

Abstract
Background
Gabapentin is commonly used to treat neuropathic pain (pain due to nerve damage). This review updates a review published in 2014, and previous reviews published in 2011, 2005 and 2000.
Objectives
To assess the analgesic efficacy and adverse effects of gabapentin in chronic neuropathic pain in adults.
Search methods
For this update we searched CENTRAL), MEDLINE, and Embase for randomised controlled trials from January 2014 to January 2017. We also searched the reference lists of retrieved studies and reviews, and online clinical trials registries.
Selection criteria
We included randomised, double‐blind trials of two weeks' duration or longer, comparing gabapentin (any route of administration) with placebo or another active treatment for neuropathic pain, with participant‐reported pain assessment.
Data collection and analysis
Two review authors independently extracted data and assessed trial quality and potential bias. Primary outcomes were participants with substantial pain relief (at least 50% pain relief over baseline or very much improved on Patient Global Impression of Change scale (PGIC)), or moderate pain relief (at least 30% pain relief over baseline or much or very much improved on PGIC). We performed a pooled analysis for any substantial or moderate benefit. Where pooled analysis was possible, we used dichotomous data to calculate risk ratio (RR) and number needed to treat for an additional beneficial outcome (NNT) or harmful outcome (NNH). We assessed the quality of the evidence using GRADE and created 'Summary of findings' tables.
Main results
We included four new studies (530 participants), and excluded three previously included studies (126 participants). In all, 37 studies provided information on 5914 participants. Most studies used oral gabapentin or gabapentin encarbil at doses of 1200 mg or more daily in different neuropathic pain conditions, predominantly postherpetic neuralgia and painful diabetic neuropathy. Study duration was typically four to 12 weeks. Not all studies reported important outcomes of interest. High risk of bias occurred mainly due to small size (especially in cross‐over studies), and handling of data after study withdrawal.
In postherpetic neuralgia, more participants (32%) had substantial benefit (at least 50% pain relief or PGIC very much improved) with gabapentin at 1200 mg daily or greater than with placebo (17%) (RR 1.8 (95% CI 1.5 to 2.1); NNT 6.7 (5.4 to 8.7); 8 studies, 2260 participants, moderate‐quality evidence). More participants (46%) had moderate benefit (at least 30% pain relief or PGIC much or very much improved) with gabapentin at 1200 mg daily or greater than with placebo (25%) (RR 1.8 (95% CI 1.6 to 2.0); NNT 4.8 (4.1 to 6.0); 8 studies, 2260 participants, moderate‐quality evidence).
In painful diabetic neuropathy, more participants (38%) had substantial benefit (at least 50% pain relief or PGIC very much improved) with gabapentin at 1200 mg daily or greater than with placebo (23%) (RR 1.7 (95% CI 1.4 to 2.0); NNT 6.6 (5.0 to 10); 6 studies, 1331 participants, moderate‐quality evidence). More participants (52%) had moderate benefit (at least 30% pain relief or PGIC much or very much improved) with gabapentin at 1200 mg daily or greater than with placebo (37%) (RR 1.4 (95% CI 1.3 to 1.6); NNT 6.6 (4.9 to 9.9); 7 studies, 1439 participants, moderate‐quality evidence).
For all conditions combined, adverse event withdrawals were more common with gabapentin (11%) than with placebo (8.2%) (RR 1.4 (95% CI 1.1 to 1.7); NNH 30 (20 to 65); 22 studies, 4346 participants, high‐quality evidence). Serious adverse events were no more common with gabapentin (3.2%) than with placebo (2.8%) (RR 1.2 (95% CI 0.8 to 1.7); 19 studies, 3948 participants, moderate‐quality evidence); there were eight deaths (very low‐quality evidence). Participants experiencing at least one adverse event were more common with gabapentin (63%) than with placebo (49%) (RR 1.3 (95% CI 1.2 to 1.4); NNH 7.5 (6.1 to 9.6); 18 studies, 4279 participants, moderate‐quality evidence). Individual adverse events occurred significantly more often with gabapentin. Participants taking gabapentin experienced dizziness (19%), somnolence (14%), peripheral oedema (7%), and gait disturbance (14%).
Authors' conclusions
Gabapentin at doses of 1800 mg to 3600 mg daily (1200 mg to 3600 mg gabapentin encarbil) can provide good levels of pain relief to some people with postherpetic neuralgia and peripheral diabetic neuropathy. Evidence for other types of neuropathic pain is very limited. The outcome of at least 50% pain intensity reduction is regarded as a useful outcome of treatment by patients, and the achievement of this degree of pain relief is associated with important beneficial effects on sleep interference, fatigue, and depression, as well as quality of life, function, and work. Around 3 or 4 out of 10 participants achieved this degree of pain relief with gabapentin, compared with 1 or 2 out of 10 for placebo. Over half of those treated with gabapentin will not have worthwhile pain relief but may experience adverse events. Conclusions have not changed since the previous update of this review.
Plain language summary
Gabapentin for chronic neuropathic pain in adults
Bottom line
There is moderate‐quality evidence that oral gabapentin at doses of 1200 mg daily or more has an important effect on pain in some people with moderate or severe neuropathic pain after shingles or due to diabetes.
Background
Neuropathic pain comes from damaged nerves. It is different from pain messages that are carried along healthy nerves from damaged tissue (for example, from a fall or cut, or arthritic knee). Neuropathic pain is often treated by different medicines (drugs) to those used for pain from damaged tissue, which we often think of as painkillers. Medicines that are sometimes used to treat depression or epilepsy can be effective in some people with neuropathic pain. One of these is gabapentin. Our definition of a good result was someone with a high level of pain relief and able to keep taking the medicine without side effects making them stop.
Study characteristics
In January 2017 we searched for clinical trials in which gabapentin was used to treat neuropathic pain in adults. We found 37 studies that satisfied the inclusion criteria, randomising 5914 participants to treatment with gabapentin, placebo, or other drugs. Studies lasted 4 to 12 weeks. Most studies reported beneficial outcomes that people with neuropathic pain think are important. Results were mainly in pain after shingles and pain resulting from nerve damage in diabetes.
Key results
In pain after shingles, 3 in 10 people had pain reduced by half or more with gabapentin and 2 in 10 with placebo. Pain was reduced by a third or more for 5 in 10 with gabapentin and 3 in 10 with placebo. In pain caused by diabetes, 4 in 10 people had pain reduced by half or more with gabapentin and 2 in 10 with placebo. Pain was reduced by a third or more for 5 in 10 with gabapentin and 4 in 10 with placebo. There was no reliable evidence for any other type of neuropathic pain.
Side effects were more common with gabapentin (6 in 10) than with placebo (5 in 10). Dizziness, sleepiness, water retention, and problems with walking each occurred in about 1 in 10 people who took gabapentin. Serious side effects were uncommon, and not different between gabapentin and placebo. Slightly more people taking gabapentin stopped taking it because of side effects.
Gabapentin is helpful for some people with chronic neuropathic pain. It is not possible to know beforehand who will benefit and who will not. Current knowledge suggests that a short trial is the best way of telling.
Quality of the evidence
The evidence was mostly of moderate quality. This means that the research provides a good indication of the likely effect. The likelihood that the effect will be substantially different is moderate.
Summary of findings
Background
This is an update of a Cochrane Review titled 'Gabapentin for chronic neuropathic pain and fibromyalgia in adults', published in 2014 (Moore 2014a). The review has now been split and this update will consider only neuropathic pain. A separate updated review of gabapentin for fibromyalgia has been published (Cooper 2017).
Earlier versions of this review include 'Gabapentin for chronic neuropathic pain and fibromyalgia in adults' (Moore 2011a), and 'Gabapentin for acute and chronic pain' (Wiffen 2005). That was itself split out of a review previously published in the Cochrane Library on 'Anticonvulsant drugs for acute and chronic pain' (Wiffen 2000), an update of yet an older systematic review (McQuay 1995).
At a meeting in Oxford in early 2009 with Cochrane's Editor‐in‐Chief, it was decided to create separate chronic pain and acute pain reviews from the then current review on acute and chronic pain together (Wiffen 2005). The meeting was in response to controversy in the USA over the effectiveness of gabapentin as an analgesic (Landefeld 2009), together with calls for the 2005 review to be updated with the inclusion of unpublished information made available through litigation (Vedula 2009). It was agreed to update the 2005 review by splitting the earlier one into two components: one review looking at the role of gabapentin in chronic neuropathic pain (including neuropathic pain of any cause, and fibromyalgia), and a second one to determine the effects of gabapentin in acute postoperative pain. Other reviews may examine gabapentin in chronic musculoskeletal pain. The unpublished data were included in the 2011 review on chronic neuropathic pain and fibromyalgia (Moore 2011a), and in established acute postoperative pain (Straube 2010).
This latest update is based on a template for drugs to treat neuropathic pain, using current standards for Cochrane Reviews, including assessment of the reliability of the evidence with GRADE, and based on criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Moore 2013a; Appendix 1).
Description of the condition
Neuropathic pain is a consequence of a pathological maladaptive response of the nervous system to 'damage' from a wide variety of potential causes (Colloca 2017). It is characterised by pain in the absence of an noxious stimulus, or where minor or moderate nociceptive stimuli evoke exaggerated levels of pain. Neuropathic pain may be spontaneous (continuous or paroxysmal) in its temporal characteristics or be evoked by sensory stimuli (dynamic mechanical allodynia where pain is evoked by light touch of the skin).
Neuropathic pain is heterogeneous in etiology, pathophysiology, and clinical appearance. The 2011 International Association for the Study of Pain definition of neuropathic pain is "pain caused by a lesion or disease of the somatosensory system" (Jensen 2011), based on a definition agreed at an earlier consensus meeting (Treede 2008). Neuropathic pain is associated with a variety of sensory loss (numbness) and sensory gain (allodynia) clinical phenomena, the exact patterns of which vary between people and disease, perhaps reflecting different pain mechanisms operating in an individual person and, therefore, potentially predictive of response to treatment (Demant 2014; Helfert 2015; von Hehn 2012). A new approach of subgrouping people with peripheral neuropathic pain of different etiologies according to intrinsic sensory profiles has generated three profiles that may be related to pathophysiological mechanisms and may be useful in clinical trial design to enrich the study population for treatment responders (Baron 2017).
Pre‐clinical research hypothesises a bewildering array of possible pain mechanisms that may operate in people with neuropathic pain, which largely reflect pathophysiological responses in both the central and peripheral nervous systems, including neuronal interactions with immune cells (Baron 2012; Calvo 2012; von Hehn 2012). Overall, the treatment gains in neuropathic pain, to even the most effective of available drugs, are modest (Finnerup 2015; Moore 2013b), and a robust classification of neuropathic pain is not yet available (Finnerup 2013).
Neuropathic pain is usually classified according to the cause of nerve injury. There may be many causes, but some common causes of neuropathic pain include diabetes (painful diabetic neuropathy (PDN)), shingles (postherpetic neuralgia (PHN)), amputation (stump and phantom limb pain), neuropathic pain after surgery or trauma, stroke or spinal cord injury, trigeminal neuralgia, and HIV infection. Sometimes the cause is unknown.
Many people with neuropathic pain conditions are significantly disabled with moderate or severe pain for many years. Chronic pain conditions comprised five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life and employment, and increased healthcare costs (Moore 2014a). A US study found the healthcare costs were threefold higher for people with neuropathic pain than matched control subjects (Berger 2004). A UK study and a German study showed a two‐ to threefold higher level of use of healthcare services in people with neuropathic pain than those without (Berger 2012; Berger 2009). For PHN, for example, studies demonstrate large loss of quality of life and substantial costs (Scott 2006; Van Hoek 2009).
In systematic reviews, the overall prevalence of neuropathic pain in the general population is reported to be between 7% and 10% (Van Hecke 2014), and about 7% in a systematic review of studies published since 2000 (Moore 2014a). In individual countries, prevalence rates have been reported as 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), and up to 8% in the UK (Torrance 2006). Some forms of neuropathic pain, such as PDN and post‐surgical chronic pain (which is often neuropathic in origin), are increasing (Hall 2008). The prevalence of PHN is likely to fall if vaccination against the herpes virus becomes widespread.
Estimates of incidence vary between individual studies for particular origins of neuropathic pain, often because of small numbers of cases. In primary care in the UK, between 2002 and 2005, the incidences (per 100,000 person‐years' observation) were 28 (95% confidence interval (CI), 27 to 30) for PHN, 27 (95% CI, 26 to 29) for trigeminal neuralgia, 0.8 (95% CI, 0.6 to 1.1) for phantom limb pain, and 21 (95% CI, 20 to 22) for PDN (Hall 2008). Other studies have estimated an incidence of 4 in 100,000 per year for trigeminal neuralgia (Katusic 1991; Rappaport 1994), and 12.6 per 100,000 person‐years for trigeminal neuralgia and 3.9 per 100,000 person‐years for PHN in a study of facial pain in the Netherlands (Koopman 2009). One systematic review of chronic pain demonstrated that some neuropathic pain conditions, such as PDN, can be more common than other neuropathic pain conditions, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007). It is also the case that pains not classified as neuropathic can have neuropathic features. In a community study of recent joint pain, features of neuropathic pain were common and were present in over half of those reporting pain of at least moderate severity (Soni 2013).
Neuropathic pain is difficult to treat effectively, with only a minority of people experiencing a clinically relevant benefit from any one intervention (Kalso 2013; Moore 2013b). A multidisciplinary approach is now advocated, combining pharmacological interventions with physical or cognitive (or both) interventions. The evidence for more invasive interventional therapies such as neural blockade or intrathecal medication is very weak, or non‐existent (Dworkin 2013). Conventional analgesics such as paracetamol (acetaminophen) and nonsteroidal anti‐inflammatory drugs (NSAID) are not thought to be effective, but without evidence to support or refute that view (Moore 2015a; Wiffen 2016). Some people may derive some benefit from a topical lidocaine patch or low‐concentration topical capsaicin, although evidence about benefits is uncertain (Derry 2012; Derry 2014). High‐concentration topical capsaicin may benefit some people with PHN (Derry 2017). Treatment is often by so‐called 'unconventional analgesics' (pain modulators) such as antidepressants (duloxetine and amitriptyline; Lunn 2014; Moore 2014b; Moore 2015b; Sultan 2008), or antiepileptics (gabapentin or pregabalin; Moore 2009; Moore 2014b; Wiffen 2013). Evidence for efficacy of opioids is unconvincing (Derry 2016; Gaskell 2016; Stannard 2016; Wiffen 2015).
The proportion of people who achieve worthwhile pain relief (typically at least 50% pain intensity reduction; Moore 2014c) is small, generally only 10% to 25% more than with placebo, with numbers needed to treat for an additional beneficial outcome (NNT) usually between 4 and 10 (Kalso 2013; Moore 2013b). Neuropathic pain is not particularly different from other chronic pain conditions in that only a small proportion of trial participants have a good response to treatment (Moore 2013b).
The current National Institute for Health and Care Excellence (NICE) guidance for the pharmacological management of neuropathic pain suggests offering a choice of amitriptyline (Moore 2012b), duloxetine (Lunn 2014), gabapentin, or pregabalin (Moore 2009) as initial treatment for neuropathic pain (with the exception of trigeminal neuralgia), with switching if the first, second, or third drugs tried are not effective or not tolerated (NICE 2013). This concurs with other recent guidance (Finnerup 2015).
Description of the intervention
Gabapentin is licensed for the treatment of peripheral and central neuropathic pain in adults in the UK at doses up to 3.6 grams (3600 mg) daily. It is given orally, usually as tablets or capsules, but sometimes as an oral solution (50 mg/ml). Guidance suggests that gabapentin treatment can be started at a dose of 300 mg per day for treating neuropathic pain. Based on individual patient response and tolerability, the dosage may be increased by 300 mg per day until pain relief is experienced or adverse effects make taking the drug intolerable (EMC 2017). US marketing approval for gabapentin was granted in 2002 for postherpetic neuralgia; in Europe, the label was changed to include peripheral neuropathic pain in 2006. Gabapentin has the trade name NeurontinTM, and is also available as generic products in some parts of the world.
Gabapentin has a half‐life of five to seven hours. It is absorbed through a saturable transport system, so that absorption is not linear, and the transporter is found only in the proximal small bowel. This means that the drug needs to be administered at least three times daily, and may result in plasma trough levels. Two new formulations have attempted to improve the availability of the drug. The first is an extended release, gastro‐retentive formulation, designed to provide continuous delivery at the optimal site of absorption over 8 to 10 hours (Sang 2013). The second uses an extended‐release prodrug (gabapentin encarbil) that is absorbed through a high capacity transport system found throughout the intestine, and then undergoes rapid hydrolysis to gabapentin. It is claimed to provide sustained, dose‐proportional gabapentin exposure (Backonja 2011), and can be administered twice daily.
Gabapentin can also be formulated as an aqueous solution for injection. This formulation is not available commercially or licensed for treatment of any type of neuropathic pain or fibromyalgia.
Gabapentin misuse has been reported, and the consequences documented and systematically reviewed (Evoy 2017; Quintero 2017).
How the intervention might work
Gabapentin's mechanism of action is primarily attributed to its effect on calcium channels located throughout the peripheral and central nervous systems, which modify the release of neurotransmitters and reduce excitability of nerve cells (Boyle 2014; Chang 2014). This mode of action confers antiepileptic, analgesic, and sedative effects. Research also indicates that gabapentin acts by blocking new synapse formation (Eroglu 2009).
Why it is important to do this review
Some, but not all, antiepileptics can reduce neuropathic pain (Wiffen 2010). Gabapentin is an antiepileptic widely prescribed for neuropathic pain, and it is common practice in some countries to aim for the maximum tolerated dose. There is growing controversy over whether this practice is justified by experimental evidence from double‐blind randomised trials. Guidance on prescribing typically puts gabapentin amongst the first‐line agents (Finnerup 2015; NICE 2013). Despite this guidance based on good evidence, prescribing for neuropathic pain often involves paracetamol or paracetamol combined with opioids (Hall 2013), for which there is no evidence of efficacy (Wiffen 2016).
The standards used to assess evidence in chronic pain trials have evolved substantially in recent years, with particular attention being paid to trial duration, withdrawals, and statistical imputation following withdrawal, all of which can substantially alter estimates of efficacy (Appendix 1). The most important change is the move from using mean pain scores, or mean change in pain scores, to the number of people who have a large decrease in pain (by at least 50%) and who continue in treatment, ideally in trials of 8 to 12 weeks' duration or longer. Pain intensity reduction of 50% or more correlates with improvements in co‐morbid symptoms, function, and quality of life. These standards are set out in the PaPaS Author and Referee Guidance for pain studies of Cochrane Pain, Palliative and Supportive Care (PaPaS 2012).
This Cochrane Review assesses the evidence using methods that make both statistical and clinical sense, and uses developing criteria for what constitutes reliable evidence in chronic pain (Moore 2010a). Trials included and analysed meet a minimum of reporting quality (blinding, randomisation), validity (duration, dose and timing, diagnosis, outcomes, etc), and size (ideally at least 500 participants in a comparison in which the NNT is 4 or above; Moore 1998). This approach sets high standards for the demonstration of efficacy and marks a departure from how reviews were conducted previously.
Objectives
To assess the analgesic efficacy and adverse effects of gabapentin in chronic neuropathic pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with double‐blind (participant and observers) assessment of participant‐reported outcomes, following two weeks of treatment or longer, although the emphasis of the review was on studies of eight weeks or longer. We required full journal publication, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials, and abstracts with sufficient data for analysis.
We did not include short abstracts (usually meeting reports with inadequate or no reporting of data). We excluded studies of experimental pain, case reports, and clinical observations.
Types of participants
We included adult participants aged 18 years and above, with one or more chronic neuropathic pain condition including (but not limited to):
cancer‐related neuropathy;
central neuropathic pain;
complex regional pain syndrome (CRPS) Type II;
HIV neuropathy;
painful diabetic neuropathy;
phantom limb pain;
postherpetic neuralgia;
postoperative or traumatic neuropathic pain;
spinal cord injury;
trigeminal neuralgia.
Where we included studies with more than one type of neuropathic pain, we analysed results according to the primary condition if identifiable.
Types of interventions
Gabapentin in any dose, by any route, administered for the relief of neuropathic pain and compared to placebo or any other active comparator.
Types of outcome measures
We anticipated that studies would use a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity or pain relief, or both. We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These were defined as:
at least 30% pain relief over baseline (moderate);
at least 50% pain relief over baseline (substantial);
much or very much improved on Patient Global Impression of Change scale (PGIC; moderate);
very much improved on PGIC (substantial).
These outcomes concentrate on dichotomous outcomes where pain responses do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50% pain intensity reduction, and ideally having no worse than mild pain (Moore 2013c; O'Brien 2010).
Primary outcomes
Participant‐reported pain intensity reduction of 30% or greater
Participant‐reported pain intensity reduction of 50% or greater
Patient‐reported global impression of clinical change (PGIC) much or very much improved
Patient‐reported global impression of clinical change (PGIC) very much improved
Secondary outcomes
Any pain‐related outcome indicating some improvement.
Withdrawals due to lack of efficacy, adverse events, and for any cause.
Participants experiencing any adverse event.
Participants experiencing any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, is an 'important medical event' that may jeopardise the patient, or may require an intervention to prevent one of the above characteristics or consequences.
Specific adverse events, particularly somnolence and dizziness.
Search methods for identification of studies
Electronic searches
For this update we searched the following databases, without language restrictions:
Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO), 1 January 2014 to 16 January 2017;
MEDLINE via Ovid, 1 January 2014 to 16 January 2017;
Embase via Ovid, 1 January 2014 to 16 January 2017.
See Appendix 2 for the CENTRAL search strategy, Appendix 3 for the MEDLINE search strategy, and Appendix 4 for the Embase search strategy.
Searching other resources
We reviewed the bibliographies of any RCTs identified and review articles, and searched clinical trial databases (ClinicalTrials.gov (ClinicalTrials.gov) and World Health Organization (WHO) International Clinical Trials Registry Platform (ICTTRP) (apps.who.int/trialsearch/)) to identify additional published or unpublished data. We did not contact investigators or study sponsors.
Data collection and analysis
We performed separate efficacy analyses according to particular neuropathic pain conditions, and combined different neuropathic pain conditions in analyses for adverse events and withdrawals only.
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria, and we obtained full copies of the remaining studies. Two review authors made the decisions. Two review authors (RAM, SD) then read these studies independently and reached agreement by discussion. We did not anonymise the studies in any way before assessment. We have provided a PRISMA flow chart to illustrate the flow of studies (Moher 2009) (Figure 1).
1.
Study flow diagram
Data extraction and management
Three review authors (RAM, PW, SD) extracted data independently, using a standard data extraction form, and agreed data before entry into Review Manager (RevMan) 5 (RevMan 2014) or any other analysis method. We included information about the pain condition and number of participants treated, drug and dosing regimen, study design, study duration and follow‐up, analgesic outcome measures and results, withdrawals and adverse events (participants experiencing any adverse event, particular adverse events, or a serious adverse event).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind as a minimum.
Two review authors (SD, PW) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 8, Higgins 2011), and adapted from those used by Cochrane Pregnancy and Childbirth, with any disagreements resolved by discussion. We assessed the following for each study:
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process: random number table or computer random‐number generator); unclear risk of bias (when the method used to generate the sequence was not clearly stated). We excluded studies at a high risk of bias that used a non‐random process (odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (telephone or central randomisation; consecutively‐numbered, sealed, opaque envelopes); unclear risk of bias (when method not clearly stated). We excluded studies that did not conceal allocation and were therefore at a high risk of bias (open list).
Blinding of participants and personnel (checking for possible performance bias), and blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study personnel and participants (all outcomes were self‐assessed) from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, for example, identical tablets, matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies at a high risk of bias that were not double‐blind.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (fewer than 10% of participants did not complete the study or used 'baseline observation carried forward' (BOCF) analysis, or both); unclear risk of bias (used 'last observation carried forward' (LOCF) analysis); or high risk of bias (used 'completer' analysis).
Size of study (checking for possible biases confounded by small size (Dechartres 2013; Dechartres 2014; Moore 1998; Nüesch 2010; Thorlund 2011)). We assessed studies as being at low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); or high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
We calculated the number needed to treat for an additional beneficial outcome (NNT) as the reciprocal of the absolute risk reduction (ARR) (McQuay 1998). For unwanted effects, the NNT becomes the number needed to treat for an additional harmful outcome (NNH) and was calculated in the same manner. We used dichotomous data to calculate risk ratio (RR) with 95% confidence intervals (CI) using a fixed‐effect model unless significant statistical heterogeneity was found (see below). We did not use continuous data in analyses.
Unit of analysis issues
The unit of analysis was the individual participant. For cross‐over studies we planned to use first period data where possible, but otherwise to use available data and consider any potential bias that this study design presented.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis where the ITT population consisted of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one post‐baseline assessment. We assigned zero improvement (baseline observation carried forward (BOCF)) to missing participants wherever possible.
We paid particular attention to methods used for imputation of missing data due to withdrawals for adverse events and lack of efficacy.
Assessment of heterogeneity
We dealt with clinical heterogeneity by combining studies that examined similar conditions. We assessed statistical heterogeneity visually (L'Abbé 1987) and with the use of the I2 statistic (Higgins 2003). When the I² value was greater than 50%, we considered possible reasons for this.
Assessment of reporting biases
The aim of this review was to use dichotomous outcomes of known utility and of value to people with neuropathic pain (Hoffman 2010; Moore 2010a; Moore 2010b; Moore 2010c; Moore 2014c). The review did not depend on what the authors of the original studies chose to report or not, and studies that did not report dichotomous results for an outcome did not contribute to pooled analyses for that outcome. We extracted and used continuous data, which probably reflect efficacy and utility poorly, for illustrative purposes only.
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean a NNT of 10 or higher in this condition; Moore 2008).
We looked for effects of possible enrichment, either complete or partial, in enrolment of participants into the studies. Enrichment typically means including participants known to respond to a therapy, and excluding those known not to respond, or to suffer unacceptable adverse effects, though for gabapentin no significant effects have been shown from partial enrichment (Straube 2008). Enriched enrolment randomised withdrawal studies, known to produce higher estimates of efficacy, would not be pooled (McQuay 2008).
Data synthesis
We used a fixed‐effect model for meta‐analysis, unless there was significant clinical heterogeneity and it was still considered appropriate to combine studies. In such cases we would use a random‐effects model.
Quality of evidence
Quality of the evidence
We used the GRADE system to assess the quality of the evidence related to the key outcomes listed in Types of outcome measures, as appropriate (Appendix 6). Two review authors (RAM, SD) independently rated the quality of the evidence for each outcome.
We paid particular attention to inconsistency, where point estimates varied widely across studies or confidence intervals (CIs) of studies showed minimal or no overlap (Guyatt 2011), and potential for publication bias, based on the amount of unpublished data required to make the result clinically irrelevant (Moore 2008).
In addition, there may be circumstances where the overall rating for a particular outcome needs to be adjusted as recommended by GRADE guidelines (Guyatt 2013a). For example, where there were so few data that the results were highly susceptible to the random play of chance, or if a study used last observation carried forward (LOCF) imputation in circumstances where there were substantial differences in adverse event withdrawals, one would have no confidence in the result, and would need to downgrade the quality of the evidence by three levels, to very low quality. In circumstances where there were no data reported for an outcome, we would have reported the level of evidence as very low quality (Guyatt 2013b).
In addition, we are aware that many Cochrane Reviews are based largely or wholly on small underpowered studies, and the danger of making conclusive assessments of evidence based on inadequate information (AlBalawi 2013; Brok 2009; Roberts 2015; Turner 2013).
'Summary of findings' table
We have included a 'Summary of findings' table as set out in the PaPaS author guide (PaPaS 2012), and recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 11, Schünemann 2011a). The table includes, where possible, outcomes equivalent to moderate or substantial benefit of at least 30% and at least 50% pain intensity reduction, PGIC (possibly at least substantial improvement and at least moderate improvement) (Dworkin 2008), withdrawals due to lack of efficacy, withdrawals due to adverse events, serious adverse events, and death (a particular serious adverse event).
For the 'Summary of findings' table we used the following descriptors for levels of evidence (EPOC 2015):
High: this research provides a very good indication of the likely effect. The likelihood that the effect will be substantially different† is low.
Moderate: this research provides a good indication of the likely effect. The likelihood that the effect will be substantially different† is moderate.
Low: this research provides some indication of the likely effect. However, the likelihood that it will be substantially different† is high.
Very low: this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different† is very high.
† Substantially different: a large enough difference that it might affect a decision.
Subgroup analysis and investigation of heterogeneity
We planned for all analyses to be according to individual painful condition, because placebo response rates with the same outcome can vary between conditions, as can the drug‐specific effects (Moore 2009). We also planned subgroup analysis according to dose of gabapentin, and duration of study if sufficient data were available.
Sensitivity analysis
In the 2014 review we considered a sensitivity analysis for formulation of gabapentin (standard, gastroretentive, slow‐release), but there were insufficient data for meaningful analysis, and there were no additional data for these formulations. We planned no other sensitivity analyses because the evidence base was known to be too small to allow reliable analysis. Performing analyses that might inform on which patients were most likely to benefit from gabapentin treatment would require efficacy data together with detailed assessment of the exact nature and type of neuropathic pain at the individual participant level (Tölle 2013). No such data were expected to be available.
Results
Description of studies
Results of the search
In the previous version of this review we considered 36 studies in 37 reports examining oral gabapentin, involving 5483 participants with chronic neuropathic pain in various different conditions, mainly PHN, PDN, or mixed neuropathic pain.
Updated database searches from January 2014 to 17 January 2017 identified 107 potentially relevant reports in CENTRAL, 237 in MEDLINE, and 484 in Embase. No additional studies were identified in clinical trials registries or reference lists of included studies or reviews.
After de‐duplication and screening of titles and abstracts, we obtained the full text of seven reports. Of these, we included three new studies, with 468 participants (Atkinson 2016; Cohen 2015; Gong 2008). We also identified one report that was a secondary analysis of a study that was already included (Calkins 2016, see Zhang 2013), and two reports of pooled analyses of two studies that were already included (Freeman 2015 and Metha 2016, see Sang 2013; Wallace 2010).
One study that was previously ongoing has now completed. We could not identify a published article for this study, but we did find a synopsis with some results on the pharmaceutical company's website (NCT00904202). The study satisfied our inclusion criteria and was therefore included in this review (62 participants). We could not find any updated information on the remaining three ongoing studies (Fleckstein 2009; IRCT201212019014N14; NCT00674687).
We reassessed and excluded one study that had been included in the earlier review (Ho 2009). This small (18 participants) cross‐over study in small fibre sensory neuropathy used a one‐week titration period, followed by one week at the maximum dose and one week of wash‐out, then crossed over to repeat the sequence with the other treatment. We excluded it because of the very short treatment periods (only one week at a stable dose), there was some uncertainty about the dosing schedule (although the maximum dose was clearly stated), and participants could take additional gabapentin to a maximum of 1200 mg daily if they required rescue medication and paracetamol was inadequate. There was no information about the use of this additional gabapentin, or how data from participants using it were analysed. Two further studies from the previous review are in conditions not now considered neuropathic pain (Kimos 2007; Van de Vusse 2004).
Figure 1 illustrates the flow of studies for this update.
Included studies
This update therefore includes four additional studies involving 530 participants, bringing the total for the review to 37 studies involving 5914 participants, although not all of the participants took all the study medication, and not all the participants were included in results.
The majority of studies involved participants with PHN and PDN. Other neuropathic pain conditions studied were spinal cord injury, phantom limb pain, cancer, nerve injury pain, CRPS, HIV, and radicular leg pain. Four studies enrolled participants with a mixture of types of neuropathic pain.
Four studies (Irving 2009; Sandercock 2012; Sang 2013; Wallace 2010) used a gastroretentive, extended‐release formulation of gabapentin, and four others (Backonja 2011; Harden 2013; Rauck 2013a; Zhang 2013) used an extended‐release prodrug, gabapentin encarbil.
Twenty‐five studies had a parallel‐group design and 12 had a cross‐over design (Bone 2002; Gilron 2005; Gilron 2009; Gordh 2008; Gorson 1999; Harden 2013; Levendoglu 2004; Morello 1999; Rao 2007; Rintala 2007; Smith 2005; Tai 2002). We used whatever data were available from the cross‐over studies, including first period or multiple periods, though there are major issues with what constitutes the ITT denominator where there are significant withdrawals.
Parallel‐group trials were larger than cross‐over trials. The 25 parallel‐group studies involved 5298 participants (mean 204, median 162 participants, range 26 to 452), while the 12 cross‐over studies involved 621 participants (mean 48, median 40 participants, range 14 to 120). Not all studies reported the results on an ITT basis, and this was particularly the case for cross‐over studies with multiple comparisons.
Twenty‐eight studies either described enrolment processes that were not enriched, or had no exclusion criteria that would raise the possibility of enrichment (Straube 2008). Seven studies were partially enriched (Caraceni 2004; Irving 2009; Rice 2001; Sang 2013; Serpell 2002) or excluded participants with previous inadequate response to treatment with gabapentin or pregabalin as an exclusion criterion, which may have led to enrichment (Cohen 2015; Wallace 2010). Two studies enriched for tolerance to gabapentin, but not response (Backonja 2011; Harden 2013), which is probably equivalent to partial enrichment. Participants in these two studies were treated with gabapentin encarbil, a prodrug of gabapentin; these are analysed alongside the other studies, but with a view to sensitivity analysis.
Three studies reported using baseline observation carried forward (BOCF) imputation for the primary outcome (Sandercock 2012; Sang 2013; Wallace 2010), sometimes alongside last observation carried forward (LOCF) analyses, and one reported using BOCF imputation for the responder analyses (Rauck 2013b). Thirty‐one studies either made no mention of an imputation method for missing data (19) or declared use of LOCF (12). Others performed analyses on completers only (Atkinson 2016 (for responder analysis); Rintala 2007), and one presented results without imputation (Rao 2007).
Details of all eligible studies are given in the 'Characteristics of included studies' table.
Excluded studies
We excluded 25 studies from this review. The earlier review excluded 21 studies because they were open‐label studies, were studies in chronic conditions not considered for this review, investigated related acute conditions or preventive strategies, or did not have an appropriate comparator.
We excluded one new study because it did not have an appropriate comparator and did not appear to be blinded (Ding 2014). We reassessed and excluded three previously included studies, one because of its short duration, use of gabapentin as rescue medication, and unclear methods of analysis (Ho 2009), and two because definitions of chronic neuropathic pain had changed, and these two were now outside the current definitions (Kimos 2007; Van de Vusse 2004).
Risk of bias in included studies
The risk of bias assessments identified that adequate sequence generation and allocation concealment were often inadequately reported. Additional risk of bias also derived from studies being small, and rarely describing how efficacy data were handled on withdrawal (Figure 2).
2.
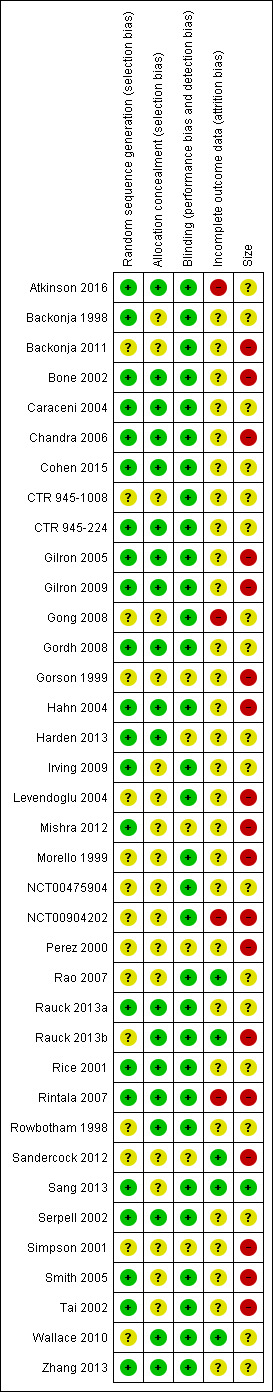
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
All studies were described as randomised, but only 22 adequately described the method used to generate the random sequence. Only 19 adequately described how the sequence was concealed. We judged studies with inadequate descriptions at unclear risk, although in most cases the methods were probably adequate but not reported.
Blinding
All studies were described as double‐blind (participants, who also assessed outcomes, and personnel), but six did not adequately describe the method used to achieve and maintain blinding (Gorson 1999; Harden 2013; Mishra 2012; Perez 2000; Sandercock 2012; Simpson 2001). We judged studies with inadequate descriptions at unclear risk, although in most cases the methods were probably adequate but not reported.
Incomplete outcome data
We judged four studies at high risk of bias because they reported only on participants who completed treatment phases (Atkinson 2016; Rintala 2007), did not report groups or reasons for withdrawal and used LOCF imputation where there was 7% attrition (Gong 2008), or did not report all expected outcomes in the results synopsis and used LOCF imputation (NCT00904202). We judged five studies at low risk of bias for this domain (Rao 2007; Rauck 2013b; Sandercock 2012; Sang 2013; Wallace 2010), and the remaining 28 at unclear risk, mainly because they used LOCF imputation for early withdrawals.
Other potential sources of bias
We judged one study to be at low risk of bias due to study size (more than 200 participants each treatment arm; Sang 2013), 18 at unknown risk, with between 50 and 200 participants per treatment arm, and 18 of the included studies at high risk of bias due to study size smaller than 50 participants per treatment arm.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. Gabapentin compared with placebo for postherpetic neuralgia: efficacy.
| Gabapentin compared with placebo for postherpetic neuralgia: efficacy | ||||||
|
Patient or population: adults with postherpetic neuralgia Settings: community Intervention: gabapentin ≥ 1800 mg daily or gabapentin encarbil 1200 mg daily Comparison: placebo | ||||||
| Outcome | Probable outcome with gabapentin | Probable outcome with placebo |
RR and NNT (95% CI) |
Number of studies, participants | Certainty of the evidence (GRADE) | Comments |
| At least 50% reduction in pain or equivalent | 330 per 1000 | 190 per 1000 | RR 1.7 (1.4 to 2.0) NNT 6.9 (5.5 to 9.4) |
7 studies 2031 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| IMMPACT definition ‐ any substantial pain benefit | 320 per 1000 | 170 per 1000 | RR 1.8 (1.5 to 2.1) NNT 6.7 (5.4 to 8.7) |
8 studies 2260 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| Patient Global Impression of Change much or very much improved | 390 per 1000 | 290 per 1000 | RR1.3 (1.2 to 1.5) NNT 9.7 (6.9 to 16) |
7 studies 2013 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| IMMPACT definition ‐ any at least moderate pain benefit (includes Gong 2008 at 25% pain relief) |
46 per 1000 | 25 per 1000 | RR 1.8 (1.6 to 2.0) NNT 4.8 (4.1 to 6.0) |
8 studies 2260 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| CI: confidence interval; IMMPACT: Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials; NNT: number needed to treat for an additional beneficial outcome; RR: risk ratio | ||||||
| Descriptors for levels of evidence (EPOC 2015):
High quality: this research provides a very good indication of the likely effect. The likelihood that the effect will be substantially different† is low.
Moderate quality: this research provides a good indication of the likely effect. The likelihood that the effect will be substantially different† is moderate.
Low quality: this research provides some indication of the likely effect. However, the likelihood that it will be substantially different† is high.
Very low quality: this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different† is very high. †Substantially different: a large enough difference that it might affect a decision. | ||||||
Summary of findings 2. Gabapentin compared with placebo for peripheral diabetic neuropathy: efficacy.
| Gabapentin compared with placebo for peripheral diabetic neuropathy: efficacy | ||||||
|
Patient or population: adults with peripheral diabetic neuropathy Settings: community Intervention: ≥ 1800 mg daily or gabapentin encarbil 1200 mg daily Comparison: placebo | ||||||
| Outcome | Probable outcome with gabapentin | Probable outcome with placebo |
RR and NNT (95% CI) |
Number of studies, participants | Certainty of the evidence (GRADE) | Comments |
| At least 50% pain intensity reduction | 380 per 1000 | 230 per 1000 | RR 1.7 (1.4 to 2.0) NNT 6.6 (5.0 to 9.7) |
6 studies 1331 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| Any definition of substantial benefit (at least 50% pain intensity reduction or PGIC very much improved) | 380 per 1000 | 230 per 1000 | RR 1.7 (1.4 to 2.0) NNT 6.6 (5.0 to 9.7) |
6 studies 1331 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| PGIC much or very much improved | 500 per 1000 | 300 per 1000 | RR 1.7 (1.4 to 2.0) NNT 4.9 (3.6 to 7.6) |
5 studies 695 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| Any definition of moderate benefit (at least 30% pain intensity reduction or PGIC much or very much improved) | 520 per 1000 | 370 per 1000 | RR 1.4 (1.3 to 1.6) NNT 6.6 (4.9 to 9.9) |
7 studies 1439 participants |
Moderate | Downgraded because of issues around dosing, formulation, and imputation |
| CI: confidence interval; IMMPACT: Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials; NNT: number needed to treat for an additional beneficial outcome; RR: risk ratio | ||||||
| Descriptors for levels of evidence (EPOC 2015):
High quality: this research provides a very good indication of the likely effect. The likelihood that the effect will be substantially different† is low.
Moderate quality: this research provides a good indication of the likely effect. The likelihood that the effect will be substantially different† is moderate.
Low quality: this research provides some indication of the likely effect. However, the likelihood that it will be substantially different† is high.
Very low quality: this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different† is very high. † Substantially different: a large enough difference that it might affect a decision. | ||||||
Summary of findings 3. Gabapentin compared with placebo for neuropathic pain (all conditions pooled): adverse events and withdrawals.
| Gabapentin compared with placebo for neuropathic pain (all conditions pooled): adverse events and withdrawals | ||||||
|
Patient or population: adults with neuropathic pain Settings: community Intervention: gabapentin 1800 mg to 3600 mg daily (gabapentin encarbil 1200 mg to 3600 mg daily) Comparison: placebo | ||||||
| Outcome | Probable outcome with gabapentin | Probable outcome with placebo |
RR and NNH (95% CI) |
Number of studies, participants | Certainty of the evidence (GRADE) | Comments |
| Participants experiencing at least one adverse event | 630 per 1000 | 490 per 1000 | RR 1.3 (1.2 to 1.4) NNH 7.5 (6.1 to 9.6) |
18 studies 4279 participants |
Moderate | Many events. Unlikely new research would change this finding |
| Adverse event withdrawals | 110 in 1000 | 82 in 1000 | RR 1.4 (1.1 to 1.7) NNH 30 (20 to 66) |
22 studies 4346 participants |
High | Unlikely new research would change this finding |
| Serious adverse events | 32 in 1000 | 28 in 1000 | RR 1.2 (0.83 to 1.7) NNH not calculated |
19 studies 3948 participants |
Moderate | Small number of events but no suggestion of difference |
| Death | 3 in max 3603 exposed | 5 in max 2377 exposed | Not calculated | Not calculated | Very low | Few events, relatively short duration for drug possibly taken over periods of years |
| CI: confidence interval; NNH: number needed to treat for an additional harmful outcome; RR: risk ratio | ||||||
| Descriptors for levels of evidence (EPOC 2015):
High quality: this research provides a very good indication of the likely effect. The likelihood that the effect will be substantially different† is low.
Moderate quality: this research provides a good indication of the likely effect. The likelihood that the effect will be substantially different† is moderate.
Low quality: this research provides some indication of the likely effect. However, the likelihood that it will be substantially different† is high.
Very low quality: this research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different† is very high. † Substantially different: a large enough difference that it might affect a decision. | ||||||
Appendix 7 contains details of withdrawals, efficacy, and adverse events in the individual studies.
Efficacy
We report efficacy results where data were available, or where there was sufficient information to justify analysis, defined as information from 200 participants or more, ideally from at least two studies.
Analyses 1.1 to 1.5 show results for the following outcomes: at least 50% reduction in pain (Analysis 1.1; Figure 3); Patient Global Impression of Change (PGIC) very much improved (Analysis 1.2; Figure 4); PGIC much or very much improved (Analysis 1.3; Figure 5); IMMPACT outcome of substantial improvement in pain (Analysis 1.4; Figure 6); IMMPACT outcome of at least moderate improvement in pain (Analysis 1.5; Figure 7).
1.1. Analysis.
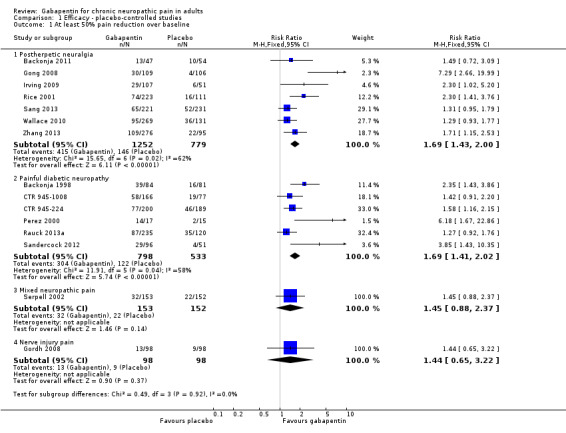
Comparison 1 Efficacy ‐ placebo‐controlled studies, Outcome 1 At least 50% pain reduction over baseline.
3.
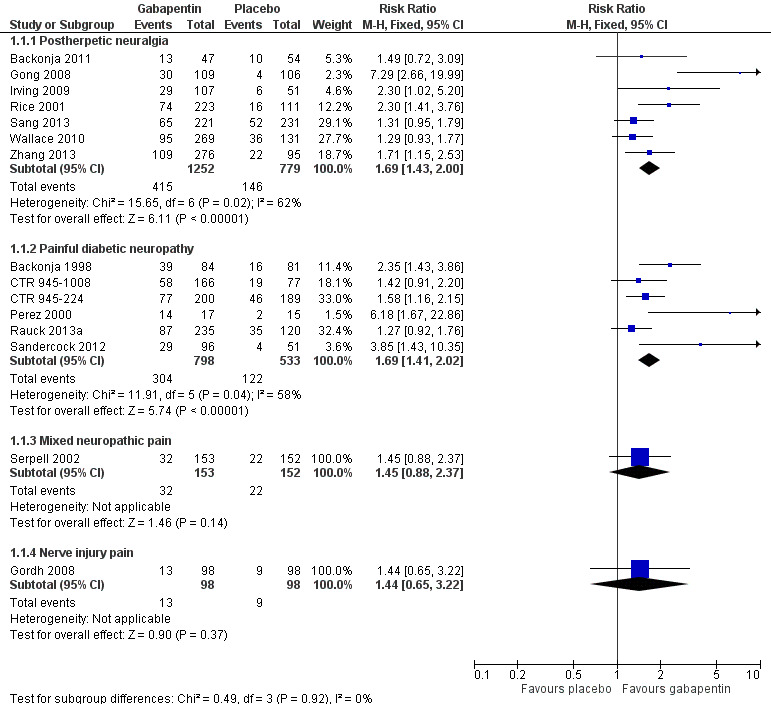
Forest plot of comparison: 1 All placebo‐controlled studies, outcome: 1.1 At least 50% pain reduction over baseline.
1.2. Analysis.
Comparison 1 Efficacy ‐ placebo‐controlled studies, Outcome 2 Very much improved.
4.
Forest plot of comparison: 1 All placebo‐controlled studies, outcome: 1.2 Very much improved.
1.3. Analysis.
Comparison 1 Efficacy ‐ placebo‐controlled studies, Outcome 3 Much or very much improved.
5.
Forest plot of comparison: 1 All placebo‐controlled studies, outcome: 1.3 Much or very much improved.
1.4. Analysis.
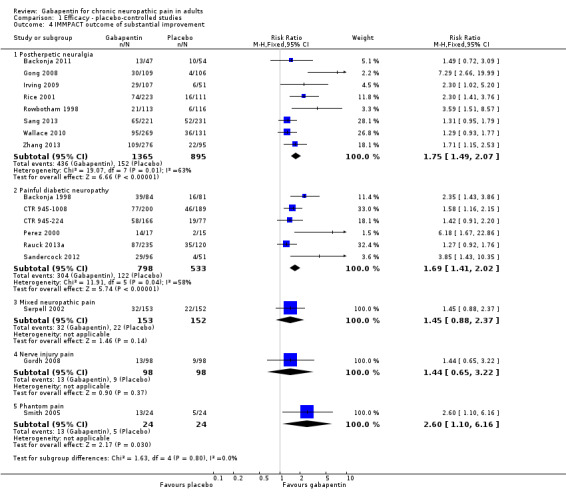
Comparison 1 Efficacy ‐ placebo‐controlled studies, Outcome 4 IMMPACT outcome of substantial improvement.
6.
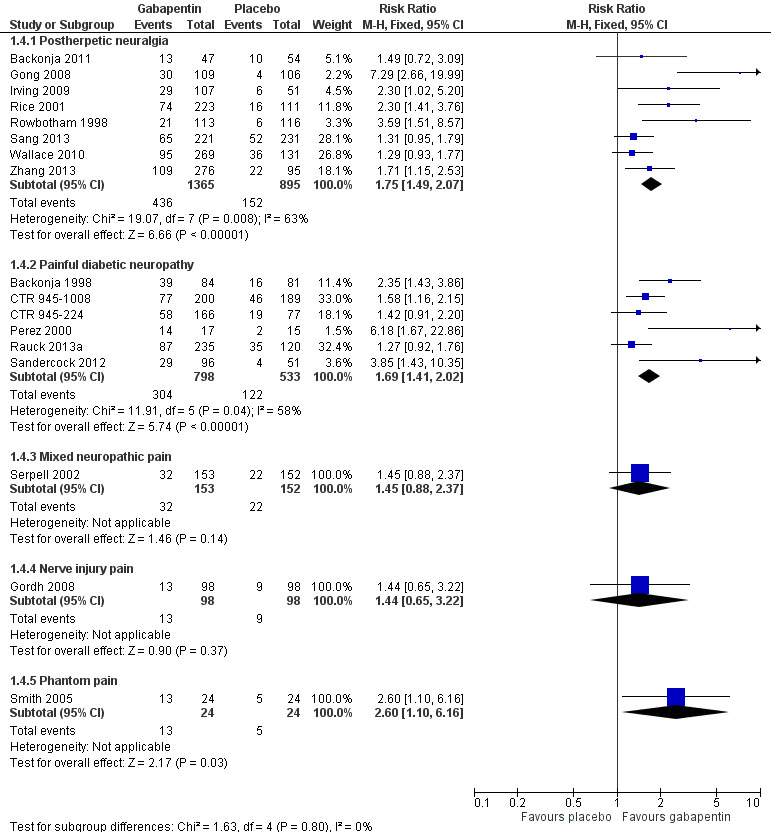
Forest plot of comparison: 1 All placebo‐controlled studies, outcome: 1.4 IMMPACT outcome of substantial improvement.
1.5. Analysis.
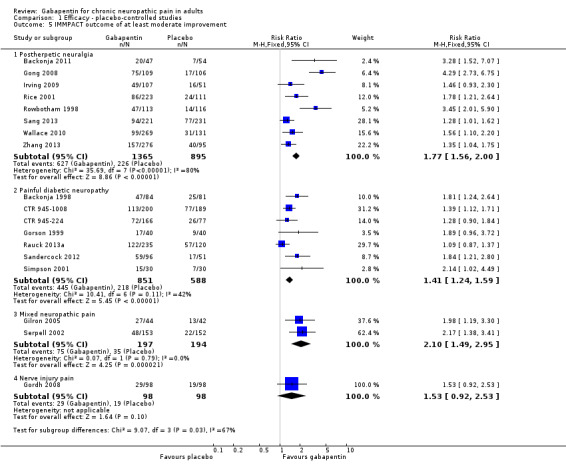
Comparison 1 Efficacy ‐ placebo‐controlled studies, Outcome 5 IMMPACT outcome of at least moderate improvement.
7.
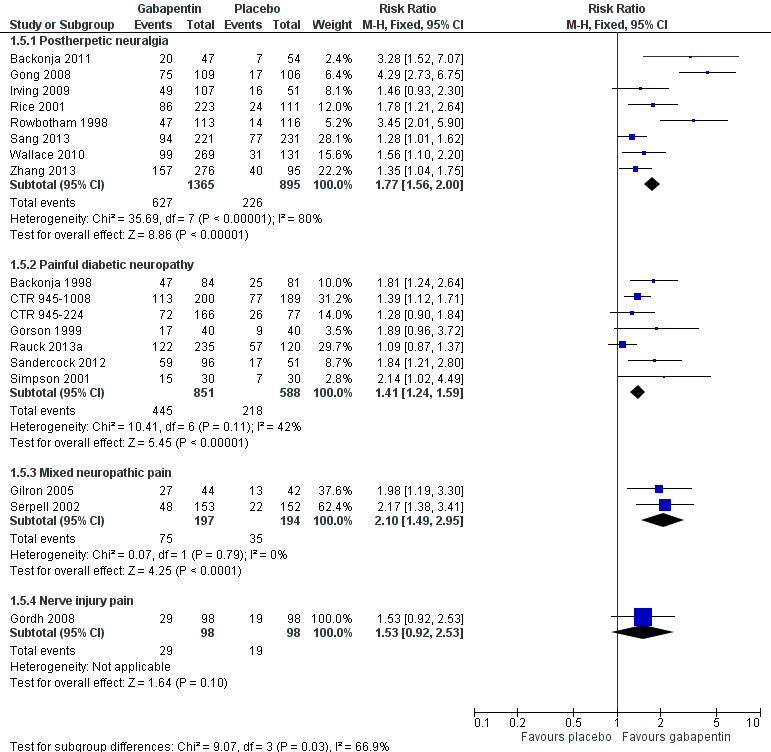
Forest plot of comparison: 1 All placebo‐controlled studies, outcome: 1.5 IMMPACT outcome of at least moderate improvement.
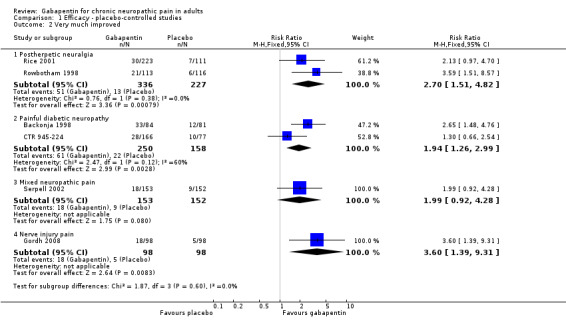
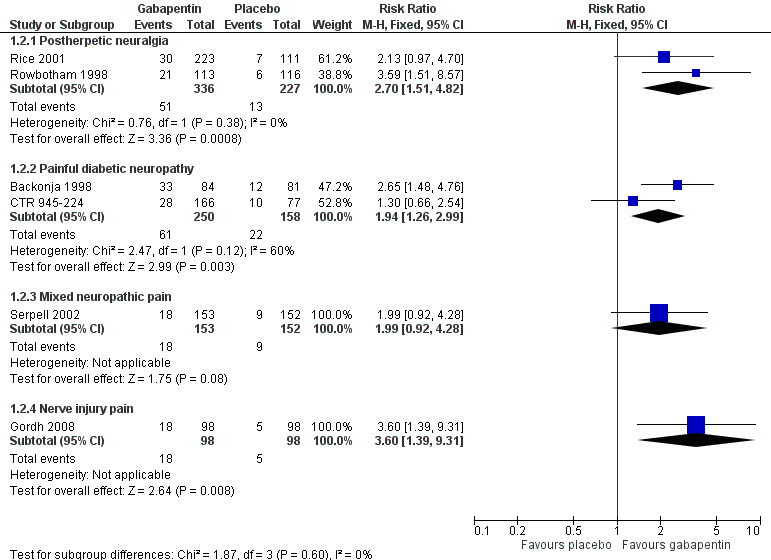
Postherpetic neuralgia (PHN)
Of the 11 studies in PHN, nine (Backonja 2011; Gong 2008; Irving 2009; NCT00475904; Rice 2001; Rowbotham 1998; Sang 2013; Wallace 2010; Zhang 2013) had a placebo control, and two (Chandra 2006; Harden 2013) an active control only. All nine placebo‐controlled studies had a parallel‐group design, with study duration of four to 12 weeks; daily gabapentin doses varied between 1800 mg and 3600 mg, while the dose of gabapentin encarbil was 1200 mg to 3600 mg daily.
In seven studies reporting the outcome, at least 50% pain intensity reduction occurred in 33% of participants given gabapentin and 19% of those given placebo by the end of the study, with considerable consistency between studies (Summary of results A; Figure 8). Available data on dosing regimens were too sparse to establish a dose‐response relationship. A number of outcomes consistent with IMMPACT recommendations for substantial and moderate benefit were reported in two or more placebo‐controlled studies, and the results showed gabapentin at doses of 1800 mg daily or more, or gabapentin encarbil at 1200 mg daily, to be more effective than placebo (Summary of results A). For a PGIC of much or very much improved, 39% of participants achieved this level of improvement with gabapentin and 29% with placebo. Other outcomes are reported in Summary of results A.
8.
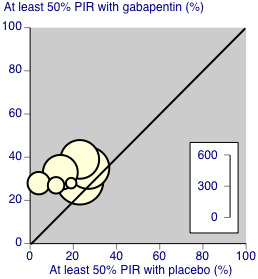
Postherpetic neuralgia: percentage of participants achieving at least 50% pain intensity reduction (PIR) over baseline with gabapentin 1200 mg‐3600 mg daily, or placebo
Only two of these studies (Gong 2008; Rice 2001; 24% of participants) used a standard formulation of gabapentin, and removing them from the analysis did not significantly change the result. Similarly, removing the two studies using gabapentin encarbil (Backonja 2011; Zhang 2013; 21% of participants) did not affect the result. There were insufficient data for subgroup analyses based on dose or duration of studies.
We assessed the quality of evidence as moderate. Results were consistent between studies, but there were uncertainties and differences between dosing and dosing schedules, formulation, and imputation methods used.
Summary of results A. Efficacy outcomes with gabapentin in postherpetic neuralgia (PHN)
| Number of | Percent with outcome | |||||
| Outcome | Studies | Participants | Gabapentin | Placebo | Risk ratio (95% CI) | NNT (95% CI) |
| Substantial benefit | ||||||
| At least 50% pain intensity reduction | 7 | 2031 | 33 | 19 | 1.7 (1.4 to 2.0) | 6.9 (5.5 to 9.4) |
| PGIC very much improved | 2 | 563 | 15 | 6 | 2.7 (1.5 to 4.8) | 11 (7.0 to 22) |
| Any definition of substantial benefit (at least 50% pain intensity reduction or PGIC very much improved) | 8 | 2260 | 32 | 17 | 1.8 (1.5 to 2.1) | 6.7 (5.4 to 8.7) |
| Moderate benefit | ||||||
| At least 30% pain intensity reduction | 2 | 529 | 54 | 38 | 1.4 (1.1 to 1.7) | 6.5 (4.0 to 16) |
| PGIC much or very much improved | 7 | 2013 | 39 | 29 | 1.3 (1.2 to 1.5) | 9.7 (6.9 to 16) |
| Any definition of moderate benefit (at least 25% pain intensity reduction or PGIC much or very much improved) | 8 | 2260 | 46 | 25 | 1.8 (1.6 to 2.0) | 4.8 (4.1 to 6.0) |
In the active controlled study involving 76 participants, gabapentin at doses of up to 2700 mg daily was compared to nortriptyline at doses of up to 150 mg daily over nine weeks. At least 50% improvement in pain over baseline using a VAS pain scale was achieved by 13/38 (34%) with gabapentin and 14/38 (37%) with nortriptyline, broadly in line with event rates in placebo‐controlled studies (Chandra 2006). Harden 2013 compared two dosing regimens of gabapentin encarbil in previous low dose treatment failures and found that about 13% did respond at the 50% pain reduction level.
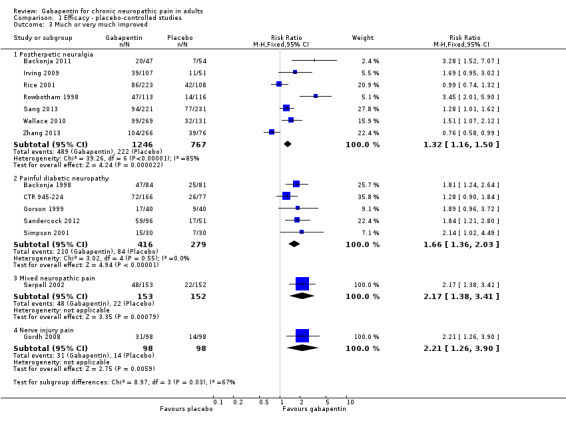
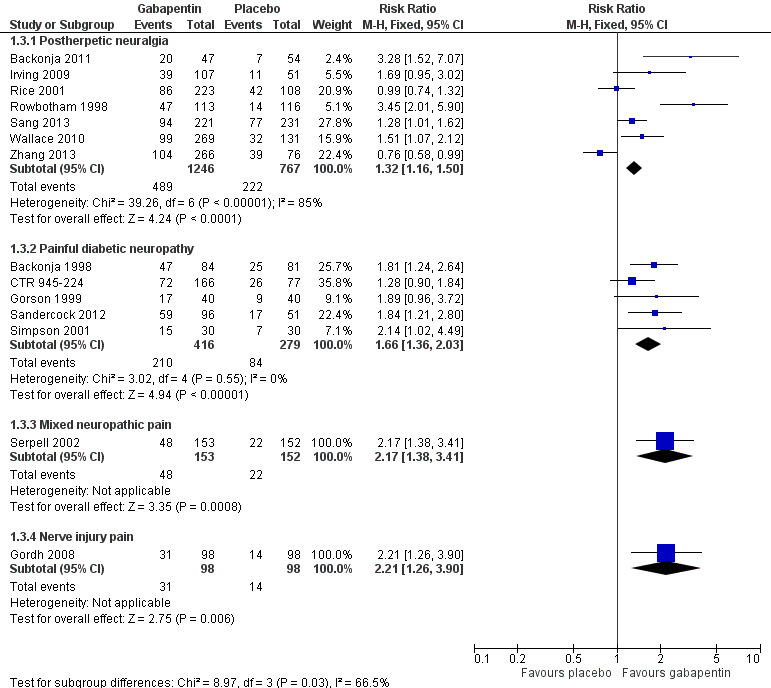
Painful diabetic neuropathy (PDN)
Seven of the nine studies in PDN were of parallel‐group design (Backonja 1998; CTR 945‐1008; CTR 945‐224; Perez 2000; Rauck 2013a; Sandercock 2012; Simpson 2001); two had a cross‐over design (Gorson 1999; Morello 1999). Eight had a placebo comparator, while one (Morello 1999) had an active control only. Seven placebo‐controlled parallel‐group studies had a study duration between four and 14 weeks; all but one (Sandercock 2012) of seven weeks or longer. Daily gabapentin doses varied between 600 mg and 3600 mg; doses below 1200 mg were used in two studies, 900 mg daily as the only gabapentin dose in one (Gorson 1999), and 600 mg daily in one arm of another (CTR 945‐224). Gabapentin encarbil at doses of 1200 and 3600 mg daily was compared with pregabalin 300 mg daily and placebo in one study (Rauck 2013a).
At least 50% pain intensity reduction occurred in 38% of participants given gabapentin and 23% of those given placebo by the end of the study, with considerable consistency between studies (Summary of results B; Figure 9). Available data on dosing regimens were too sparse to establish a dose‐response relationship. A number of outcomes consistent with IMMPACT recommendations for substantial and moderate benefit were reported in two or more placebo‐controlled studies, and the results showed gabapentin at doses of 1200 mg daily or more to be more effective than placebo (Summary of results B). For PGIC much or very much improved; 50% of participants achieved this level of improvement with gabapentin and 30% with placebo. We obtained very similar results when we omitted data from Simpson 2001 because of concerns one peer reviewer expressed about this study in a previous version of the review; no other efficacy outcome data were included from this study. Other outcomes are reported in Summary of results B.
9.
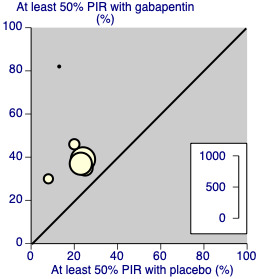
Painful diabetic neuropathy: percentage of participants achieving at least 50% pain intensity reduction (PIR) over baseline with gabapentin 1200‐3600 mg daily, or placebo
Two studies (Rauck 2013a; Sandercock 2012; 35% of participants) used the gabapentin encarbil or gastroretentive formulations. Removing these from the analysis did not change the result. There were insufficient data for subgroup analyses based on dose or duration of studies.
We assessed the quality of evidence as moderate. Results were consistent between studies, but there were uncertainties and differences between dosing and dosing schedules, formulation, and imputation methods used.
Summary of results B. Efficacy outcomes with gabapentin in painful diabetic neuropathy (PDN) (1200 mg daily or greater)
| Number of | Percent with outcome | |||||
| Outcome | Studies | Participants | Gabapentin | Placebo | Risk ratio (95% CI) | NNT (95% CI) |
| Substantial benefit | ||||||
| At least 50% pain intensity reduction | 6 | 1331 | 38 | 23 | 1.7 (1.4 to 2.0) | 6.6 (5.0 to 9.7) |
| PGIC very much improved | 2 | 408 | 24 | 14 | 1.9 (1.3 to 3.0) | 9.6 (5.5 to 35) |
| Any definition of substantial benefit (at least 50% pain intensity reduction or PGIC very much improved) | 6 | 1331 | 38 | 23 | 1.7 (1.4 to 2.0) | 6.6 (5.0 to 9.7) |
| Moderate benefit | ||||||
| At least 30% pain intensity reduction | 2 | 744 | 54 | 43 | 1.2 (1.1 to 1.5) | 9.4 (5.6 to 29) |
| PGIC much or very much improved | 5 | 695 | 50 | 30 | 1.7 (1.4 to 2.0) | 4.9 (3.6 to 7.6) |
| PGIC much or very much improved (excluding Simpson 2001) | 4 | 635 | 51 | 31 | 1.6 (1.3 to 2.0) | 5.1 (3.7 to 8.3) |
| Any definition of moderate benefit (at least 30% pain intensity reduction or PGIC much or very much improved) | 7 | 1439 | 52 | 37 | 1.4 (1.3 to 1.6) | 6.6 (4.9 to 9.9) |
Gabapentin 600 mg daily produced lesser effects than 1200 mg and 2400 mg daily in a study that compared them (CTR 945‐224). In one placebo‐controlled cross‐over study involving 40 randomised participants, moderate or excellent pain intensity reduction was achieved by 17/40 (43%) with gabapentin 900 mg daily over six weeks, compared with 9/40 (23%) with placebo (Gorson 1999).
In one active‐controlled study involving 25 participants, gabapentin at 1800 mg daily was compared to amitriptyline 75 mg daily over six weeks. Complete or a lot of pain relief was achieved by 6/21 (29%) with gabapentin and 5/21 (24%) with amitriptyline (Morello 1999).
Mixed neuropathic pain
One exploratory study (Rauck 2013b) examined the effects of intrathecal gabapentin in participants with chronic, intractable non cancer pain, the majority (147/170; 86%) of whom were classified as having pain of neuropathic or mixed types. Three different doses (1 mg, 6 mg, and 30 mg daily) were compared with placebo. There was no significant reduction in group mean pain scores within and between groups over the 22 day treatment period. The number of participants experiencing at least 30% reduction in pain was 4/42, 4/41, 1/41, and 2/44 for the 1 mg, 6 mg, 30 mg, and placebo groups, respectively.
Four studies examined the effects of oral gabapentin in mixed neuropathic painful conditions (Gilron 2005; Gilron 2009; NCT00904202; Serpell 2002); two included participants with PHN and PDN (Gilron 2005; Gilron 2009); in another the most common conditions were CRPS and PHN (Serpell 2002); and the fourth study enrolled participants with PHN, DN, CRPS, carpel tunnel syndrome, HIV neuropathy, idiopathic sensory neuropathy, and other peripheral neuropathy (proportions not reported, NCT00904202). One had a parallel‐group comparison with placebo over eight weeks (Serpell 2002), and one had a parallel‐group comparison with placebo, lidocaine patch, and gabapentin in combination with lidocaine patch over five weeks (NCT00904202). The others had cross‐over designs that included placebo and morphine alone and in combination with gabapentin over five weeks (Gilron 2005), and nortriptyline alone or in combination with gabapentin over six weeks (Gilron 2009).
One parallel‐group comparison with placebo used gabapentin titrated to a maximum of 2400 mg daily in 305 participants (Serpell 2002). Only for the PGIC outcome of much or very much improved was there a significant benefit of gabapentin (Summary of results C).
Summary of results C. Efficacy outcomes with gabapentin in mixed neuropathic pain (Serpell 2002)
| Number of | Percent with outcome | |||||
| Outcome | Studies | Participants | Gabapentin | Placebo | Risk ratio (95% CI) | NNT (95% CI) |
| At least 50% pain intensity reduction | 1 | 305 | 21 | 14 | 1.5 (0.9 to 2.4) | not calculated |
| PGIC very much improved | 1 | 305 | 12 | 6 | 2.0 (0.9 to 4.3) | not calculated |
| PGIC much or very much improved | 1 | 305 | 31 | 14 | 2.2 (1.4 to 3.4) | not calculated |
The other parallel‐group comparison used gabapentin titrated to 1800 mg daily over one week in 62 participants (NCT00904202), and did not report any of our specified efficacy outcomes. It did report the group mean change in pain intensity at the end of the study as 44% for gabapentin alone, 39% for lidocaine patch alone, 50% for the combination, and 26% for placebo (ITT analysis assumed). The number of participants who were satisfied or very satisfied with treatment were 65% for gabapentin alone, 69% for lidocaine patch alone, 69% for the combination, and 64% for placebo. There were no statistically significant differences between treatment groups.
One placebo‐controlled cross‐over study (Gilron 2005) over five weeks provided results for moderate pain relief for participants who completed a given treatment period. Gabapentin alone (target dose 3200 mg daily), morphine alone (target dose 120 mg daily), and the combination (target dose gabapentin 2400 mg plus 60 mg morphine daily) were significantly better than placebo (Summary of results D). These results were calculated from the numbers and percentages with a moderate response. The total was larger than the 57 randomised, because some participated in more than one treatment arm.
Summary of results D. Efficacy outcomes with gabapentin in mixed neuropathic pain (Gilron 2005)
| Number of | Percent with outcome | |||||
| At least moderate pain relief | Studies | Participants | Gabapentin, morphine, of their combination | Placebo | Risk ratio (95% CI) | NNT (95% CI) |
| Gabapentin alone | 1 | 96 | 61 | 25 | 2.5 (1.5 to 4.2) | not calculated |
| Morphine alone | 1 | 96 | 80 | 25 | 3.2 (1.9 to 5.2) | not calculated |
| Gabapentin plus morphine | 1 | 93 | 78 | 25 | 3.1 (1.9 to 5.1) | not calculated |
The other cross‐over study compared gabapentin alone (target dose 3600 mg daily), nortriptyline (target dose 100 mg daily) and the combination (target dose 3600 mg gabapentin plus 100 mg nortriptyline daily) over six weeks (Gilron 2009). Pain intensity was significantly lower with the combination, by less than 1 point out of 10 on a numerical rating pain scale.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Radicular leg pain
One study compared gabapentin, titrated to a maximum of 3600 mg daily, with placebo over 12 weeks in 108 participants, 46 of whom had radicular pain (Atkinson 2016). Although results were not reported separately for these participants, the investigators did report that there was no difference between those with and without radicular pain. In an exploratory analysis of completers, 36% of participants in both groups reported a 30% or more decrease in pain intensity, and 26% and 29% reported a 50% or more decrease with gabapentin (34 participants) and placebo (38 participants), respectively. There was also no difference between groups for 'patient estimation of pain improvement' at the end of the study.
Another study compared gabapentin, titrated to a target of 1800 to 3600 mg daily, with epidural steroid over three months (Cohen 2015). The study reported only group mean decreases in average and worst leg pain at the end of treatment, which ranged from 1.6 to 2.7, with large variation within groups. There were no significant differences between the groups.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Spinal cord injury
The efficacy of gabapentin in spinal cord injury pain at maximum doses of 1800 mg or 3600 mg daily was compared with placebo in three cross‐over trials (Levendoglu 2004; Rintala 2007; Tai 2002) over periods of four and eight weeks. None of the studies reported dichotomous outcomes equivalent to moderate or substantial pain relief.
One eight‐week study randomised 20 participants to a maximum of 3600 mg gabapentin daily or placebo over eight weeks (Levendoglu 2004) and reported a 62% average fall in pain with gabapentin compared with a 13% fall with placebo.
A second eight‐week study randomised 38 participants to a maximum of 3600 mg gabapentin daily, amitriptyline 150 mg daily, or placebo over eight weeks (Rintala 2007). It claimed statistical superiority for amitriptyline for the 22 participants completing all three phases, and no benefit of gabapentin over placebo.
The final study comparing gabapentin with placebo over four weeks in seven participants had no interpretable results (Tai 2002).
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Nerve injury pain
A single cross‐over study evaluated the efficacy of gabapentin at a maximum of 2400 mg daily compared with placebo over five‐week treatment periods (Gordh 2008). Among the 98 participants of the 120 randomised who completed both treatment periods, at least 50% pain intensity reduction was achieved by 13 (13%) with gabapentin and 9 (9%) with placebo, which did not reach statistical significance. At least 30% pain intensity reduction was achieved by 29 (29%) with gabapentin and 19 (19%) with placebo, which did not reach statistical significance.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Phantom limb pain
Two cross‐over studies evaluated the efficacy of gabapentin compared with placebo in phantom limb pain (Bone 2002; Smith 2005). Bone 2002 randomised 19 participants to a maximum of 2400 mg gabapentin daily, or the maximum tolerated dose, with six‐week treatment periods. Using an ITT approach, weekly VAS pain scores were lower at week six only with gabapentin, but not at any other time, nor with categorical pain measures. Smith 2005 randomised 24 participants to gabapentin titrated to a maximum daily dose of 3600 mg. A "meaningful decrease in pain" (the top of a five‐point scale) was achieved by 13 participants (54%) with gabapentin and 5 (21%) with placebo.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Cancer‐related neuropathic pain
Three studies examined gabapentin in the short term in cancer‐related neuropathic pain (Caraceni 2004; Mishra 2012; Rao 2007). A parallel‐group study (Caraceni 2004) randomised 121 participants to titration to a maximum of gabapentin 1800 mg daily or placebo, with 10 days of treatment. The average pain intensity was somewhat lower with gabapentin than with placebo, but the number of participants described as having pain under control was very similar with both treatments after six days, with 50% to 60% with pain under control over six to 10 days. A cross‐over study (Rao 2007) compared gabapentin titrated to 2700 mg daily with placebo in chemotherapy‐induced neuropathic pain over three weeks. There was no significant difference between gabapentin and placebo, but the study did recruit participants both with pain or sensory loss or paraesthesia, and baseline pain scores were only about 4/10 on a numerical rating scale. The study probably lacked sensitivity to detect any difference.
The third study compared gabapentin 1800 mg daily with pregabalin 600 mg daily and amitriptyline 100 mg daily for a total of four weeks (Mishra 2012). No dichotomous data were reported; a decrease in pain scores in all groups in all weeks was reported, together with a morphine‐sparing effect and improvement in functional capacity. Morphine‐sparing and functional capacity were significantly better with pregabalin than the other treatments.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
HIV‐associated sensory neuropathies
A single parallel‐group study compared gabapentin titrated to 2400 mg daily with placebo over four weeks in 24 participants with painful HIV‐associated neuropathies (Hahn 2004). On average, pain and sleep improved substantially with both gabapentin and placebo, though the time courses differed. After four weeks, there was no difference in median pain scores, though the placebo response had an unusual time course in 11 participants.
We assessed the quality of evidence as very low with a small number of studies, participants, and events.
Withdrawals (see Summary of results E)
We pooled data from participants with different types of neuropathic pain for analyses of withdrawals.
All‐cause withdrawals
Twenty‐two studies with 4617 participants reported on withdrawals for any cause, which occurred in 20% of participants with gabapentin at daily doses of 1200 mg or more, and in 19% with placebo (Analysis 2.1). The risk ratio was 1.0 (0.92 to 1.2). The NNH was not calculated.
2.1. Analysis.
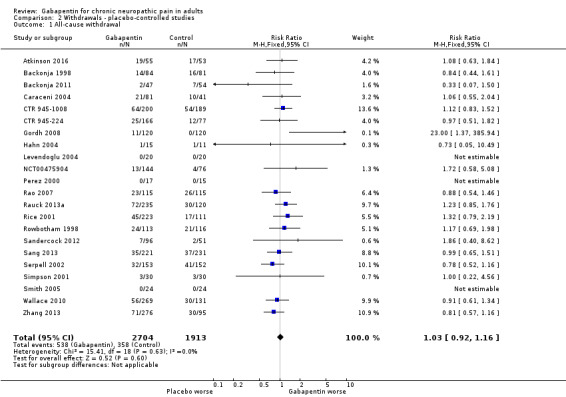
Comparison 2 Withdrawals ‐ placebo‐controlled studies, Outcome 1 All‐cause withdrawal.
Adverse event withdrawals
Twenty‐two studies with 4346 participants reported on adverse event withdrawals, which occurred in 11% of participants with gabapentin at daily doses of 1200 mg or more, and in 8.2% with placebo (Analysis 2.2). The risk ratio was 1.4 (1.1 to 1.7), and the NNH was 30 (20 to 66).
2.2. Analysis.
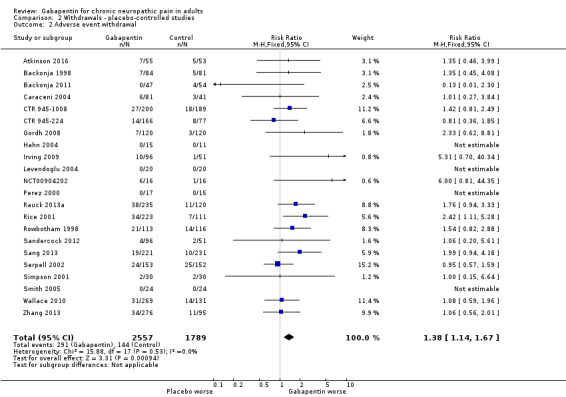
Comparison 2 Withdrawals ‐ placebo‐controlled studies, Outcome 2 Adverse event withdrawal.
Lack of efficacy withdrawals
Fifteen studies with 3559 participants reported on lack of efficacy withdrawals, which occurred in 1.9% of participants with gabapentin at daily doses of 1200 mg or more, and in 3.3% with placebo (Analysis 2.3). The risk ratio was 0.57 (0.37 to 0.88), and the number needed to treat to prevent one withdrawal (NNTp) NNTp was 73 (41 to 360).
2.3. Analysis.
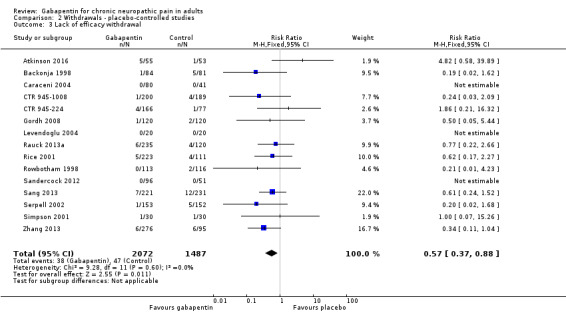
Comparison 2 Withdrawals ‐ placebo‐controlled studies, Outcome 3 Lack of efficacy withdrawal.
We assessed the quality of evidence for withdrawals as high, based on a reasonable number of events and generally good reporting.
Adverse events (see Summary of results E)
We pooled data from participants with different types of neuropathic pain for analyses of adverse events.
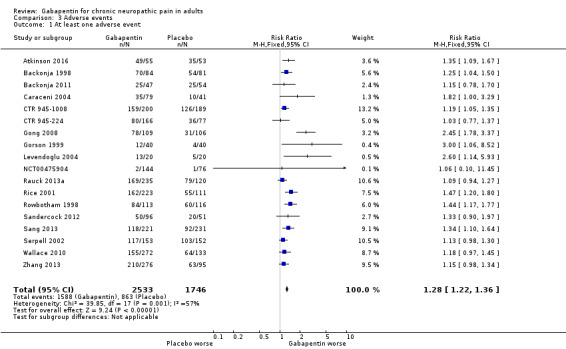
Participants experiencing at least one adverse event
Eighteen studies with 4279 participants reported on participants experiencing at least one adverse event, which occurred in 63% of participants with gabapentin at daily doses of 1200 mg or more, and in 49% with placebo (Analysis 3.1). The risk ratio was 1.3 (1.2 to 1.4), and the NNH was 7.5 (6.1 to 9.6). We assessed the quality of evidence as moderate, based on a reasonable number of events and consistency, but limited quality of reporting adverse events.
3.1. Analysis.
Comparison 3 Adverse events, Outcome 1 At least one adverse event.
Serious adverse events
NIneteen studies reported on 3948 participants experiencing a serious adverse event, which occurred in 3.2% of participants with gabapentin at daily doses of 1200 mg or more, and in 2.8% with placebo (Analysis 3.2). The risk ratio was 1.2 (0.83 to 1.7). The NNH was not calculated. We assessed the quality of evidence as moderate due to the limited number of events.
3.2. Analysis.
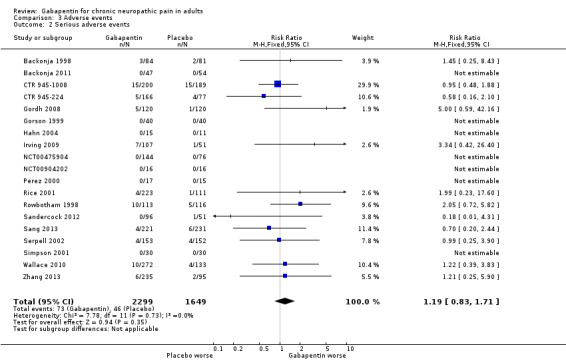
Comparison 3 Adverse events, Outcome 2 Serious adverse events.
Particular adverse events
Somnolence, drowsiness, or sedation was reported as an adverse event in 20 studies with 4288 participants, and it occurred in 14% of participants with gabapentin at doses of 1200 mg daily or more, and in 5.2% with placebo (Analysis 3.3). The risk ratio was 2.8 (2.3 to 3.5), and the NNH was 11 (9.4 to 14).
3.3. Analysis.
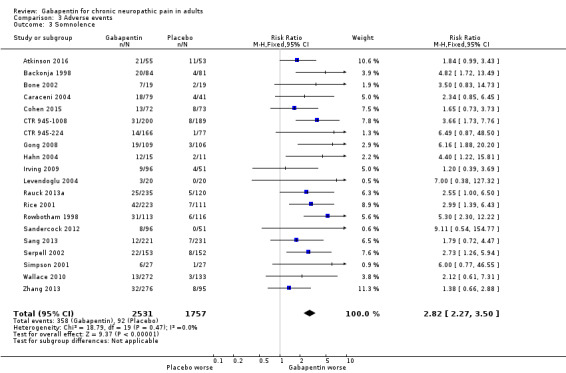
Comparison 3 Adverse events, Outcome 3 Somnolence.
Dizziness was reported as an adverse event in 21 studies with 4739 participants, and it occurred in 19% of participants with gabapentin at doses of 1200 mg daily or more, and in 6.6% with placebo (Analysis 3.4). The risk ratio was 2.9 (2.4 to 3.4), and the NNH was 8.0 (7.0 to 9.4).
3.4. Analysis.
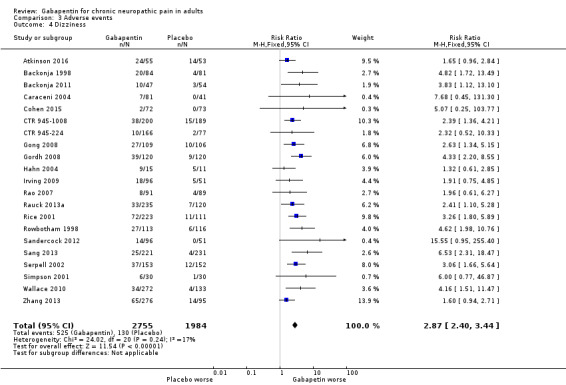
Comparison 3 Adverse events, Outcome 4 Dizziness.
Peripheral oedema was reported as an adverse event in 12 studies with 3325 participants, and it occurred in 6.7% of participants with gabapentin at doses of 1200 mg daily or more, and in 1.7% with placebo (Analysis 3.5). The risk ratio was 4.1 (2.7 to 6.4), and the NNH was 20 (16 to 27).
3.5. Analysis.
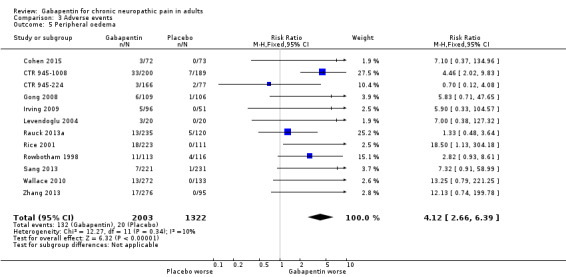
Comparison 3 Adverse events, Outcome 5 Peripheral oedema.
We assessed the quality of evidence for these outcomes as moderate. While there was a reasonable number of events, definitions of adverse events and reporting was not consistent.
Ataxia or gait disturbance was reported as an adverse event in four studies with 510 participants. It occurred in 14% of participants with gabapentin at doses of 1200 mg daily or more, and in 2.6% with placebo (Analysis 3.6). The risk ratio was 5.5 (2.5 to 12), and the NNH was 8.5 (6.1 to 14).
3.6. Analysis.
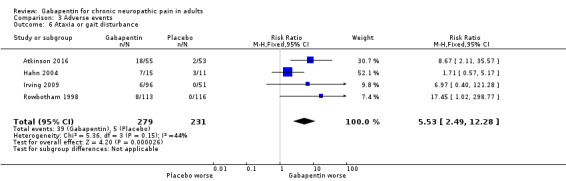
Comparison 3 Adverse events, Outcome 6 Ataxia or gait disturbance.
We assessed the quality of evidence for ataxia as low. There was a small number of studies and events.
Summary of results E: Withdrawals and adverse events with gabapentin (1200 mg daily or more) compared with placebo
| Number of | Percent with outcome | |||||
| Outcome | Studies | Participants | Gabapentin | Placebo | Risk ratio (95% CI) | NNH (95% CI) |
| Withdrawal ‐ all‐cause | 22 | 4617 | 20 | 19 | 1.0 (0.91 to 1.2) | Not calculated |
| Withdrawal due to adverse events | 22 | 4346 | 11 | 8.2 | 1.4 (1.1 to 1.7) | 30 (20 to 66) |
| At least one adverse event | 18 | 4279 | 63 | 49 | 1.3 (1.2 to 1.4) | 7.5 (6.1 to 9.6) |
| Serious adverse event | 19 | 3948 | 3.2 | 2.8 | 1.2 (0.83 to 1.7) | Not calculated |
| Somnolence/drowsiness | 20 | 4288 | 14 | 5.2 | 2.8 (2.3 to 3.5) | 11 (9.4 to 14) |
| Dizziness | 21 | 4739 | 19 | 6.6 | 2.9 (2.4 to 3.4) | 8.0 (7.0 to 9.4) |
| Peripheral oedema | 12 | 3325 | 6.7 | 1.7 | 4.1 (2.7 to 6.4) | 20 (16 to 27) |
| Ataxia/gait disturbance | 4 | 510 | 14 | 2.6 | 5.5 (2.5 to 12) | 8.5 (6.1 to 14) |
| Outcome | Studies | Participants | Gabapentin | Placebo | Risk ratio (95% CI) | NNTp (95% CI) |
| Withdrawal ‐ lack of efficacy | 15 | 3559 | 1.9 | 3.3 | 0.57 (0.37 to 0.88) | 73 (41 to 360) |
Death
Deaths were rare in these studies. Five deaths occurred in PHN studies; three with placebo: one in 231 participants (Sang 2013), one in 116 (Rowbotham 1998) and one in 133 (Wallace 2010); two with gabapentin: one in 223 participants (Rice 2001), and one in 107 (Irving 2009). An unpublished study (CTR 945‐1008) reported two deaths: one of 200 participants treated with gabapentin, and one of 189 treated with placebo. A further study reported two deaths in 152 participants taking placebo (Serpell 2002). Overall, three deaths occurred with gabapentin and five with placebo. We assessed the quality of evidence as very low due to the very small number of events.
Discussion
Summary of main results
Gabapentin is a reasonably effective treatment for a variety of neuropathic pain conditions. It has been demonstrated to be better than placebo across all studies for IMMPACT outcomes of substantial and at least moderate improvement, producing almost identical results for all trials and those in parallel‐group studies lasting six weeks or longer. Numbers needed to treat for an additional beneficial outcome (NNTs) were between 5 and 7 for substantial and at least moderate improvement in PHN and PDN (moderate‐quality evidence). Results were consistent across the major neuropathic pain conditions tested, though gabapentin was tested only in small numbers in uncommon neuropathic pain conditions. The review concentrated on doses of gabapentin of 1200 mg daily or greater, though a wide range of fixed doses and dose titration regimens were used.
Gabapentin was tested in nine different chronic pain conditions generally considered to be neuropathic in origin. For only three neuropathic pain conditions was there sufficient information to be confident that it worked satisfactorily, namely PHN, PDN, and mixed neuropathic pain, itself principally, though not exclusively, PHN and PDN.
Benefit was balanced by more withdrawals due to adverse events (high‐quality evidence), and participants taking gabapentin experienced more adverse events (high‐quality evidence), including somnolence, dizziness, peripheral oedema, and gait disturbance than did those taking placebo (moderate‐quality evidence). Serious adverse events were no more common with gabapentin than placebo (moderate‐quality evidence), and death was an uncommon finding in these studies (very low‐quality evidence).
Overall completeness and applicability of evidence
Efficacy and adverse event outcomes were not consistently reported across the studies, and this limited the analyses to some extent. However, for the most important efficacy and adverse event outcomes, analyses across all conditions were mostly based on between 1000 and about 4700 participants. All the larger studies (typically those with more than 100 participants) reported some efficacy outcome equivalent to one or both of the IMMPACT outcomes of at least moderate or substantial benefit. Clearly, analysis at the level of the individual participant would facilitate a more robust estimate (Moore 2013a). Such analysis can also demonstrate a link between benefit in terms of pain and benefit in other outcomes, including quality of life (Hoffman 2010).
Possible sources of bias that could have affected the results of the review included the following.
Duration ‐ NNT estimates of efficacy in chronic pain studies tend to increase (get worse) with increasing duration (Moore 2010e). However, limiting studies to those of six weeks or longer did not change the main efficacy outcomes, mainly because most participants were in longer‐duration studies.
Outcomes may affect estimates of efficacy, but the efficacy outcomes chosen were of participants achieving the equivalent of IMMPACT‐defined moderate or substantial improvement, and it is likely that lesser benefits, such as 'any benefit' or 'any improvement', are potentially related to lesser outcomes, though this remains to be clarified.
The dose of gabapentin used differed between studies, in terms of maximum allowable dose, and whether the dose was fixed, titrated to effect, or titrated up to the maximum irrespective of beneficial or adverse effects. We chose to pool data irrespective of dose, within broad limits, because it was the only practical way to deal with dose in a pooled analysis, and because of a lack of good evidence of any clear dose‐response effect for gabapentin in neuropathic pain.
In some circumstances cross‐over trials have been shown to exaggerate treatment effects in comparison with parallel‐group designs (Khan 1996), but the extent is unclear, and it is unlikely to be the source of major bias (Elbourne 2002). Withdrawals from cross‐over studies meant that any results were likely to be per protocol for completers rather than a true ITT analysis. Parallel‐group studies were larger than cross‐over studies, and dominated the analyses in terms of number of participants. The 25 parallel‐group studies involved 5298 participants (median 204), while the 12 cross‐over studies involved 621 participants (median 40 participants). Additionally, few cross‐over studies reported outcomes that could be used in the analyses.
The absence of publication bias (unpublished trials showing no benefit of gabapentin over placebo) can never be proven. However, we can calculate the number of participants in studies of zero benefit (risk ratio of 1) required for the absolute benefit to reduce beneficial effects to a negligible amount (Moore 2008). If an NNT of 10 were considered a level that would make gabapentin clinically irrelevant, then the number of participants with zero benefit would be 2448 for a moderate response and 1113 for a substantial response in PHN, and 741 for a moderate response and 887 for a substantial response in PDN. With median study size for parallel‐group studies of about 200 participants, this would require a minimum of seven unavailable studies in PHN and four in PDN. While not impossible, this seems unlikely given the paucity of new data in the last three years.
There is one important unknown for most studies, namely whether the definition of response in the trials included only participants who had both an analgesic response and were able to take gabapentin. If response included an LOCF assessment of efficacy from those who discontinued, this could have affected the results (Moore 2012a). LOCF tends to overestimate treatment effects when adverse event withdrawals with drug are higher than that with placebo. For gabapentin, the excess adverse withdrawal over placebo was about 3%. This is not likely to result in significant overestimation in treatment effect (Moore 2012a). In a similar situation, duloxetine produced little different NNTs using LOCF and BOCF in four different chronic pain conditions (Moore 2014b).
Another issue is how to deal with relatively short term, small, multiple cross‐over studies that intensively study participants on a daily basis (Gilron 2005; Gilron 2009), and do not report outcomes of clinical relevance (participants with adequate pain relief), but rather average pain scores, whose relevance has been questioned because of underlying skewed distributions (McQuay 1996; Moore 2010a; Moore 2013a). This study design can provide useful and clinically relevant information, such as the relatively rapid onset of effect of therapies in neuropathic pain, or how individual patients respond to several different drugs. However, they are difficult to include in pooled analyses, and their small size and brevity come with significant potential biases. Small size has become a particular issue, with increasing association of small study size with positive bias (Dechartres 2013; Dechartres 2014; Fanelli 2017; Nguyen 2017). Cochrane Reviews have received criticism for being overly confident with inadequate data (AlBalawi 2013; Brok 2009; Roberts 2015; Turner 2013).
There were almost no data for direct comparisons with other active treatments. It is questionable how important direct comparisons may be; they compare average efficacy rates between different active therapies, but individual people may respond to one drug, but not another (Moore 2013b).
Finally, there was no way to incorporate into the review important observations on the timing and consistency of analgesia with gabapentin in neuropathic pain. In PHN, individual participant‐level pooled analyses of several large trials have demonstrated that, judged by the proportion of participants with a 1 out of 10 point pain intensity reduction, around 20 to 40 days is needed for effects to be seen (Rauck 2013c). Early response, defined as a 30% pain intensity reduction or greater, was predictive of response after 10 weeks, while pain intensity reduction of less than 10% at week 5 was the best early predictor of lack of response at week 10 (Jensen 2012).
While there was considerable information about withdrawals and adverse events, rare but serious adverse events could not be addressed in these studies. We are aware that erectile dysfunction has been a cause for concern for younger men treated with antiepileptic drugs for epilepsy (Smalldone 2004), and anorgasmia has been reported with gabapentin (Perloff 2011). Adverse event reporting of erectile dysfunction or anorgasmia in these trials was sparse or not present, and the effects of gabapentin on sexual function may not be well represented. Moreover, the included studies did not address gabapentin misuse (Evoy 2017; Quintero 2017).
Quality of the evidence
The studies included in this review covered a large number of different painful conditions. The main quality issues involve reporting of outcomes of interest, particularly dichotomous outcomes equivalent to IMMPACT, appropriate analysis of data for participants who withdrew, and better reporting of adverse events. The earliest study was published in 1998, and the past decade or so has seen major changes in clinical trial reporting. The studies themselves appear to be well‐conducted, and individual participant analysis could overcome some of the shortcomings of reporting.
Potential biases in the review process
We know of no potential biases in the review process.
Agreements and disagreements with other studies or reviews
The results for neuropathic pain in this review have not changed noticeably since the last updates in 2014 and 2011.
Summary of results F. Comparison of NNTs (95% CI) from previous and present reviews
| 2005 review | 2011 update | 2014 Update | 2017 update | ||||
| Outcomes | Any improvement |
IMMPACT moderate benefit |
IMMPACT substantial benefit |
IMMPACT moderate benefit |
IMMPACT substantial benefit |
IMMPACT moderate benefit |
IMMPACT substantial benefit |
| All studies | 4.3 (3.5 to 5.7) | 5.8 (4.8 to 7.2) | 6.8 (5.6 to 8.7) | Not calculated in this review | Not calculated in this review | ||
| PHN | 3.9 (3.0 to 5.7) | 5.5 (4.3 to 7.7) | 7.5 (5.2 to 14) | 5.7 (4.6 to 7.5) | 6.8 (5.4 to 9.3) | 5 (5 to 6) | 7 (6 to 9) |
| PDN | 2.9 (2.2 to 4.3) | 8.1 (4.7 to 28) | 5.8 (4.3 to 9.0) | 6.6 (4.9 to 9.9) | 5.9 (4.6 to 8.3) | 7 (5 to 10) | 7 (5 to 10) |
Other systematic reviews
A number of guidelines based on systematic reviews have concluded that gabapentin is helpful in neuropathic pain (Finnerup 2015; Moulin 2014; NICE 2013; SIGN 2013). In PHN, a systematic review found that higher gabapentin doses may not provide greater benefit, but may increase the risk of adverse events (Wang 2017).
One other review has provided NNTs for gabapentin in different neuropathic pain conditions based on 50% pain relief, quoting NNTs of 4.7 and 4.3 for neuropathic pain and peripheral pain, and 4.6 for PHN and 3.9 for PDN (Finnerup 2005). A systematic review of therapies for PHN considered gabapentin effective, with an NNT of 4.6 (Hempenstall 2005). These efficacy estimates are more optimistic than NNTs for the IMMPACT substantial benefit calculated for this review, and more optimistic than NNTs calculated for the same outcome of at least 50% pain relief for PHN of 5.7 and PDN of 5.8. The use of more stringent criteria for efficacy, and availability of more information from longer duration studies has led to more conservative efficacy results. Both pregabalin and duloxetine have NNTs in the region of 5 to 6 for at least 50% pain relief over eight to 12 weeks compared with placebo in PHN and PDN (Lunn 2009; Moore 2009; Sultan 2008).
A number of other systematic reviews have examined the efficacy of gabapentin in neuropathic pain. Systematic reviews of gabapentin for neuropathic pain in spinal cord injury (Tzellos 2008) and fibromyalgia (Hauser 2009; Tzellos 2010) found no more studies than those reported here. An examination of the effects of enriched enrolment found no more studies, and gave similar results for withdrawals and adverse events based on a more limited data set (Straube 2008). A review comparing gabapentin and duloxetine in PDN was limited to two gabapentin studies, was statistical in nature, and restricted to average changes in some efficacy parameters (Quilici 2009). The most directly relevant review was a comparison between gabapentin and tricyclic antidepressants (Chou 2009), in which a meta‐analysis of six placebo‐controlled gabapentin studies in PHN, PDN, and mixed neuropathic pain was performed. Using a mixture of outcomes the relative benefit compared with placebo was 2.2, similar to the benefits found for the 'all studies' analysis and for analyses for PHN, PDN, and mixed neuropathic pain in this review. Phillips 2010 examined the same single study of gabapentin (Hahn 2004) as part of a wider review of pharmacological interventions for HIV neuropathy and came to similar conclusions. The UK NICE guidance on pharmacological management of neuropathic pain has gabapentin as one of four drugs to try initially, with early switching if pain relief is not forthcoming (NICE 2013).
One further review in the public domain (Perry 2008) was performed as part of a legal case in the USA ending in 2009. Perry 2008 considered similar outcomes to this review; NRS or VAS pain score was given hierarchical priority between 50% or greater reduction in pain score (higher priority) and PGIC (lower priority) mainly because it was the pre‐defined primary end point in almost all studies, and for some studies it was difficult to determine how the secondary endpoints were manipulated during post hoc changes in statistical analysis plans. The Perry conclusions are very similar to those of the present review. The likely real differences would lie in the fact that Perry excluded Perez 2000 and Simpson 2001, and did not have access to Sandercock 2012, Irving 2009, and Wallace 2010.
Perry's conclusion on effectiveness was a clinical judgement based on balancing NNH against NNT, using the Cochrane Glossary definition of effectiveness, and presuming that inherent biases in the studies (enrichment, exclusion of many typical real world patients) implied that on balance the benefit of gabapentin use on average does not exceed the harm, which is a somewhat different issue than addressed by this Cochrane Review.
Authors' conclusions
Implications for practice.
For people with neuropathic pain
Gabapentin at a dose of 1800 to 3600 mg daily (1200 to 3600 mg gabapentin encarbil) can provide good levels of pain relief to some people with postherpetic neuralgia and peripheral diabetic neuropathy. Evidence for other types of neuropathic pain is very limited. The outcome of at least 50% pain intensity reduction is regarded as a useful outcome of treatment by people with chronic neuropathic pain, and the achievement of this degree of pain relief is associated with important beneficial effects on sleep interference, fatigue, and depression, as well as quality of life, function, and work. Around 3 to 4 out of 10 achieved this degree of pain relief with gabapentin, compared with 1 to 2 out of 10 for placebo. Over half of those treated with gabapentin will not have worthwhile pain relief.
For clinicians
Gabapentin at a dose of 1800 to 3600 mg daily (1200 to 3600 mg gabapentin encarbil) can provide good levels of pain relief to some people with postherpetic neuralgia and peripheral diabetic neuropathy. Evidence for other types of neuropathic pain is very limited. No evidence regarding a dose‐response effect was available for doses above 1200 mg daily, but limited evidence suggested that doses lower than 1200 mg daily were less effective. Over half of those treated with gabapentin will not have worthwhile pain relief.
For policy makers
Gabapentin at a dose of 1800 to 3600 mg daily (1200 to 3600 mg gabapentin encarbil) can provide good levels of pain relief to some people with postherpetic neuralgia and peripheral diabetic neuropathy. Evidence for other types of neuropathic pain is very limited. The level of efficacy found for gabapentin is consistent with the efficacy estimates for other drug therapies in these conditions. Over half of those treated with gabapentin will not have worthwhile pain relief.
For funders
Gabapentin at a dose of 1800 to 3600 mg daily (1200 to 3600 mg gabapentin encarbil) can provide good levels of pain relief to some people with postherpetic neuralgia and peripheral diabetic neuropathy. Evidence for other types of neuropathic pain is very limited. The outcome of at least 50% pain intensity reduction is regarded as a useful outcome of treatment by people with chronic neuropathic pain, and the achievement of this degree of pain relief is associated with important beneficial effects on sleep interference, fatigue, and depression, as well as quality of life, function, and work. Around 3 to 4 out of 10 achieved this degree of pain relief with gabapentin, compared with 1 to 2 out of 10 for placebo. Over half of those treated with gabapentin will not have worthwhile pain relief. The level of efficacy found for gabapentin is consistent with the efficacy estimates for other drug therapies in these conditions.
Implications for research.
General
The design of studies in neuropathic pain, and the outcomes, are well understood, but as the number of people experiencing good pain relief with gabapentin over the longer term (12 weeks) is likely to be small, an enriched‐enrolment randomised‐withdrawal (EERW) design might provide the highest sensitivity to detect a signal (Moore 2015c). Since combination therapy for neuropathic pain has been reported to be more effective than monotherapy with any drug (Chaparro 2012), and combination therapy is common clinical practice, studies examining gabapentin in combination with an antidepressant could be of interest. Combinations with strong opioids are likely to be used less, owing to their limited efficacy and known harms. More interesting might be the combined use of gabapentin with tricyclic antidepressants, weak opioids, or tramadol, and examination of the timings and sequencing of these drugs with gabapentin.
More research is warranted into the efficacy of gabapentin in painful neuropathic pain conditions where there is currently inadequate information. These conditions tend to be uncommon, and studies can be difficult, with few possible participants.
Design
Reporting of clinically relevant outcomes using appropriate imputation for withdrawal would improve the relevance of the findings for clinical practice. The use of EERW designs for comparison with classic trial designs indicates that good quality EERW designs of long duration may be appropriate for neuropathic pain.
Stratification by phenotype might be an interesting possibility for future studies (Baron 2017), as well as the possibility of measuring pain scores with activity (including dynamic tactile allodynia) versus at rest or on average/worst/best over prior 24 hours. Participant‐level data might be of importance in identifying responder clusters and characteristics.
While pain is important, other outcomes relating to function, sleep, fatigue, and quality of life are also important, and are probably closely linked (Hoffman 2010). Participant‐level data could shed light on these relationships.
The main issue, though, is not whether gabapentin is effective, but how best to use it in clinical practice to generate the best results for most people with a chronic neuropathic pain condition, in the shortest time, and at the lowest cost. New study designs have been proposed to examine this (Moore 2010f).
Measurement (endpoints)
Assessment of neuropathic pain and other symptoms should be based on dichotomous participant‐reported outcomes of proven clinical utility.
Comparison between active treatments
There seems little point in comparing gabapentin directly with other treatments; the issue is what works for whom.
Feedback
Feedback submitted 2015, 29 May 2015
Summary
Date of Submission: 29‐May‐2015
Name: Michael Chan BSc(Pharm); Danielle Ghag BSc(Pharm); Aaron Tejani PharmD
Affiliation: UBC
Role: Pharmacist
Comment: Written by Michael Chan BSc(Pharm), Danielle Ghag BSc(Pharm), Aaron Tejani PharmD
Dear Cochrane Review Team,
We read with great interest the systematic review of Gabapentin for chronic neuropathic pain and fibromyalgia in adults by Moore 2014. Although this systematic review has taken on the arduous task of ascertaining the highest level of available evidence, it is made difficult by the inherent bias that plagues the trials in the literature. This was evidenced upon further analysis of the 6 trials that were included in outcome 1.1, “At least 50% pain reduction over baseline”. The results of this outcome were subject to the limitations of the methodology in these studies that were not adequately accounted for in this review article.
The five‐point Oxford Scale was included for each study to assess the risk of bias. This scale has been shown to provide unreliable validity assessments and its use is discouraged because it does not address important biases such as allocation concealment. Moreover, since gabapentin has a profound side effect profile, participants may have correctly anticipated which treatment they received. Thus, we feel that blinding is not adequately assessed through the Oxford scale, as points are allocated for double blinding without considering whether blinding was maintained throughout the study. In these cases, the risk of bias due to blinding may be better represented as an unclear risk or as some may argue, high risk. This would lead to reclassification of Sang 2013, Wallace 2010 and Zhang 2013 from low risk to unclear or high risk of bias, which may impact our interpretation of outcome 1.1. Furthermore, the effect size of gabapentin may be an overestimation as compromised blinding may account for an exaggerated effect of 13% (Savović 2012).
The aforementioned risk of bias due to blinding may be exacerbated by partial enrichment of the population that was enrolled. Studies by Sang 2013, Wallace 2010 and Zhang 2013 included patients who had previously responded to gabapentin, and excluded those who did not respond or tolerate gabapentin. This subset of participants who have already received the active drug, may be able to determine which drug they are receiving based on their knowledge of its anticipated effects, therefore jeopardizing blinding. Thus, enrichment can introduce performance and selection bias, which falsely inflates the proportion of patients who respond to active treatment.
This review assumed that treatment effects were not significantly affected by partial enrichment based on the results of the systematic review by Straube 2008, which examined the effects of enrichment in 21 trials of gabapentin or pregabalin. Of the 12 studies that examined gabapentin specifically, 10 were not enriched and 2 were partially enriched. A limitation of Straube 2008 was that the 2 partially enriched studies did not provide the proportion of patients taking gabapentin at baseline. This makes it difficult to determine the degree and implications of enrichment. Also, Straube 2008 stated it was difficult to make meaningful comparisons between trials using different doses of gabapentin and enrolment strategies.
The issue of enriched enrolment is exemplified by the poorly described baseline characteristics in most of the studies for outcome 1.1. Although, Sang 2013 specified that 43.6% and 39.6% of those in the gabapentin and placebo groups respectively had received gabapentin or pregabalin prior to enrolment, other studies did not disclose this information. Due to the uncertainty surrounding the impact that enrichment has on the treatment effects of gabapentin, we believe that a subgroup analysis may be appropriate to analyze enriched and non‐enriched studies independently. The impact of enrichment may jeopardize internal and external validity, which we feel were not adequately addressed in the “Overall Completeness and Applicability of the Evidence”.
The majority of the included studies reported in outcome 1.1 did not disclose the proportion of patients receiving tricyclic antidepressants concomitantly or specify whether the dose was altered during the study. Since there is uncertainty surrounding the maintenance of blinding, this could lead to researchers favoring the gabapentin group by altering TCAs or other analgesics accordingly.
The review article stated that a fixed‐effects model would be used if statistically significant heterogeneity was found. Despite this, even though there was statistically significant heterogeneity for outcomes 1.2.2 and 1.3.1, a fixed effects model was still used. Moreover, the review did not provide an assessment of possible reasons for heterogeneity. A random‐effects model meta‐analyses would be a more conservative approach to address the heterogeneity to provide a more meaningful conclusion (Higgins 2011).
For outcome 1.1 Baseline Observation Carried Forward (BOCF) was utilized to address attrition in two of the six studies, which accounted for over half of the weight. Although deemed a conservative approach, it can lead to an overestimation or underestimation of the number of patients with greater than 50% improvement from baseline. For example, the BOCF may indirectly overestimate the treatment effect of gabapentin by not taking into account the proportion of those receiving placebo who experienced a 50% improvement. This is of particular concern since we believe that blinding may have been compromised in these trials as described above. This unclear risk of bias is not captured in the summary tables which classifies BOCF as low risk. Moreover, the Summary of Findings Table for Main Comparisons for postherpetic neuralgia states that “Imputation method used [was] (LOCF) and small study size could influence results to reduce gabapentin efficacy”. This statement is not entirely accurate as Sang 2013 and Wallace 2010, which account for approximately 58.1% of the weight of outcome 1.1, use BOCF. Even so, we disagree with the fact that the Last Observation Carried Forward (LOCF) would reduce the treatment as it may in fact increase or decrease it. Despite our best efforts to postulate whether or not LOCF and BOCF would alter treatment effects, the best approach would be delving into the individual studies and contacting the authors for missing information.
One possible intervention to increase the confidence of the results in this review would be to conduct a sensitivity analysis. We would have liked to see a sensitivity analysis performed regardless of the number of studies available. Sensitivity analysis would help to characterize the impact of methodological limitations on the results of the systematic review.
Best Regards,
Michael Chan BSc(Pharm),
Danielle Ghag BSc(Pharm) and
Aaron M Tejani PharmD
References:
1. Zhang L, Rainka M, Freeman R, Harden RN, Bell CF, Chen C, et al. A Randomized, Double‐Blind, Placebo‐Controlled Trial to Assess the Efficacy and Safety of Gabapentin Enacarbil in Subjects With Neuropathic Pain Associated With Postherpetic Neuralgia (PXN110748). J Pain. 2013 Jun;14(6):590–603.
2. Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. Br J Clin Pharmacol. 2008 Aug;66(2):266–75.
3. Wallace MS, Irving G, Cowles VE. Gabapentin Extended‐Release Tablets for the Treatment of Patients with Postherpetic Neuralgia. Clin Drug Investig. 2010;30(11):765–76.
4. Moore RA, Wiffen PJ, Derry S, Toelle T, Rice ASC. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. In: The Cochrane Collaboration, editor. Cochrane Database of Systematic Reviews [Internet]. Chichester, UK: John Wiley & Sons, Ltd; 2014 [cited 2015 May 29]. Available from: http://doi.wiley.com/10.1002/14651858.CD007938.pub3
5. Rice ASC, Maton S, Group1UK PNS, others. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain. 2001;94(2):215–24.
6. Sang CN, Sathyanarayana R, Sweeney M, Investigators D‐1796 S, others. Gastroretentive gabapentin (G‐GR) formulation reduces intensity of pain associated with postherpetic neuralgia (PHN). Clin J Pain. 2013;29(4):281–8.
7. Savović J, Jones H, Altman D, Harris R, Jűni P, Pildal J et al. Influence of reported study design characteristics on intervention effect estimates from randomised controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment. 2012;16(35). .
8. Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
I agree with the conflict of interest statement below:
I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Reply
Chan and colleagues begin by suggesting that the presence of adverse events with an active drug may compromise an overall blinding of the trial by an external observer, as they would anticipate that a person with adverse events had had an active drug, while those without had placebo. That would be even when, as in the three studies you mentioned, there was a matched placebo so that neither patients nor observers were aware of the allocation initially.
Of course, for the individual patient, who cannot see the overall picture, that would not be the case. And since the individual patient makes their own judgment about pain and other outcomes, the position of the outside observer is irrelevant. Moreover, when you look at the actual event rates for adverse events in these three trials, and overall, there is a rather low increase in adverse event rates (RR 1.25 overall). Wallace and Zhang showed no difference in event rates between gabapentin and placebo, which makes it especially hard to see how this suggested bias would act.
In this circumstance, it is hard to see what justification they can have for their statements, unless supported with empirical evidence from elsewhere. We have been looking for some years now, as we have an interest in the methodology of systematic reviews and sources of bias, and are aware of none.
In passing, we use both the Oxford Quality Score (to help justify inclusion and exclusion – studies must be randomised and double blind to be accepted) and a version of Risk of Bias. The OQS now has well over 10,000 citations, and is validated. Cochrane RoB omits several important, and possibly crucial, sources of bias. Neither is perfect, but when detecting bias we need all the tools at our disposal.
They also make a point about partial enrichment. The situation right now is that there is zero empirical evidence that partial enrichment makes any difference to results of clinical trials in neuropathic pain. It may well be, as they say, that some residual bias is not accounted for, but that is speculation, and not fact. The fact is that the three studies that they seem to be concerned about are not out of line with others in the analyses, and one of them, for analysis of PGIC, was not different from placebo.
Chan and colleagues are also concerned with patients receiving TCAs. Actually, it is very unlikely that TCA prescribing changes affected the results. Most trials indicated that any concomitant therapies would not be changed during the course of the trial. It is an interesting speculation, but since tricyclic efficacy is as low as all others in NP (based on the rather inadequate evidence we have, as well as clinical experience), one would really need to push this to an extreme to explain any result. Is there any evidence that increasing doses of TCAs has any dramatic effect on analgesia? We know of none, and we also know that most people do not respond to TCAs while many suffer adverse events, which often make them desist. It is a hard argument to maintain.
Issues around statistics refer to situations with only a handful of studies, or where one study (Zhang) gave a result favouring placebo. Random effects models are more appropriate where there is clinical heterogeneity, which we try to avoid. Changing to random effects does not change the result, but we might revisit this. Actually RE is more appropriate where there are a number of small studies, which is where heterogeneity can occur – but there are number of issues intertwined here, so it isn't simple. For example, examples of fraudulent research often show high degrees of homogeneity, and heterogeneity tests can be used to detect fraud. We may need to reword the methods and revisit thinking on this.
We found their point about imputation rather difficult to understand. We cannot see why that should be because the imputation is applied equally to both active and placebo. In several individual patient level calculations that have used LOCF and BOCF there has been little effect of imputation method on placebo, only on active treatments where there is a large adverse event withdrawal rate, as we pointed out in our analysis in Pain. And there is good evidence of potentially very large positive bias for opioids in chronic non‐cancer pain.
We are sorry Chan and colleagues disagree with the current evidence on imputation method. We use BOCF to produce a result where patients who are able to remain on treatment with tolerable adverse events have a high degree of pain relief. That makes clinical sense, and is what systematic reviews tell us that patients want. It also makes sound economic sense. Using LOCF to impute results where up to 65% of patients drop out over 12 weeks (as in opioid studies in chronic non‐cancer pain) might be of some statistical interest, and might produce significant results where BOCF does not, but it takes some explaining as to its relevance to the real world. Unless and until that is explained to us and supported with empirical evidence, we are more than happy to stick to our guns on this.
As to contacting authors, we have done – or rather had discussions with pharmaceutical companies about the possibility of obtaining individual patient level data for gabapentin. This will not be possible. It is a shame, because in other circumstances where we could obtain patient level data we have been able to make some interesting and important methodological advances, even though you appear not to agree with them.
We find it hard to understand why Chan and colleagues would want sensitivity analysis with inadequate data. What we know is that small studies, and small numbers of small studies, can give us the wrong answer. This has been evident for at least 20 years, and is supported by several recent major studies, often in pain topics. To use unreliable evidence on which to base judgments like that seems retrograde.
Andrew Moore, Sheena Derry, Phil Wiffen
Editorial note: this review will be assessed for updating in 2019, and may then be split into two reviews: neuropathic pain, and fibromyalgia.
Contributors
Feedback Editor Kate Seers, Managing Editor Anna Hobson, and review authors.
Feedback submitted, 26 March 2019
Summary
Date of Submission: 27‐Mar‐2019 Name: Aron Nenninger Email Address: Aron.Nenninger@northernhealth.ca Affiliation: British Columbia PAD Provincial Academic Detailing Service, Canada Role: Academic Detailing Pharmacist
I am writing to pass along what appears to be a data transcription error within the recent Cochrane Review, Gabapentin for Chronic Neuropathic Pain in Adults, CD007938, published in June 2017. While reading one of the articles included in the Cochrane Review, referenced as Rauck 2013a (1), it appears there is a data transcription error in the Cochrane analysis in which data from the pregabalin active control group seems to have been used in place of data from the placebo control group. It looks like the data in question are reported from Rauck’s table 7. In the Cochrane Analysis 1.1, At least 50% pain reduction over baseline, the placebo group from Rauck is reported as 14/66 which seems to be data from the pregabalin group in the original paper. The placebo group in Rauck table 7 should be reported as 35/120. Similarly in Analysis 1.4, IMMPACT outcome of substantial improvement, it appears the same data was used from Rauck as in analysis 1.1, so a similar issue exists with pregabalin results (14/66) being reported as the placebo (35/120). In the rest of the Cochrane analyses, the placebo group from Rauck is reported with a denominator of 120, and appears accurate. Thank you for your ongoing work on the Cochrane reviews and in how outcomes are reported. They are consistently very useful in trying to sort out the clinical utility of these medications, and in informing frameworks of how to approach pain conditions.
1. Rauck R, Makumi CW, Schwartz S, Graff O, Meno‐Tetang G, Bell CF, et al. A randomized, controlled trialof gabapentin enacarbil in subjects with neuropathic painassociated with diabetic peripheral neuropathy. PainPractice 2013;13(6):485–96. CTG: NCT02074267; DOI:10.1111/papr. 12014
Reply
Thank you for spotting this. You are of course correct. We have made the correction, and the result is that, compared with placebo and for at least 50% pain relief, the RR falls from 1.9 (1.5 to 2.3) to 1.7 (1.4 to 2.0), while the NNT increases from 5.9 (4.6 to 8.3) to 6.6 (5.0 to 9.7); the number in the analyses increases slightly. The same change was made to the IMMPACT substantial outcome, that uses identical data. That's analyses 1.1 and 1.4 (Analysis 1.1; Analysis 1.4).
We have also made the changes in the Abstract, Results section (including a new version of Figure 9), SoF tables (Table 1; Table 2; Table 3), and in the Discussion, where relevant.
As you point out, the correction makes no clinical difference.
While it is annoying that a mistake occurred, it is terrific in a living document like this that it can be corrected as soon as it is spotted and verified.
Andrew Moore
Contributors
Feedback Editor Hayley Barnes, Co‐ordinating Editor Christopher Eccleston, and Managing Editor Anna Erskine.
What's new
| Date | Event | Description |
|---|---|---|
| 18 February 2020 | Amended | Clarification added to Declarations of interest. |
| 11 October 2017 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 3, 2009 Review first published: Issue 3, 2011
| Date | Event | Description |
|---|---|---|
| 28 May 2019 | Amended | Contact details updated. |
| 27 March 2019 | Feedback has been incorporated | See Feedback 2. |
| 28 April 2017 | New search has been performed | This review has been updated to include the results of a new search on 17 January 2017 |
| 25 April 2017 | New citation required but conclusions have not changed | New search resulting in four additional studies (530 participants). Modified inclusion and exclusion criteria, mainly concerned with newer definitions of neuropathic pain resulting in exclusion of three previously included studies |
| 23 July 2015 | Amended | This review is being split; see Published notes |
| 6 July 2015 | Feedback has been incorporated | See Feedback section for details. |
| 19 May 2014 | Amended | Mistake in Summary of findings table corrected |
| 28 April 2014 | Review declared as stable | This review will be assessed for updating in 2019. |
| 17 March 2014 | New citation required but conclusions have not changed | Additional studies did not change efficacy or harm estimates in any clinically significant way |
| 17 March 2014 | New search has been performed | New searches. New studies added. Minor methodological amendments made, in line with current standards. The original chronic pain review included 14 studies with 1392 participants in 13 reports. The 2011 update involved 29 studies in 29 reports with 3571 participants. In this update we consider 33 studies in 34 reports, involving 4388 participants taking oral gabapentin. We have added seven new studies of oral gabapentin with 1919 participants (Backonja 2011; Harden 2013; Mishra 2012; NCT00475904; Rauck 2013a; Sang 2013; Zhang 2013) and another new publication (Sandercock 2012) that provided results for a study that was already included but did not provide usable data (Sandercock 2009). We also identified a small study, with 170 participants, using an experimental formulation of injected (intrathecal) gabapentin (Rauck 2013b). |
Notes
No new studies likely to change the conclusions are expected. Therefore, this review has now been stabilised until 2022 following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
We wish to thank Dr Thomas Perry for directing us to clinical trial reports and synopses of published and unpublished studies of gabapentin, and Dr Mike Clarke and colleagues for their advice and support relating to the 2011 update. Professor Henry McQuay was an author and contributor on previous versions and provided considerable help and guidance throughout.
We are extremely grateful to Ke Deng for timely help with translation and data extraction for one of the included studies (Gong 2008).
The review followed the agreed template for neuropathic pain, which was developed in collaboration with Cochrane Musculoskeletal and Cochrane Neuromuscular Diseases. The editorial process was managed by Cochrane Pain, Palliative and Supportive Care.
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to Cochrane Pain, Palliative and Supportive Care (PaPaS). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several changes in how the efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with 'any improvement'. Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be of longer duration, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for the inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. To summarise some of the recent insights that must be considered in this new review:
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011b), back pain (Moore 2010d), and arthritis (Moore 2010e), as well as in fibromyalgia (Straube 2010); in all cases average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable.
As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials of less than 12 weeks' duration, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2010d); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of people with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis to 30% in fibromyalgia (Moore 2009; Moore 2010d; Moore 2010e; Moore 2013b; Moore 2014b; Straube 2008; Sultan 2008). One Cochrane Review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good reasons for doing so.
Individual patient analyses indicate that people who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010c; Moore 2014b).
Imputation methods such as last observation carried forward (LOCF), used when participants withdraw from clinical trials, can overstate drug efficacy especially when adverse event withdrawals with drug are greater than those with placebo (Moore 2012a).
Appendix 2. CENTRAL search strategy
(gabapentin* or neurontin* or neurotonin*):TI,AB,KY (1184)
MESH DESCRIPTOR Neuralgia EXPLODE ALL TREES (718)
MESH DESCRIPTOR Peripheral Nervous System Diseases EXPLODE ALL TREES (2963)
MESH DESCRIPTOR Somatosensory Disorders EXPLODE ALL TREES (796)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)):TI,AB,KY (3931)
((neur* or nerv*) adj6 (compress* or damag*)):TI,AB,KY (732)
2 OR 3 OR 4 OR 5 OR 6 (7377)
1 AND 7 (215)
01/01/2014 TO 16/01/2017:CD (269940)
8 AND 9 (107)
Appendix 3. MEDLINE (via OVID) search strategy
(gabapentin* or neurontin* or neurotonin*).mp. (5327)
exp NEURALGIA/ (18298)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (142504)
exp SOMATOSENSORY DISORDERS/ (20859)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (50119)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (59288)
2 or 3 or 4 or 5 or 6
randomized controlled trial.pt. (484826)
controlled clinical trial.pt. (97360)
randomized.ab. (371343)
placebo.ab. (182076)
drug therapy.fs (2092559)
randomly.ab. (255301)
trial.ti. (167136)
groups.ab (1572017)
8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 (3937255)
1 and 7 and 16 (1363)
limit 17 to yr="2014 ‐Current" (237)
Appendix 4. Embase (via OVID) search strategy
gabapentin/ (25201)
(gabapentin* or neurontin* or neurotonin*).mp. (35892)
1 or 2 (25892)
exp neuralgia/ (93025)
exp peripheral neuropathy/ (62561)
exp somatosensory disorder/ (83900)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (94901)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (81121)
4 or 5 or 6 or 7 or 8 (330939)
clinical trial/ (1025277)
controlled clinical trial/ (468659)
randomized controlled trial/ (469523)
double‐blind procedure/ (140238)
(clin* adj25 trial*).mp. (1445057)
((doubl* or trebl* or tripl*) adj25 (blind* or mask*)). mp. (222270)
placebo*.ti.ab. (252536)
random*.ti,ab. (1172190)
10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 (2247471)
3 and 9 and 18 (2998)
limit 19 to yr="2014 ‐Current" (484)
Appendix 5. Potential sources of bias in studies of chronic pain used in the 'Risk of bias' table
| Item | High | Unclear | Low |
| Randomisation | Not randomised | Claims randomisation, but no method given | Randomised by adequate method |
| Allocation concealment | Not reported | Reported but not described | Allocation undertaken independently and blind to investigator |
| Blinding | Not double‐blind | Claims double‐blind, but no method | Convincingly double‐blind |
| Duration | 2 weeks or less | 3 to 6 weeks | 7 weeks or more |
| Outcome | Anything less than 30% pain intensity reduction Pain state ≥ 50/100 mm or equivalent or undefined | Responder: pain intensity reduction of ≥ 30% from baseline State: final pain intensity < 50/100 mm, or equivalent | Responder: pain intensity reduction of ≥ 50% from baseline State: final pain intensity < 30/100 mm, or equivalent State: no worse than mild pain |
| Incomplete outcome assessment | Average results only | Responder or state with last observation carried forward or imputation method for missing data or after withdrawal not stated | Responder or state response, using baseline observation carried forward (zero improvement after withdrawal) |
| Size | < 50 participants per treatment arm | 50 to 199 participants per treatment arm | ≥ 200 participants per treatment arm |
Appendix 6. GRADE: criteria for assigning grade of evidence
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Cochrane Handbook of Systematic Reviews of Interventions, Chapter 12, Schünemann 2011b).
High: randomised trials; or double‐upgraded observational studies
Moderate: downgraded randomised trials; or upgraded observational studies
Low: double‐downgraded randomised trials; or observational studies
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports
Factors that may decrease the quality level of a body of evidence are:
limitations in the design and implementation of available studies suggesting high likelihood of bias;
indirectness of evidence (indirect population, intervention, control, outcomes);
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
imprecision of results (wide confidence intervals);
high probability of publication bias.
Factors that may increase the quality level of a body of evidence are:
large magnitude of effect;
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
dose‐response gradient.
Appendix 7. Summary of outcomes in individual studies
| Study | Withdrawals | Efficacy | Adverse events (general) | Adverse events (specific) |
| Postherpetic neuralgia | ||||
| Backonja 2011 | All‐cause withdrawal
GabaEn 2/47
Placebo 7/54 AE withdrawal GabaEn 0/47 Placebo 4/54 (inc pain 2, dizziness 1, noncardiac chest pain 1) No LoE withdrawals in double‐blind phase Gabapentin phase (titration) AE withdrawal 3/116 (4 later: dizziness 2, lethargy 1, tachycardia 1) LoE withdrawal 1/116 |
At least 30% reduction in pain GabaEn 26/47 Placebo 15/54 At least 50% reduction in pain GabaEn 13/47 Placebo 10/54 PGIC much and very much improved GabaEn 20/47 Placebo 7/54 | At least one AE GabaEn 25/47 Placebo 25/47 No SAE No deaths In gabapentin phase 48/115 had ≥ one AE | Dizziness GabaEn 10/47 Placebo 3/54 Nausea GabaEn 5/47 Placebo 2.54 Headache GabaEn 4/47 Placebo 4/54 Diarrhoea GabaEn 3/47 Placebo 1/54 Fatigue GabaEn 2/47 Placebo 4/54 In gabapentin phase dizziness 16/115, nausea 2, headache 5, diarrhoea 4, fatigue 2 |
| Chandra 2006 | All‐cause withdrawal Gabapentin 3/38 Nortriptyline 2/38 AE withdrawal Gabapentin 0/38 Nortriptyline 1/38 LoE withdrawal Gabapentin 0/38 Nortriptyline 1/38 | At least 50% improvement over baseline pain (Likert) Gabapentin 7/38 Nortriptyline 9/38 At least 50% improvement over baseline pain (VAS) Gabapentin 13/38 Nortriptyline 14/38 | No serious AE reported No deaths reported | Sleepiness Gabapentin 4/38 Nortriptyline 6/38 Giddiness Gabapentin 1/38 Nortriptyline 0/38 |
| Gong 2008 | 16 participants withdrew because of AEs, lack of analgesic effect, or other reasons No further details | 25% to ≤ 50% pain relief Gabapentin 45/109 Placebo 13/106 ≥ 50% pain relief Gabapentin 30/109 Placebo 4/106 "Patient evaluation" "Mild effective" Gabapentin 37/109 Placebo 28/106 "Excellent" Gabapentin 48/109 Placebo 5/106 | At least one AE Gabapentin 78/109 Placebo 31/106 Most appeared in the first week, and symptoms relieved gradually as the treatment continued |
Dizziness Gabapentin 27/109 Placebo 10/106 Somnolence Gabapentin 19/109 Placebo 3/106 Peripheral oedema Gabapentin 6/109 Placebo 1/106 |
| Harden 2013 | All‐cause GabaEn 1200 12/91 GabeEn 3600 3/85 GabaEn 2400 1 (cross‐over) AE withdrawal GabaEn 1200 3/91 GabaEn 3600 0/85 LoE withdrawal GabaEn 1200 4/91 GabaEn 3600 0/85 | ≥ 50% red in PI At end of period 1 GabaEn 1200 7/49 GabaEn 3600 5/44 At end of period 2 GabaEn 1200 8/41 GabaEn 3600 11/41 ≥ 30% red in PI At end of period 1 GabaEn 1200 13/49 GabaEn 360013/44 At end of period 2 GabaEn 120015/41 GabaEn 3600 19/41 PGIC much or v much improved GabaEn 1200 17/63 GabaEn 3600 28/61 (paper says ITT ‐ not sure where denominator comes from; summary says "at last week of treatment" ‐ completer?) | Overall incidence of AEs and changes in safety parameters were small and similar between doses One SAE during down titration (auditory hallucination) At least 1 AE B'line Gabapentin 1800 2/94 GabaEn 1200 15/91 GabaEn 2400 2.82 GabaEn 3600 14/85 Down‐titration 2/80 | Dizziness
GabaEn 1200 0/91
GabaEn 3600 3/85 Somnolence GabaEn 1200 3/91 GabaEn 3600 2/85 Peripheral oedema GabaEn 1200 1/91 GabaEn 3600 1/85 |
| Irving 2009 | All‐cause withdrawal 15 total AE withdrawal Gabapentin 1800 single dose 4/44 Gabapentin 1800 split dose 6/52 Placebo 1/51 | At least 50% reduction in pain score Gabapentin 1800 single dose 14/55 Gabapentin 1800 split dose 15/52 Placebo 6/51 At least 30% reduction in pain score Gabapentin 1800 single dose 24/55 Gabapentin 1800 split dose 25/52 Placebo 16/51 PGIC very much or much improved Gabapentin 1800 single dose 18/55 Gabapentin 1800 split dose 21/52 Placebo 11/5 Significantly better sleep with gabapentin compared with placebo | Serious AE Gabapentin 1800 single dose 4/55 Gabapentin 1800 split dose 3/52 Placebo 1/51 Deaths Gabapentin 1800 single dose 0/55 Gabapentin 1800 split dose 1/52 Placebo 0/51 | Somnolence Gabapentin 1800 single dose: 5/55 Gabapentin 1800 split dose: 4/52 Placebo: 4/51 Dizziness Gabapentin 1800 single dose: 12/55 Gabapentin 1800 split dose: 6/52 Placebo: 5/51 Gait disturbance Gabapentin 1800 single dose: 4/55 Gabapentin 1800 split dose: 2/52 Placebo: 0/51 Peripheral oedema Gabapentin 1800 single dose: 4/55 Gabapentin 1800 split dose: 1/52 Placebo: 0/51 |
| NCT00475904 | All‐cause Gabapentin 13/144 A+K cream 15/140 Placebo 4/76 No reasons for withdrawal given | Reduction in PI from baseline Mean data only No significant difference between Gabapentin and cream Cream marginally better than placebo Note ‐ claims ITT analysis with LOCF, but numbers analysed are fewer than randomised (Gabapentin 6, Cream 5) | At least one AE Gabapentin 2/144 Cream 7/144 Placebo 1/76 No SAE Assume no deaths | Vertigo Gabapentin 2/144 Cream 7/144 Placebo 1/76 No other AEs reported |
| Rice 2001 | Gabapentin 1800 mg All‐cause 22 AE 15 LoE 4 Gabapentin 2400 mg All‐cause 23 AE 19 LoE 1 Placebo All‐cause 17 AE 7 LoE 4 | At least 50% reduction in mean pain score Gabapentin 1800: 37/115 Gabapentin 2400: 37/108 Placebo: 16/111 PGIC very much or much improved Gabapentin 1800: 44/115 Gabapentin 2400: 42/108 Placebo: 24/111 PGIC very much improved (CTR) Gabapentin 1800: 18/115 Gabapentin 2400: 12/108 Placebo: 7/111 PGIC much improved (CTR) Gabapentin 1800: 26/115 Gabapentin 2400: 30/108 Placebo: 17/111 Some significant differences in QoL measures and sleep | At least one AE Gabapentin 1800: 81/115 Gabapentin 2400: 81/108 Placebo: 55/111 SAE Gabapentin 1800: 3/115 Gabapentin 2400: 1/108 Placebo: 1/111 Death: Gabapentin 1800: 0/115 Gabapentin 2400: 1/108 Placebo: 0/111 | Somnolence Gabapentin 1800: 20/115 Gabapentin 2400: 22/108 Placebo: 7/111 Dizziness Gabapentin 1800: 36/115 Gabapentin 2400: 36/108 Placebo: 11/111 Asthenia Gabapentin 1800: 7/115 Gabapentin 2400: 6/108 Placebo: 4/111 Peripheral oedema Gabapentin 1800: 6/115 Gabapentin 2400: 12/108 Placebo: 0/111 |
| Rowbotham 1998 | Gabapentin All‐cause 24 AE 21 LoE 0 Placebo All‐cause 21 AE 14 LoE 2 | PGIC moderate or much improved Gabapentin: 47/113 Placebo: 14/116 PGIC CTR much improved Gabapentin: 21/113 Placebo: 6/116 PGIC CTR moderately improved Gabapentin: 26/113 Placebo: 8/116 No change in pain 60% placebo, 23% gabapentin No change or worse in pain 68% placebo, 26% gabapentin Significant improvement over placebo in 5/9 SF‐36 QoL and 5/7 mood states | At least one AE Gabapentin 84/113 Placebo 60/116 Minor AE (treatment‐related) Gabapentin: 62/113 Placebo: 32/116 SAE (treatment‐related) Gabapentin: 0/113 (10/113 CTR) Placebo: 0/116 (5/116 CTR) Death: Gabapentin: 0/113 Placebo: 1/116 | Somnolence Gabapentin: 31/113 Placebo: 6/116 Dizziness Gabapentin: 27/113 Placebo: 6/116 Ataxia Gabapentin: 8/113 Placebo: 0/116 Peripheral oedema Gabapentin: 11/113 Placebo: 4/116 |
| Sang 2013 | All‐cause withdrawal Gabapentin 35/221 Placebo 37/231 AE withdrawal Gabapentin 19/221 Placebo 10/231 LoE withdrawal Gabapentin 7/221 Placebo 12/231 | At least 50% reduction in pain Gabapentin 65/221 Placebo 52/231 PGIC very much or much improved Gabapentin 94/221 Placebo 77/231 | At least one AE Gabapentin 118/221 Placebo 92/231 Serious AE Gabapentin 4/221 Placebo 6/231 none attributed to study drug Deaths Gabapentin 0/221 Placebo 1/231 | Dizziness Gabapentin 25/221 Placebo 4/231 Somnolence Gabapentin 12/221 Placebo 7/231 Headache Gabapentin 10/221 Placebo 9.231 Nausea Gabapentin 10/221 Placebo 7/231 Peripheral oedema Gabapentin 7/221 Placebo 1/231 Nasopharyngitis Gabapentin 5/221 Placebo 6/231 |
|
Wallace 2010 y |
All‐cause withdrawal Gabapentin 56/269 Placebo 30/131 AE withdrawal Gabapentin 31/269 Placebo 14/131 | At least 50% improvement over baseline pain (Likert)
Gabapentin 95/269
Placebo 36/131 Much or very much improved on PGIC Gabapentin 99/269 Placebo 32/131 |
At least one AE Gabapentin 155/272 Placebo 64/133 Serious AE Gabapentin 10/272 Placebo 4/133 Deaths Gabapentin 0/272 Placebo 1/133 | Dizziness
Gabapentin 34/272
Placebo 4/133 Somnolence Gabapentin 13/272 Placebo 3/133 Peripheral oedema Gabapentin 13/272 Placebo 0/133 |
| Zhang 2013 | To end of maintenance phase All‐cause withdrawal GabaEr1200 20/107 GabaEr 2400 21/82 GabaEr 3600 30/87 Placebo 30/95 AE withdrawal GabaEr1200 6/107 GabaEr 2400 12/82 GabaEr 3600 16/87 Placebo 11/95 LoE GabaEr1200 1/107 GabaEr 2400 1/82 GabaEr 3600 4/87 Placebo 6/95 Withdrawal of consent and protocol deviation most common other reasons | At least 50% reduction in pain by end maintenance GabaEr1200 44/107 GabaEr 2400 28/82 GabaEr 3600 37/87 Placebo 22/95 At least 30% reduction in pain by end maintenance GabaEr1200 57/107 GabaEr 2400 48/82 GabaEr 3600 52/87 Placebo 40/95 PGIC much and very much improved GabaEr1200 24/85 GabaEr 2400 45/103 GabaEr 3600 35/78 Placebo 39/76 Note not ITT | At least 1 AE GabaEr1200 75/107 GabaEr 2400 64/82 GabaEr 3600 71/87 Placebo 63/95 SAE: GabaEr1200 0/107 GabaEr 2400 4/82 GabaEr 3600 2/87 Placebo 2/95 No deaths | Dizziness
GabaEr120018/107
GabaEr 2400 21/82
GabaEr 3600 26/87
Placebo 14/95 Somnolence GabaEr120011/107 GabaEr 2400 9/82 GabaEr 3600 12/87 Placebo 8/95 Peripheral oedema GabaEr1200 6/107 GabaEr 2400 6/82 GabaEr 3600 5/87 Placebo 0/95 Other AEs in ≥ 5% reported |
| Painful diabetic neuropathy | ||||
| Backonja 1998 | All‐cause withdrawal Gabapentin 14/84 Placebo 16/81 AE withdrawal Gabapentin 7/84 Placebo 5/81 LoE withdrawal Gabapentin 1/84 Placebo 5/81 | PGIC much or moderately improved Gabapentin 47/84 Placebo 25/81 At least 50% reduction in pain (CTR) Gabapentin 39/84 Placebo 16/81 PGIC much improved (CTR) Gabapentin 33/84 Placebo 12/81 PGIC moderately or much improved (CTR) Gabapentin 47/84 Placebo 25/81 | At least one AE Gabapentin 70/84 Placebo 54/81 Serious AE Gabapentin 3/84 Placebo 2/81 Deaths Gabapentin 0/84 Placebo 0/81 | Dizziness Gabapentin 20/84 Placebo 4/81 Somnolence Gabapentin 19/84 Placebo 5/81 |
| CTR 945‐224 | All‐cause withdrawal
Gabapentin 600 12/82
Gabapentin 1200 6/82
Gabapentin 2400 19/84
Placebo 12/77
AE withdrawal
Gabapentin 600 8/82
Gabapentin 1200 3/82
Gabapentin 2400 11/84
Placebo 8/77 LoE withdrawal Gabapentin 600 0/82 Gabapentin 1200 0/82 Gabapentin 2400 4/84 Placebo 1/77 |
At least 50% reduction in pain score Gabapentin 600 13/82 Gabapentin 1200 33/82 Gabapentin 2400 25/84 Placebo 19/77 PGIC very much improved Gabapentin 600 9/82 Gabapentin 1200 14/82 Gabapentin 2400 14/84 Placebo 10/77 PGIC much or very much improved Gabapentin 600 22/82 Gabapentin 1200 36/82 Gabapentin 2400 36/84 Placebo 26/77 | At least 1 AE Gabapentin 600 40/82 Gabapentin 1200 35/82 Gabapentin 2400 45/84 Placebo 36/77 Serious AE Gabapentin 600 5/82 Gabapentin 1200 2/82 Gabapentin 2400 3/84 Placebo 4/77 There were no deaths | Somnolence Gabapentin 600 4/82 Gabapentin 1200 3/82 Gabapentin 2400 11/84 Placebo 1/77 Dizziness Gabapentin 600 7/82 Gabapentin 1200 4/82 Gabapentin 2400 6/84 Placebo 2/77 Peripheral oedema Gabapentin 600 4/82 Gabapentin 1200 1/82 Gabapentin 2400 2/84 Placebo 2/77 |
| CTR 945‐1008 Multicentre | All‐cause withdrawal Gabapentin 64/200 Placebo 54/189 AE withdrawal Gabapentin 27/200 Placebo 18/189 LoE withdrawal Gabapentin 1/200 Placebo 4/189 | At least 30% reduction in pain Gabapentin 113/200 Placebo 77/189 At least 50% reduction in pain Gabapentin 77/200 Placebo 46/189 | At least one AE Gabapentin 159/200 Placebo 126/189 Serious AE Gabapentin 15/200 Placebo 15/189 Deaths Gabapentin 1/200 Placebo 1/189 | Somnolence Gabapentin 31/200 Placebo 8/189 Dizziness Gabapentin 38/200 Placebo 15/189 Asthenia Gabapentin 22/200 Placebo 8/189 Peripheral oedema Gabapentin 33/200 Placebo 7/189 |
|
Gorson 1999 Gorson et al. J Neurol, Neurosurg Psych 1999 66:251‐252 |
Moderate or excellent pain relief (both phases) Gabapentin 17/40 Placebo 9/40 | At least one AE Gabapentin 12/40 Placebo 4/40 Serious AE Gabapentin 0/40 Placebo 0/40 Deaths (inferred) Gabapentin 0/40 Placebo 0/40 | ||
| Morello 1999 | All‐cause withdrawal/early cross‐over Gabapentin 3/25 Amitriptyline 4/25 AE withdrawal/early cross‐over Gabapentin 2/25 Amitriptyline 3/25 LoE withdrawal/early cross‐over Gabapentin 0/25 Amitriptyline 1/25 | No significant difference at end of treatment Pain relief at end of treatment (6‐point global score), complete, a lot Gabapentin 6/21 Amitriptyline 5/21 Pain relief at end of treatment (global score), at least moderate Gabapentin 11/21 Amitriptyline 14/21 | At least one AE Gabapentin 18/23 Amitriptyline 17/24 No serious AEs or deaths noted | Sedation Gabapentin 12/23 Amitriptyline 8/24 Dizziness Gabapentin 7/23 Amitriptyline 2/24 Ataxia Gabapentin 5/23 Amitriptyline 2/24 Peripheral oedema Gabapentin 3/23 Amitriptyline 2/24 |
| Perez 2000 | No withdrawals apparent | At least 50% reduction in pain by 4 weeks Gabapentin 14/17 Placebo 2/15 | No major side effects reported for gabapentin group | No data |
| Rauck 2013a | All‐cause withdrawal GabaEn1200 15/62 GabaEn 2400 19/56 GabaEn 3600 38/117 Pregab 300 19/66 Placebo 30/120 AE withdrawal GabaEn1200 5/62 GabaEn 2400 12/56 GabaEn 3600 21/117 Pregab 300 6/66 Placebo 11/120 LoE withdrawal GabaEn1200 2/62 GabaEn 2400 0/56 GabaEn 3600 4/117 Pregab 300 3/66 Placebo 4/120 Protocol deviation most common other cause for not completing | At least 50% reduction in pain by end M week 12 GabaEn1200 26/62 GabaEn 2400 15/56 GabaEn 3600 46/117 Pregab 300 14/66 Placebo 35/120 At least 30% reduction in pain by end M week 12 GabaEn1200 31/62 GabaEn 2400 25/56 GabaEn 3600 66/117 Pregab 300 28/66 Placebo 57/120 | At least 1 AE GabaEn1200 45/62 GabaEn 2400 38/56 GabaEn 3600 86/117 Pregab 300 47/66 Placebo 79/120 SAE: 22 participants reported 29 nonfatal SAEs ‐ no clear differences between groups No deaths | Dizziness
GabaEn1200 9/62
GabaEn 2400 8/56
GabaEn 3600 16/117
Pregab 300 9/66
Placebo 7/120 Somnolence GabaEn1200 2/62 GabaEn 2400 7/56 GabaEn 3600 16/117 Pregab 300 9/66 Placebo 5/120 Peripheral oedema GabaEn1200 2/62 GabaEn 2400 0/56 GabaEn 3600 11/117 Pregab 300 3/66 Placebo 5/120 Details of other AEs occurring in at least 5% of any group |
| Sandercock 2012 | All‐cause withdrawal Gabapentin (1) 4/46 Gabapentin (2) 3/50 Placebo 2/51 Adverse event Gabapentin (1) 2/46 Gabapentin (2) 2/50 Placebo 2/51 No lack of efficacy withdrawals ‐ remaining 3 were protocol violation (1) and withdrew consent (2) | At least 50% reduction in pain from baseline to week 4 (BOCF) Gabapentin (1) 34.8% = 16/46 Gabapentin (2) 26.0% = 13/50 Placebo 7.8% = 4/51 PGIC much or very much improved Gabapentin (1) 55.3% = 25/45 Gabapentin (2) 67.4% = 34/50 Placebo 34% = 17/51 Similar results for sleep interference | At least one AE Gabapentin (1) 27/47 Gabapentin (2) 23/49 Placebo 20/51 Serious AE Gabapentin (1) 0/47 Gabapentin (2) 0/49 Placebo 1/51 (judged not related) No deaths | Dizziness
Gabapentin (1) 8/47
Gabapentin (2) 6/49
Placebo 0/51 Somnolence Gabapentin (1) 6/47 Gabapentin (2) 2/49 Placebo 0/51 Nausea Gabapentin (1) 2/47 Gabapentin (2) 3/49 Placebo 0/51 Headache Gabapentin (1) 2/47 Gabapentin (2) 3/49 Placebo 2/51 |
| Simpson 2001 | All‐cause withdrawal Gabapentin 3/30 Placebo 3/30 Lack of efficacy Gabapentin 1/30 Placebo 1/30 Adverse event Gabapentin 2/30 Placebo 2/30 | PGIC moderate or much improved Gabapentin: 15/30 Placebo: 7/30 | No deaths reported, and no serious adverse events reported | Somnolence Gabapentin 6/27 Placebo 1/27 Dizziness Gabapentin 6/27 Placebo 1/28 |
| Mixed neuropathic pain | ||||
| NCT00904202 | AE withdrawals Gabapentin 6/16 Lidocaine 3/14 Combination 6/16 Placebo 1/16 | No statistically significant differences between groups for any efficacy parameter % pain relief (mean change at end of study): Gabapentin 43.6% Lidocaine 39.2% Combo 50% Placebo 26.4% Satisfaction with treatment (satisfied and very satisfied): Gabapentin 65% Lidocaine 69% Combo 69% Placebo 64% |
Overall 61/62 participants reported at least one AE No deaths 1 SAE (lidocaine (haemorrhagic stroke) | Most frequent AEs were fatigue, dizziness (excluding vertigo), weakness, somnolence, decreased appetite, nausea, confusion, vision blurred Occurred at similar frequencies in each group except blurred vision (gabapentin 12/16, lidocaine 2/14, combination 8/16, placebo 8/16)) |
| Gilron 2005 | 16 withdrawals during treatment | At least moderate pain relief (5‐point scale) for those completing a given treatment: Placebo 13/42 Gabapentin 27/44 Morphine 35/44 gabapentin/morphine 32/41 | Not interpretable | Not interpretable |
| Gilron 2009 | All‐cause withdrawals Gabapentin 8/54 Nortriptyline 2/52 Combination 1/52 AE withdrawals Gabapentin 7/54 Nortriptyline 1/52 Combination 1/52 | Pain significantly lower with combination than either drug alone, by < 1/10 points | No serious AE recorded | Individual AE reporting showed higher incidence during titration than at maximum tolerated dose |
| Serpell 2002 | All‐cause withdrawals Gabapentin 32/153 Placebo 41/152 AE withdrawals Gabapentin 24/153 Placebo 25/152 LoE withdrawals Gabapentin 1/153 Placebo 5/152 | At least 50% reduction in pain Gabapentin 32/153 Placebo 22/152 PGIC very much or much improved Gabapentin 48/153 Placebo 22/152 PGIC very much improved CTR Gabapentin 18/153 Placebo 9/152 PGIC much improved CTR Gabapentin 30/153 Placebo 13/152 | At least one AE Gabapentin 117/153 Placebo 103/152 Serious AE Gabapentin 4/153 Placebo 4/152 Deaths Gabapentin 0/153 Placebo 2/152 | Somnolence Gabapentin 22/153 Placebo 8/152 Dizziness Gabapentin 37/153 Placebo 12/152 |
| Radicular leg pain | ||||
| Atkinson 2016 | All‐cause withdrawal Gabapentin 19/55 Placebo 17/53 AE withdrawal Gabapentin 7/55 Placebo 5/53 LoE withdrawal Gabapentin 5/55 Placebo 1/53 Other (?Lost to follow‐up): Gabapentin 7/55 Placebo 11/53 | ≥ 30% decrease in pain intensity
Gabapentin 36% (n = 34)
Placebo 36% (n = 38)
≥ 50% decrease in pain intensity
Gabapentin 26% (n = 34)
Placebo 29% (n = 38)
No significant differences between those with (n = 46) and without (n = 62) radiating pain
Participant estimation of pain improvement at exit ≥ 30% Gabapentin 43% (probably n =34) Placebo 38% (probably n =38) ≥ 50% Gabapentin 23% (probably n =34) Placebo 20% (probably n =38) |
At least one AE (possibly or probably attributed) Gabapentin 49/55 Placebo 35/53 Most mild or moderate | Dizziness Gabapentin 24/55 Placebo 14/53 Fatigue/Asthenia Gabapentin 27/55 Placebo 15/53 Somnolence (inc sleep) Gabapentin 21/55 Placebo 11/53 Loss of balance Gabapentin 18/55 Placebo 2/53 Accomodation disturbance Gabapentin 19/55 Placebo 3/53 Concentration difficulties Gabapentin 21/55 Placebo 6/53 Dry mouth Gabapentin 22/55 Placebo 10/53 |
| Cohen 2015 | All‐cause withdrawal Gabapentin 40/72 Steroid 30/73 "Negative outcome" Gabapentin 39/72 Steroid 23/73 "Withdrew" Gabapentin 0/72 Steroid 7/73 Lost to follow‐up Gabapentin 1/72 Steroid 2/73 | Decrease in average leg pain at 3 months
Gabapentin 1.6 (SD 2.7)
Steroid 2.0 (SD 2.6) Decrease in worst leg pain at 3 months Gabapentin 2.3 (SD 3.5) Steroid 2.7 (SD 3.2) |
At least one AE (injection‐related)
Gabapentin 7/72
Steroid 6/73 At least one AE (drug‐related) Gabapentin 37/72 Steroid 30/73 |
Sedation/fatigue Gabapentin 13/72 Placebo 8/73 Cognition Gabapentin 7/72 Placebo 5/73 Swelling (oedema?) Gabapentin 3/72 Placebo 0/73 Dizziness Gabapentin 2/72 Placebo 0/73 Dry mouth Gabapentin 2/72 Placebo 0/73 |
| Spinal cord injury | ||||
| Tai 2002 | Discontinuations All‐cause 7/14 Urinary retention 1/14 | Not interpretable | No data "No significant side effects noted at the maximum dosage" | No data |
| Levendoglu 2004 | All completed | Average fall in pain 62% with gabapentin, 13% with placebo Mean scores without SD. No dichotomous results | All‐cause AE Gabapentin 13/20 Placebo 5/20 | Sedation Gabapentin 3/20 Placebo 0/20 Oedema Gabapentin 3/20 Placebo 0/20 |
| Rintala 2007 | 16/38 withdrew | No dichotomous data. The paper claims statistical superiority of amitriptyline over gabapentin using paired t‐tests for 22 participants completing all 3 phases. It also claims no benefit of gabapentin over placebo | No dichotomous data | No dichotomous data |
| Nerve injury pain | ||||
| Gordh 2008 | All‐cause withdrawal Gabapentin 11/120 Placebo 11/120 AE withdrawal Gabapentin 7/120 Placebo 3/120 LoE withdrawal Gabapentin 1/120 Placebo 2/120 | Marked pain relief Gabapentin 18/98 Placebo 5/98 Marked or moderate pain relief Gabapentin 31/98 Placebo 14/98 No pain relief Gabapentin 54/98 Placebo 70/98 At least 50% pain relief Gabapentin 11 13/98 Placebo 7 9/98 At least 30% pain relief Gabapentin 20 29/98 Placebo 10 19/98 Benefits from gabapentin over placebo for sleep and some aspects of quality of life | Serious AE Gabapentin 5/120 Placebo 1/120 | Dizziness Gabapentin 39/120 Placebo 9/120 |
| Phantom | ||||
| Smith 2005 | No apparent withdrawals | "Meaningful decrease in pain" (top of 5‐point scale) Gabapentin 13/24 Placebo 5/24 | No data | No data |
| Bone 2002 | No data on where withdrawals occurred | No dichotomous data Significant benefit for gabapentin by week 6 for pain | No data | Somnolence Gabapentin 7/19 Placebo 2/19 Dizziness Gabapentin 2/19 Placebo 1/19 |
| Cancer‐associated neuropathic pain | ||||
| Caraceni 2004 | All‐cause withdrawal Gabapentin 21/80 Placebo 10/41 AE withdrawal Gabapentin 6/80 Placebo 3/41 LoE withdrawal Gabapentin 0/80 Placebo 0/41 | Somewhat better pain responses with gabapentin than placebo | No data Any AE Gabapentin 35/79 Placebo 10/41 | Somnolence Gabapentin 18/79 Placebo 4/41 Dizziness Gabapentin 7/89 Placebo 0/41 |
| Rao 2007 | All‐cause withdrawal Gabapentin 23/115 Placebo 26/115 | No significant difference between gabapentin and placebo, but pain scores were low and the study may have lacked sensitivity | No data | Dizziness Gabapentin 8/91 Placebo 4/89 |
| HIV | ||||
| Hahn 2004 | All‐cause withdrawal Gabapentin 1/15 Placebo 1/11 AE withdrawal Gabapentin 1/15 Placebo 0/11 | Improvement in pain and sleep interference with gabapentin and placebo, with sustained difference in sleep but not pain | No serious AE or deaths reported | Somnolence Gabapentin 12/15 Placebo 2/11 Dizziness Gabapentin 9/15 Placebo 5/11 Disturbed gait Gabapentin 7/15 Placebo 3/11 |
| AE: adverse event; CTR: clinical trial report; GabaEn: gabapentin encarbil; GabaEr: gabapentin extended‐release; LoE: lack of efficacy; PGIC: Patient Global Impression of Change; QoL: quality of life; SAE: serious adverse event; VAS: visual analogue scale; | ||||
Data and analyses
Comparison 1. Efficacy ‐ placebo‐controlled studies.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At least 50% pain reduction over baseline | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Postherpetic neuralgia | 7 | 2031 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [1.43, 2.00] |
| 1.2 Painful diabetic neuropathy | 6 | 1331 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [1.41, 2.02] |
| 1.3 Mixed neuropathic pain | 1 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.88, 2.37] |
| 1.4 Nerve injury pain | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.65, 3.22] |
| 2 Very much improved | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Postherpetic neuralgia | 2 | 563 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.70 [1.51, 4.82] |
| 2.2 Painful diabetic neuropathy | 2 | 408 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [1.26, 2.99] |
| 2.3 Mixed neuropathic pain | 1 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [0.92, 4.28] |
| 2.4 Nerve injury pain | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.6 [1.39, 9.31] |
| 3 Much or very much improved | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Postherpetic neuralgia | 7 | 2013 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [1.16, 1.50] |
| 3.2 Painful diabetic neuropathy | 5 | 695 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.36, 2.03] |
| 3.3 Mixed neuropathic pain | 1 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [1.38, 3.41] |
| 3.4 Nerve injury pain | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.26, 3.90] |
| 4 IMMPACT outcome of substantial improvement | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Postherpetic neuralgia | 8 | 2260 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.75 [1.49, 2.07] |
| 4.2 Painful diabetic neuropathy | 6 | 1331 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [1.41, 2.02] |
| 4.3 Mixed neuropathic pain | 1 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.88, 2.37] |
| 4.4 Nerve injury pain | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.65, 3.22] |
| 4.5 Phantom pain | 1 | 48 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.6 [1.10, 6.16] |
| 5 IMMPACT outcome of at least moderate improvement | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Postherpetic neuralgia | 8 | 2260 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [1.56, 2.00] |
| 5.2 Painful diabetic neuropathy | 7 | 1439 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [1.24, 1.59] |
| 5.3 Mixed neuropathic pain | 2 | 391 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.10 [1.49, 2.95] |
| 5.4 Nerve injury pain | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.92, 2.53] |
Comparison 2. Withdrawals ‐ placebo‐controlled studies.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause withdrawal | 22 | 4617 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.92, 1.16] |
| 2 Adverse event withdrawal | 22 | 4346 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.14, 1.67] |
| 3 Lack of efficacy withdrawal | 15 | 3559 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.37, 0.88] |
Comparison 3. Adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At least one adverse event | 18 | 4279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [1.22, 1.36] |
| 2 Serious adverse events | 19 | 3948 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.83, 1.71] |
| 3 Somnolence | 20 | 4288 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.82 [2.27, 3.50] |
| 4 Dizziness | 21 | 4739 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.87 [2.40, 3.44] |
| 5 Peripheral oedema | 12 | 3325 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.12 [2.66, 6.39] |
| 6 Ataxia or gait disturbance | 4 | 510 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.53 [2.49, 12.28] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Atkinson 2016.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, not enriched Forced titration to target or maximum tolerated dose over 4 weeks, then stable to 12 weeks |
|
| Participants | Non‐specific back pain with and without a radiating component, back pain primarily in lumbar region. Pain present on daily basis for ≥ 6 months, PI ≥ 2/10, impact on ≥ 2 aspects of daily life
Excluded: major coexisting illness, coexisting pain condition due to other disorders, contraindication to study medication, recent or planned surgery, history of alcohol or substance abuse, major depression, history of psychosis, cognitive impairment, pregnant or lactating N = 108 Mean age 56 years, 23% women Initial pain intensity "moderate" Impact on everyday function "mild to moderate" Mean duration of back pain 17 (± 15) years |
|
| Interventions | Gabapentin to maximum 3600 mg daily, n = 55 Placebo, n = 53 All muscle relaxants, antidepressants, opioids, discontinued ≥ 2 weeks before baseline assessment; NSAIDs permitted |
|
| Outcomes | ≥ 30% and ≥ 50% reduction in PI Participants estimation of pain improvement at exit (≥ 30%, ≥ 50%) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 US VA sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated randomization tables, stratified by site" |
| Allocation concealment (selection bias) | Low risk | Remote allocation from central pharmacy; "sequentially numbered opaque sealed envelope" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Gabapentin 300 mg over‐capsulated and identical placebo capsules |
| Incomplete outcome data (attrition bias) Efficacy | High risk | Used random‐effects regression models for primary outcome, but reported only on completers (33% attrition) |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (55, 53) |
Backonja 1998.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, not enriched, LOCF Titration to maximum tolerated dose or 3600 mg daily over 4 weeks, then stable dose for 4 weeks (8 weeks in total) |
|
| Participants | PDN. Pain duration > 3 months before treatment, PI ≥ 40/100 at randomisation N = 165 Mean age 53 years, 40% women Initial mean pain score 6.4/10 |
|
| Interventions | Gabapentin 3600 mg daily (max), n = 84 Placebo, n = 81 Medication for diabetes control remained stable during study. Paracetamol (max 3 g daily) allowed |
|
| Outcomes | PGIC much or moderately improved ≥ 50% reduction in pain (CTR) PGIC much improved (CTR) PGIC moderately or much improved (CTR) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Parke‐Davies/Pfizer sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "supplied in identical capsules in blinded fashion". "All participants were supplied with an equal number of capsules". |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (81, 84) |
Backonja 2011.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, enriched for tolerance (but not response), LOCF Open‐label titration with gabapentin from 300 mg at night to maximum 600 mg 3 times daily (1800 mg/d) over 4 days, maintained on maximum tolerated dose for 7 days, then randomised to double blind treatment with 600 mg gabapentin encarbil twice daily or placebo for 2 weeks |
|
| Participants | PHN. Pain > 3 months after healing of skin rash. PI at randomisation ≥ 40/100 N = 102 in double‐blind phase, and 116 in open‐label phase Mean age 65 years, 51% women Initial average daily pain score 6.1/10, and 4.5 before randomisation |
|
| Interventions | Gabapentin encarbil 1200 mg daily, n = 47 (equivalent to 624 mg gabapentin, given as divided dose) Placebo, n = 54 Antiepileptic medication discontinued ≥ 7 days before open label phase. Antidepressant and narcotic analgesics continued if stable > 1 month |
|
| Outcomes | ≥ 50% reduction in pain ≥ 30% reduction in pain PGIC much and very much improved Withdrawals Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 XenoPort sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "matching placebo" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | High risk | < 50 participants per treatment arm |
Bone 2002.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. No imputation method mentioned Titration to maximum tolerated dose or 2400 mg daily over 1 week, then stable dose for 5 weeks (6 weeks total); 1‐week washout, then cross‐over |
|
| Participants | Established phantom limb pain ≥ 6 months. PI before treatment > 3/10 N = 19 (14 completed both treatment periods) Mean age 56 years, 21% women Initial pain score 6.4/10 |
|
| Interventions | Gabapentin 2400 mg daily (max) Placebo Paracetamol + codeine 500 mg/30 mg (max 12 tablets daily) allowed as rescue medication. Stable, low doses of TCAs continued |
|
| Outcomes | No dichotomous efficacy data Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Pfizer Pharmaceuticals supplied gabapentin and placebo capsules. No other funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | "The hospital pharmacists were also responsible for issuing identical, coded medication bottles containing identical tablets of gabapentin or placebo" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical, coded medication bottles containing identical tablets of gabapentin or placebo" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | No imputation mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (19 randomised, 14 completed both phases) |
Caraceni 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment. No imputation method mentioned Titration to pain ≤ 3/10 or limit of tolerability, or maximum 1800 mg daily (10 days in total) |
|
| Participants | Neuropathic cancer pain despite regular systemic opioid therapy. Pain at randomisation ≥ 5/10 N = 121 Mean age 60 years, 56% women Initial pain intensity 7.3/10 |
|
| Interventions | Gabapentin 1800 mg daily (max), n = 80 Placebo, n = 41 Any previous analgesics continued unchanged. One additional dose of opioid allowed for rescue medication |
|
| Outcomes | No dichotomous efficacy data Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Sponsored/Pfizer Italy and Spain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block of three randomisation list |
| Allocation concealment (selection bias) | Low risk | Remote pharmacy department provided numbered containers |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | Unclear risk | < 50 participants per treatment arm (41, 80) |
Chandra 2006.
| Methods | Randomised, double‐blind, active‐controlled, parallel groups, no enrichment Dose escalation every 2 weeks until adequate pain relief obtained or limit of tolerability, to maximum nortriptyline 150 mg daily or gabapentin 2700 mg daily by 4 weeks, then stable dose for 5 weeks (9 weeks in total) |
|
| Participants | PHN. Pain > 2 months after healing of skin rash. PI at randomisation ≥ 40/100 N = 76 Mean age 54 years, 50% women Initial average daily pain score 5.7/10 |
|
| Interventions | Gabapentin 2700 mg daily (max), n = 38 Nortriptyline 150 mg daily (max), n = 38 Of 'responders' ˜ 80% gabapentin took 2700 mg daily, ˜ 66% nortriptyline took 75 mg daily |
|
| Outcomes | ≥ 50% pain relief over baseline pain ≥ 50% pain relief over (VAS) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Sponsored Pfizer/independent |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "block‐of‐three randomization list was used" |
| Allocation concealment (selection bias) | Low risk | "code supplied in sealed envelopes, opened at time of enrolment", "drugs dispensed in sealed envelopes" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "drugs placed in identical capsules", "matching placebo of nortriptyline" to blind different dosing schedules |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm |
Cohen 2015.
| Methods | Multicentre, randomised, double‐blind (double‐dummy), active‐controlled, parallel groups, partial enrichment. Titration to maximum tolerated dose (1800 mg to 3600 mg daily) over 15‐24 days (3 months total) | |
| Participants | Radicular leg pain for ≥ 6 weeks and < 4 years, PI ≥ 4/10 or ≥ 3/10 if leg pain ≥ back pain, symptoms of lumbosacral radicular pain. Findings of herniated disc or spinal stenosis on MRI concordant with presentation. Age ≥ 17 years
Excluded: neuropathic pain > 4 years, previous failed trial with gabapentin or pregabalin, steroid injections ≤ 3 years, cauda equina syndrome, planned surgery N = 145 Mean age 43 years, 26% women 18% had pain ≤ 3 months, 25% taking opioids Initial PI: worst leg 7.8/10; average leg 5.4/10 |
|
| Interventions | Gabapentin capsule + saline injection, n = 72 Depomethylprednisolone 60 mg injection + 1 ml 0.25% bupivacaine + placebo capsule, n = 73 Gabapentin titrated to 1800 mg to 3600 mg/day (3 divided doses) over 15‐24 days, but ≥ 5 days before follow‐up Steroid injected into epidural space (interlaminar or transforaminal), saline injected into posterior ligaments Tramadol and NSAIDs "as needed" for rescue medication, or opioids increased by ≥ 20% for those taking them. No other co‐interventions |
|
| Outcomes | Mean PI, for average and worst leg and back pain Global evaluation (non‐standard scales) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Congressional grant from the Center for Rehabilitation Sciences Research, Bethesda |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation tables, stratified by site. Central pharmacy |
| Allocation concealment (selection bias) | Low risk | "sequentially numbered opaque sealed envelope" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "A central research pharmacy over‐capsulated 300 mg gabapentin and placebo capsules to appear identical". Participants "visually shielded from the image screen" during injections", had no further contact with physician |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm |
CTR 945‐1008.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, no obvious enrichment, LOCF Titration from 300 mg daily to maximum tolerated dose or 3600 mg daily over 3 weeks, then stable dose for 12 weeks (15 weeks total) |
|
| Participants | PDN. Pain duration > 3 months, PI at randomisation ≥ 40/100 N = 389 Mean age 58 years, "more men than women" |
|
| Interventions | Gabapentin 3600 mg daily (max), n = 200 Placebo, n = 189 |
|
| Outcomes | ≥ 30% reduction in pain ≥ 50% reduction in pain Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 Pfizer sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Matching placebo |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (189, 200) |
CTR 945‐224.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, no enrichment, probably LOCF Titration over 3 weeks to 600, 1200, or 2400 mg daily, then stable dose to 4 weeks (7 weeks total) |
|
| Participants | PDN for 1‐5 years. PI at randomisation ≥ 40/100 N = 325 Mean age 60 years, 44% women Initial pain score 6.2/10 |
|
| Interventions | Gabapentin 600 mg, n = 82 Gabapentin 1200 mg, n = 82 Gabapentin 2400 mg, n = 84 Placebo, n = 77 |
|
| Outcomes | ≥ 50% reduction in pain score PGIC very much improved PGIC much or very much improved Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Parke‐Davis/Pfizer sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation code |
| Allocation concealment (selection bias) | Low risk | Randomisation code broken after last participant completed |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Matching placebo |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Probably LOCF |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (77‐84) |
Gilron 2005.
| Methods | Randomised, double‐blind, placebo‐controlled 4‐period cross‐over, no enrichment. No imputation method mentioned (but if half of scores missing, outcome considered missing) Titration to target doses or limit of tolerability over 3 weeks, then stable dose for 1 week, and tapered dose for 1 week (5 weeks in total); 3‐day washout and cross‐over to next treatment |
|
| Participants | PDN and PHN. Pain ≥ moderate for 3 months N = 57 Median age 62 years, 44% women Initial mean pain score 5.8/10 |
|
| Interventions | Gabapentin 3200 mg daily (max) Morphine 120 mg daily (max) Gabapentin plus morphine 2400 mg/60 mg daily (max) Placebo (lorazepam) 1.6 mg Mean maximum tolerated doses: gabapentin alone 2207 ± 89 mg, morphine alone 45.3 ± 3.9 mg, gabapentin + morphine 1705 ± 83 + 34.4 ± 2.6 mg |
|
| Outcomes | Pain relief for those completing a given treatment (5‐point scale) Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Canadian Institutes of Health Research grant. Pharma supplied medicines |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced Latin‐square cross‐over design |
| Allocation concealment (selection bias) | Low risk | "concealed allocation schedule" prepared remotely |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical appearing blue and grey capsules .... in accord with a double‐dummy design" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (data available 40‐44 completing a given treatment) |
Gilron 2009.
| Methods | Randomised, double‐blind, placebo‐controlled 3‐period cross‐over, no enrichment. No imputation method mentioned Titration to target doses or limit of tolerability over 24 days, then stable dose for 1 week, and tapered dose for 1 week (6 weeks in total); 6‐day washout and cross‐over to next treatment |
|
| Participants | PDN and PHN. Pain ≥ moderate for 6 months N = 56 Median age 64 years, 40% women Initial mean pain score 5.4/10 |
|
| Interventions | Gabapentin 3600 mg daily (max) Nortriptyline 100 mg daily (max) Gabapentin plus nortriptyline 3600 mg/100 mg daily (max) Mean (SE) maximum tolerated doses: gabapentin alone 2433 ± 106 mg, nortriptyline alone 62 ± 3.6 mg, gabapentin + nortriptyline 2180 ± 108 + 50 ± 3.5 mg |
|
| Outcomes | Pain relief (mean) Withdrawals Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Canadian Institutes of Health Research grant. Study drugs from pharma |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced Latin‐square cross‐over design |
| Allocation concealment (selection bias) | Low risk | "concealed allocation" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "double dummy" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | Reporting on < 50 completing 2 periods |
Gong 2008.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups. No enrichment or imputation method mentioned Forced titration from 300 mg daily to 1800 mg daily over 8 days, then stable dose to 6 weeks |
|
| Participants | PHN N = 231 Mean age 66 years (± 12), 43% women Mean baseline PI: 6.2/10 (± 1.3) |
|
| Interventions | Gabapentin 1800 mg daily, n = 109 Placebo, n = 106 Rescue medication: 2 x 100 mg tramadol if required 3 days after reaching maximum dose of gabapentin |
|
| Outcomes | ≥ 25% and ≥ 50% pain relief PGIC ("mild effective" and "excellent") Sleep Quality of life Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 0, Total = 4 Unknown funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not adequately described. "Patients were randomised to different groups according to their recruitment order", but then refers to "pre‐determined code" |
| Allocation concealment (selection bias) | Unclear risk | Not adequately described. "Researchers allocated the treatments according to the pre‐determined code" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Identical‐appearing capsules containing placebo were used to blind the patients" |
| Incomplete outcome data (attrition bias) Efficacy | High risk | Withdrawals (7%) and reasons for withdrawal not given per treatment group. No information about how data from withdrawals contributed to analyses |
| Size Efficacy | Unclear risk | 50‐199 participants per treatment arm (106, 109) |
Gordh 2008.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. No imputation method mentioned Titration over 2 weeks from 300 mg to maximum pain relief at a tolerable dose or 2400 mg daily, then stable dose for 3 weeks (5 weeks total); 3‐week washout, then cross‐over |
|
| Participants | Peripheral nerve injury with pain ≥ 6 months. PI at randomisation > 30/100 N = 120 (efficacy analysis based on 98 who completed both treatment periods) Mean age 49 years, 53% women Initial pain intensity 53/100 |
|
| Interventions | Gabapentin 2400 mg daily (max) Placebo Mean daily dose of gabapentin 2243 mg ± 402 mg Paracetamol ± codeine and dextropropoxyphene permitted as rescue medication Analgesics and NSAIDs used by ˜ 50% during study |
|
| Outcomes | ≥ 50% pain relief (weekly mean pain score) ≥ 30% pain relief Marked pain relief (5‐point scale) Marked or moderate pain relief (5‐point scale) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Sponsored by Parke‐Davis AB, later Pfizer AB |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list was generated by the Clinical Pharmaceutical Operation Center in Freiburg |
| Allocation concealment (selection bias) | Low risk | Central, remote allocation, "sealed code envelope" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "capsules that were identical in appearance" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (98 completed both periods and included in efficacy analysis) |
Gorson 1999.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. No imputation method mentioned Titration over 3 days to 900 mg, then fixed dose for remainder of 6‐week period; 3 week washout, then cross‐over |
|
| Participants | PDN 1‐5 years, pain ≥ moderate for over 3 months. Pain intensity at randomisation ≥ 40/100 N = 40 Mean age 62 years, 23% women Initial pain intensity not reported |
|
| Interventions | Gabapentin 900 mg, n = 19 (first phase) Placebo, n = 21 (first phase) Medication for diabetes control remained stable during study. Stable doses of NSAID or narcotics allowed |
|
| Outcomes | Pain relief at end of treatment (4‐point global score) moderate or excellent Adverse events |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 0, Total = 3 Sponsored by Warner Lambert/Parke‐Davis Note: no separate data for first period, small group sizes, non standard global scale |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (19, 21) |
Hahn 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, not enriched. No imputation method mentioned Titration over 2 weeks to adequate pain relief or 2400 mg daily, then stable dose for 2 weeks (4 weeks in total) |
|
| Participants | Painful HIV sensory neuropathy by standard definitions. Pain at any level including mild pain at randomisation N = 26 Mean age 45 years, 23% women Initial mean pain score 4.9/10 (lower limit of range 1.5) |
|
| Interventions | Gabapentin 2400 mg daily (max), n = 15 (10 participants took max dose) Placebo, n = 11 |
|
| Outcomes | No dichotomous efficacy data Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Pfizer grant |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was performed by producing a randomisation schedule that assigned each patient to GBP or a matching placebo" |
| Allocation concealment (selection bias) | Low risk | Remote allocation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identically appearing capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (11, 15) |
Harden 2013.
| Methods | Randomised, double blind, cross‐over, dose‐comparison. Two 4‐week treatments plus 4 day washout | |
| Participants | PHN for at least 3 months after rash healing, with inadequate response to gabapentin 1800 mg daily, but no response to either gabapentin or pregabalin N = 93 Mean age 63 years, 39% women Mean baseline pain 6/10 |
|
| Interventions | Gabapentin encarbil at two different dose ranges | |
| Outcomes | ≥ 50% and ≥ 30% pain reduction at end of treatment periods Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 1, W = 1, Total = 5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated |
| Allocation concealment (selection bias) | Low risk | Remote allocation |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No details |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (93) |
Irving 2009.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment, LOCF, extended‐release formulation Gradual titration to 1800 mg over 2 weeks, then stable for 2 weeks (4 weeks in total) |
|
| Participants | PHN. Pain > 3 months after healing of skin rash, PI at randomisation ≥ 4/10 N = 158, mean age 70 years, 53% women Initial average daily pain score 6.5/10 |
|
| Interventions | Gabapentin ER 1800 mg daily, n = 55 Gabapentin ER 1800 mg daily in split doses, n = 52 Placebo, n = 51 Rescue with paracetamol up to 4000 mg daily, or paracetamol plus hydrocodone 500 mg/5 mg up to 8 tablets daily |
|
| Outcomes | ≥ 50% reduction in pain score ≥ 30% reduction in pain score PGIC much or very much improved Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Sponsored by Depomed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated list |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐dummy method |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (51‐55) |
Levendoglu 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. No imputation method mentioned Titration to limit of tolerability or maximum of 3600 mg over 4 weeks, then stable dose for remainder of 8‐week period; 2‐week washout then cross‐over |
|
| Participants | Complete traumatic SCI at lumbar or thoracic level. Pain duration before treatment ≥ 6 months, PI at randomisation > 4/10 N = 20 Mean age 36 years, 35% women Initial average daily pain 9/10 |
|
| Interventions | Gabapentin 3600 mg daily (max) Placebo Mean max tolerated dose of gabapentin 2850 ± 751 mg No concurrent analgesics allowed |
|
| Outcomes | Pain reduction (mean data only) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 No funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identically appearing capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (20) |
Mishra 2012.
| Methods | Randomised, double‐blind, active‐ and placebo‐controlled, parallel groups. Not enriched. No imputation method mentioned Three active treatments, with low starting dose and increases at start of weeks 2 and 3. Total duration 4 weeks Gabapentin 900 mg daily (divided x 2) increasing to 1800 mg daily (divided x 3) Pregabalin 150 mg daily (divided x 2) increasing to 600 mg daily (divided x 2) Amitriptyline 50 mg/d increasing to 100 mg/d at bedtime |
|
| Participants | Cancer with neuropathic pain N = 120 Age and sex distribution not reported Baseline pain 7.6/10 |
|
| Interventions | Gabapentin 1800 mg daily, n = 30 Pregabalin 600 mg daily, n = 30 Amitriptyline 100 mg daily, n = 30 Placebo, n = 30 |
|
| Outcomes | Mean changes for pain functional capacity and opioid sparing | |
| Notes | Oxford Quality Score: R = 2, DB = 1, W = 0, Total = 3 Funding from Institute Research Grant of All India Institute of Medical Sciences |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised random list |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | All drugs encapsulated, but no mention of equal numbers and regimen or double‐dummy method |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (30) |
Morello 1999.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. No imputation method mentioned Titration over 2 days and adjusted thereafter until adequate pain relief obtained or limit of tolerability to maximum 1800 mg gabapentin or 75 mg amitriptyline daily, then stable dose for remainder of 6‐week period; 1‐week washout, then cross‐over |
|
| Participants | PDN. Pain duration > 3 months before treatment, no initial PI at inclusion N = 25 (19 completed 6 weeks with both study drugs) Mean age 60 years, 4% women Initial pain intensity mild/moderate |
|
| Interventions | Gabapentin 1800 mg daily (max) Amitriptyline 75 mg daily (max) Paracetamol allowed as rescue medication (max 1300 mg daily) |
|
| Outcomes | Pain relief at end of treatment (6‐point global score), complete or a lot Pain relief at end of treatment (6‐point global score), at least moderate Adverse events Withdrawal |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 No funding mentioned Note: no separate data for first period, small group sizes, non standard global scale |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported (all except clinical research pharmacist remained blinded until study termination) |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "all capsules were identical in taste, color, size, and shape" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (25 randomised, 19 completed both periods |
NCT00475904.
| Methods | Randomised, double‐blind, double‐dummy, placebo‐controlled, parallel groups, 4 weeks, not enriched | |
| Participants | PHN ≥ 3 months after healing of rash. Age ≥ 18 years N = 360 Mean age 53 years, 38% women |
|
| Interventions | Gabapentin 1800 mg daily, n = 144 Topical cream with amitriptyline and ketamine, n = 140 Placebo for oral and topical cream, n = 76 |
|
| Outcomes | Mean reduction in PI from baseline | |
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐dummy method |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (76 to 144) |
NCT00904202.
| Methods | Randomised, double‐blinded, double‐dummy, parallel groups, not enriched. Forced titration over 1 week Duration of treatment: 5 weeks |
|
| Participants | Various peripheral neuropathic pain conditions (diagnosis of: PHN, PDN, CRPS, carpel tunnel syndrome, HIV neuropathy, idiopathic sensory neuropathy, other peripheral neuropathy). Age ≥ 18 years N = 62 Age not reported, % women not reported Baseline PI 4/10 |
|
| Interventions | Gabapentin titrated to 1800 mg daily over first week + placebo patch, n = 16 Lidocaine patch 5% (up to 4 patches) applied once daily + placebo capsules, n = 14 Gabapentin 1800 mg + lidocaine 5% patch daily, n = 16 Placebo capsules + placebo patch, n = 16 |
|
| Outcomes | Average daily pain intensity (BPS questions 3, 4, 5, 6) PGIC Patient satisfaction Percent pain relief (BPI question 8) Adverse events, dermal assessment |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 (from synopsis) Endo Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not reported |
| Allocation concealment (selection bias) | Unclear risk | Method not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐dummy method |
| Incomplete outcome data (attrition bias) Efficacy | High risk | 2 participants did not provide efficacy data (1 lidocaine, 1 placebo). LOCF for early discontinuation. All outcomes not reported (synopsis) |
| Size Efficacy | High risk | < 50 participants per treatment arm (14‐16) |
Perez 2000.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, not obviously enriched. No imputation method mentioned Dose adjusted on successive visits to clinic, "based on clinical symptoms", to a maximum of 1200 mg daily (12 weeks total) |
|
| Participants | PDN. Conventional treatment unsuccessful. PI ≥ 60/100 at randomisation N = 32 Mean age 54 years, 53% women |
|
| Interventions | Gabapentin 1200 mg daily (max), n = 17 Placebo, n = 15 All participants continued with non‐opioid analgesia |
|
| Outcomes | ≥ 50% pain reduction | |
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 0, Total = 2 No funding mentioned Published as letter, some details confirmed by correspondence |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (15, 17) |
Rao 2007.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, not enriched. Missing data handled in a number of ways, and results presented without imputation Titration over 3 weeks to limit of tolerability or 2700 mg daily, then stable dose for 3 weeks (6 weeks total); then 2‐week weaning‐off and washout, and cross‐over |
|
| Participants | Chemotherapy‐induced peripheral neuropathy lasting ≥ 1 month. PI at randomisation ≥ 4/10 N = 115 Mean age 59 years, 73% women Initial average daily pain 4/10 |
|
| Interventions | Gabapentin 2700 mg daily (max) Placebo Usual cancer therapy continued |
|
| Outcomes | No dichotomous data Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 No funding mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical placebo capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Low risk | Results presented without imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (115) |
Rauck 2013a.
| Methods | Randomised, double‐blind (double‐dummy), placebo‐ and active‐controlled, parallel groups, not enriched. Screening 4 weeks, baseline 1 week, up titration 1 week, maintenance 12 weeks, down titration 1 week | |
| Participants | PDN. Pain ≥ 6 months, ≥18 years, PI ≥ 4/10 N = 421 Mean age 59 years, 41% women Baseline PI 6.5/10 |
|
| Interventions | Gabapentin encarbil 1200 mg daily, n = 62 Gabapentin encarbil 2400 mg daily, n = 56 Gabapentin encarbil 3600 mg daily, n 117 Pregabalin 300 mg daily, n = 66 Placebo, n = 120 |
|
| Outcomes | Pain intensity reduction of at least 50% and at least 30% end of maintenance over baseline Adverse events | |
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 GSK sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated |
| Allocation concealment (selection bias) | Low risk | Third party pharmacist |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐dummy method |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm |
Rauck 2013b.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, not enriched. Both LOCF and BOCF imputation methods used in analyses Intrathecal drug delivery system implanted and filled with saline until randomisation. Fixed dose of gabapentin (1 mg, 6 mg or 30 mg/d) or placebo for 22 days, followed by 7‐day taper |
|
| Participants | Chronic intractable pain below neck for ≥ 1 year (86% classified as neuropathic or mixed). PI at screening 5/10 N = 170 Mean age 50 years, 58% women Baseline PI ≥ 7.5/10 |
|
| Interventions | Gabapentin injection 1 mg, 6 mg, 30 mg daily, n = 42, 41, 43 respectively Placebo (saline) injection, n = 44 |
|
| Outcomes | Pain intensity reduction Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 Medtronic |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Low risk | "coded drug syringe labels, stored in sealed, sequentially numbered randomization envelopes". Pharmacist took next sequential envelope, prepared assigned drug, and attached coded label before sending to clinic |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Both treatments were clear liquids. Saline (placebo) "seemed identical to gabapentin" |
| Incomplete outcome data (attrition bias) Efficacy | Low risk | BOCF analysis reported alongside LOCF |
| Size Efficacy | High risk | < 50 participants per treatment group (41‐44) |
Rice 2001.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment, LOCF 4‐day forced titration, then further titration over 2 weeks to target dose, and stable dose for 4 weeks (7 weeks in total). Participants unable to tolerate dosing regimen were withdrawn |
|
| Participants | PHN. Pain > 3 months after healing of rash, PI ≥ 40/100 at randomisation N = 334 Median age 75 years, 59% women Initial average daily pain 6.5/10 |
|
| Interventions | Gabapentin 1800 mg daily, n = 115 Gabapentin 2400 mg daily, n = 108 Placebo, n = 111 |
|
| Outcomes | ≥ 50% reduction in mean pain score PGIC much or very much improved PGIC much and very much improved (CTR) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Pfizer sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated randomisation list" |
| Allocation concealment (selection bias) | Low risk | List held securely and released only after study completion |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical‐appearing capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (108‐115) |
Rintala 2007.
| Methods | Randomised, double‐blind, placebo‐controlled, 3‐way cross‐over, not enriched. No imputation method mentioned Titration over 4 weeks to pain control, limit of tolerability, or maximum amitriptyline 150 mg daily, gabapentin 3600 mg daily, then stable dose for remainder of 8‐week period; 1‐week washout then cross‐over Analysis for completers only |
|
| Participants | SCI at any level and degree of completeness. Pain duration before treatment > 6 months, PI at randomisation > 5/10 N = 38, only 22 participants completed all 3 cross‐overs Mean age 43 years, 9% women Initial pain intensity 5.6/10 |
|
| Interventions | Amitriptyline 150 mg daily (max) Gabapentin 3600 mg daily (max) Placebo (diphenhydramine) 75 mg daily Oxycodone + paracetamol 5/325 mg (max 8 tablets daily) allowed for rescue medication |
|
| Outcomes | No dichotomous data for efficacy or harm Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Department of Veterans Affairs grant |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "table of random numbers" |
| Allocation concealment (selection bias) | Low risk | Prepared, packaged and labelled by remote, commercial compounding pharmacy |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical capsules" |
| Incomplete outcome data (attrition bias) Efficacy | High risk | Completers only |
| Size Efficacy | High risk | < 50 participants per treatment arm (38 randomised, 22 completed 3 phases) |
Rowbotham 1998.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, no enrichment, LOCF 4‐week titration to maximum tolerated dose, or 3600 mg then stable dose for 4 weeks (8 weeks in total) |
|
| Participants | PHN. Pain > 3 months after healing of rash, PI at randomisation ≥ 40/100 N = 229 Median age 73 years, 48% women Initial average daily pain 6.4/10 |
|
| Interventions | Gabapentin 3600 mg daily (max), n = 113; (83% had ≥ 2400 mg daily) Placebo, n = 116 |
|
| Outcomes | PGIC moderate or much improved PGIC CTR moderate and much improved No change in pain SF36 and QoL Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 3 Parke‐Davies sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Low risk | "subject‐specific bottles based on randomisation schedule" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identically appearing capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (113, 116) |
Sandercock 2012.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, no obvious enrichment Gabapentin titrated over 2 weeks to 3000 mg daily, then stable dose for 2 weeks (4 weeks total) |
|
| Participants | PDN. PI at randomisation ≥ 4/10 N = 147 Mean age 59 years, 45% women Initial PI 6.8/10 |
|
| Interventions | Gabapentin ER, 3000 mg daily (as single dose), n = 46 Gabapentin ER, 3000 mg daily (as divided dose), n = 50 Placebo, n = 51 |
|
| Outcomes | ≥ 50% decrease in average daily pain PGIC much or very much improved Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 No obvious funding, but one author from what may be a pharmaceutical company Full publication of study previously partially published as letter (Sandercock 2009) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "All patients received an appropriate combination of active and placebo tablets to achieve the required dosing and maintain the study blind ‐ implies active and placebo were indistinguishable" |
| Incomplete outcome data (attrition bias) Efficacy | Low risk | BOFC analysis provided for primary outcome |
| Size Efficacy | High risk | < 50 participants per treatment arm (46‐51) |
Sang 2013.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment, BOCF 2‐week titration to maximum tolerated dose, or 3600 mg then stable dose for 8 weeks (10 weeks in total), then 1 week taper |
|
| Participants | PHN. Pain > 6 months and < 5 years after healing of rash, PI at randomisation ≥ 40/100 N = 452 Mean age 65 years, 63% women Initial average daily pain 6.5/10 |
|
| Interventions | Gabapentin ER, 1800 mg daily (as single dose), n = 221 Placebo, n = 231 |
|
| Outcomes | ≥ 50% reduction in pain | |
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Supported by Depomed Inc |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "electronic randomization scheme that was stratified by site" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "matched placebo" |
| Incomplete outcome data (attrition bias) Efficacy | Low risk | BOCF for primary endpoint |
| Size Efficacy | Low risk | > 200 participants per treatment group (221, 231) |
Serpell 2002.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment. No imputation method mentioned. Participants withdrawing due to lack of efficacy were defined as non‐responders (n = 6), but treatment of substantial AE withdrawals (n = 49) and all‐cause withdrawals (n = 73) not reported Titration over 5 weeks from 900 mg daily until pain controlled, or to maximum of 2400 mg daily, then fixed dose (8 weeks in total) |
|
| Participants | Mixed neuropathic pain, most common conditions were CRPS (28%), PHN (14%). PI at randomisation ≥ 4/10 Excluded: individuals who had previously failed to respond to gabapentin at ≥ 900 mg daily, or had experienced intolerable side effects at any dose N = 305 Median age 57 years, 53% women Initial mean pain score 7.2/10 |
|
| Interventions | Gabapentin 2400 mg daily (max), n = 153 Placebo, n = 152 101 took 2400 mg, 189 took 1800 mg, 27 took 900 mg Stable antidepressant therapy and NSAID/opioid therapy for other conditions allowed Paracetamol 500 mg/codeine 30 mg or paracetamol 500 mg (max 8 tablets daily) allowed as rescue medication |
|
| Outcomes | ≥ 50% reduction in pain PGIC much or very much improved PGIC much improved and very much improved (CTR) Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Parke‐Davies sponsored |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list |
| Allocation concealment (selection bias) | Low risk | Randomisation list centrally held ‐ remote allocation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "identical capsules" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | Unclear risk | 50‐200 participants per treatment arm (152, 153) |
Simpson 2001.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, not obviously enriched (part 1 of study only) Titration over 4 weeks to maximum tolerated dose, then stable dose for 4 weeks (8 weeks in total) |
|
| Participants | PDN. Pain duration > 3 months before treatment, PI ≥ 40/100 at randomisation N = 60 Mean age 50 years, 40% women Initial pain score 6.5/10 |
|
| Interventions | Gabapentin 3600 mg daily (max), n = 30 Placebo, n = 30 |
|
| Outcomes | PGIC moderate or much improved Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 1, W = 1, Total = 3 No funding mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (30) |
Smith 2005.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, no enrichment. No imputation method mentioned Titration in 300 mg increments every 2‐3 days until pain intensity of 0 or uncomfortable side effects, or maximum 3600 mg daily, then stable dose for remainder of 6‐week treatment period, followed by titration off medication in week 7; 5‐week washout, then cross‐over |
|
| Participants | Phantom limb pain and residual limb pain. Time since amputation ≥ 6 months, PI before randomisation > 3/10 N = 24 Mean age 52 years, 25% women Initial pain intensity 4.4/10 |
|
| Interventions | Gabapentin 3600 mg daily (max), (19/24 took max dose) Placebo |
|
| Outcomes | Meaningful decrease in pain (5‐point scale) | |
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 0, Total = 4 No funding mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "capsules that were identical in appearance" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (24) |
Tai 2002.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over, no enrichment. No imputation method mentioned Titration to limit of tolerability or maximum 1800 mg over 3 weeks, then stable for remainder of 4‐week period; 2‐week washout then cross‐over |
|
| Participants | Traumatic spinal cord injury > 30 days. PI before treatment > 4/10 N = 14 (7 participants with data) Age 27‐48 years, 1/7 women |
|
| Interventions | Gabapentin 1800 mg daily (max) Placebo NSAID, TCA and narcotics allowed for rescue medication as needed |
|
| Outcomes | Withdrawals | |
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Grants from American Academy of Physical Medicine and Rehabilitation and Eastern Paralyzed Veterans Association |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random distribution table |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Capsules with "identical shape and colour" |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | Imputation not mentioned |
| Size Efficacy | High risk | < 50 participants per treatment arm (7/14 with data) |
Wallace 2010.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups, partial enrichment, with exclusion of participants known not to respond to gabapentin or pregabalin, or who experienced dose‐limiting adverse events with gabapentin Gabapentin extended‐release given in fixed doses of 1800 mg, either as a single morning dose, or divided between 600 mg morning plus 1200 mg evening. No titration. Total duration 10 weeks |
|
| Participants | PHN. Pain at least 3 months after healing of acute herpes zoster skin rash. Initial pain ≥ 4/10 N = 405 Mean age 66 years, 52% women Mean initial pain 6.5/10 |
|
| Interventions | Gabapentin ER 1800 mg daily, n = 272 Placebo, n = 133 |
|
| Outcomes | A range of pain measures were used, but main results reported on numeric 0‐10 rating scale, as well as PGIC Adverse events Withdrawals |
|
| Notes | Oxford Quality Score: R = 1, DB = 2, W = 1, Total = 4 Sponsored by Depomed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Low risk | Use of blinded medication carton |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical blister packs |
| Incomplete outcome data (attrition bias) Efficacy | Low risk | BOCF used for main results, with LOCF also |
| Size Efficacy | Unclear risk | 50‐200 per treatment arm (133, 272) |
Zhang 2013.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel groups. Screening 4 weeks, baseline 1 week, up titration 1 week, maintenance 12 weeks, down titration 1 week | |
| Participants | PHN ≥ 3 months after healing of rash, PI ≥ 4/10, age ≥ 18 years N = 371 Mean age 62 years, 48% women Baseline PI 6/10 |
|
| Interventions | Gabapentin encarbil 1200 mg daily, n = 107 Gabapentin encarbil 2400 mg daily, n = 82 Gabapentin encarbil 3600 mg daily, n = 87 Placebo, n = 95 |
|
| Outcomes | At least 50% and at least 30% pain intensity reduction by end of maintenance over baseline PGIC much or very much improved Adverse events |
|
| Notes | Oxford Quality Score: R = 2, DB = 2, W = 1, Total = 5 Sponsored by GSK XenoPort |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Matching placebo |
| Incomplete outcome data (attrition bias) Efficacy | Unclear risk | LOCF imputation |
| Size Efficacy | Unclear risk | 50‐200 per group treatment arm (82‐107) |
ACR = American College of Rheumatology; AE = adverse event; BOCF = baseline observation carried forward; CRPS = complex regional pain syndrome; CTR = clinical trial report; DB = double‐blinding; ER = extended release; IASP = International Association for the Study of Pain; LOCF = last observation carried forward; max: maximum; NSAID = non‐steroidal anti‐inflammatory drug; OTC = over the counter; PDN = painful diabetic neuropathy; PGIC = Patient Global Impression of Change; PDN = painful diabetic neuropathy; PHN = postherpetic neuralgia; PI = pain intensity; QoL = quality of life; R = randomisation; SCI = spinal cord injury; TCA = tricyclic antidepressants; VAS: visual analogue scale; W = withdrawals.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arai 2010 | No mention of blinding of therapies in gabapentin plus imipramine additions to opioids in cancer pain |
| Berry 2005 | Single dose of gabapentin for treatment of acute herpes zoster |
| Dallocchio 2000 | Painful diabetic neuropathy, open comparison of gabapentin and amitriptyline |
| Ding 2014 | Not indexed as blinded. Gabapentin assessed as add‐on therapy to transdermal fentanyl |
| Dworkin 2009 | Study for acute herpes zoster pain |
| Ho 2009 | Short duration (1 week at stable dose), potential use of significant dose of gabapentin as rescue medication |
| Jean 2005 | Postherpetic neuralgia, with open administration of gabapentin |
| Kasimcan 2010 | Acute and chronic radicular pain, with open administration of gabapentin |
| Keskinbora 2007 | Neuropathic cancer pain, with open administration of gabapentin |
| Kimos 2007 | Condition (CRPS 1) not now defined as neuropathic pain |
| Ko 2010 | Open comparison of gabapentin and tramadol/paracetamol in painful diabetic neuropathy |
| McCleane 2001 | Low back pain, not specifically neuropathic |
| NCT00634543 | Open label study |
| NCT01263132 | No active or placebo comparator, randomised for B vitamins not gabapentin |
| NCT01623271 | Single group cohort without comparator |
| Nikolajsen 2006 | Trial of gabapentin in surgery to test whether use in surgery prevents development of phantom pain. There was no beneficial effect |
| Pandey 2002 | Guillain‐Barré syndrome |
| Pandey 2005 | Guillain‐Barré syndrome |
| Salvaggio 2008 | Facial pain, open administration of gabapentin plus tramadol |
| Sator‐Katzenschlager 2005 | Chronic pelvic pain, with open administration of gabapentin |
| Tanenberg 2011 | Open label study |
| Van de Vusse 2004 | Condition (masticatory pain) not now defined as neuropathic pain |
| Yaksi 2007 | Lumbar spinal stenosis, with open administration of gabapentin |
| Yelland 2009 | No‐of‐1 study with short treatment periods of 2 weeks in chronic neuropathic pain, and with high withdrawal rate. Study design highly unusual and difficult to interpret |
| Yildrim 2003 | Not double‐blind |
Characteristics of ongoing studies [ordered by study ID]
Fleckstein 2009.
| Trial name or title | Acupuncture in acute herpes zoster pain therapy (ACUZoster) – design and protocol of a randomised controlled trial |
| Methods | Double blinded, randomised controlled trial, parallel groups |
| Participants | Confirmed diagnosis of acute herpes zoster, pain intensity > 30 mm on a visual analogue scale (VAS 0 – 100 mm), standardised antiviral therapy. Men and women, ≥ 18 years old |
| Interventions | Semi‐standardised acupuncture, sham laser acupuncture, gabapentin with individualised dosage between 900 mg and 3600 mg daily |
| Outcomes | Alteration of pain intensity before and 1 week after treatment sessions |
| Starting date | Recruitment for the trial started in November 2008 |
| Contact information | dominik.irnich@med.uni‐muenchen.de |
| Notes | NCT00885586 ‐ "still recruiting"; record verified February 2017 |
IRCT201212019014N14.
| Trial name or title | Effect of gabapentin on heart rate variability in diabetic painful peripheral neuropathy: a double blinded randomised clinical trial |
| Methods | Double‐blinded, randomised controlled trial, parallel groups |
| Participants | Diabetic painful peripheral neuropathy. Men and women, ≥ 18 years old |
| Interventions | Gabapentin capsule 100 mg in the first day, 200 mg in the second day, and 300 mg daily from third day for 3 months plus moisturizing cream (as placebo) with a phalanx size 3 times a day for three months Capsule like gabapentin including starch (as placebo) daily for 3 months plus Kapsycin cream for reducing pain with a phalanx size 3 times a day for 3 months |
| Outcomes | Standard deviation of "N‐N (SDNN)" using 24 hours Holter monitoring device Orthostatic hypotension Resting tachycardia Any adverse events |
| Starting date | Recruitment stared 21 December 2012, expected to end March 2013 |
| Contact information | m.vasheghani@umsha.ac.ir |
| Notes | Recruitment complete. No further update by February 2017 |
NCT00674687.
| Trial name or title | A study of the efficacy of gabapentin in neuropathic pain patients as measured by quantitative sensory testing |
| Methods | Randomised, double‐blind, cross‐over |
| Participants | Men and women, ≥ 18 years old. Neuropathic pain of peripheral origin as a consequence of either postherpetic neuralgia or post‐traumatic neuropathic pain. Pain ≥ 4/10 for von Frey filament‐evoked allodynia at the skin area |
| Interventions | Gabapentin titrated to 1800 mg daily, placebo |
| Outcomes | Presence/intensity of punctate allodynia (von Frey filament) |
| Starting date | July 2004, completed 2006 |
| Contact information | Director, Clinical Trial Disclosure Group, Pfizer, Inc. |
| Notes | Possible exclude as response to evoked pain, but inadequate information to judge; 23 enrolled No further update by February 2017, and no further information on Pfizer Clinical Study Results Synopses |
VAS: visual analogue scale.
Differences between protocol and review
The protocol for the original gabapentin review (Wiffen 2005) was superceded and split, and an updated protocol produced for the 2011 review (Moore 2011a), to reflect, at least in part, the more recent developments in understanding of potential biases in chronic pain trials, and new outcomes of direct relevance to people with neuropathic pain. The main difference between the original review and the updated protocol was more emphasis being given to a set of core outcomes, although all of those outcomes were included in the updated protocol.
In the 2014 update we emphasised the difference between first tier and second tier evidence, and also emphasised the differences between conditions now defined as neuropathic pain, and other conditions such as masticatory pain, complex regional pain syndrome‐1, and fibromyalgia.
In the 2017 update, we have removed tiers of evidence as these are now largely superceded by GRADE. We have set a minimum study duration of two weeks for this chronic pain condition, in keeping with other reviews in this area that now use only longer duration studies. We are using newer definitions for what constitutes neuropathic pain.
Contributions of authors
For the 2011 update: PW, RAM, and SD wrote the 2011 protocol; PW, SD, and RAM carried out searches, assessed inclusion of papers, and extracted data. RAM wrote up the 2011 review and all authors contributed to the final draft and approved the published version.
For the 2014 update: RAM and SD carried out searches, selected studies, and added new data to the review. TRT and AR commented on clinical aspects relating to gabapentin. All authors contributed to the final draft and approved the published version.
For the 2017 update: RAM and SD carried out searches, selected studies, and added new data to the review. All authors contributed to the final draft and approved the published version.
PW will be responsible for the update.
Sources of support
Internal sources
-
Oxford Pain Relief Trust, UK.
General institutional support
External sources
NHS Cochrane Collaboration Programme Grant Scheme, UK.
European Union Biomed 2 Grant no. BMH4 CT95 0172, UK.
-
The National Institute for Health Research (NIHR), UK.
NIHR Cochrane Programme Grant: 13/89/29 ‐ Addressing the unmet need of chronic pain: providing the evidence for treatments of pain.
Declarations of interest
PW: none known
SD: none known
RFB: none known. RFB is a retired specialist pain physician who has managed patients with neuropathic pain.
ASCR: undertakes consultancy and advisory board work for Imperial College Consultants ‐ since June 2013 this has included remunerated work for: Spinifex, Abide, Astellas, Neusentis, Merck, Medivir, Mitsubishi, Aquilas, Asahi Kasei, Relmada, Novartis, and Orion. All consultancy activity relates to consultancy advice on the preclinical/clinical development of drugs for neuropathic pain. Neusentis was a subsidiary of Pfizer. He owned share options in Spinifex Pharmaceuticals which was acquired by Novartis in July 2015. ASCR was a Principal Investigator in the EuroPain consortium. EuroPain has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement number 115007, resources for which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/20072013) and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies (www.imieuropain.org). Specifically, research funding for ASCR's laboratory has been received by Imperial College from Pfizer (manufacturer of gabapentin) and Astellas ‐ both these grants were for projects related to improving the validity of animal models of neuropathic pain. ASCR is a site investigator for the Neuropain project, funded by Pfizer via Kiel University ‐ Chief Investigator Prof Ralf Baron. He is Vice‐Chair of the International Association for the Study of Pain (IASP) Special Interest Group on Neuropathic Pain (www.neupsig.org) and serves on the Executive Committee of ACTTION (Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks; www.acttion.org).
TRT is a site investigator for the Neuropain project, funded by Pfizer. Since 2014 TRT has consulted with or received lecture fees from pharmaceutical companies related to chronic pain and analgesics: Astellas, Eli Lilly, Grünenthal, Pfizer, and Mundipharma.
TP: none known. TP is a specialist pain physician who has managed patients with neuropathic pain.
RAM has received grant support from Grünenthal relating to individual participant‐level analyses of trial data regarding tapentadol in osteoarthritis and back pain (2015) and from Novartis for network meta‐analyses in acute pain. He has received honoraria for attending boards with Menarini concerning methods of analgesic trial design (2014), with Novartis (2014) about the design of network meta‐analyses, and RB on understanding pharmacokinetics of drug uptake (2015). He has received honoraria from Omega Pharma (2016) and Futura Pharma (2016) for providing advice on trial and data analysis methods.
This review was identified in a 2019 audit as not meeting the current definition of the Cochrane Commercial Sponsorship policy. At the time of its publication it was compliant with the interpretation of the existing policy. As with all reviews, new and updated, at update this review will be revised according to 2020 policy update.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Atkinson 2016 {published data only}
- Atkinson JH, Slater MA, Capparelli EV, Patel SM, Wolfson T, Gamst A, et al. A randomized controlled trial of gabapentin for chronic low back pain with and without a radiating component. Pain 2016;157(7):1499‐507. [DOI: 10.1097/j.pain.0000000000000554] [DOI] [PMC free article] [PubMed] [Google Scholar]
Backonja 1998 {published data only}
- Trial summary ‐ Parke Davis/Pfizer 945‐210. Unpublished report.
- Backonja M, Beydoun A, Edwards KR, Schwartz SL, Fonseca V, Hes M, et al. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial. JAMA 1998;280(21):1831‐6. [DOI: 10.1001/jama.280.21.1831] [DOI] [PubMed] [Google Scholar]
Backonja 2011 {published data only}
- Backonja MM, Canafax DM, Cundy KC. Efficacy of gabapentin enacarbil vs placebo in patients with postherpetic neuralgia and a pharmacokinetic comparison with oral gabapentin. Pain Medicine 2011;12(7):1098‐108. [DOI: 10.1111/j.1526-4637.2011.01139.x] [DOI] [PubMed] [Google Scholar]
Bone 2002 {published data only}
- Bone M, Critchley P, Buggy DJ. Gabapentin in postamputation phantom limb pain: a randomized, double‐blind, placebo‐controlled, cross‐over study. Regional Anesthesia and Pain Medicine 2002;27(5):481‐6. [DOI: 10.1053/rapm.2002.35169] [DOI] [PubMed] [Google Scholar]
Caraceni 2004 {published data only}
- Caraceni A, Zecca E, Bonezzi C, Arcuri E, Yaya Tur R, et al. Gabapentin for neuropathic cancer pain: a randomized controlled trial from the Gabapentin Cancer Pain Study Group. Journal of Clinical Oncology 2004;22(14):2909‐17. [DOI: 10.1200/JCO.2004.08.141] [DOI] [PubMed] [Google Scholar]
Chandra 2006 {published data only}
- Chandra K, Shafiq N, Pandhi P, Gupta S, Malhotra S. Gabapentin versus nortriptyline in post‐herpetic neuralgia patients: a randomized, double‐blind clinical trial ‐ the GONIP Trial. International Journal of Clinical Pharmacology and Therapeutics 2006;44(8):358‐63. [PUBMED: 16961166] [DOI] [PubMed] [Google Scholar]
Cohen 2015 {published data only}
- Cohen SP, Hanling S, Bicket MC, White RL, Veizi E, Kurihara C, et al. Epidural steroid injections compared with gabapentin for lumbosacral radicular pain: multicenter randomized double blind comparative efficacy study. BMJ 2015;350:h1748. [CTG: NCT01495923; DOI: 10.1136/bmj.h1748] [DOI] [PMC free article] [PubMed] [Google Scholar]
CTR 945‐1008 {unpublished data only}
- Protocol A9451008. A 15 week, randomized, double‐blind, placebo‐controlled, parallel‐group, multi‐center study of Neurontin (gabapentin) for efficacy and quality of life in patients with painful diabetic peripheral neuropathy. PhrmaWebSynopsis ‐ Final 2 June 2005.
CTR 945‐224 {unpublished data only}
- Protocol 945‐224. A double‐blind placebo‐controlled trial with 3 doses of gabapentin for treatment of painful diabetic peripheral neuropathy. May 29, 1998 through September 7, 1999. Unpublished.
Gilron 2005 {published data only}
- Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. New England Journal of Medicine 2005;352(13):1324‐34. [DOI: 10.1056/NEJMoa042580] [DOI] [PubMed] [Google Scholar]
Gilron 2009 {published data only}
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double‐blind, randomised controlled crossover trial. Lancet 2009;374(9697):1252‐61. [DOI: 10.1016/s0140-6736(09)61081-3] [DOI] [PubMed] [Google Scholar]
Gong 2008 {published data only}
- Gong Z‐Y, Ye T‐H, Hao R‐R, Shi Y‐X, Xiong L‐Z, Wang Q‐S, et al. The efficacy and safety of gabapentin in postherpetic neuralgia. Chinese Journal of Pain Medicine 2008;2. [Google Scholar]
Gordh 2008 {published data only}
- Gordh TE, Stubhaug A, Jensen TS, Arner S, Biber B, Boivie J, et al. Gabapentin in traumatic nerve injury pain: a randomized, double‐blind, placebo‐controlled, cross‐over, multi‐center study. Pain 2008;138(2):255‐66. [DOI: 10.1016/j.pain.2007.12.011] [DOI] [PubMed] [Google Scholar]
Gorson 1999 {published data only}
- Gorson KC, Schott C, Herman R, Ropper AH. Gabapentin in the treatment of painful diabetic neuropathy: a placebo controlled, double blind, crossover trial. Journal of Neurology, Neurosurgery and Psychiatry 1999;66:251‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hahn 2004 {published data only}
- Hahn K, Arendt G, Braun JS, Giesen HJ, Husstedt IW, et al. German Neuro‐AIDS Working Group. A placebo‐controlled trial of gabapentin for painful HIV‐associated sensory neuropathies. Journal of Neurology 2004;251(10):1260‐6. [DOI: 10.1007/s00415-004-0529-6] [DOI] [PubMed] [Google Scholar]
Harden 2013 {published data only}
- Harden RN, Freeman R, Rainka M, Zhang L, Bell C, Berges A, et al. A phase 2a, randomized, crossover trial of gabapentin enacarbil for the treatment of postherpetic neuralgia in gabapentin inadequate responders. Pain Medicine 2013;14(12):1918‐32. [DOI: 10.1111/pme.12227] [DOI] [PubMed] [Google Scholar]
Irving 2009 {published data only}
- Irving G, Jensen M, Cramer M, Wu J, Chiang YK, Tark M, et al. Efficacy and tolerability of gastric‐retentive gabapentin for the treatment of postherpetic neuralgia: results of a double‐blind, randomized, placebo‐controlled clinical trial. Clinical Journal of Pain 2009;25(3):185‐92. [DOI: 10.1097/AJP.0b013e3181934276] [DOI] [PubMed] [Google Scholar]
- Jensen MP, Chiang YK, Wu J. Assessment of pain quality in a clinical trial of gabapentin extended release for postherpetic neuralgia. Clinical Journal of Pain 2009;25(4):286‐92. [DOI: 10.1097/AJP.0b013e318192bf87] [DOI] [PubMed] [Google Scholar]
Levendoglu 2004 {published data only}
- Levendoglu F, Ogun CO, Ozerbil O, Ogun TC, Ugurlu H. Gabapentin is a first line drug for the treatment of neuropathic pain in spinal cord injury. Spine 2004;29(7):743‐51. [DOI: 10.1097/01.BRS.0000112068.16108.3A] [DOI] [PubMed] [Google Scholar]
Mishra 2012 {published data only}
- Mishra S, Bhatnagar S, Goyal GN, Rana SP, Upadhya SP. A comparative efficacy of amitriptyline, gabapentin, and pregabalin in neuropathic cancer pain: a prospective randomized double‐blind placebo‐controlled study. American Journal of Hospice and Palliative Care 2012;29(3):177‐82. [DOI: 10.1177/1049909111412539] [DOI] [PubMed] [Google Scholar]
Morello 1999 {published data only}
- Morello CM, Leckband SG, Stoner CP, Moorhouse DF, Sahagian GA. Randomized double‐blind study comparing the efficacy of gabapentin with amitriptyline on diabetic peripheral neuropathy pain. Archives of Internal Medicine 1999;159(16):1931‐7. [PUBMED: 10493324] [DOI] [PubMed] [Google Scholar]
NCT00475904 {published data only}
- A Phase II, double‐blind, randomized, placebo‐controlled non‐inferiority trial of EpiCept™ NP‐1 topical cream (2% ketamine/4% amitriptyline) vs. oral gabapentin in postherpetic neuralgia (PHN). clinicaltrials.gov/ct2/show/NCT00475904 (accessed 13 February 2017). [CTG: NCT00475904]
NCT00904202 {published data only}
- A study of lidocaine patch 5% alone, gabapentin alone, and lidocaine patch 5% and gabapentin in combination for the relief of pain in patients with diverse peripheral neuropathic pain conditions. clinicaltrials.gov/ct2/show/NCT00904202 (accessed 13 February 2017). [CTG: NCT00904202]
- Clinical trial results summary: a multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study of lidocaine patch 5% alone, gabapentin alone, and lidocaine patch 5% and gabapentin in combination for the relief of pain in patients with diverse peripheral neuropathic pain conditions. endo.com/home/search?k=EN3220‐009 (accessed 13 February 2017).
Perez 2000 {published data only}
- Perez HET, Sanchez GF. Gabapentin therapy for diabetic neuropathic pain. Journal of Medicine 2000;108:689. [DOI: 10.1016/S0002-9343(00)00398-3] [DOI] [PubMed] [Google Scholar]
Rao 2007 {published data only}
- Rao RD, Michalak JC, Sloan JA, Loprinzi CL, Soori GS, Nikcevich DA, et al. North Central Cancer Treatment Group. Efficacy of gabapentin in the management of chemotherapy‐induced peripheral neuropathy: a phase 3 randomized, double‐blind, placebo‐controlled, crossover trial (N00C3). Cancer 2007;110(9):2110‐8. [CTG: NCT00027963; DOI: 10.1002/cncr.23008] [DOI] [PubMed] [Google Scholar]
Rauck 2013a {published data only}
- Rauck R, Makumi CW, Schwartz S, Graff O, Meno‐Tetang G, Bell CF, et al. A randomized, controlled trial of gabapentin enacarbil in subjects with neuropathic pain associated with diabetic peripheral neuropathy. Pain Practice 2013;13(6):485‐96. [CTG: NCT02074267; DOI: 10.1111/papr.12014] [DOI] [PubMed] [Google Scholar]
Rauck 2013b {published data only}
- Rauck R, Coffey RJ, Schultz DM, Wallace MS, Webster LR, McCarville SE, et al. Intrathecal gabapentin to treat chronic intractable noncancer pain. Anesthesiology 2013;119(3):675‐86. [CTG: NCT00414466; DOI: 10.1097/ALN.0b013e3182a10fbf] [DOI] [PubMed] [Google Scholar]
Rice 2001 {published data only}
- Study detail and analysis, Parke‐Davis 945‐295. Unpublished Report No. RR‐430‐00124 2000.
- Rice AS, Maton S, Postherpetic Neuralgia Study Group. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain 2001;94(2):215‐24. [DOI: 10.1016/S0304-3959(01)00407-9] [DOI] [PubMed] [Google Scholar]
Rintala 2007 {published data only}
- Rintala DH, Holmes SA, Courtade D, Fiess RN, Tastard LV, Loubser PG. Comparison of the effectiveness of amitriptyline and gabapentin on chronic neuropathic pain in persons with spinal cord injury. Archives of Physical Medicine and Rehabilitation 2007;88(12):1547‐60. [DOI: 10.1016/j.apmr.2007.07.038] [DOI] [PubMed] [Google Scholar]
Rowbotham 1998 {published data only}
- Detailed summary, study No.4, Parke‐Davis 945‐211. Unpublished Report No. RR‐995‐00070 1998.
- Rowbotham M, Harden N, Stacey B, Bernstein P, Magnus‐Miller L. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA 1998;280(21):1837‐42. [DOI: 10.1001/jama.280.21.1837] [DOI] [PubMed] [Google Scholar]
Sandercock 2012 {published data only}
- Sandercock D, Cramer M, Biton V, Cowles VE. A gastroretentive gabapentin formulation for the treatment of painful diabetic peripheral neuropathy: efficacy and tolerability in a double‐blind, randomized, controlled clinical trial. Diabetes Research and Clinical Practice 2012;97(3):438‐45. [DOI: 10.1016/j.diabres.2012.03.010] [DOI] [PubMed] [Google Scholar]
- Sandercock D, Cramer M, Wu J, Chiang YK, Biton V, Heritier M. Gabapentin extended release for the treatment of painful diabetic peripheral neuropathy: efficacy and tolerability in a double‐blind, randomized, controlled clinical trial. Diabetes Care 2009;32(2):e20. [CTG: NCT00712439; DOI: 10.2337/dc08-1450] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sang 2013 {published data only}
- Freeman R, Wallace MS, Sweeney M, Backonja MM. Relationships among pain quality, pain impact, and overall improvement in patients with postherpetic neuralgia treated with gastroretentive gabapentin. Pain Medicine (Malden, Mass.) 2015;16(10):2000‐11. [DOI: 10.1111/pme.12791] [DOI] [PubMed] [Google Scholar]
- Mehta N, Bucior I, Bujanover S, Shah R, Gulati A. Relationship between pain relief, reduction in pain‐associated sleep interference, and overall impression of improvement in patients with postherpetic neuralgia treated with extended‐release gabapentin. Health and Quality of Life Outcomes 2016;14:54. [DOI: 10.1186/s12955-016-0456-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang CN, Sathyanarayana R, Sweeney M, DM‐1796 Study Investigators. Gastroretentive gabapentin (G‐GR) formulation reduces intensity of pain associated with postherpetic neuralgia (PHN). Clinical Journal of Pain 2013;29(4):281‐8. [DOI: 10.1097/AJP.0b013e318258993e] [DOI] [PubMed] [Google Scholar]
Serpell 2002 {published data only}
- Parke Davis/Pfizer. 945‐430‐306. Unpublished 2000.
- Serpell MG, Neuropathic pain study group. Gabapentin in neuropathic pain syndromes: a randomised, double‐blind, placebo‐controlled trial. Pain 2002;99(3):557‐66. [DOI: 10.1016/S0304-3959(02)00255-5] [DOI] [PubMed] [Google Scholar]
Simpson 2001 {published data only}
- Simpson DA. Gabapentin and venlafaxine for the treatment of painful diabetic neuropathy. Journal of Clinical Neuromuscular Disease 2001;3(2):53‐62. [PUBMED: 19078655] [DOI] [PubMed] [Google Scholar]
Smith 2005 {published data only}
- Smith DG, Ehde DM, Hanley MA, Campbell KM, Jensen MP, Hoffman AJ, et al. Efficacy of gabapentin in treating chronic phantom limb and residual limb pain. Journal of Rehabilitation Research and Development 2005;42(5):645‐54. [DOI: 10.1682/JRRD.2005.05.0082] [DOI] [PubMed] [Google Scholar]
Tai 2002 {published data only}
- Tai Q, Kirshblum S, Chen B, Millis S, Johnston M, DeLisa JA. Gabapentin in the treatment of neuropathic pain after spinal cord injury: a prospective, randomized, double‐blind, crossover trial. Journal of Spinal Cord Medicine 2002;25(2):100‐5. [PUBMED: 12137213] [DOI] [PubMed] [Google Scholar]
Wallace 2010 {published data only}
- Freeman R, Wallace MS, Sweeney M, Backonja MM. Relationships among pain quality, pain impact, and overall improvement in patients with postherpetic neuralgia treated with gastroretentive gabapentin. Pain Medicine 2015;16(10):2000‐11. [DOI: 10.1111/pme.12791] [DOI] [PubMed] [Google Scholar]
- Mehta N, Bucior I, Bujanover S, Shah R, Gulati A. Relationship between pain relief, reduction in pain‐associated sleep interference, and overall impression of improvement in patients with postherpetic neuralgia treated with extended‐release gabapentin. Health and Quality of Life Outcomes 2016;14:54. [DOI: 10.1186/s12955-016-0456-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MS, Irving G, Cowles VE. Gabapentin extended‐release tablets for the treatment of patients with postherpetic neuralgia: a randomized, double‐blind, placebo‐controlled, multicentre study. Clinical Drug Investigation 2010;30(11):765‐76. [DOI: 10.2165/11539520-000000000-00000] [DOI] [PubMed] [Google Scholar]
Zhang 2013 {published data only}
- Calkins AM, Gudin J, Gidal B, Jaros MJ, Kim R, Shang G. Impact of data imputation methodology on pain assessment over 24 hours in a randomized, placebo‐controlled study of gabapentin enacarbil in patients with neuropathic pain associated with postherpetic neuralgia. Pain Medicine 2016;17(4):728‐36. [DOI: 10.1093/pm/pnv072] [DOI] [PubMed] [Google Scholar]
- Zhang L, Rainka M, Freeman R, Harden RN, Bell CF, Chen C, et al. A randomized, double‐blind, placebo‐controlled trial to assess the efficacy and safety of gabapentin enacarbil in subjects with neuropathic pain associated with postherpetic neuralgia (PXN110748). Journal of Pain 2013;14(6):590‐603. [CTG: NCT00619476; DOI: 10.1016/j.jpain.2013.01.768] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arai 2010 {published data only}
- Arai YC, Matsubara T, Shimo K, Suetomi K, Nishihara M, Ushida T, et al. Low‐dose gabapentin as useful adjuvant to opioids for neuropathic cancer pain when combined with low‐dose imipramine. Journal of Anesthesia 2010;24(3):407‐10. [DOI: 10.1007/s00540-010-0913-6] [DOI] [PubMed] [Google Scholar]
Berry 2005 {published data only}
- Berry JD, Petersen KL. A single dose of gabapentin reduces acute pain and allodynia in patients with herpes zoster. Neurology 2005;65(3):444‐7. [DOI: 10.1212/01.wnl.0000168259.94991.8a] [DOI] [PubMed] [Google Scholar]
Dallocchio 2000 {published data only}
- Dallocchio C, Buffa C, Mazzarello P, Chiroli S. Gabapentin vs. amitriptyline in painful diabetic neuropathy: an open‐label pilot study. Journal of Pain and Symptom Management 2000;20(4):280‐5. [DOI: 10.1016/S0885-3924(00)00181-0] [DOI] [PubMed] [Google Scholar]
Ding 2014 {published data only}
- Ding Y, Yao P, Lan P, Ma J, Wang Z, Hong T, et al. Assessment of transdermal fentanyl combined with gabapentin for malignant neuropathic pain treatment. Chinese Journal of Clinical Oncology 2014;41(20):1307‐11. [DOI: 10.3969/j.issn.1000-8179.20141212] [DOI] [Google Scholar]
Dworkin 2009 {published data only}
- Dworkin RH, Barbano RL, Tyring SK, Betts RF, McDermott MP, Pennella‐Vaughan J, et al. A randomized, placebo‐controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain 2009;142(3):209‐17. [DOI: 10.1016/j.pain.2008.12.022] [DOI] [PubMed] [Google Scholar]
Ho 2009 {published data only}
- Ho TW, Backonja M, Ma J, Leibensperger H, Froman S, Polydefkis M. Efficient assessment of neuropathic pain drugs in patients with small fiber sensory neuropathies. Pain 2009;14(1‐2):19‐24. [DOI: 10.1016/j.pain.2008.07.013] [DOI] [PubMed] [Google Scholar]
Jean 2005 {published data only}
- Jean WH, Wu CC, Mok MS, Sun WZ. Starting dose of gabapentin for patients with post‐herpetic neuralgia‐‐a dose‐response study. Acta Anaesthesiologica Taiwan 2005;43(2):73‐7. [PUBMED: 16060401] [PubMed] [Google Scholar]
Kasimcan 2010 {published data only}
- Kasimcan O, Kaptan H. Efficacy of gabapentin for radiculopathy caused by lumbar spinal stenosis and lumbar disk hernia. Neurologia Medico Chirurgica (Tokyo) 2010;50(12):1070‐3. [DOI: 10.2176/nmc.50.1070] [DOI] [PubMed] [Google Scholar]
Keskinbora 2007 {published data only}
- Keskinbora K, Pekel AF, Aydinli I. Gabapentin and an opioid combination versus opioid alone for the management of neuropathic cancer pain: a randomized open trial. Journal of Pain Symptom Management 2007;34(2):183‐9. [10.1016/j.jpainsymman.2006.11.013] [DOI] [PubMed] [Google Scholar]
Kimos 2007 {published data only}
- Kimos P, Biggs C, Mah J, Heo G, Rashiq S, Thie NM, et al. Analgesic action of gabapentin on chronic pain in the masticatory muscles: a randomized controlled trial. Pain 2007;127(1‐2):151‐60. [DOI: 10.1016/j.pain.2006.08.028] [DOI] [PubMed] [Google Scholar]
Ko 2010 {published data only}
- Ko SH, Kwon HS, Yu JM, Baik SH, Park IB, Lee JH, et al. Comparison of the efficacy and safety of tramadol/acetaminophen combination therapy and gabapentin in the treatment of painful diabetic neuropathy. Diabetes Medicine 2010;27(9):1033‐40. [DOI: 10.1111/j.1464-5491.2010.03054.x] [DOI] [PubMed] [Google Scholar]
McCleane 2001 {published data only}
- McCleane GJ. Does gabapentin have an analgesic effect on background, movement and referred pain? A randomised, double‐blind, placebo controlled study. The Pain Clinic 2001;13:103. [Google Scholar]
NCT00634543 {published data only}
- A study to compare safety and efficacy of tramadol hydrochloride/acetaminophen with gabapentin in participants with diabetic neuropathy. clinicaltrials.gov/ct2/show/NCT00634543 (accessed 13 February 2017). [CTG: NCT00634543]
NCT01263132 {published data only}
- Neuropathic pain treatment using F0434 vs. gabapentin in patients with chronic distal diabetic polyneuropathy: a randomized, controlled, double‐blind study. clinicaltrials.gov/ct2/show/NCT01263132 (accessed 13 February 2017). [CTG: NCT01263132]
NCT01623271 {published data only}
- Treatment of complex regional pain syndrome with once daily gastric‐retentive gabapentin (Gralise). clinicaltrials.gov/ct2/show/NCT01623271 (accessed 13 February 2017). [CTG: NCT01623271]
Nikolajsen 2006 {published data only}
- Nikolajsen L, Finnerup NB, Kramp S, Vimtrup AS, Keller J, Jensen TS. A randomized study of the effects of gabapentin on postamputation pain. Anesthesiology 2006;105(5):1008‐15. [PUBMED: 17065896] [DOI] [PubMed] [Google Scholar]
Pandey 2002 {published data only}
- Pandey CK, Bose N, Garg G, Singh N, Baronia A, Agarwal A, et al. Gabapentin for the treatment of pain in Guillain‐Barre syndrome: a double‐blinded, placebo‐controlled, crossover study. Anesthesia and Analgesia 2002;95(6):1719‐23. [DOI: 10.1097/00000539-200212000-00046] [DOI] [PubMed] [Google Scholar]
Pandey 2005 {published data only}
- Pandey CK, Raza M, Tripathi M, Navkar DV, Kumar A, Singh UK. The comparative evaluation of gabapentin and carbamazepine for pain management in Guillain‐Barre syndrome patients in the intensive care unit. Anesthesia and Analgesia 2005;101(1):220‐5. [DOI: 10.1213/01.ANE.0000152186.89020.36] [DOI] [PubMed] [Google Scholar]
Salvaggio 2008 {published data only}
- Salvaggio I, Adducci E, Dell'Aquila L, Rinaldi S, Marini M, Zappia L, et al. Facial pain: a possible therapy with stellate ganglion block. Pain Medicine 2008;9(7):958‐62. [DOI: 10.1111/j.1526-4637.2008.00515.x] [DOI] [PubMed] [Google Scholar]
Sator‐Katzenschlager 2005 {published data only}
- Sator‐Katzenschlager SM, Scharbert G, Kress HG, Frickey N, Ellend A, Gleiss A, et al. Chronic pelvic pain treated with gabapentin and amitriptyline: a randomized controlled pilot study. Wiener Klinische Wochenschrift 2005;117(21‐22):761‐8. [DOI: 10.1007/s00508-005-0464-2] [DOI] [PubMed] [Google Scholar]
Tanenberg 2011 {published data only}
- Tanenberg RJ, Irving GA, Risser RC, Ahl J, Robinson MJ, Skljarevski V, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open‐label, randomized, noninferiority comparison. Mayo Clinic proceedings 2011;86(7):615‐26. [CTG: NCT00385671; DOI: 10.4065/mcp.2010.0681] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van de Vusse 2004 {published data only}
- Vusse AC, Stomp‐van den Berg SG, Kessels AH, Weber WE. Randomised controlled trial of gabapentin in Complex Regional Pain Syndrome type 1 [ISRCTN84121379]. BMC Neurology 2004;4:13. [DOI: 10.1186/1471-2377-4-13] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yaksi 2007 {published data only}
- Yaksi A, Ozgonenel L, Ozgonenel B. The efficiency of gabapentin therapy in patients with lumbar spinal stenosis. Spine 2007;32(9):939‐42. [DOI: 10.1097/01.brs.0000261029.29170.e6] [DOI] [PubMed] [Google Scholar]
Yelland 2009 {published data only}
- Yelland MJ, Poulos CJ, Pillans PI, Bashford GM, Nikles CJ, Sturtevant JM, et al. N‐of‐1 randomized trials to assess the efficacy of gabapentin for chronic neuropathic pain. Pain Medicine 2009;10(4):754‐61. [DOI: 10.1111/j.1526-4637.2009.00615.x] [DOI] [PubMed] [Google Scholar]
Yildrim 2003 {published data only}
- Yildirim K, Siseciolglu M, Karatay S, Erdal A, Levent A, Ugur M. The effectiveness of gabapentin in patients with chronic radiculopathy. The Pain Clinic 2003;15(3):213‐8. [Google Scholar]
References to ongoing studies
Fleckstein 2009 {published data only}
- Fleckenstein J, Kramer S, Hoffrogge P, Thoma S, Lang PM, Lehmeyer L, et al. Acupuncture in acute herpes zoster pain therapy (ACUZoster) ‐ design and protocol of a randomised controlled trial. BMC Complementary and Alternative Medicine 2009;9:31. [CTG: NCT00885586; DOI: 10.1186/1472-6882-9-31] [DOI] [PMC free article] [PubMed] [Google Scholar]
IRCT201212019014N14 {published data only}
- Vasheghani M (contact person). Effect of gabapentin on heart rate variability in diabetic painful peripheral neuropathy: a double blinded randomized clinical trial. apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT201212019014N14 (accessed 13 February 2017).
NCT00674687 {published data only}
- A study of the efficacy of gabapentin in neuropathic pain patients as measured by quantitative sensory testing. clinicaltrials.gov/ct2/show/NCT00674687 (accessed 13 February 2017). [NCT: NCT00674687]
Additional references
AlBalawi 2013
- AlBalawi Z, McAlister FA, Thorlund K, Wong M, Wetterslev J. Random error in cardiovascular meta‐analyses: how common are false positive and false negative results?. International Journal of Cardiology 2013;168(2):1102‐7. [DOI: 10.1016/j.ijcard.2012.11.048] [DOI] [PubMed] [Google Scholar]
Baron 2012
- Baron R, Wasner G, Binder A. Chronic pain: genes, plasticity, and phenotypes. Lancet Neurology 2012;11(1):19‐21. [DOI: 10.1016/S1474-4422(11)70281-2] [DOI] [PubMed] [Google Scholar]
Baron 2017
- Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism‐related organizing principle based on sensory profiles. Pain 2017;158(2):261‐72. [DOI: 10.1097/j.pain.0000000000000753] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berger 2004
- Berger A, Dukes EM, Oster G. Clinical characteristics and economic costs of patients with painful neuropathic disorders. Journal of Pain 2004;5(3):143‐9. [DOI: 10.1016/j.jpain.2003.12.004] [DOI] [PubMed] [Google Scholar]
Berger 2009
- Berger A, Toelle T, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical and economic characteristics of patients with painful neuropathic disorders in Germany. Pain Practice 2009;9(1):8‐17. [DOI: 10.1111/j.1533-2500.2008.00244.x] [DOI] [PubMed] [Google Scholar]
Berger 2012
- Berger A, Sadosky A, Dukes E, Edelsberg J, Oster G. Clinical characteristics and patterns of healthcare utilization in patients with painful neuropathic disorders in UK general practice: a retrospective cohort study. BMC Neurology 2012;12:8. [DOI: 10.1186/1471-2377-12-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lantéri‐Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136(3):380‐7. [DOI] [PubMed] [Google Scholar]
Boyle 2014
- Boyle Y, Fernando D, Kurz H, Miller SR, Zucchetto M, Storey J. The effect of a combination of gabapentin and donepezil in an experimental pain model in health volunteers: results of a randomised controlled trial. Pain 2014;155(12):2510‐6. [DOI: 10.1016/j.pain.2014.09.003] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive‐‐Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [DOI: 10.1093/ije/dyn188] [DOI] [PubMed] [Google Scholar]
Calvo 2012
- Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurology 2012;11(7):629‐42. [DOI: 10.1016/S1474-4422(12)70134-5] [DOI] [PubMed] [Google Scholar]
Chang 2014
- Chang CY, Challa CK, Shah J, Eloy JD. Gabapentin in acute postoperative pain management. Biomed Research International 2014;2014:631756. [DOI: 10.1155/2014/631756] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chaparro 2012
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD008943.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chou 2009
- Chou R, Carson S, Chan BK. Gabapentin versus tricyclic antidepressants for diabetic neuropathy and post‐herpetic neuralgia: discrepancies between direct and indirect meta‐analyses of randomized controlled trials. Journal of General Internal Medicine 2009;24(2):178‐88. [DOI: 10.1007/s11606-008-0877-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Colloca 2017
- Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nature Reviews Disease Primers 2017;3:17002. [DOI: 10.1038/nrdp.2017.2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cooper 2017
- Cooper TE, Derry S, Wiffen PJ, Moore RA. Gabapentin for fibromyalgia pain in adults. Cochrane Database of Systematic Reviews 2017, Issue 1. [DOI: 10.1002/14651858.CD012188.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2013
- Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta‐epidemiological study. BMJ 2013;346:f2304. [DOI: 10.1136/bmj.f2304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2014
- Dechartres A, Altman DG, Trinquart L, Boutron I, Ravaud P. Association between analytic strategy and estimates of treatment outcomes in meta‐analyses. JAMA 2014;312:623‐30. [DOI: 10.1001/jama.2014.8166] [DOI] [PubMed] [Google Scholar]
Demant 2014
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double‐blind, placebo‐controlled phenotype‐stratified study. Pain 2014;155(11):2263‐73. [DOI: 10.1016/j.pain.2014.08.014] [DOI] [PubMed] [Google Scholar]
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2014
- Derry S, Wiffen PJ, Moore RA, Quinlan J. Topical lidocaine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD010958.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2016
- Derry S, Stannard C, Cole P, Wiffen PJ, Knaggs R, Aldington D, et al. Fentanyl for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 10. [DOI: 10.1002/14651858.CD011605.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2017
- Derry S, Rice ASC, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2017, Issue 1. [DOI: 10.1002/14651858.CD007393.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Dworkin 2013
- Dworkin RH, O'Connor AB, Kent J, Mackey SC, Raja SN, Stacey BR, et al. Interventional management of neuropathic pain: NeuPSIG recommendations. Pain 2013;154(11):2249‐61. [DOI: 10.1016/j.pain.2013.06.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
EMC 2017
- Electronic Medicines Compendium. emc.medicines.org.uk/ (accessed 13 February 2017).
EPOC 2015
- Effective Practice, Organisation of Care (EPOC). 23. Worksheets for preparing a Summary of Findings using GRADE. Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services. Available at: epoc.cochrane.org/epoc‐specific‐resources‐review‐authors (accessed 13 February 2017).
Eroglu 2009
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009;139(2):380‐92. [DOI: 10.1016/j.cell.2009.09.025] [DOI] [PMC free article] [PubMed] [Google Scholar]
Evoy 2017
- Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs 2017;77(4):403‐26. [DOI: 10.1007/s40265-017-0700-x] [DOI] [PubMed] [Google Scholar]
Fanelli 2017
- Fanelli D, Costas R, Ioannidis JP. Meta‐assessment of bias in science. PNAS 2017;114:3714‐9. [DOI: 10.1073/pnas.1618569114] [DOI] [PMC free article] [PubMed] [Google Scholar]
Finnerup 2005
- Finnerup NB, Otto M, McQuay HJ, Jensen TS, Sindrup SH. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain 2005;118(3):289‐305. [DOI: 10.1016/j.pain.2005.08.013] [DOI] [PubMed] [Google Scholar]
Finnerup 2013
- Finnerup NB, Scholz J, Attal N, Baron R, Haanpää M, Hansson P, et al. Neuropathic pain needs systematic classification. European Journal of Pain 2013;17(7):953‐6. [DOI: 10.1002/j.1532-2149.2012.00282.x] [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurology 2015;14(2):162‐73. [DOI: 10.1016/S1474-4422(14)70251-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gaskell 2016
- Gaskell H, Derry S, Stannard C, Moore RA. Oxycodone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 7. [DOI: 10.1002/14651858.CD010692.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self‐reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52(1):132‐6. [DOI: 10.1111/j.1399-6576.2007.01486.x] [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence‐‐inconsistency. Journal of Cinical Epidemiology 2011;64(12):1294‐302. [DOI: 10.1016/j.jclinepi.2011.03.017] [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso‐Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66:151‐7. [DOI: 10.1016/j.jclinepi.2012.01.006] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables‐binary outcomes. Journal of Clinical Epidemiology 2013;66:158‐72. [DOI: 10.1016/j.jclinepi.2012.01.012] [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002‐2005. BMC Family Practice 2008;9:26. [DOI: 10.1186/1471-2296-9-26] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hall 2013
- Hall GC, Morant SV, Carroll D, Gabriel ZL, McQuay HJ. An observational descriptive study of the epidemiology and treatment of neuropathic pain in a UK general population. BMC Family Practice 2013;14:28. [DOI: 10.1186/1471-2296-14-28] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hauser 2009
- Haüser W, Bernardy K, Uceyler N, Sommer C. Treatment of fibromyalgia syndrome with gabapentin and pregabalin ‐ a meta‐analysis of randomized controlled trials. Pain 2009;145(1‐2):69‐81. [DOI: 10.1016/j.pain.2009.05.014] [DOI] [PubMed] [Google Scholar]
Helfert 2015
- Helfert SM, Reimer M, Höper J, Baron R. Individualized pharmacological treatment of neuropathic pain. Clinical Pharmacology and Therapeutics 2015;97(2):135‐42. [DOI: 10.1002/cpt.19] [DOI] [PubMed] [Google Scholar]
Hempenstall 2005
- Hempenstall K, Nurmikko TJ, Johnson RW, A'Hern RP, Rice AS. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Medicine 2005;2(7):e164. [DOI: 10.1371/journal.pmed.0020164] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org. The Cochrane Collaboration.
Hoffman 2010
- Hoffman DL, Sadosky A, Dukes EM, Alvir J. How do changes in pain severity levels correspond to changes in health status 3 and function in patients with painful diabetic peripheral neuropathy?. Pain 2010;149(2):194‐201. [DOI: 10.1016/j.pain.2009.09.017] [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJM, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI: 10.1016/0197-2456(95)00134-4] [DOI] [PubMed] [Google Scholar]
Jensen 2011
Jensen 2012
- Jensen MP, Hsu PH, Vanhove GF. Early pain reduction can predict treatment response: results of integrated efficacy analyses of a once‐daily gastroretentive formulation of gabapentin in patients with postherpetic neuralgia. Pain Medicine 2012;13(8):1059‐66. [DOI: 10.1111/j.1526-4637.2012.01427.x.] [DOI] [PubMed] [Google Scholar]
Kalso 2013
- Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013;347:f7339. [DOI: 10.1136/bmj.f7339] [DOI] [PubMed] [Google Scholar]
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota, 1945‐1984. Neuroepidemiology 1991;10(5‐6):276‐81. [DOI: 10.1159/000110284] [DOI] [PubMed] [Google Scholar]
Khan 1996
- Khan KS, Daya S, Jadad A. The importance of quality of primary studies in producing unbiased systematic reviews. Archives of Internal Medicine 1996;156(6):661‐6. [DOI: 10.1001/archinte.1996.00440060089011] [DOI] [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147(1‐3):122‐7. [DOI: 10.1016/j.pain.2009.08.023] [DOI] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107(2):224‐33. [DOI: 10.7326/0003-4819-107-2-224] [DOI] [PubMed] [Google Scholar]
Landefeld 2009
- Landefeld CS, Steinman MA. The Neurontin legacy ‐ marketing through misinformation and manipulation. New England Journal of Medicine 2009;360(2):103‐6. [DOI: 10.1056/NEJMp0808659] [DOI] [PubMed] [Google Scholar]
Lunn 2009
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy or chronic pain. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007115.pub2] [DOI] [PubMed] [Google Scholar]
Lunn 2014
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD007115.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1995
- McQuay H, Carroll D, Jadad AR, Wiffen P, Moore A. Anticonvulsant drugs for management of pain: a systematic review. BMJ 1995;311(7012):1047‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1996
- McQuay H, Carroll D, Moore A. Variation in the placebo effect in randomised controlled trials of analgesics: all is as blind as it seems. Pain 1996;64(2):331‐5. [DOI] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An evidence‐based resource for pain relief. An evidence‐based resource for pain relief. Oxford: Oxford University Press, 1998. [ISBN: 0‐19‐263048‐2] [Google Scholar]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic Pain. In: Stevens A, Raftery J, Mant J, Simpson S editor(s). Health Care Needs Assessment: The Epidemiologically Based Needs Assessment Reviews: Third Series. Oxford: Radcliffe Publishing, 2007. [ISBN: 978‐1‐84619‐063‐6] [Google Scholar]
McQuay 2008
- McQuay HJ, Derry S, Moore RA, Poulain P, Legout V. Enriched enrolment with randomised withdrawal (EERW): time for a new look at clinical trial design in chronic pain. Pain 2008;135(3):217‐220. [DOI: 10.1016/j.pain.2008.01.014] [DOI] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15‐24. [ISBN: 978‐0‐931092‐69‐5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, McQuay H, ACTINPAIN Writing Group of the IASP Special Interest Group on Systematic Reviews in Pain Relief, Cochrane Pain, Palliative and Supportive Care Systematic Review Group Editors. "Evidence" in chronic pain‐‐establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Straube S, Derry S, McQuay HJ. Topical review: chronic low back pain analgesic studies ‐ a methodological minefield. Pain 2010;149(3):431‐4. [DOI: 10.1016/j.pain.2010.02.032] [DOI] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses ‐ do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI: 10.1016/j.pain.2010.07.013] [DOI] [PubMed] [Google Scholar]
Moore 2010e
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010f
- Moore RA, Derry S, McQuay HJ, Straube S, Aldington D, Wiffen P, et al. for the ACTINPAIN writing group of the IASP Special Interest Group (SIG) on Systematic Reviews in Pain Relief. Clinical effectiveness: an approach to clinical trial design more relevant to clinical practice, acknowledging the importance of individual differences. Pain 2010;149(2):173‐6. [DOI: 10.1016/j.pain.2009.08.007] [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI: 10.1016/j.pain.2010.11.030] [DOI] [PubMed] [Google Scholar]
Moore 2012a
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI: 10.1016/j.pain.2011.10.004] [DOI] [PubMed] [Google Scholar]
Moore 2012b
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Moore 2013a
- Moore RA. What works for whom? Determining the efficacy and harm of treatments for pain. Pain 2013;154(Supplement 1):S77‐S86. [DOI: 10.1016/j.pain.2013.03.024] [DOI] [PubMed] [Google Scholar]
Moore 2013b
- Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ 2013;346:f2690. [DOI: 10.1136/bmj.f2690] [DOI] [PubMed] [Google Scholar]
Moore 2013c
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI: 10.1111/anae.12148] [DOI] [PubMed] [Google Scholar]
Moore 2014b
- Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions ‐ individual patient data responder analysis. European Journal of Pain 2014;18(1):67‐75. [DOI: 10.1002/j.1532-2149.2013.00341.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014c
- Moore RA, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non‐cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79‐94. [DOI: 10.1111/papr.12050] [DOI] [PubMed] [Google Scholar]
Moore 2015a
- Moore RA, Chi CC, Wiffen PJ, Derry S, Rice ASC. Oral nonsteroidal anti‐inflammatory drugs for neuropathic pain. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD010902.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015b
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 7. [DOI: 10.1002/14651858.CD008242.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015c
- Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain 2015;156(8):1382‐95. [DOI: 10.1097/j.pain.0000000000000088] [DOI] [PubMed] [Google Scholar]
Moulin 2014
- Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, et al. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Research & Management 2014;19(6):328‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nguyen 2017
- Nguyen TL, Collins GS, Lamy A, Devereaux PJ, Daurès JP, Landais P, et al. Simple randomization did not protect against bias in smaller trials. Journal of Clinical Epidemiology 2017;84:105‐13. [DOI: 10.1016/j.jclinepi.2017.02.010] [DOI] [PubMed] [Google Scholar]
NICE 2013
- National Institute for Health and Care Excellence (NICE). Neuropathic pain ‐ pharmacological management: the pharmacological management of neuropathic pain in adults in non‐specialist settings, 2013. www.nice.org.uk/guidance/cg173 (accessed 13 February 2017). [PubMed]
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment: outcomes in chronic pain. Pain Medicine 2010;11:6‐15. [DOI: 10.1111/j.1526-4637.2009.00685.x] [DOI] [PubMed] [Google Scholar]
PaPaS 2012
- Cochrane Pain, Palliative and Supportive Care Group. PaPaS Author and Referee Guidance. papas.cochrane.org/papas‐documents (accessed 7 February 2017).
Perloff 2011
- Perloff MD, Thaler DE, Otis JA. Anorgasmia with gabapentin may be common in older patients. American Journal of Geriatric Pharmacotherapy 2011;9(3):199‐203. [DOI: 10.1016/j.amjopharm.2011.04.007] [DOI] [PubMed] [Google Scholar]
Perry 2008
- Perry T. Neurontin ‐ expert opinion on efficacy and effectiveness for pain. industrydocumentslibrary.ucsf.edu/drug/docs/#id=fhhw0217 (accessed 13 February 2017).
Phillips 2010
- Phillips TJ, Cherry CL, Cox S, Marshall SJ, Rice AS. Pharmacological treatment of painful HIV‐associated sensory neuropathy: a systematic review and meta‐analysis of randomised controlled trials. PLOS One 2010;5(12):e14433. [DOI: 10.1371/journal.pone.0014433] [DOI] [PMC free article] [PubMed] [Google Scholar]
Quilici 2009
- Quilici S, Chancellor J, Lothgren M, Simon D, Said G, Le TK, et al. Meta‐analysis of duloxetine vs. pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurology 2009;9:6. [DOI: 10.1186/1471-2377-9-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Quintero 2017
- Quintero GC. Review about gabapentin misuse, interactions, contraindications and side effects. Journal of Experimental Pharmacology 2017;9:13‐21. [10.2147/JEP.S124391. eCollection 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self‐sustaining discharge in the trigeminal ganglion. Pain 1994;56(2):127‐38. [DOI: 10.1016/0304-3959(94)90086-8] [DOI] [PubMed] [Google Scholar]
Rauck 2013c
- Rauck RL, Irving GA, Wallace MS, Vanhove GF, Sweeney M. Once‐daily gastroretentive gabapentin for postherpetic neuralgia: integrated efficacy, time to onset of pain relief and safety analyses of data from two phase 3, multicenter, randomized, double‐blind, placebo‐controlled studies. Journal of Pain and Symptom Management 2013;46(2):219‐28. [DOI: 10.1016/j.jpainsymman.2012.07.011] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roberts 2015
- Roberts I, Ker K, Edwards P, Beecher D, Manno D, Sydenham E. The knowledge system underpinning healthcare is not fit for purpose and must change. BMJ 2015;350:h2463. [DOI: 10.1136/bmj.h2463] [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Scott 2006
- Scott FT, Johnson RW, Leedham‐Green M, Davies E, Edmunds WJ, Breuer J. The burden of herpes zoster: a prospective population based study. Vaccine 2006;24(9):1308‐14. [DOI: 10.1016/j.vaccine.2005.09.026] [DOI] [PubMed] [Google Scholar]
SIGN 2013
- Scottish Intercollegiate Guidelines Network. SIGN Guideline 136: Management of Chronic Pain. sign.ac.uk/guidelines/fulltext/136/contents.html 2013; Vol. (accessed 1 March 2017).
Smalldone 2004
- Smaldone M, Sukkarieh T, Reda A, Khan A. Epilepsy and erectile dysfunction: a review. Seizure 2004;13(7):453‐9. [DOI: 10.1016/j.seizure.2003.12.006] [DOI] [PubMed] [Google Scholar]
Soni 2013
- Soni A, Batra R, Gwilym S, Spector T, Hart D, Arden N, et al. Neuropathic features of joint pain: a community‐based study. Arthritis & Rheumatism 2013;April 1:Epub ahead of print. [DOI: 10.1002/art.37962] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stannard 2016
- Stannard C, Gaskell H, Derry S, Aldington D, Cole P, Cooper TE, et al. Hydromorphone for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD011604.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrollment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75. [DOI: 10.1111/j.1365-2125.2008.03200.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Wiffen PJ, McQuay HJ. Single dose oral gabapentin for established acute postoperative pain in adults. Cochrane Database of Systematic Reviews 2010, Issue 5. [DOI: 10.1002/14651858.CD008183.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-29] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, et al. The number of patients and events required to limit the risk of overestimation of intervention effects in meta‐analysis‐‐a simulation study. PLoS One 2011;6(10):e25491. [DOI: 10.1371/journal.pone.0025491] [DOI] [PMC free article] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7(4):281‐9. [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐5. [DOI: 10.1212/01.wnl.0000282763.29778.59] [DOI] [PubMed] [Google Scholar]
Turner 2013
- Turner RM, Bird SM, Higgins JP. The impact of study size on meta‐analyses: examination of underpowered studies in Cochrane Reviews. PLoS One 2013;8(3):e59202. [DOI: 10.1371/journal.pone.0059202] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tzellos 2008
- Tzellos TG, Papazisis G, Amaniti E, Kouvelas D. Efficacy of pregabalin and gabapentin for neuropathic pain in spinal‐cord injury: an evidence‐based evaluation of the literature. European Journal of Clinical Pharmacology 2008;64(9):851‐8. [DOI: 10.1007/s00228-008-0523-5] [DOI] [PubMed] [Google Scholar]
Tzellos 2010
- Tzellos TG, Toulis KA, Goulis DG, Papazisis G, Zampeli VA, Vakfari A, et al. Gabapentin and pregabalin in the treatment of fibromyalgia: a systematic review and a meta‐analysis. Journal of Clinical Pharmacology and Therapeutics 2010;35(6):369‐56. [DOI: 10.1111/j.1365-2710.2009.01144.x] [DOI] [PubMed] [Google Scholar]
Tölle 2013
- Tölle TR, Backonja M. Pharmacological Therapy for Neuropathic Pain. In: McMahon SB, Koltenburg M, Tracey I, Turk DC editor(s). Wall and Melzack's Textbook of Pain. 5. Philadelphia: Elsevier, 2013:1003‐1011. [ISBN 978‐0‐7020‐4059‐7] [Google Scholar]
Van Hecke 2014
- Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 2014;155(4):654‐62. [DOI: 10.1016/j.pain.2013.11.013] [DOI] [PubMed] [Google Scholar]
Van Hoek 2009
- Hoek AJ, Gay N, Melegaro A, Opstelten W, Edmunds WJ. Estimating the cost‐effectiveness of vaccination against herpes zoster in England and Wales. Vaccine 2009;27(9):1454‐67. [DOI: 10.1016/j.vaccine.2008.12.024] [DOI] [PubMed] [Google Scholar]
Vedula 2009
- Vedula SS, Bero L, Scherer RW, Dickersin K. Outcome reporting in industry‐sponsored trials of gabapentin for off‐label use. New England Journal of Medicine 2009;361(20):1963‐71. [DOI: 10.1056/NEJMsa0906126] [DOI] [PubMed] [Google Scholar]
von Hehn 2012
- Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012;73(4):638‐52. [DOI: 10.1016/j.neuron.2012.02.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI: 10.1016/S0140-6736(12)61729-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2017
- Wang J, Zhu Y. Different doses of gabapentin formulations for postherpetic neuralgia: A systematical review and meta‐analysis of randomized controlled trials. Journal of Dermatology Treatment 2017;28(1):65‐77. [DOI: 10.3109/09546634.2016.1163315] [DOI] [PubMed] [Google Scholar]
Wiffen 2010
- Wiffen PJ, Collins S, McQuay HJ, Carroll D, Jadad A, Moore RA. Anticonvulsant drugs for acute and chronic pain. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD001133.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA, Aldington D, Cole P, Rice ASC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia ‐ an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD010567.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2015
- Wiffen PJ, Derry S, Moore RA, Stannard C, Aldington D, Cole P, et al. Buprenorphine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011603.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2016
- Wiffen PJ, Knaggs R, Derry S, Cole P, Phillips T, Moore RA. Paracetamol (acetaminophen) with or without codeine or dihydrocodeine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012227.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Moore 2011a
- Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2011, Issue 3. [DOI: 10.1002/14651858.CD007938.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014a
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice ASC. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD007938.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2000
- Wiffen P, Collins S, McQuay H, Carroll D, Jadad A, Moore A. Anticonvulsant drugs for acute and chronic pain. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD001133.pub2] [DOI] [PubMed] [Google Scholar]
Wiffen 2005
- Wiffen PJ, McQuay HJ, Edwards JE, Moore RA. Gabapentin for acute and chronic pain. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005452] [DOI] [PubMed] [Google Scholar]