

Abstract
Allergic diseases have in common a dysfunctional epithelial barrier, which allows the penetration of allergens and microbes, leading to the release of type 2 cytokines that drive allergic inflammation. The accessibility of skin, compared with lung or gastrointestinal tissue, has facilitated detailed investigations into mechanisms underlying epithelial barrier dysfunction in atopic dermatitis (AD). This Review describes the formation of the skin barrier and analyzes the link between altered skin barrier formation and the pathogenesis of AD. The keratinocyte differentiation process is under tight regulation. During epidermal differentiation, keratinocytes sequentially switch gene expression programs, resulting in terminal differentiation and the formation of a mature stratum corneum, which is essential for the skin to prevent allergen or microbial invasion. Abnormalities in keratinocyte differentiation in AD skin result in hyperproliferation of the basal layer of epidermis, inhibition of markers of terminal differentiation, and barrier lipid abnormalities, compromising skin barrier and antimicrobial function. There is also compelling evidence for epithelial dysregulation in asthma, food allergy, eosinophilic esophagitis, and allergic rhinosinusitis. This Review examines current epithelial barrier repair strategies as an approach for allergy prevention or intervention.
Allergic diseases such as atopic dermatitis (AD), food allergy (FA), asthma, and allergic rhinitis affect more than 30% of the population (1–3). These diseases have in common a dysfunctional epithelial barrier, resulting in the penetration of allergens and microbes, accompanied by the release of epithelial-derived cytokines (e.g., thymic stromal lymphopoietin [TSLP], IL-25, IL-33), which drive type 2 immune responses. Although other immune pathways can modify the course of illness, cytokines including IL-4, IL-13, IL-31, TSLP, and IL-33 play a key role in allergic diseases (4–8), eliciting local tissue injury and repair (5, 9–14).
AD is the most prevalent chronic inflammatory skin disease (3, 15–19). In a subset of allergic patients, it is thought that AD-related skin epithelial dysfunction contributes to the atopic march, which starts with AD and often leads directly to FA (20–23). The link from AD to respiratory allergy is more controversial; however, atopic march progression is facilitated in patients who develop IgE to both food and inhalant allergens (24).
In this Review, we describe formation of the skin barrier, review the link between altered skin barrier formation and AD, discuss evidence for epithelial barrier dysfunction in other allergic diseases, and explore epithelial barrier intervention/repair strategies with the goal of preventing AD and the atopic march.
The cornified envelope and keratinocyte differentiation in the skin
The skin’s barrier function primarily depends on the outermost epidermal layer, stratum corneum (SC). To form SC, keratinocytes pass through a tightly regulated differentiation program and sequentially form stratum basale (SB), stratum spinosum (SS), stratum granulosum (SG), and SC layers of the skin (Figure 1). In human skin, keratinocytes irreversibly exit the cell cycle after mitosis in the basal layer and differentiate progressively across the epidermis toward the SC. Each layer is defined by its expression of characteristic morphological and biochemical features indicating its state of differentiation (25–27). The keratins KRT5 and KRT14 are predominantly expressed by basal keratinocytes. The markers of early differentiation KRT1 and KRT10 are made in SS. The cornified envelope precursors involucrin (IVL) and transglutaminase-1 (TGM1) are also present in SS. Late differentiation markers including the cornified envelope protein loricrin (LOR) and the precursor of the keratin cross-linking protein filaggrin (FLG) are expressed in SG keratinocytes. TGM1 cross-links IVL, LOR, and other structural proteins to form the cornified envelope.
Figure 1. Structure of human epidermis.

Human epidermis is composed of SB, SS, SG, and SC. In SC, corneocytes (flattened and denucleated keratinocytes) and intercellular lipids released from lamellar bodies form the “brick and mortar” structures. The cornified envelope, a highly cross-linked layer of insoluble proteins, forms under the corneocyte cell membrane, anchored by extracellular lipids. Components of the cornified envelope (keratohyalin granules, a source of filaggrin) and lamellar bodies (containing lipids, lipid-processing enzymes, corneodesmosin, proteases, and protease inhibitors) are formed in SG. The surface of the SC is shed off by degradation of corneodesmosomes via the activity of several proteases.
Calcium forms a steep gradient across the human epidermis, increasing from the SB (5 μM) to the outer SG (>20 μM) (27, 28). Importantly, there is cell-to-cell heterogeneity in average Ca2+ concentrations in the epidermis (28). Formation of the Ca2+ gradient coincides with key developmental milestones of skin barrier formation and differentiation into the SC.
Primary human keratinocyte cultures are an excellent model for studying epidermal differentiation, as they recapitulate the steps of epidermal cell differentiation (29). When cultured at low external calcium ([Ca2+]o) (~0.06 mM), epidermal keratinocytes proliferate rapidly and express a basal cell phenotype. Raising [Ca2+]o above 1.3 mM (i.e., calcium switch) promotes cell differentiation, as indicated by adherens junction and desmosome formation, along with cell layer stratification and cornification. After calcium switch, changes in gene expression occur with a defined temporal order: first KRT1/10, followed by IVL and TGM1, and finally profilaggrin and LOR (30), reflecting the sequence of epidermal differentiation.
p63 transcriptional factor, described as the master regulator of skin development (31, 32), plays an essential role in maintaining self-renewing populations. p63 deficiency causes keratinocyte senescence (33), and p63-deficient keratinocytes display differentiation defects (34). Keratinocyte development in SS is selectively controlled by the Notch pathway (35–38). Notch activity in keratinocytes results in withdrawal from the cell cycle and induces expression of early differentiation markers (39) like KRT1/10, which are required for maintenance of epidermal integrity (40). KRT10-deficient mice demonstrate increased transepidermal water loss (TEWL), delayed barrier repair after barrier disruption, reduced acid sphingomyelinase (SMase) activity, and reduced ratio of ceramides to total lipids (41). Skin fragility in KRT10-deficient mice may be the consequence of two complementary mechanisms: a decrease of normal KRT1/10 filaments and an increase in KRT6/16, with a poor filament-forming capacity (42). KRT1-deficient mice also demonstrate increased TEWL and cell fragility. Transcriptome profiling revealed a KRT1-mediated gene expression signature resembling AD skin, with upregulation of S100A8, S100A9, and TSLP, compensatory increases in KRT5/14 filaments in basal epithelium, and increased KRT6/16 filaments in suprabasal epithelium (43). Skin-specific ablation of Notch produces severe AD (44, 45) and disrupted keratinocyte differentiation (46, 47). The absence of Notch activity allows Wnt/β-catenin signaling to persist in a tissue where it is normally repressed (36, 48), supporting hyperproliferation.
Filaggrin.
SG keratinocytes produce filaggrin (FLG) as a large precursor, profilaggrin, the major component of keratohyalin granules. Profilaggrin comprises 10–12 FLG repeats flanked by single N- and C-terminal domains. At the SG-to-SC transition, profilaggrin’s rapid dephosphorylation and cleavage generate positively charged FLG monomers. Profilaggrin’s N-terminus translocates into the nucleus, where it may be involved in nuclear breakdown. FLG monomers, on the other hand, associate with keratin filaments and form the corneocyte fibrous matrix. In SC, after deimination by peptidylarginine deiminases, FLG becomes neutrally charged, enabling its disassociation from keratins and degradation into free amino acids, mediated by several proteases, including caspase-14, bleomycin hydrolase, and calpain-1. These amino acids (pyrrolidone carboxylic acid, urocanic acid) constitute natural moisturizing factor (NMF) and are essential for regulation of skin hydration (49), pH (50), photoprotection (50), and immune modulation (50–54).
Formation of lipid lamellae in keratinocytes.
Lamellar bodies formed in the SS layers are organelles derived from the Golgi apparatus and contain phospholipids, glycosylceramides, sphingomyelin, and cholesterol. During the SG-to-SC transition, at the apical surface of SG cells, lamellar bodies secrete their contents (including various proteases, protease inhibitors, and lipids such as acylceramides) into the extracellular space between SG and lower SC. Once secreted from SG cells, acylceramides become part of the cornified envelope, forming repeated sheets of lipid lamellae that serve as an impermeable barrier in the SC (55).
Epidermal ceramides are critical for skin barrier function (56). Acylceramides (EOS, EOH, EOP, and EODS) account for about 12% of SC ceramides (57). Although normal ceramides contain two hydrophobic chains (sphingoid base and fatty acid), acylceramides have three hydrophobic chains (sphingoid base, an ω-OH ultra-long-chain fatty acid [ULCFA] with 30–36 carbon-chain length, and linoleic acid [C18:2-COOH]). Acylceramides serve as precursors to protein-bound ceramides for skin barrier formation. Several unique enzymes are involved in acylceramide synthesis (refs. 56–60 and Figure 2).
Figure 2. Lamellar body formation and generation of acylceramides and protein-bound ceramides in human epidermis.

The enzymes and reactions responsible for creating acylceramides and protein-bound ceramides are shown. The schematics represent acylceramide production, lamellar body assembly and secretion in SG, and formation of the protein-bound ceramides in SC. Acylceramides are formed in the ER and secreted through the Golgi apparatus. ω-OH protein-bound ceramides are formed at the cell membrane. Sequential actions of the fatty acid elongases ELOVL1 and ELOVL4 generate ULCFAs of up to C26 and C28 carbon-chain-lengths, respectively. The cytochrome P450 enzyme CYP4F22 then ω-hydroxylates these ULCFAs, generating ω-OH ULCFAs. Next, the ceramide synthase CERS3 uses ω-OH ULCFAs for ω-OH ceramide synthesis. Finally, the transacylase PNPLA1 forms an ester linkage between the 18:2n-6 fatty acid taken from triglycerides and the ω-OH group of ω-OH ceramide to create an acylceramide. Each enzyme involved in acylceramide production is localized in the ER, indicating that acylceramide production takes place there. Once produced, the UDP-glucose ceramide glucosyltransferase UGCG glycosylates acylceramides in the Golgi apparatus, followed by ABCA12-mediated transport into lamellar bodies. In the course of protein-bound ceramide production, the 18:2n-6 fatty acid portions of acylceramides are subjected to peroxidation by the lipoxygenases ALOX12B and ALOXE3, followed by deglycosylation by β-glucosylceramidase (GBA). Transglutaminase then cross-links the exposed ω-OH group with cornified envelope proteins such as involucrin, envoplakin, and periplakin. Cer, ceramide; GlcCer, glucosylceramide; G, glycosyl group.
Failure of terminal keratinocyte differentiation in AD skin
AD skin is characterized by broad defects in terminal keratinocyte differentiation (ref. 61 and Figure 3), expansion of cells in the SB layer, and concomitant reduction in cells of the SS and SG layers (62, 63). Hyperproliferating epithelium is associated with KRT6/16 overexpression (64). Consistent with a block in terminal keratinocyte differentiation, AD skin has reduced expression of skin barrier proteins including FLG, IVL, and LOR, antimicrobial peptides, and β-defensins (65–67).
Figure 3. Epidermal differentiation pattern in normal and AD skin.

(A and B) Epidermal differentiation pattern in normal (A) and AD (B) skin. The keratinocyte differentiation process is under tight regulation. Cells proliferate in the basal layer of the epidermis. As basal layer keratinocytes detach from the basement membrane and migrate into the first suprabasal layer in the spinous layer, they irreversibly exit the cell cycle and switch from KRT5/KRT14 to KRT1/KRT10 production. During epidermal differentiation, keratinocytes sequentially switch gene expression programs and express the granular layer differentiation markers FLG, LOR, and TGM1. The Wnt/β-catenin pathway is active in the proliferating epidermis, whereas keratinocyte differentiation in the spinous layer is under the control of the Notch pathway. Changes in extracellular Ca2+ and lipid metabolism trigger protein kinase C (PKC) pathway activation and regulate the transcription of FLG, LOR, IVL, and TGM1. In AD skin, abnormalities in the differentiation of keratinocytes result in hyperproliferation of the basal layer, reduction of the spinous layer, and inhibition of markers of terminal differentiation, all of which compromise skin barrier and antimicrobial function.
Studies have linked FLG loss-of-function mutations to the structural abnormalities that underlie AD pathogenesis (17, 51). FLG-null mutations are the strongest known genetic risk factor for AD. Presence of FLG mutation increases AD risk 3-fold in comparison with the general population and predisposes to earlier disease onset, prolonged duration, and increased disease severity. FLG mutations are the most common among Northern Europeans, with a prevalence of approximately 10%, the main mutations being R501X and 2282del4 (17). There are ethnic differences in AD-associated FLG mutations: in Asian populations, FLG P478S and C3321delA variants, uncommon in European populations, are associated with increased AD risk (68–71). In African American children, FLG2 loss-of-function mutations are associated with increased AD risk (72). Several uncommon FLG mutations in African American AD patients were described recently (73).
A number of additional factors play a role in regulating FLG expression in the skin (Table 1). For example, type 2 cytokines decrease FLG expression in AD skin, even in subjects without FLG mutations (52, 65). FLG intragenic copy number (typically 10–12 repeats) is an independent risk factor: increased copy number is protective, with each additional FLG repeat equating to a 12% risk reduction for AD (74). A recent paper reported that DNA methylation of the CpG site in the FLG gene region significantly increased AD risk (75). FLG is only one of approximately 45 genes within the epidermal differentiation complex (EDC) on chromosome 1q21, many of which may contribute to AD. For instance, levels of hornerin and other FLG-like proteins are decreased in AD skin (76, 77). However, FLG deficiency likely has the greatest impact in AD.
Table 1. Factors that influence filaggrin expression in the skin.
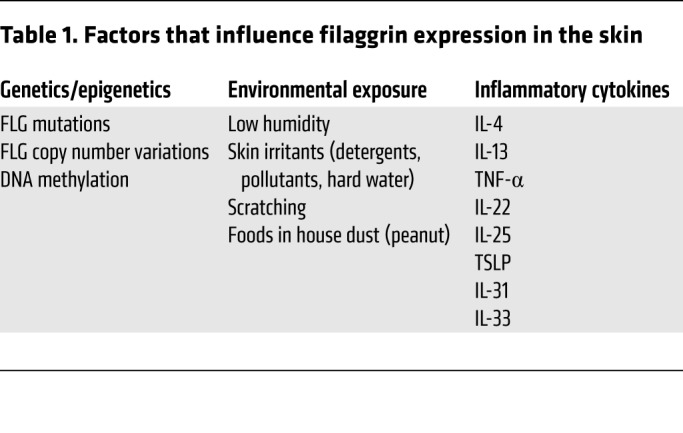
Environmental factors that adversely impact on skin barrier integrity enhance AD risk and severity. Mechanical damage, repetitive scratching, use of detergents, humidity, exposure to exogenous proteases, and air pollution also negatively impact FLG expression (78).
FLG-deficient mouse models exhibit enhanced percutaneous microbial and allergen penetration (79, 80). Similarly, examination of human skin samples determined that FLG mutations impair epidermal permeability barrier function (81). Knockdown of FLG expression in keratinocytes led to increased permeability in human keratinocyte organotypic cultures (82). Epithelial damage leads to innate immune activation, including release of proinflammatory cytokines and chemokines by keratinocytes (83), and activation of antigen presentation by skin-resident Langerhans cells and dermal DCs (84).
Electron microscopy studies illustrate that changes in epidermal FLG expression converge on the lamellar body secretory system to provoke skin barrier abnormality (81). Patients with FLG mutations demonstrate a retraction of cytosolic keratin filaments into a perinuclear shell around nuclei. This cytoskeletal abnormality appears to affect two cellular processes. First, it results in incomplete loading of cargo, including lipids, into nascent lamellar bodies, evidenced by empty microvesicles within these organelles. The resulting deposition of nonlamellar contents in the intercellular spaces then leads to focal defects in the extracellular lamellar bilayer system. Second, the cytoskeletal abnormality impairs secretion of lamellar bodies (85). FLG-deficient organotypic 3D skin cultures demonstrate disordered lipid lamellae (86). Another study reported decreased expression of FLG-related proteins (FLG2 and hornerin), lower expression of the FLG-degrading enzyme bleomycin hydrolase, and lower activity of another enzyme, caspase-14, in 3D cultures of keratinocytes with inhibited FLG expression (82). The mechanism for the feedback between FLG expression and expression/activity of FLG-processing enzymes has not been established.
Decrease in FLG expression is reflected in reduced production of FLG breakdown products, which are involved in skin acidification. This raises skin pH, thereby activating proteases (87). Activated serine skin kallikrein proteases (KLKs) interact with protease-activated type 2 receptor (PAR2), expressed by SG keratinocytes (88). PAR2 activation has been shown to inhibit lamellar body secretion in the skin (88–90) and induce the pro-Th2 cytokine TSLP (91).
Tight junctions in the SG form an additional component of the skin barrier, limiting allergen and microbe penetration, facilitating paracellular passage of soluble mediators, and regulating TEWL. Tight junctions are composed of transmembrane proteins including claudin-1 (CLDN1) that are essential for skin barrier function (92). CLDN1 levels are reduced in the skin of AD patients (93). In vitro studies have demonstrated that inhibiting CLDN1 expression enhances skin penetration (93). CLDN1 knockdown in keratinocytes enhances herpes simplex virus 1 (HSV-1) infectivity (94), particularly in patients with a history of eczema herpeticum (94).
S100A7, S100A8, and S100S9 proteins are upregulated in AD (95, 96), and their secretion amplifies skin inflammation. For instance, S100A9-activated keratinocytes selectively increase IL-33 production (95). Th2 cytokines inhibit S100A11 protein expression, which is required for the regulation of skin barrier integrity and innate immune responses (97).
Mutations in genes involved in acylceramide production (CERS3, ELOVL4, CYP4F22, UGCG, DEGS1, PNPLA1, DGAT2, ALOX12B, ABCA12) cause severe skin barrier defects in human patients and mouse models, and are often associated with ichthyosis symptoms (12, 56). Skin barrier lipids and proteins require coordinated assembly, and lipids may be critical to trigger profilaggrin-to-filaggrin processing (98).
Role of immune activation in keratinocyte dysfunction.
Decreased expression of EDC proteins (including FLG, LOR, and IVL), disruption of lipid lamellar body secretion, decreased antimicrobial peptide production (human β-defensins 2 and 3, and increased Staphylococcus aureus colonization can be attributable to the IL-4/IL-13 environment in AD skin (17, 64–67, 97, 99–103). Recently, clinical studies involving dupilumab, a monoclonal antibody that blocks signaling by IL-4 and IL-13, demonstrated significant clinical improvement in more than 70% of adults with moderate to severe AD (104, 105). These studies support the central role of Th2 cytokines in AD skin pathology.
IL-22, also overexpressed in AD skin, inhibits epidermal skin barrier function and suppress skin expression of FLG (106); however, in contrast to type 2 cytokines, it does not inhibit keratinocyte antimicrobial peptide production (107). Despite similar inhibitory effects on epidermal barrier function, IL-4/IL-13 and IL-22 utilize distinct receptors and transcriptional factors (IL-4Rα, IL-13Rα, common γ chain, and STAT6 versus IL-22R1, IL-10R2, and STAT3, respectively) (108, 109). Proinflammatory cytokines like TNF-α (110) and IL-33 (111) also inhibit FLG expression by keratinocytes.
Role of microbiome in keratinocyte dysfunction in AD skin.
Abnormalities in the skin microbiome are common in AD. IL-4 and IL-13 promote S. aureus invasion/colonization in AD skin (67, 112–115) by inhibiting epidermal barrier function (97, 116), increasing S. aureus skin binding sites (e.g., fibronectin), inhibiting TLR function, and decreasing antimicrobial peptide production (67, 100, 103). Levels of staphylococcal lipoteichoic acid (LTA) from lesional AD skin correlate with the Eczema Area and Severity Index (117). Superantigen-producing S. aureus colonization correlates with serum IL-4 (19), and superantigens are strong activators of IL-4, IL-13, and IL-22 production in AD (118).
Longitudinal studies indicate that S. aureus colonization emerges during the onset of AD (119, 120). Studies emphasize a bidirectional dialogue between skin bacteria and the host organism, with microbiota activating and educating host immunity and vice versa (119, 121).
Advances in next-generation sequencing provide a more comprehensive picture of skin microbiome (122). 16s RNA sequencing and metagenomic analysis of bacterial DNA collected from AD skin documented increased S. aureus colonization and decreased bacterial diversity (120, 123). Specific S. aureus strains have been associated with AD severity (123). S. aureus clones identified in severe AD patients were enriched for the expression of toxin genes and genes involved in antibiotic resistance. Murine skin colonization models demonstrated S. aureus strain–specific differences in eliciting skin inflammation and immune signatures characteristic of AD patients. Specifically, S. aureus isolates from AD patients with more severe flares induced epidermal thickening and expansion of cutaneous Th2 and Th17 cells, suggesting that functional differences between staphylococcal strains may contribute to AD disease complexity (123).
Functions of normal skin microbiota include enhancement of innate immunity (124–126), limiting of pathogen invasion (126, 127), and control of Treg function (128). Birth cohort studies indicate that the presence of Staphylococcus species other than S. aureus at 2 months of life might protect infants against later development of AD (129). In contrast, early-life skin colonization with S. aureus may actively contribute to clinical AD onset in infancy (130). Several virulence factors, e.g., lytic toxins, enterotoxins, and proteases, produced by S. aureus contribute to AD pathogenesis or exacerbation through mechanisms acting on keratinocytes (cell lysis, proinflammatory cytokines production, inhibition of keratinocyte differentiation program) and immune cells (T cell activation, production of proinflammatory cytokines) (116, 131–134). S. aureus induces detrimental effects on keratinocytes, exploiting AD epidermal barrier defects to trigger cytokine expression (135). Activation of serine proteases is essential for S. aureus penetration into the skin (136).
IL-4/IL-13 overproduction provides a permissive environment for S. aureus growth (112, 137). Acidification of the skin by FLG breakdown products was shown to reduce expression of S. aureus virulence factors (138). In addition, FLG breakdown products appear to inhibit S. aureus expression of iron-regulated surface determinant A (138). Reduced levels of FLG breakdown products in AD skin may support S. aureus colonization. Th2 cytokines can also enhance the effects of staphylococcal products. For instance, compared with normal keratinocytes, AD keratinocytes have increased sensitivity to α-toxin, a cytolytic toxin produced by S. aureus. Differentiated keratinocytes are protected from cell death, whereas IL-4/IL-13 treatment increases sensitivity to α-toxin–induced lethality (113, 116). The combination of IL-4/IL-13 induces biochemical changes that decrease levels of acid SMase, an enzyme that cleaves the α-toxin ligand sphingomyelin (113). SMase and its enzymatic product, phosphocholine, prevent IL-4/IL-13–mediated increases in α-toxin–induced cell death (113).
S.
aureus infection may predispose the host toward disseminated viral infections. We have demonstrated that sublytic staphylococcal α-toxin increases viral loads and promotes entry of HSV-1 and vaccinia virus (VV) into keratinocytes. VV load was significantly greater in murine skin inoculated with an α-toxin–producing S. aureus strain compared with murine skin inoculated with the isogenic α-toxin–deleted strain. α-Toxin’s viral-enhancing effect, mediated by ADAM10, was associated with its pore-forming property (133).
We recently demonstrated an interplay between the S. aureus cell wall component LTA and IL-4/IL-13 in inhibiting wound healing in AD skin (139). We found that keratinocytes are highly responsive to LTA, documenting changes in expression of genes involved in regulating epidermal development, wound responses, keratinocyte proliferation, cell differentiation, and Notch signaling pathways (134). Staphylococcal LTA inhibits expression of early keratinocyte differentiation markers, including KRT1/10, and desmoglein 1 (DSG1), which are essential for skin barrier function. LTA-mediated inhibition was found to be p63 dependent (134).
Lipid abnormalities predominate in AD skin.
Brown and colleagues stratified the analysis of AD skin transcriptome based on FLG gene mutations and found that patients with normal FLG genotype have significant changes in the expression of enzymes involved in lipid metabolism and synthesis (140). This emphasizes the importance of lipid metabolism in AD independent of FLG genotype. Extracellular lipids account for up to 10% of SC mass (141–143). Several skin lipids have antiinflammatory and antimicrobial properties (85, 144). Free fatty acids and sphingoid bases have documented antimicrobial activity (145–148). Several free fatty acids serve as natural agonists for PPAR transcriptional factors (149), which are essential in regulating lipid metabolism enzymes in the skin (150).
Two independent research groups demonstrated reduced ceramide levels in AD skin in parallel with a decline in free fatty acid chain length (151–153). These changes in skin lipid composition resulted in aberrant lipid organization and positively correlated with the degree of TEWL in AD skin (85, 142). Notably, changes in ceramide levels and free fatty acid chain length distribution did not correlate with FLG genotype, but correlated with AD severity and levels of FLG breakdown products (151, 153). This emphasizes the importance of skin inflammation in suppressing FLG and skin barrier lipids.
Concurrent reduction in ceramide and free fatty acid chain length in the SC of AD suggests alterations in a common synthetic pathway for ceramides and free fatty acids (143, 151, 153). We hypothesized that type 2 immune response alters AD skin lipid metabolism (154). Mass spectrometric analysis of lesional SC from AD subjects and IL-13–transgenic mice revealed an increased proportion of short-chain (N-14:0-24:0) NS-ceramides (non-hydroxy fatty acid sphingosine ceramides), sphingomyelins, and 14:0-22:0-lysophosphatidylcholines (14:0-22:0-LPCs) with simultaneous decline in the proportion of corresponding long-chain species (N-26:0-32:0 sphingolipids and 24:0-30:0-LPCs) in comparison with healthy controls. An increase in short-chain LPC species was also observed in nonlesional AD skin. Similar changes were observed in IL-4/IL-13–treated Ca2+-differentiated human keratinocytes in vitro, and all increases were blocked by siRNA-mediated silencing of STAT6, a master regulator of IL-4/IL-13 signaling. RNA sequencing analysis of AD versus healthy SC identified decreased expression of the fatty acid elongases ELOVL3 and ELOVL6, which contributed to observed changes in atopic skin lipids. Thus, our data strongly support the pathogenic role of type 2 immune activation in AD skin lipid metabolism (154). Recently, Danso et al. confirmed that alterations in the expression of key enzymes involved in SC lipid synthesis contribute to changes in the lipid composition in AD skin (155).
Epithelial barrier dysfunction in other allergic diseases
There is compelling evidence for epithelial barrier dysfunction in other allergic diseases (10, 156). Polymorphisms in the IL1RL1/IL18R1 locus and the IL33 and TSLP genes linking epithelium-derived cytokines to type 2 inflammation in asthma have been documented (157). In addition, several polymorphisms in genes associated with epithelial homeostasis and differentiation have been identified (157–160). Increased number of goblet cells with increased mucin gene expression, increased MUC5AC protein relative to MUC5B (161–163), and reductions in ciliated cell numbers were shown in asthmatics, along with increased expression of EGFR (164) and type 2 cytokines, including IL-13, IL-5, and IL-9 (165, 166). Studies report disruption of airway epithelium in asthmatics, including loss of tight junction and reduction in adherens junction proteins. Functional studies indicate increased sensitivity of asthmatic airway epithelia to environmental stressors and oxidative stress, reducing the threshold for epithelial damage (167–169). Increased barrier permeability in asthma has been shown to promote allergic sensitization, reduce the threshold for epithelial damage, and activate type 2 responses. Changes in microbial diversity of asthmatic airways have been reported (170–173). Finally, impaired epithelial barrier repair in asthmatics leads to failure to resolve inflammatory responses (9, 10, 174).
Eosinophilic esophagitis (EoE) is considered a type 2 immune disease and often coexists with AD, asthma, and FA (13). The mucosa of the human esophagus is lined by the multilayer squamous nonkeratinized epithelium, which provides a protective barrier against environmental insults. Epithelial responses drive the majority of transcriptional changes in EoE (13, 175–177). The EoE transcriptome revealed a significant downregulation of multiple structural genes, including FLG, IVL, and the small proline-rich (SPRR) gene family (178, 179). IL-13 stimulation of esophageal epithelial cells induced disease-associated pathways, which overlapped with those observed in the esophageal mucosa of EoE patients (175, 180). Impaired barrier function is a hallmark of allergic inflammation in EoE, with inhibition of tight junction proteins (claudin-1, claudin-7, occludin), adherens junction proteins (E-cadherin), and desmosomal proteins (DSG1) compromising esophageal epithelium integrity. GWAS documented that most genes associated with increased susceptibility to EoE are expressed in the esophageal epithelium, substantiating the role of epithelial responses in EoE pathogenesis (13, 181–183). EoE-specific dysregulation is highly enriched in genes associated with protease-related activities: serine peptidase inhibitors from the SERPIN family; serine protease inhibitors, Kazal-type (SPINKs); and the protease calpain-14 (13). These proteases and their inhibitors not only regulate epithelial barrier function but are also implicated directly in inducing type 2 immune responses (13).
Altered epithelial barrier function has also been documented in allergic rhinitis, including alterations in epithelial physical barrier, mucus production, antimicrobial defense, microbiome, and immune response (184).
Epithelial-mesenchymal transition is a process during which epithelia lose many epithelial characteristics, including tight junctions, while acquiring properties of mesenchymal cells, including motility, loose cell adhesion, and depolarized cytoskeletal arrangements. Epithelial-mesenchymal transition facilitates the development of tissue fibrosis in response to injury and chronic inflammation (185). Although not described in AD, it plays an important role in asthma (186, 187) and EoE (188).
AD is often associated with sensitization to multiple foods, including peanuts, egg, milk, and tree nuts (15). A recent meta-analysis of 66 studies concluded that peanut allergy is often preceded by AD (23). Cutaneous food allergen exposure may promote sensitization (189, 190). Inflammatory cytokines released by the skin epithelium, including IL-25, IL-33, and TSLP, act on dendritic cells and other innate immune cells, leading to type 2 immune allergic response rather than tolerogenic responses (191–196). Animal studies demonstrated an essential role for TSLP in the epicutaneous induction of FA with AD-like skin lesions. Increased TSLP in the epidermis elicits the accumulation of basophils into the skin that promote type 2 cytokine responses (192). In addition, TSLP signaling on epidermal Langerhans cells may be important for IgE production during the epicutaneous sensitization to food allergens (197). In a murine model, sensitization to food allergens through an AD-like skin lesion was associated with expansion of TSLP-elicited basophils in the skin, supporting antigen-specific Th2 cytokine responses, increased antigen-specific IgE levels, and mast cell accumulation in the intestine. Notably, disruption of TSLP responses or basophil depletion reduced susceptibility to intestinal FA in this model system, whereas transferring TSLP-elicited basophils into intact skin promoted disease (193).
Importantly, several studies support the concept that children become allergic to peanuts through environmental exposure to peanut protein in household dust, especially if the infant has a disrupted skin barrier (e.g., FLG loss-of-function mutations or a history of eczema) (198, 199). Peanut protein found in the dust of the infant’s home environment is biologically active and stimulates dose-dependent activation of basophils from peanut-allergic children (200).
Interestingly, a recent study (21) revealed that increased TEWL in the newborn period, even without concurrent development of AD, predicts FA occurrence at 2 years of age. Using a novel skin tape stripping (STS) technique, we found that epidermal TSLP expression occurs at 2 months of age prior to the onset of AD at 24 months of age (201). The combination of family history and increased epidermal TSLP gave an odds ratio higher than 20 for AD development.
Strategies to improve the epithelial barrier and prevent allergy
The approaches for improving the epithelial barrier are summarized in Table 2 and detailed below.
Table 2. Strategies to repair epithelial barrier function.

Prevention.
Several birth cohort studies report that the use of emollients to improve the skin barrier can prevent eczema occurrence (202, 203). In these studies, skin emollients reduced AD occurrence by approximately 50%. Transcriptomic studies found that nonlesional, normal-appearing skin of AD patients is distinct from the skin of healthy control subjects, with evidence of type 2 inflammatory responses and reduced skin barrier function in nonlesional AD (64, 140, 154, 204). It would be interesting to determine whether addition of intermittent proactive topical antiinflammatory therapies, including low-potency topical corticosteroids (205), which are known to reduce eczema relapse down to 3 months of age, further reduces AD and FA occurrence. Application of emollients in the PEBBLES study showed a trend for decreased food sensitization at 6 and 12 months of age, but the study was not powered to measure clinical FA outcomes. Prevention of food sensitization in the PEBBLES study was more effective with early introduction (<2 weeks after birth) and frequent use (≥5 days per week) of the lipid-rich emollient (206).
Proactive therapy.
The concomitant allergic inflammation accompanying AD can also reduce skin barrier function, and thus drive IgE responses to skin-penetrating foods. Therefore, FA prevention may require proactive skin barrier and antiinflammatory therapy to reduce type 2 immune responses to epicutaneous allergen sensitization. Alternatively, skin barrier dysfunction is well established in patients with severe eczema and type 2 immune activation. Antagonism of type 2 cytokines such as IL-4/IL-13, TSLP, or IL-33 may enhance epithelial function and reduce allergen sensitization in patients with established AD (NCT03389893 by the Atopic Dermatitis Research Network, ClinicalTrials.gov; and refs. 104, 207, 208).
Environmental control.
Since low humidity, skin irritants (detergents), pollutants, hard water, and environmental allergens are known to induce eczema, avoidance of these environmental factors may prevent epithelial barrier dysfunction.
Microbiome.
S. aureus colonization breaks down the skin barrier and predates eczema development (130, 135). Studies suggest that S. aureus colonization is increased on AD skin as a result of loss of commensal bacteria (127). Studies in the AD Research Network are examining whether targeted transplantation of S. hominis can reduce S. aureus colonization in AD (NCT03151148). An additional clinical study is evaluating whether topical application of Gram-negative coccobacillus Roseomonas mucosa can be used to treat AD (NCT03151148).
Alternative strategies.
Vitamin D regulates FLG, LOR, IVL, and lipid lamellae formation. Maintaining normal vitamin D levels is therefore essential for development of the skin barrier (209). Moisturizers, occlusive agents, and humectants are used in AD to soften skin, create a physical barrier, and retain water (210). Experimental FLG replacement therapies currently under investigation include read-through drugs that lead to skipping of nonsense FLG mutations, drugs that regulate FLG production, FLG monomers, and FLG metabolites (12).
Noninvasive approaches for assessing skin function
TEWL measurement is widely used to assess skin barrier function (211). During this procedure, sensors are placed in contact with the skin surface to measure water evaporating from the skin. In addition to TEWL measurements at the skin surface, TEWL measurements can be combined with STS to measure skin barrier integrity. With STS, the uppermost layers of the skin are peeled away using adhesive discs (154, 204). Skin with compromised skin barrier exhibits greater changes in TEWL. The area under the curve for TEWL measurements over a defined number of STS reflects the overall integrity of the SC.
Our research group has pioneered novel methods to profile skin through minimally invasive and scarless STS analysis combined with transcriptomics, lipidomics, and proteomics (102, 201, 212). Using an STS protein mass spectrometry analysis, AD skin exhibits significantly lower expression of skin barrier proteins (FLG2, corneodesmosin, DSG1, DSC1, and TGM3) and enzymes (arginase-1, caspase-14, and γ-glutamyl cyclotransferase) involved in generating NMF (212, 213).
We have now extended this STS noninvasive technology to whole transcriptome sequencing together with lipidomics and proteomics. This has proven a powerful technique for agnostic examination of genomic, lipidomic, and proteomic expression profiles in nonlesional and lesional AD skin. Using these techniques, we are starting to develop new insights about AD endotypes, which include epidermal type 2 inflammation, AD patients with FA, eczema herpeticum, genetically determined deficiencies in skin cornification, and skin lipid barrier dysfunction in AD (154, 204).
Conclusions
Clinical manifestations and skin pathology in AD are driven by impaired skin barrier and type 2–skewed immune responses.
(a) Impaired skin barrier function is caused by changes in the expression of key structural cornified barrier proteins and skin barrier lipids. FLG mutations are the most profound single-gene defects involved in AD (17). FLG deficiency promotes inflammation and inflammatory cell infiltration in the skin.
(b) Changes in FLG expression alter skin acidification, which, in turn, supports activation of skin proteases that alter skin barrier homeostasis by interfering with lipid lamellae assembly and support the onset of type 2 inflammatory response through TSLP and IL-33 activation (85).
(c) Type 2–skewed immune responses in AD favor epidermal barrier disruption by inhibiting the expression of FLG and other structural protein in skin (100). Th2 cytokines also inhibit production of skin barrier lipids in the skin (154). These changes are already present in nonlesional, normal-appearing AD skin (64, 154, 204), and are further aggravated in AD lesional skin.
(d) Other factors contributing to AD include S. aureus overgrowth and skin microbiota dysbiosis, IgE-mediated sensitization, and chronic itch. AD pathobiology is a complex interaction of epidermal barrier disruption, type 2 immune response, and imbalanced skin microbiota. Improved understanding of underlying pathology in AD will allow development of target therapies and optimize inhibition of inflammation in AD and other allergic diseases.
(e) Importantly, in a large birth cohort study, neonatal skin barrier dysfunction at 2 days of life predates the development of AD and FA later in childhood (21). This supports the notion that compromised skin barrier is critical for allergic sensitization.
(f) The interplay between genetic predisposition, microbial colonization, and type 2 inflammatory responses is important for the development of epidermal barrier abnormalities and onset of allergic responses. The key challenge now is identifying interventions to protect skin barrier function in early infancy and prevent onset of type 2 inflammatory responses and development of allergy.
(g) There is compelling evidence for epithelial dysregulation in other allergic diseases, including asthma, FA, EoE, and allergic rhinosinusitis. Through translational approaches that restore epithelial barrier homeostasis, it may be possible to prevent or modify the course of multiple allergic disorders and intervene at timeframes close to the origin of allergic diseases.
Acknowledgments
This project was funded in part by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01 AR41256, NIH/National Institute of Allergy and Infectious Diseases grant U19 AI117673, NIH/National Center for Research Resources grant UL1 RR025780, and the Edelstein Family Chair of Pediatric Allergy-Immunology at National Jewish Health.
Version 1. 02/18/2019
Electronic publication
Version 2. 04/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019 American Society for Clinical Investigation
Reference information: J Clin Invest. 2019;129(4):1463–1474.https://doi.org/10.1172/JCI124608.
Contributor Information
Elena Goleva, Email: golevae@njhealth.org.
Evgeny Berdyshev, Email: berdysheve@njhealth.org.
Donald Y.M. Leung, Email: leungd@njhealth.org.
References
- 1.Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers. 2015;1:15025. doi: 10.1038/nrdp.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renz H, et al. Food allergy. Nat Rev Dis Primers. 2018;4:17098. doi: 10.1038/nrdp.2017.98. [DOI] [PubMed] [Google Scholar]
- 3.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. doi: 10.1038/s41572-018-0001-z. [DOI] [PubMed] [Google Scholar]
- 4.Brunner PM, Guttman-Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol. 2017;139(4S):S65–S76. doi: 10.1016/j.jaci.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu W, Freeland DMH, Nadeau KC. Food allergy: immune mechanisms, diagnosis and immunotherapy. Nat Rev Immunol. 2016;16(12):751–765. doi: 10.1038/nri.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11(4):289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10(2):103–110. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 8.Licona-Limón P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. 2013;14(6):536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 9.Grainge CL, Davies DE. Epithelial injury and repair in airways diseases. Chest. 2013;144(6):1906–1912. doi: 10.1378/chest.12-1944. [DOI] [PubMed] [Google Scholar]
- 10.Loxham M, Davies DE. Phenotypic and genetic aspects of epithelial barrier function in asthmatic patients. J Allergy Clin Immunol. 2017;139(6):1736–1751. doi: 10.1016/j.jaci.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farhadi A, Banan A, Fields J, Keshavarzian A. Intestinal barrier: an interface between health and disease. J Gastroenterol Hepatol. 2003;18(5):479–497. doi: 10.1046/j.1440-1746.2003.03032.x. [DOI] [PubMed] [Google Scholar]
- 12.Egawa G, Kabashima K. Barrier dysfunction in the skin allergy. Allergol Int. 2018;67(1):3–11. doi: 10.1016/j.alit.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Rochman M, Azouz NP, Rothenberg ME. Epithelial origin of eosinophilic esophagitis. J Allergy Clin Immunol. 2018;142(1):10–23. doi: 10.1016/j.jaci.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson HA, O’Mahony L, Burks AW, Plaut M, Lack G, Akdis CA. Mechanisms of food allergy. J Allergy Clin Immunol. 2018;141(1):11–19. doi: 10.1016/j.jaci.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Leung DY, Guttman-Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol. 2014;134(4):769–779. doi: 10.1016/j.jaci.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest. 2004;113(5):651–657. doi: 10.1172/JCI21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315–1327. doi: 10.1056/NEJMra1011040. [DOI] [PubMed] [Google Scholar]
- 18.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 19.Czarnowicki T, Krueger JG, Guttman-Yassky E. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol Pract. 2014;2(4):371–379. doi: 10.1016/j.jaip.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Roduit C, et al. Phenotypes of atopic dermatitis depending on the timing of onset and progression in childhood. JAMA Pediatr. 2017;171(7):655–662. doi: 10.1001/jamapediatrics.2017.0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelleher MM, et al. Skin barrier impairment at birth predicts food allergy at 2 years of age. J Allergy Clin Immunol. 2016;137(4):1111–1116.e8. doi: 10.1016/j.jaci.2015.12.1312. [DOI] [PubMed] [Google Scholar]
- 22.Hill DA, Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. 2018;120(2):131–137. doi: 10.1016/j.anai.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsakok T, et al. Does atopic dermatitis cause food allergy? A systematic review. J Allergy Clin Immunol. 2016;137(4):1071–1078. doi: 10.1016/j.jaci.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 24.Tran MM, et al. Predicting the atopic march: results from the Canadian Healthy Infant Longitudinal Development Study. J Allergy Clin Immunol. 2018;141(2):601–607.e8. doi: 10.1016/j.jaci.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs E. Epidermal differentiation: the bare essentials. J Cell Biol. 1990;111(6 pt 2):2807–2814. doi: 10.1083/jcb.111.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6(4):328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 27.Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab. 2012;7(4):461–472. doi: 10.1586/eem.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Celli A, Sanchez S, Behne M, Hazlett T, Gratton E, Mauro T. The epidermal Ca(2+) gradient: measurement using the phasor representation of fluorescent lifetime imaging. Biophys J. 2010;98(5):911–921. doi: 10.1016/j.bpj.2009.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckert RL, Crish JF, Robinson NA. The epidermal keratinocyte as a model for the study of gene regulation and cell differentiation. Physiol Rev. 1997;77(2):397–424. doi: 10.1152/physrev.1997.77.2.397. [DOI] [PubMed] [Google Scholar]
- 30.Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109(3):1207–1217. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang A, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 32.Koster MI, et al. p63 induces key target genes required for epidermal morphogenesis. Proc Natl Acad Sci U S A. 2007;104(9):3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su X, Cho MS, Gi YJ, Ayanga BA, Sherr CJ, Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28(13):1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20(22):3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanpain C, Lowry WE, Pasolli HA, Fuchs E. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006;20(21):3022–3035. doi: 10.1101/gad.1477606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collu GM, Hidalgo-Sastre A, Brennan K. Wnt-Notch signalling crosstalk in development and disease. Cell Mol Life Sci. 2014;71(18):3553–3567. doi: 10.1007/s00018-014-1644-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefort K, Dotto GP. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol. 2004;14(5):374–386. doi: 10.1016/j.semcancer.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 38.Moriyama M, et al. Multiple roles of Notch signaling in the regulation of epidermal development. Dev Cell. 2008;14(4):594–604. doi: 10.1016/j.devcel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Watt FM, Estrach S, Ambler CA. Epidermal Notch signalling: differentiation, cancer and adhesion. Curr Opin Cell Biol. 2008;20(2):171–179. doi: 10.1016/j.ceb.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace L, Roberts-Thompson L, Reichelt J. Deletion of K1/K10 does not impair epidermal stratification but affects desmosomal structure and nuclear integrity. J Cell Sci. 2012;125(pt 7):1750–1758. doi: 10.1242/jcs.097139. [DOI] [PubMed] [Google Scholar]
- 41.Jensen JM, Schütze S, Neumann C, Proksch E. Impaired cutaneous permeability barrier function, skin hydration, and sphingomyelinase activity in keratin 10 deficient mice. J Invest Dermatol. 2000;115(4):708–713. doi: 10.1046/j.1523-1747.2000.00103.x. [DOI] [PubMed] [Google Scholar]
- 42.Reichelt J, Bauer C, Porter R, Lane E, Magin V. Out of balance: consequences of a partial keratin 10 knockout. J Cell Sci. 1997;110(pt 18):2175–2186. doi: 10.1242/jcs.110.18.2175. [DOI] [PubMed] [Google Scholar]
- 43.Roth W, et al. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J Cell Sci. 2012;125(pt 22):5269–5279. doi: 10.1242/jcs.116574. [DOI] [PubMed] [Google Scholar]
- 44.Dumortier A, et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS One. 2010;5(2):e9258. doi: 10.1371/journal.pone.0009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murthy A, Shao YW, Narala SR, Molyneux SD, Zúñiga-Pflücker JC, Khokha R. Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity. 2012;36(1):105–119. doi: 10.1016/j.immuni.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Rangarajan A, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20(13):3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33(3):416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 48.Panelos J, Massi D. Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol Ther. 2009;8(21):1986–1993. doi: 10.4161/cbt.8.21.9921. [DOI] [PubMed] [Google Scholar]
- 49.Rawlings AV, Harding CR. Moisturization and skin barrier function. Dermatol Ther. 2004;17(suppl 1):43–48. doi: 10.1111/j.1396-0296.2004.04s1005.x. [DOI] [PubMed] [Google Scholar]
- 50.Gibbs NK, Tye J, Norval M. Recent advances in urocanic acid photochemistry, photobiology and photoimmunology. Photochem Photobiol Sci. 2008;7(6):655–667. doi: 10.1039/b717398a. [DOI] [PubMed] [Google Scholar]
- 51.McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol. 2013;131(2):280–291. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- 52.Kezic S, et al. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy. 2011;66(7):934–940. doi: 10.1111/j.1398-9995.2010.02540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(pt 9):1285–1294. doi: 10.1242/jcs.033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walterscheid JP, et al. Cis-urocanic acid, a sunlight-induced immunosuppressive factor, activates immune suppression via the 5-HT2A receptor. Proc Natl Acad Sci U S A. 2006;103(46):17420–17425. doi: 10.1073/pnas.0603119103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsui T, Amagai M. Dissecting the formation, structure and barrier function of the stratum corneum. Int Immunol. 2015;27(6):269–280. doi: 10.1093/intimm/dxv013. [DOI] [PubMed] [Google Scholar]
- 56.Kihara A. Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res. 2016;63:50–69. doi: 10.1016/j.plipres.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 57.Breiden B, Sandhoff K. The role of sphingolipid metabolism in cutaneous permeability barrier formation. Biochim Biophys Acta. 2014;1841(3):441–452. doi: 10.1016/j.bbalip.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Hirabayashi T, et al. PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nat Commun. 2017;8:14609. doi: 10.1038/ncomms14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohno Y, Kamiyama N, Nakamichi S, Kihara A. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω-O-acylceramide. Nat Commun. 2017;8:14610. doi: 10.1038/ncomms14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohno Y, et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci U S A. 2015;112(25):7707–7712. doi: 10.1073/pnas.1503491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guttman-Yassky E, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009;124(6):1235–1244.e58. doi: 10.1016/j.jaci.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 62.Jensen JM, et al. Impaired sphingomyelinase activity and epidermal differentiation in atopic dermatitis. J Invest Dermatol. 2004;122(6):1423–1431. doi: 10.1111/j.0022-202X.2004.22621.x. [DOI] [PubMed] [Google Scholar]
- 63.Totsuka A, Omori-Miyake M, Kawashima M, Yagi J, Tsunemi Y. Expression of keratin 1, keratin 10, desmoglein 1 and desmocollin 1 in the epidermis: possible downregulation by interleukin-4 and interleukin-13 in atopic dermatitis. Eur J Dermatol. 2017;27(3):247–253. doi: 10.1684/ejd.2017.2985. [DOI] [PubMed] [Google Scholar]
- 64.Suárez-Fariñas M, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127(4):954–964.e1. doi: 10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howell MD, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120(1):150–155. doi: 10.1016/j.jaci.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126(3):332–337. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ong PY, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 68.Meng L, et al. Filaggrin gene mutation c.3321delA is associated with various clinical features of atopic dermatitis in the Chinese Han population. PLoS One. 2014;9(5):e98235. doi: 10.1371/journal.pone.0098235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu HS, et al. Mutations in the filaggrin are predisposing factor in Korean children with atopic dermatitis. Allergy Asthma Immunol Res. 2013;5(4):211–215. doi: 10.4168/aair.2013.5.4.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cai SC, et al. Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Br J Dermatol. 2012;166(1):200–203. doi: 10.1111/j.1365-2133.2011.10541.x. [DOI] [PubMed] [Google Scholar]
- 71.Wang IJ, Lin TJ, Kuo CF, Lin SL, Lee YL, Chen PC. Filaggrin polymorphism P478S, IgE level, and atopic phenotypes. Br J Dermatol. 2011;164(4):791–796. doi: 10.1111/j.1365-2133.2011.10212.x. [DOI] [PubMed] [Google Scholar]
- 72.Margolis DJ, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol. 2014;133(3):784–789. doi: 10.1016/j.jaci.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Margolis DJ, et al. Uncommon filaggrin variants are associated with persistent atopic dermatitis in African Americans. J Invest Dermatol. 2018;138(7):1501–1506. doi: 10.1016/j.jid.2018.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown SJ, et al. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Invest Dermatol. 2012;132(1):98–104. doi: 10.1038/jid.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ziyab AH, Karmaus W, Holloway JW, Zhang H, Ewart S, Arshad SH. DNA methylation of the filaggrin gene adds to the risk of eczema associated with loss-of-function variants. J Eur Acad Dermatol Venereol. 2013;27(3):e420–e423. doi: 10.1111/jdv.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pellerin L, et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J Allergy Clin Immunol. 2013;131(4):1094–1102. doi: 10.1016/j.jaci.2012.12.1566. [DOI] [PubMed] [Google Scholar]
- 77.Pendaries V, et al. In a three-dimensional reconstructed human epidermis filaggrin-2 is essential for proper cornification. Cell Death Dis. 2015;6:e1656. doi: 10.1038/cddis.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thyssen JP, Kezic S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol. 2014;134(4):792–799. doi: 10.1016/j.jaci.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 79.Scharschmidt TC, et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J Allergy Clin Immunol. 2009;124(3):496–506, 506.e1. doi: 10.1016/j.jaci.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fallon PG, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41(5):602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gruber R, et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol. 2011;178(5):2252–2263. doi: 10.1016/j.ajpath.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pendaries V, et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J Invest Dermatol. 2014;134(12):2938–2946. doi: 10.1038/jid.2014.259. [DOI] [PubMed] [Google Scholar]
- 83.Kezic S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129(4):1031–1039.e1. doi: 10.1016/j.jaci.2011.12.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshida K, et al. Distinct behavior of human Langerhans cells and inflammatory dendritic epidermal cells at tight junctions in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134(4):856–864. doi: 10.1016/j.jaci.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 85.Elias PM, Wakefield JS. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134(4):781–791.e1. doi: 10.1016/j.jaci.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vávrová K, et al. Filaggrin deficiency leads to impaired lipid profile and altered acidification pathways in a 3D skin construct. J Invest Dermatol. 2014;134(3):746–753. doi: 10.1038/jid.2013.402. [DOI] [PubMed] [Google Scholar]
- 87.Elias PM. Lipid abnormalities and lipid-based repair strategies in atopic dermatitis. Biochim Biophys Acta. 2014;1841(3):323–330. doi: 10.1016/j.bbalip.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hachem JP, et al. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol. 2006;126(7):1609–1621. doi: 10.1038/sj.jid.5700288. [DOI] [PubMed] [Google Scholar]
- 89.Hachem JP, et al. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol. 2006;126(9):2074–2086. doi: 10.1038/sj.jid.5700351. [DOI] [PubMed] [Google Scholar]
- 90.Hachem JP, et al. Sustained serine proteases activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol. 2005;125(3):510–520. doi: 10.1111/j.0022-202X.2005.23838.x. [DOI] [PubMed] [Google Scholar]
- 91.Briot A, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Furuse M, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156(6):1099–1111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Benedetto A, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3):773–786.e1. doi: 10.1016/j.jaci.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Benedetto A, et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol. 2011;128(1):242–246.e5. doi: 10.1016/j.jaci.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin S, et al. DAMP molecules S100A9 and S100A8 activated by IL-17A and house-dust mites are increased in atopic dermatitis. Exp Dermatol. 2014;23(12):938–941. doi: 10.1111/exd.12563. [DOI] [PubMed] [Google Scholar]
- 96.Gittler JK, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130(6):1344–1354. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Howell MD, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol. 2008;128(9):2248–2258. doi: 10.1038/jid.2008.74. [DOI] [PubMed] [Google Scholar]
- 98.Epp N, et al. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177(1):173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suárez-Fariñas M, et al. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132(2):361–370. doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Howell MD, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24(3):341–348. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 101.Sehra S, et al. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol. 2010;184(6):3186–3190. doi: 10.4049/jimmunol.0901860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Broccardo CJ, Mahaffey SB, Strand M, Reisdorph NA, Leung DY. Peeling off the layers: skin taping and a novel proteomics approach to study atopic dermatitis. J Allergy Clin Immunol. 2009;124(5):1113–1115.e1. doi: 10.1016/j.jaci.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nomura I, et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. 2003;171(6):3262–3269. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 104.Beck LA, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–139. doi: 10.1056/NEJMoa1314768. [DOI] [PubMed] [Google Scholar]
- 105.Hamilton JD, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol. 2014;134(6):1293–1300. doi: 10.1016/j.jaci.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 106.Wolk K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36(5):1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 107.Nograles KE, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159(5):1092–1102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13(1):21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 109.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15(5):271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 110.Kim BE, et al. TNF-α downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-α antagonists to improve skin barrier. J Invest Dermatol. 2011;131(6):1272–1279. doi: 10.1038/jid.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ryu WI, Lee H, Bae HC, Ryu HJ, Son SW. IL-33 down-regulates filaggrin expression by inducing STAT3 and ERK phosphorylation in human keratinocytes. J Dermatol Sci. 2016;82(2):131–134. doi: 10.1016/j.jdermsci.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 112.Cho SH, Strickland I, Boguniewicz M, Leung DY. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol. 2001;108(2):269–274. doi: 10.1067/mai.2001.117455. [DOI] [PubMed] [Google Scholar]
- 113.Brauweiler AM, Goleva E, Leung DYM. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6) J Invest Dermatol. 2014;134(8):2114–2121. doi: 10.1038/jid.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kisich KO, Carspecken CW, Fiéve S, Boguniewicz M, Leung DY. Defective killing of Staphylococcus aureus in atopic dermatitis is associated with reduced mobilization of human beta-defensin-3. J Allergy Clin Immunol. 2008;122(1):62–68. doi: 10.1016/j.jaci.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 115.Kisich KO, Howell MD, Boguniewicz M, Heizer HR, Watson NU, Leung DY. The constitutive capacity of human keratinocytes to kill Staphylococcus aureus is dependent on beta-defensin 3. J Invest Dermatol. 2007;127(10):2368–2380. doi: 10.1038/sj.jid.5700861. [DOI] [PubMed] [Google Scholar]
- 116.Brauweiler AM, et al. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin-induced keratinocyte death. J Allergy Clin Immunol. 2013;131(2):421–427.e1. doi: 10.1016/j.jaci.2012.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Travers JB, et al. Infected atopic dermatitis lesions contain pharmacologic amounts of lipoteichoic acid. J Allergy Clin Immunol. 2010;125(1):146–152.e1. doi: 10.1016/j.jaci.2009.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Niebuhr M, Scharonow H, Gathmann M, Mamerow D, Werfel T. Staphylococcal exotoxins are strong inducers of IL-22: a potential role in atopic dermatitis. J Allergy Clin Immunol. 2010;126(6):1176–1183.e4. doi: 10.1016/j.jaci.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 119.Kobayashi T, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42(4):756–766. doi: 10.1016/j.immuni.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kong HH, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oh J, et al. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013;23(12):2103–2114. doi: 10.1101/gr.159467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16(3):143–155. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 123.Byrd AL, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med. 2017;9(397):eaal4651. doi: 10.1126/scitranslmed.aal4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lai Y, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15(12):1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Linehan JL, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell. 2018;172(4):784–796.e18. doi: 10.1016/j.cell.2017.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Naik S, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520(7545):104–108. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakatsuji T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 2017;9(378):eaah4680. doi: 10.1126/scitranslmed.aah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scharschmidt TC, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. 2015;43(5):1011–1021. doi: 10.1016/j.immuni.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kennedy EA, et al. Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol. 2017;139(1):166–172. doi: 10.1016/j.jaci.2016.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meylan P, et al. Skin colonization by Staphylococcus aureus precedes the clinical diagnosis of atopic dermatitis in infancy. J Invest Dermatol. 2017;137(12):2497–2504. doi: 10.1016/j.jid.2017.07.834. [DOI] [PubMed] [Google Scholar]
- 131.Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26(3):422–447. doi: 10.1128/CMR.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Foster TJ, Geoghegan JA, Ganesh VK, Höök M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol. 2014;12(1):49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bin L, et al. Staphylococcus aureus α-toxin modulates skin host response to viral infection. J Allergy Clin Immunol. 2012;130(3):683–691.e2. doi: 10.1016/j.jaci.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brauweiler AM, Hall CF, Goleva E, Leung DYM. Staphylococcus aureus lipoteichoic acid inhibits keratinocyte differentiation through a p63-mediated pathway. J Invest Dermatol. 2017;137(9):2030–2033. doi: 10.1016/j.jid.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nakatsuji T, et al. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol. 2016;136(11):2192–2200. doi: 10.1016/j.jid.2016.05.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus induces increased serine protease activity in keratinocytes. J Invest Dermatol. 2017;137(2):377–384. doi: 10.1016/j.jid.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol. 2001;116(5):658–663. doi: 10.1046/j.0022-202x.2001.01331.x. [DOI] [PubMed] [Google Scholar]
- 138.Miajlovic H, Fallon PG, Irvine AD, Foster TJ. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J Allergy Clin Immunol. 2010;126(6):1184–1190.e3. doi: 10.1016/j.jaci.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brauweiler AM, Goleva E, Hall CF, Leung DYM. Th2 cytokines suppress lipoteichoic acid-induced matrix metalloproteinase expression and keratinocyte migration in response to wounding. J Invest Dermatol. 2015;135(10):2550–2553. doi: 10.1038/jid.2015.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cole C, et al. Filaggrin-stratified transcriptomic analysis of pediatric skin identifies mechanistic pathways in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134(1):82–91. doi: 10.1016/j.jaci.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feingold KR, Elias PM. Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta. 2014;1841(3):280–294. doi: 10.1016/j.bbalip.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 142.Sahle FF, Gebre-Mariam T, Dobner B, Wohlrab J, Neubert RH. Skin diseases associated with the depletion of stratum corneum lipids and stratum corneum lipid substitution therapy. Skin Pharmacol Physiol. 2015;28(1):42–55. doi: 10.1159/000360009. [DOI] [PubMed] [Google Scholar]
- 143.van Smeden J, Bouwstra JA. Stratum corneum lipids: their role for the skin barrier function in healthy subjects and atopic dermatitis patients. Curr Probl Dermatol. 2016;49:8–26. doi: 10.1159/000441540. [DOI] [PubMed] [Google Scholar]
- 144.Drake DR, Brogden KA, Dawson DV, Wertz PW. Thematic review series: skin lipids. Antimicrobial lipids at the skin surface. J Lipid Res. 2008;49(1):4–11. doi: 10.1194/jlr.R700016-JLR200. [DOI] [PubMed] [Google Scholar]
- 145.Arikawa J, Ishibashi M, Kawashima M, Takagi Y, Ichikawa Y, Imokawa G. Decreased levels of sphingosine, a natural antimicrobial agent, may be associated with vulnerability of the stratum corneum from patients with atopic dermatitis to colonization by Staphylococcus aureus. J Invest Dermatol. 2002;119(2):433–439. doi: 10.1046/j.1523-1747.2002.01846.x. [DOI] [PubMed] [Google Scholar]
- 146.Chen CH, et al. An innate bactericidal oleic acid effective against skin infection of methicillin-resistant Staphylococcus aureus: a therapy concordant with evolutionary medicine. J Microbiol Biotechnol. 2011;21(4):391–399. [PubMed] [Google Scholar]
- 147.Jang H, et al. Linoleic acid salt with ultrapure soft water as an antibacterial combination against dermato-pathogenic Staphylococcus spp. J Appl Microbiol. 2016;120(2):280–288. doi: 10.1111/jam.13012. [DOI] [PubMed] [Google Scholar]
- 148.Takigawa H, Nakagawa H, Kuzukawa M, Mori H, Imokawa G. Deficient production of hexadecenoic acid in the skin is associated in part with the vulnerability of atopic dermatitis patients to colonization by Staphylococcus aureus. Dermatology (Basel) 2005;211(3):240–248. doi: 10.1159/000087018. [DOI] [PubMed] [Google Scholar]
- 149.Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series: skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res. 2008;49(3):499–509. doi: 10.1194/jlr.R800001-JLR200. [DOI] [PubMed] [Google Scholar]
- 151.Janssens M, et al. Increase in short-chain ceramides correlates with an altered lipid organization and decreased barrier function in atopic eczema patients. J Lipid Res. 2012;53(12):2755–2766. doi: 10.1194/jlr.P030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ishikawa J, et al. Changes in the ceramide profile of atopic dermatitis patients. J Invest Dermatol. 2010;130(10):2511–2514. doi: 10.1038/jid.2010.161. [DOI] [PubMed] [Google Scholar]
- 153.van Smeden J, et al. The importance of free fatty acid chain length for the skin barrier function in atopic eczema patients. Exp Dermatol. 2014;23(1):45–52. doi: 10.1111/exd.12293. [DOI] [PubMed] [Google Scholar]
- 154.Berdyshev E, et al. Lipid abnormalities in atopic skin are driven by type 2 cytokines. JCI Insight. 2018;3(4):e98006. doi: 10.1172/jci.insight.98006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Danso M, et al. Altered expression of epidermal lipid bio-synthesis enzymes in atopic dermatitis skin is accompanied by changes in stratum corneum lipid composition. J Dermatol Sci. 2017;88(1):57–66. doi: 10.1016/j.jdermsci.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 156.Cookson W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol. 2004;4(12):978–988. doi: 10.1038/nri1500. [DOI] [PubMed] [Google Scholar]
- 157.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Koppelman GH, et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2009;180(10):929–935. doi: 10.1164/rccm.200810-1621OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bønnelykke K, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(1):51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 160.Laitinen T, et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304(5668):300–304. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 161.Lachowicz-Scroggins ME, et al. Abnormalities in MUC5AC and MUC5B protein in airway mucus in asthma. Am J Respir Crit Care Med. 2016;194(10):1296–1299. doi: 10.1164/rccm.201603-0526LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fahy JV. Goblet cell and mucin gene abnormalities in asthma. Chest. 2002;122(6 suppl):320S–326S. doi: 10.1378/chest.122.6_suppl.320s. [DOI] [PubMed] [Google Scholar]
- 163.Woodruff PG, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to goblet cell production in human bronchi. Am J Respir Crit Care Med. 2001;163(2):511–516. doi: 10.1164/ajrccm.163.2.2001038. [DOI] [PubMed] [Google Scholar]
- 165.Kondo M, Tamaoki J, Takeyama K, Nakata J, Nagai A. Interleukin-13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am J Respir Cell Mol Biol. 2002;27(5):536–541. doi: 10.1165/rcmb.4682. [DOI] [PubMed] [Google Scholar]
- 166.Vermeer PD, Harson R, Einwalter LA, Moninger T, Zabner J. Interleukin-9 induces goblet cell hyperplasia during repair of human airway epithelia. Am J Respir Cell Mol Biol. 2003;28(3):286–295. doi: 10.1165/rcmb.4887. [DOI] [PubMed] [Google Scholar]
- 167.Erzurum SC. New insights in oxidant biology in asthma. Ann Am Thorac Soc. 2016;13(suppl 1):S35–S39. doi: 10.1513/AnnalsATS.201506-385MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schroer KT, et al. Downregulation of glutathione S-transferase pi in asthma contributes to enhanced oxidative stress. J Allergy Clin Immunol. 2011;128(3):539–548. doi: 10.1016/j.jaci.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bucchieri F, et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol. 2002;27(2):179–185. doi: 10.1165/ajrcmb.27.2.4699. [DOI] [PubMed] [Google Scholar]
- 170.Durack J, et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140(1):63–75. doi: 10.1016/j.jaci.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huang YJ, et al. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136(4):874–884. doi: 10.1016/j.jaci.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Goleva E, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188(10):1193–1201. doi: 10.1164/rccm.201304-0775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242(1):205–219. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- 175.Blanchard C, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol. 2007;120(6):1292–1300. doi: 10.1016/j.jaci.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 176.Sherrill JD, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun. 2014;15(6):361–369. doi: 10.1038/gene.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wen T, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology. 2013;145(6):1289–1299. doi: 10.1053/j.gastro.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rochman M, et al. Profound loss of esophageal tissue differentiation in patients with eosinophilic esophagitis. J Allergy Clin Immunol. 2017;140(3):738–749.e3. doi: 10.1016/j.jaci.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Blanchard C, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006;116(2):536–547. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Blanchard C, et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol. 2010;184(7):4033–4041. doi: 10.4049/jimmunol.0903069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kottyan LC, et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet. 2014;46(8):895–900. doi: 10.1038/ng.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Martin LJ, et al. Eosinophilic esophagitis (EoE) genetic susceptibility is mediated by synergistic interactions between EoE-specific and general atopic disease loci. J Allergy Clin Immunol. 2018;141(5):1690–1698. doi: 10.1016/j.jaci.2017.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sleiman PM, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun. 2014;5:5593. doi: 10.1038/ncomms6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang N, Van Crombruggen K, Gevaert E, Bachert C. Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy. 2016;71(3):295–307. doi: 10.1111/all.12809. [DOI] [PubMed] [Google Scholar]
- 185.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Loffredo LF, Abdala-Valencia H, Anekalla KR, Cuervo-Pardo L, Gottardi CJ, Berdnikovs S. Beyond epithelial-to-mesenchymal transition: Common suppression of differentiation programs underlies epithelial barrier dysfunction in mild, moderate, and severe asthma. Allergy. 2017;72(12):1988–2004. doi: 10.1111/all.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pain M, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. 2014;23(131):118–130. doi: 10.1183/09059180.00004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kagalwalla AF, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129(5):1387–1396.e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lack G. Update on risk factors for food allergy. J Allergy Clin Immunol. 2012;129(5):1187–1197. doi: 10.1016/j.jaci.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 190.Tordesillas L, et al. Skin exposure promotes a Th2-dependent sensitization to peanut allergens. J Clin Invest. 2014;124(11):4965–4975. doi: 10.1172/JCI75660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Oyoshi MK, Oettgen HC, Chatila TA, Geha RS, Bryce PJ. Food allergy: insights into etiology, prevention, and treatment provided by murine models. J Allergy Clin Immunol. 2014;133(2):309–317. doi: 10.1016/j.jaci.2013.12.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hussain M, et al. Basophil-derived IL-4 promotes epicutaneous antigen sensitization concomitant with the development of food allergy. J Allergy Clin Immunol. 2018;141(1):223–234.e5. doi: 10.1016/j.jaci.2017.02.035. [DOI] [PubMed] [Google Scholar]
- 193.Noti M, et al. Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J Allergy Clin Immunol. 2014;133(5):1390–1399, 1399.e1. doi: 10.1016/j.jaci.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Han H, Roan F, Johnston LK, Smith DE, Bryce PJ, Ziegler SF. IL-33 promotes gastrointestinal allergy in a TSLP-independent manner. Mucosal Immunol. 2018;11(2):394–403. doi: 10.1038/mi.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Han H, Roan F, Ziegler SF. The atopic march: current insights into skin barrier dysfunction and epithelial cell-derived cytokines. Immunol Rev. 2017;278(1):116–130. doi: 10.1111/imr.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Han H, Thelen TD, Comeau MR, Ziegler SF. Thymic stromal lymphopoietin-mediated epicutaneous inflammation promotes acute diarrhea and anaphylaxis. J Clin Invest. 2014;124(12):5442–5452. doi: 10.1172/JCI77798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nakajima S, et al. Langerhans cells are critical in epicutaneous sensitization with protein antigen via thymic stromal lymphopoietin receptor signaling. J Allergy Clin Immunol. 2012;129(4):1048–1055.e6. doi: 10.1016/j.jaci.2012.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Brough HA, Lack G. The role of environmental exposure to peanut and the development of peanut allergy. Ann Allergy Asthma Immunol. 2018;120(3):232–233. doi: 10.1016/j.anai.2017.10.033. [DOI] [PubMed] [Google Scholar]
- 199.Brough HA, et al. Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J Allergy Clin Immunol. 2015;135(1):164–170. doi: 10.1016/j.jaci.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Brough HA, et al. Peanut protein in household dust is related to household peanut consumption and is biologically active. J Allergy Clin Immunol. 2013;132(3):630–638. doi: 10.1016/j.jaci.2013.02.034. [DOI] [PubMed] [Google Scholar]
- 201.Kim J, et al. Epidermal thymic stromal lymphopoietin predicts the development of atopic dermatitis during infancy. J Allergy Clin Immunol. 2016;137(4):1282–1285.e4. doi: 10.1016/j.jaci.2015.12.1306. [DOI] [PubMed] [Google Scholar]
- 202.Simpson EL, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134(4):818–823. doi: 10.1016/j.jaci.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Horimukai K, et al. Application of moisturizer to neonates prevents development of atopic dermatitis. J Allergy Clin Immunol. 2014;134(4):824–830.e6. doi: 10.1016/j.jaci.2014.07.060. [DOI] [PubMed] [Google Scholar]
- 204.Dyjack N, et al. Minimally invasive skin tape strip RNA sequencing identifies novel characteristics of the type 2-high atopic dermatitis disease endotype. J Allergy Clin Immunol. 2018;141(4):1298–1309. doi: 10.1016/j.jaci.2017.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lio PA, Lee M, LeBovidge J, Timmons KG, Schneider L. Clinical management of atopic dermatitis: practical highlights and updates from the atopic dermatitis practice parameter 2012. J Allergy Clin Immunol Pract. 2014;2(4):361–369. doi: 10.1016/j.jaip.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 206.Lowe AJ, et al. A randomized trial of a barrier lipid replacement strategy for the prevention of atopic dermatitis and allergic sensitization: the PEBBLES pilot study. Br J Dermatol. 2018;178(1):e19–e21. doi: 10.1111/bjd.15747. [DOI] [PubMed] [Google Scholar]
- 207.Simpson EL, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–2348. doi: 10.1056/NEJMoa1610020. [DOI] [PubMed] [Google Scholar]
- 208.Thaçi D, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387(10013):40–52. doi: 10.1016/S0140-6736(15)00388-8. [DOI] [PubMed] [Google Scholar]
- 209.Bikle DD, et al. 25 Hydroxyvitamin D 1 α-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. J Invest Dermatol. 2004;122(4):984–992. doi: 10.1111/j.0022-202X.2004.22424.x. [DOI] [PubMed] [Google Scholar]
- 210.Elias PM, Sugarman J. Does moisturizing the skin equate with barrier repair therapy? Ann Allergy Asthma Immunol. 2018;121(6):653–656.e2. doi: 10.1016/j.anai.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Alexander H, Brown S, Danby S, Flohr C. Research techniques made simple: transepidermal water loss measurement as a research tool. J Invest Dermatol. 2018;138(11):2295–2300.e1. doi: 10.1016/j.jid.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 212.Broccardo CJ, et al. Comparative proteomic profiling of patients with atopic dermatitis based on history of eczema herpeticum infection and Staphylococcus aureus colonization. J Allergy Clin Immunol. 2011;127(1):186–193, 193.e1. doi: 10.1016/j.jaci.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Winget JM, et al. Quantitative proteomic analysis of stratum corneum dysfunction in adult chronic atopic dermatitis. J Invest Dermatol. 2016;136(8):1732–1735. doi: 10.1016/j.jid.2016.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]