

Abstract
Fibrodysplasia ossificans progressiva (FOP), a congenital heterotopic ossification (HO) syndrome caused by gain-of-function mutations of bone morphogenetic protein (BMP) type I receptor ACVR1, manifests with progressive ossification of skeletal muscles, tendons, ligaments, and joints. HO can occur in discrete flares, often triggered by injury or inflammation, or may progress incrementally without identified triggers. Mice harboring an Acvr1R206H knock-in allele recapitulated the phenotypic spectrum of FOP, including injury-responsive intramuscular HO, and spontaneous articular, tendon and ligament ossification. HO in these diverse tissues could be compartmentalized into two lineages: an Scx+ tendon-derived progenitor that mediates endochondral HO of ligaments and joints without exogenous injury, and a muscle-resident interstitial Mx1+ population that mediates intramuscular, injury-dependent endochondral HO. Expression of Acvr1R206H in either lineage conferred aberrant gain of BMP signaling and chondrogenic differentiation in response to Activin A, and gave rise to mutation-expressing hypertrophic chondrocytes within HO lesions. Expression of the ligand-independent ACVR1Q207D mutant accelerated and increased the penetrance of all observed phenotypes, but did not abrogate the need for antecedent injury in muscle HO, suggesting the need for an ligand-independent injury factor. Both injury-dependent intramuscular and spontaneous ligament HO in Acvr1R206H knock-in mice were effectively controlled by the selective ACVR1 inhibitor LDN-212854. The diverse phenotypes of HO found in FOP are rooted in cell-autonomous effects of dysregulated ACVR1 signaling in multiple non-overlapping tissue-resident progenitors, with implications for strategies to modify their recruitment or plasticity.
One Sentence Summary:
Tissue-specific manifestations of the congenital bone forming syndrome FOP are mediated by multiple tissue-resident stem cell populations.
Introduction
Heterotopic ossification (HO) broadly describes the formation of ectopic endochondral bone in muscles, tendons, ligaments and other soft tissues. HO is a debilitating complication of fractures, joint replacement surgery and other soft tissue trauma, suggesting a process of disordered injury repair. Fibrodysplasia ossificans progressiva (FOP) is a congenital HO syndrome in which individuals have minor skeletal abnormalities at birth, but develop progressive HO during childhood and young adulthood culminating in severe immobilization and reduced life expectancy due to restrictive lung disease and traumatic injuries (1). Progression may occur in episodic flares, which can follow accidental trauma, surgery, intramuscular immunization, inflammation, or viral prodromes. Recently it has been recognized that significant progression can occur gradually without known flares, antecedent injury or triggers, but it is unclear if such activity is mechanistically distinct from flare-related episodes (2). FOP arises from gain-of-function mutations in bone morphogenetic protein (BMP) type I receptor ACVR1 (a.k.a., ALK2), with approximately 97% of individuals harboring a classic ACVR1R206H variant (3–6). Using genetically engineered mice harboring this variant, we recently found that ACVR1R206H drives HO in FOP by conferring to cells aberrant activation of the BMP signaling pathway by Activin ligands (7), a signaling defect also observed in mesenchymal stem cells derived from patient-derived iPSCs (8). As maladaptive BMP/Activin/TGF-β family ligand signaling is a shared property of both genetic and acquired forms of HO (9–15), it has been suggested that FOP and HO are mediated by common effector and progenitor cells. However, the identity and niche of these progenitors as well as their mechanistic relationship to either triggered or spontaneous HO have yet to be determined.
Previous approaches sought to identify cell populations contributing to HO lesions via immune histology or genetic marking techniques in animal models of HO caused by exogenous BMP ligands. These studies explored the role of diverse tissue-resident mesenchymal, vascular, circulating, hematopoietic, and bone marrow-derived populations, demonstrating their participation, but not identifying the populations which are sufficient to initiate this process and manifest the cell autonomous effects of dysregulated BMP signaling. Here we used tissue-targeted expression of a ligand-responsive and a constitutively-active ACVR1 mutant to identify two non-overlapping tissue-resident populations that appear to be responsible for different anatomic phenotypes of FOP. The differential effects of these mutations reveal potentially distinct ligand-regulated and ligand-independent aspects of these phenotypes. Thus, these studies define the cell progenitors that manifest the effects of dysregulated Activin ligand signaling and reveal a requirement for injury in intramuscular HO that is independent of ligand.
Results
FOP exhibits diverse spatiotemporal phenotypes
FOP manifests with HO in multiple soft tissue compartments, including skeletal muscle, intra- and peri-articular tissues, ligaments, fascia, and tendon, as demonstrated in radiographs obtained from individuals with classic FOP due to ACVR1R206H (Fig. 1A–D). This spectrum of phenotypes manifests in distinct tissues with varying natural histories and functional consequences (2, 6, 16). Intramuscular HO in FOP is frequently preceded by local trauma, or symptoms of myositis or swelling constituting a “flare”, and infiltrates the muscle to cause altered mechanics, pain and reduced range-of-motion that progresses to immobilization (Fig. 1A–B) (6, 17). HO may also affect peri- and intra-articular structures (Fig. 1B–C), including ossification of articular cartilage, fascia, ligaments and tendons, with direct impact on joint mobility, as well as exostosis or osteochondroma formation on long bones (Fig. 1D). Less is known about the triggers for bone formation in the non-muscle tissues, including the role, if any, of injury, or of physiologic vs. pathophysiologic mechanical loading. Moreover, a significant proportion of disease progression in FOP occurs in the absence of known triggers or myositis prodromes (2, 6, 18).
Fig 1. The classic FOP-causing ACVR1R206H allele is associated with intramuscular, peri-articular, ligament and tendon ossification in man and mouse.
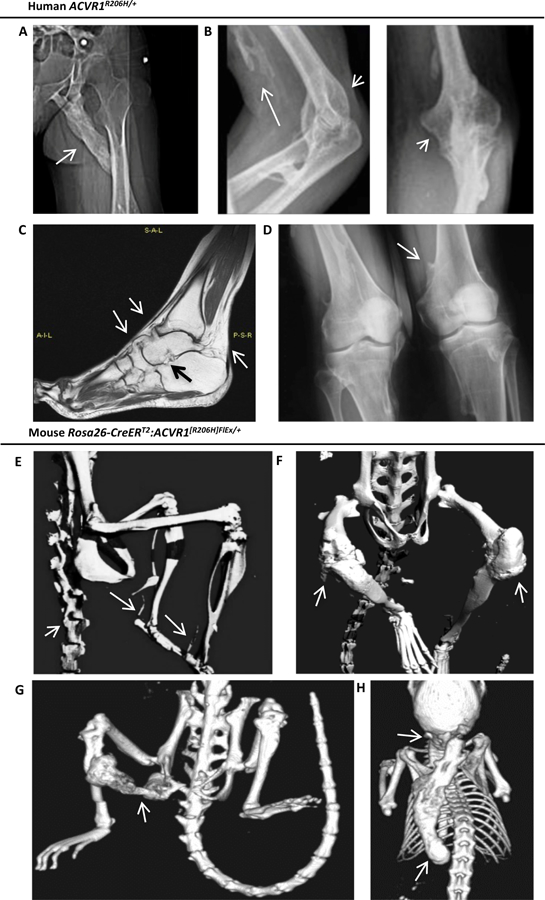
(A-D) Radiographic manifestations in patients with ACVR1R206H/+ genotype-confirmed classic FOP. (A) Female patient with intramuscular ossification (arrow) bridging the ischium and left femoral shaft, infiltrating the gluteus and hamstring. (B) Male patient with intramuscular ossification infiltrating the biceps (long arrow) and dense peri-articular ossification surrounding the olecranon and coronoid fossae (short arrows). (C) T1 TSE MRI of a female patient reveals ossification of tibiotalar, talo-navicular and naviculo-calcaneal ligaments, and the Achilles tendon insertion (arrows) resulting in permanent plantar flexion. (D) A male patient with osteochondroma (arrow) of the distal humerus. (E-H) Micro-CT imaging of Rosa-CreERT2:Acvr1[R206H]FlEx/+ mice treated with tamoxifen for 10–12 wks reveals diffuse axial and appendicular heterotopic ossification after 4 wks, involving the paraspinal ligaments and Achilles tendons (E), peri-articular ossification of the knees (F), sporadic intramuscular ossification infiltrating hamstring and gastrocnemius, bridging ischium, femur and tibia (G), and of the interscapular muscles (H) with frequent handling for examination.
Global postnatal expression of Acvr1R206H in mice recapitulates human FOP phenotypes
Since germline expression of Acvr1R206H in knock-in mice results in prenatal lethality (19), a conditional-on Acvr1[R206H]FlEx, knock-in allele (7) was expressed globally via the tamoxifen inducible Rosa-CreERT2 transgene in 10–12 week old mice, resulting in spontaneous ligamentous, tendon, peri-articular, and intra-articular HO diffusely throughout the axial and appendicular skeleton within 8–12 weeks, mimicking the pattern seen in human disease (Fig. 1E–H). Sporadic intramuscular HO was observed in a portion of mice, with approximately 15% of mice developing HO in hindlimb muscles (Fig. 1G–I) reminiscent of intramuscular disease in humans (Fig. 1A–B), and infrascapular soft tissues (Fig. 1H), also a frequent site of flare related-activity in patients (2, 16), particularly when handled frequently for examination. Following tamoxifen-induced global expression of the mutant allele, Acvr1[R206H]FlEx/+ mice demonstrated a spectrum of injury-mediated and spontaneous FOP phenotypes reminiscent of human disease.
Scx+ progenitor lineages are sufficient for spontaneous ligament, tendon and joint HO
Seeking to determine the lineage(s) sufficient for the cell-autonomous effects of mutant ACVR1 in various tissues, we postulated that tendon-derived stem cells marked by the transcription factor Scleraxis (Scx) (20), and their analogous populations in ligaments and fascia might account for tendon and ligament HO. Expression of the conditional Acvr1R206H allele via the Scleraxis (Scx)-Cre transgene (Fig. 2A) resulted in progressive and spontaneous ligament, tendon, and periarticular ossification, affecting distal hindlimbs at the tibialis anterior and patellar ligaments, Achilles tendon, and knee joints (Fig. 2A–B), as well as diffuse involvement of forelimb, shoulder, costochondral, and hip joints, and paraspinal ligaments (Fig. S1), consistent with the pattern of expression of an Scx-GFP reporter allele. Significant HO in these mice was first evident between 8 and 18 weeks of age, and progressed over 52 weeks (Fig. 2A, Fig. S2). Similar to human disease, severe HO was sporadic, with an overall penetrance of 25–50% in each hindlimb within the first 26 weeks (Fig. S2).
Fig 2. Expression of mutant ACVR1R206H or ACVR1Q207D alleles in Scx-lineage tendon-resident progenitor cells results in spontaneous peri-articular, ligament, and tendon ossification.
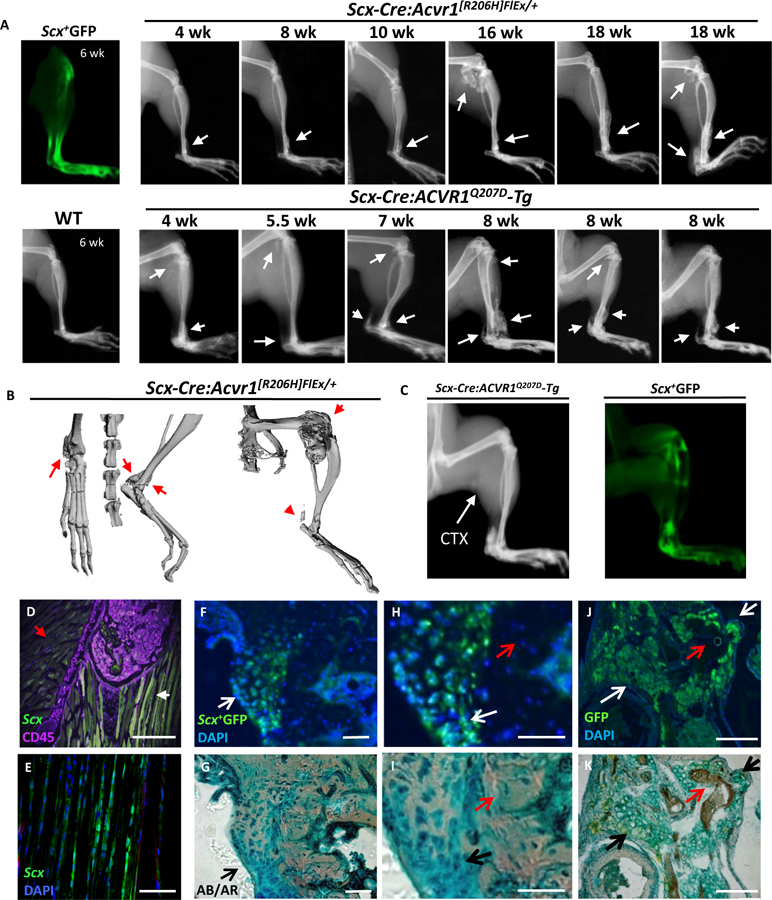
(A) Scleraxis expression is localized to tibiotalar ligament, patellar and Achilles tendons as observed by ex vivo fluorescence in Scx-GFP transgenic mice. Spontaneous ossification of tibiotalar ligament, patellar tendon and Achilles tendon progresses slowly in Scx-Cre:ACVR1R206H mice from 4 through 18 wks, with rapid progression in Scx-Cre:ACVR1Q207D-Tg mice from 4 through 8 weeks (B) Micro-CT of Scx-Cre:ACVR1R206H mice reveals distinct ossification of the tibiotalar ligament, Achilles tendon, and periarticular ossification, but no intramuscular ossification. (C-D) Scx-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice injected with CTX (P21, gastrocnemius) develop typical tendon and ligament ossification, as seen by x-ray (P42) corresponding to areas of Scleraxis expression via Scx+YFP fluorescence but lack evidence of any intramuscular ossification (0/5 transgenic mice injected). (E) Cryosections of fibular head counterstained for CD45 (magenta) reveal Scleraxis expression (green) at ligamentous insertions (white arrow) but not in adjacent skeletal muscle (red arrow) in Scx-GFP transgenic mice. The HO lesions infiltrating the (F) talonavicular ligament, (G) talonavicular ligament in higher magnification, and (H) patellar tendon in Scx-Cre:ACVR1Q207D-Tg;Rosa26-YFP mice reflect the contribution of Scx+YFP+ cells to nearly all Alcian Blue (AB)-stained hypertrophic chondrocytes and heterotopic cartilage (white arrows) in these lesions, but essentially no contribution to osteocytes or mineralized matrix (red arrows) stained with Alizarin Red (AR), with DAPI as a nuclear counter-stain.
To test whether or not ligand-sensitivity of the Acvr1R206H allele might account for its variable penetrance, the ligand-independent, constitutively-active ACVR1Q207D transgene was expressed in Scx+ lineages. Scx-Cre:ACVR1Q207D-Tg mice exhibited substantial HO of the Achilles tendon, tibialis ligaments, knee and costochondral joints with 100% penetrance by 8 weeks (Fig. 2A). Importantly, all anatomic phenotypes observed in Acvr1[R206H]FlEx/+ mice were also seen in ACVR1Q207D-Tg mice , albeit with greater penetrance, severity, and more rapid onset (Fig. S1 and Fig. S2). These effects were restricted to tendon and ligaments, as direct muscle injury with cardiotoxin (CTX) injection did not cause muscle ossification nor did it exacerbate tendon ossification (Fig. 2C). Scx-GFP immunofluorescence confirmed the presence of Scx+ cells at tendinous insertions of the patella, but not in adjoining medialis or gastrocnemius muscle or bone marrow hematopoietic (CD45+) cells (Fig. 2D–E).
Histology revealed replacement of ligamentous and tendon structures with nascent and mineralized chondrogenic matrix and bone marrow (Fig. 2F–H), reflecting endochondral ossification rather than the non-endochondral calcification seen in calcifying tendinopathies (21). The talus, distal tibia and patellar bones featured prominent exostoses contiguous with bridging ligaments (Fig. 2F–H), similar to those seen in human FOP disease (Fig. 1C). Rosa26-YFP reporter activity revealed an Scx+ lineage origin of essentially 100% (164/164 nuclei counted) of hypertrophic chondrocytes in HO lesions, but no significant contribution to osteocytes or mineralized cortex (0/64 nuclei counted) in HO lesions of the talonavicular, tibialis anterior, and patellar ligaments (Fig. 2F–H), suggesting that mutant ACVR1 induces the chondrogenic differentiation of Scx+ lineages to provide a substrate for endochondral bone.
Muscle-resident Mx1+ progenitor lineages are sufficient for intramuscular HO
It was previously shown that bone marrow-derived Mx1+ lineages serve as a reservoir of endochondral progenitors in fracture healing (22). Following injections with pIpC at ages P7-P21, Mx1-Cre mice exhibited high frequency recombination via the Rosa26-mTmG and Rosa26-YFP reporter alleles in microvascular endothelial cells co-staining with vWF, >90% of CD45+ bone marrow cells, and 30–50% of muscle interstitial cells located outside of laminin-stained myofibers (Fig. 3A–D, Fig. S3, Fig. S4). Mx1-Cre:Acvr1[R206H]FlEx/+ mice did not exhibit spontaneous HO, but when subjected to CTX-induced muscle injury at P21, robust intramuscular HO that spared tendons and ligaments was observed (Fig. 3E, Fig. S3A). As noted in human disease (Fig. 1A), intramuscular HO in these mice led to fused long bones and bridged joints (Fig. 3F), restricting hindlimb mobility. Thus, in contrast to the Scx+ tendon and ligament phenotype, expression of mutant ACVR1 in the Mx1+ population resulted in a non-overlapping intramuscular HO phenotype that required extrinsic injury to manifest. Similar to Scx-Cre, the HO seen in Mx1-Cre:Acvr1[R206H]FlEx/+ mice was sporadic and incompletely penetrant, with 30–50% of injured hindlimbs developing severe HO at 60 days (Fig. S3).
Fig 3. Expression of mutant ACVR1R206H or ACVR1Q207D alleles in Mx1-lineages results in injury-dependent intramuscular heterotopic ossification.
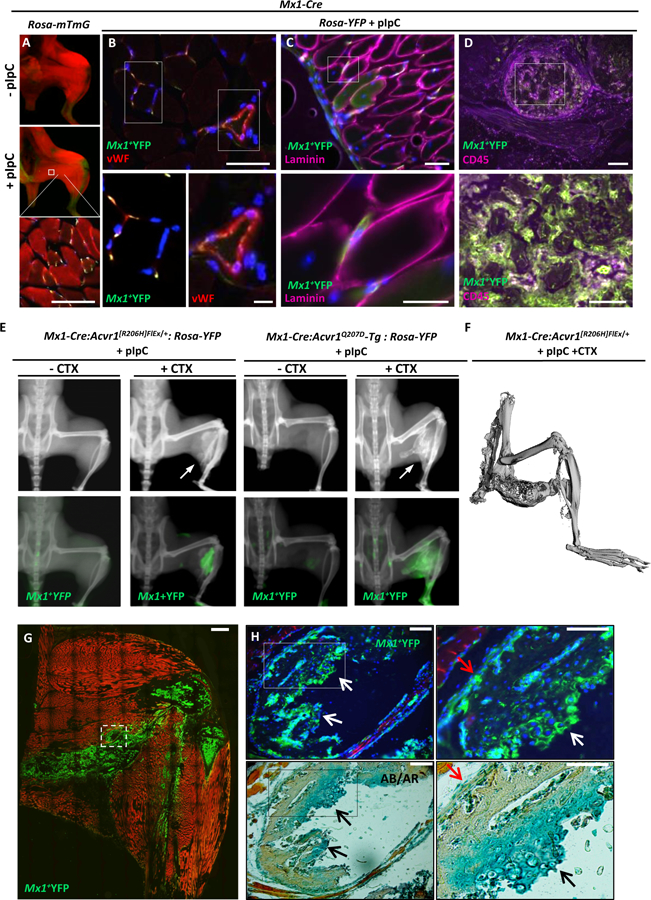
(A) Treatment of Mx1-Cre:Rosa26-mTmG (mTmG) mice with pIpC activates GFP expression within skeletal muscle interstitial cells (red=mTomato, yellow=mGFP). (B-D) Gastrocnemius muscles of pIpC treated Mx1-Cre:Rosa26-YFP mice reveals a portion of Mx1+ lineage (YFP, green) cells in the interstitium and microvascular endothelium co-staining with DAPI (blue) and vWF (red) and shown at higher magnification (B, and inset panels below). Mx1+ cells were located consistently outside of myofiber basement membranes (C), based on laminin staining (magenta), and accounted for a high percentage of bone marrow cells based on co-staining with CD45 (magenta, D). (E) Treatment of Mx1-Cre:Acvr1[R206H]FlEx/+:Rosa26-YFP and Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice with pIpC results in injury-dependent ossification of hindlimb muscles following cardiotoxin (CTX) mediated injury, with YFP-marked intramuscular bone. (F) Micro-CT imaging of Mx1-Cre:Acvr1[R206H]FlEx/+ mice treated with pIpC and CTX reveals ossification infiltrating hamstring and gastrocnemius, fusing ischium and tibia without articular involvement. (G) Intramuscular HO in Mx1-Cre:ACVR1Q207D-Tg:Rosa26-mTmG mice is derived from GFP-marked Mx1+ cells, which also marks heterotopic bone marrow, with detail of lesion structure shown (G) and at higher magnification (H) revealing a mutant Mx1+ origin for nearly 100% of Alcian Blue (AB)-stained hypertrophic chondrocytes, and periosteal cells, in contrast to no contribution to Alizarin Red (AR)-stained mineralized matrix or associated osteocytes.
To test whether or not ligand-sensitivity of the Acvr1R206H allele might account for the need for extrinsic injury or variable penetrance, Mx1-Cre was used to express the constitutively-active ACVR1Q207D transgene. Surprisingly, no spontaneous HO occurred when ACVR1Q207D was expressed via Mx1-Cre following pIpC injections (Fig. S3). Following CTX-induced injury, however, aggressive HO occurred in 100% of treated mice by 60 days. While ligand-independent activity of ACVR1Q207D led to more highly penetrant HO than ligand-sensitive Acvr1R206H in Scx+ or Mx1+ populations, the requirement for antecedent injury for intramuscular HO with both mutants suggested that enhanced receptor signaling is necessary but not sufficient for intramuscular HO.
Immunochemical staining revealed ossified lesions to contain hypertrophic chondrocytes, marrow, cartilage and heterotopic growth plates, consistent with an endochondral process (Fig. 3G–H). Sequential fluorescent imaging and Alcian blue/Alizarin red staining of sections revealed that nearly 100% of the hypertrophic chondrocytes of intramuscular heterotopic lesions (202/204 nuclei counted) were derived from Mx1+ lineage YFP-marked tissues (Fig. 3H), whereas few heterotopic osteocytes (1/24 nuclei counted), and occasional periosteal cells were derived from YFP-marked lineages.
Since Mx1-Cre marked muscle interstitial, bone marrow, and microvascular endothelial lineages (Fig. 3B), additional studies were performed to determine the sub-compartment(s) responsible for this phenotype. Given the high frequency of recombination in the bone marrow, including CD45+ cells (Fig. 3D, Fig. S4), and the prior observation that bone marrow Mx1+ cells are an important osteoprogenitor lineage (22), the role of bone marrow was tested by reciprocal bone marrow transplants between WT and mutant reporter mice (Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP). Mutant mice using the highly penetrant ACVR1Q207D allele were used rather than the incompletely penetrant Acvr1[R206H]FlEx, as detecting a negative result with a power of 95% and an α=0.05 would require at least 30 transplants based on a penetrance of 30% for Acvr1[R206H]FlEx (Figs. S2, S3). When mutant reporter donor or recipient mice were injected with pIpC from P7-P21, consistently high frequency (>90%) recombination of bone marrow was observed based on reporter fluorescence (Fig. 4A). When neonatal WT mice that had been conditioned prenatally with busulfan were injected at P2 with bone marrow obtained from mutant reporter mice previously treated with pIpC (P7-P21), replacement of bone marrow with 78.2% ± 15.9% (n=5) YFP+ cells was observed by P21, approaching frequencies seen in donor mice (Fig. 4B–C). Despite high-efficiency engraftment with mutant ACVR1 marrow, no HO was observed with or without intramuscular CTX treatment in any of these mice (0/5). Conversely, when mutant reporter mice conditioned prenatally with busulfan were injected at P2 with bone marrow obtained from WT mice at P21, the frequency of YFP+ cells in bone marrow following pIpC was markedly decreased (0.5–5%; n=11), consistent with near total replacement of marrow with WT cells (Fig. 4D), yet no attenuation of HO was observed following CTX treatment in any of these mice. These results suggested that cell-autonomous effects of mutant ACVR1 are not mediated by transplantable bone marrow lineages.
Fig 4. Mx1-lineage muscle interstitial but not bone marrow cells are sufficient for intramuscular HO.
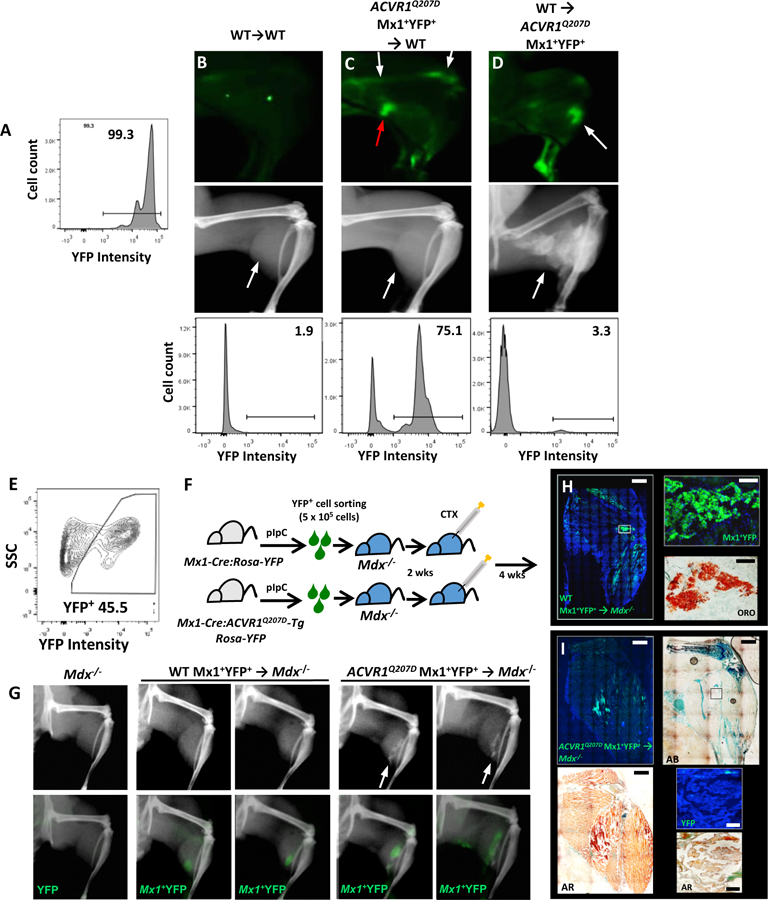
(A-D) Reciprocal bone marrow transplant experiments demonstrate medullary expression of mutant ACVR1 is dispensable for HO. (A) Control Mx1-Cre:Rosa26-YFP or mutant Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice treated with pIpC exhibited >90% YFP labeling of marrow cells. Control or mutant mice were preconditioned antenatally with busulfan (E16, maternal i.p. injection), and engrafted at P2 with total bone marrow cells (5 ×105 cells i.p.) harvested at P21 from WT or mutant donor mice previously treated with pIpC (P7-P21). In contrast to WT mice engrafted with WT marrow (B), WT mice engrafted with mutant marrow (C) exhibited a high percentage of YFP+ bone marrow comparable to mutant donor mice (A) by flow cytometry 78.2% ± 15.9% (n=5), and ex vivo fluorescence (white arrows), but did not exhibit injury-dependent ossification following CTX, despite the presence of infiltrating YFP+ cells due to CTX-induced inflammation (red arrow). Conversely, Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice engrafted with WT marrow (D) exhibited very low frequencies of residual YFP+ bone marrow by flow cytometry (bottom panel, 0.5–5%; n=11), but exhibited robust CTX-induced intramuscular ossification with YFP+ lesions (white arrows, top and middle panel). (E-I) Mx1+ lineage muscle interstitial cell engraftment studies demonstrate Mx1+ lineage interstitial cells are sufficient for injury-dependent intramuscular HO. Mx1+YFP+ (5×105) cells sorted from the muscles of P21 control Mx1-Cre:Rosa26-YFP or mutant Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice previously treated with pIpC (P7-P19) were transplanted into gastrocnemius muscles of Dmdmdx−5cv:Rag1null (Mdx−/−) mice (P21) in Matrigel (E-F). In comparison to Mdx−/− control mice injected with Matrigel only (G, left), Mdx−/− mice injected with WT Mx1+YFP+ cells exhibited engraftment after 6 wks based on YFP fluorescence but no heterotopic ossification with or without injury (G, middle panel, no lesions seen in 5 treated mice), whereas Mdx−/− mice injected with mutant Mx1+YFP+ cells exhibited engraftment and developed intramuscular ossification following CTX treatment (G, right panel, lesions seen in 3/5 mice treated). (H) Histological analysis of mice injected with WT Mx1+YFP+ control cells demonstrated engraftment of YFP+ cells interspersed in gastrocnemius and popliteal fossa, all of which stain with Oil Red O (ORO). (I) Mice engrafted with ACVR1Q207D Mx1+YFP+ cells demonstrated engraftment of YFP+ cells throughout HO lesions of the gastrocnemius, with mineralization evident by Alizarin Red (AR), formation of ectopic cartilage demonstrated by Alcian Blue (AB), but were notable for the absence of YFP fluorescence in heterotopic marrow, shown by fluorescence and DAPI counter-staining (inset top panel), and the co-localization of YFP fluorescence with mineralized areas (AR, inset bottom panel).
To test the sufficiency of the interstitial muscle cell population, muscle-resident Mx1+ lineages were marked by pIpC treatment (P7-P21) in Mx1-Cre:Rosa26-YFP (control) or Mx1-Cre: ACVR1Q207D-Tg: Rosa26-YFP (mutant) mice, dispersed by mechanical and enzymatic dissociation and isolated by FACS, and then injected into the muscles of Dmdmdx−5cv:Rag1−/− recipients. Recipient mice injected with labeled control cells demonstrated engraftment of YFP+ cells in muscle, but did not demonstrate HO with or without CTX injury (0/5 mice tested, Fig. 4G). Rather, engrafted YFP+ cells were observed to stain with Oil Red O, consistent with an adipogenic fate following injury (Fig. 4H). In contrast, recipients engrafted with mutant reporter cells reliably demonstrated HO following CTX injury (3/5 mice tested). The histological examination of engrafted mutant cells revealed co-localization with areas of mineralization and chondrogenic differentiation stained by Alizarin red/Alcian blue, but the absence of YFP+ cells in heterotopic marrow (Fig. 4I). These findings suggested that expression of mutant ACVR1 in Mx1+ muscle-resident interstitial lineages mediates HO by skewing a normally adipogenic population towards an endochondral fate, while expression of mutant ACVR1 in the bone marrow is dispensable for HO.
Endothelial, bone marrow, pericyte, and smooth muscle expression of mutant ACVR1 are insufficient for HO
To corroborate results obtained by cell transplantation studies, the contribution of various lineages was tested directly by mating a panel of lineage-specific Cre strains against conditional ACVR1 mutant mice. As in cell transplantation experiments, mice expressing the highly penetrant ACVR1Q207D-Tg were used for these studies, since detecting a negative result with 95% power and an α of 0.05 for each lineage-targeting experiment with would require observations from at least 30 compound heterozygous mice based on a 30% penetrance of Acvr1R206H (Figs. S2 and S3). Thus conditional ACVR1Q207D-Tg mice expressing the Rosa-YFP reporter allele to confirm lineage-specific recombination (23, 24) were mated with Myf6-Cre, SM22α-Cre, Vav1-Cre, Cadh5-CreERT2, Cspg4-CreERT2, and Pax7-Cre-ERT2 strains to target expression of ACVR1Q207D to skeletal myofiber, smooth muscle, bone marrow, vascular endothelial, pericyte, and satellite cell compartments (Table 1). These matings yielded viable progeny in Mendelian ratios. However, activation of mutant ACVR1 expression during development or following tamoxifen injection (P7 – P21) failed to generate spontaneous or cardiotoxin (CTX) injury-induced HO by x-ray (Table 1) within 90 days, despite confirmation of efficient recombination in target tissues.
Table 1.
Cre-recombinase expressing mouse strains used for targeting mutant ACVR1
| Transgenic strain |
Lineages targeted | Temporal expression |
Phenotype | Compound heterozygotes/live births |
HO/n tested |
|---|---|---|---|---|---|
| Pax7-Cre (57) | Satellite cell; Myoblast; Myofiber |
Embryonal – maturity |
Embryonal lethality | 0/>30 | − |
|
Pax7-CreERT2
(57) |
Satellite cell | Inducible (tamoxifen) |
No spontaneous or injury- induced HO |
Mendelian | 0/10 |
| Myf6-Cre (56) | Myofiber and late myoprogenitor |
Embryonal – maturity |
No spontaneous or injury- induced HO |
Mendelian | 0/10 |
|
SM-MHC-Cre
(51) |
Vascular smooth muscle |
Embryonal – maturity |
Embryonal lethality | 0/>30 | − |
|
SM22α-Cre
(55) |
Vascular smooth muscle; Pericyte subset |
Embryonal – maturity |
No spontaneous or injury- induced HO |
Mendelian | 0/13 |
| Tie2-Cre (59) | Vascular endothelium; Myeloid subset; circulating endothelial progenitor |
Embryonal – maturity |
Embryonal lethality | 0/>25 | − |
|
Cadh5- CreERT2 (58) |
VE-Cadherin expressing mature endothelial cells |
Inducible (tamoxifen) |
No spontaneous or injury- induced HO |
Mendelian | 0/11 |
|
Cspg4- CreERT2 (52) |
NG2-expressing pericytes |
Inducible (tamoxifen) |
No spontaneous or injury- induced HO |
Mendelian | 0/17 |
| Vav1-Cre (24) | Bone marrow hematopoietic and endothelial |
Embryonal – maturity |
No spontaneous or injury- induced HO |
Mendelian | 0/14 |
| Mx1-Cre (50) | Bone marrow; spleen; thymus; liver |
Inducible (pIpC) |
Injury-induced intramuscular HO |
Mendelian | 0/20 −CTX; >30/30 +CTX |
| Scx-Cre (20) | Tendon progenitor cells |
Embryonal – maturity |
Spontaneous joint
and ligament HO |
Mendelian | >30/30 |
Various tissue promoter-specific Cre-expressing mouse strains were mated with homozygous ACVR1Q207D-Tg/Q207D-Tg mice and tested for the capacity to generate live compound transgenic or compound knock-in and transgenic offspring. No live compound mutant mice were produced as a result of mating ACVR1Q207D-Tg/Q207D-Tg mice with Pax7-Cre, SM-MHC-Cre, or Tie2-Cre strains among the stated number of live births per mating strategy. In matings with Myf6-Cre, SM22α-Cre, and Vav1-Cre, compound mutant mice were produced in expected Mendelian ratios, however, no spontaneous HO was observed in a minimum of 10 live compound mutants when observed for up to 16 weeks of age, nor did any HO occur with intramuscular cardiotoxin injection at P10-P14 in a minimum of 5 injected compound mutants. When offspring of ACVR1Q207D-Tg/Q207D-Tg and Pax7-CreERT2 or Cadh5-CreERT2, Cspg4-CreERT2 matings were injected with tamoxifen (50 mg/kg i.p daily P14-P21), high efficiency recombination was observed among sublaminar satellite cells and vascular endothelial cells, respectively. However, no spontaneous or cardiotoxin-induced HO was observed among a minimum of 10 compound mutant mice following tamoxifen injection, and observation up to 16 weeks of age.
Acvr1R206H confers Activin A-induced SMAD1/5/8 activation and endochondral differentiation to Mx1+ and Scx+ lineages
Similar to previous observations (7, 8), expression of Acvr1R206H in myofibroblasts did not increase basal activation of BMP receptor-associated SMADs 1/5/8, but conferred an ability to activate SMAD1/5/8 in response to Activin A, a property not seen in WT cells, and moderately enhanced sensitivity to BMP4 (Fig. S5A), whereas activation of SMAD3 by Activin A was essentially unchanged in mutant vs. WT cells. Similarly, Mx1+YFP+cells isolated from the skeletal muscle interstitium of pIpC-activated Mx1-Cre:Acvr1[R206H]FlEx/+:Rosa26-YFP mice did not exhibit baseline changes in alkaline phosphatase activity as compared to wild-type Mx1+ cells (Fig. 5A), but exhibited markedly enhanced sensitivity to ligand-induced differentiation in response to Activin A and BMP4, but not BMP6 (Fig. 5B–D). Consistent with these observations, the baseline expression of a panel of endochondral genes was not altered with expression of Acvr1R206H, however, challenge with Activin A induced expression of the endochondral genes Fibromodulin and Col1a2 (Fig. S5B–C). Consistent with the earlier finding that Activin A is a critical mediator of HO in Acvr1[R206H]FlEx/+ mice (7), these findings demonstrate Acvr1R206H confers aberrant Activin A-mediated signaling and endochondral differentiation in muscle-resident Mx1+ lineages.
Fig 5. Expression of ACVR1R206H in Mx1 and Scx-lineage cells modifies their osteo-chondrogenic differentiation potential in a ligand-dependent manner.
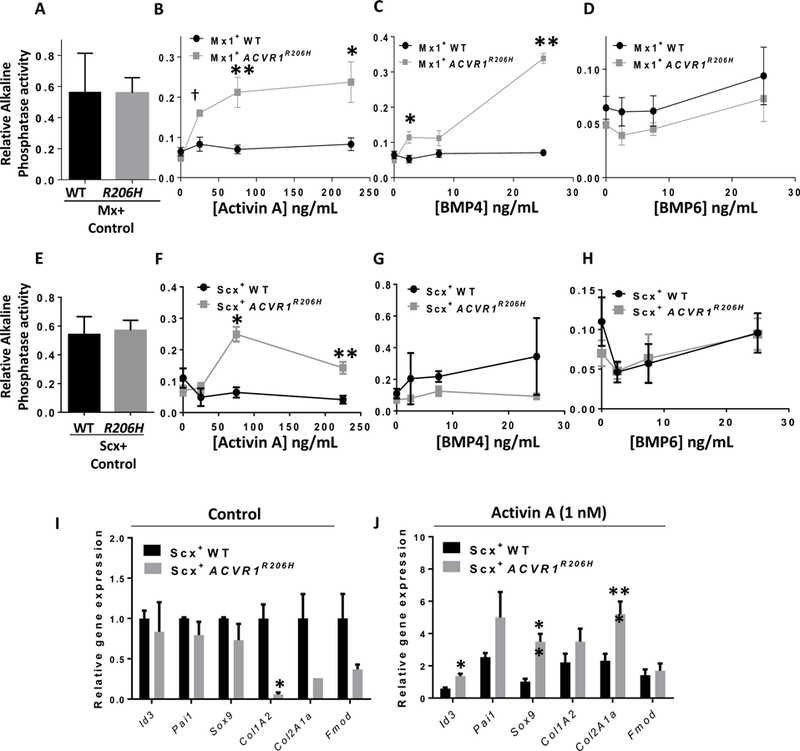
(A) Spontaneous (n=8) and (B-D) ligand-induced alkaline phosphatase expression in freshly isolated interstitial Mx1+ACVR1R206H and Mx1+ control cells obtained from quiescent, non-injured muscles following culture in the presence or absence of ligand for 4 d (n=4; p-values from left to right: 5B. †0.006,**0.01, *0.03; 5C. *0.02, **2×10−6). (E) Spontaneous (n=4) and (F-H) ligand-induced alkaline phosphatase expression in freshly isolated YFP+ cells from 2-week old Scx-Cre:ACVR1R206H-Tg:Rosa-YFP (Scx+ACVR1R206H) and Scx-Cre:Rosa26-YFP (Scx+WT) mice following culture in the presence or absence of ligand for 4 d (n=4; p-values from left to right: *1.3×10−6, **0.0001). Expression of BMP and TGF-β transcriptional targets and endochondral genes in Scx+ACVR1R206H and Scx+WT cells without (I) and with (J) Activin A (1 nM) treatment for 48 h; (n=4; *p<0.02, **p<0.02, ***p<0.003). Unpaired Student’s t-test was used for statistical analysis.
Similar to Mx1+ cells, Acvr1R206H Scx+ cells did not exhibit enhanced endochondral potential in the absence of exogenous ligand (Fig. 5E), but acquired sensitivity to Activin A, which elicited alkaline phosphatase activity in mutant but not wild-type Scx+ cells (Fig. 5F–H). In the absence of exogenous ligand, Acvr1R206H did not induce the activation of endochondral genes (Fig. 5I), but decreased the expression of Col1A2, Col2A1a and Fibromodulin, apparently modulating tendon phenotype. Treatment with Activin A induced the expression of Id3, Sox9 and Col2A1a in ACVR1R206H as compared to WT Scx+ cells, consistent with Activin-mediated chondrogenic differentiation (Fig. 5J).
To analyze subpopulations of Scx+ lineages that might contribute to tissue-specific HO, multiparameter FACS using surface lineage markers was combined with Scx-regulated expression of Rosa26-YFP (Fig. S6A–B). Total Scx+YFP+ cells accounted for ≤5% of tendon derived mononuclear cells (Fig. S6A). Acvr1R206H/+ but not WT Scx+ cells exhibited enhanced osteogenic and chondrogenic differentiation in response to Activin A, but exhibited decreased adipogenic differentiation in the presence or absence of exogenous ligands (Fig.S7A–C). These results suggested Acvr1R206H exerts ligand-dependent and possibly ligand-independent effects on the fate of Scx+ fibroadipogenic progenitor subpopulations. To test the impact of enhanced, ligand-independent ACVR1 signaling on Scx+ fibroadipogenic progenitors with a highly penetrant mutation, PDGFRα+ and PDGFRα- fractions of Scx+ cells were fractionated from tendons of Scx-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice and cultured in specific osteogenic, chondrogenic, and adipogenic media (Fig. S7D–F). Constitutive activation of ACVR1 signaling in these lineages revealed markedly enhanced osteogenic and chondrogenic potential among the PDGFRα+ subset, as compared to PDGFRα- or unfractionated Scx+ cells, suggesting an important role of this fibroadipogenic progenitor population in mediating the effects of mutant ACVR1.
A similar analysis of Mx1+ subpopulations was performed. Since muscle-resident Mx1+YFP+ cells accounted for a sizable (30–50%) fraction of mononuclear muscle interstitial cells (Fig. S6B), these cells were fractionated further for analysis. Wild-type Mx1+YFP+ cells lacking CD31 and CD45 (Lin-) and enriched for Sca1+ demonstrated spontaneous and high frequency adipogenic differentiation, which was markedly diminished in Acvr1R206H/+ cells (Fig. S8A), suggesting that Acvr1R206H alters the potential of a fibroadipogenic Mx1+ subset. To test the impact of enhanced, ligand-independent ACVR1 signaling on Mx1+ subpopulations with a highly penetrant mutation, interstitial muscle cells were fractionated from hindlimbs of Mx1-Cre:ACVR1Q207D-Tg:Rosa26-YFP mice. The enhanced osteogenic and chondrogenic potential of these mutant cells was enriched in CD31-, CD45-, and Sca1+ fractions (Fig. S8B). Similarly, when Mx1+ cells expressing ACVR1Q207D were compared to wild-type cells, enhanced osteogenic and chondrogenic potential was enriched in CD31-, CD45-, and Lin-Sca1+PDGFRα+ subsets (Fig. S8C).
ACVR1 inhibition prevents intramuscular and joint HO in Acvr1[R206H]FlEx/+ mice
A selective, ACVR1-biased BMP type I receptor inhibitor LDN-212854 has been shown to inhibit intramuscular HO in the adenovirus Cre-triggered ACVR1Q207D-Tg mouse model (14). This ACVR1-selective inhibitor was tested in the Rosa26-CreERT2:Acvr1[R206H]FlEx/+ mouse, to determine whether or not spontaneous tendon and ligament HO respond differently to ACVR1 inhibition than injury-induced intramuscular HO, and to determine whether or not ACVR1 inhibition is equally effective in an Activin ligand-mediated model (7) as compared to the previously tested ligand-independent model. Administration of LDN-212854 essentially abrogated spontaneous joint and ligamentous HO, as well as the sporadic and handling-induced intramuscular HO seen in mice when administered for 4 wks following tamoxifen administration (Fig. 6). Taken together, these data demonstrate that selective inhibition of ACVR1 in vivo is sufficient to abrogate the cell-autonomous and Activin-mediated effects of mutant ACVR1 in tendon-derived and muscle resident interstitial populations when administered prophylactically.
Fig 6. Treatment with an ACVR1-selective kinase inhibitor prevents HO in global knock-in Acvr1R206H mice in vivo.
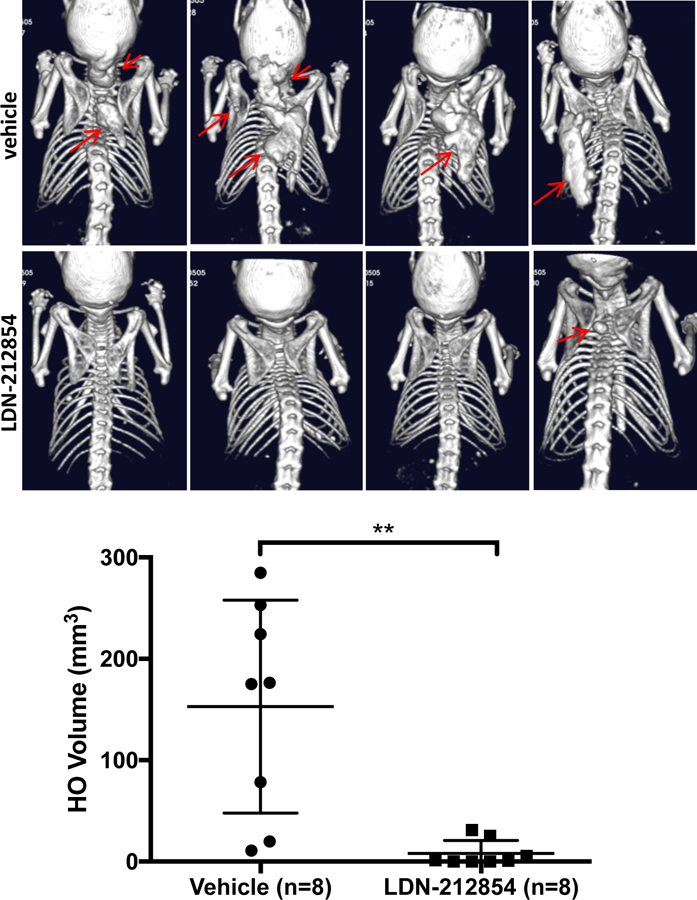
Rosa-CreERT2:Acvr1[R206H]FlEx/+mice treated with tamoxifen for 10–12 wks, followed by treatment with vehicle for 4 wks developed spontaneous joint and ligamentous HO, as well as prominent interscapular HO at sites of handling and examination (red arrows, top panels), whereas animals treated with the ACVR1-selective inhibitor LDN-212854 (2.5 mg/kg twice daily s.c.) demonstrated near-complete inhibition of joint, ligamentous, and interscapular HO (red arrow, lower panel).
Discussion
Two distinct tissue-resident progenitor lineages were identified that drive muscle vs. tendon and ligament HO: an Mx1+ interstitial lineage in muscle that gives rise to injury-dependent intramuscular HO, and an Scx+ lineage that gives rise to apparently spontaneous HO of tendons and ligaments. Expression of mutant ACVR1 in either of these lineages in vivo exerted chondrogenic effects, as the cartilage and hypertrophic chondrocytes of heterotopic bone lesions were entirely derived from mutant cells. Expression of Acvr1R206H in these lineages also conferred aberrant Activin A-mediated endochondral differentiation, consistent with previous observations in surrogate cell lineages and in vivo in conditional knock-in mice (7, 8). Our findings are consistent with earlier studies demonstrating chondrogenic effects of gain-of-function ACVR1 mutants, shown in embryonic fibroblasts from Acvr1R206H/+ knock-in mice, micromass cultures induced to express ACVR1R206H, and developing chick limb buds induced to express ACVR1Q207D (25–27). These data suggest gain-of-function ACVR1 mutations promote ectopic chondrogenesis as a substrate for heterotopic endochondral ossification, which we now confirm as the mechanism of action within two tissue-resident mesenchymal progenitor populations.
In contrast to previous approaches that analyzed various cell populations contributing to HO lesions via immunohistochemistry and lineage tracing, the current study identified cell lineages that are sufficient to initiate HO due to the cell-autonomous effects of ACVR1 mutations. Previous studies implicated Glast-expressing muscle interstitial and perivascular lineages, CD56+PDGFRα+ muscle interstitial cells, Tie2-expressing endothelial-related cells, and marrow-derived Col1+CD45+ osteoprogenitors as potential contributors to HO lesions in humans, or induced by BMP injection or overexpression in mice (19, 28–33). These studies demonstrate that a wide variety of lineages may be recruited to HO lesions, but do not identify the lineages sufficient to initiate HO. Consistent with this notion, it was shown that chimeric Acvr1R206H knock-in mice obtained from blastocyst injection develop HO lesions that incorporate both mutation-positive and mutation-negative cells (19). The current study similarly demonstrates heterogeneity of mutation-positive and negative cells within lesions, but goes further to show that mutant ACVR1 initiates this process via endochondral ossification in two anatomically distinct progenitor lineages.
A prior study examining lesions from a BM-chimeric FOP patient engrafted with wild-type BM as treatment for aplastic anemia, and HO lesions in BM chimeric mice revealed the participation of donor-derived hematopoietic cells in early pre-osseous lesions, but not in mature heterotopic bone (34). Despite complete engraftment of the FOP patient with wild-type BM, FOP disease continued to progress, indicating that expression of mutant ACVR1 in transplantable BM cells is dispensable. In the current study using reciprocal BM transplantation as well as hematopoietic and vascular-targeted expression via the Vav1 promoter, we eliminate hematopoietic and other transplantable BM-derived lineages as necessary or sufficient sites of mutant ACVR1 signaling in HO. The possibility of a cell-autonomous BM effect was important to evaluate, as the Mx1-Cre knock-in allele, introduced at the interferon-responsive myxovirus resistance locus, has been used to mark hematopoietic BM-derived cells (35), a subset of which function as primitive osteoprogenitors that migrate to sites of injury to mediate fracture repair (22). The BM-derived Mx1+Lin-Sca1+PDGFRα+ cells of that study share a similar surface phenotype to the muscle-localized Mx1+ cells of the current study; however, in contrast, we show that the muscle interstitial Mx1+ population functions independently of the BM-derived population, and can engraft wild-type muscle to initiate HO.
In addition to marking a subset of BM, Vav1-Cre and Mx1-Cre also mark microvascular endothelium (22) as confirmed in the present study. We exclude the possibility that mature endothelial cells are sufficient to account for the Mx1 phenotype using both Cadh5-CreERT2 and Vav1-Cre mice, but have not ruled out the possibility that postnatal administration of pIpC to Mx1-Cre mice could have marked Cadh5- immature EC progenitors that might contribute. Following a report that Tie2-lineage cells may contribute to HO (33), a recent study demonstrated that a muscle-resident, Tie2-lineage, Lin- Sca1+PDGFRα+ population contributes to BMP-induced heterotopic bone (32). It is likely that the interstitial Mx1+Lin-Sca1+PDGFRα+ population identified in the current study overlaps significantly with these non-endothelial Tie2-lineage interstitial cells; however, the present study extends these concepts by showing that this population is sufficient to initiate HO in vivo via the chondrogenic effects of mutant ACVR1, that this population exhibits Activin A gain-of-function, in contrast to FOP-mutation expressing iPSC-derived endothelial cells (36), that mature Cadh5-lineage ECs cannot initiate HO, and that muscle-resident CD31+ cells do not manifest the chondrogenic or osteogenic effects of mutant ACVR1.
Our observations that Mx1+ Lin-Sca1+PDGFRα+ interstitial cells exhibit markedly enhanced chondrogenic potential and decreased adipogenic potential with mutant ACVR1, and that in muscle engraftment studies the default adipogenic fate of wild-type Mx1+ interstitial cells was modified by mutant ACVR1 to form bone led us to postulate that Mx1+ HO progenitors represent ‘reprogrammed’ fibroadipogenic progenitor cells. Fibroadipogenic progenitors, defined similarly as muscle resident Lin-Sca1+PDGFRα+ cells, play physiologic roles in muscle injury repair and tissue homeostasis, contribute to fatty infiltration and fibrosis of muscles in settings of injury or myopathy, and are suggested to contribute to HO (30, 37–39). These cells may overlap with PW1+ muscle interstitial cells (PIC), which express PDGFRα and Sca1 early in ontogeny and also exhibit fibro-adipogenic potential (40).
While Scleraxis-expressing lineages have been implicated previously in the normal development and maintenance of tendon and ligament tissues (20, 41), less is known about their contribution to disease, or the impact of BMP and TGFβ family signaling upon their function. Scleraxis lineage tendon-derived progenitors have been described to lack hematopoietic lineage markers and express Sca1 (42), consistent with current findings. While PDGFRα has been used to segregate subpopulations of tendon-derived progenitor cells, its relationship to tendon regenerative, fibroadipogenic or osteogenic function was previously unexplored. We found that Scx+ cells and particularly the PDGFRα+ subfraction exhibited enhanced chondrogenic potential with expression of mutant ACVR1 even in the absence of ligand, consistent with the spontaneous HO of tendons and ligaments observed in two mutant ACVR1 mouse strains when activated by Scx-Cre. When constitutively-active, ligand-insensitive ACVR1Q207D was expressed in Scx+ lineages, these same phenotypes occurred with greater penetrance, suggesting that Activin expression may be a regulator of HO in those tissues, and its limiting presence may be a regulator of penetrance. It is possible that injury-induced factors required to activate Mx1+ derived lineages are constitutively produced in the milieu of mechanosensitive tendon and ligament tissues, thereby eliminating the need of experimentally-induced injury. Since Scleraxis-derived tissues function in force-transmission and are regulated by signals mediated by physiologic and pathophysiologic loading (41), Scx+ progenitors may be triggered by mechanical loading in the presence of mutant ACVR1 to undergo osteogenic and chondrogenic differentiation, yielding spontaneous endochondral lesions in the absence of known extrinsic stimuli.
ACVR1Q207D causes constitutive ligand-independent activation of BMP signaling, while Acvr1R206H confers enhanced sensitivity to various ligands including Activin A (5, 27, 43–45). Expression of the knock-in Acvr1R206H allele in Mx1+ and Scx+ lineages sensitized cells to Activin A-induced SMAD1/5/8 signaling and chondrogenic differentiation not seen in wild-type cells, extending our recent findings (7). Activin A and related ligands are known to be induced by injury in muscle and other tissues, suggesting a possible contributing mechanism for HO following muscle injury in mice expressing mutant ACVR1 via Mx1-Cre. Since Activin ligands are effectors of muscle homeostasis and regenerative responses following injury (46, 47), the functions gained by Activin A in mutant Mx1+ are potential mechanisms for their contribution to HO. The persistent need for muscle injury with ligand-insensitive ACVR1Q207D Mx1+ cells suggests however that ligand-independent injury-mediated factors may be required to prime an endochondral differentiation program.
A limitation of this study is that to overcome embryonic lethality caused by global expression of mutant ACVR1 in mice (19), tissue-specific and/or postnatal expression of mutant ACVR1 alleles was driven using various Cre mouse strains. Postnatal expression of ACVR1 did not replicate some developmental phenotypes associated with FOP, including skeletal malformations such as hallux valgus and osteochondroma. Moroever none of the Cre-targeting strategies, including Rosa26-CreERT2, would reveal the impact of expressing this mutation in all cells from conception as occurs in affected humans. In Scx-Cre mice, mutant ACVR1 was expressed developmentally in tendon-related lineages, while in Mx1-Cre mice expression was activated by pIpC injection only after birth, making functional comparisons of these lineages not strictly equivalent. The observation that intramuscular HO occurs sporadically without intentional injury in a minority (15%) of Acvr1[R206H]FlEx mutant mice driven by Rosa26-CreERT2, but occurs in Mx1-Cre driven ACVR1Q207D or Acvr1R206H only with muscle injury suggests that Rosa-CreERT2, being nearly universal in its expression, may impact a wider array of muscle associated progenitors, or may target the same populations with greater efficiency than Mx1-Cre. While expression of Acvr1R206H in Mx1+ and Scx+ lineages together account for much of the phenotypic spectrum of FOP in humans, the identification of these lineages does not rule out a distinct or redundant contribution of other lineages within these or other tissue compartments. One might extrapolate from these results that non-genetic HO may derive from similar lineages, to the extent that FOP and acquired HO share biological mechanisms; however, this concept will need to be tested rigorously, as Acvr1R206H progenitor cells and mice exhibit Activin gain-of-function not seen in acquired forms of HO, and as a consequence, the cellular progenitors and signals which recruit HO in each disorder could be distinct.
The identification of two distinct HO-driving progenitors has implications for the development of therapy. Local therapies neutralizing a single population in a given tissue are unlikely to provide satisfactory treatment for these syndromes, whereas systemic strategies targeting shared phenotypes, signaling, recruitment, or function of multiple tissue-resident progenitors are more likely to be effective—perhaps via the common marker PDGFRα or its ligands. Strategies to neutralize Activin A could be effective in FOP, based on the gained function of Activin A in these two distinct lineages and our previous findings in the Acvr1R206H model (7), but the impact of this therapy on the constitutive functions of Activin in these tissues will need to be considered, and the contribution of muscle injury appears to be not only independent of ligand but independent of ACVR1 signaling. The distinction raised here between injury-induced vs. injury-independent phenotypes has implications for trial design, as recent clinical trials for the treatment of FOP have been designed to intercept flare-associated events (48), yet controlling the significant portion of disease progression that occurs in the absence of flares or injury (2) might require chronic therapy. In the absence of injury or acute flares, and particularly in tendons, ligaments, and fascia, hypotheses regarding the underlying trigger(s) such as ligands, inflammation, force-transduction, and pathophysiologic stress will need to be tested directly, and are likely to reveal how tissue homeostasis and regeneration are regulated in these tissues.
Materials and Methods
Study design
This study was designed to ascertain whether the tissue-specific expression of hyperactive mutant ACVR1 alleles targeted to specific progenitor compartments could replicate tissue-specific phenotypes of FOP and possibly HO. These studies used a conditional knock-in mouse expressing the FOP-causing Acvr1R206H allele (7), as well as a previously described conditional transgenic mouse expressing the constitutively active ACVR1Q207D mutant allele (15). Under various promoters, expression of the ACVR1Q207D transgene yielded similar results to that of the Acvr1R206H knock-in allele, with essentially identical anatomic distribution but significant differences in phenotypic severity attributable to known differences in biochemical activity of these mutants (5, 27, 44, 45), and potentially the relative levels of expression. For transplantation experiments, Cre-mediated expression, and identification of functionally relevant subcompartments, use of the ACVR1Q207D-Tg strain was employed for its high sensitivity for in vivo and cellular phenotypes, whereas replication in the Acvr1[R206H]FlEx model was used to confirm that these findings were not artifacts due to ectopic expression or the high level of signaling activation mediated by ACVR1Q207D. The study’s primary objectives were to test the sufficiency of defined lineages and their subpopulations for initiating HO. Mice were assigned to different experimental groups based on their genotypes; specifically the expression of mutant ACVR1 knock-in or transgenic alleles, various Cre deleter alleles, and the Rosa26-YFP reporter. Animals expressing all three alleles were assigned to the mutant (test) group, while those expressing only Cre and Rosa26-YFP were assigned to WT (control) group. Gross, radiographic, and histomorphometric analyses for these targeting experiments were performed in a blinded fashion with respect to genotype or treatment. In Mx1-Cre experiments, pIpC was administered to all mice regardless of genotype. The number of observations in vivo were confirmed at a minimum in 5 compound transgenic or knock-in mice of a given genotype and treatment condition, or in at least 5 BM-transplanted or muscle-engrafted mice, unless otherwise noted in individual experiments. All other assays, including cell differentiation and flow cytometric measurements were performed with a minimum of three experimental or biological replicates, unless otherwise noted, and data are represented as mean ± standard error of the mean based on these replicate measurements.
Mouse breeding, genotyping, and conditional expression
Mice were maintained in accordance with Institutional Animal Care and Use Committee guidelines under approved experimental protocols. Cre-inducible constitutively-active ACVR1Q207D transgenic mice, and inducible Acvr1[R206H]FlEx/+ knock-in mice were as previously described (7, 49). Both ACVR1Q207D-Tg and Acvr1[R206H]FlEx/+ mice were bred onto C57BL/6 backgrounds for >8 generations, bred with Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo (Rosa-mTmG) or Gt(ROSA)26Sortm1(EYFP)Cos (Rosa-YFP) reporter mice (23, 24), and with various promoter-specific Cre transgenic or Cre knock-in mice to yield compound transgenic and/or knock-in mice expressing tissue specific Cre and inducible reporter alleles. In the case of Mx1-Cre mice, Cre recombinase expression was induced by administering polyinosinic-polycytidylic acid (pIpC, Sigma), 4.2 µg/g i.p. every other day to mice from ages P7-P19 for a total of 7 doses. Fluorescent, recombined cells obtained from Mx1-Cre:ACVR1Q207D-Tg:Rosa-YFP, Mx1-Cre:Acvr1[R206H]FlEx/+:Rosa-YFP, and Mx1-Cre:Rosa-YFP controls are abbreviated as ACVR1Q207D Mx1+YFP+, ACVR1R206H Mx1+YFP+, and WT Mx1+YFP+ cells. Muscle injury was induced by i.m. injection of 2 µg cardiotoxin (Sigma) into the popliteal fossa on P21. Mx1-Cre knock-in (50), SMMHC-Cre knock-in (51), Vav1-Cre (24), Cspg4-CreERT2 (52) and Gt(ROSA)26Sortm1(cre/ERT2)Tyj (Rosa-CreERT2) (53) mouse strains were obtained from Jackson. C57BL/6 Dmdmdx−5cv:Rag1null mice (54), SM22α-Cre (55), Scx-Cre, Scx-GFP (20), Myf6-Cre (56), Pax7-Cre, Pax7-CreERT2 (57), and VE-Cadherin-CreERT2 mice (58) were obtained from their labs of origin. For tamoxifen-inducible strains, P14-P28 mice were treated with tamoxifen 50 mg/kg i.p. daily (Sigma) in peanut oil from P14-P21 to induce Cre recombinase activity. Rosa-CreERT2:Acvr1[R206H]FlEx/+ mice maintained on mixed C57BL/6NTac-129S6/SvEvTac background were treated with 40 mg/kg of tamoxifen i.p. daily for 8 d to induce Cre activity. Starting concurrently, mice were injected s.c. with LDN-212854, synthesized as described (14), at 3 mg/kg (n=8) or vehicle (n=8) twice daily for 4 weeks.
Imaging
Assessment of YFP reporter fluorescence, and radiographic assessment of HO were analyzed in whole mice directly following euthanasia (MS FX In-Vivo Pro, Bruker). Micro-computed tomography (micro-CT) imaging was carried out on samples fixed overnight in 1% paraformaldehyde followed by scanning (µCT35, ScanCo). For the serial assessment of heterotopic bone, mice were anesthetized by isofluorane and scanned with a field of view at 60 mm x120 mm, using an in vivo micro-CT system (Quantum FX, PerkinElmer) set to a current of 160 μA, voltage of 90 kVp, with a voxel size at 120 or 240 μm.
BM transplantation
BM was harvested by flushing the long bones (femur, pelvis) with ice-cold PBS, followed by RBC lysis (BD, Lyse). Live cells were counted by Trypan blue staining. WT C57BL/6 dams were injected on E19 with busulfan (15 mg/kg) i.p. and resulting pups were injected i.p. on P2 with 5 × 106 Mx1+YFP+ BM cells isolated from P21 pIpC-treated Mx1-Cre:ACVR1Q207D-Tg:Rosa-YFP donors. Alternatively, E19 dams carrying Mx1-Cre:ACVR1Q207D-Tg:Rosa-YFP pups were treated with busulfan, and resulting pups injected i.p. on P2 with 5 × 106 bone marrow cells from P21 WT or Rosa-mTmG donors. Recipient pups were treated with 4.2 µg/g pIpC i.p. every other day from P14-P26. Mice were treated with cardiotoxin on P30 and assessed at P60 for HO and engraftment of isogeneic BM.
Histology and immunofluorescence
Hind limbs were fixed overnight in 1% PFA at 4°C followed by decalcification in saturated EDTA for 1 week. Samples were then equilibrated in 30% sucrose overnight before embedding in OCT (Tissue-Tek, 4583) and stored in −80°C. The CryoJane tape-transfer system (Leica Biosystems) was used to obtain 10 µm sections. For histochemical staining, cryosections were post-fixed in 4oC methanol for 5 min and air dried, followed by staining with freshly prepared hematoxylin and eosin solutions, Movat’s modified pentachrome, BM Purple (Roche), Alcian Blue (3%), or Alizarin Red (1%) solutions. For fluorescence, PBS-hydrated cryosections were analyzed for YFP or Tomato fluorescence without further fixation. For immunofluorescence, sections were blocked with 5% goat serum in PBS for 45 min at 25oC, followed by incubation with primary antibodies in blocking solution overnight at 4oC. For intracellular antigens, permeabilization was carried out with 0.1% Triton X-100 in blocking solution. Alexa Flour 488, Cy3 or Cy5-linked secondary antibodies were used to stain sections for 45 min at 25oC. DAPI containing mounting media (Vector Shield) was used to mount sections. An Olympus BX63 and CellSens Dimension software were used for image acquisition and analysis (Olympus).
Isolation and flow cytometry analysis of Mx1+ and Scx+ cells
Mx1-Cre:Rosa-YFP or Mx1-Cre:ACVR1Q207D-Tg:Rosa-YFP mice were sacrificed 48 h after the last pIpC dose or 48 h after cardiotoxin injection. Mx1+ interstitial cells were isolated by mechanical and enzymatic digestion of the gastrocnemius muscle using the muscle cell isolation kit (Miltenyi), as per manufacturer’s instructions. The muscle dissociation protocol on a gentleMACS dissociator (Miltenyi) was used for mechanical and enzymatic disruption. An interstitial cell pellet was obtained after RBC lysis and filtered through a 40 µm strainer (BD Biosciences). Cell surface marker analysis was carried out on the LSR II (BD Biosciences) and cell sorting was carried out on the MoFlo (Beckmann Coulter) or FACS Aria (BD Biosciences). DAPI was used as a viability marker for cell sorting. Following antibodies were used: CD45-PE or APC (eBioscience), CD31-PE (BD Pharmingen) or CD31-Pacific Blue (eBioscience), Sca1-PE-Cy7 (eBioscience), PDGFRα-PE (eBioscience), CD11b-PerCP-Cy5.5 (Biolegend), c-kit-Pacific Blue (Biolegend), CD105-APC (Biolegend), CD90-Pacific Blue (Biolegend), Tie2-APC (Biolegend).
Two wk old Scx-Cre:Rosa-YFP or Scx-Cre:ACVR1Q207D-Tg:Rosa-YFP mice were sacrificed, and muscles and tendons of the hind limb were mechanically dissected from the long bones, followed by enzymatic digestion with a mix of collagenase II (Gibco, 3 mg/ml) and dispase (Gibco, 4 mg/ml), prepared in DMEM for 45 min at 37oC. Intermittent tissue mincing was carried out using the tumor 4 program on the gentleMACS dissociator. Scx+YFP+ cells were sorted by FACS, using a similar panel of antibodies used to analyze Mx1+ populations. Freshly sorted Mx1+and Scx+ cells were either frozen immediately for RNA studies or seeded in their respective growth media (and on plates coated with Matrigel (Corning) in case of Mx1+ cells) for differentiation assays. Mx1+ cells were expanded in DMEM supplemented with 20% FBS (Invitrogen), 10% horse serum (Invitrogen), 2.5 ng/ml bFGF (Sigma) and antibiotics (Sigma), while Scx+ cells were seeded in DMEM containing 10% FBS, 100 µM β-mercaptoethanol (Gibco) and antibiotics.
Adoptive transfer of muscle interstitial cells
Mx1-Cre:ACVR1Q207D-Tg:Rosa-YFP or Mx1-Cre:Rosa-YFP mice were injected with pIpC on alternating days from P7-P19. Interstitial cells were isolated from dispersed muscles on P21, viable YFP+ cells sorted and collected by FACS, with enrichment or depletion of various markers. YFP+ cells (5 × 105) were resuspended in 40 µL of ice cold high concentration Matrigel, and injected with a 31 g needle i.m. into the popliteal fossa of P14 Dmdmdx−5cv:Rag1null (a.k.a. Mdx−/−) C57BL/6 background recipients. Cells were allowed to engraft for two wks, at which point they received the standard dose of cardiotoxin. After 4 wks, cardiotoxin injected recipient mice were scanned for HO by x-ray and YFP imaging.
In vitro differentiation assays
Equal numbers (500–1000 cells per well) of sorted cells were seeded in growth media onto Matrigel-coated 96-well plates. Cells were seeded in quadruplicate for osteo-, chondro- and adipogenic differentiation analysis in regular growth media, or alternatively in specific differentiation-inducing media. For the former experiments, cells were expanded in bFGF-supplemented growth media for 2 wk, followed by analysis of spontaneous differentiation. For the latter experiments, following expansion in growth media, cells were transferred to specific differentiation media. All differentiation media utilized basal DMEM, supplemented for osteogenic differentiation with 0.1 µM dexamethasone, 50 µg/ml ascorbic acid, and 10 mM β-glycerophosphate, supplemented for chondrogenic differentiation with 50 µg/ml ascorbic acid, 1 ng/ml TGF-β1, and 1 µM dexamethasone, and supplemented for adipogenic differentiation using 5 µg/ml insulin, 0.5 µM IBMX, 60 µM indomethacin, and 1µM dexamethasone (all from Sigma), and grown for an additional 3 wks. For experiments testing the effects of ligands upon cell differentiation, cells were treated with media containing reduced fetal calf serum (2%) supplementation, and varying concentrations of BMP or Activin ligands based on empirically determined EC50 for these endpoints.
Analysis of multi-lineage differentiation
Relative cell counts of cultured grown in 96 well plates were determined based on Hoechst 33342 (10 µg/ml at 37oC for 45 min, Sigma) fluorescence (Ex350/Em450 nm) fluorescence measured with a microplate reader (FLUOstar Omega, BMG) before analysis of differentiation. Alkaline phosphatase activity was assayed as a surrogate of spontaneous endochondral differentiation, by lysing cells in 1% Triton X-100, reacting with pNPP substrate (Sigma, RT x 15’−30’) and measuring absorbance at 405 nm. Alkaline phosphatase activity in fixed cells was measured using BM Purple stain (Roche). For analysis of mineralization, as well as chondrogenic and adipogenic differentiation, cells were fixed with 0.5% glutaraldehyde (Sigma) for 20 min, followed by staining with alizarin red (2% in dH2O), alcian blue (1% in 3% acetic acid), or Oil Red O (in 60% isopropanol). Mineralization was quantified by solubilization in 10% formic acid and measuring absorbance at 450 nm; Alcian blue staining was quantified by solubilization with 6M guanidinium HCl, and measuring absorbance at 595 nm; Oil Red O stained lipid droplets were solubilized with isopropanol, and absorbance measured at 500 nm.
Analysis of gene expression
Freshly sorted Mx1+ or Scx+ cells were centrifuged at 400 g x 10 min and pellets were resuspended in Trizol (Sigma), and manufacturer’s instructions were followed for RNA isolation. 1 µg RNA was used for cDNA synthesis, using a high capacity cDNA synthesis kit (KAPA Biosystems). Abundance was determined by quantitative RT-PCR using SYBR Green master mix (KAPA Biosystems) with an Eppendorf realplex2 cycler. Primer pairs are described in Supplemental Table 1
Myofibroblast cultures and ligand-mediated signaling
For studies examining the impact of Acvr1R206H upon ligand-mediated signaling, primary lung myofibroblast cells were isolated from Acvr1[R206H]FlEx/+ mice using the lung dissociation kit and gentleMACS dissociator (Miltenyi), and cultured in RPMI supplemented with 10% FCS and antibiotics. Following expansion and 2 sequential passages, cells were subjected to infection with adenovirus expressing Cre recombinase (Ad-Cre) or Ad-RFP (20 MOI), and tested by qPCR to confirm high frequency (>90%) recombination of the Acvr1[R206H]FlEx allele. Cells were stimulated with varying concentrations of recombinant BMP, TGF-β, Activin and GDF ligands (R&D Systems) based on empirically determined EC50 for 30 min at 37oC, extracts separated by SDS-PAGE, and immunoblotted for phosphorylated SMAD1/5 and SMAD3 using a rabbit monoclonal Ab recognizing both proteins at different molecular weights (clone EP823Y/ab52903, Epitomics/Abcam), or a rabbit monoclonal antibody recognizing SMAD1/5/8 (clone 13820, Cell Signaling).
Statistical analysis
Unpaired two-tailed Student’s t-test with Welch’s correction was used to determine the significance of differences observed between wild-type and mutants, for in vitro differentiation assays, gene expression studies, ligand and drug treatments. Data have been presented as mean ± SEM to represent the variation within a group. GraphPad Prism software and Microsoft Excel were used for data analysis.
Supplementary Material
Acknowledgments:
The authors thank Drs. Y. Mishina, E. Gussoni, and A. Aliprantis for providing critical reagents and experimental advice. We thank G. Buruzula from the Flow Core Facility, Joslin Diabetes Center for technical assistance in FACS experiments, H. Nakajima for technical assistance with cryosectioning and microscopy, and Dr D. Xia for assistance in x-ray analysis of mouse phenotypes. Cre-inducible constitutively-active ACVR1Q207D transgenic mice were a kind gift from Dr. Yuji Mishina. C57BL/6 Dmdmdx−5cv:Rag1null mice were a kind gift from Dr. Emanuela Gussoni. SM22α-Cre transgenic mice were kindly provided by Dr. John Lepore, Scx-Cre and Scx-GFP mice were a kind gift from Dr. Cliff Tabin, and VE-Cadherin-CreERT2 mice were provided by Dr. Luisa Iruela-Arispe.
Funding: This work was supported by US National Institutes of Health Grants HL079943 (PBY), AR057374 (PBY), HL007604 (BWH Cardiovascular Division T32 supporting DD), AR066261 (NIAMS P30 Center for Skeletal Research Core), a Department of Defense Grant MR140072 (PBY), a Harvard Stem Cell Institute Seed Award (PBY), a Pulmonary Hypertension Association Clinician Scientist Career Development Award (PBY), a Leducq Foundation Transatlantic Network of Excellence Award (PBY), a Howard Hughes Medical Institute Early Career Physician-Scientist Award (PBY), and a Massachusetts Technology Transfer Award (PBY). AJW received support from the NIH (HL100402).
Footnotes
Competing interests: ANE, SJH and LH are employees of Regeneron Pharmaceuticals, Inc. and hold stock in the company. PBY and Partners Healthcare hold patents on small molecules that inhibit BMP receptor signaling and may be entitled to royalties.
Data and materials availability: All non-commercially available materials described in this paper (including the Acvr1[R206H]FlEx/+ knock-in mouse) may be obtained through MTA.
References:
- 1.Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM, Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 22, 191–205 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, Kaplan FS, The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J Bone Miner Res 31, 650–656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Brown MA, Kaplan FS, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38, 525–527 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM, Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Human mutation 30, 379–390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, Endo K, Awakura T, Fukushi J, Nakashima Y, Chiyonobu T, Kawara A, Nishida Y, Wada I, Akita M, Komori T, Nakayama K, Nanba A, Maruki Y, Yoda T, Tomoda H, Yu PB, Shore EM, Kaplan FS, Miyazono K, Matsuoka M, Ikebuchi K, Ohtake A, Oda H, Jimi E, Owan I, Okazaki Y, Katagiri T, Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 284, 7149–7156 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W, Zhang K, Song L, Pang J, Ma H, Shore EM, Kaplan FS, Wang P, The phenotype and genotype of fibrodysplasia ossificans progressiva in China: a report of 72 cases. Bone 57, 386–391 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN, ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 7, 303ra137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J, Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 112, 15438–15443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toom A, Arend A, Gunnarsson D, Ulfsparre R, Suutre S, Haviko T, Selstam G, Bone formation zones in heterotopic ossifications: histologic findings and increased expression of bone morphogenetic protein 2 and transforming growth factors beta2 and beta3. Calcif Tissue Int 80, 259–267 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Lories RJ, Derese I, Luyten FP, Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. The Journal of clinical investigation 115, 1571–1579 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber FE, Schmokel H, Oelgeschlager M, Nickel J, Maly FE, Hortschansky P, Gratz KW, Deletion mutants of BMP folding variants act as BMP antagonists and are efficient inhibitors for heterotopic ossification. J Bone Miner Res 18, 2142–2151 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Hannallah D, Peng H, Young B, Usas A, Gearhart B, Huard J, Retroviral delivery of Noggin inhibits the formation of heterotopic ossification induced by BMP-4, demineralized bone matrix, and trauma in an animal model. J Bone Joint Surg Am 86-A, 80–91 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Peterson JR, De La Rosa S, Eboda O, Cilwa KE, Agarwal S, Buchman SR, Cederna PS, Xi C, Morris MD, Herndon DN, Xiao W, Tompkins RG, Krebsbach PH, Wang SC, Levi B, Treatment of heterotopic ossification through remote ATP hydrolysis. Sci Transl Med 6, 255ra132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB, Development of an ALK2-Biased BMP Type I Receptor Kinase Inhibitor. ACS Chem Biol 8, 1291–1302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD, BMP type I receptor inhibition reduces heterotopic ossification. Nat Med 14, 1363–1369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS, The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am 75, 215–219 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Huning I, Gillessen-Kaesbach G, Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation. Mol Syndromol 5, 201–211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith R, Athanasou NA, Vipond SE, Fibrodysplasia (myositis) ossificans progressiva: clinicopathological features and natural history. QJM 89, 445–446 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM, An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res 27, 1746–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pryce BA, Brent AE, Murchison ND, Tabin CJ, Schweitzer R, Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Developmental dynamics : an official publication of the American Association of Anatomists 236, 1677–1682 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Archer RS, Bayley JI, Archer CW, Ali SY, Cell and matrix changes associated with pathological calcification of the human rotator cuff tendons. J Anat 182 ( Pt 1), 1–11 (1993). [PMC free article] [PubMed] [Google Scholar]
- 22.Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT, Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L, A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007). [DOI] [PubMed] [Google Scholar]
- 24.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D, Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33, 314–325 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM, Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells 32, 1289–1300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ, ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J Bone Miner Res 18, 1593–1604 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, Shore EM, The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. The Journal of clinical investigation 119, 3462–3472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suda RK, Billings PC, Egan KP, Kim JH, McCarrick-Walmsley R, Glaser DL, Porter DL, Shore EM, Pignolo RJ, Circulating Osteogenic Precursor Cells in Heterotopic Bone Formation. Stem Cells, (2009). [DOI] [PMC free article] [PubMed]
- 29.Kan L, Peng CY, McGuire TL, Kessler JA, Glast-expressing progenitor cells contribute to heterotopic ossification. Bone 53, 194–203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oishi T, Uezumi A, Kanaji A, Yamamoto N, Yamaguchi A, Yamada H, Tsuchida K, Osteogenic differentiation capacity of human skeletal muscle-derived progenitor cells. PLoS One 8, e56641 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS, Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 91, 652–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ, Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res 27, 1004–1017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR, Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16, 1400–1406 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, Senitzer D, Forman SJ, Emerson SG, Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg Am 89, 347–357 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, Orkin SH, Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes & development 18, 2336–2341 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barruet E, Morales BM, Lwin W, White MP, Theodoris CV, Kim H, Urrutia A, Wong SA, Srivastava D, Hsiao EC, The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem cell research & therapy 7, 115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penton CM, Thomas-Ahner JM, Johnson EK, McAllister C, Montanaro F, Muscle side population cells from dystrophic or injured muscle adopt a fibro-adipogenic fate. PLoS One 8, e54553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe-Suzuki Y, Takeda S, Tsujikawa K, Tsuchida K, Yamamoto H, Fukada S, Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. Journal of cell science 124, 3654–3664 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K, Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12, 143–152 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA, Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol 12, 257–266 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Murchison ND, Price BA, Conner DA, Keene DR, Olson EN, Tabin CJ, Schweitzer R, Regulation of tendon differentiation by scleraxis distinguishes force-transmitting tendons from muscle-anchoring tendons. Development 134, 2697–2708 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Mienaltowski MJ, Adams SM, Birk DE, Regional differences in stem cell/progenitor cell populations from the mouse achilles tendon. Tissue Eng Part A 19, 199–210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagarova J, Vonner AJ, Armstrong KA, Borgermann J, Lai CS, Deng DY, Beppu H, Alfano I, Filippakopoulos P, Morrell NW, Bullock AN, Knaus P, Mishina Y, Yu PB, Constitutively active ALK2 receptor mutants require type II receptor cooperation. Molecular and cellular biology 33, 2413–2424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN, Structure of the BMP receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 287, 36990–36998 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le VQ, Wharton KA, Hyperactive BMP signaling induced by ALK2(R206H) requires type II receptor function in a Drosophila model for classic fibrodysplasia ossificans progressiva. Dev Dyn 241, 200–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaden BC, Wang YX, Wilson JM, Culver AE, Milner A, Datta-Mannan A, Shetler P, Croy JE, Dai G, Krishnan V, Inhibition of activin A ameliorates skeletal muscle injury and rescues contractile properties by inducing efficient remodeling in female mice. Am J Pathol 184, 1152–1166 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Zhu J, Li Y, Lu A, Gharaibeh B, Ma J, Kobayashi T, Quintero AJ, Huard J, Follistatin improves skeletal muscle healing after injury and disease through an interaction with muscle regeneration, angiogenesis, and fibrosis. Am J Pathol 179, 915–930 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.C. P. Inc., An Efficacy and Safety Study of Palovarotene to Treat Preosseous Flare-ups in FOP Subjects 2014.
- 49.Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y, Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44, 159–167 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, Lafave LM, Koche R, Shih AH, Guryanova OA, Kim E, Li S, Pandey S, Shin JY, Telis L, Liu J, Bhatt PK, Monette S, Zhao X, Mason CE, Park CY, Bernstein BE, Aifantis I, Levine RL, Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 210, 2641–2659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen M, Herring BP, Regulation of microRNAs by Brahma-related gene 1 (Brg1) in smooth muscle cells. J Biol Chem 288, 6397–6408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A, Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T, Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Lawlor MW, Alexander MS, Viola MG, Meng H, Joubert R, Gupta V, Motohashi N, Manfready RA, Hsu CP, Huang P, Buj-Bello A, Kunkel LM, Beggs AH, Gussoni E, Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am J Pathol 181, 961–968 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS, High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis 41, 179–184 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR, Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes & development 18, 2614–2626 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishijo K, Hosoyama T, Bjornson CR, Schaffer BS, Prajapati SI, Bahadur AN, Hansen MS, Blandford MC, McCleish AT, Rubin BP, Epstein JA, Rando TA, Capecchi MR, Keller C, Biomarker system for studying muscle, stem cells, and cancer in vivo. Faseb J, (2009). [DOI] [PMC free article] [PubMed]
- 58.Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF, Iruela-Arispe ML, VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn 235, 3413–3422 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA, Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193, 741–754 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.