

Abstract
Background:
Mutations or truncation of the ligand-binding domain (LBD) of androgen receptor (AR) underlie treatment resistance for prostate cancer (PCa). Thus, targeting the AR N-terminal domain (NTD) could overcome such resistance.
Methods:
Luciferase reporter assays after transient transfection of various DNA constructs were used to assess effects of E1A proteins on AR-mediated transcription. Immunofluorescence microscopy and subcellular fractionation were applied to assess intracellular protein localization. Immunoprecipitation and mammalian two-hybrid assays were used to detect protein-protein interactions. qRT-PCR was employed to determine RNA levels. Western blotting was used to detect protein expression in cells. Effects of adenoviruses on prostate cancer cell survival were evaluated with CellTiter-Glo assays.
Results:
Adenovirus 12 E1A (E1A12) binds specifically to the AR. Interestingly, the full-length E1A12 (266 aa) preferentially binds to full-length AR, while the small E1A12 variant (235 aa) interacts more strongly with AR-V7. E1A12 promotes AR nuclear translocation, likely through mediating intramolecular AR NTD-LBD interactions. In the nucleus, AR and E1A12 co-expression in AR-null PCa cells results in E1A12 redistribution from CBX4 foci, suggesting a preferential AR-E1A12 interaction over other E1A12 interactors. E1A12 represses AR-mediated transcription in reporter gene assays and endogenous AR target genes such as ATAD2 and MYC in AR-expressing PCa cells. AR-expressing PCa cells are more sensitive to death induced by a recombinant adenovirus expressing E1A12 (Ad-E1A12) than AR-deficient PCa cells, which could be attributed to the increased viral replication promoted by androgen stimulation. Targeting the AR by E1A12 promotes apoptosis in PCa cells that express the full-length AR or C-terminally truncated AR variants. Importantly, inhibition of mTOR signaling that blocks the expression of anti-apoptotic proteins markedly augments Ad-E1A12-induced apoptosis of AR-expressing cells. Mechanistically, Ad-E1A12 infection triggers apoptotic response while activating the PI3K-AKT-mTOR signaling; thus, mTOR inhibition enhances apoptosis in AR-expressing PCa cells infected by Ad-E1A12.
Conclusion:
Ad12 E1A inhibits AR-mediated transcription and suppresses PCa cell survival, suggesting that targeting the AR by E1A12 might have therapeutic potential for treating advanced PCa with heightened AR signaling.
Keywords: Androgen receptor, AR splice variants (AR-Vs), PI3K-kinase (PI3K)-Akt-mTOR signaling, transcriptional regulation, prostate cancer gene therapy
1. Introduction
Prostate cancer (PCa), a form of adenocarcinoma developed in prostate epithelium, is the most commonly diagnosed male malignancy and the second leading-cause of cancer-related death in men (1). Androgen receptor (AR), a member of the nuclear receptor (NR) superfamily, is an androgen-dependent transcriptional factor that regulates gene expression required for the survival and proliferation of prostate epithelial cells, thereby promoting prostatic development and, ironically, driving prostatic tumorigenesis and PCa progression (2,3). AR is a modular protein with several defined structural domains with distinct functions. The long and intrinsically unstructured N-terminal domain (NTD) is required for transcription activation (4), probably through recruiting coregulators such as CBP (CREBBP) and p300 (EP300) (5,6). The short DNA-binding domain (DBD) folds into a defined structure that recognizes specific DNA sequences known as androgen-responsive elements (AREs) (7). The structured C-terminal domain, linked to DBD by an unstructured hinge region, contains the ligand-binding pocket that specifically binds androgens, thus termed ligand-binding domain (LBD) (2,8).
Because androgens are critical for AR nuclear translocation and transactivation of AR target genes, androgen deprivation (ADT) and antiandrogen therapies through either surgical or medical castration have been widely used for PCa therapies (9). These therapies work well initially, but in recurring PCa, the cancer cells become more aggressive and castration-resistant. Once metastatic diseases develop from castration-resistant PCa (CRPC), which occurs near the end stages of the disease, the hope of patient survival diminishes dramatically with a median survival of only about 18 months. Surprisingly, the transcriptional activity of AR remains crucial for the survival and proliferation of PCa cells even after they become castration-resistant (10). Indeed, in model prostate xenografts when tumor becomes castration resistant, gene expression analysis revealed that the AR signaling pathways are consistently upregulated (11). In addition, targeting CYP17, an enzyme key to androgen and estrogen biosynthesis, with the specific inhibitor abiraterone acetate (Zytiga), improves CRPC patient survival, indicating continued dependence of CRPC on AR signaling pathways (12). Beneficial clinical responses were also observed with the second-generation AR antagonist MDV3100 (enzalutamide) for CRPC (13). A decline of prostate-specific antigen (PSA) was used as the primary surrogate indicator of antitumor activity in early clinical trials using both Zytiga and MDV3100 (10). PSA expression is driven predominantly by the upstream ARE-containing enhancer. PSA levels eventually increase and are associated with disease progression for the majority of patients receiving treatment with Zytiga and MDV3100, suggesting that AR reactivation may underpin the therapeutic resistance (10). Acquired AR LBD mutations and increased intratumoral androgen biosynthesis have been shown to contribute to therapeutic resistance to MDV3100 (14) and Zytiga (15), respectively. The elevated expression of C-terminally truncated AR variants (AR-Vs) has been observed in CRPC and correlates with poor patient prognosis (16–20). AR-Vs contain NTD and DBD but not LBD, and thus are capable of directing AR target gene expression (21). Indeed, AR-Vs are ligand-independent and constitutively active transcriptional activators (22,23). These independent mechanisms of AR reactivation highlight the challenges of using current hormone therapies to achieve durable inhibition of AR signaling. Thus, alternative strategies to suppress AR reactivation are needed to attain durable AR inhibition for effectively treating CRPC.
The early region 1A (E1A) of the adenovirus genome expresses several alternatively spliced mRNAs encoding various E1A isoforms (24). The two widely studied Ad5 E1A splice variants encode for the 289-residue (herein referred to as E1A5) and the 243-residue (e1a5) proteins. These E1A proteins contain several conserved regions (CR1–4). E1A is intrinsically unstructured with the exception for a zinc-finger structure encoded by CR3 that is deleted in the e1a5 isoform (25). E1A5 is a powerful transcriptional activator owing to its strong interactions with the transcriptional machinery and coregulators such as p300/CBP (26). E1A does not directly bind to DNA, but CR3 along with adjacent sequences have been shown to interact with diverse DNA-binding transcription factors (TFs) (27,28). The 243-residue e1a5 does not bind to TFs (27,28), although it can modulate transcription, for example, by sequestering coregulators. Indeed, e1a5 seems to inhibit AR-mediated transcription, presumably through sequestering coactivators p300/CBP (5,29). Interestingly, the N-terminal domain of E1As contains a conserved sequence motif that resembles the hydrophobic NR-interacting sequence found in corepressors (CoRs) such as NCORs. Through this motif, E1As can compete against CoRs for binding to NRs, thereby promoting NR activation (30,31).
There are >50 serotypes of human adenoviruses. These viruses display marked genetic diversity and clinical phenotypes (32,33). Although CR1–4 are retained in E1As of all human adenoviruses, they exhibit significant variations in other regions of the proteins. Consequently, E1As of different serotypes may exert considerable functional differences. Indeed, distinct functions have been observed for Ad12 E1A (herein referred to as E1A12) (34,35). For example, E1A12 but not E1A5 can enhance the transcriptional repression function of the orphan NR COUP-TFII (also known as NR2F2) (34). E1A12, but not E1A5, has been reported to inhibit the phosphorylation of the p65 subunit of NF-κB by the protein kinase A catalytic subunit (PKAc), resulting in the suppression of NF-κB-mediated transactivation (35,36). We report here that E1A12 binds to the AR and suppresses the transactivation function of AR. Furthermore, a recombinant adenovirus expressing E1A12 (Ad-E1A12) seems to kill PCa cells through inhibition of AR signaling.
2. Materials and methods
2.1. Cell lines and antibodies.
Human PCa cell lines LNCaP, DU145 and PC-3, human embryonic kidney cell line HEK293 and human osteosarcoma cell line Saos-2 were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM). R1-AD1 is a subline of PCa cell line CWR-R1. R1-I567 and R1-D567 are engineered derivatives of R1-AD1 that express C-terminally truncated AR (AR-Vs) (23). Media were supplemented with penicillin to 10 units/mL, and streptomycin to 10 μg/mL, 10% bovine calf serum (BCS), fetal bovine serum (FBS), or charcoal-stripped FBS (CSS), as indicated. All cells were cultured in a 37°C incubator with 5% carbon dioxide (CO2). Cell transfection was done with Lipofectamine® 2000 (Life Technologies).
Anti-AR (sc-815 and sc-7305), anti-normal rabbit IgG (sc-2027), anti-Ad5 E1A (M73), anti-PSA (sc-7316), anti-p53 (SC-126), lamin A/C (sc-7292), and lamin B (sc-6217) antibodies were purchased from Santa Cruz Biotechnology. Anti-pS473 AKT1 (2118–1) and anti-PCNA (2714–1) antibodies were from Epitomics. Anti-FLAG (M2, F1804) and anti-α-tubulin (T5168) mouse monoclonal antibodies were obtained from Sigma. Anti-pS235/236 S6 (2211), anti-Survivin (2808), anti-acetyl-CoA synthetase (ACSS2, 3658), anti-ACC1 (ACACA, 3676), and anti-AR (5153) antibodies were from Cell Signaling Technology. Anti-H2AX (A300–083A) and anti-γ−H2AX (A300–081A) were from Bethyl Laboratories. Anti-FAS (FASN, 10624–2-AP) antibody was from ProteinTech. Anti-HSP60 (H99020), CtBP1 (612042) and anti-p21 (556431) antibodies were obtained from BD Biosciences. Anti-GFP mouse monoclonal antibody was from Babco. Anti-adenovirus type 5 DNA-binding protein (DBP) antibody (B6–8) was as described previously (37).
2.2. DNA constructs and recombinant adenoviruses.
The expression plasmids for wt E1A12 and various mutants are reported previously (38). The full-length AR (GFP-AR, plasmid # 28235) and AR-V7 (GFP-AR-V7, plasmid # 86856) fused to GFP were obtained from Addgene. The mCherry-AR construct was made by inserting the full-length AR cDNA fragment from the GFP-AR plasmid at the 3’ end of the mCherry coding sequence. FLAG-tagged AR NTD (1–566) and AR NTD-DBD-Hinge (1–802) were generated by PCR using the GFP-AR as template and cloned into pExchange-3B. FLAG-tagged full-length AR and constructs with a specific deletion within the AR NTD were described (39). Ad-E1A12 and other viruses with mutated E1A12 were constructed as reported previously (38). Recombinant viruses were generated through homologous recombination between pShuttle-CMV carrying the E1A12 expression cassette and pAdEasy-1, an E1 and E3-deleted Ad vector (Agilent Technologies) (40). Ad-E1A12 was packaged in HEK293 cells. The recombinant viruses expressing a mutated E1A12 were constructed similarly. A large-scale preparation of purified Ad-E1A12 viral particles was done by ViraQuest, Inc. Ad-eGFP and wild-type (wt) Ad5 were purchased from ViraQuest, Inc.
2.3. Luciferase reporter gene assay.
The AR NTD (aa 1–566) was fused to the C-terminus of Gal4 DNA-binding domain (Gal4-BD). The firefly luciferase reporter is under the control of an artificial promoter containing five copies of Gal4-binding sites upstream of the Ad5 E4 core promoter consisting of a TATA box and an initiator element (41). This reporter and the sea pansy (Renilla) luciferase reporter were co-transfected into Saos-2 or DU145 cells along with indicated combinations of expression plasmids in triplicate. At 24 h after transfection, cells were washed twice with phosphate-buffered saline (PBS) and then lysed for the dual luciferase assay according to manufacturer’s protocol (Promega). The firefly luminescence readouts were normalized against the Renilla luciferase readouts in each transfection. For mammalian two-hybrid assays, the AR LBD (aa 690–919) was fused to Gal4-BD and the AR NTD (aa 1–566) was fused to the C-terminus of VP16 activation domain. Transfections and luciferase assays were performed similarly as above.
2.4. Cell viability assay.
Cells were seeded in triplicate in a 96-well plate. At 24 h after seeding, viruses were added to cell cultures. At 2 h after viral infection, vehicle (DMSO) or a specific inhibitor was added to the cell cultures. At 96 h after adding adenoviruses, cell viability assays were performed using CellTiter-Glo reagent (Promega) essentially as reported previously (42). The luminescence readouts were subsequently averaged and normalized against a relevant control.
2.5. Quantitative real-time RT-PCR.
LNCaP or R1-AD1 cells were uninfected or infected with Ad-eGFP, or Ad-E1A12 (1,000 vps/cell). At 48 h post-infection, RNAs were extracted using the RNeasy kit (Qiagen). cDNAs were synthesized from total RNAs using MultiScribe reverse transcriptase kit (Applied Biosystems), which were used as templates for real-time PCR with the SYBR-green detection method. Quantification was as described previously (38). The PCR primers were: AR (5’- CAGTGGATGGGCTGAAAAAT-3’ and 5’-GGAGCTTGGTGAGCTGGTAG-3’); FKBP5 (5’- AGGAGGGAAGAGTCCCAGTG-3’ and 5’-TGGGAAGCTACTGGTTTTGC-3’); ATAD2 (5’- TCAGGCTCCATTGGAAAAAC-3’ and 5’-CCTGCGGAAGATAATCGGTA-3’); MYC (5’- AGCGACTCTGAGGAGGAACA-3’ and 5’-CTCTGACCTTTTGCCAGGAG-3’); GLUD1 (5’- GGAGGTTCACCATGGAGCTA-3’ and 5’-CCTATGGTGCTGGCATAGGT-3’); TFRC (5’- AAAATCCGGTGTAGGCACAG-3’ and 5’-CACCAACCGATCCAAAGTCT-3’); and ACTB (5’- GCTCCTCCTGAGCGC AAGTACTC-3’ and 5’ - GTGGACAGCGAGGCCAGGAT-3’).
2.6. Western blotting.
Cells were seeded and cultured in multi-well plates. At 24 h after seeding, adenovirus alone or together with a specific inhibitor was added (drug was added 2h after viral infection to avoid possible interference with viral entry). At 24 h after adding adenovirus, both floating and adherent cells were lysed with 1×Passive Lysis Buffer (Promega). Lysates were frozen at −80°C overnight and thawed at room temperature. Protein samples in 1×SDS sample buffer were heated at 95°C for 5 min. The samples were loaded on an SDS-polyacrylamide gel. The proteins were then blotted onto a membrane (Immobilon-P, Millipore), and incubated with a primary antibody at 4 °C overnight with rotation. After washes, the membrane was incubated with a proper secondary antibody at room temperature for 45 min. Proteins were detected using the Immobilon Western Chemiluminescent kit (Millipore).
2.7. Immunoprecipitation (IP).
LNCaP or R1-AD1 cells were infected with Ad-E1A12 at the MOI of 100 vps/cell. The infected cells were collected at 48 h post infection by scraping. 293T or Saos-2 cells cultured in 10-cm or 6-well plates were transfected with various combinations of expression plasmids. The transfected cells were harvested by trypsinization 24 h after transfection. Cell pellets were washed twice with cold PBS and then lysed with the RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) along with a protease inhibitor cocktail (P8340, Sigma). Lysates were frozen at −80 °C overnight and thawed at room temperature. Lysates were rotated at 4 °C for 30 min. The freeze-thaw cycle was repeated once. Cell debris was cleared by centrifugation, and 10% of the lysates were reserved as input samples. The remaining lysates were incubated with either a proper antibody or the control anti-IgG antibody at 4 °C overnight with rotation. Protein G-agarose slurry (50 μl) was added to the cell lysate-antibody mixture and was rotated for 2 h at 4 °C. The mixture was then centrifuged and washed four times with cold RIPA buffer. The beads with bound proteins were suspended in 50 μL RIPA buffer plus 10 μL of 6 × SDS sample buffer. The samples were heated at 95°C for 5 min and analyzed by Western blotting.
2.8. Immunofluorescence microscopy (IF).
PC-3, DU145 or Saos-2 cells were seeded directly on a culture plate or on glass coverslips. At 24h after seeding, the cells were transfected with various DNA constructs. Cells were cultured in medium with regular bovine serum or CSS. For cells cultured in medium with CSS, vehicle (ethanol) or R1881 (1 nM final concentration) was added. Live cell imaging was conducted using a Leica DM IRBE inverted microscope at various times after R1881 addition. For cells cultured on coverslips, the transfected cells were fixed with 4% paraformaldehyde at 24h after transfection and were subjected to an antibody staining protocol as described (43).
2.9. Cell fractionation.
LNCaP cells cultured in CSS medium were exposed to the vehicle control (ethanol) or R1881 (10 nM), or infected with Ad-eGFP) or Ad-E1A12). At 48 h after treatments, cells were harvested using a cell lifter. Cells were pelleted by centrifugation at 700 × g for 10 min at 4°C. The pellets were washed once with PBS, and pelleted again. Cells were resuspended in ice-cold hypotonic buffer (10 mM Tris-HCl, pH 8.0, 1 mM KCl, 1.5 mM MgCl2 and 1 mM DTT) containing 100-fold diluted protease inhibitor cocktail (Sigma P8340). The cell suspension was incubated for 30 min on ice. The intact nuclei were pelleted by centrifugation at 4°C at 10,000 × g for 10 min. The supernatant (the cytoplasmic fraction) was collected. The pellet (the nuclear fraction) was washed once with ice-cold 1 × TE buffer (10 mM Tris HCl and 1 mM EDTA, pH 8), and dissolved with a protein lysis buffer (50 mM Tris-HCl pH 7.5, 0.5% Igepal-CA630, 5% glycerol, 150 mM NaCl, 1.5 mM MgCl2, and 25 mM NaF). Protein concentrations of each subcellular fraction were determined using the Bradford method and an equal amount of proteins was loaded for SDS-PAGE and immunoblotting.
3. Results
3.1. E1A12 inhibits AR NTD-mediated transactivation.
Previous studies indicated that the 243-residue isoform of Ad5 E1A (ela5) interferes with AR activation, possibly through sequestration of transcriptional coactivators p300/CBP (5). To assess potential effects of the large E1A isoforms of various Ad species (serotypes), we conducted luciferase reporter gene assays using AR NTD fused to Gal4-BD. AR NTD increased the luciferase activity by ~20-fold, similar to c-Jun-mediated activation (Fig. 1A). Both Ad12 (E1A12) and Ad5 (E1A5) E1A did not significantly affect the reporter activity when coexpressed with Gal4-BD (Fig. 1A), suggesting that E1A is not recruited to this artificial promoter. Surprisingly, E1A12 expression markedly inhibited AR NTD-mediated transactivation (Fig. 1A lane 9 and Fig. 1B lane 10). In contrast, coexpression of E1A5 dramatically increased AR NTD-mediated transactivation (Fig. 1A, lane 10 and Fig. 1B lane 9). The expression levels of the transfected constructs were assessed using Western blotting. The E1A12 and E1A5 expression levels were similar (note that the polyclonal anti-E1A12 antibody recognizes both E1A12 and E1A5), although AR NTD levels were slightly reduced when either E1A5 or E1A12 were coexpressed (Fig. 1A). Because E1A12 and E1A5 displayed opposite effects on AR NTD-mediated transcription, the functional impact of E1As on AR must not be due to the moderate reduction of AR NTD expression. Indeed, similar effects on AR-mediated transactivation by E1A12 (repression) and E1A5 (activation) were seen, even though the levels of AR NTD proteins were largely unchanged (Fig. 1B lanes 9 and 10). We also examined the effects of E1As from other Ad species on AR NTD-mediated transactivation. We found that E1As from Ad3, Ad4, Ad9 and Ad40 all suppressed AR NTD-mediated transcription (Fig. 1B). Surprisingly, the levels of AR NTD fusion construct with Gal4-BD were markedly reduced when coexpressed with these E1A constructs (compare lanes 11–14 to lane 2, the lower panel of Fig. 1B). Thus, it is possible that these E1A constructs might trigger degradation of AR NTD, which could also contribute to the inhibition of AR NTD-mediated transactivation.
Figure 1. E1A12 represses the AR-mediated transactivation.
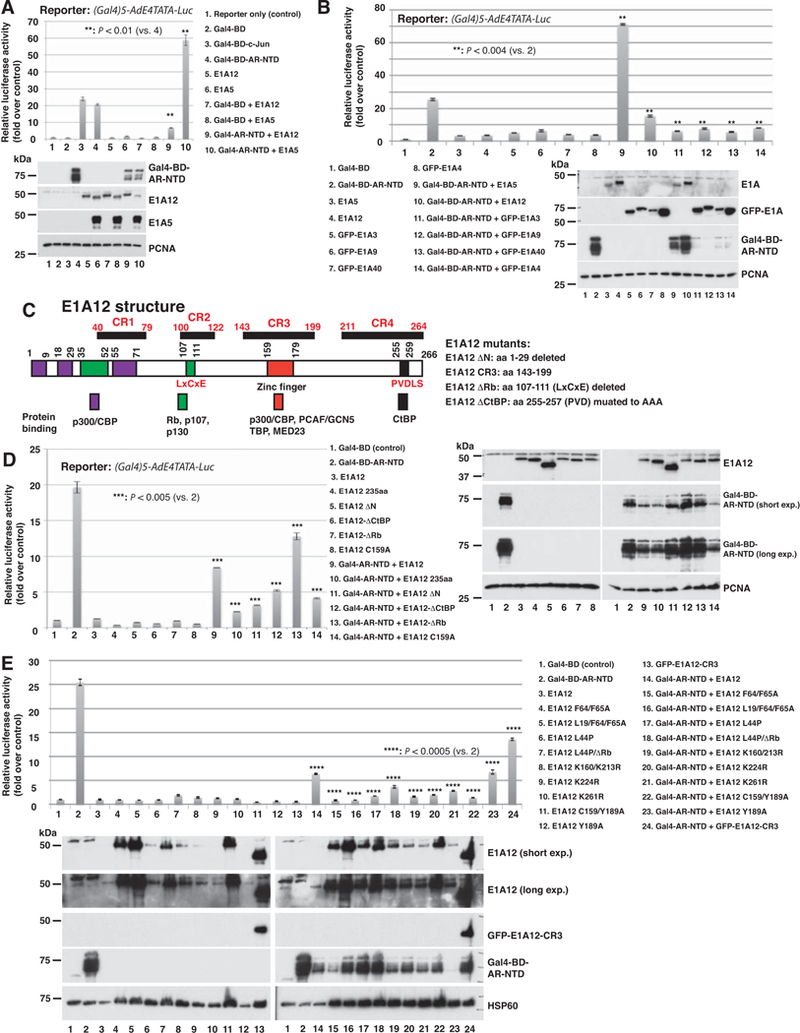
(A) E1A12 inhibits, but Ad5 E1A (E1A5) activates, AR NTD-mediated transactivation. The luciferase reporter containing five tandem Gal4-binding sites was cotransfected with the indicated constructs or the combinations thereof into Saos-2 cells. Luciferase activity was measured 24 h after transfection. Shown are the average values of relative reporter activity from three independent transfections along with the standard error of the mean (SEM). Lower panel: the expression of transfected constructs was detected with Western blotting. (B) The large isoforms of E1A from Ad3 (E1A3), Ad9 (E1A9), Ad40 (E1A40) and Ad4 (E1A4) also suppress the AR NTD. Luciferase reporter assays were done as in (A). Western blotting data of the transfected cells are shown in the lower panel. The Ad5 and Ad12 E1As were detected with an antiserum raised against Ad12 E1A, whereas the E1As from other Ad species were detected with an anti-GFP antibody. (C) The domain structure of E1A12. The conserved regions (CR1-CR4) along with sequences that bind to cellular proteins are depicted. The numbers refer to the positions of amino acid residues. The mutant constructs of E1A12 with specific point mutations or deletions that were used for reporter assays are also shown. (D and E) Effects of E1A12 mutations on AR NTD-mediated transcription. The reporter assays were done as in (A). Western blotting data of the transfected constructs are also shown.
E1A proteins interact with diverse cellular proteins through their conserved regions (CR1–4, Fig. 1C). We thus assessed the effects of various E1A12 mutants with point mutations or short deletions on the AR transactivation function. Among the E1A12 mutants we have tested, all suppressed AR NTD-mediated transactivation, although some mutants were either more potent or less so than wt E1A12 (Fig. 1D and1E). Notably, N-terminal deletion (E1A12-ΔN with the deletion of aa 1–29) or point mutations of the hydrophobic residues that are involved in binding the TAZ2 domain of CBP/p300 (F64/65A) (44) seem to enhance E1A12-mediated repression of AR NTD (Fig. 1D lane 11; Fig. 1E lanes 15 and 16), suggesting that E1A-p300/CBP interaction might antagonize E1A12-mediated inhibition of AR. Interestingly, changing C159 that is presumably a Zn-binding residue in the Zn-finger region to Ala also appears to intensify AR repression (Fig. 1D lane 14 and Fig. 1E lane 22). Thus, the integrity of Zn-finger in CR3 seems dispensable for AR repression. By contrast, the deletion of the high-affinity Rb-binding site (known as LxCxE motif) in E1A12 (E1A12-ΔRb) reduced repression of AR NTD (Fig. 1D lane 13). Finally, the E1A12 CR3 region (aa 143–199) alone was sufficient to inhibit AR NTD-mediated transactivation, although less potent than the wt E1A12 (Fig. 1E lane 24). E1A12 also inhibited transactivation mediated by the full-length AR in DU145 cells (Fig. S1). Additionally, the E1A12 splice variant lacking CR3 (E1A12 235aa) also suppressed AR NTD-mediated transactivation (Fig. 1D lane 10). Note that E1A12 235aa significantly inhibited the reporter in the absence of AR NTD (Fig. 1D lane 4). We detected the expression of the various transfected constructs (Fig. 1D right panel and Fig. 1E lower panel). Notably, several E1A12 constructs were expressed at higher levels (E1A12-ΔN, lanes 5 and 11 in Fig. 1D right panel; E1A12 F64/F65A, L19A/F64/F65A, and C159/Y189A, lanes 4, 5, 11, 15, 16 and 22 in Fig. 1E lower panel). The levels of Gal4-BD-AR-NTD construct were variable and in general the protein levels of this construct did not correlate with its repression by the E1A12 constructs. Taken together, these results indicate that E1A12 proteins (both 266 aa and 235 aa splice variants) employ a novel mechanism to interfere with AR-mediated transcription, probably through a direct physical interaction of the AR (see below).
3.2. E1A12 binds to the AR.
Inhibition of AR-mediated transactivation by E1A12 could stem from coactivator sequestration or direct physical interactions between the AR and E1A12. To assess whether E1A12 binds to AR, we conducted immunoprecipitation (IP) experiments. In transfected 293T cells, both the 266 aa and 235 aa variants of E1A12 interacted with the full-length AR and AR-V7 (Fig. 2A). Interestingly, the large E1A12 variant (266 aa) exhibited a higher affinity to the full-length AR than to AR-V7 (compare lane 3 to 2 in Fig. 2A). In contrast, the small E1A12 variant (235 aa) appeared to bind AR-V7 more strongly than to the full-length AR (compare lane 6 to 5 in Fig. 2A). These data suggest that both the AR NTD and LBD may contribute to binding the large E1A12 variant, while the AR NTD may be the primary binding site for the small E1A12 variant.
Figure 2. E1A12 266 aa and 235 aa interact with the AR.
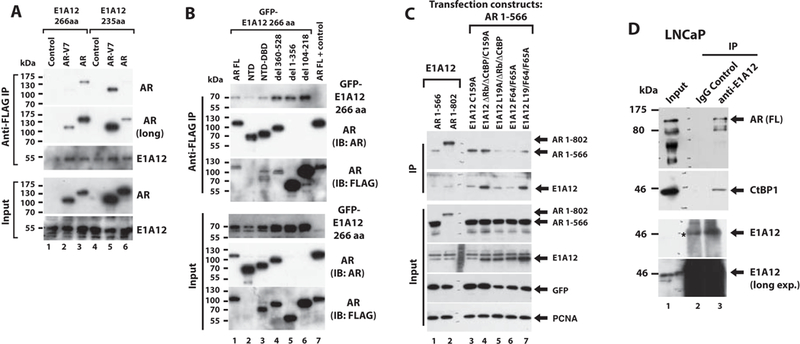
(A) Co-IP of E1A12 266 aa and 235 aa with the full-length AR or AR-V7. 293T cells were transfected with vectors expressing FLAG-E1A12 (266 aa or 235 aa), mCherry-full-length AR, or GFP-AR-V7 as indicated. The lysates of the transfected cells were subject to anti-FLAG IP. (B) The indicated FLAG-AR constructs (full-length (FL), NTD (1–566), NTD-DBD (aa 1–623), del 360–528, del 1–356 and del 104–218) along with GFP-E1A12 266 aa were transfected into 293T cells. Cotransfection of the FLAG-AR FL along with a control plasmid serves as a control (lane 7). anti-FLAG IP was done as in (A). Note that the AR mAb (Cell Signaling Technology #5153) used for Western blotting did not bind to the AR del 1–356 and AR del 104–216. The anti-FLAG mAb M2 was used to detect these constructs. (C) Interactions of the E1A12 mutants and the AR NTD. The indicated constructs were transfected into Saos-2 cells, and the extracts of the transfected cells were subjected to IP with an anti-E1A12 antibody and the co-precipitated proteins were detected with an anti-AR NTD antibody. The GFP construct was included as a control for transfection and PCNA was detected as a loading control. (D) E1A12 binds to the endogenous AR. LNCaP cells were infected with Ad-E1A12 and the extracts of the infected cells were subjected to IP with a polyclonal anti-E1A12 antibody. The control IP was done with a rabbit IgG antibody. The coprecipitated materials were analyzed by SDS-PAGE and Western blotting with a mouse monoclonal anti-AR antibody. The known E1A-binding protein CtBP1 was detected as a positive control. Note that E1A12 (lane 3) appeared at the same position as a cross-reactive band in the IgG (control) lane (denoted with *, lane 2).
In transfected 293T cells, the AR NTD was sufficient to bind E1A12 266 aa variant, albeit at a lower affinity (Fig. 2B, lane 2). The AR NTD-DBD fragment (Fig. 2B lane 3) and AR constructs with a specific deletion in the NTD (del 360–528, lane 4; del 1–356, lane 5; del 104–218, lane 6, Fig. 2B) were able to bind E1A12 266 aa. We then assessed the interaction of AR with various mutated forms of E1A12. Again, E1A12 266 aa interacted efficiently with the AR NTD (1–566) and AR NTD-DBD-Hinge (1–802) (Fig. 2C lanes 1 and 2). Nonetheless, the AR NTD-DBD-hinge construct seemed to bind to E1A12 more efficiently than the AR NTD (compare lane 2 with lane 1 in the IP panels of Fig. 2C), suggesting that the AR DBD and/or the hinge region might stabilize the AR-E1A12 interactions. The deletion of the Rb and CtBP-binding sites or the mutation of C159 in the Zn-finger of E1A12 did not affect the AR-E1A12 interactions (Fig. 2C). Likewise, mutation of CBP/p300-binding residues in the N-terminal domain of E1A12 did not impact the AR-E1A12 interactions. Thus, the AR-E1A12 interaction is not likely mediated through a cellular protein that binds to E1A’s CRs. Endogenous full-length AR was efficiently co-precipitated with E1A12 in LNCaP cells (Fig. 2D). As a positive control, CtBP1 was also precipitated in the anti-E1A12 immunocomplexes (Fig. 2D). Similarly, E1A12 also coprecipitated with the endogenous AR in the prostate cancer cell line R1-AD1 (data not shown).
3.3. E1A12 promotes androgen-independent nuclear translocation of AR.
AR is sequestered in the cytoplasm in the absence of androgens. Upon ligand binding, AR undergoes conformational changes and translocates to the nucleus. As E1A12 interacted with AR in both nucleus and cytoplasm (Fig. 2), we wondered whether the E1A12-AR interaction would affect intracellular localization of AR. To test this, we used live cell imaging to track AR localization. We used the osteosarcoma Saos-2 cell line for this purpose, due to its lack of endogenous AR expression and the superior performance for fluorescence microscopy studies. AR was exclusively in the cytoplasm (97.6% of the transfected cells) in the absence of androgens in Saos-2 cells (Fig. 3A panels 1 and 2 and Fig. 3B). Both the 266aa and 235aa isoforms of E1A12 were exclusively or predominantly nuclear in the absence (panels 3–6, Fig. 3A) or presence of the androgen analog R1881 (panels 17–20 in Fig. 3A). Strikingly, coexpression of E1A12 266aa with AR resulted in marked nuclear accumulation of AR in the absence of androgens (Fig. 3A panels 7–10). In contrast, the 235aa isoform failed to promote AR nuclear translocation (panels 11–14). Quantitatively, 97.2% of the cells cotransfected with mCherry-AR and GFP-E1A12 266aa showed higher levels of nuclear AR than AR in the cytoplasm (Fig. 3B). In contrast, 96.5% of the cells coexpressing mCherry-AR and GFP-E1A12 235aa showed cytoplasmic AR localization in the absence of R1881 (Fig. 3B). As described above, E1A12 266 aa seemed to preferentially bind the full-length AR, while E1A12 235 aa interacted more strongly with the AR NTD (Fig. 2). These data suggest that physical interaction between E1A12 266 aa and AR might induce a conformational change of the AR to promote AR nuclear translocation in the absence of androgens. Up to 90 min after the addition of R1881, AR in a substantial fraction of the transfected cells (42.3%) remained in the cytoplasm (Fig. 3A panels 15, 16, 29, and 30 and Fig. 3B right panel). It is clear that AR nuclear localization increased steadily with concomitant reduction of cytoplasmic AR when E1A12 266 aa was coexpressed in the presence of R1881 (Fig. 3A panels 21–24 and 35–38). Intriguingly, the 235 aa E1A12 protein facilitated AR nuclear translocation in the presence of R1881 (Fig. 3A panels 25–28 and 39–42). In fact, at 30 min after R1881 addition, 98.9% of the cells expressing both mCherry-AR and GFP-E1A12 235 aa exhibited higher levels of the nuclear AR than the cytoplasmic AR, whereas AR remained predominantly in the cytoplasm in the majority (65%) of the transfected cells expressing mCherry-AR alone (Fig. 3B middle panel). We further confirmed that E1A12 also enabled AR nuclear localization in the absence of androgens in PC-3 cells (Fig. S2). Quantitatively, nearly 100% of the transfected cells showed higher levels of the nuclear AR than the cytoplasmic AR. Notably, R1881-mediated AR nuclear import was more efficient in PC-3 cells than in Saos-2, in which AR remained predominantly in the cytoplasm at 30 min after the addition of R1881 in 65% of the transfected Saos-2 cells (Fig. 3A panels 15 and 16 and Fig. 3B middle panel). Even at 90 min after the addition of R1881, 42.3% of the transfected Saos-2 cells still showed higher levels of AR in the cytoplasm than in the nucleus (Fig. 3B right panel). However, AR nuclear translocation was complete at 24 h after R1881 addition (Fig. S3). By contrast, R1881 promotes rapid AR nuclear import in PC-3 cells. We observed the AR nuclear accumulation at 10 min after R1881 addition; 82% of the transfected PC-3 cells exhibited higher AR levels in the nucleus than in the cytoplasm (Fig. S4). Taken together, these data demonstrate that AR-E1A12 interaction promotes AR nuclear translocation in an androgen-independent manner.
Figure 3. E1A12 promotes AR nuclear entry independently of androgens.
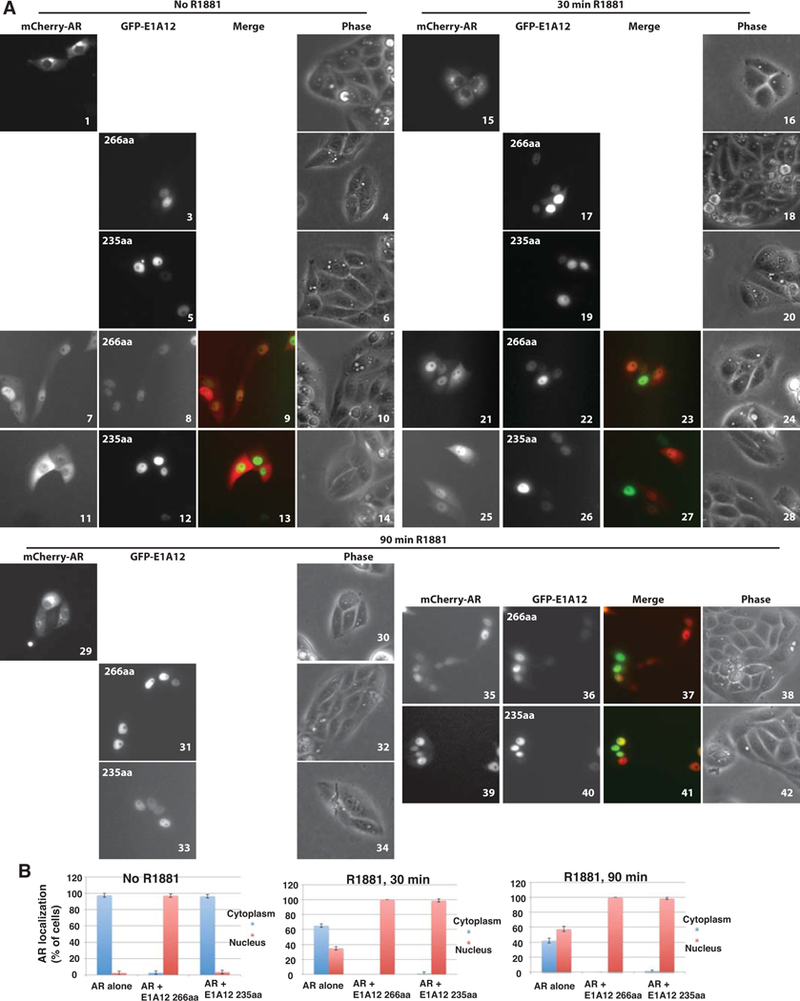
(A) Saos-2 cells were transfected with GFP-E1A12 266aa, 235aa, or mCherry-AR alone, or a combination thereof. At 8 h after transfection, the culture medium was removed and cells were washed with PBS. The DMEM medium supplemented with 10% CSS was then added to the culture plates. Cells were imaged at 24 h after transfection. Immediately after the initial imaging, the androgen analog R1881 was added to the cell cultures to the final concentration of 1 nM. The cells were then imaged at the indicated times after R1881 addition. Representative images are shown. (B) Quantification of AR subcellular localization. Cells exhibiting exclusive or predominant nuclear AR signal are grouped as cells with nuclear AR; likewise, cells with exclusive or predominant cytoplasmic AR signal are shown as cells with AR in the cytoplasm. For each transfection experiment, over 100 transfected cells in five random microscopic fields were quantified. The average percent values of the five fields along with SEM are shown.
To explore whether E1A12 could also promote nuclear translocation of the endogenous AR, LNCaP cells were cultured in CSS medium. The cells were exposed to the vehicle control (ethanol) or R1881, or infected with the control virus (Ad-eGFP) or a recombinant adenovirus expressing the E1A12 266 aa variant (Ad-E1A12). Cells were then fractionated into the cytoplasmic or nuclear fractions. As shown in Fig. 4A and B, R1881 markedly stimulated AR nuclear translocation. Compared to the cells infected with Ad-eGFP, the ratio of nuclear to cytoplasmic AR was clearly higher in cells infected with Ad-E1A12 (Fig. 4A and B). Of note, the viral DBP was detected as a surrogate marker of Ad-E1A12 infection, as the DBP expression strictly requires E1A12 expression (38). Thus, E1A12 266 aa was able to promote the nuclear entry of endogenous AR, although the potency to stimulate AR nuclear translocation by E1A12 266 aa was notably weaker that R1881 (Fig. 4B).
Figure 4. E1A12 enhances the nuclear translocation of endogenous AR.
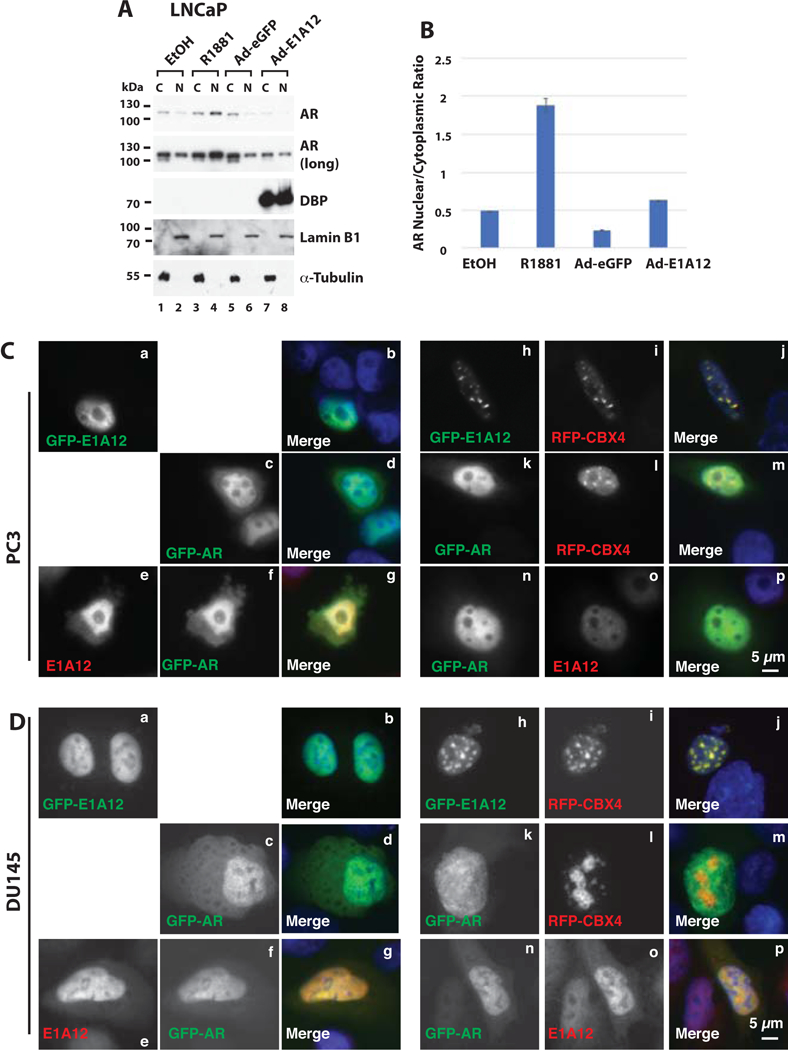
(A) LNCaP cells were cultured with DMEM medium with 10% CSS in a in 6-well plate. Cells were then exposed to ethanol, R1881 (10 nM), Ad-eGFP, or Ad-E1A12 (MOI at about 1,000 viral particles per cells) as indicated. At 48 h after treatment, cells were fractionated into the cytoplasmic (C) and nuclear (N) fractions. An equal amount of proteins in each fraction was loaded for SDS-PAGE and Western blotting with antibodies against the indicated proteins. (B) Quantification of AR levels in the cytoplasm and nucleus. The protein levels shown in (A) were quantified with the ImageJ software. The images of the blots after exposure for different lengths of time were analyzed and the mean band intensities were averaged. Shown is a bar graph of the ratio of the nuclear AR band intensity to that of cytoplasmic AR along with the calculated standard deviations. (C and D) Co-localization of E1A12 266aa and AR. E1A12 266aa, GFP-AR, RFP-CBX4 were expressed alone or in various combinations in PC-3 cells (C) and DU-145 (D) via transient transfection. Cells were fixed 24 h after transfection. In the cells transfected with E1A12 266aa and GFP-AR, E1A12 266aa was detected with a rabbit polyclonal antibody and goat anti-rabbit IgG conjugated to Alexa Fluor 594 fluorescent dye. AR and CBX4 were detected through the GFP and RFP tag, respectively. Cells shown in panels n to p in both C and D were also transfected with an untagged CBX4 construct that was not detected.
To further examine AR-E1A12 interactions, E1A12 and GFP-AR were expressed alone or together in the AR-negative PC-3 (Fig. 4C) or DU145 (Fig. 4D) cells. As shown in Fig. 4C and D, E1A12 localized exclusively in the nucleus when expressed alone (Fig. 4C and D panels a and b). GFP-AR was found in both the nucleus and cytoplasm, although the nuclear signal of GFP-AR was clearly much higher (panels c and d in Fig. 4C and D). By contrast, in cells co-expressing both E1A12 and GFP-AR, although predominantly localized to the nucleus, substantial presence of both AR and E1A12 in the cytoplasm became apparent (panels e-g, Fig. 4C and D), suggesting that E1A12 interacts with AR in both the cytoplasm and the nucleus. Furthermore, we tested whether AR could induce redistribution of E1A12 from its binding partners. To this end, we co-expressed CBX4 (also known as PC-2), a component of the Polycomb group (PcG) PRC1 complex that we found to colocalize with E1A12 in PcG nuclear bodies (panels h-j in Fig. 4C and D). CBX4 did not obviously colocalize with GFP-AR (panels k-m, Fig. 4C and D). However, when E1A12, GFP-AR and RFP-CBX4 were co-expressed, E1A12 was radically redistributed from PcG bodies to the nucleoplasm, similar to the intracellular distribution of GFP-AR (panels n-p, Fig. 4C and D), suggesting that AR expression forces E1A12 to dissociate from PcG bodies due to stronger AR-E1A12 interactions. Together, these data suggest that E1A12-AR interaction is quite strong, probably dominant over E1A12’s associations with other proteins.
3.4. E1A12 266aa bridges an interaction between AR NTD and LBD.
Androgens have been shown to promote biologically relevant inter- or intramolecular interactions between the AR N- and C-terminal domains (45,46). The AR N-terminal FXXLF motif is thought to interact with a hydrophobic binding pocket in LBD upon ligand binding. The AR N/C interaction has been suggested to promote AR nuclear translocation, to slow the dissociation of agonists, and to stabilize AR (46,47). We showed that E1A12 266 aa could promote the AR nuclear translocation in the absence of androgens (Figs. 3 and 4), which could be attributed to the ability of E1A12 266 aa to mediate an AR N/C interaction. To test this hypothesis, we conducted mammalian two-hybrid assays using Gal4-BD-LBD (aa 690–919) and NTD (aa 1–566)-VP16 hybrid constructs in transfected cells. Data presented in Fig. 5 indicate that the expression of E1A12 or the E1A12-ΔN mutant activated luciferase reporter mediated by the Gal4-BD-LBD and NTD-VP16 hybrids. Notably, E1A12 265 aa, E1A12 235 aa as well as E1A5 increased the reporter activity when cotransfected with Gal4-BD. However, the two latter E1A constructs did not enhance the reporter activity when coexpressed with Gal4-BD-LBD and NTD-VP16, indicating that E1A12 specifically mediates an NTD-LBD interaction. Furthermore, E1A12-ΔN construct by itself did not stimulate the reporter output when coexpressed with Gal4-BD or Gal4-BD-LBD, yet it markedly enhanced the reporter activity when coexpressed with Gal4-BD-LBD and NTD-VP16 constructs (Fig. 5A lane 13). Thus, the presence of the E1A12 CR3 domain is critical for bridging an AR NTD-LBD interaction (Fig. 5), and a direct recruitment of NTD-VP16 to the reporter by E1A12 could be excluded. These observations are consistent with the finding that E1A12 266 aa preferentially binds with the full-length AR (Fig. 2A). Since the AR DBD and hinge region are not included in our two-hybrid constructs, E1A12-mediated AR N/C interaction appears biochemically distinct from the androgen-mediated N/C interaction, which depends on the presence of a longer AR LBD fragment (aa 658–919) capable of high-affinity ligand-binding (46). These results demonstrate that E1A12 can bridge a unique AR N/C interaction (Fig. 5B).
Figure 5. The E1A12 266 aa variant bridges the AR N/C interaction.
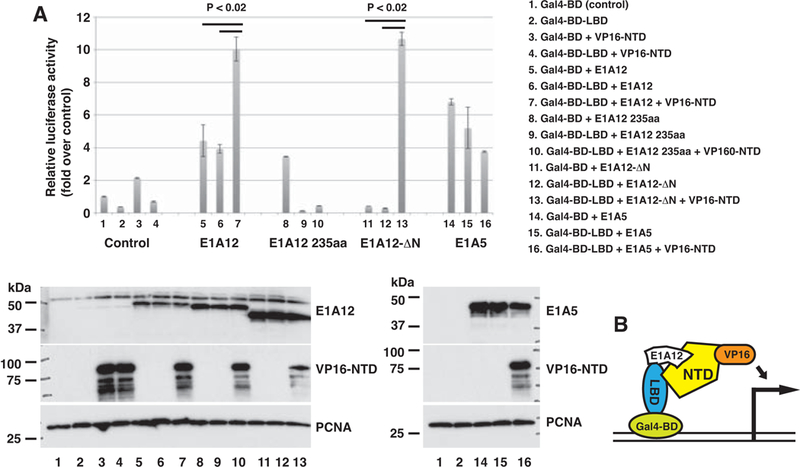
(A) The AR LBD (690–919) was fused to the Gal4-BD and the AR NTD (1–566) was attached to the VP16 activation domain. The luciferase reporter used in Fig. 1 along with a sea pansy (Renilla) luciferase reporter were cotransfected with the indicated plasmids into Saos-2 cells. Dual luciferase assays were conducted as in Fig. 1. Shown are average values along with SEM of three experiments. The P values were calculated based on Student’s t test. The expression of the transfected constructs is detected in a Western blot (lower panels). (B) A cartoon showing an E1A12 266aa-mediated AR NTD-LBD interaction.
3.5. E1A12 266 aa inhibits the expression of AR target genes.
To assess functional impact of E1A12 266 aa on AR-mediated gene expression, LNCaP and R1-AD1 cells were mock treated or infected with Ad-eGFP or Ad-E1A12. As shown in Fig. 6, Ad-E1A12 infection resulted in reduced expression of AR and classical AR target genes FKBP5 and ATAD2. MYC expression is regulated by AR (48). Ad-E1A12 also downregulated MYC as well as MYC target genes GLUD1 encoding glutamate dehydrogenase 1 (49), and TFRC encoding transferrin receptor protein 1 (50), which has been explored as a tractable biomarker for monitoring MYC inhibition in a PCa therapeutic setting (51). These results indicate that E1A12 266 aa can repress the expression of certain AR target genes.
Figure 6. Downregulation of AR target genes by E1A12 266aa.
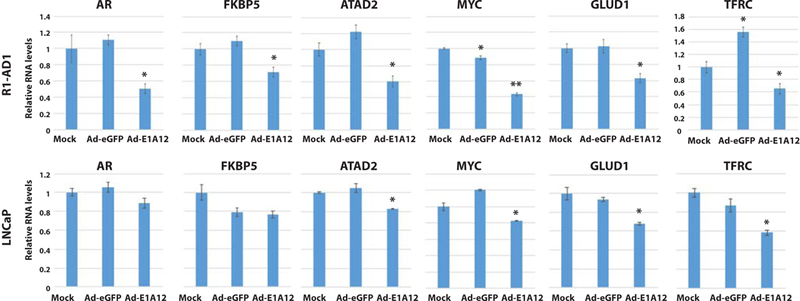
R1-AD1 and LNCaP cells were mock treated or infected with Ad-eGFP or Ad-E1A12 as in Fig. 4A. At 48 h post infection, RNAs were isolated and subjected to qRT-PCR with primers specific to the indicated genes. The mRNA levels were normalized against that of ACTB. Shown are average values of the relative RNA levels to the control (mock) along with SEM. *: P <0.05; **: P <0.01 (Student’s t test).
3.6. AR-expressing PCa cells display heightened sensitivity to Ad-E1A12-mediated killing.
Inhibition of AR by E1A12 may compromise the survival for PCa cells, in particular those depending on AR for growth. To test this, we infected PCa cell lines with Ad-E1A12. The viability of Ad-E1A12 infected cells decreased in a dose-dependent manner in LNCaP and R1-AD1 cells, and control Ad-eGFP did not significantly affect cell viability [the loss of viability of R1-AD1 line was seen at a high multiplicity of infection (MOI) of Ad-eGFP] (Fig. 7). Notably, PC-3 cells were more resistant to Ad-E1A12 than LNCaP and R1-AD1 cells; at the MOI of 1,000 Ad-E1A12 viral particles per cell (vps/cell), 82% of infected PC-3 cells were viable compared to 55%, and 35% of viable cells in Ad-E1A12-infected R1-AD1 and LNCaP cells, respectively (Fig. 7).
Figure 7. Ad-E1A12 in combination with pharmacological mTOR inhibition effectively kills PCa cells with AR expression.
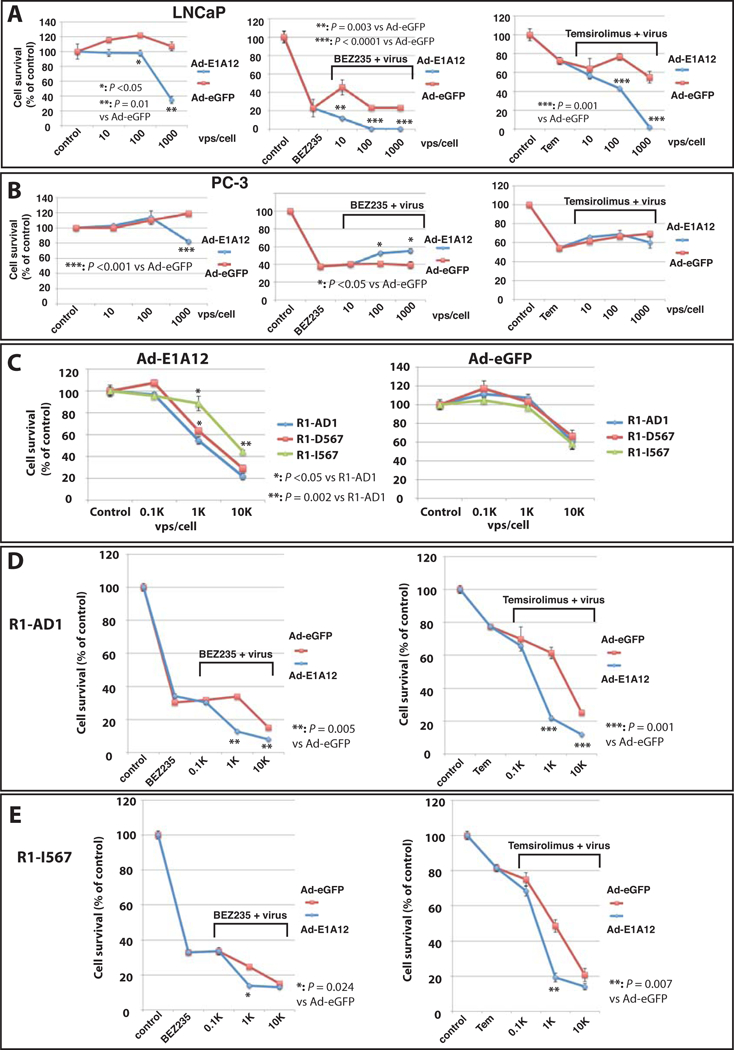
PCa cell lines LNCaP (A), PC-3 (B) and R1-AD1 and its derivatives (C, D and E) were infected with Ad-eGFP or Ad-E1A12 at the indicated MOIs. These cell lines were also treated with DMSO (control), an indicated mTOR inhibitor at 0.1 μM alone, or together with the viruses at various MOIs as indicated. Cell viability assays were conducted at 96 h post infection with CellTiter-Glo assay kit. Shown are average values along with SEM of three experiments. Tem: temsirolimus. The P values were calculated using Student’s t test.
The cross-inhibitory effects between the PI3K-AKT pathway and the AR signaling pathway have been observed. Inhibition of one pathway resulted in enhanced signaling of the other (52,53). Ad infection can activate the PI3K-AKT-mTOR pathway (54); we reported recently that Ad-E1A12 infection markedly activates this pathway (38). We thus hypothesized that pharmacological inhibition of the PI3K-AKT-mTOR pathway could synergize with Ad-E1A12 to kill PCa cells. Both LNCaP and PC-3 were sensitive to the dual PI3K/mTOR kinase inhibitor BEZ235 (55) and the allosteric mTORC1 inhibitor temsirolimus (Fig. 7). Cotreatment of LNCaP cells with Ad-E1A12 and BEZ235 or temsirolimus markedly increased cell death (Fig. 7A). Interestingly, neither BEZ235 nor temsirolimus sensitized Ad-E1A12-infected PC-3 to undergo cell death (Fig. 7B). In fact, Ad-E1A12 actually increased viability of BEZ235-treated PC-3 cells (Fig. 7B). Phenotypically, Ad-E1A12 infection of LNCaP cells with or without an mTOR inhibitor resulted in detachment from culture plates see (Fig. S5), whereas Ad-E1A12 infected PC-3 cells were not detached. Thus, E1A-mediated suppression of AR signaling in conjunction with the blockade of the PI3K-AKT-mTOR pathway greatly undermines the survival of PCa cells expressing AR.
PCa cells expressing AR variants lacking the LBD (AR-Vs) have been shown to acquire androgen-independent growth (18,23,56). Since E1A12 binds to the AR NTD (Fig. 2), we reason that PCa cells expressing AR-Vs should also be susceptible to E1A12 expression. We infected the engineered R1-AD1 cells only expressing AR-Vs (R1-D567 and R1-I567). The parental cell line expressing full-length AR (R1-AD1) was more sensitive to Ad-E1A12, compared to R1-D567 and R1-I567 (Fig. 7C). However, both R1-D567 and R1-I567 were also sensitive to Ad-E1A12-mediated killing, although R1-I567 was notably more resistant to Ad-E1A12 infection (Fig. 7C). In contrast, all three lines displayed similar survival profiles upon infection with the control Ad-eGFP virus (Fig. 7C). Similar to LNCaP cells, both BEZ235 and temsirolimus, albeit moderately, enhanced Ad-E1A12-induced apoptosis in both R1-AD1 (Fig. 7D) and R1-I567 (Fig. 7E) cell lines. Taken together, these observations support the idea that targeting AR by E1A12 represents a major mechanism for killing PCa cells, and that pharmacological inhibition of the mTOR signaling augments the potency of Ad-E1A12 to kill AR-expressing PCa cells.
3.7. Molecular events upon Ad-E1A12-infected cells.
We assessed potential molecular mechanisms that may contribute to the synergistic killing of cancer cells by Ad-E1A12 and mTOR inhibition. The infection of LNCaP cells with Ad-E1A12 (Fig. 8A lane 3) or other viruses with mutated E1A12 (Ad-E1A12-L19A/ΔRb, Ad-E1A12-L19A, and Ad-E1A12-ΔN) (lanes 4–6), as well as wt Ad5 (lane 7) markedly increased the phosphorylation of AKT and ribosomal protein S6 (Fig. 8A), consistent with previous findings that Ad and Ad-E1A12 activates both PI3K-AKT and mTOR pathways (38,54). Note that Ad-eGFP infection see (Fig. S6) failed to activate Akt and S6K, which could be attributed to the lack of expression of E4ORF1 and other E4 proteins that are critical for activating the PI3K-AKT-mTOR signaling (54), as E1A is essential for E4 gene expression in infected cells (26). BEZ235 dramatically suppressed the phosphorylation of both AKT and S6, although the levels of phosphorylated AKT were still detectable in cells infected with Ad-E1A12 or mutants (Fig. 8A lanes 8–14). Ad infection did not affect AR protein levels (Fig. 8A), and reduced the level of AR mRNA only slightly in LNCaP cells (Fig. 6). Consistent with previous observations by others (53), BEZ235 treatment moderately increased AR levels (compared lanes 7–14 with lane 1, and lane 23 with lane 15 in the AR panels of Fig. 8A). Interestingly, AR protein levels were markedly reduced in wt Ad5-infected cells in the presence of BEZ235 (lanes 14). To assess whether AR downregulation was due to proteasome-mediated degradation, the proteasome inhibitor MG132was added to the infected cells. However, MG132 did not restore AR expression (lane 28 of Fig. 8A). The expression of PSA was not obviously altered by viral infection or BEZ235 treatment (lanes 1–7 in the PSA panel in Fig. 8A). However, exposure of LNCaP cells to both BEZ235 and MG132 reduced PSA expression, especially in cells also infected with viruses (Fig. 8A lanes 24–28 of the PSA panel). The inability of Ad-E1A12 to suppress PSA expression is possibly due to the activation of AKT in virus-infected PCa cells (38), as AKT activation drives AR-mediated PSA expression (57).
Figure 8. Functional impact of Ad-E1A12 on AR and cell death.
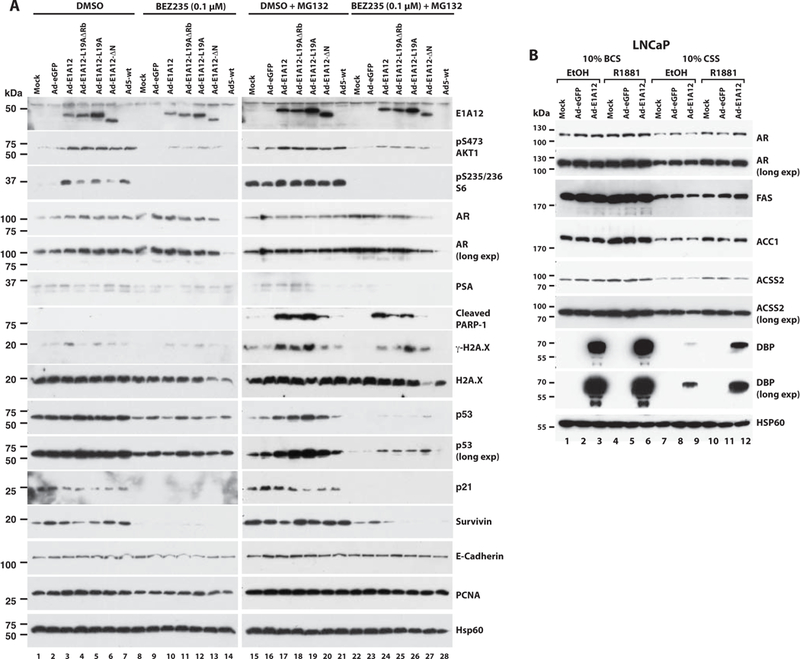
(A) LNCaP cells were uninfected (mock) or infected with an indicated virus (100 viral particles/cell). At 2 h after adding virus, the cells were either exposed to the solvent (DMSO) or BEZ235 (0.1 μM). In a separate set of cell cultures, the proteasome inhibitor MG132 was also added 6 h before cell lysis to the final concentration of 27 μM. At 24 h post infection, cells were harvested for Western blotting analysis with antibodies against the indicated proteins. E-Cadherin, PCNA and Hsp60 were detected as loading controls. (B) LNCaP cells were cultured in DMEM medium with 10% of a regular serum (bovine calf serum, BCS) or 10% CSS as indicated. Cells were treated with ethanol, or R1881 (10 nM), and then infected with Ad-eGFP or Ad-E1A12. Cell lysates were subject to SDS-PAGE and Western blotting as in A.
Notably, BEZ235 treatment of LNCaP cells markedly reduced the expression of p53, p21 and Survivin (encoded by BIRC5), and proteasome inhibition with MG132 was largely ineffective to impact the expression of these proteins (Fig. 8A). Interestingly, the expression of E1A12 or its mutants resulted in higher levels of p53 in the presence of MG132 (Fig. 8A lanes 17–20 and 24–27 of the p53 panels). However, whether this resulted in p53 activation is unclear, as increased p53 did not lead to increased expression of the p53 target gene p21 (Fig. 8A lanes 17–20 and 24–27 of the p21 panels). Remarkably, MG132 treatment dramatically increased the levels of the cleaved form of PARP-1 in cells infected with Ad-E1A12, Ad-E1A12-L19A/ΔRb, and Ad-E1A12-L19A (lanes 17–19 and 24–26 of the cleaved PARP panel in Fig. 8A), and to a lesser extent in cells infected with Ad-E1A12-ΔN (lanes 20 and 27). The PARP-1 cleavage is a hallmark event during apoptosis. Thus, E1A12 infection induces apoptosis in PCa cells. Furthermore, the levels of the phosphorylated form of histone H2A.X (γ−H2A.X) were also increased upon infection with Ad-E1A12, or a virus with a mutant E1A12, especially in the presence of MG132 (lanes 17–19 of the γ-H2A.X panel in Fig. 8A), indicative of increased nuclear apoptotic signaling (58–60). Notably, the increased levels of cleaved PARP-1 and γ-H2A.X directly correlated with increased levels of p53 (Fig. 8A lanes 17–20 and 24–27), which depended on E1A12 expression, as these proteins were not induced in cells infected with Ad-eGFP or wt Ad5 (Fig. 8A). This effect was independent of Rb binding by E1A, as an E1A12 mutant lacking the high affinity Rb-binding site was active to increase the levels of PARP-1, γ-H2A.X and p53 (Fig. 8A lanes 18 and 25). However, the E1A12 N-terminal sequence was important for this effect (Fig. 8A lanes 20 and 27). Notably, mTOR signaling inhibition by BEZ235 resulted in marked suppression of Survivin levels (Fig. 8A lanes 8–14 and 22–28), while γ-H2A.X and cleaved PARP-1 expression remained in Ad-E1A12-infected cells in the presence of the proteasome inhibitor MG132 (Fig. 8A), providing an explanation for increased cell death in cells cotreated with Ad-E1A12 and BEZ235 (Fig. 7). Taken together, these results indicate that Ad-E1A12 triggers a number of tumor suppressive mechanisms in infected cells, while activating the PI3K-AKT-mTOR cell survival signaling. Thus, when combined with pharmacological inhibition of the PI3K-AKT-mTOR pathway, Ad-E1A12 can potently kill PCa cells.
To further assess effects of E1A12 on AR signaling, LNCaP cells were infected with Ad-E1A12 in the presence or the absence of an androgen. Consistent with data shown in Fig. 8A, AR protein levels were not impacted by E1A12 irrespective of androgen stimulation (Fig. 8B). Genes involved in lipid metabolism have been shown to be regulated by the AR in PCa cells (61). Indeed, the fatty acid synthase (FAS) and acetyl-CoA carboxylase 1 (ACC1) were markedly upregulated upon androgen stimulation (Fig. 8B). However, Ad-E1A12 infection did not affect the expression of FAS or ACC1. Interestingly, R1881 also stimulated the expression of acetyl-coenzyme A synthetase (ACSS2), and Ad-E1A12 infection reduced ACSS2 levels in LNCaP cells exposed to R1881 (compare lanes 6 with 4 and 12 with 10 in Fig. 8B), suggesting E1A12 interferes with androgen-stimulated ACSS2 expression.
Interestingly, the expression of the adenoviral DBP was markedly increased upon R1881 stimulation in LNCaP cells infected with Ad-E1A12. DBP is required for Ad replication (62). As shown previously (38), E1A12 expression was essential for DBP expression as DBP was not expressed in LNCaP cells infected with Ad-eGFP (Fig. 8B). These results suggest that E1A12 deploys AR to stimulate viral gene expression for efficient viral replication, potentially resulting in enhanced oncolytic effects of Ad-E1A12. This provides a potential explanation for the increased sensitivity of AR-expressing PCa cells to Ad-E1A12 infection.
4. Discussion
Herein we show that E1A12 binds to the AR. The AR-E1A12 interaction impacts AR signaling, viral replication, and PCa cell survival. Furthermore, the E1A12 266 aa variant appears to mediate the AR N/C interaction. Interestingly, the E1A12 266 aa variant binds preferentially to the full-length AR (Fig. 2). Thus, E1A12 might contain binding sites for the AR NTD and LBD in order to bridge the AR N/C interaction. E1A12 harbors several LxxLL-like motifs. In particular, two motifs in the N-terminal domain of E1A12, 23-VDNFF-27 and 61-VNEFF-65, could tightly bind to the hydrophobic cleft in the AR LBD known as activation function 2 (AF-2) that interacts with the LxxLL motifs of the AR NTD and several AR coregulators (2). Ligand binding to LBD induces a conformational change to make the AF-2 hydrophobic groove accessible for coregulators. However, androgens were not required for E1A12 to mediate the AR N/C interaction, as the N/C interaction was also detected in mammalian two-hybrid assays in the absence of androgens (data not shown). Furthermore, E1A12 promotes AR nuclear translocation in the absence of androgens (Figs. 3 and 4). Nonetheless, there is no experimental evidence yet as to whether E1A12 indeed binds to AF-2. Alternatively, E1A12 could also contact other sites in LBD, which might enable the FQNLF motif in the AR NTD to bind the AF-2 groove. Regardless, whereas the bridging of the AR NTD/LBD interaction by E1A12 can facilitate AR nuclear translocation, the E1A12-AR interaction could block the recruitment of coregulators to the AR NTD and LBD by occupying the interaction interfaces between AR and coregulators, resulting in inhibition of AR-mediated transcription.
Because AR is critical for PCa cells to survive and proliferate, inhibition of AR remains the major goal for treating advanced PCa (63). AR antagonists such as enzalutamide as well as drugs that block the intratumoral androgen biosynthesis such as abiraterone acetate have been in clinical use for treating metastatic PCa. Resistance to such therapies occurs rapidly due to AR reactivation (14,15,63). Mutations in LBD and the expression of AR-Vs lacking LBD contribute to AR reactivation. Thus, targeting the AR NTD could lead to new therapies for treating advanced PCa. Although small molecule inhibitors that interact with the AR NTD have been reported (64), the intrinsically disordered structure of NTD presents a formidable obstacle for stable interactions of the AR NTD with small compounds (4). However, macromolecules can stably interact with the AR NTD because of much larger interfaces for binding interactions between the AR NTD and proteins. A number of AR coregulators such as p300/CBP form stable complexes with the AR NTD (5,6). We show here that E1A12 also binds to the AR NTD. This interaction at least partially suppresses AR transactivation function. Furthermore, E1A12 expression undermines the survival of AR-expressing PCa cells. The direct and specific suppression of the AR NTD by E1A12 provides a new means to block AR activation in PCa. We have developed a novel Ad vector to express E1A12 (Ad-E1A12) (38). This virus was highly effective to kill PCa cells expressing full-length AR and AR-Vs (Fig. 7). Remarkably, androgen stimulation appears to enhance Ad-E1A12 replication (Fig. 8B). Thus, Ad-E1A12 could be a powerful oncolytic virus for anti-PCa gene therapy. Although anticancer virotherapy based on oncolytic viruses have been tested for treating advanced solid tumors including CRPC with a curative potential, oncolytic Ad vectors developed thus far are based on Ad5 backbone. The ability of E1A12 to bind specifically to the AR and inhibit its transactivation function suggests that Ad-E1A12 might have an unprecedented therapeutic potency for treating advanced PCa.
Notably, we observed that Ad-E1A12 infection hyperactivated the PI3K-AKT-mTOR pathway (Fig. 8A) (38). This can be attributed to Ad-intrinsic factors that trigger the independent activation of both PI3K-AKT and mTOR pathways upon Ad infection (54). Additionally, AR inhibition by E1A12 could further activate the PI3K-AKT signaling, as cross-inhibitory effects between the PI3K-AKT and AR signaling pathways have been observed (52,53); thus, inhibition of one pathway resulted in enhanced signaling of the other. Activation of the PI3K-AKT signaling pathway is a hallmark of CRPC. Loss of PTEN or activation of PI3K occurs in up to 70% of late stage PCa cases (65), leading to PCa cell proliferation, survival, and invasion and androgen-independent growth phenotype. Therefore, cotargeting both pathways is expected to enhance therapeutic efficacy against CRPC (52,53). We demonstrated that Ad-E1A12 infection in combination with pharmacological inhibition of the PI3K-AKT-mTOR pathway is synthetically lethal to LNCaP cells (Fig. 7). Thus, Ad-E1A12 in conjunction with inhibitors of the PI3K-AKT-mTOR pathway such as various mTOR kinase inhibitors might be a particular powerful modality for treating CRPC. Alternatively, the deletion of E4 ORFs in Ad-E1A12 vector could remove the viral factors that hyperactivate AKT and mTOR, thereby bypassing the cell surviving mechanisms. Such a vector is expected to be lethal in killing PCa cells without AKT/mTOR inhibitors.
Active suppression of the host DNA damage response (DDR) is important for efficient production of the linear double-stranded Ad genome (66). The histone-like Ad core protein VII associates with host chromatins and inhibits ATM-mediated phosphorylation of H2AX, resulting in reduced levels of γ-H2A.X (66). Interestingly, Ad-E1A12 infection increased the level of γ-H2A.X (Fig. 8A lane 3), which was further elevated by proteasomal inhibition (Fig. 8A lane 17), probably due to reduced proteasomal degradation of H2AX mediated by the E3 ubiquitin ligase HUWE1 (67). It has been shown that γ-H2A.X is critical for apoptotic cell death (58–60). Thus, it appears likely that γ-H2A.X accumulation may be an important event leading to apoptosis in Ad-E1A12-infected cells as evidenced by the appearance of cleaved PARP-1 (Fig. 8A). Of note, p53 expression appears to depend on AKT signaling in LNCaP cells (Fig. 8A). Nonetheless, p53 levels did not strictly correlate with the appearance of cleaved PARP-1 (Fig. 8A). Further studies will be required to ascertain if p53 contributes to Ad-E1A12-mediated apoptosis. It is worth noting that inhibition of the mTOR signaling markedly suppressed the expression of the anti-apoptotic protein Survivin, while γ-H2A.X and cleaved PARP-1 expression persisted in Ad-E1A12-infected cells in the presence of the proteasome inhibitor MG132. Thus, the complementary mechanisms of action of mTOR inhibition and Ad-E1A12 infection may operate to increase PCa cell death. Our data also suggest that a combination of Ad-E1A12, mTOR blockade and proteasome inhibition could be very effective for killing PCa cells, which can be tested in future studies.
5. Conclusion
Both E1A12 266 aa and 235 aa proteins interact with the AR. The E1A12 266 aa variant bridges an interaction between the AR NTD and LBD in the absence of an androgen, thereby promoting AR nuclear translocation. The E1A12-AR interactions inhibit AR function. Androgen stimulation enhances Ad-E1A12 replication. Infection of PCa cells with Ad-E1A12 results in cell death, which was enhanced by inhibiting the PI3K-AKT-mTOR signaling. Our findings suggest that the Ad-E1A12 might have therapeutic potential for treating advanced PCa with heightened AR signaling.
Supplementary Material
Acknowledgement:
We thank Arnie Berk, Hancheng Guan, Jianrong Lu and David Ornelles for providing reagents. The work was supported by grants from Bankhead-Coley Cancer Research Program, and James and Esther King Biomedical Research Program, Florida Department of Health (09BB-11, 09BW-05, 4BF02, 6BC03, and 6JK03 to D. Liao), the Florida Breast Cancer Foundation (to D. Liao), the National Natural Science Foundation of China (Grant No. 81502213, to D. Li), KU Leuven grant GOA/15/017 (to FC), and the National Cancer Institute (CA092236, to D. Liao). Dawei Li and Jia Wang were supported by a scholarship from China Scholarship Council (CSC).
Footnotes
CONFLICTS OF INTEREST
There are no conflicts of interest to disclose.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63(1):11–30. [DOI] [PubMed] [Google Scholar]
- 2.van de Wijngaart DJ, Dubbink HJ, van Royen ME, Trapman J, Jenster G. Androgen receptor coregulators: recruitment via the coactivator binding groove. Mol Cell Endocrinol 2012;352(1–2):57–69. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 2007;27(3):380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McEwan IJ. Intrinsic disorder in the androgen receptor: identification, characterisation and drugability. Mol Biosyst 2012;8(1):82–90. [DOI] [PubMed] [Google Scholar]
- 5.Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein in androgen receptor-mediated signaling. Proc Natl Acad Sci U S A 1998;95(5):2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J Biol Chem 1998;273(48):31853–31859. [DOI] [PubMed] [Google Scholar]
- 7.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A 2004;101(14):4758–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He B, Gampe RT Jr., Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell 2004;16(3):425–438. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008;8(4):440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res 2011;17(7):1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10(1):33–39. [DOI] [PubMed] [Google Scholar]
- 12.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, de Bono JS. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 2009;27(23):3742–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet 2010;375(9724):1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013;2:e00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011;71(20):6503–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009;69(6):2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009;69(1):16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 2010;120(8):2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonarakis ES, Armstrong AJ, Dehm SM, Luo J. Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate cancer and prostatic diseases 2016;19(3):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, Silberstein JL, Taylor MN, Maughan BL, Denmeade SR, Pienta KJ, Paller CJ, Carducci MA, Eisenberger MA, Luo J. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol 2017;35(19):2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer 2011;18(5):R183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem 2012;287(23):19736–19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, Voytas DF, Dehm SM. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci U S A 2013;110(43):17492–17497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller MS, Pelka P, Fonseca GJ, Cohen MJ, Kelly JN, Barr SD, Grand RJ, Turnell AS, Whyte P, Mymryk JS. Characterization of the 55-residue protein encoded by the 9S E1A mRNA of species C adenovirus. J Virol 2012;86(8):4222–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J Virol 2008;82(15):7252–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005;24(52):7673–7685. [DOI] [PubMed] [Google Scholar]
- 27.Liu F, Green MR. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell 1990;61(7):1217–1224. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Green MR. Promoter targeting by adenovirus E1a through interaction with different cellular DNA-binding domains. Nature 1994;368(6471):520–525. [DOI] [PubMed] [Google Scholar]
- 29.Hoti N, Li Y, Chen CL, Chowdhury WH, Johns DC, Xia Q, Kabul A, Hsieh JT, Berg M, Ketner G, Lupold SE, Rodriguez R. Androgen receptor attenuation of Ad5 replication: implications for the development of conditionally replication competent adenoviruses. Mol Ther 2007;15(8):1495–1503. [DOI] [PubMed] [Google Scholar]
- 30.Meng X, Webb P, Yang YF, Shuen M, Yousef AF, Baxter JD, Mymryk JS, Walfish PG. E1A and a nuclear receptor corepressor splice variant (N-CoRI) are thyroid hormone receptor coactivators that bind in the corepressor mode. Proc Natl Acad Sci U S A 2005;102(18):6267–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato Y, Ding A, Heimeier RA, Yousef AF, Mymryk JS, Walfish PG, Shi YB. The adenoviral E1A protein displaces corepressors and relieves gene repression by unliganded thyroid hormone receptors in vivo. Cell Res 2009;19(6):783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avvakumov N, Kajon AE, Hoeben RC, Mymryk JS. Comprehensive sequence analysis of the E1A proteins of human and simian adenoviruses. Virology 2004;329(2):477–492. [DOI] [PubMed] [Google Scholar]
- 33.Robinson CM, Singh G, Henquell C, Walsh MP, Peigue-Lafeuille H, Seto D, Jones MS, Dyer DW, Chodosh J. Computational analysis and identification of an emergent human adenovirus pathogen implicated in a respiratory fatality. Virology 2011;409(2):141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao B, Ricciardi RP. E1A is the component of the MHC class I enhancer complex that mediates HDAC chromatin repression in adenovirus-12 tumorigenic cells. Virology 2006;352(2):338–344. [DOI] [PubMed] [Google Scholar]
- 35.Jiao J, Guan H, Lippa AM, Ricciardi RP. The N terminus of adenovirus type 12 E1A inhibits major histocompatibility complex class I expression by preventing phosphorylation of NF-kappaB p65 Ser276 through direct binding. J Virol 2010;84(15):7668–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan H, Jiao J, Ricciardi RP. Tumorigenic adenovirus type 12 E1A inhibits phosphorylation of NF-kappaB by PKAc, causing loss of DNA binding and transactivation. J Virol 2008;82(1):40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang H, Zheng Z, Zhao LY, Li Q, Liao D. Downregulation of Mdm2 and Mdm4 enhances viral gene expression during adenovirus infection. Cell Cycle 2012;11(3):582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Li D, Luo J, Tian G, Zhao LY, Liao D. Intrinsic cellular signaling mechanisms determine the sensitivity of cancer cells to virus-induced apoptosis. Sci Rep 2016;6:37213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res 2006;66(1):543–553. [DOI] [PubMed] [Google Scholar]
- 40.Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2007;2(5):1236–1247. [DOI] [PubMed] [Google Scholar]
- 41.Santiago A, Godsey AC, Hossain J, Zhao LY, Liao D. Identification of two independent SUMO-interacting motifs in Daxx: evolutionary conservation from Drosophila to humans and their biochemical functions. Cell Cycle 2009;8(1):76–87. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, Jahn SC, Saldanha SA, Planck J, Geary KR, Ma H, Law BK, Roush WR, Hodder P, Liao D. Small-Molecule Inhibitors of Acetyltransferase p300 Identified by High-Throughput Screening Are Potent Anticancer Agents. Mol Cancer Ther 2013;12(5):610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santiago A, Li D, Zhao LY, Godsey A, Liao D. p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol Biol Cell 2013;24(17):2739–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci U S A 2009;106(32):13260–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langley E, Kemppainen JA, Wilson EM. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J Biol Chem 1998;273(1):92–101. [DOI] [PubMed] [Google Scholar]
- 46.Wilson EM. Analysis of interdomain interactions of the androgen receptor. Methods Mol Biol 2011;776:113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J Biol Chem 1994;269(18):13115–13123. [PubMed] [Google Scholar]
- 48.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu YM, Cao X, Qin ZS, Wang S, Feng FY, Chinnaiyan AM. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510(7504):278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, Dang CV, Wei CL. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A 2006;103(47):17834–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, Dang CV. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol 2006;26(6):2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aggarwal R, Behr SC, Paris PL, Truillet C, Parker MFL, Huynh LT, Wei J, Hann B, Youngren J, Huang J, Premasekharan G, Ranatunga N, Chang E, Gao KT, Ryan CJ, Small EJ, Evans MJ. Real-Time Transferrin-Based PET Detects MYC-Positive Prostate Cancer. Mol Cancer Res 2017;15(9):1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011;19(6):792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011;19(5):575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J 2005;24(6):1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008;7(7):1851–1863. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013;73(2):483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SO, Lou W, Hou M, Onate SA, Gao AC. Interleukin-4 enhances prostate-specific antigen expression by activation of the androgen receptor and Akt pathway. Oncogene 2003;22(39):7981–7988. [DOI] [PubMed] [Google Scholar]
- 58.Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell 2006;23(1):121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 2000;275(13):9390–9395. [DOI] [PubMed] [Google Scholar]
- 60.Solier S, Pommier Y. The nuclear gamma-H2AX apoptotic ring: implications for cancers and autoimmune diseases. Cell Mol Life Sci 2014;71(12):2289–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 2011;30(13):2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Breukelen B, Brenkman AB, Holthuizen PE, van der Vliet PC. Adenovirus Type 5 DNA Binding Protein Stimulates Binding of DNA Polymerase to the Replication Origin. Journal of Virology 2003;77(2):915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010;17(6):535–546. [DOI] [PubMed] [Google Scholar]
- 65.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Avgousti DC, Della Fera AN, Otter CJ, Herrmann C, Pancholi NJ, Weitzman MD. Adenovirus core protein VII down-regulates the DNA damage response on the host genome. J Virol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H, Teraoka H, Yoshioka K. ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient gammaH2AX Foci Formation. Cell reports 2015;13(12):2728–2740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.