

Francisella tularensis is a Gram-negative, facultative intracellular pathogen and the causative agent of tularemia. Previous studies with the attenuated live vaccine strain (LVS) identified a role for the outer membrane protein TolC in modulation of host cell responses during infection and virulence in the mouse model of tularemia.
KEYWORDS: Francisella tularensis, Schu S4, TolC, tularemia, virulence
ABSTRACT
Francisella tularensis is a Gram-negative, facultative intracellular pathogen and the causative agent of tularemia. Previous studies with the attenuated live vaccine strain (LVS) identified a role for the outer membrane protein TolC in modulation of host cell responses during infection and virulence in the mouse model of tularemia. TolC is an integral part of efflux pumps that export small molecules and type I secretion systems that export a range of bacterial virulence factors. In this study, we analyzed TolC and its two orthologs, FtlC and SilC, present in the fully virulent F. tularensis Schu S4 strain for their contributions to multidrug efflux, suppression of innate immune responses, and virulence. We found that each TolC ortholog participated in multidrug efflux, with overlapping substrate specificities for TolC and FtlC and a distinct substrate profile for SilC. In contrast to their shared roles in drug efflux, only TolC functioned in the modulation of macrophage apoptotic and proinflammatory responses to Schu S4 infection, consistent with a role in virulence factor delivery to host cells. In agreement with previous results with the LVS, the Schu S4 ΔtolC mutant was highly attenuated for virulence in mice by both the intranasal and intradermal routes of infection. Unexpectedly, FtlC was also critical for Schu S4 virulence, but only by the intradermal route. Our data demonstrate a conserved and critical role for TolC in modulation of host immune responses and Francisella virulence and also highlight strain- and route-dependent differences in the pathogenesis of tularemia.
INTRODUCTION
Francisella tularensis is a highly virulent, Gram-negative zoonotic pathogen and the causative agent of tularemia (1). Clinical manifestations depend on the route of inoculation, with the most common form being ulceroglandular tularemia contracted by insect bite or handling contaminated materials. The most lethal form of tularemia occurs following pneumonic exposure, with inhalation of as few as 10 bacteria sufficient to cause severe disease (2, 3). Due to its low infectious dose, ease of aerosolization, and prior use as a bioweapon and the high morbidity and mortality rates associated with infection, F. tularensis is designated a tier 1 select agent by the U.S. Centers for Disease Control and Prevention (1, 4, 5). There are two clinically relevant subspecies of F. tularensis: Francisella tularensis subsp. tularensis (type A), and Francisella tularensis subsp. holarctica (type B) (1, 2, 4). F. tularensis subsp. tularensis is highly virulent and causes the most severe form of tularemia, while F. tularensis subsp. holarctica is comparably less virulent and causes a milder disease. The F. tularensis live vaccine strain (LVS) is an attenuated derivative of an F. tularensis subsp. holarctica isolate (4). The LVS has proven useful as an experimental F. tularensis strain, as it causes a lethal infection in mice that mimics human tularemia (4). An additional species of Francisella, F. novicida, has also proven useful as a laboratory strain (6). Despite the utility of both the LVS and F. novicida as attenuated models, the mechanisms and strategies critical for virulence of human pathogenic F. tularensis are not well understood. Therefore, experimentation with fully virulent F. tularensis subsp. tularensis strains, such as the Schu S4 strain, is critical for a more complete understanding of the pathogenesis of tularemia.
A hallmark of F. tularensis virulence is the organism’s ability to interfere with host innate immune responses during infection, with robust proinflammatory responses not observed until 2 to 3 days postinfection (7, 8). F. tularensis is a facultative intracellular pathogen that can invade and replicate within a variety of host cells, although macrophages are the primary replicative niche in vivo (9, 10). Following phagocytosis, F. tularensis rapidly escapes the phagosome and enters the cytosol (11). Once in the cytosol, F. tularensis replicates robustly, eventually triggering host cell death, which contributes to bacterial release, dissemination, and infection of additional host cells (9). Early during infection, F. tularensis actively downmodulates the host’s proinflammatory responses (12–17). In addition, F. tularensis actively dampens host programmed cell death responses during infection, presumably to preserve intracellular replicative niches (18–24). Eventual death of F. tularensis-infected host cells occurs primarily via caspase-1-dependent pyroptosis and caspase-3-dependent apoptosis, with fully virulent strains primarily triggering the latter (25–27). However, many questions remain regarding the precise molecular mechanisms behind the immunomodulatory capacity of F. tularensis.
TolC is the prototypical outer membrane (OM) channel component of Gram-negative tripartite efflux pumps (28). Bacterial tripartite efflux pumps include both multidrug exporters and type I secretion systems (T1SS) (29). Multidrug efflux pumps confer resistance to a wide variety of antibiotics, detergents, dyes, and other harmful small molecules (29). T1SS facilitate the export of many virulence factors directly from the cytoplasm to the extracellular milieu in a single, ATP-dependent step, including α-hemolysin (Escherichia coli), alkaline protease (Pseudomonas aeruginosa), and adenylate cyclase toxin (Bordetella pertussis) (29). The Francisella genome contains three TolC orthologs that are conserved among different Francisella species (Table 1) (30, 31). Two of these orthologs, tolC (FTT1724) and ftlC (FTT1095), encode proteins related to the E. coli TolC protein involved in hemolysin secretion (30). The third ortholog, silC (FTT1258), encodes a protein more closely related to the CusC family of TolC-related proteins involved in efflux of metal ions and other small molecules (31–34).
TABLE 1.
TolC orthologs encoded in Francisella genomes
| Protein |
F. novicida U112 gene (% ID)a |
F. tularensis LVS gene (% ID) |
F. tularensis Schu S4 gene |
|---|---|---|---|
| TolC | FTN_1703 (99) | FTL_1865 (99) | FTT1724 |
| FtlC | FTN_0779 (99) | FTL_1107 (99) | FTT1095 |
| SilC | FTN_1277 (99) | FTL_0686 (99) | FTT1258 |
a% ID, amino acid sequence identity to corresponding Schu S4 protein.
In previous studies with the LVS, we found that both TolC and FtlC participate in multidrug efflux but that only TolC contributes to virulence (24, 30, 35). We subsequently showed that the LVS actively delays induction of macrophage apoptosis and blocks host proinflammatory responses in a TolC-dependent manner and that TolC is required for optimal bacterial replication within macrophages (24, 35). These studies identify a specialized role for TolC in modulating innate immune responses to LVS infection, suggesting that TolC functions as part of a T1SS for virulence factor delivery to host cells. However, minimal information is available regarding TolC function in fully virulent strains of F. tularensis. A single study by Kadzhaev et al. revealed that a tolC transposon insertion mutant in the F. tularensis Schu S4 strain had only a marginal attenuation in virulence, as determined using the mouse intradermal infection model (36). Minimal information is also available regarding FtlC and SilC function in Francisella. SilC has been characterized as an OM protein and protective antigen in the Schu S4 strain (31, 37). Additionally, a recent study with the LVS identified roles for SilC in multidrug efflux, resistance to oxidative stress, and virulence in mice (38). Overall, there is a need for improved understanding of the contributions of TolC orthologs to F. tularensis virulence, particularly in human pathogenic strains.
In the current study, we constructed unmarked ΔtolC, ΔftlC, and ΔsilC deletion mutants and their complements in the human pathogenic F. tularensis Schu S4 strain. Using these constructs, we determined the contributions of TolC, FtlC, and SilC to Schu S4 multidrug resistance, cytotoxicity toward macrophages, suppression of proinflammatory responses during macrophage infection, and virulence in mice. We found that all three proteins participate in resistance to a variety of drugs and antibiotics. However, and similar to our finding with the LVS, only TolC was important for Schu S4 modulation of macrophage responses to infection. Finally, virulence studies with mice revealed that the Schu S4 ΔtolC mutant is highly attenuated for virulence via the intranasal and intradermal infection routes, whereas the ΔftlC mutant is attenuated via the intradermal route only. In contrast, SilC makes minimal contributions to Schu S4 virulence. Taken together, our results demonstrate that TolC performs a specialized role during intracellular infection of host cells and is a critical Schu S4 virulence factor. Our findings also reveal a novel role for FtlC as a critical Schu S4 virulence factor by the intradermal route, highlighting differences between the LVS and fully virulent F. tularensis, as well as route-dependent requirements for pathogenesis.
RESULTS
TolC, FtlC, and SilC contribute to Schu S4 multidrug resistance.
To investigate contributions of the three TolC orthologs—TolC, FtlC, and SilC (Table 1)—to Schu S4 multidrug resistance, intracellular pathogenesis, and virulence, we constructed unmarked chromosomal deletions of each gene using an allelic-exchange protocol (39). None of the mutants exhibited growth defects in rich or defined media (see Fig. S1 in the supplemental material). Additionally, we constructed unmarked, chromosomally complemented strains for each deletion mutant by inserting the tolC, ftlC, or silC open reading frames, under the control of the constitutive F. tularensis groE promoter, at the att7 chromosomal integration site (39).
To determine roles of the TolC orthologs in Schu S4 multidrug resistance, we tested each mutant strain for susceptibility to a variety of drugs and antibiotics using a disc diffusion assay. Our results (Tables 2 and 3) showed that loss of TolC, FtlC, or SilC caused increased susceptibility to selected antimicrobial compounds compared to findings with the wild-type (WT) Schu S4 strain, indicating that all three proteins participate as components of Schu S4 efflux machineries important for resistance to different compounds. In agreement with our previous analysis with the LVS, the Schu S4 ΔtolC and ΔftlC mutants had overlapping substrate specificities, exhibiting increased sensitivity to SDS and erythromycin and with a trend toward increased susceptibility to chloramphenicol (30). The Schu S4 ΔsilC mutant also exhibited increased sensitivity to SDS but in addition was more susceptible to carbonyl cyanide m-chlorophenylhydrazone (CCCP), nalidixic acid, and silver nitrate (Tables 2 and 3). This matches the reported sensitivity of a LVS ΔsilC mutant to nalidixic acid and silver nitrate (38) and the role of CusC-related proteins in heavy metal ion export (32). Complementation of all three OM channel protein mutants resulted in restoration of WT sensitivities (Table 3). Furthermore, and in agreement with previous findings with the LVS, we found that SilC is important for resistance to tert-butyl hydroperoxide (tBh), a radical-producing compound, but not hydrogen peroxide (38). These data indicate that in Schu S4, SilC may also be important for detoxifying certain radical oxygen species.
TABLE 2.
Drug sensitivities of Schu S4 ΔtolC, ΔftlC, and ΔsilC mutant strains
| Compound | Concn, μg/disc | Zone of inhibition, mm (mean ± SD), for strain with indicated genotypea |
|||
|---|---|---|---|---|---|
| WT | ΔtolC | ΔftlC | ΔsilC | ||
| SDS | 188 | 6 ± 1 | 14 ± 2 | 13 ± 0 | 10 ± 0 |
| Streptomycin | 2.5 | 15 ± 1 | 15 ± 3 | 14 ± 2 | 17 ± 1 |
| Chloramphenicol | 1.25 | 10 ± 4 | 15 ± 2 | 14 ± 3 | 11 ± 4 |
| Erythromycin | 5 | 29 ± 1 | 39 ± 2 | 33 ± 1 | 29 ± 1 |
| Ampicillin | 2.5 | 6 ± 0 | 6 ± 0 | 6 ± 0 | 6 ± 0 |
| Polymyxin B | 100 | 6 ± 0 | 6 ± 0 | 6 ± 0 | 6 ± 0 |
| CCCP | 10 | 9 ± 0 | ND | ND | 13 ± 1 |
| Nalidixic acid | 5 | 20 ± 1 | ND | ND | 26 ± 1 |
| Silver nitrate | 500 | 6 ± 1 | ND | ND | 11 ± 1 |
| Hydrogen peroxide | 1.8 | 8 ± 1 | 8 ± 1 | 8 ± 1 | 8 ± 1 |
| 3.6 | 19 ± 0 | 19 ± 1 | 19 ± 1 | 19 ± 1 | |
| t-Butyl hydroperoxide | 9 | 8 ± 2 | 7 ± 1 | 8 ± 1 | 14 ± 2 |
| 18 | 8 ± 1 | 8 ± 1 | 8 ± 2 | 19 ± 1 | |
Zones of growth inhibition (including the 6-mm filter disc) are shown. Values shown in bold are significantly different from corresponding WT values by Student t test. ND, not determined.
TABLE 3.
Drug sensitivities of Schu S4 ΔtolC, ΔftlC, and ΔsilC mutant and complemented strains
| Compound | Concn, μg/disc | Zone of inhibition, mm (mean ± SD), for strain with indicated genotypea |
||||||
|---|---|---|---|---|---|---|---|---|
| WT | ΔtolC | ΔtolC::tolC | ΔftlC | ΔftlC::ftlC | ΔsilC | ΔsilC::silC | ||
| SDS | 188 | 6 ± 1 | 14 ± 2 | 9 ± 0 | 13 ± 0 | 6 ± 1 | 10 ± 0 | 8 ± 0 |
| Chloramphenicol | 1.25 | 10 ± 4 | 15 ± 2 | 10 ± 4 | 14 ± 3 | 11 ± 4 | 11 ± 4 | 10 ± 3 |
| Nalidixic acid | 5 | 22 ± 1 | 23 ± 1 | 22 ± 1 | 24 ± 2 | 19 ± 1 | 25 ± 1 | 24 ± 2 |
| t-Butyl hydroperoxide | 9 | 7 ± 2 | ND | ND | ND | ND | 14 ± 2 | 10 ± 0 |
| 18 | 8 ± 1 | ND | ND | ND | ND | 19 ± 1 | 13 ± 1 | |
Zones of growth inhibition (including the 6-mm filter disc) are shown. Values shown in bold are significantly different from corresponding WT values by Student t test. ND, not determined.
The Schu S4 ΔtolC mutant is hypercytotoxic to macrophages compared to the WT strain.
In previous studies with the LVS, we found that TolC plays a specialized role during infection of host cells not shared by FtlC (24, 30, 35). Namely, the LVS ΔtolC mutant triggers premature apoptotic death in macrophages during infection compared to that with the WT strain, resulting in a loss of the intracellular niche and reduced replication (24). Using a lactate dehydrogenase (LDH) release assay, we found that infection of murine bone marrow-derived macrophages (BMDM) with the Schu S4 ΔtolC mutant resulted in increased toxicity compared to that in infection with WT Schu S4 (Fig. 1A). Note that we used a multiplicity of infection (MOI) of 500 for the macrophage infections; this higher MOI was used to adjust for the fact that under biosafety level 3 (BSL3) conditions, we did not centrifuge the plates to facilitate bacterial contact with the macrophages. In contrast to that with the ΔtolC mutant, infection with the Schu S4 ΔftlC and ΔsilC mutants induced levels of cytotoxicity similar to those with the WT strain (Fig. 1A). Complementation of the ΔtolC mutant reduced cytotoxicity back to WT levels (Fig. 1A). The Schu S4 ΔtolC mutant, but not the ΔftlC and ΔsilC mutants, also exhibited increased cytotoxicity toward human monocyte-derived macrophages (huMDM) compared to that of WT Schu S4 (Fig. 1C). These data show that the Schu S4 ΔtolC mutant is hypercytotoxic to both murine and human macrophages, and they further suggest that the function of TolC in Schu S4 during infection of host cells is distinct from the roles of FtlC and SilC.
FIG 1.
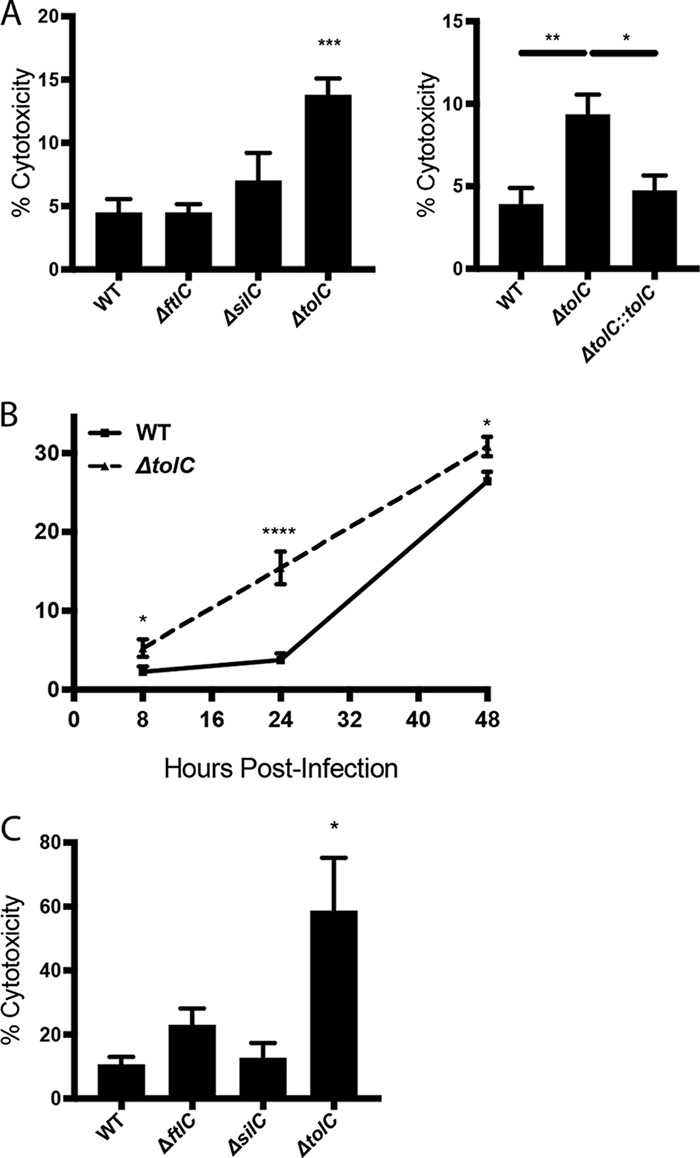
Cytotoxicity of the Schu S4 ΔtolC, ΔftlC, and ΔsilC mutants toward murine and human macrophages. (A and B) BMDM were infected with either the WT Schu S4 or indicated mutant or complemented strain at an MOI of 500 for 2 h, treated with gentamicin for 1 h, and then incubated in gentamicin-free medium until the indicated time points. (A) At 24 h postinfection, BMDM culture supernatants were collected and assayed for levels of LDH release. (B) At 8, 24, or 48 h postinfection, BMDM culture supernatants were collected and assayed for levels of LDH release. (C) huMDM were infected with either the WT Schu S4 or indicated mutant strain at an MOI of 25. At 24 h postinfection, culture supernatants were collected and assayed for levels of LDH release. Data represent means ± SEMs from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; calculated by one-way ANOVA with Tukey’s multiple-comparison posttest (A and C) or unpaired Student t test (B).
To more closely examine the role of TolC in the ability of Schu S4 to modulate host cell death during infection, we infected BMDM with WT Schu S4 or the ΔtolC mutant and examined LDH release at various times postinfection (8, 24, or 48 h). The ΔtolC mutant was hypercytotoxic to BMDM as early as 8 h postinfection, a phenotype that persisted through 24 h postinfection (Fig. 1B). By 48 h postinfection, however, cytotoxicity in WT-infected BMDM was similar to that in ΔtolC-infected BMDM (Fig. 1B). These experiments demonstrate that the Schu S4 ΔtolC mutant is hypercytotoxic to BMDM at early times postinfection and indicate that Schu S4 delays macrophage death in a TolC-dependent manner.
Next, we determined the contributions of TolC, FtlC, and SilC to the ability of Schu S4 to invade and replicate within macrophages. At early times (3 h) after infection of BMDM, intracellular CFU of the ΔtolC, ΔftlC, and ΔsilC mutants were similar to those of WT Schu S4, indicating that none of the TolC orthologs affects initial bacterial uptake or escape from the phagosome (Fig. 2). At later stages of infection (24 h), intracellular replication of the ΔftlC and ΔsilC mutants was similar to that of the WT strain (Fig. 2). In contrast, the ΔtolC mutant replicated to lower CFU than those of the WT or the complemented mutant (Fig. 2). Thus, as found for the LVS, TolC is required for maximal intracellular replication of Schu S4 following initial infection of macrophages (24). Together with the increased cytotoxicity of the Schu S4 ΔtolC mutant, this suggests that the TolC-dependent delay in host cell death is required for F. tularensis Schu S4 to maintain its intracellular niche and thereby maximize replication.
FIG 2.

Intracellular replication of the Schu S4 ΔtolC, ΔftlC, and ΔsilC mutants. BMDM were infected as described for Fig. 1 with either the WT Schu S4 or indicated mutant or complemented strain at an MOI of 500. At 3 or 24 h postinfection, cells were washed and then lysed in DMEM plus 0.1% DOC and serially diluted to determine levels of intracellular bacteria by plating for CFU. Data represent means ± SEMs from at least 3 independent experiments. *, P < 0.05, calculated by one-way ANOVA with Tukey’s multiple-comparison posttest.
F. tularensis Schu S4 actively delays macrophage apoptosis in a TolC-dependent manner.
Loss of TolC in some bacteria causes pleiotropic effects on cell metabolism, gene expression, and structural integrity (40, 41). However, in previous studies, we found no detectable effect of tolC deletion on gene expression or envelope integrity of the LVS (24, 30, 35). This is important, as the hypercytotoxic phenotype of a number of F. tularensis mutants is due to passive structural defects and intracellular lysis, leading to activation of the AIM2 inflammasome and caspase-1-dependent pyroptosis (42, 43). To determine if the increased cytotoxicity of the Schu S4 ΔtolC mutant was due to increased intracellular lysis and resulting pyroptosis, we infected BMDM with WT Schu S4 or the ΔtolC mutant and examined cleavage of caspase-1 at 24 h postinfection. Neither the WT nor the mutant strain induced detectable levels of caspase-1 cleavage (Fig. 3A), suggesting that the increased cytotoxicity of the Schu S4 ΔtolC mutant is not due to alterations of the OM or defects in envelope integrity. In agreement with this, we found that the Schu S4 ΔtolC mutant is not more sensitive to reactive oxygen species (ROS) or the membrane-targeting antimicrobial peptide polymyxin B (Table 2).
FIG 3.
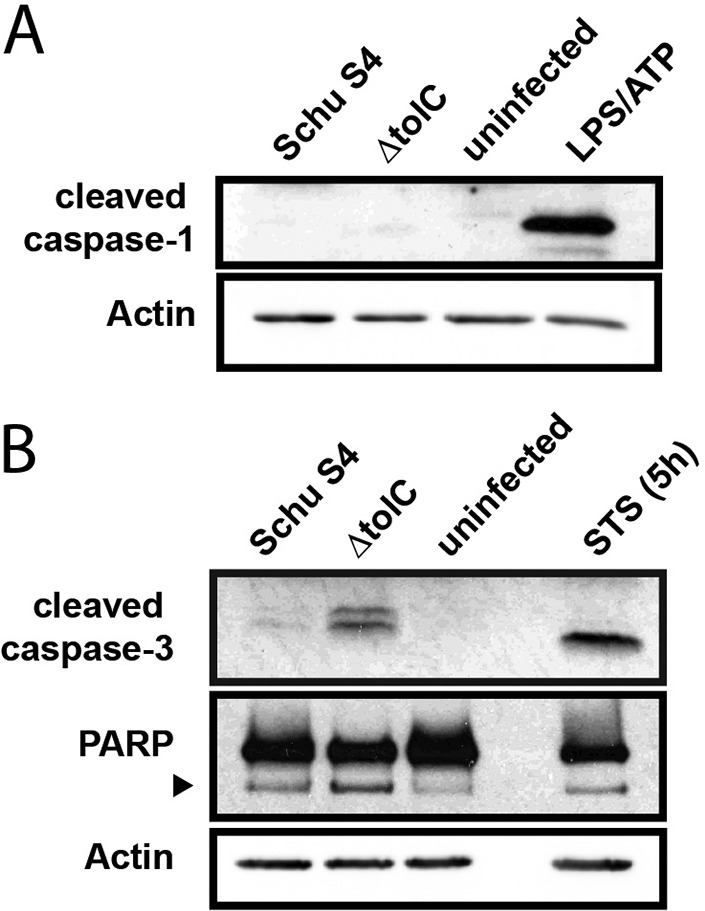
Analysis of apoptotic and pyroptotic markers in response to Schu S4 ΔtolC infection of BMDM. BMDM were infected with either WT Schu S4 or the ΔtolC mutant at an MOI of 500. At 24 h postinfection, cells were collected and lysed. Activation of either caspase-1 (A) or caspase-3 and PARP (B) was examined via SDS-PAGE and immunoblotting. As a positive control for apoptosis, cells were treated with staurosporine (STS; 125 ng/ml) for 5 h. The black arrowhead in panel B indicates the 89-kDa PARP cleavage fragment. Results are representative of those from 2 independent experiments.
To determine if Schu S4 actively inhibits macrophage apoptosis in a TolC-dependent manner, we examined LDH released from BMDM at 24 h after infection with the WT Schu S4 or ΔtolC mutant alone or from cells coinfected with the ΔtolC mutant (MOI: 500) and increasing amounts of the WT strain (MOI: 100 to 500). As expected, the single-strain control infections showed that the ΔtolC mutant is hypercytotoxic to BMDM compared to the WT strain (Fig. 4). However, the cytotoxicity caused by infection with the ΔtolC mutant was reduced as BMDM were coinfected with increasing amounts of WT Schu S4. In fact, coinfection of BMDM with equal numbers of WT and ΔtolC Schu S4 organisms completely blocked the increased cytotoxicity caused by infection with the mutant alone (Fig. 4). These results argue against the hypercytotoxicity of the ΔtolC mutant being due to a passive structural defect and support the hypothesis that F. tularensis actively inhibits host cell death during infection, possibly via a TolC-secreted effector.
FIG 4.
Coinfection of BMDM with WT and ΔtolC Schu S4. BMDM were infected singly with either WT Schu S4 or the ΔtolC mutant at an MOI of 500 or coinfected with the indicated MOI of WT Schu S4 and the ΔtolC mutant. At 24 h postinfection, culture supernatants were collected and assayed for the levels of LDH release. Data represent means ± SEMs from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; calculated by one-way ANOVA with Tukey’s multiple-comparison posttest.
We next tested if the hypercytotoxicity of the Schu S4 ΔtolC mutant was due to increased activation of apoptosis in infected cells. Infection of BMDM with the ΔtolC mutant resulted in substantial cleavage of caspase-3 at 24 h postinfection, whereas minimal caspase-3 cleavage was observed in cells infected with the WT Schu S4 strain (Fig. 3B). Additionally, we observed increased cleavage of poly(ADP-ribose) polymerase (PARP) in ΔtolC mutant-infected BMDM (Fig. 3B). PARP is a DNA repair enzyme that is cleaved by activated caspase-3 during apoptosis (44). Therefore, in agreement with previous findings with the LVS (24), these results suggest that F. tularensis Schu S4 actively delays apoptotic host cell death in a TolC-dependent manner.
F. tularensis Schu S4 modulates macrophage proinflammatory responses in a TolC-dependent manner.
In addition to delaying host cell death, the LVS modulates host proinflammatory responses during infection via TolC (35). To assess this aspect of TolC function in fully virulent F. tularensis, we analyzed levels of several cytokines released from BMDM infected with either WT or ΔtolC Schu S4 or left uninfected as a control. In response to infection with the ΔtolC mutant, BMDM released elevated levels of the chemokines CXCL1 and CXCL2 compared to those in infection with WT Schu S4 (Fig. 5). These chemokines are released from monocytes for recruitment of neutrophils to sites of infection (45, 46). In addition, BMDM infected with the ΔtolC mutant released elevated levels of interleukin 6 (IL-6) (Fig. 5), another proinflammatory cytokine secreted by macrophages to heighten immune responses to infection (47). Increased levels of released IL-1β were also detected in response to BMDM infection with the Schu S4 ΔtolC mutant. However, the levels detected in this case were extremely low and may have been the result of uncleaved IL-1β released from dying BMDM. Levels of IL-10 detected for BMDM infected with the ΔtolC mutant were also very low, and no changes were observed in release of IL-17. Taken together, these data demonstrate that in addition to apoptosis, Schu S4 suppresses host proinflammatory responses during infection in a TolC-dependent manner.
FIG 5.
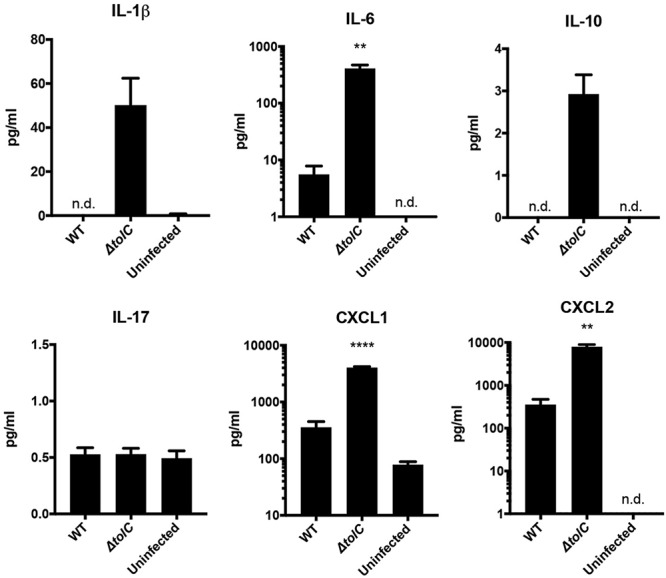
Proinflammatory responses elicited by the Schu S4 ΔtolC mutant during infection of BMDM. BMDM were infected with either WT Schu S4 or the ΔtolC mutant at an MOI of 500 for 24 h. Culture supernatants were collected and assayed for levels of the indicted cytokines. Data represent means ± SEMs from at least three independent experiments. **, P < 0.01; ****, P < 0.0001; calculated by one-way ANOVA with Tukey’s multiple-comparison posttest (for CXCL1) or unpaired Student t test (for IL-6 and CXCL2, comparing the WT with the ΔtolC mutant).
TolC and FtlC both contribute to Schu S4 virulence in mice.
To evaluate contributions of the three TolC orthologs to the virulence of F. tularensis Schu S4, we compared the ΔtolC, ΔftlC, and ΔsilC mutants to the WT strain during infection of C3H/HeN mice by both the intranasal and intradermal routes. In agreement with the extreme virulence of the Schu S4 strain, all mice infected with 10 CFU of the WT strain succumbed to infection within 5 days by both routes (Fig. 6 and 7). In comparison, the ΔftlC and ΔsilC mutants exhibited partial attenuation by the intranasal route, with ∼50% of mice infected with 10 CFU surviving to the end of the study (day 21 postinfection) (Fig. 6B and C). All mice infected with 100 CFU of the ΔftlC and ΔsilC mutants by the intranasal route succumbed to infection with kinetics similar to those in mice infected with WT Schu S4 (Fig. 6B and C). Notably, mice infected intranasally with the ΔtolC mutant exhibited complete (10 and 50 CFU) or partial (100 CFU) survival or a delayed time to death (1,000 CFU) compared to WT-infected mice (Fig. 6A). Complementation restored virulence of the ΔtolC mutant by the intranasal route (Fig. 6D). Similar experiments using the intradermal route of infection revealed a complete loss of virulence for the ΔtolC and ΔftlC mutants under the study conditions, as mice infected with up to 1,000 CFU of the ΔftlC mutant or up to 10,000 CFU of the ΔtolC mutant survived throughout the 21-day course of infection (Fig. 7A and B). Importantly, this attenuation of both the ΔtolC and ΔftlC mutants was rescued in the complemented strains (Fig. 7D). In contrast, the ΔsilC mutant remained just as virulent as the WT Schu S4 strain via the intradermal infection route, with all mice infected with 10 CFU of the ΔsilC mutant succumbing to infection with kinetics identical to those of the WT strain (Fig. 7C).
FIG 6.

Contributions of TolC orthologs to F. tularensis Schu S4 virulence in mice via the intranasal route. Mice were infected intranasally with the indicated doses of either WT Schu S4 or the ΔtolC mutant (A), ΔftlC mutant (B), or ΔsilC mutant (C) and monitored for survival for 21 days (for all strains, n = 10 mice). (D) Mice were infected with 50 CFU of the WT, the ΔtolC mutant, or the complemented Schu S4 strain and monitored for survival for 21 days (for WT-infected mice, n = 3; for all others, n = 5). **, P < 0.01; ***, P < 0.001; calculated by log rank test.
FIG 7.

Contributions of TolC orthologs to F. tularensis Schu S4 virulence in mice via the intradermal route. Mice were infected intradermally with the indicated doses of either WT Schu S4 or the ΔtolC mutant (A), ΔftlC mutant (B), or ΔsilC mutant (C) and monitored for survival for 21 days (for all strains, n = 10 mice). (D) Mice were infected with 50 CFU of WT Schu S4 or the indicated mutant or complemented strain and monitored for survival for 21 days (for WT-infected mice, n = 3; for all others, n = 5). ***, P < 0.001; calculated by log rank test.
Based on these results, we estimated 50% lethal doses (LD50) for each Schu S4 strain (Table 4) (48). The LD50 for WT Schu S4 was <10 CFU by either the intradermal or intranasal route of infection. Of the Schu S4 ΔtolC, ΔftlC and ΔsilC mutants, only the ΔtolC mutant was highly attenuated for virulence by both routes of infection, consistent with a specialized role for TolC as an F. tularensis virulence factor. The attenuation of the ΔtolC mutant was greater by the intradermal route (LD50 > 10,000 CFU) than by the intranasal route (LD50 = 200 CFU). In addition to the ΔtolC mutant, the ΔftlC mutant was also highly attenuated, but only by the intradermal route (LD50 > 1,000 CFU). These results identify both TolC and FtlC as critical Schu S4 virulence factors by the intradermal route, suggesting that this route has more stringent requirements for TolC and FtlC function, possibly via their shared roles in small-molecule efflux (Table 2). In contrast, the ΔsilC mutant exhibited no alteration in virulence compared to WT Schu S4 by the intradermal route (LD50 < 10 CFU), and both the ΔsilC and ΔftlC mutants had only minor decreases in virulence (LD50 = 10 CFU) by the intranasal route.
TABLE 4.
Estimated LD50 values for Schu S4 strains
|
Schu S4 strain |
LD50 (CFU) |
|
|---|---|---|
| Intranasal | Intradermal | |
| WT | <10 | <10 |
| ΔtolC mutant | 200 | >10,000 |
| ΔftlC mutant | 10 | >1000 |
| ΔsilC mutant | 10 | <10 |
DISCUSSION
F. tularensis is a highly virulent Gram-negative human pathogen and potential bioterrorism agent. A hallmark of F. tularensis infection is the organism’s ability to interfere with host immune responses during infection. However, many of the bacterial factors responsible for immune evasion and virulence remain unclear (9). TolC is the canonical OM channel protein involved in type I protein secretion and multidrug efflux from Gram-negative bacteria (29). We previously described a role for TolC in the ability of the F. tularensis LVS to delay host cell death during infection and facilitate bacterial virulence in mice (24, 30, 35). As the LVS is an attenuated derivative of F. tularensis subsp. holarctica, we sought in this study to understand the contribution of TolC to pathogenesis of fully virulent F. tularensis. This is important, as phenotypes for attenuated strains of Francisella do not always recapitulate in fully virulent strains (36, 49, 50). Therefore, it is critical to analyze the role for TolC in fully virulent, human-pathogenic strains such as the F. tularensis subsp. tularensis (type A) Schu S4 strain.
The Schu S4 genome encodes three homologs of the E. coli TolC protein: FtlC, SilC, and TolC (30). In this study, we constructed ftlC, silC, and tolC deletion mutations in the Schu S4 background and determined the impact of the mutations on multidrug efflux, modulation of macrophage responses to infection, and virulence in the mouse model of tularemia. Our results demonstrate that all three TolC orthologs contribute to Schu S4 multidrug resistance. Additionally, we found that the Schu S4 ΔtolC mutant is hypercytotoxic to macrophages and has a reduced ability to replicate intracellularly compared to that of the WT strain. The ΔftlC and ΔsilC mutants did not display altered cytotoxicity or intracellular replication, identifying a specialized role for TolC in the ability of F. tularensis Schu S4 to modulate host responses during infection. Furthermore, the ΔtolC mutant elicited greater proinflammatory responses from macrophages than did WT Schu S4. Finally, we found that while SilC has minimal impact on Schu S4 virulence in mice, both FtlC and TolC are critical virulence factors, with the ΔtolC mutant attenuated by both the intranasal and intradermal routes of infection and the ΔftlC mutant attenuated only by the intradermal route.
Bacterial pathogens have evolved various mechanisms to counteract the harmful effects of antimicrobial compounds, including the use of multidrug efflux systems (28, 29). Multidrug efflux systems reliant on TolC orthologs are conserved across bacteria and contribute to bacterial virulence (29, 51–54). Previous work identified roles for all three TolC orthologs in resistance of the F. tularensis LVS to a variety of antibiotics and other toxic chemicals (30, 38). Consistent with these previous studies, we found that all three TolC orthologs contribute to multidrug resistance in the F. tularensis Schu S4 strain. TolC and FtlC make overlapping contributions to Schu S4 resistance to diverse compounds, including SDS, chloramphenicol, and erythromycin. In contrast, we found that SilC participates in efflux of a distinct set of molecules (Tables 2 and 3). The Schu S4 ΔsilC mutant had increased sensitivity to CCCP, nalidixic acid, and silver nitrate compared to that of the parental Schu S4 strain. Furthermore, and in agreement with previous analysis with the LVS, the Schu S4 ΔsilC mutant had increased sensitivity to the ROS-generating agent tBh. SilC belongs to the CusC family of TolC-related proteins (32), and the silC gene is part of an operon together with the genes encoding the EmrA1and EmrB efflux transporter components (38, 55). The three components in this operon thus likely function together in the efflux of a specific set of molecules. In contrast, the tolC and ftlC genes are located individually in different genomic regions. Thus, TolC and FltC may both function as OM channels for multiple efflux systems in Francisella (29, 56), providing overlapping functionalities for small-molecule export and multidrug resistance.
In contrast to the common roles of TolC, FtlC, and SilC in multidrug resistance, our findings identify a unique role for TolC in the ability of F. tularensis Schu S4 to suppress host cell death and preserve its intracellular replicative niche. Matching previous observations with the LVS (24), the Schu S4 ΔtolC mutant was unable to delay activation of host apoptotic pathways during infection of BMDM, and this correlated with decreased intracellular replication of the mutant compared to that of WT Schu S4. Importantly, neither the Schu S4 ΔftlC nor ΔsilC mutant exhibited increased toxicity during macrophage infection, and both replicated to intracellular levels similar to those of the WT strain. Similarly, only the Schu S4 ΔtolC mutant showed increased toxicity to huMDM. These results support a model wherein F. tularensis suppresses host cell death pathways in a TolC-dependent manner to allow for maximal replication in the protected intracellular niche (24). In addition to increased host cell death, we observed heightened proinflammatory responses of BMDM during infection with the ΔtolC mutant compared to those with WT Schu S4. The ΔtolC mutant elicited increased release of CXCL1 and CXCL2, which are potent chemokines (45, 46), and the proinflammatory cytokine IL-6 (47). Taking these findings together, we propose that in addition to its role in multidrug efflux, TolC also functions as part of a T1SS for the delivery of F. tularensis effectors that alter host innate immune responses during infection.
In other bacteria, such as E. coli, loss of TolC leads to alterations in various aspects of bacterial physiology, including membrane integrity, sensitivity to ROS, and metabolic defects (57). However, while E. coli encodes a single TolC protein, the Francisella genome encodes three TolC orthologs (30, 31, 38). We propose that these additional orthologs compensate for any physiological effects that might be caused by loss of TolC in the F. tularensis OM. Previous studies with the LVS found no evidence for alterations in envelope integrity for the ΔtolC mutant and, in contrast, found that the LVS actively inhibited host cell death in a TolC-dependent manner, consistent with a role for TolC in the delivery of virulence factors (24, 35). In agreement with this, several lines of evidence support maintenance of normal cellular integrity in the Schu S4 ΔtolC mutant and an active role for TolC in suppressing host cell responses to infection. Neither the Schu S4 ΔtolC nor the other deletion mutants exhibited in vitro growth defects, and all were capable of replicating intracellularly in BMDM (Fig. 2), indicating that they maintained the ability to survive within the macrophage phagosome and escape to the cytoplasm. Moreover, the Schu S4 ΔtolC mutant did not display increased sensitivity to ROS or the antimicrobial peptide polymyxin B, and we found no evidence for caspase-1 activation during macrophage infection with the ΔtolC mutant. The latter point is crucial, as hypercytotoxicity of Francisella mutants may be a result of compromised structural integrity and activation of caspase-1-dependent pyroptosis (42, 43). Finally, we found that coinfection of BMDM with both WT and ΔtolC Schu S4 suppressed the increased cytotoxicity observed upon infection with the mutant alone. This result is inconsistent with TolC performing a passive, structural role and instead supports a model wherein F. tularensis actively delivers effectors via TolC to dampen host cell responses during infection.
Using the mouse intranasal and intradermal models of tularemia, we found differing contributions of TolC, FtlC, and SilC to Schu S4 virulence, with TolC critical for virulence by both routes of infection and FtlC critical by the intradermal route only. These results are noteworthy given that the extreme virulence of Schu S4 in mice (LD50 < 10 CFU by both infection routes) makes it difficult to observe phenotypes and dissect virulence mechanisms. Our results are in contrast to a previous screen by Kadzhaev and colleagues for attenuated Schu S4 mutants via the intradermal route of infection (36). In that study, a transposon insertion mutation in tolC had only a modest effect on Schu S4 virulence, with mice infected with 1,000 CFU of the mutant exhibiting only a delay in time to death. Our experiments were performed with a clean deletion mutation of tolC, whereas Kadzhaev et al. analyzed a transposon insertion mutation (36); the most likely explanation for the differences in our findings is that the transposon insertion did not fully disable TolC function. In addition, we were able to complement the mutant phenotypes in our various in vitro cell culture and in vivo assays, confirming the specificity of our Schu S4 ΔtolC mutation. An alternative explanation for the differences between our results is that Kadzhaev et al. used BALB/c mice for their infections, whereas we used C3H/HeN mice. We view this explanation as unlikely, as the Schu S4 ΔtolC mutant exhibited the same increased cytotoxicity and decreased intracellular replication during infection of BMDM from both BALB/c and C3H/HeN mice (unpublished data) as well as during infection of huMDM (Fig. 1C). In addition, the phenotype of the LVS ΔtolC mutant is similar in BMDM from different mouse strains (C3H/HeN, C57BL/6, and BALB/c) as well as in huMDM (reference 35 and unpublished data).
Although we found that TolC, FtlC, and SilC each participated in Schu S4 multidrug efflux, only TolC functioned in dampening host cell death responses and only TolC was critical for Schu S4 virulence by both the intranasal and intradermal routes. This implies a specialized role for TolC in F. tularensis virulence that is independent of, or in addition to, its role in multidrug efflux and is likely related to modulation of host innate immune responses. It is important to note, however, that both the ΔftlC and ΔsilC mutants exhibited small but significant attenuation in virulence by the intranasal route compared to WT Schu S4, with LD50 values for the mutants increasing to 10 CFU, compared to <10 CFU for the WT strain. In addition, the Schu S4 ΔftlC mutant was highly attenuated for virulence in mice by the intradermal route. These data highlight route-dependent differences in the pathogenesis of tularemia and suggest that the common function of TolC and FtlC in multidrug efflux may be essential for successful Schu S4 infection via the intradermal route. These data also highlight differences between the fully virulent F. tularensis subsp. tularensis Schu S4 strain and attenuated F. tularensis subsp. holarctica LVS, as TolC, but not FtlC, was found to be important for LVS virulence in mice by the intradermal route (30).
In conclusion, we show that the F. tularensis Schu S4 TolC orthologs contribute to multidrug efflux, modulation of host cell responses, and virulence. The data presented here reinforce the hypothesis that TolC has a specialized role in the secretion of virulence factors aimed at disabling host responses during infection and that TolC makes unique contributions to F. tularensis virulence. We also identify a novel role for FtlC in Schu S4 virulence by the intradermal route. These studies lay the groundwork for future efforts directed toward identifying TolC-secreted effectors and understanding the molecular basis for the extreme virulence of F. tularensis.
MATERIALS AND METHODS
Bacteria and growth conditions.
Unless otherwise indicated, the F. tularensis Schu S4 strain (BEI Research Resources Repository) and its derivatives were grown on chocolate II agar plates (BD Biosciences) or in modified Mueller-Hinton broth (MHB; Mueller-Hinton broth [BD Biosciences] containing 1% glucose, 0.025% ferric pyrophosphate, and 0.05% l-cysteine hydrochloride) (39), unless otherwise noted. All Schu S4 mutants and complemented strains are described below. All growth and manipulations of the Schu S4 strain were performed under BSL3 containment conditions.
Genetic manipulation of bacteria.
The Schu S4 ΔtolC, ΔftlC, and ΔsilC deletion mutants were made using a two-step allelic-exchange protocol developed by LoVullo et al. (39). All primers used in strain construction are listed in Table S1 in the supplemental material. Briefly, PCR products corresponding to regions of chromosomal DNA directly upstream and downstream of tolC (FTT1724), ftlC (FTT1095c), or silC (FTT1258) were amplified, ligated using GC overlap PCR (58), and cloned into the pMP812 suicide vector to make pMP812-ΔtolC, pMP812-ΔftlC, and pMP812-ΔsilC. These plasmids were electroporated into electrocompetent Schu S4, prepared as previously described (59). Transformants were selected on kanamycin (5 μg/ml), grown overnight in the absence of antibiotics, and then plated onto solid media containing 8% sucrose. Sucrose-resistant colonies were screened via colony PCR for the presence or absence of tolC, ftlC, or silC (Fig. S2A).
For unmarked chromosomal complementation of the Schu S4 ΔtolC, ΔftlC, and ΔsilC deletion strains, each gene was fused to the groE promoter and inserted into the chromosome of the cognate deletion mutant strain at the attTn7 site downstream of the conserved glmS gene (39). Briefly, the transposase-encoding shuttle plasmid pMP720 was electroporated into each deletion mutant and transformants were selected on hygromycin (200 μg/ml). The tolC, ftlC, and silC genes were amplified, placed downstream of the groE promoter, and separately cloned into pMP749 to make pMP749-tolC+, pMP749-ftlC+, and pMP749-silC+. The pMP749-based plasmids were electroporated into the corresponding deletion mutant containing pMP720, and transformants were selected on kanamycin. For each complementation, a single kanamycin-resistant colony was grown overnight in the absence of antibiotics and then plated onto kanamycin. Kanamycin-resistant colonies were then screened for hygromycin sensitivity via replica plating, indicating loss of pMP720. These colonies were then screened via colony PCR to confirm reinsertion of tolC, ftlC, or silC into the chromosome. Finally, the aphA-1 kanamycin resistance marker was removed from each strain by electroporation of a resolvase-encoding plasmid, pMP672 (encoding hygromycin resistance). The complemented strains containing pMP672 were plated on hygromycin, grown overnight in the absence of antibiotics to cure pMP672, and then plated on kanamycin. Kanamycin-sensitive, hygromycin-sensitive colonies were screened via colony PCR for the presence of the tolC, ftlC, or silC gene (Fig. S2B). One colony was selected for each gene and maintained as the Schu S4 ΔtolC::tolC, ΔftlC::ftlC, or ΔsilC::silC complemented strain. The WT Schu S4, ΔtolC mutant, ΔftlC mutant, and their complemented strains were compared for growth in MHB, brain heart infusion broth (BHI [pH 6.8]; BD Biosciences), and Chamberlain’s defined medium (CDM) (60).
Disc diffusion assay.
Lawns of the WT Schu S4, ΔtolC, ΔftlC, ΔsilC, ΔtolC::tolC+, ΔftlC::ftlC+, or ΔsilC::silC+ strain were plated from frozen stocks. Prior to incubation of plates, solutions of the following compounds (at the indicated stock concentrations) were diluted in sterile water: SDS (44 mM), streptomycin (290 μM), chloramphenicol (260 μM), erythromycin (460 μM), ampicillin (480 μM), CCCP (3.25 mM), nalidixic acid (1.45 mM), silver nitrate (196 mM), H2O2 (8.8 M), and tBh (7.8 M). To examine sensitivity of the Schu S4 strains to the compounds listed above, a 15-μl aliquot of each desired dilution was added to 6-mm paper discs (BD Biosciences) and the saturated discs were placed onto the bacterial lawns. Following incubation for 72 h, zones of growth inhibition were measured and recorded as diameters (in millimeters), including the 6-mm disc.
Preparation of murine macrophages.
Bone marrow-derived macrophages (BMDM) were generated from the femurs of C3H/HeN mice as previously described (35). Bone marrow-derived cells were cultured for 6 days in bone marrow medium (BMM; Dulbecco modified Eagle medium [DMEM] with GlutaMax [Gibco] supplemented with 30% L929 cell supernatant, 20% fetal bovine serum [FBS; HyClone], and 1 mM sodium pyruvate). For cytotoxicity and intracellular replication experiments, BMDM were seeded in 24-well plates at a concentration of 1.5 × 105/well in 1 ml of BMM. For immunoblotting experiments, BMDM were seeded in 6-well plates at a concentration of 1 × 106/well in 3 ml of BMM. BMDM were allowed to adhere to plates overnight and then were washed with phosphate-buffered saline (PBS) prior to infection with the desired F. tularensis strains resuspended in bone marrow infection medium (BMIM; DMEM with GlutaMax [Gibco] supplemented with 15% L929 cell supernatant, 5% FBS, and 1 mM sodium pyruvate).
All protocols involving animals were approved by the Institutional Animal Care and Use Committee of Stony Brook University.
BMDM infections.
For immunoblotting, intracellular replication, and cytotoxicity experiments, BMDM were seeded as described above and infected with WT Schu S4, the ΔtolC, ΔftlC, or ΔsilC mutant, or the complemented strain at an MOI of 500. Plates were not centrifuged following addition of bacteria, necessitating the high MOI. For coinfection cytotoxicity experiments, BMDM were seeded and infected as described above with the WT Schu S4 or ΔtolC mutant alone at an MOI of 500 or with a mixture of the ΔtolC mutant (MOI = 500) and WT Schu S4 (MOI = 100, 200, 400, or 500). BMDM were incubated with the bacteria for 2 h, washed with PBS, and incubated for an additional 1 h with gentamicin (10 μg/ml) to kill extracellular bacteria. After gentamicin treatment, cells were washed with PBS and then incubated with fresh BMIM (lacking gentamicin) until the desired time points.
Cytotoxicity assays.
BMDM were cultured and infected as described above. At the desired times postinfection, supernatants from infected BMDM were collected and analyzed for the presence of LDH using the CytoTox 96 nonradioactive cytotoxicity assay (Promega) according to the manufacturer’s protocol. Background LDH release was quantified by examining supernatants from uninfected BMDM, and maximum LDH release was quantified from cells that were lysed via a single −80°C/54°C freeze-thaw cycle. Percent LDH release was calculated by subtracting background LDH release from all experimental sample values, dividing by the maximum LDH release minus background LDH release, and multiplying by 100.
Intracellular replication assays.
BMDM were cultured and infected as described above. At the desired times postinfection, cells were washed with PBS and lysed in DMEM plus 0.1% deoxycholate (DOC) for 10 min at room temperature (61). Lysates were serially diluted, plated, and incubated for 3 days prior to enumeration of CFU.
Immunoblotting.
For analysis of pyroptosis and apoptosis during infection, BMDM were cultured and infected as described above. At 24 h postinfection, cells were scraped and the supernatant and cell mixtures were collected. For positive apoptosis controls, cells were treated with 1 μM staurosporine (Invitrogen) for 5 h. For positive pyroptosis controls, cells were treated with E. coli lipopolysaccharide (LPS; 100 ng/ml; Sigma) for 4 h, followed by ATP (2.5 mM; Roche) for 1 h. Cells were pelleted, washed once with PBS, and then lysed in radioimmunoprecipitation assay (RIPA) buffer containing 1× protease inhibitor cocktail (Roche). Ten micrograms of each protein sample was separated via SDS-PAGE. After separation, proteins were transferred to nitrocellulose membranes at 110 V for 90 min. Membranes were blocked with 3% bovine serum albumin (BSA) in TBST (Tris-buffered saline [TBS] with 0.1% Tween 20) at room temperature for 2 h. Membranes were then incubated with primary polyclonal antibodies recognizing PARP (Cell Signaling; no. 9542; 1:1,000 dilution), caspase-1 (laboratory collection of J. B. Bliska), caspase-3 (Cell Signaling; no. 9661; 1:1,000 dilution), or β-actin (Sigma; A3854; 1:50,000 dilution), overnight at 4°C. Membranes were washed with TBST four times (15, 5, 5, and 5 min) and incubated with a horseradish peroxidase-conjugated mouse anti-rabbit IgG secondary antibody (Cell Signaling; no. 7074, 1:2,000 dilution) for 1 h at room temperature. Membranes were washed as described above, and bands were visualized by enhanced chemiluminescence (GE Life Sciences).
Luminex assay.
BMDM were cultured and infected as described above. At the desired times postinfection, supernatants from infected BMDM were collected and analyzed for the levels of the cytokines IL-1β, IL-6, IL-10, IL-17, CXCL1, and CXCL2 using a MILLIPLEX MAP mouse cytokine/chemokine magnetic bead panel (Millipore) and a Luminex 200 analyzer.
Isolation and infection of human monocyte-derived macrophages.
Human monocytes were isolated from venous blood of consenting healthy donors as described previously (62). Blood was collected using 0.12% disodium EDTA as an anticoagulant. Mononuclear cells were fractionated using a gradient overlay of Lymphoprep (STEMCELL Technologies). The monocyte fraction was collected and washed. Monocytes were isolated by negative selection using the Monocyte Isolation Kit II (Miltenyi Biotec) according to the manufacturer’s instructions. Cells were plated in 24-well plates at concentration of 2 × 105 per well and cultured for 5 days in RPMI medium (Gibco) supplemented with 10% FBS and 50 ng/ml of recombinant human macrophage colony-stimulating factor (M-CSF; R&D Systems). On day 6, cells were infected with bacteria at an MOI of 25 in RPMI medium (Gibco) supplemented with 10% FBS and 50 ng/ml of M-CSF as described for BMDM infection.
Infection of mice.
For all mouse infections, inoculums were prepared by growing bacteria for 72 h on plates, inoculating overnight cultures, and then diluting the resulting cultures to the desired final concentrations. Intranasal infections were performed by administering a 10-μl inoculum into each naris (20 μl total). Intradermal infections were performed by injecting a 50-μl inoculum into the pinnae. Female C3H/HeN mice (6 to 8 weeks old; Charles River Labs) were used for all infections. Actual inocula were determined by retrospective CFU counts. All mice were monitored for survival for 21 days following infection. All mouse infections with were performed at the Rutgers Regional Biocontainment Laboratory under animal BSL3 conditions.
Statistical analysis.
For the drug sensitivity assays, one-tailed P values were calculated by one-way analysis of variance (ANOVA). For 24-h LDH release assays and coinfection experiments, one-tailed P values were calculated by one-way ANOVA with Tukey’s multiple-comparison posttest. For the LDH release time course experiment and the cytokine release data, one-tailed P values were calculated using the unpaired Student t test. All mouse survival studies were analyzed using the log rank test. Statistical calculations were performed using GraphPad Prism 6 software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeronimo Cello (Stony Brook University) for assistance with all BSL3-related experiments and training. We thank David Perlin, Steven Park, and Juanita Vakerich (Rutgers Regional Biocontainment Laboratory) for performing animal BSL3 infections. We thank Liise-anne Pirofski (Albert Einstein College of Medicine) for assistance with the Luminex bead array assay. We thank Indra Jayatilaka (Stony Brook University) for assistance with human blood collection. We thank James Bliska (Dartmouth Geisel School of Medicine) for providing the anti-caspase-1 antibody. The following reagent was obtained through BEI Resources, NIAID, NIH: Francisella tularensis subsp. tularensis, strain Schu S4 (FSC237), NR-643.
This work was supported by NIAID, NIH, awards R21 AI115069 (D.G.T.), F31 AI124570 (E.J.K.), and T32 AI007539 (E.J.K.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00823-18.
REFERENCES
- 1.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Tonat K. Working Group on Civilian Biodefense. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 2.Ellis J, Oyston PC, Green M, Titball RW. 2002. Tularemia. Clin Microbiol Rev 15:631–646. doi: 10.1128/CMR.15.4.631-646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldman KA, Enscore RE, Lathrop SL, Matyas BT, McGuill M, Schriefer ME, Stiles-Enos D, Dennis DT, Petersen LR, Hayes EB. 2001. An outbreak of primary pneumonic tularemia on Martha’s Vineyard. N Engl J Med 345:1601–1606. doi: 10.1056/NEJMoa011374. [DOI] [PubMed] [Google Scholar]
- 4.Oyston PC, Sjostedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol 2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 5.Hirschmann JV. 2018. From squirrels to biological weapons: the early history of tularemia. Am J Med Sci 356:319–328. doi: 10.1016/j.amjms.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Kingry LC, Petersen JM. 2014. Comparative review of Francisella tularensis and Francisella novicida. Front Cell Infect Microbiol 4:35. doi: 10.3389/fcimb.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma J, Mares CA, Li Q, Morris EG, Teale JM. 2011. Features of sepsis caused by pulmonary infection with Francisella tularensis type A strain. Microb Pathog 51:39–47. doi: 10.1016/j.micpath.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. 2008. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect Immun 76:3001–3010. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones BD, Faron M, Rasmussen JA, Fletcher JR. 2014. Uncovering the components of the Francisella tularensis virulence stealth strategy. Front Cell Infect Microbiol 4:32. doi: 10.3389/fcimb.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts LM, Tuladhar S, Steele SP, Riebe KJ, Chen CJ, Cumming RI, Seay S, Frothingham R, Sempowski GD, Kawula TH, Frelinger JA. 2014. Identification of early interactions between Francisella and the host. Infect Immun 82:2504–2510. doi: 10.1128/IAI.01654-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Celli J, Zahrt TC. 2013. Mechanisms of Francisella tularensis intracellular pathogenesis. Cold Spring Harb Perspect Med 3:a010314. doi: 10.1101/cshperspect.a010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabrik I, Link M, Putzova D, Plzakova L, Lubovska Z, Philimonenko V, Pavkova I, Rehulka P, Krocova Z, Hozak P, Santic M, Stulik J. 2018. The early dendritic cell signaling induced by virulent Francisella tularensis strain occurs in phases and involves the activation of extracellular signal-regulated kinases (ERKs) and p38 in the later stage. Mol Cell Proteomics 17:81–94. doi: 10.1074/mcp.RA117.000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Putzova D, Panda S, Hartlova A, Stulik J, Gekara NO. 2017. Subversion of innate immune responses by Francisella involves the disruption of TRAF3 and TRAF6 signalling complexes. Cell Microbiol 19:e12769. doi: 10.1111/cmi.12769. [DOI] [PubMed] [Google Scholar]
- 14.Krocova Z, Macela A, Kubelkova K. 2017. Innate immune recognition: implications for the interaction of Francisella tularensis with the host immune system. Front Cell Infect Microbiol 7:446. doi: 10.3389/fcimb.2017.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dotson RJ, Rabadi SM, Westcott EL, Bradley S, Catlett SV, Banik S, Harton JA, Bakshi CS, Malik M. 2013. Repression of inflammasome by Francisella tularensis during early stages of infection. J Biol Chem 288:23844–23857. doi: 10.1074/jbc.M113.490086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melillo AA, Bakshi CS, Melendez JA. 2010. Francisella tularensis antioxidants harness reactive oxygen species to restrict macrophage signaling and cytokine production. J Biol Chem 285:27553–27560. doi: 10.1074/jbc.M110.144394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melillo AA, Mahawar M, Sellati TJ, Malik M, Metzger DW, Melendez JA, Bakshi CS. 2009. Identification of Francisella tularensis live vaccine strain CuZn superoxide dismutase as critical for resistance to extracellularly generated reactive oxygen species. J Bacteriol 191:6447–6456. doi: 10.1128/JB.00534-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinkead LC, Allen LA. 2016. Multifaceted effects of Francisella tularensis on human neutrophil function and lifespan. Immunol Rev 273:266–281. doi: 10.1111/imr.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinkead LC, Fayram DC, Allen LH. 2017. Francisella novicida inhibits spontaneous apoptosis and extends human neutrophil lifespan. J Leukoc Biol 102:815–828. doi: 10.1189/jlb.4MA0117-014R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai XH, Sjostedt A. 2003. Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect Immun 71:4642–4646. doi: 10.1128/IAI.71.8.4642-4646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCracken JM, Kinkead LC, McCaffrey RL, Allen LA. 2016. Francisella tularensis modulates a distinct subset of regulatory factors and sustains mitochondrial integrity to impair human neutrophil apoptosis. J Innate Immun 8:299–313. doi: 10.1159/000443882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santic M, Pavokovic G, Jones S, Asare R, Kwaik YA. 2010. Regulation of apoptosis and anti-apoptosis signalling by Francisella tularensis. Microbes Infect 12:126–134. doi: 10.1016/j.micinf.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen LA. 2012. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immunol 188:3351–3363. doi: 10.4049/jimmunol.1102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doyle CR, Pan JA, Mena P, Zong WX, Thanassi DG. 2014. TolC-dependent modulation of host cell death by the Francisella tularensis live vaccine strain. Infect Immun 82:2068–2078. doi: 10.1128/IAI.00044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bokhari SM, Kim KJ, Pinson DM, Slusser J, Yeh HW, Parmely MJ. 2008. NK cells and gamma interferon coordinate the formation and function of hepatic granulomas in mice infected with the Francisella tularensis live vaccine strain. Infect Immun 76:1379–1389. doi: 10.1128/IAI.00745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parmely MJ, Fischer JL, Pinson DM. 2009. Programmed cell death and the pathogenesis of tissue injury induced by type A Francisella tularensis. FEMS Microbiol Lett 301:1–11. doi: 10.1111/j.1574-6968.2009.01791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wickstrum JR, Bokhari SM, Fischer JL, Pinson DM, Yeh HW, Horvat RT, Parmely MJ. 2009. Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect Immun 77:4827–4836. doi: 10.1128/IAI.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koronakis V, Eswaran J, Hughes C. 2004. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu Rev Biochem 73:467–489. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- 29.Hinchliffe P, Symmons MF, Hughes C, Koronakis V. 2013. Structure and operation of bacterial tripartite pumps. Annu Rev Microbiol 67:221–242. doi: 10.1146/annurev-micro-092412-155718. [DOI] [PubMed] [Google Scholar]
- 30.Gil H, Platz GJ, Forestal CA, Monfett M, Bakshi CS, Sellati TJ, Furie MB, Benach JL, Thanassi DG. 2006. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc Natl Acad Sci U S A 103:12897–12902. doi: 10.1073/pnas.0602582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huntley JF, Conley PG, Hagman KE, Norgard MV. 2007. Characterization of Francisella tularensis outer membrane proteins. J Bacteriol 189:561–574. doi: 10.1128/JB.01505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rensing C, Grass G. 2003. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev 27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 33.Delmar JA, Su CC, Yu EW. 2014. Bacterial multidrug efflux transporters. Annu Rev Biophys 43:93–117. doi: 10.1146/annurev-biophys-051013-022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conroy O, Kim EH, McEvoy MM, Rensing C. 2010. Differing ability to transport nonmetal substrates by two RND-type metal exporters. FEMS Microbiol Lett 308:115–122. doi: 10.1111/j.1574-6968.2010.02006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Platz GJ, Bublitz DC, Mena P, Benach JL, Furie MB, Thanassi DG. 2010. A tolC mutant of Francisella tularensis is hypercytotoxic compared to the wild type and elicits increased proinflammatory responses from host cells. Infect Immun 78:1022–1031. doi: 10.1128/IAI.00992-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadzhaev K, Zingmark C, Golovliov I, Bolanowski M, Shen H, Conlan W, Sjostedt A. 2009. Identification of genes contributing to the virulence of Francisella tularensis SCHU S4 in a mouse intradermal infection model. PLoS One 4:e5463. doi: 10.1371/journal.pone.0005463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huntley JF, Conley PG, Rasko DA, Hagman KE, Apicella MA, Norgard MV. 2008. Native outer membrane proteins protect mice against pulmonary challenge with virulent type A Francisella tularensis. Infect Immun 76:3664–3671. doi: 10.1128/IAI.00374-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alqahtani M, Ma Z, Ketkar H, Suresh RV, Malik M, Bakshi CS. 2018. Characterization of a unique outer membrane protein required for oxidative stress resistance and virulence of Francisella tularensis. J Bacteriol 200:693–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LoVullo ED, Molins-Schneekloth CR, Schweizer HP, Pavelka MS Jr. 2009. Single-copy chromosomal integration systems for Francisella tularensis. Microbiology 155:1152–1163. doi: 10.1099/mic.0.022491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhamdhere G, Zgurskaya HI. 2010. Metabolic shutdown in Escherichia coli cells lacking the outer membrane channel TolC. Mol Microbiol 77:743–754. doi: 10.1111/j.1365-2958.2010.07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos MR, Cosme AM, Becker JD, Medeiros JM, Mata MF, Moreira LM. 2010. Absence of functional TolC protein causes increased stress response gene expression in Sinorhizobium meliloti. BMC Microbiol 10:180. doi: 10.1186/1471-2180-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng K, Broz P, Jones J, Joubert LM, Monack D. 2011. Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell Microbiol 13:1586–1600. doi: 10.1111/j.1462-5822.2011.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LA, Jones BD. 2011. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect Immun 79:581–594. doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. 2005. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J Biol Chem 280:857–860. doi: 10.1074/jbc.C400538200. [DOI] [PubMed] [Google Scholar]
- 45.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. 1989. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A 86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haskill S, Peace A, Morris J, Sporn SA, Anisowicz A, Lee SW, Smith T, Martin G, Ralph P, Sager R. 1990. Identification of three related human GRO genes encoding cytokine functions. Proc Natl Acad Sci U S A 87:7732–7736. doi: 10.1073/pnas.87.19.7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka T, Narazaki M, Kishimoto T. 2014. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed LJ, Muench H. 1938. A simple method for estimating fifty percent endpoints. Am J Hyg 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 49.Lindgren H, Shen H, Zingmark C, Golovliov I, Conlan W, Sjostedt A. 2007. Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect Immun 75:1303–1309. doi: 10.1128/IAI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin A, Scott DW, Mann BJ. 2008. Francisella tularensis subsp. tularensis Schu S4 disulfide bond formation protein B, but not an RND-type efflux pump, is required for virulence. Infect Immun 76:3086–3092. doi: 10.1128/IAI.00363-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. doi: 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buckley AM, Webber MA, Cooles S, Randall LP, La Ragione RM, Woodward MJ, Piddock LJ. 2006. The AcrAB-TolC efflux system of Salmonella enterica serovar Typhimurium plays a role in pathogenesis. Cell Microbiol 8:847–856. doi: 10.1111/j.1462-5822.2005.00671.x. [DOI] [PubMed] [Google Scholar]
- 53.Spaniol V, Bernhard S, Aebi C. 2015. Moraxella catarrhalis AcrAB-OprM efflux pump contributes to antimicrobial resistance and is enhanced during cold shock response. Antimicrob Agents Chemother 59:1886–1894. doi: 10.1128/AAC.03727-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alcalde-Rico M, Hernando-Amado S, Blanco P, Martínez JL. 2016. Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front Microbiol 7:1483. doi: 10.3389/fmicb.2016.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Z, Banik S, Rane H, Mora VT, Rabadi SM, Doyle CR, Thanassi DG, Bakshi CS, Malik M. 2014. EmrA1 membrane fusion protein of Francisella tularensis LVS is required for resistance to oxidative stress, intramacrophage survival and virulence in mice. Mol Microbiol 91:976–995. doi: 10.1111/mmi.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atkins HS, Dassa E, Walker NJ, Griffin KF, Harland DN, Taylor RR, Duffield ML, Titball RW. 2006. The identification and evaluation of ATP binding cassette systems in the intracellular bacterium Francisella tularensis. Res Microbiol 157:593–604. doi: 10.1016/j.resmic.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S. 2011. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front Microbiol 2:189. doi: 10.3389/fmicb.2011.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cha-Aim K, Fukunaga T, Hoshida H, Akada R. 2009. Reliable fusion PCR mediated by GC-rich overlap sequences. Gene 434:43–49. doi: 10.1016/j.gene.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 59.Qin A, Mann BJ. 2006. Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol 6:69. doi: 10.1186/1471-2180-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chamberlain RE. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl Microbiol 13:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chalabaev S, Anderson CA, Onderdonk AB, Kasper DL. 2011. Sensitivity of Francisella tularensis to ultrapure water and deoxycholate: implications for bacterial intracellular growth assay in macrophages. J Microbiol Methods 85:230–232. doi: 10.1016/j.mimet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noah CE, Malik M, Bublitz DC, Camenares D, Sellati TJ, Benach JL, Furie MB. 2010. GroEL and lipopolysaccharide from Francisella tularensis live vaccine strain synergistically activate human macrophages. Infect Immun 78:1797–1806. doi: 10.1128/IAI.01135-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.