

Abstract
Expression of the metastasis suppressor NME1 in melanoma is associated with reduced cellular motility, invasion, and metastasis, but mechanisms underlying these activities are not completely understood. Herein we report a novel mechanism through which NME1 drives formation of large, stable focal adhesions (FAs) in melanoma cells via induction of integrin β3 (ITGβ3), and in one cell line, concomitant suppression of integrin β1 (ITGβ1) transcripts. Forced expression of NME1 resulted in a strong activation of the promoter region (−301 to +13) of the ITGB3 gene. Chromatin immunoprecipitation (ChIP) analysis revealed the transcriptional induction was associated with direct recruitment of NME1 and an increase in the epigenetic activation mark, acetylation of histone 3 on lysine 27 (H3K27Ac) to a 1 kb stretch of 5’-flanking sequence of the ITGB3 gene. Unexpectedly, NME1 did not affect the amount either ITGβ1 or ITGβ3 proteins were internalized and recycled, processes commonly associated with regulating expression of integrins at the cell surface. The ability of NME1 to suppress motile and invasive phenotypes of melanoma cells was dependent on its induction of ITGβ3. Expression of ITGβ3 mRNA was associated with increased disease-free survival time in melanoma patients of the TCGA collection, consistent with its potential role as an effector of the metastasis suppressor function of NME1. Together, these data indicate metastasis suppressor activity of NME1 in melanoma is mediated by induction of ITGB3 gene transcription, with NME1-driven enrichment of ITGβ3 protein at the cell membrane resulting in attenuated cell motility through the stabilization of large focal adhesions.
Keywords: NME1, transcription, ITGβ3, melanoma, cell motility, metastasis
INTRODUCTION
Melanoma is the most deadly skin cancer, and its incidence is increasing in the United States and around the world1. Metastasis suppressor genes selectively inhibit cancer metastasis, offering novel insights into potential therapeutic targets and prognostic markers for advanced melanoma and other malignancies2. NME1 (previously termed NM23-H1, or nucleoside diphosphate kinase-A/NDPK-A) was the first metastasis suppressor described3, with suppressor activity demonstrated in melanoma, breast carcinoma, and other cancers4. The mechanisms through which NME1 inhibits metastatic potential, however, are not fully understood. Motility-suppressing activity of NME15–7 has been attributed to interactions with signaling cascades8–10 and modulation of RNA expression11, 12.
Cell motility requires intricate contacts between the cell and its microenvironment, a process mediated by integrin proteins. In humans, eighteen α and eight β integrin subunits form 24 combinations of covalently-linked αβ dimers 13. αβ integrins are transmembrane receptors that bind selectively to ligands in the extracellular matrix (ECM). Cell surface expression of integrins is deregulated in a variety of cancers14. Overexpression of the integrin beta 1 (ITGβ1) subunit often defines the invading front of skin tumors15, and hyperactivation of ITGβ1 can drive melanoma metastasis16. Downregulation of ITGβ3 in melanoma17–19 and other cancer types20–22 can impair tumor cell motility, suggesting a metastasis-promoting function23. In the context of a triple-negative breast cancer cell line (4T1), however, forced ITGβ3 expression alone was not sufficient to drive lung metastasis24. Moreover, ITGβ3 induces oncogene-induced senescence in human fibroblasts by activating TGFβ-mediated signaling pathways25. Together, these studies indicate that the impact of ITGβ3 on growth and metastatic phenotypes can be highly context-dependent.
We showed previously in melanoma cells that NME1 induces expression and extracellular deposition of fibronectin (FN)26, a key ECM ligand of ITGβ3 and ITGβ3. Herein, we observe that NME1 induces a switch in cell surface expression from predominantly ITGβ1 to that of ITGβ3, and show these effects are not mediated by the canonical integrin trafficking pathways demonstrated by others27, 28.29 Instead, NME1 upregulates expression of ITGβ3 via a novel mechanism involving induction of ITGB3 gene transcription. We further demonstrate that the NME1-ITGβ3 regulatory axis is obligatory for motility- and invasion-suppressing activities of NME1.
Materials and Methods
Cell lines and culture conditions
Melanoma cell lines WM793, 1205LU and WM1158 (gifts of Dr. Meenhard Herlyn, Wistar Institute, Philadelphia, PA, USA) were cultured at 37ºC and 5% CO2 in Tu2% media, composed of: MCDB:Leibovitz-15 medium (4:1, v/v; Sigma-Aldrich, St. Louis, MO, USA), 2 mM CaCl2, 2.5 µg/ml insulin and 2% fetal bovine serum (Life Technologies, Grand Island, NY, USA). 1205LU-derived cell lines stably transfected with empty or NME1 expression plasmid have been described previously 30. MDA-MB-435s/M14 cells (referred to as M14 in this text) were obtained from R. Plattner (University of Kentucky, Lexington, KY, USA). Mouse embryonic fibroblasts (MEFs) were isolated from wild-type or NME1−/− C57BL/6 mice (E13.5). M14 and MEF cells were maintained at 37ºC and 10% CO2 in DMEM, 10% fetal bovine serum, 4.5 g/l D-glucose, L-glutamine, and 110 mg/l sodium pyruvate. RNA knockdown was achieved by lentiviral delivery of Mission shRNA (Sigma-Aldrich) against ITGβ3 (TRCN0000003236, ITGβ3 shRNA-a; TRCN0000318546, ITGβ3 shRNA-b; TRCN0000003235, ITGβ3 shRNA-c) or a non-targeted shRNA (SHC002, Sigma-Aldrich) as a negative control. For assessment of focal adhesions, 1205LU cells were transduced with a retrovirus vector encoding a DsRed-paxillin fusion protein (provided by Cai Huang, University of Kentucky). Forced NME1 expression was achieved by infection with a lentiviral NME1 expression vector that concomitantly expresses the fluorescent tag, eGFP and fluorescence-activated cell sorting (FACS) for GFP-positive cells.
TIRF Microscopy
For TIRF microscopy, cells were plated onto glass-bottom chamber slides, cultured for 2 days in complete media, then serum-starved for 24 hours. Microscopy was conducted with a Nikon Ti-series inverted microscope equipped with a 60x APO TIRF objective. Time-lapse videos were captured over a total of 2 hours with image acquisition every 15 minutes.
Cell adhesion assay
Cell adhesion assays were conducted essentially as described 31, with the following modifications. 96-well plates were coated with FN variants at 37°C for 1h, washed twice with 0.1% BSA (Millipore-Sigma), and blocked for 1 hour at 37°C with 10 mg/mL BSA. Plates were chilled on ice until addition of cells. Cells were dissociated using enzyme-free Cell Dissociation Buffer (Gibco) and counted. Cells were plated in a total volume of 100 µl per well and then incubated at 37°C for 15 or 30 minutes. Media and unattached cells were aspirated and wells washed three times with PBS. Cells were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and cell adhesion quantified by measuring absorbance at 550 nm.
Cell Surface Protein Measurement
For analysis of alpha integrin subunits on the cell surface, WM793 cells were isolated using enzyme-free Cell Dissociation Buffer (Gibco, Gaithersburg, MD, USA), resuspended in 100 µL of media, and incubated with anti-integrin antibodies at 37°C for 30 minutes. Cells were washed twice with ice-cold media and incubated with APC-conjugated isotype specific secondary antibodies on ice for 25 minutes. Labeled cells were resuspended in PBS and subjected to FACS (FACS Calibur Flow Cytometer, Becton-Dickinson, Mountain View, CA).
Beta integrins were measured via surface biotinylation. Cells at 90% confluency were treated with 80 µM Dynasore (Millipore-Sigma, St. Louis, MO, USA) or vehicle for 1 hour. Cells were isolated using enzyme-free Cell Dissociation Buffer and washed three times with ice-cold PBS (pH 8.0). Cells (2.5×107) were biotinylated by incubation with 2 mM Sulfo-NHS-LC-Biotin (ThermoFisher Scientific, Rockford, IL, USA) on ice for 1 hour. Reactions were quenched with three washes of PBS containing 100 mM glycine, pelleting of cells, and lysis in RIPA buffer. Cell lysates were subjected to pulldown with Neutravidin beads (ThermoFisher Scientific) at 4°C overnight, and biotinylated proteins eluted with 100 µL of 0.1 M glycine-HCl, pH 2.8 (10 minutes, room temperature). Eluted samples were neutralized with 15 µL of 1 M (Tris, pH 8.5) and subjected to immunoblot analysis.
Immunoblot analysis
Whole cell lysates were generated by lysis in RIPA buffer containing EDTA-free HALT protease inhibitors (Thermo Scientific, Rockford, IL). Lysates were subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Proteins of interest were detected via chemiluminescence after exposure to primary antibodies against NME1 (610247, BD Biosciences, San Jose, CA), ITGβ3 (AB1932, Millipore-Sigma), ITGβ1 (AB1952, Millipore-Sigma), ITGαv (AB1930, Millipore-Sigma), histone 3 (clone D1H12, Cell Signaling), p-FAK (KAP-TK131, Enzo Life Sciences, Farmingdale, NY), FAK (clone 12G4 Enzo), p-SRC (2101S, Cell Signaling), SRC (clone 36D10, Cell Signaling), p-p130cas (4011s, Cell Signaling), p130cas (Sana Cruz), and β-tubulin (clone 9F3, Cell Signaling) followed by isotype-specific HRP-conjugated secondary antibodies.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from cells using the RNeasy kit as per manufacturer’s instructions (Qiagen, Valencia,CA). Complementary DNA was generated with random hexamer primers and the TaqMan RT reagents (Applied Biosystems, Carlsbad, CA). SYBR Green based qRT-PCR was conducted for the genes of interest and normalized to RPL13a using the 2-ΔΔCT method. RNA-specific PCR primer sequences are provided in Supplementary Table 1.
Luciferase Assay
The ITGβ1-luc reporter has been described (gifted by Dr. Sean Colgan, University of Denver Colorado)32. The ITGβ3 promoter-luciferase reporter plasmid was generated by PCR amplification of the −301 to +13 region from human genomic DNA, followed by purification and ligation of the fragment into the pGL3-Basic (promoter-less) plasmid containing a firefly luciferase reporter mini-gene (Promega, Madison, WI) with T4 ligase (New England Bioscience, Ipswich, MA). The ITGβ3 promoter fragment was ligated into KpnI/NheI-linearized pGL3-Basic with T4 ligase (New England Bioscience, Ipswich, MA). Cells were transfected with 100 ng of ITGB3 reporter plasmid using Fugene 6 transfection reagent (Promega). To control for transfection efficiency, transfections included 50 ng of a Renilla luciferase expression vector (gifted by Dr. Richard Eckert, University of Maryland, Baltimore). Luciferase assays were performed on cell lysates prepared 72h post-transfection using the Dual-Luciferase Reporter 1,000 Assay System (Promega). Luciferase activity of the experimental reporter was normalized to Renilla luciferase activity as measured in relative light units (RLU).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using the ChIP-IT Express Kit (Active Motif, Carlsbad, CA). Twenty-five μg of sheared chromatin was pre-cleared with Protein G magnetic beads (Cell Signaling) and isotype control IgG for 1 hour at 4°C, then subjected to immunoprecipitation at 4°C (14–16h) with 2 μg of the indicated antibody. Anti-NME1 (scNM301) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) while anti-Histone 3 lysine 27 acetylation (H3K27ac, clone D5E4), rabbit IgG (clone DA1E) and mouse IgG (clone G3A1) were from Cell Signaling. A portion (10%) of sheared chromatin was used as input DNA. DNA was purified using the QiaQuick PCR Purification Kit (Qiagen, Valencia, CA, USA) prior to qPCR.
Wound Healing and Single-cell Three-dimensional Cell Invasion Assays
For wound-healing assays, WM1158 cells were transduced twice (24 and 48 hours post-seeding), with shRNA-expressing viruses. Twenty-four hours post-transduction, scratches were created on cell monolayers with a 200 μl pipette tip, followed by rinsing and replenishment with Tu media (0.2% FBS). Phase-contrast images were taken after wounding (0 and 24 hour) with an EVOS inverted microscope (Thermo-Fisher). Individual migrating cells were enumerated using the particle counting function in ImageJ (NIH, Bethesda, MD, USA).
For 3-dimensional invasion assays, WM1158 cells were transduced with lentivirus shRNA vectors in two sequential infections (MOI 104/infection). Twenty-four hours post-transduction, cell aggregates were generated by dissociating cell monolayers and re-plating 104 cells/well in TU2% in a 96-well dish coated with 0.75% agarose. Cell aggregates were formed overnight at 37°C in 5% CO2 and embedded (1/well) in 100µl of a 50:50 mixture of Matrigel and TU2% on ice; 8 aggregates per condition were plated in each independent experiment. Gels were solidified at 37°C, 5% CO2 for 30min prior, then imaged at 0 and 24h after plating. Invading cells, defined as cells penetrating past the border of the initial cell aggregate, were quantified at using ImageJ.
Statistical analysis
Results were analyzed with SigmaStat v3.5 statistical software (Systat Software, Chicago, IL). Unless stated otherwise, all data are representative of n≥3 independent experiments. Additional details for statistical analyses are provided in figure legends.
TCGA analyses
TCGA mRNA expression data from Skin Cutaneous Melanoma (SKCM) samples was obtained through GDAC FireHose (v01–28-2016). Results are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. The dataset was run through the TCGA IlluminaHiSeq_RNASeqV2 pipeline. mRNA expression data was imported into RStudio and normalized using the voom function in the LIMMA package (v1.0.136, RStudio Inc., Boston, MA, USA). Clinical information for SKCM patients was obtained through FireHose version 01–28-2016 and imported into RStudio. To measure correlations between gene expression and clinical outcome, SKCM patient identifiers from clinical and z-score information were matched. Patients were grouped into “High” and “Low” expression groups based on the median for each gene of interest and analyzed for alterations in overall survival and disease-free survival33. Kaplan-Meier survival analysis was completed through the survival package in RStudio.
RESULTS
Suppression of melanoma cell motility by NME1 is associated with formation of large, slow-cycling focal adhesions
Focal adhesions (FAs) contain clusters of integrin receptors that associate with cytoskeletal and signaling molecules to regulate cell migration and invasion. The impact of NME1 on FA dynamics was measured in two variants of the 1205Lu melanoma cell line, generated by stable transfection with empty expression vector or NME1 expression plasmid as previously described 30. The 1205Lu cell line was considered appropriate for these studies as it exhibits a highly motile phenotype in culture that is potently suppressed by NME1 overexpression30. FAs were visualized by transient retroviral vector-mediated expression of a dsRed-labeled form of the FA-associated protein, paxillin. Empty plasmid vector-transfected control 1205Lu cells (VEC) were motile, displaying bipolar morphology and abundant small adhesions at leading and trailing edges (Figure 1a). In contrast, forced NME1 expression elicited cell spreading and large, radially-distributed FAs at leading and trailing edges. NME1-expressing cells lacked clear polarity, and most failed to migrate during observation. Time-lapse TIRF microscopy revealed rapid turnover of small adhesions at leading and trailing edges in control cells (Figure 1b, subpanels ii and iii), while larger, radially-distributed FAs induced by forced NME1 expression exhibited slow, centripetal turnover (subpanels iv-vi; video files, Supplementary Figure 1).
Figure 1.
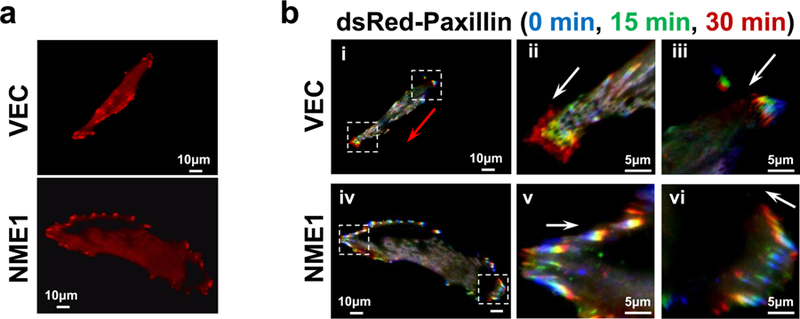
NME1 promotes the formation of large, slow-cycling focal adhesions in melanoma cells. (a) Visualization of focal adhesions (FAs) in serum-starved 1205LU cells (VEC, empty vector-transfected; NME1, forced NME1 expression) after retrovirus-mediated expression of dsRed-labeled paxillin. The NME1-transfected cell line expresses 3-fold higher levels of NME1 than the control VEC line 30. (b) TIRF time-lapse microscopy of cells in panel (a), with paxillin-labeled structures monitored and color-coded for the indicated time points (0, 15 and 30 min). “Leading” and “trailing” edges of cells are outlined with boxes in subpanels i and iv; leading edges are magnified in subpanels ii and v, and trailing edges in iii and vi. Red arrows depict net migration of the entire cell; white arrows depict movement of focal adhesions within a cell. Results shown are representative of at least three cells monitored per cell line.
NME1 expression promotes melanoma cell adhesion by modulating balance of integrin beta subunits at the cell surface
To address whether NME1-induced stabilization of large FAs was mediated by altered FN-integrin interactions at the cell surface, impact of NME1 on cell adhesion to FN substrates of varying integrin-binding affinities was measured in the 1205Lu cell line. The analyses were also conducted in M14 cells, another metastatic melanoma-derived line whose motility is strongly suppressed by NME126. Forced NME1 expression strongly enhanced adhesion to full length FN and a 110kDa proteolytic fragment containing the integrin-binding domain (Figure 2a). NME1 had no impact on adhesion to a FN fragment containing the transglutaminase 2 (TG2) binding domain but lacking integrin-binding sites. Thus, NME1 expression enhances integrin-mediated adhesion of melanoma cells to FN.
Figure 2.

NME1 promotes integrin-mediated adhesion to fibronectin (FN) via regulation of integrin beta subunit expression at the cell surface. (a) Adhesion assays were conducted as described 31, with minor modifications (Materials and Methods). Diagram depicts domains of the FN molecule; those associated with binding to integrins (ITG-BD) and transglutaminase 2 (TG2-BD) are indicated with brackets. Regions corresponding to FN fragments used in cell adhesion studies are identified with orange lines above the molecule (42 and 110 kDa, respectively). Bar graph summarizes adhesion of 1205Lu cells stably transfected with either empty or NME1 expression vector. Cells were incubated for 15 min on plates coated with full length (FL) fibronectin (FN) or FN fragments containing either the ITG-BD (110 kD) or TG2-BD (42 kD). *p≤0.05 by Student’s t-test. (b) Cell surface expression of the indicated alpha integrin subunits (α4, α5 and αv) was quantified in 1205Lu cells (−/+ forced NME1 expression) by flow cytometry. (c) NME1 expression induces a switch in cell surface expression from predominantly ITGβ1 to ITGβ3. Cell surface expression of beta integrins (“Surface”) was determined in the indicated M14- and 1205Lu-derived cell lines by surface biotinylation and immunoblot analysis, as described (Materials and Methods). Expression of NME1 and β-tubulin (β-tub) were measured in the “intracellular” compartment by immunoblot analysis. (d) Impact of forced NME1 expression on expression of total and activated forms of focal adhesion kinase (FAK), SRC and p130Cas was determined in M14 cells by immunoblot analysis. (e) Cell surface and intracellular expression of ITGβ1, ITGβ3 and ITGαv was measured in M14 cells (−/+ forced NME1 expression) following a 1h incubation with either vehicle (“DMSO”) or the dynamin inhibitor dynasore (“Dyn”; 80 mM).
NME1 expression did not regulate surface expression of integrin subunits α4, α5 or αv (Figure 2b), but inhibited surface expression of ITGβ1 in both M14 and 1205Lu melanoma cell lines, and increased that of ITGβ3 (Figure 2c). NME1 also inhibited activation of three intracellular effectors of integrin signaling, FAK, SRC and p130Cas (Figure 2d).
Impact of NME1 on levels of αv, ITGβ1 and ITGβ3 were compared between cell surface and whole cell preparations. Forced expression of NME1 was associated with reduced surface expression of ITGβ1 and an increase in that of ITGβ3(Figure 2e), with minimal effect on ITGαv. Within cell lysates, control vector-transfected cells exhibited a predominant 140 kDa species consistent with the mature cell surface form and lesser amounts of a smaller protein (120 kDa). NME1 expression was associated with absence of mature ITGβ1 and appearance of two smaller ITGβ1 species (110 and 90 kDa), consistent with a prior report of cytoplasmic, immature precursors of ITGβ134. Consistent with this interpretation, the 90 kDa form co-migrated precisely with enzymatically de-glycosylated ITGβ1 (Figure S1). These data suggest NME1 (or indirectly via induction of ITGβ3) may interfere with surface expression of ITGβ1 by inhibiting maturation in ER/Golgi compartments. In contrast, intracellular ITGβ3 and ITGαv were present in their mature forms. NME1 expression produced a more robust increase in total ITGβ3 than seen at the surface. Total ITGαv was modestly induced by NME1, contrasting with a marginal change at the surface.
Surface expression of integrins is regulated via rates of internalization and sorting within endosomes for degradation or recycling to the plasma membrane 35. The nucleoside diphosphate kinase (NDPK) activity of NME1 fuels endocytosis by supplying dynamin with GTP36, suggesting the NME1-induced switch in cell surface expression of ITGβ1 and ITGβ3 was mediated via differential effects of NME1 on dynamin-dependent trafficking. In control cells, a dynamin inhibitor (Dynasore) increased surface expression of ITGβ1, consistent with ITGβ1 internalization via dynamin-dependent endocytosis. However, Dynasore failed to reverse the NME1-mediated reduction in cell surface ITGβ1, indicating ITGβ1 downregulation is independent of dynamin-fueling activity of NME1.
Dynasore did not affect surface or total ITGβ3 in control cells, consistent with dynamin-independent endocytosis. With forced NME1 expression, however, Dynasore induced ITGβ3 expression within both cellular compartments. These modest Dynasore-induced increases, however, are consistent with dynamin-driven endocytosis of ITGβ3 that would reduce rather than increase surface ITGβ3 expression in response to NME1. Thus, NME1 regulates surface expression of ITGβ1 and ITGβ3 via impacts on their overall steady-state concentrations.
NME1 induces ITGβ3 gene transcription via direct physical interactions with the promoter
Impacts of NME1 on expression of ITGβ1 and ITGβ3 mRNAs were measured in three human melanoma-derived cell lines (M14, WM1158 and WM793). Both spliced and unspliced forms of ITGβ1 RNA were strongly reduced in response to NME1 expression in M14 cells (Figure 3a), consistent with repression of ITGB1 gene transcription. Moreover, transcriptional activity of a 1 kb fragment of the ITGβ1 promoter was inhibited strongly by forced NME1 expression (Figure 3b, left). NME1 failed to regulate expression of ITGβ1 transcripts in WM793 and WM1158 cells, however, nor was ITGβ1 mRNA affected by ablation of the NME1 locus37 in mouse embryo fibroblasts (MEFs) (Figure 3a).
Figure 3.

NME1 activates transcription of the ITGβ3 gene via direct binding to the promoter region. (a) Expression of mRNA encoding ITGβ3 was measured by quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR) in the indicated melanoma (M14, WM1158, and WM793; −/+ forced NME1 expression) and mouse embryo fibroblast (wild-type C57BL/6, “WT”; NME1 knockout, “NME1ko”) cell lines. Expression of ITGβ3 in the respective control conditions of each panel (vector or WT) is normalized to a value of 1. (b) Shown at top is a schematic representation of a promoter-reporter cassette containing the ITGB3 promoter (“ITGB3-P”; −301 to +13) in linkage with a firefly luciferase reporter mini-gene. Summarized below are amounts of luciferase activity obtained after transient transfection of the indicated melanoma cell lines with the ITGB3 promoter-luciferase plasmid in the absence or presence of forced NME1 expression. Activity is expressed as relative luciferase activity (“Rel. luc. activity”), with activity obtained in the absence of forced NME1 expression assigned a value of 1. *Denotes p < 0.05 by Student’s t-test. (c). Impact of forced NME1 expression on occupancy of the ITGB3 promoter by transcriptionally active chromatin and NME1 was assessed in M14 cells by chromatin immunoprecipitation assay (ChIP). ChIP reactions were conducted with antibodies directed to IgG, acetylated histone 3 (at lysine 27, or “H3K27ac”), or NME1 as indicated. Immunoprecipitated DNA was analyzed by qPCR with a series of five amplicons (150– 200 bp) spanning the ITGB3 promoter from −−958 to +15, as shown. *denotes p < 0.05 by Student’s t-test.
In contrast, ITGβ3 mRNA was induced by NME1 in all three melanoma cell lines, and significantly reduced in mouse embryo fibroblasts (MEFs) from NME1 knockout mice (Figure 3a). To assess impact of NME1 on ITGB3 transcription, a ITGβ3 promoter fragment (−301/+13) was cloned from human genomic DNA and inserted into a luciferase reporter vector (pGL3-basic, Promega). NME1 expression induced ITGβ3 promoter activity by approximately 8-fold (Figure 3b, left) in M14 cells, and to a significant extent in WM1158 cells (Figure 3b, right).
To measure impact of NME1 on the endogenous ITGβ3 promoter, levels of an epigenetic mark associated with transcription activation (histone 3 acetylation on lysine 27; H3K27Ac) were assessed by chromatin immunoprecipitation (ChIP). In the absence of NME1 overexpression, modest levels of H3K27Ac were associated with the −150/+15 subregion of the ITGβ3 promoter, and upstream within a −956/−877 subregion (Fig. 3c, middle). Forced NME1 expression elicited greater association of H3K27Ac with the −150/+15 and −956/−877 subregions, with contact also detected in the 333/−143 subregion.
While NMEs have been suggested to function as DNA-binding transcription factors 38, this function had not been carefully demonstrated in living cells. In the absence of NME1 overexpression, ChIP performed with anti-NME1 antibody revealed interactions of endogenous NME1 across the ITGβ3 promoter (−956/+15) except within the −645/−316 subregion (Figure 3c, right), and strongest binding was seen with the GC-rich proximal promoter (−150/+15). Forced NME1 expression enhanced association of NME1 with the proximal promoter (−150 to +15) and three subregions further upstream (−333/−143, −645/−316 and −956/−877). Co-enrichment of NME1 with the H3K27Ac modification within the ITGβ3 promoter region demonstrates NME1 induces ITGβ3 transcription via its DNA-binding function.
ITGβ3 mediates motility- and invasion-suppressing activities of NME1
To determine whether ITGβ3 mediates the ability of NME1 to suppress cell motility and invasion, expression of NME1 and ITGβ3 was modulated in the metastatic WM1158 cell line. WM1158 cells were particularly suitable for analyzing impacts of the NME1-ITGβ3 regulatory loop on cellular motility and invasion activities in a number of respects. Their strong motile and invasive characteristics provided a high threshold of activity upon which individual and combined effects of NME1 and ITGβ3 could be detected. In addition, as a prototype of a metastatic cell line expressing very low levels of the metastasis suppressor NME1, they were ideal for assessing impacts of forced NME1 expression. Moreover, forced NME1 expression induced ITGβ3 expression in robust fashion (Figure 3), facilitating analysis of the specific contributions of ITGβ3 using an shRNA approach.
Transduction with lentiviral NME1 expression vector resulted in robust induction of NME1 expression (Figure 4a). Expression of two distinct shRNAs blocked expression of ITGβ3 protein in control cells, and reduced the expression of ITGβ3 in NME1-expressing cells to levels observed in the absence of forced NME1 expression. Silencing of ITGβ3 expression with either shRNA sequence also resulted in a slight induction in NME1 expression. A time-course analysis following pulse administration of the protein synthesis inhibitor cycloheximide revealed the effect of ITGβ3 silencing was due to stabilization of NME1 protein (Figure S2). Using the scratch assay approach, NME1 was shown to inhibit migration of individual cells (Figure 4b and 4c). Silencing of ITGβ3 in control cells inhibited motility, strongly suggesting a motility-driving activity for ITGβ3 in the context of low NME1 expression. Nevertheless, the possibility cannot be excluded that the small induction in NME1 expression elicited by ITGβ3 silencing (Figure 4a) may have contributed to the reduction in motility. In NME1-overexpressing cells, however, ITGβ3 silencing clearly ablated the motility suppressing-function of NME1. Thus, at higher levels of NME1 and ITGβ3 expression, ITGβ3 is a motility-suppressing effector of NME1.
Figure 4.
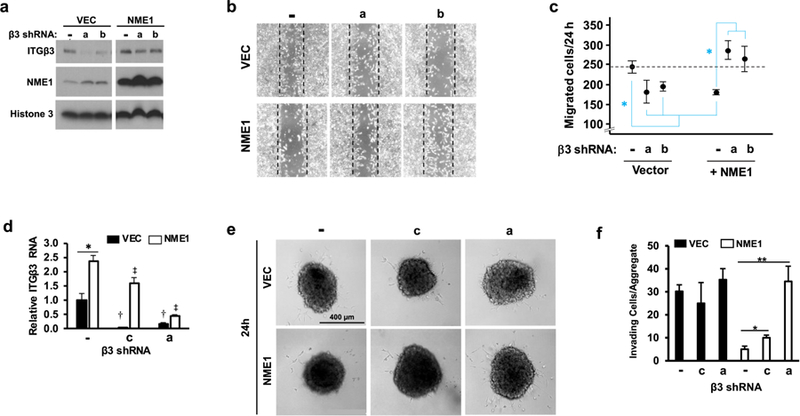
NME1 suppresses motile and invasive potential of WM1158 melanoma cells by upregulating ITGβ3 expression. (a) Expression of NME1 and ITGβ3 proteins was measured by immunoblot analysis in cells receiving the indicated combinations of forced NME1 expression and shRNAs directed to ITGβ3. shRNA treatments consisted a non-targeting control shRNA (−) or one of two shRNAs specific for ITGβ3 (“a” or “b”). (b) Representative images of wound/scratch assays were acquired 24 h after wound induction in cells receiving the indicated combinations of forced NME1 expression and shRNA-mediated silencing of ITGβ3. Dotted boxes depict borders of original wounds. (c) Graph provides a quantitative analysis of individual cells migrating into wounds. Closed circles and error bars represent means (+/− SEM) derived from 3– 4 independent experiments. Asterisks denote means that are significantly different (*p≤0.05 by ANOVA with Holm-Sidak pairwise testing). (d) Expression of ITGβ3 mRNA was measured in cells used for measurement of single cell invasion activity in 3-dimensional sphere assays after the indicated combination of forced NME1 expression and shRNA sequences (-, non-targeting control; ITGβ3-directed shRNAs “c” and “a”). The shRNA sequence “c” was employed instead of sequence “b” used in panel a, as sequence “b” strongly disrupted spheroid formation. Cultures were co-infected with a lentiviral vector for expression of eGFP for enhanced imaging of invading cells. *, comparison between vector- and NME1-treated; †, denotes significant silencing of ITGβ3 RNA in absence of forced NME1 expression (black bars); ‡, denotes significant silencing of ITGβ3 RNA in presence of forced NME1 expression (white bars). p < 0.05. (e) Representative images of spheroids and invading cells in response to NME1 and shRNA treatments (24 and 72h post-infection). (f) Single cells detached from spheroids were counted as described in Materials and Methods. Asterisks denote means that are significantly different (*p≤0.05, **p≤0.01 by ANOVA with Holm-Sidak pairwise testing).
Impacts of NME1 and ITGβ3 on invasive potential were measured with a 3-dimensional Matrigel-embedded sphere assay. As previously observed (Figure 3), NME1 induced expression of ITGβ3 (2.4-fold) (Figure 4d). In the absence of NME1 overexpression, robust ITGβ3 knockdown was achieved with two distinct shRNA sequences, “a” and “c” (>90% and 70%, respectively; Figure 4d). When NME1 was overexpressed, ITGβ3 was strongly silenced with shRNA sequence “a” (>80%), and to a significant degree with “c”. NME1 expression strongly inhibited the number of invasive single cells (Figure 4e and f). Under conditions of low NME1 expression, silencing of ITGβ3 expression had no impact on invasive activity, contrasting with the motility-driving activity of ITGβ3 at low NME1 levels (Figure 4b and c). In NME1-overexpressing cells, however, ITGβ3 silencing strongly induced cell invasiveness. Together, these studies demonstrate ITGβ3 mediates both the motility- and invasion-suppressing activities of NME1.
ITGβ3 mRNA expression is associated with extended survival of melanoma patients
Having demonstrated that motility- and invasion-suppressing activity of NME1 involves induction of integrin-beta transcripts in melanoma cells, we posited that expression of ITGβ1 and/or ITGβ3 RNA would be predictive of survival in melanoma patients. Patients from the Skin Cutaneous Melanoma (SKCM) dataset of The Cancer Genome Atlas (TCGA) were divided into “high” and “low” expression groups for ITGβ1 and ITGβ3 (Materials and Methods) to measure associations with overall and progression-free survival. While ITGβ1 mRNA was not correlated with overall nor progression-free survival, ITGβ3 expression was strongly predictive of prolonged progression-free survival (p < 0.001), and approached significance for overall survival (p < 0.067). Categorization into two populations based on expression of both ITGβ1 and ITGβ3 (high ITGβ1/low ITGβ3 versus low ITGβ1/high ITGβ3) provided no advantage over ITGβ3 alone (data not shown). NME1 mRNA expression correlated with neither progression-free nor overall survival. This was anticipated, in light of our demonstration that downregulation of NME1 protein results from lysosomal degradation and not NME1 transcripts39. These findings identify ITGβ3 RNA as a predictor of prolonged survival in melanoma patients, and suggest a key role for the NME1-ITGβ3 regulatory axis in melanoma progression.
Discussion
Herein, NME1 expression was shown to induce expression of ITGβ3 at the surface of melanoma cells, driven by direct enhancement of ITGB3 transcription. NME1-induced ITGB3 transcription was accompanied by potent suppression of ITGB1 transcription in one of the melanoma cell lines studied (M14). These transcription-directed mechanisms were unexpected and novel, in light of more-recognized roles of trafficking processes for regulating cell surface integrins35. NME1-mediated regulation of RNA and protecontext-depein for ITGβ3, and sometimes ITGβ1, was sufficiently robust to regulate surface expression by mass action throughout endocytosis and recycling.
NME1 was shown to activate ITGβ3 transcription directly through its DNA binding function. Binding of NME1 was detected at multiple sites within the ITGB3 promoter, and was augmented by forced NME1 expression. Binding of NME1 to the ITGB3 promoter was associated with increased levels of the transcriptional activation mark, H3K27Ac. Studies have demonstrated binding of NME1 to a variety of DNA sequences40, but relevance to transcriptional function was not established. Analyses of DNA binding with recombinant NME1 in vitro from our laboratory41 revealed a preference for binding to single-stranded DNA without a strictly-defined consensus binding sequence, suggesting binding is directed to single-stranded and/or non-B form DNA 42. The ITGB3 promoter (−150 to +15), which was the most highly enriched for NME1 binding, is highly GC-rich and prone to formation of such non-B form DNA structures. A parallel is suggested by the vascular smooth muscle α-actin gene promoter, regulated through competition between transcription-repressing factors with affinity for non-B form (e.g. Purα/Purβ) and transcription-enhancing factors that bind double-stranded DNA (e.g. TEF-1, SRF and Sp1)43. Such a model could address how NME1 can both induce (ITGB3) and repress (ITGB1) transcriptional activity, or downregulate both genes in other cell types 10, depending on proteins and signaling pathways operative within a given cellular context.
NME1 represses activity of the ITGAV promoter in B cell lines44 through a transcriptional corepressor (i.e. non-DNA binding) function, with repression reversed by the Epstein-Barr virus-encoded protein, EBNA3C. This corepressor function was proposed to be implemented via physical interactions of NME1 with GATA-1 and EBNA3C. Evidence has been provided for coregulatory (non-DNA binding) functions of NME1 in other settings as well 45, 46. Together, our studies provide strong evidence that expression of integrins is regulated at the level of gene transcription.
NME1-mediated induction in surface ITGβ3 was associated with formation of large and stable focal adhesions, enhanced cell adhesion to FN, and reduced activation of three FA-associated mediators of intracellular signaling (FAK, SRC and CAS). Our findings indicate the effects of NME1 on FA dynamics are driven by higher ITGβ3 (and sometimes lower ITGβ1) content within FA complexes. Motility-regulating activity of ITGβ3 was context-dependent, with ITGβ3 expression suppressing motility when NME1 expression was high, but promoting motility when NME1 expression was low. This context-dependence could result from interactions of ITGβ3 with NME1-induced alterations in expression of other genes and/or signaling pathways. NME1 regulates a broad spectrum of RNAs in melanoma47, 48 and breast carcinoma11 cell lines, as well as intracellular signaling pathways that could cooperate with altered integrin expression 49. The context-dependence of ITGβ3 actions is also consistent with the biphasic (i.e. nonlinear) relationship between cell adhesion to substrate and migration rates. Weak cell adhesion is associated with slow migration, intermediate adhesion with accelerated migration, and strong adhesion with migration arrest50. In this regard, a shift in integrin beta subunits towards the ITGβ3 isoform induces a switch in downstream integrin-mediated signaling and a reduction in RhoA activation required for directional migration 51, 52. Also, αvβ3-FN complexes are more resistant to dissociation in response to allosteric binding of Ca+2 in vitro than α5β1-FN complexes 53. Our observations may explain discordance in prior studies of ITGβ3 17–19 that did not take NME1 expression into account. They also suggest loss of NME1 in advanced melanoma 4, 54 converts ITGβ3 to a metastasis driver.
NME1 and its intrinsic NDPK activity fuel dynamin with GTP for endocytosis of cell surface receptors for ligands such as transferrin, interleukin-2β subunit and EGF 36. We observe, however, that impacts of NME1 on surface ITGβ1 and ITGβ3 are not mediated by its dynamin-fueling activity. This could be explained by differences in the endocytic cargo or cell lines under study. Our analyses do not exclude the possibility that NME1 regulates cell surface expression of ITGβ1 and ITGβ3 to a minor degree through dynamin-independent mechanisms.
ITGβ3 RNA expression was associated with prolonged survival of melanoma patients, and was a stronger predictor of survival than the NME1 transcript itself. This is not surprising, in light of the observation that downregulation of NME1 in metastatic melanoma cell lines is not mediated via regulation of the NME1 transcript, but rather by proteolysis in lysosomes 39. It will be of interest to determine whether the power of ITGβ3 RNA as a prognostic marker is enhanced in conjunction with NME1 protein expression, or within a larger expression signature.
Our study demonstrates that NME1 is a key transcriptional regulator of the ITGB3 gene, leading to suppression of motility and invasion activity. These findings also show that ITGβ1 and ITGβ3 expression is not regulated wholly through their intracellular trafficking (e.g. endocytosis, targeting to lysosomes and recycling to the cell surface). The study also illustrates how the metastasis suppressor NME1 can be exploited as a means for discovery of novel mechanisms and prognostic markers operative during melanoma progression.
Supplementary Material
Figure 5.
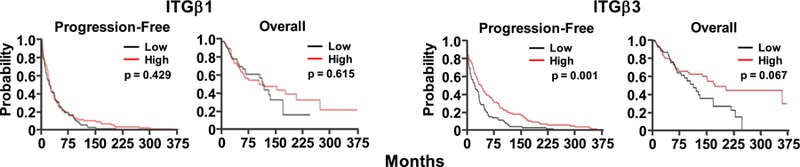
Expression of ITGβ3 RNA is associated with prolonged survival expression in skin cutaneous melanoma (SKCM) patients of The Cancer Gene Atlas (TCGA). A total of 234 patients were grouped into subpopulations (117 patients per subpopulation) representing either “High” or “Low” expression of RNAs encoding ITGβ1 or ITGβ3, relative to the median level of expression for each. Shown are Kaplan-Meier plots of survival (overall and disease-free) for the various groups.
Acknowledgments
This work was supported by the National Institutes of Health/National Cancer Institute through research grants (CA83237, CA159871, CA159871-S1), a training grant (T32 CA154274), and an education grant (R25 GM055036). The study was also supported by a research grant from the Maryland Stem Cell Research Foundation (MSCRFI-1638). The authors express their sincere appreciation to Dr. Alexey Belkin for his helpful advice throughout the project, and to Dr. Laurence Magder for his assistance with statistical analysis.
Abbreviations:
- FA
focal adhesions
- ITGβ1
integrin beta 1
- ITGβ3
integrin beta 3
- ChIP
chromatin immunoprecipitation
- TCGA
The Cancer Genome Atlas
- SKCM
skin cutaneous melanoma
- ECM
extracellular matrix
- TIRF
total internal reflection fluorescence
- ITGαv
integrin alpha v
- NDPK
nucleoside diphosphate kinase
- MEF
mouse embryonic fibroblast
- H3K27Ac
histone 3 lysine 27 acetylation
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has beenThis is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.SEER cancer statistics review In: Horner MJ, Krapcho M, Neyman N, et al. , eds. Bethesda, MD, 2006. [Google Scholar]
- 2.Smith SC, Theodorescu D. Learning therapeutic lessons from metastasis suppressor proteins. Nat Rev Cancer 2009;9:253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, Sobel ME. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 1988;80:200–4. [DOI] [PubMed] [Google Scholar]
- 4.Hartsough MT, Steeg PS. Nm23/nucleoside diphosphate kinase in human cancers. J Bioenerg Biomembr 2000;32:301–08. [DOI] [PubMed] [Google Scholar]
- 5.Hsu S, Huang F, Ossowski L, Friedman E. Colon carcinoma cells with inactive nm23 show increased motility and response to motility factors. Carcinogenesis 1995;16:2259–62. [DOI] [PubMed] [Google Scholar]
- 6.Kantor JD, McCormick B, Steeg PS, Zetter BR. Inhibition of cell motility after nm23 transfection of human and murine tumor cells. Cancer Res 1993;53:1971–73. [PubMed] [Google Scholar]
- 7.Lee HY, Lee H. Inhibitory activity of nm23-H1 on invasion and colonization of human prostate carcinoma cells is not mediated by its NDP kinase activity. Cancer Lett 1999;145:93–9. [DOI] [PubMed] [Google Scholar]
- 8.Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K, Sugimura H. Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc Natl Acad Sci U S A 2001;98:4385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami M, Meneses PI, Lan K, Robertson ES. The suppressor of metastasis Nm23-H1 interacts with the Cdc42 Rho family member and the pleckstrin homology domain of oncoprotein Dbl-1 to suppress cell migration. Cancer Biol Ther 2008;7:677–88. [DOI] [PubMed] [Google Scholar]
- 10.Li MQ, Shao J, Meng YH, Mei J, Wang Y, Li H, Zhang L, Chang KK, Wang XQ, Zhu XY, Li DJ. NME1 suppression promotes growth, adhesion and implantation of endometrial stromal cells via Akt and MAPK/Erk1/2 signal pathways in the endometriotic milieu. Hum Reprod 2013;28:2822–31. [DOI] [PubMed] [Google Scholar]
- 11.Horak CE, Lee JH, Elkahloun AG, Boissan M, Dumont S, Maga TK, Arnaud-Dabernat S, Palmieri D, Stetler-Stevenson WG, Lacombe ML, Meltzer PS, Steeg PS. Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res 2007;67:7238–46. [DOI] [PubMed] [Google Scholar]
- 12.Puts GS, Leonard MK, Pamidimukkala NV, Snyder DE, Kaetzel DM. Nuclear functions of NME proteins. Lab Invest 2018;98:211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature Rev Cancer 2010;10:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato T, Enomoto A, Watanabe T, Haga H, Ishida S, Kondo Y, Furukawa K, Urano T, Mii S, Weng L, Ishida-Takagishi M, Asai M, Asai N, Kaibuchi K, Murakumo Y, Takahashi M. TRIM27/MRTF-B-dependent integrin beta1 expression defines leading cells in cancer cell collectives. Cell Rep 2014;7:1156–67. [DOI] [PubMed] [Google Scholar]
- 15.Kato H, Liao Z, Mitsios JV, Wang HY, Deryugina EI, Varner JA, Quigley JP, Shattil SJ. The primacy of beta1 integrin activation in the metastatic cascade. PLoS ONE 2012;7:e46576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller DW, Bosserhoff AK. Integrin beta 3 expression is regulated by let-7a miRNA in malignant melanoma. Oncogene 2008;27:6698–706. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Regezi J, Ross FP, Blystone S, Ilic D, Leong SP, Ramos DM. Integrin alphavbeta3 mediates K1735 murine melanoma cell motility in vivo and in vitro. J Cell Sci 2001;114:2665–72. [DOI] [PubMed] [Google Scholar]
- 18.Aznavoorian S, Stracke ML, Parsons J, McClanahan J, Liotta LA. Integrin alphavbeta3 mediates chemotactic and haptotactic motility in human melanoma cells through different signaling pathways. J Biol Chem 1996;271:3247–54. [DOI] [PubMed] [Google Scholar]
- 19.Hong SK, Park JR, Kwon OS, Kim KT, Bae GY, Cha HJ. Induction of integrin beta3 by sustained ERK activity promotes the invasiveness of TGFbeta-induced mesenchymal tumor cells. Cancer Lett 2016;376:339–46. [DOI] [PubMed] [Google Scholar]
- 20.Li W, Liu C, Zhao C, Zhai L, Lv S. Downregulation of beta3 integrin by miR-30a-5p modulates cell adhesion and invasion by interrupting Erk/Ets1 network in triple-negative breast cancer. Int J Oncol 2016;48:1155–64. [DOI] [PubMed] [Google Scholar]
- 21.Liu Z, Han L, Dong Y, Tan Y, Li Y, Zhao M, Xie H, Ju H, Wang H, Zhao Y, Zheng Q, Wang Q, Su J, Fang C, Fu S, Jiang T, Liu J, Li X, Kang C, Ren H. EGFRvIII/integrin beta3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 2016;7:4680–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of beta1 integrin induces beta3 integrin switching, which drives breast cancer metastasis by TGF-beta. Mol Biol Cell 2013;24:3449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-beta Pathway. Cell Rep 2017;18:2480–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE, Balcioglu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, Meerman JH, van de Water B, Danen EH. beta1 integrin inhibition elicits a prometastatic switch through the TGFbeta-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal 2014;7:ra15. [DOI] [PubMed] [Google Scholar]
- 25.Novak M, Leonard MK, Yang XH, Kowluru A, Belkin AM, Kaetzel DM. Metastasis suppressor NME1 regulates melanoma cell morphology, self-adhesion and motility via induction of fibronectin expression. Exp Dermatol 2015;24:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Worth DC, Hodivala-Dilke K, Robinson SD, King SJ, Morton PE, Gertler FB, Humphries MJ, Parsons M. Alpha v beta3 integrin spatially regulates VASP and RIAM to control adhesion dynamics and migration. J Cell Biol 2010;189:369–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan MR, Hamidi H, Bass MD, Warwood S, Ballestrem C, Humphries MJ. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev Cell 2013;24:472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keely S, Glover LE, MacManus CF, Campbell EL, Scully MM, Furuta GT, Colgan SP. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J 2009;23:1338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parnell LD, Lindenbaum P, Shameer K, Dall’Olio GM, Swan DC, Jensen LJ, Cockell SJ, Pedersen BS, Mangan ME, Miller CA, Albert I. BioStar: an online question & answer resource for the bioinformatics community. PLoS Comput Biol 2011;7:e1002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Franceschi N, Hamidi H, Alanko J, Sahgal P, Ivaska J. Integrin traffic - the update. J Cell Sci 2015;128:839–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boissan M, Montagnac G, Shen Q, Griparic L, Guitton J, Romao M, Sauvonnet N, Lagache T, Lascu I, Raposo G, Desbourdes C, Schlattner U, Lacombe ML, Polo S, van der Bliek AM, Roux A, Chavrier P. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014;344:1510–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnaud-Dabernat S, Bourbon PM, Dierich A, Le Meur M, Daniel J-Y. Knockout mice as model systems for studying nm23/NDP kinase gene functions. Application to the nm23-M1 gene. J Bioenerg Biomembr 2003;35:19–30. [DOI] [PubMed] [Google Scholar]
- 33.Ma D, Xing Z, Liu B, Pedigo N, Zimmer S, Bai Z, Postel E, Kaetzel DM. NM23-H1 cleaves and represses transcriptional activity of nuclease-hypersensitive elements in the PDGF-A promoter. J Biol Chem 2002;277:1560–67. [DOI] [PubMed] [Google Scholar]
- 34.Fiore LS, Ganguly SS, Sledziona J, Cibull ML, Wang C, Richards DL, Neltner JM, Beach C, McCorkle JR, Kaetzel DM, Plattner R. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene 2014;33:4508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cervoni L, Egistelli L, Eufemi M, d’Abusco AS, Altieri F, Lascu I, Turano C, Giartosio A. DNA sequences acting as binding sites for NM23/NDPK proteins in melanoma M14 cells. J Cell Biochem 2006;98:421–28. [DOI] [PubMed] [Google Scholar]
- 36.Ma D, Xing Z, Liu B, Pedigo NG, Zimmer SG, Bai Z, Postel EH, Kaetzel DM. NM23-H1 and NM23-H2 repress transcriptional activities of nuclease-hypersensitive elements in the platelet-derived growth factor-A promoter. J Biol Chem 2002;277:1560–7. [DOI] [PubMed] [Google Scholar]
- 37.Postel E, Berberich SJ, Rooney JW, Kaetzel DM. Human NM23/nucleoside diphosphate kinase regulates gene expression through DNA binding to nuclease-hypersensitive elements. J Bioenerg Biomembr 2000;32:277–84. [DOI] [PubMed] [Google Scholar]
- 38.Knapp AM, Ramsey JE, Wang SX, Strauch AR, Kelm RJ Jr., Structure-function analysis of mouse Pur beta II. Conformation altering mutations disrupt single-stranded DNA and protein interactions crucial to smooth muscle alpha-actin gene repression. J Biol Chem 2007;282:35899–909. [DOI] [PubMed] [Google Scholar]
- 39.Choudhuri T, Verma SC, Lan K, Robertson ES. Expression of alpha V integrin is modulated by Epstein-Barr virus nuclear antigen 3C and the metastasis suppressor Nm23-H1 through interaction with the GATA-1 and Sp1 transcription factors. Virology 2006;351:58–72. [DOI] [PubMed] [Google Scholar]
- 40.Choudhuri T, Murakami M, Kaul R, Sahu SK, Mohanty S, Verma SC, Kumar P, Robertson ES. Nm23-H1 can induce cell cycle arrest and apoptosis in B cells. Cancer Biol Ther 2010;9:1065–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rayner K, Chen YX, Hibbert B, White D, Miller H, Postel EH, O’Brien ER. Discovery of NM23-H2 as an estrogen receptor beta-associated protein: role in estrogen-induced gene transcription and cell migration. J Steroid Biochem Mol Biol 2008;108:72–81. [DOI] [PubMed] [Google Scholar]
- 42.McCorkle JR, Leonard MK, Kraner SD, Blalock EM, Ma D, Zimmer SG, Kaetzel DM. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer Genomics Proteomics 2014;11:175–94. [PMC free article] [PubMed] [Google Scholar]
- 43.Leonard MK, McCorkle JR, Snyder DE, Novak M, Zhang Q, Shetty AC, Mahurkar AA, Kaetzel DM. Identification of a gene expression signature associated with the metastasis suppressor function of NME1: prognostic value in human melanoma. Lab Invest 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steeg PS, Palmieri D, Ouatas T, Salerno M. Histidine kinases and histidine phosphorylated proteins in mammalian cell biology, signal transduction and cancer. Cancer Lett 2003;190:1–12. [DOI] [PubMed] [Google Scholar]
- 45.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 1997;385:537–40. [DOI] [PubMed] [Google Scholar]
- 46.Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J Cell Biol 2002;159:1071–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huveneers S, Truong H, Fassler R, Sonnenberg A, Danen EH. Binding of soluble fibronectin to integrin alpha5 beta1 - link to focal adhesion redistribution and contractile shape. J Cell Sci 2008;121:2452–62. [DOI] [PubMed] [Google Scholar]
- 48.Mould AP, Craig SE, Byron SK, Humphries MJ, Jowitt TA. Disruption of integrin-fibronectin complexes by allosteric but not ligand-mimetic inhibitors. Biochem J 2014;464:301–13. [DOI] [PubMed] [Google Scholar]
- 49.Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, Avril MF, Ortiz Romero PL, Robert T, Balacescu O, Eggermont AM, Lenoir G, Sarasin A, Tursz T, van den Oord JJ, Spatz A. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst 2006;98:472–82. [DOI] [PubMed] [Google Scholar]
- 50.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF, Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness, Nature 385(1997) 537–540. [DOI] [PubMed] [Google Scholar]
- 51.Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A, The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading,organization of cell matrix adhesions, and fibronectin fi- brillogenesis, J. Cell Biol 159 (2002) 1071–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huveneers S, Truong H, Fassler R, Sonnenberg A, Danen EH, Binding of soluble fibronectin to integrin alpha5 beta1 - link to focal adhesion redistribution and contractile shape, J. Cell Sci 121 (2008) 2452–2462. [DOI] [PubMed] [Google Scholar]
- 53.Mould AP, Craig SE, Byron SK, Humphries MJ, Jowitt TA, Disruption of integrin-fibronectin complexes by allosteric but not ligand-mimetic inhibitors, Biochem. J 464 (2014) 301–313. [DOI] [PubMed] [Google Scholar]
- 54.Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, Avril MF, Ortiz Romero PL, Robert T, Balacescu O, Eggermont AM, Lenoir G, Sarasin A, Tursz T, van den Oord JJ, Spatz A, Gene expression profiling of primary cutaneous melanoma and clinical outcome, J. Natl. Cancer Inst 98 (2006) 472–482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.