

Abstract
Horizontal gene transfer (HGT) involves the transmission of genetic material between distinct evolutionary lineages and can be an important source of biological innovation. Reports of interkingdom HGT to eukaryotic microbial pathogens have accumulated over recent years. Verticillium dahliae is a notorious plant pathogen that causes vascular wilt disease on hundreds of plant species, resulting in high economic losses every year. Previously, the effector gene Ave1 and a glucosyltransferase-encoding gene were identified as virulence factor-encoding genes that were proposed to be horizontally acquired from a plant and a bacterial donor, respectively. However, to what extent HGT contributed to the overall genome composition of V. dahliae remained elusive. Here, we systematically searched for evidence of interkingdom HGT events in the genome of V. dahliae and provide evidence for extensive horizontal gene acquisition from bacterial origin.
Keywords: horizontal gene transfer, Verticillium, fungus, ascomycete, bacteria
Introduction
Genetic information is generally vertically transferred from parents to their offspring. However, genetic information can also be transmitted laterally between reproductively isolated species, often referred to as horizontal gene transfer (HGT). It has been well established that HGT plays a significant role in the adaptive evolution of prokaryotic species (Eisen 2000; Koonin et al. 2001; Bapteste et al. 2009). Three well-characterized mechanisms contribute to DNA uptake by prokaryotes, namely transformation, conjugation, and transduction. With transformation, a DNA fragment from a dead, degraded bacterium or another donor enters a competent recipient bacterium (Johnston et al. 2014), whereas conjugation is the active DNA transfer between prokaryotic cells by direct cell-to-cell contact or by a bridgelike connection between two cells (Norman et al. 2009). Finally, transduction involves the transfer of a DNA fragment from one prokaryotic cell to another by a virus or viral vector. HGT in prokaryotes takes place at all taxonomic levels: from individuals of the same population up to interkingdom transfers.
HGT also contributes to the evolution of eukaryotes, although it occurs much less frequent than in prokaryotes (Kurland et al. 2003; Bock 2010). HGT has played an important role in the evolution of pathogenic traits in plant pathogens (Soanes and Richards 2014). For instance, the wheat pathogenic fungi Pyrenophora tritici-repentis and Phaeosphaeria nodorum both contain the near-identical effector gene ToxA that encodes a host-specific toxin that acts as pathogenicity factor (Friesen et al. 2006). Compelling evidence revealed that ToxA was acquired by Pyrenophoratritici-repentis from Phaeosphaerianodorum via HGT, giving rise to pathogenicity of the former species on wheat plants (Friesen et al. 2006). Interestingly, ToxA was similarly found in the genome of yet another wheat pathogen, Bipolaris sorokiniana (McDonald et al. 2018).
Although knowledge on mechanisms of HGT in eukaryotes remains limited, HGT in filamentous plant pathogens is thought to occur more often between closely related species because donor and recipient species share similar genomic architectures (Mehrabi et al. 2011). For the filamentous plant pathogenic fungus Fusarium oxysporum, an entire chromosome can be transferred from one strain to another through coincubation under laboratory conditions in vitro (Ma et al. 2010; van Dam et al. 2017).
Intriguingly, interkingdom HGT also contributed to the genome evolution of filamentous plant pathogens despite the evolutionary distant relation with the donor. For example, phylogenetic analyses of plant pathogenic oomycete species revealed 34 gene families that have undergone HGT between fungi and oomycetes (Richards et al. 2011). The repertoire of HGT candidates includes genes encoding proteins with the capacity to break down plant cell walls and effector proteins to support interaction with host plants (Richards et al. 2011). Furthermore, genomic and phylogenetic analyses of the fungus Fusarium pseudograminearum, the major cause of crown and root rot of barley and wheat in Australia, have revealed that a novel virulence gene was horizontally acquired from a bacterial species (Gardiner et al. 2012).
The fungal genus Verticillium contains nine haploid species plus the allodiploid V. longisporum (Inderbitzin et al. 2011; Depotter et al. 2017). These ten species are phylogenetically subdivided into two clades: Flavexudans and Flavnonexudans (Inderbitzin et al. 2011; Shi-Kunne et al. 2018). The Flavnonexudans clade comprises Verticilliumnubilum, Verticilliumalfalfae, Verticilliumnonalfalfae, Verticilliumdahliae, and V. longisporum, whereas the Flavexudans clade comprises Verticilliumalbo-atrum, Verticilliumisaacii, Verticilliumtricorpus, Verticilliumklebahnii, and Verticilliumzaregamsianum (Inderbitzin et al. 2011). Among these Verticillium spp., V. dahliae is the most notorious plant pathogen that is able to cause disease in hundreds of plant species (Fradin and Thomma 2006; Inderbitzin and Subbarao 2014). Furthermore, Verticilliumalbo-atrum, V. alfalfae, V. nonalfalfae, and V. longisporum are pathogenic, albeit with narrower host ranges (Inderbitzin and Subbarao 2014). Although the remaining species Verticilliumtricorpus, Verticilliumzaregamsianum, Verticilliumnubilum, Verticilliumisaacii, and Verticilliumklebahnii have incidentally been reported as plant pathogens too, they are mostly considered saprophytes that thrive on dead organic material and their incidental infections should be seen as opportunistic (Ebihara et al. 2003; Inderbitzin et al. 2011; Gurung et al. 2015). Verticillium spp. are considered to be strictly asexual, yet various mechanisms contributing to the genomic diversity of V. dahliae have been reported (Seidl and Thomma 2014, 2017; Faino et al. 2016), including HGT (Klosterman et al. 2011; de Jonge et al. 2012). Previously, Ave1 and a glucosyltransferase-encoding gene were found to be acquired from a plant and a bacterial donor, respectively, both of which were found to contribute to V. dahliae virulence during plant infection (Klosterman et al. 2011; de Jonge et al. 2012). These two interkingdom HGT events inspired us to study the extent and potential impact of interkingdom HGT to V.dahliae.
Materials and Methods
Construction of Local Protein Database and Protein BLAST
The protein database used for BLAST analyses was generated using the reference proteomes from UniProtKB excluding viruses (downloaded July 7, 2016). Entries from UniProt were renamed to include their UniProt ID and NCBI taxonomy ID. The proteome database contains proteomes of 5,074 species, including 4,305 prokaryotes and 769 eukaryotes. The prokaryotic species of this database comprise 4,123 bacteria and 182 archaea. The bacterial species comprise 1,737 Terrabacteria, 1,383 Proteobacteria, 368 Sphingobacteria, and 57 Plancterbacteria. The eukaryotic species of the database comprise 373 fungi and 14 oomycete species. Among the fungi species are 257 ascomycete and 84 basidiomycete species, respectively. Three Verticillium species are included in this database, namely V. dahliae (strain VdLs17), V. alfalfae (strain VaMs102), and V. longisporum (strain VL1) (Klosterman et al. 2011). Sequences shorter than ten amino acids were removed from the database. All predicted proteins sequences from V. dahliae strain JR2 were queried to search homologs in the local protein database (UniProtKB) using BlastP (E-value = 0.001, max target seqs = 1,000). We searched for the presence of the HGT candidates in one strain per Verticillium species that were studied previously (Depotter et al. 2017; Shi-Kunne et al. 2018). Protein sequences of each HGT candidate were queried against the proteome of each strain using BlastP (with default settings).
Identification of HGT Candidates
Alien Index (AI) scores were calculated using a custom-made Python script which applied the following formula: , where bbhG and bbhO are the E-values (<10−3) of the best BLAST hit from the ingroup and outgroup, respectively. Best BLAST hits were determined by the highest bit score. Genes with AI score >1 were classified as positive AI genes. For each AI positive gene, homologs were selected based on the BLAST criteria that at least 70% of query length is covered by at least 60% of hit sequence using a custom-made Python script. These homologs were then aligned using MAFFT with default setting (Katoh and Standley 2013). Alignments were subsequently curated using Gblocks (Castresana 2000) with nonstringent parameters. Phylogenetic trees of the curated alignments were constructed using FastTree (Price et al. 2010) with gamma likelihood and WAG amino acid substitution matrix. Phylogenetic trees were automatically evaluated using a custom-made Python script that removes phylogenetic trees with candidate genes that are directly adjacent to non-Verticillium fungal species (ingroup species) rather than nonfungal species (outgroup species). Subsequently, new phylogenetic trees (for manual inspection) were reconstructed using RAxML (Stamatakis 2014) with automatic substitution model determination for branching values.
Gene Expression Analysis
To obtain RNA-Seq data for V. dahliae grown in culture medium, isolate JR2 was grown for 3 days in potato dextrose broth with three biological replicates. To obtain RNA-Seq data from V. dahliae grown in planta, the roots of 3-week-old uprooted Arabidopsisthaliana (Col-0) plants were dipped in a suspension of 106 conidiospores per ml of water for 5 min and replanted in soil. After root inoculation, plants were grown in individual pots in a greenhouse under a cycle of 16 h of light and 8 h of darkness, with temperatures maintained between 20 and 22 °C during the day and a minimum of 15 °C overnight. At 21 days postinoculation, three samples per treatment containing ten stem fragments of 3 cm length, harvested above the soil line, and all the flowering stems from ten plants were pooled. Plant material was freeze dried for 24 h and total RNA was extracted based on TRIzol RNA extraction (Simms et al. 1993). cDNA synthesis, library preparation (TruSeq RNA-Seq short insert library), and Illumina sequencing (single-end 50 bp) was performed at the Beijing Genome Institute (Hong Kong, China). In total, ∼2 and ∼1.5 Gb of filtered reads were obtained for the V. dahliae samples grown in culture medium and in planta, respectively. RNA-Seq data were submitted to the Sequence Read Archive database under the accession number: SRP149060 (accession: PRJNA473305).
The RNA sequencing reads were mapped to their previously assembled genomes using the Rsubread package in R (Liao et al. 2013). The comparative transcriptomic analysis was performed with the package edgeR in R (v3.4.3) (Robinson et al. 2010; McCarthy et al. 2012). Genes are considered differently expressed when P value <0.05 with a log 2-fold-change ≥1. P values were corrected for multiple comparisons according to Benjamini and Hochberg (1995).
Results
Interkingdom HGT to V. dahliae and Other Ascomycete Fungi
To systematically search for genes in the V. dahliae genome that are derived from interkingdom HGT, we downloaded a recent UniProtKB proteome database that contains proteomes of 5,074 species, including 4,123 bacteria, 182 archaea, and 769 eukaryotes (fig. 1). The database contains proteomes of 373 fungal species, including V. dahliae (strain VdLs17), V. alfalfae (strain VaMs102), and V. longisporum (strain VL1) (Klosterman et al. 2011). In our analyses, we focused on the complete telomere-to-telomere genome assembly of V. dahliae strain JR2 (Faino et al. 2015). We first queried each of the 11,430 protein sequences of V. dahliae strain JR2 using BLAST against the aforementioned UniProtKB protein database. Subsequently, we utilized the AI method (Gladyshev et al. 2008; Alexander et al. 2016) that generates AI scores for each V. dahliae gene based on the comparison of best BLAST hits with ingroup and outgroup species; in this case non-Verticillium fungal species and nonfungal species, respectively (fig. 2A). In this manner, we queried for HGT candidates that were acquired only by Verticillium spp., or that were acquired by ancestral fungal species but retained only by Verticillium spp. during evolution. When the best BLAST hit of a gene within the outgroup is better than the best hit within the ingroup, the AI score is positive (i.e., AI > 1) and this gene is considered a potential HGT candidate. This AI analysis yielded 42 HGT candidates with AI positive scores (AI > 1) for V. dahliae JR2 (fig. 2B), which were further analyzed. To this end, we aligned protein sequences of all the homologs for each candidate (from ingroups and outgroups) and constructed phylogenetic trees that were automatically evaluated to remove phylogenetic trees with candidate genes that are directly adjacent to non-Verticillium fungal species (ingroup species) rather than nonfungal species (outgroup species). However, candidate genes that are closely related to the homologs of outgroup species in a phylogenetic tree can also result from duplication followed by multiple gene losses in the ingroup species. Typically, in such a phylogenetic tree, paralogs of the candidate gene cluster with genes of non-Verticillium fungal species (ingroup species) and form separate branches from the branch of the candidate gene. Thus, we removed the candidates with phylogenetic trees that can be explained by gene losses in multiple species. In the end, we obtained two HGT candidates (HGT-1 and HGT-2) that were acquired only by Verticillium spp., or that were acquired by ancestral fungal species but kept only by Verticillium spp. during evolution (table 1). To check whether these two V. dahliae candidates are also present in other Verticillium spp., we queried them using BLAST against the proteomes of previously published Verticillium spp. (Depotter et al. 2017; Shi-Kunne et al. 2018). We found that candidate VDAG_JR2_Chr4g10560 (HGT-1) is present in all Verticillium species and that VDAG_JR2_Chr8g03340 (HGT-2) is only present in V. alfalfae, V. nonalfalfae, and V. longisporum (figs. 3 and 4).
Fig. 1.

—Phylogenetic tree of the species in the selected local database. Different colors represent different taxonomical groups in the UniProtKB protein database. The number of species (spp.) on each branch is indicated. The phylogenetic position of Verticillium spp. is indicated on the right.
Fig. 2.

—AI-based HGT detection method for detecting HGT candidates. (A) Simplified phylogenies of ingroup and outgroup. In AI-A setting, ingroup and outgroup species are non-Verticillium fungal species and nonfungal species, respectively. In AI-B setting, ingroup and outgroup species are nonascomycete fungal species and nonfungal species, respectively. (B) Distribution of AI sores. AI scores from two settings were calculated for every gene in the genome of Verticillium dahliae strain JR2 and they were ordered by decreasing AI score. (C) Numbers of genes with AI positive scores from two different settings.
Table 1.
Information on HGT Candidates
| HGT ID | Gene ID | Putative Donor | Putative Gene Product | Functional Categorya | Secreted | In Planta Expression |
|---|---|---|---|---|---|---|
| HGT-1 | Chr4g10560 | Bacterial | Unknown | Unknown | No | No |
| HGT-2 | Chr8g03340 | Bacterial | Pyridoxal-dependent decarboxylase | Pyridoxal phosphate binding | No | No |
| HGT-3 | Chr1g00380 | Bacterial | Glycosyl hydrolase | Carbohydrate metabolism | Yes | No |
| HGT-4 | Chr1g00710 | Bacterial | Unknown | Unknown | Yes | No |
| HGT-5 | Chr1g17450 | Bacterial | Glycosyl hydrolase (GH27) | Carbohydrate metabolism | Yes | No |
| HGT-6 | Chr1g24620 | Bacterial | Luciferase-like monooxygenase | Oxidoreductase activity | No | No |
| HGT-7 | Chr1g24680 | Bacterial | Glutathione-dependent formaldehyde-activating enzyme | Detoxification of formaldehyde | No | Yes |
| HGT-8 | Chr1g25610 | Bacterial | Glutathione S-transferase | Detoxification of reactive Electrophillic compounds | No | No |
| HGT-9 | Chr1g28650 | Bacterial | Glycosyl hydrolases | Carbohydrate metabolism | No | No |
| HGT-10 | Chr2g00370 | Bacterial | Unknown | Unknown | No | No |
| HGT-11 | Chr2g02230 | Bacterial | Carbohydrate esterase (CE1) | Carbohydrate metabolism | No | No |
| HGT-12 | Chr2g02370 | Bacterial | 3-Demethylubiquinone-9 3-methyltransferase | Unknown | No | Yes |
| HGT-13 | Chr2g02510 | Bacterial | Methyltransferase | Unknown | No | No |
| HGT-14b | Chr2g04700 | Bacterial | Glycosyl transferase family group 2 | Carbohydrate metabolism | No | No |
| HGT-15 | Chr2g07220 | Bacterial | Pyridine nucleotide-disulfide oxidoreductase | Oxidoreductase activity | No | No |
| HGT-16 | Chr2g08740 | Bacterial | Enolase | Carbohydrate metabolism | No | Yes |
| HGT-17 | Chr3g05430 | Bacterial | Chondroitin AC/alginate lyase | Glycosaminoglycans degradation | Yes | Yes |
| HGT-18 | Chr3g06830 | Bacterial | Acetyltransferase (GNAT) family | Transcriptional regulation | No | No |
| HGT-19 | Chr3g12010 | Bacterial | Glycosyl hydrolase (GH74) | Hydrolase activity | Yes | Yes |
| HGT-20 | Chr3g13080 | Bacterial | Phosphoinositide phospholipase | Hydrolase activity | Yes | Yes |
| HGT-21 | Chr4g04370 | Bacterial | Membrane protein | Unknown | No | No |
| HGT-22 | Chr4g11370 | Bacterial | Glycosyl hydrolase (GH74) | Carbohydrate metabolism | No | No |
| HGT-23 | Chr4g11740 | Bacterial | Amidohydrolase | Hydrolase activity | Yes | No |
| HGT-24 | Chr4g11940 | Bacterial | Carbohydrate esterases (CE1) | Carbohydrate metabolism | Yes | No |
| HGT-25 | Chr5g01710 | Bacterial | l-Asparaginase | Hydrolase activity | No | No |
| HGT-26c | Chr5g02170 | Plant | Plant natriuretic peptide | Virulence factor | Yes | Yes |
| HGT-27 | Chr5g05890 | Bacterial | Glycosyl hydrolases (GH43/29) | Carbohydrate metabolism | Yes | No |
| HGT-28 | Chr5g09290 | Bacterial | Protein of unknown function | Unknown | No | Yes |
| HGT-29 | Chr6g02080 | Bacterial | Bacteriocin-protection, YdeI or OmpD-associated protein | Oxidase activity | No | No |
| HGT-30 | Chr6g03000 | Bacterial | Pyridoxal phosphate–dependent enzyme | Metabolic activity | No | No |
| HGT-31 | Chr6g05320 | Bacterial | OsmC-like protein | Oxidative stress regulation | No | No |
| HGT-32 | Chr6g05650 | Bacterial | Luciferase-like monooxygenase | Oxidoreductase activity | No | Yes |
| HGT-33 | Chr6g10680 | Bacterial | Acetyltransferase (GNAT) family | N-Acetyltransferase activity | No | Yes |
| HGT-34 | Chr7g01340 | Bacterial | M6 family metalloprotease | Peptidase activity | No | No |
| HGT-35 | Chr7g01630 | Bacterial | Glycosyl hydrolases (GH136) | Carbohydrate metabolism | No | No |
| HGT-36 | Chr7g03210 | Bacterial | Aminotransferases | Catalytic activity | No | No |
| HGT-37 | Chr7g03340 | Bacterial | Alpha/beta hydrolase | Hydrolytic activity | No | Yes |
| HGT-38 | Chr7g03590 | Bacterial | Insecticide toxin TcdB | Unknown | No | No |
| HGT-39 | Chr8g00620 | Bacterial | Glycerate kinase | Glycerate kinase activity | No | No |
| HGT-40 | Chr8g06130 | Bacterial | Unknown | Unknown | No | No |
| HGT-41 | Chr8g08880 | Bacterial | Nucleotidyltransferase | DNA repair | No | No |
| HGT-42 | Chr8g11000 | Bacterial | Glycosyl hydrolase (GH67) | Carbohydrate metabolism | Yes | Yes |
| HGT-43 | Chr8g11270 | Bacterial | Glycosyl hydrolases (GH43/26), (CBM42) | Carbohydrate metabolism | Yes | No |
| HGT-44 | Chr8g01080 | Bacterial | Peptidase m20 | Peptidase activity | No | No |
Functional annotation was performed by searching for conserved domains in each of these proteins using Interproscan (Jones et al. 2014).
Previously identified candidate (Klosterman et al. 2011).
Previously identified candidate (de Jonge et al. 2012).
Fig. 3.

—Evolutionary relationship of HGT-1 homologs. Protein sequences of HGT-1 homologs were aligned (using MAFFT) and the resulting alignment was used to infer a maximum-likelihood phylogeny (using RAxML). The phylogeny suggests that HGT-1 is transferred from a bacterial species. Different colors depict different groups or species. A more detailed part of the tree that contains Verticillium species is shown on the right. Orange squares indicate branches with bootstrap values ≥60.
Fig. 4.

—Evolutionary relationship of HGT-2 homologs. Protein sequences of HGT-2 homologs were aligned (using MAFFT) and the resulting alignment was used to infer a maximum-likelihood phylogeny (using RAxML). Different colors depict different groups or species. The phylogeny suggests that HGT-2 is transferred from a bacterial species. A more detailed part of the tree that contains Verticillium species is shown on the right. Orange squares indicate branches with bootstrap values ≥60.
In order to search for HGT candidates that are not only present in Verticillium spp. but also may be present in other ascomycete species, we again used the AI method, albeit with adjusted AI group settings. In this case, we set nonfungal species and nonascomycete fungal species as outgroups and ingroups, respectively. AI scanning resulted in 603 genes with AI positive scores (AI > 1), which were selected for further assessment through phylogenetic tree analysis. After assessing phylogenetic trees, we identified 43 additional HGT candidates with nonfungal origins that are also present in other fungal ascomycete species (table 1). Among these candidates, two appeared as paralogs in the phylogenetic tree. The clusters of these two genes are close to each other in the tree, which indicates that the two paralogs are likely the result of a duplication and not of independent acquisitions. Therefore, we only considered 42 HGT candidates for further analyses. Of these, one is of plant origin and 41 are of bacterial origin (table 1). The candidate of plant origin is the previously identified HGT gene, Ave1 (fig. 5) (de Jonge et al. 2012). The previously reported glucosyltransferase gene (Klosterman et al. 2011) likewise was identified as HGT event in our study (fig. 6). Among these 42 candidates, 32 are present in all of the Verticillium species. Eight of the remaining ten are only found in the Verticillium species of the Flavnonexudans clade (fig. 7). Subsequently, we queried the protein sequence of each of the HGT candidates against the NCBI database using TBlastN (web version). We found that the top best hits for each of the candidates, except for Ave1, are from bacterial species, further confirming that the HGT candidates are likely acquired from bacteria (table 2).
Fig. 5.

—Evolutionary relationship of Ave1 homologs. Protein sequences of Ave1 homologs were aligned (using MAFFT) and the resulting alignment was used to infer a maximum-likelihood phylogeny (using RAxML). Different colors depict different groups or species. A more detailed part of the tree that contains Verticillium species is shown on the right. Orange squares indicate branches with bootstrap values ≥60 or above.
Fig. 6.

—Evolutionary relationship of Verticillium dahliae glucosyltransferase homologs. Homologs of glucosyltransferase were aligned (using MAFFT) and the resulting alignment was used to infer a maximum-likelihood phylogeny (using RAxML). Different colors depict different groups or species. A more detailed part of the tree that contains Verticillium species is shown on the right. Orange squares indicate branches with bootstrap values ≥60.
Fig. 7.

—Presence and absence of each of the HGT candidates in each of the Verticillium species. Presence of the HGT candidates was assessed in one strain for each of the species that was studied previously (Depotter et al., 2017; Shi-Kunne et al. 2018) using BlastP (with default settings). Black dots indicate the presence of an HGT candidate in the corresponding Verticillium species.
Table 2.
Overview of the Most Significant (Nonascomycete) BLAST Hits
| HGT ID | Gene ID | Best (Nonascomycete) BLAST Hita |
||
|---|---|---|---|---|
| Species | E-Value | Accession No. | ||
| HGT-1 | Chr4g10560 | Pseudomonas fluorescens | 8.00E-31 | CP012830.1 |
| HGT-2 | Chr8g03340 | Solitalea canadensis | 9.00E-27 | CP003349.1 |
| HGT-3 | Chr1g00380 | Cellulosimicrobium cellulans | 3.00E-105 | CP020857.1 |
| HGT-4 | Chr1g00710 | Pseudarthrobacter equi | 6.00E-101 | XM_007833612.1 |
| HGT-5 | Chr1g17450 | Streptosporangium sp. | 0 | CP034463.1 |
| HGT-6 | Chr1g24620 | Bacillus sp. | 2.00E-142 | CP005586.1 |
| HGT-7 | Chr1g24680 | Azospirillum sp. | 3.00E-71 | KX488120.1 |
| HGT-8 | Chr1g25610 | Pectobacterium atrosepticum | 1.00E-54 | CP024956.1 |
| HGT-9 | Chr1g28650 | Spirosoma aerolatum | 7.00E-66 | CP020104.1 |
| HGT-10 | Chr2g00370 | Amycolatopsis albispora | 1.00E-108 | CP015163.1 |
| HGT-11 | Chr2g02230 | Gemmata sp. | 3.00E-41 | CP011271.1 |
| HGT-12 | Chr2g02370 | Janthinobacterium agaricidamnosum | 5.00E-59 | HG322949.1 |
| HGT-13 | Chr2g02510 | Paenibacillus ihbetae | 4.00E-28 | AP018227.1 |
| HGT-14 | Chr2g04700 | Neorhizobium sp. | 2.00E-128 | CP030830.1 |
| HGT-15 | Chr2g07220 | Asticcacaulis excentricus | 1.00E-104 | CP002395.1 |
| HGT-16 | Chr2g08740 | Actinoalloteichus sp. | 0 | CP016077.1 |
| HGT-17 | Chr3g05430 | Streptomyces bingchenggensis | 2.00E-135 | CP002047.1 |
| HGT-18 | Chr3g06830 | Thielavia terrestris | 3.00E-24 | XM_003652118.1 |
| HGT-19 | Chr3g12010 | Actinoplanes friuliensis | 6.00E-99 | CP006272.1 |
| HGT-20 | Chr3g13080 | Kribbella flavida | 4.00E-88 | CP001736.1 |
| HGT-21 | Chr4g04370 | Streptomyces hygroscopicus | 2.00E-38 | CP003720.1 |
| HGT-22 | Chr4g11370 | Paraburkholderia phenoliruptrix | 3.00E-104 | CP003863.1 |
| HGT-23 | Chr4g11740 | Sorangium cellulosum | 5.00E-84 | AM746676.1 |
| HGT-24 | Chr4g11940 | Pseudomonas salegens | 1.00E-106 | LT629787.1 |
| HGT-25 | Chr5g01710 | Rhizobium phaseoli | 2.00E-111 | CP013525.1 |
| HGT-26 | Chr5g02170 | Nicotiana attenuata | 2.00E-35 | XM_019410592.1 |
| HGT-27 | Chr5g05890 | Sphingopyxis sp. | 8.00E-151 | LT598653.1 |
| HGT-28 | Chr5g09290 | Comamonas testosteroni | 2.00E-52 | CP006704.1 |
| HGT-29 | Chr6g02080 | Shinella sp. | 1.00E-93 | CP015740.1 |
| HGT-30 | Chr6g03000 | Desulfitobacterium metallireducens | 1.00E-28 | CP007032.1 |
| HGT-31 | Chr6g05320 | Marmoricola scoriae | 4.00E-38 | LT629757.1 |
| HGT-32 | Chr6g05650 | Rhizobium tropici | 0 | CP004018.1 |
| HGT-33 | Chr6g10680 | Actinoplanes teichomyceticus | 2.00E-21 | CP023865.1 |
| HGT-34 | Chr7g01340 | Streptomyces sp. | 5.00E-52 | CP029541.1 |
| HGT-35 | Chr7g01630 | Streptomyces sp. | 5.00E-139 | KF386871.1 |
| HGT-36 | Chr7g03210 | Halioglobus japonicus | 8.00E-66 | CP019450.1 |
| HGT-37 | Chr7g03340 | Gluconobacter oxydans | 2.00E-24 | CP004373.1 |
| HGT-38 | Chr7g03590 | Psychrobacter sp. | 0 | CP012533.1 |
| HGT-39 | Chr8g00620 | Bacillus cereus | 4.00E-117 | CP001176.1 |
| HGT-40 | Chr8g06130 | Mesorhizobium sp. | 2.00E-38 | LN649230.1 |
| HGT-41 | Chr8g08880 | Streptomyces albidoflavus | 1.00E-72 | CP004370.1 |
| HGT-42 | Chr8g11000 | Candidatus koribacter | 0 | CP000360.1 |
| HGT-43 | Chr8g11270 | Streptomyces qaidamensis | 1.00E-146 | CP015098.1 |
| HGT-44 | Chr8g01080 | Polaromonas sp. | 0 | CP031013.1 |
HGT candidates were queried against NCBI database (web version) using TBlastN (E-value = E-06).
A High Proportion of V. dahliae HGT Candidates Encodes Secreted Proteins
Filamentous plant pathogens secrete large numbers of proteins, including plant cell wall-degrading enzymes and effector proteins, to interact with host plants (Cook et al. 2015). We used a combination of SignalP, TMHMM, and TargetP (Shi-Kunne et al. 2018) to identify HGT-derived genes that encode such secreted proteins. This analysis showed that 12 of the 44 candidates encode secreted proteins. Compared with the total number of the secreted proteins in the genome of V. dahliae strain JR2 (858 out of 11,430), HGT candidates are significantly enriched for genes that are predicted to encode secreted proteins (Fisher exact test, P < 0.0001). To know more about the putative functions of these proteins, we searched for conserved domains in each of these proteins using Interproscan (Jones et al. 2014). Among the 12 secreted proteins, 5 are glycoside hydrolases (GHs) and two are carbohydrate esterases (CEs), which are all carbohydrate-active enzymes (CAZymes). CAZymes are responsible for the synthesis and breakdown of glycoconjugates, oligo- and polysaccharides. CAZymes of plant-associated fungi usually comprise a large number of plant cell wall-degrading enzymes (Zhao et al. 2013). The remaining five are Ave1, a chondroitin AC/alginate lyase, a phosphoinositide phospholipase, an amidohydrolase, and a protein without functional annotation. Subsequently, we also searched for the functional annotation of the 30 nonsecreted proteins. Besides the previously identified glucosyltransferase and seven proteins without functional annotation, we obtained diverse predicted functions including transcription regulation, DNA repair, hydrolase, and metabolic activities. Overall, we observed that the full set of HGT candidate proteins (secreted and nonsecreted) is also enriched for GHs (10 out of 44) (Fisher exact test, P < 0.0001) when considering to the total amount of the GH genes in the genome of V. dahliae strain JR2 (265 out of 11,430).
To investigate the putative role of the HGT-derived genes in plant pathogen interactions, we compared expression patterns in planta and in vitro. It was previously shown that Ave1 is expressed in planta during interaction of V. dahliae with Nicotiana benthamiana plants (de Jonge et al. 2012). We assessed transcription (RNA-Seq) data of V. dahliae during colonization of A. thaliana and found that 12 HGT candidates showed in planta expression, one of which is Ave1. Four of these 12 were found to be induced when compared with the expression in in vitro–cultured mycelium (fig. 8). This suggests that besides Ave1, three additional HGT candidates might play a role during host colonization. Moreover, one of the three induced candidates encodes a secreted protein.
Fig. 8.
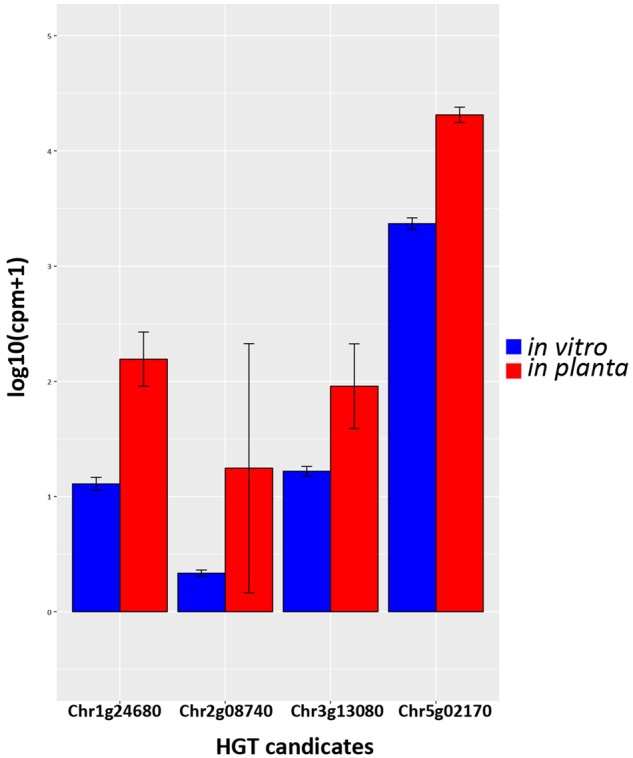
—Pair-wise comparison of HGT candidates with differential expression in vitro and in planta. Gene expressions are depicted for Verticillium dahliae strain JR2 cultured in liquid medium and upon Arabidopsis thaliana colonization, respectively. Bars represent the mean gene expression with standard deviations. The significance of difference in gene expression was calculated using t-tests relative to a threshold (TREAT) of log 2-fold-change ≥1 (McCarthy and Smyth 2009).
Verticillium dahliae HGT Candidates Preferentially Occur at Repetitive Genomic Regions
In principle, foreign DNA can be incorporated anywhere in a recipient genome as long as it does not disrupt an essential genomic element (Husnik and Mccutcheon 2017). Several studies suggest that horizontally transferred sequences are often integrated into genomic regions that are enriched in transposable elements (Gladyshev et al. 2008; Mcnulty et al. 2010; Acuña et al. 2012; Pauchet and Heckel 2013; Husnik and Mccutcheon 2017). Previously, we observed that Ave1 is located in a lineage-specific (LS) region of V. dahliae that is enriched in repeats (de Jonge et al. 2013; Faino et al. 2016). In order to assess whether the other HGT candidates are LS and associated with repeats as well, we plotted genomic repeat densities and the genomic location of all HGT candidates. Although only Ave1 is located at a genuine LS region (Depotter et al. 2018), most of the HGT candidates (34 out of 44) reside in proximity (within 1 kb) to a repetitive sequence (fig. 9). Subsequently, we compared repeat counts within 1-kb genomic windows around each of the 44 HGT candidates to the repeat counts in 1-kb genomic windows of 44 randomly picked genes (1,000 permutations), which confirmed that the HGT candidates are significantly more often adjacent to repeats than expected by chance (P = 0.0331).
Fig. 9.

—Genomic location of the HGT candidates. The outer lane represents the chromosomes of Verticillium dahliae strain JR2. The middle lane shows the relative position of the HGT candidates in each chromosome. The inner lane shows the repeat density. The red lines indicate the locations of the LS regions (Depotter et al. 2018).
Discussion
In this study, we systematically searched for evidence of interkingdom HGT events in the genome of V. dahliae using an AI based method. Subsequently, we verified HGT candidates using a phylogenetic approach. In general, there are two classical ways to identify HGT candidates: intrinsic and extrinsic methods. Intrinsic methods focus on patterns in the primary structure of genes and genome sequences and aim to find genes or genomic regions with composition patterns that differ significantly from the rest of the genome. For example, deviation in GC content and codon usage can be considered a sign for horizontal acquisition (Lawrence and Ochman 1997). In contrast, extrinsic methods focus on comparing similarity metrics between closely related and distant taxa. For example, when a gene from a species of interest (recipient) shows higher similarity (lower E-value, higher bit score in BLAST) to sequences from distantly related (donor) species than to genes of close relatives, this gene may have been horizontally acquired. Another frequently applied extrinsic approach is to assess the phylogenetic distribution of genes across a panel of diverse organisms. These phylogeny-based methods are generally thought to be more accurate (Poptsova and Gogarten 2007; Poptsova 2009). In principle, an HGT event will create a discrepancy between the gene and species trees. However, phylogeny-based methods are much more computationally intensive, because they require sequence alignments and phylogeny reconstructions (Dupont and Cox 2017). Thus, we chose to utilize an AI based method to pre-select candidates that can be further analyzed through phylogenetic analyses.
Although the AI method provided a rapid identification of HGT candidates, some limitations of the method should be kept in mind. The AI method relies on the quality of the reference database, the accuracy of the taxonomic assignment and the diversity of taxa covered by the reference database. The two HGT candidates (HGT-1 and HGT-2) that appear to be Verticillium-specific could also be explained by the absence of the homolog-containing species. In other words, the more closely related species are included in the database, the less Verticillium-specific HGT candidates might be identified. Furthermore, AI based methods rely on predefining ingroups and outgroups and can only identify HGT candidates that are absent in the ingroup species. In our case, the method is unable to identify candidates that were acquired before the formation of ascomycetes, and candidates that were also acquired by fungal nonascomycete species independently at more recent events from the same donors.
Nevertheless, the 42 HGT candidates that were also found in other ascomycete species were most likely acquired during ancient transfer events. The extent of gene transfer from prokaryotes to fungi is highly different across diverse ascomycete fungal lineages. A whole-genome study of HGT in Aspergillus fumigatus revealed that 40% of 189 transferred regions (containing 214 genes) were of bacterial origin (Mallet et al. 2010). In contrast, a study with a similar objective found only 11 genes with a convincing signature of transfer from bacterial origin in the genomes of three Colletotrichum species (Jaramillo et al. 2015). Three of the 11 Colletotrichum HGT candidates were also found in V. dahliae (strain VdLs17) and V. alfalfae (strain VaMs102) (Klosterman et al. 2011). In our study, we also found these three candidates (HGT-14, HGT-15, and HGT-44) in V. dahliae strain JR2.
We found that HGT candidates are enriched for genes encoding secreted proteins, many of which belong to a family of carbohydrate-active enzymes (CAZymes) or GHs. GHs play a fundamental role in the decomposition of plant biomass (Murphy et al. 2011), which suggests that bacterial genes might contribute to the plant-associated life styles of Verticillium species as well as of many other ascomycetes. Although these V. dahliae GH genes are not induced during infection of A. thaliana when compared with in vitro expression data, it is possible that the GH genes are induced during interaction with other host plants as V. dahliae is able to cause disease in hundreds of plant species (Fradin and Thomma 2006; Inderbitzin and Subbarao 2014). Alternatively, these proteins might play roles during life stages outside the host. Overall, when assessing transcription (RNA-Seq) data of V. dahliae-infected A. thaliana, we identified four HGT candidates (including Ave1) that were induced when compared with expression in in vitro–cultured mycelium. One of the induced candidates encodes a secreted protein that is predicted to be a phosphoinositide phospholipase, a key metabolic enzyme that is needed by all living organisms to hydrolyze phospholipids into fatty acids and lipophilic substances (Ghannoum 2000). Phospholipases are also signaling molecules that elicit stress tolerance and host immune responses in fungi (Ghannoum 2000; Soragni et al. 2001; Köhler et al. 2006). Phospholipases have been studied in some plant pathogenic fungi, such as Fusarium graminearum (Zhu et al. 2016) and Magnaporthe oryzae (Ramanujam and Naqvi 2010; Zhang et al. 2011; Yin et al. 2016), and the phospholipase (FgPLC1) regulates development, stress responses, and pathogenicity of Fusariumgraminearum (Zhu et al. 2016). Collectively, this may suggest that the V. dahliae phosphoinositide phospholipase plays an import role during interaction with host plants.
Conclusions
In this study, we applied a conservative approach combining an AI method and phylogenetic analysis to analyze interkingdom HGT to V.dahliae. Besides the previously identified effector gene Ave1 and a glucosyltransferase-encoding gene, we revealed additional HGT candidates, all of which are of bacterial origin, indicating a high number of interkingdom gene acquisitions.
Acknowledgments
Work in the laboratories of B.P.H.J.T. and M.F.S. is supported by the Research Council Earth and Life Sciences (ALW) of the Netherlands Organization of Scientific Research (NWO).
Literature Cited
- Acuña R, et al. 2012. Adaptive horizontal transfer of a bacterial gene to an invasive insect pest of coffee. Proc Natl Acad Sci U S A. 109:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander WG, Wisecaver JH, Rokas A, Hittinger CT.. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc Natl Acad Sci U S A. 113(15):4116–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, et al. 2009. Prokaryotic evolution and the tree of life are two different things. Biol Direct 4:34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57(1): 289–300. [Google Scholar]
- Bock R. 2010. The give-and-take of DNA: horizontal gene transfer in plants. Trends Plant Sci. 15(1):11–22. [DOI] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Cook DE, Mesarich CH, Thomma BPHJ.. 2015. Understanding plant immunity as a surveillance system to detect invasion. Annu Rev Phytopathol. 53:541–563. [DOI] [PubMed] [Google Scholar]
- de Jonge R, et al. 2012. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci U S A. 109(13):5110–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge R, et al. 2013. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 23(8):1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depotter JRL, Seidl MF, van den Berg GCM, Thomma BPHJ, Wood TA.. 2017. A distinct and genetically diverse lineage of the hybrid fungal pathogen Verticillium longisporum population causes stem striping in British oilseed rape. Environ Microbiol. 19(10):3997–4009. [DOI] [PubMed] [Google Scholar]
- Depotter JRL, et al. 2018. Dynamic virulence-related regions of the fungal plant pathogen Verticillium dahliae display remarkably enhanced sequence conservation. bioRxiv: https://www.biorxiv.org/content/10.1101/277558v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont P-Y, Cox MP.. 2017. Genomic data quality impacts automated detection of lateral gene transfer in fungi. G3 7(4):1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara Y, Uematsu S, Nagao H, Moriwaki J, Kimishima E.. 2003. First report of Verticillium tricorpus isolated from potato tubers in Japan. Mycoscience 44(6):481–488. [Google Scholar]
- Eisen JA. 2000. Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr Opin Genet Dev. 10(6):606–611. [DOI] [PubMed] [Google Scholar]
- Faino L, et al. 2015. Single-molecule real-time sequencing combined with optical mapping yields completely finished fungal genome. MBio 6:e00936–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faino L, et al. 2016. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 26(8):1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradin EF, Thomma BPHJ.. 2006. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol. 7(2):71–86. [DOI] [PubMed] [Google Scholar]
- Friesen TL, et al. 2006. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 38(8):953–956. [DOI] [PubMed] [Google Scholar]
- Gardiner DM, et al. 2012. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 8(9):e1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannoum MA. 2000. Potential role of phospholipases in virulence and fungal pathogenesis. Clin Mcrobiol Rev. 13(1):122–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev EA, Meselson M, Arkhipova IR.. 2008. Massive horizontal gene transfer in Bdelloid Rotifers. Science 320(5880):1210–1214. [DOI] [PubMed] [Google Scholar]
- Gurung S, et al. 2015. Host range of Verticillium isaacii and Verticillium klebahnii from artichoke, spinach, and lettuce. Plant Dis. 99(7):933–938. [DOI] [PubMed] [Google Scholar]
- Husnik F, Mccutcheon JP.. 2017. Functional horizontal gene transfer. Nat. Rev. Microbiol. 16(2):67–79. [DOI] [PubMed] [Google Scholar]
- Inderbitzin P, Davis RM, Bostock RM, Subbarao KV.. 2011. The ascomycete Verticillium longisporum is a hybrid and a plant pathogen with an expanded host range. PLoS One 6(3):e18260.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inderbitzin P, Subbarao KV.. 2014. Verticillium systematics and evolution: how confusion impedes Verticillium wilt management and how to resolve it. Phytopathology 104(6):564–574. [DOI] [PubMed] [Google Scholar]
- Inderbitzin P, et al. 2011. Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS One 6(12): e28314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo VDA, et al. 2015. Identification of horizontally transferred genes in the genus Colletotrichum reveals a steady tempo of bacterial to fungal gene transfer. BMC Genomics. 16:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C, Martin B, Fichant G, Polard P, Claverys J-P.. 2014. Bacterial transformation: distribution, shared mechanisms and divergent control. Nat Rev Microbiol. 12(3):181–196. [DOI] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosterman SJ, et al. 2011. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 7(7):e1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler GA, et al. 2006. Phospholipase A 2 and phospholipase B activities in fungi. Biochim Biophys Acta 1761(11):1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L.. 2001. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 55:709–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG, Canback B, Berg OG.. 2003. Horizontal gene transfer: a critical view. Proc Natl Acad Sci U S A. 100(17):9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H.. 1997. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 44(4):383–397. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W.. 2013. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41(10):e108.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-J, et al. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464(7287):367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet LV, Becq J, Deschavanne P.. 2010. Whole genome evaluation of horizontal transfers in the pathogenic fungus Aspergillus fumigatus. BMC Genomics. 11:171.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK.. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40(10):4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Smyth GK.. 2009. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25(6):765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MC, Ahren D, Simpfendorfer S, Milgate A, Solomon PS.. 2018. The discovery of the virulence gene ToxA in the wheat and barley pathogen Bipolaris sorokiniana. Mol Plant Pathol. 19(2):432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcnulty SN, et al. 2010. Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS One 5(6):e11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabi R, et al. 2011. Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev. 35(3):542–554. [DOI] [PubMed] [Google Scholar]
- Murphy C, Powlowski J, Wu M, Butler G, Tsang A.. 2011. Curation of characterized glycoside hydrolases of fungal origin. Database 2011: bar020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman A, Hansen LH, Sorensen SJ.. 2009. Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc B Biol Sci. 364(1527):2275–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauchet Y, Heckel DG.. 2013. The genome of the mustard leaf beetle encodes two active xylanases originally acquired from bacteria through horizontal gene transfer. Proc R Soc B 280(1763):20131021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poptsova M. 2009. Testing phylogenetic methods to identify horizontal gene transfer In: Horizontal gene transfer. Methods Mol Biol. 532:227–240. [DOI] [PubMed] [Google Scholar]
- Poptsova MS, Gogarten JP.. 2007. The power of phylogenetic approaches to detect horizontally transferred genes. BMC Evol Biol. 7:45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP.. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanujam R, Naqvi NI.. 2010. PdeH, a high-affinity cAMP phosphodiesterase, is a key regulator of asexual and pathogenic differentiation in Magnaporthe oryzae. PLoS Pathog. 6(5):e1000897.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TA, et al. 2011. Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc Natl Acad Sci U S A. 108(37):15258–15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Thomma BPHJ.. 2014. Sex or no sex: evolutionary adaptation occurs regardless. BioEssays 36(4):335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Thomma BPHJ.. 2017. Transposable elements direct the coevolution between plants and microbes. Trends Genet. 33(11):842–851. [DOI] [PubMed] [Google Scholar]
- Shi-Kunne X, Faino L, van den Berg GCM, Thomma BPHJ, Seidl MF.. 2018. Evolution within the fungal genus Verticillium is characterized by chromosomal rearrangement and gene loss. Environ Microbiol. 20:1362–1373. [DOI] [PubMed] [Google Scholar]
- Simms D, Cizdziel PE, Chomczynski P.. 1993. TRIzol: a new reagent for optimal single-step isolation of RNA. Focus 15:532–535. [Google Scholar]
- Soanes D, Richards TA.. 2014. Horizontal gene transfer in eukaryotic plant pathogens. Annu Rev Phytopathol. 52:583–614. [DOI] [PubMed] [Google Scholar]
- Soragni E, et al. 2001. A nutrient‐regulated, dual localization phospholipase A2 in the symbiotic fungus Tuber borchii. EMBO J. 20(18):5079–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam P, et al. 2017. A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci Rep. 7:9042.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, et al. 2016. Phosphodiesterase MoPdeH targets MoM ck1 of the conserved mitogen‐activated protein (MAP) kinase signalling pathway to regulate cell wall integrity in rice blast fungus Magnaporthe oryzae. Mol Plant Pathol. 17(5):654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. 2011. Two phosphodiesterase genes, PDEL and PDEH, regulate development and pathogenicity by modulating intracellular cyclic AMP levels in Magnaporthe oryzae. PLoS One 6(2):e17241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu H, Wang C, Xu J-R.. 2013. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 14(1):274.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, et al. 2016. The phospholipase C (FgPLC1) is involved in regulation of development, pathogenicity, and stress responses in Fusarium graminearum. Fungal Genet Biol. 97:1–9. [DOI] [PubMed] [Google Scholar]