36–40. [DOI] [PubMed] [Google Scholar]
- 25. Rapanelli M., Frick L., Pogorelov V., Ohtsu H., Bito H., Pittenger C., Transl. Psychiatry 2017, 7, e1013–e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunford P. J., O'Donnell N., Riley J. P., Williams K. N., Karlsson L., Thurmond R. L., J. Immunol. 2006, 176, 7062–7070. [DOI] [PubMed] [Google Scholar]
- 27. Thurmond R. L., Gelfand E. W., Dunford P. J., Nat. Rev. Drug Discovery 2008, 7, 41–53. [DOI] [PubMed] [Google Scholar]
- 28. Leurs R., Vischer H. F., Wijtmans M., de Esch I. J., Trends Pharmacol. Sci. 2011, 32, 250–257. [DOI] [PubMed] [Google Scholar]
- 29. Jablonowski J. A., Grice C. A., Chai W., Dvorak C. A., Venable J. D., Kwok A. K., Ly K. S., Wei J., Baker S. M., Desai P. J., J. Med. Chem. 2003, 46, 3957–3960. [DOI] [PubMed] [Google Scholar]
- 30. Zampeli E., Tiligada E., Br. J. Pharmacol. 2009, 157, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Staub A. M., Bovet D., Compt. Rend. Soc. de biol 1937, 125, 818. [Google Scholar]
- 32. Halpern B. N., Arch. Int. Pharmacodyn. Ther. 1942, 68, 339–343. [Google Scholar]
- 33. Marshall P. B., Br. J. Pharmacol. 1955, 10, 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durant G. J., Duncan W. A. M., Ganellin C. R., Parsons M. E., Blakemore R. C., Rasmussen A. C., Nature 1978, 276, 403–405. [DOI] [PubMed] [Google Scholar]
- 35. Durant G. J., Ganellin C. R., Hills D. W., Miles P. D., Parsons M. E., Pepper E. S., White G. R., J. Med. Chem. 1985, 28, 1414–1422. [DOI] [PubMed] [Google Scholar]
- 36. Buschauer A., J. Med. Chem. 1989, 32, 1963–1930. [DOI] [PubMed] [Google Scholar]
- 37. Parsons M. E., Blakemore R. C., Durant G. J., Ganellin C. R., Rasmussen A. C., Inflamm. Res. 1975, 5, 464–464. [DOI] [PubMed] [Google Scholar]
- 38. Lim H. D., Rijn R. M. van, Ling P., Bakker R. A., Thurmond R. L., Leurs R., J. Pharmacol. Exp. Ther. 2005, 314, 1310–1321. [DOI] [PubMed] [Google Scholar]
- 39. Eriks J. C., Goot H. van der, Sterk G. J., Timmerman H., J. Med. Chem. 1992, 35, 3239–3246. [DOI] [PubMed] [Google Scholar]
- 40. Pockes S., Wifling D., Keller M., Buschauer A., Elz S., ACS Omega 2018, 3, 2865–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kraus A., Ghorai P., Birnkammer T., Schnell D., Elz S., Seifert R., Dove S., Bernhardt G., Buschauer A., ChemMedChem 2009, 4, 232–240. [DOI] [PubMed] [Google Scholar]
- 42. Birnkammer T., Spickenreither A., Brunskole I., Lopuch M., Kagermeier N., Bernhardt G., Dove S., Seifert R., Elz S., Buschauer A., J. Med. Chem. 2012, 55, 1147–1160. [DOI] [PubMed] [Google Scholar]
- 43. Kagermeier N., Werner K., Keller M., Baumeister P., Bernhardt G., Seifert R., Buschauer A., Bioorg. Med. Chem. 2015, 23, 3957–3969. [DOI] [PubMed] [Google Scholar]
- 44. Yang R., Hey J. A., Aslanian R., Rizzo C. A., Pharmacology 2002, 66, 128–135. [DOI] [PubMed] [Google Scholar]
- 45. Igel P., Geyer R., Strasser A., Dove S., Seifert R., Buschauer A., J. Med. Chem. 2009, 52, 6297–6313. [DOI] [PubMed] [Google Scholar]
- 46. Igel P., Schneider E., Schnell D., Elz S., Seifert R., Buschauer A., J. Med. Chem. 2009, 52, 2623–2627. [DOI] [PubMed] [Google Scholar]
- 47. Ghorai P., Kraus A., Keller M., Götte C., Igel P., Schneider E., Schnell D., Bernhardt G., Dove S., Zabel M., J. Med. Chem. 2008, 51, 7193–7204. [DOI] [PubMed] [Google Scholar]
- 48. Mitsunobu O., Yamada M., Mukaiyama T., Bull. Chem. Soc. Jpn. 1967, 40, 935–939. [Google Scholar]
- 49. Dardonville C., Fernandez-Fernandez C., Gibbons S.-L., Ryan G. J., Jagerovic N., Gabilondo A. M., Meana J. J., Callado L. F., Bioorg. Med. Chem. 2006, 14, 6570–6580. [DOI] [PubMed] [Google Scholar]
- 50. Hickey S. M., Ashton T. D., Khosa S. K., Pfeffer F. M., Synlett 2012, 23, 1779–1782. [Google Scholar]
- 51. Pluym N., Brennauer A., Keller M., Ziemek R., Pop N., Bernhardt G., Buschauer A., ChemMedChem 2011, 6, 1727–1738. [DOI] [PubMed] [Google Scholar]
- 52. Buschauer A., J. Med. Chem. 1989, 32, 1963–1970. [DOI] [PubMed] [Google Scholar]
- 53.J. W. Black, G. J. Durant, J. C. Emmett, C. R. Ganellin, Thioureas, 1973, GB1305548A.
- 54.J. W. Black, G. J. Durant, J. C. Emmett, C. R. Ganellin, Isothioureas, 1973, GB1305547A.
- 55.D. B. Frennesson, D. R. Langley, M. G. Saulnier, D. M. Vyas, Methods for Preparing Macrocycles and Macrocycle Stabilized Peptides, 2012, WO2012051405A1.
- 56.W. Carling, K. Moore, Imidazolone and Oxazolone Derivatives as Dopamine Antagonists, European Patent Office, Rijswijk, 1995, WO 95/07904.
- 57. Kim K. S., Qian L., Int. J. Rapid Publ. Prelim. 1993, 34, 7677–7680. [Google Scholar]
- 58. Brimblecombe R. W., Duncan W. A. M., Durant G. J., Emmett J. C., Ganellin C. R., Parsons M. E., J. Int. Med. Res. 1975, 3, 86–92. [Google Scholar]
- 59. Brimblecombe R. W., Duncan W. A., Durant G. J., Ganellin C. R., Parsons M. E., Black J. W., Br. J. Pharmacol. 1975, 53, 435–436. [PMC free article] [PubMed] [Google Scholar]
- 60. Hough L. B., Mol. Pharmacol. 2001, 59, 415–419. [PubMed] [Google Scholar]
- 61. Ganellin C. Robin, Schwartz J. C., Stark H., Successful Drug Discovery 2018, 3, 359–381. [Google Scholar]
- 62. Baell J. B., Holloway G. A., J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- 63. Aldrich C., Bertozzi C., Georg G. I., Kiessling L., Lindsley C., Liotta D., Merz K. M., Schepartz A., Wang S., ACS Cent. Sci. 2017, 3, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Devine S. M., Mulcair M. D., Debono C. O., Leung E. W. W., Nissink J. W. M., Lim S. S., Chandrashekaran I. R., Vazirani M., Mohanty B., Simpson J. S., Baell J. B., Scammells P. J., Norton R. S., Scanlon M. J., J. Med. Chem. 2015, 58, 1205–1214. [DOI] [PubMed] [Google Scholar]
- 65. Nettekoven M., Guba W., Neidhart W., Mattei P., Pflieger P., Roche O., Taylor S., Bioorg. Med. Chem. Lett. 2005, 15, 3446–3449. [DOI] [PubMed] [Google Scholar]
- 66.V. S. Aulakh, A. Casarez, X. Lin, M. Lindvall, G. McEnroe, H. E. Moser, F. Reck, M. Tjandra, R. L. Simmons, A. Yifru, Preparation of Monobactam Organic Compounds for the Treatment of Bacterial Infections, 2015, US 20150266867A1.
- 67. Shimamura T., Shiroishi M., Weyand S., Tsujimoto H., Winter G., Katritch V., Abagyan R., Cherezov V., Liu W., Han G. W., Nature, 2011, 475, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pegoli A., She X., Wifling D., Hübner H., Bernhardt G., Gmeiner P., Keller M., J. Med. Chem. 2017, 60, 3314–3334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary