

Abstract
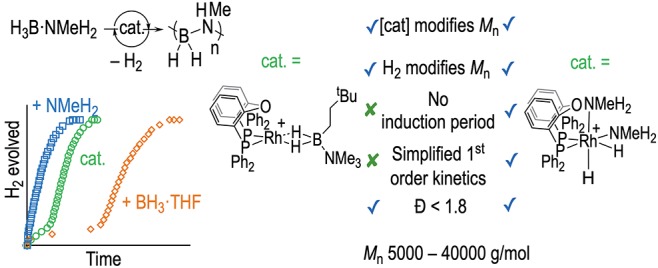
[Rh(κ2-PP-DPEphos){η2η2-H2B(NMe3)(CH2)2tBu}][BArF4] acts as an effective precatalyst for the dehydropolymerization of H3B·NMeH2 to form N-methylpolyaminoborane (H2BNMeH)n. Control of polymer molecular weight is achieved by variation of precatalyst loading (0.1–1 mol %, an inverse relationship) and use of the chain-modifying agent H2: with Mn ranging between 5 500 and 34 900 g/mol and Đ between 1.5 and 1.8. H2 evolution studies (1,2-F2C6H4 solvent) reveal an induction period that gets longer with higher precatalyst loading and complex kinetics with a noninteger order in [Rh]TOTAL. Speciation studies at 10 mol % indicate the initial formation of the amino–borane bridged dimer, [Rh2(κ2-PP-DPEphos)2(μ-H)(μ-H2BN=HMe)][BArF4], followed by the crystallographically characterized amidodiboryl complex [Rh2(cis-κ2-PP-DPEphos)2(σ,μ-(H2B)2NHMe)][BArF4]. Adding ∼2 equiv of NMeH2 in tetrahydrofuran (THF) solution to the precatalyst removes this induction period, pseudo-first-order kinetics are observed, a half-order relationship to [Rh]TOTAL is revealed with regard to dehydrogenation, and polymer molecular weights are increased (e.g., Mn = 40 000 g/mol). Speciation studies suggest that NMeH2 acts to form the precatalysts [Rh(κ2-DPEphos)(NMeH2)2][BArF4] and [Rh(κ2-DPEphos)(H)2(NMeH2)2][BArF4], which were independently synthesized and shown to follow very similar dehydrogenation kinetics, and produce polymers of molecular weight comparable with [Rh(κ2-PP-DPEphos){η2-H2B(NMe3)(CH2)2tBu}][BArF4], which has been doped with amine. This promoting effect of added amine in situ is shown to be general in other cationic Rh-based systems, and possible mechanistic scenarios are discussed.
Keywords: dehydropolymerization, rhodium, amine−borane, mechanism, DPEphos
1. Introduction
Polyaminoboranes,1−4 exemplified by N-methylpolyaminoborane (H2BNMeH)n, have alternating main-chain B–N units and are of interest as precursors to BN-based ceramics or as new unexplored materials that are isosteres of polyolefins. Since the original report of the synthesis of (H2BNMeH)n by the dehydropolymerization of H3B·NMeH2 using an Ir(POCOP)H2 catalyst (POCOP = κ3-C6H3-2,6-(OPtBu2)2),4−6 there has been significant progress in developing catalytic methodologies,7−13 as well as noncatalyzed routes.14 The accepted overarching catalytic mechanism operates via initial dehydrogenation of H3B·NMeH2 to form a transient free, or metal-bound amino–borane, which then undergoes a head-to-tail BN coupling (Scheme 1). A number of different propagation scenarios have been proposed for this latter step that show elements of chain-growth,4,10 step-growth,15 or hybrid mechanisms.16 Particularly interesting would be systems that demonstrate the potential for control17 over the polymerization process, holistically defined by degree of polymerization (as measured by Mn), dispersity (Đ), initiation/termination events, and catalyst lifetime (i.e., TON). While aspects of these performance criteria have been noted,7−10,15 there is no general approach to their optimization.
Scheme 1. Dehydropolymerization of Amine–Boranes.

We have reported cationic dehydropolymerization precatalysts based upon {Rh(Xantphos-R)}+ motifs,18,19 in which the identity of the PR2 group is changed (Scheme 2).9,10,20 When R = Ph (A), medium2 molecular weight polymer is formed (Mn = 22 700 g/mol, Đ = 2.1), a higher catalyst loading promotes lower Mn, and H2 acts to modify the polymer chain length (Mn = 2 800 g/mol, Đ = 1.8). Although detailed kinetics for H3B·NMeH2 dehydropolymerization were not reported, these observations were interpreted as signaling a coordination/insertion/chain-growth mechanism in concert with more extensive studies on H3B·NMe2H.9 There is also a significant induction period observed (∼10 min). In contrast, when R = iPr (B), H2 and catalyst loading do not significantly change Mn (9 500 g/mol, Đ ≈ 2.8), there is a negligible induction period, and a dual role11,12 for the organometallic species was proposed in which dehydrogenation/propagation occurs from different metal centers. This mechanistic switch may be influenced by the preferred ligand-coordination modes:21 Xantphos-Ph is a hemilabile ligand preferring to coordinate cis-κ2-PP and mer-κ3-POP, while Xantphos-iPr prefers mer-κ3-POP (Figure S1 compares coordination modes for crystallographically characterized Xantphos-R complexes).
Scheme 2. Comparison of Previously Reported Rh–Xantphos-Based Catalysts and Their Performance in Dehydropolymerization of H3B·NMeH2; [BArF4]− Anions Not Shown.

We now report a detailed and systematic study on the dehydropolymerization of H3B·NMeH2 using a different Rh-POP-based system: {Rh(DPEphos)}+ [DPEphos = bis(2-(diphenylphosphino)phenyl)ether]. Using this ligand, which favors cis-κ2-PP coordination (Figure S1), significant control over Mn by both catalyst loading and H2 is achieved, with Mn ranging from 5 500 to 40 000 g/mol and Đ = 1.5–1.8. These studies also reveal the formation of dimeric species, and the key role of added amine, NMeH2, in both promoting catalysis and increasing Mn/lowering Đ of the isolated polymer. Finally, combining these observations, the synthesis and evaluation in catalysis of a simple [Rh(κ2-PP-DPEphos)(NMeH2)2]+ precatalyst is reported. This positive influence of added amine is also shown to be general for other previously reported cationic Rh-based systems. The role of added amine has been recently noted with regard to increasing catalyst lifetime of Ru-based catalysts for the dehydropolymerization of H3B·NH3 by trapping BH3 formed from B–N bond cleavage,8 although the influence of amine on the characteristics of the polymer produced were not commented upon.
2. Results and Discussion
2.1. Precatalyst Synthesis
Precatalyst 2a, [Rh(κ2-P,P-DPEphos){η2η2-H2B(NMe3)(CH2)2tBu}][BArF4] (ArF = 3,5-(CF3)2C6H3), is synthesized from hydroboration of tbutylethene (TBE) by H3B·NMe3 using the NBD precursor 1a (NBD = norbornadiene), preactivated by H2 (Scheme 3). Spectroscopic data for purple 2a are similar to the previously reported Xantphos-Ph derivative, A.22 In particular, a single environment is observed in the 31P{1H} NMR spectrum [δ 40.0 ppm, J(RhP) = 180 Hz], the 3-center, 2-electron Rh···H–B groups are observed at δ −5.55 ppm (2 H) in the 1H NMR spectrum, while the 11B NMR spectrum shows a characteristically23 downfield-shifted resonance [δ 33.3 ppm], indicating a bidentate binding mode of the borane. The amine–borane in 2a is easily displaced, and the [Rh(Xantphos-Ph)]+ analogue (A) has been shown to be active for H3B·NMeH2 dehydropolymerization,9 TBE hydroboration using H3B·NMe3,24 and B–B homocoupling.22
Scheme 3. Synthesis of the {Rh(DPEphos)}+ Precatalyst 2a.

2.2. Dehydropolymerization of H3B·NMeH2: Variation of Conditions
Precatalyst 2a is an effective for dehydropolymerization, and full conversions of H3B·NMeH2 are obtained even at low loadings under a slow stream of Ar to remove H2 (e.g., 0.223 M H3B·NMeH2, [2a] = 0.1 mol %, TON = 1000, 6 h). Variation of precatalyst loadings between 0.2 and 1 mol % reveals an inverse relationship between Mn of the isolated polymer and catalyst loading (Table 1, entries 1–3, and Figure 1A). The resulting 11B NMR spectra of the reaction mixtures and isolated polymer show the characteristic2,12 broad signal at δ −6 ppm for (H2BNMeH)n and only trace (HBNMe)3 (Figure S18). The 13C{1H} NMR spectra (H8-THF) show a relatively sharp peak at δ 35.5 ppm (NMe). In contrast, at 0.1 mol % catalyst loading, Mn does not increase compared to 0.2 mol %, and there is significant 1,2-F2C6H4 insoluble polymer that is tetrahydrofuran (THF)-soluble. NMR spectroscopic analysis of this material (Figure S19) showed additional signals at δ(11B) ∼1 ppm and δ(13C{1H}) ∼35.7 ppm (br, NMe) that may signal tertiary or quaternary main-chain centers, suggesting cross-linking/chain branching.10,11,19,25 While we currently have no explanation for this change in polymer characteristics, at these very low loadings trace impurities (or products of B–N bond cleavage, vide infra) may have a disproportionate effect on the polymerization process, leading to a different product being formed. When dehydropolymerization was conducted under H2 measurement conditions (eudiometer, H2 established in the head space), or in a closed system that allows for H2 buildup, H2 likely acts as a chain-transfer/termination agent and significantly shorter polymer is isolated, for which a significantly larger signal at δ(11B) ∼−18 ppm is observed, which could be assigned to BH3 end groups15 (Figure 1B; Figure S20 shows a representative 11B NMR spectrum). Similar Đ are retained compared with the open system, as is the inverse relationship between Mn and catalyst loading (Table 1, entries 5–8). Interestingly, there is now a significant difference in Mn between 0.1 and 0.2 mol %, suggesting that H2 modifies the influence of the very low catalyst loading. A conversion versus Mn study (0.2 mol %, open system, Figure 1C) indicates that a chain-growth mechanism is operating, because at low (10%) conversions long polymer chains are observed (Mn = 24 800 g/mol, Đ = 1.2) and H3B·NMeH2 monomer dominates (Figure S21).
Table 1. GPC Characterization Data for Isolated Polyaminoboranea.
| entry | catalyst | [Rh]TOT, mol % | conditions | Mn, g/mol | Đ |
|---|---|---|---|---|---|
| 1 | 2a | 1 | open (Ar flow) | 6400 | 1.8 |
| 2 | 2a | 0.4 | open (Ar flow) | 29500 | 1.8 |
| 3 | 2a | 0.2 | open (Ar flow) | 34900 | 1.5 |
| 4 | 2a | 0.1 | open (Ar flow) | 34600 | 1.7 |
| 5 | 2a | 0.1 | H2 measurement | 29400 | 1.6 |
| 6 | 2a | 0.2 | H2 measurement | 14500 | 1.7 |
| 7 | 2a | 0.4 | H2 measurement | 10100 | 1.8 |
| 8 | 2a | 1 | H2 measurement | 5500 | 1.8 |
| 9 | 3a | 0.4 | H2 measurement | 14800 | 1.6 |
| 10 | 4b | 0.4 | H2 measurement | 15900 | 1.8 |
| 11 | 2a | 0.4 | H2 measurement/1 equiv of H3B·THF/50 μL of THF | 6600 | 1.9 |
| 12 | 2a | 0.4 | H2 measurement/10 equiv of [H2B(NMeH2)2][BArF4] | 2800 | 2.3 |
| 13 | 2a | 0.4 | H2 measurement/50 μL of THF | 11000 | 1.6 |
| 14 | 2a | 0.4 | H2 measurement/∼2 equiv of NMeH2 in 50 μL of THF | 27400 | 1.6 |
| 15 | 2a | 0.4 | open/∼2 equiv of NMeH2 in 50 μL of THF | 32100 | 1.6 |
| 16 | 5/6 | 0.2 | H2 measurement | 38900 | 1.6 |
| 17 | 5/6 | 0.4 | H2 measurement | 33200 | 1.6 |
| 18 | 5/6 | 1 | H2 measurement | 20600 | 1.5 |
| 19 | 6 | 0.8 | H2 measurement | 22500 | 1.5 |
| 20 | 2a | 0.2 | H2 measurement/∼2 equiv of NMeH2 in 50 μL of THF | 34800 | 1.5 |
| 21 | A9 | 0.2 | H2 measurement | 40500 | 1.7 |
| 22 | A9 | 0.2 | H2 measurement/∼2 equiv of NMeH2 in 50 μL of THF | 61900 | 1.6 |
| 23 | C16 | 0.2 | H2 measurement | 63100 | 1.7 |
| 24 | C16 | 0.2 | H2 measurement/∼2 equiv of NMeH2 in 50 μL of THF | 78900 | 1.6 |
All at 298 K, 0.223 M H3B·NMeH2, 1,2-F2C6H4 solvent. GPC data quoted relative to polystyrene standards (calibrated between 500 and 480 000 g/mol), triple column, RI detection, THF with 0.1 w/w% [NBu4]Br, 35 °C, sample concentration = 2 mg/cm3. Open conditions: periodic sampling by 11B NMR spectroscopy determined end point (e.g., 6 h for entry 4). Under H2 measurement conditions, the reaction was stopped when there was no significant change in H2 evolved.
Figure 1.

GPC data (relative to polystyrene standards, RI detection, THF with 0.1 w/w% [NBu4]Br, 35 °C) for (H2BNMeH)n isolated from H3B·NMeH2 dehydropolymerization (0.223 M, 1,2-F2C6H4, 20 °C) using catalyst 2a. (A) Variation of [2a] under Ar purge; (B) variation of [2a] under H2 measurement conditions (eudiometer); (C) conversion versus Mn/Đ plot, open conditions, where each individual data point is a PPh3-quenched experiment after an appropriate time.
We have previously, but briefly, reported similar control of molecular weight by catalyst loading and H2 for catalyst A and suggested a coordination/dehydrogenation/insertion/chain-growth mechanism for the dehydropolymerization, in which the same metal center both dehydrogenates an amine–borane and promotes propagation.9 This more comprehensive data with 2a supports a similar mechanism in the {Rh(DPEphos)}+ system. That H2 acts to modify the polymer chain may arise from chain-termination/transfer by hydrogenolysis of a Rh–BH2(polymeryl) or Rh–NMeH(polymeryl) bond. The use of H2 as a chain-termination agent in olefin polymerization is well-established, operating through sigma-bond metathesis of [M]-CH2-polymeryl with H2 to form a metal hydride and free polymer.26 The inverse relationship between Mn and catalyst loading suggests dehydropolymerization at a single metal center, as lower catalyst loadings lead to less propagating sites for the concomitantly formed H2B=NMeH. Interestingly, this relationship between Mn and initiating sites is also reminiscent of a classical radical polymerization mechanism where the net order in initiator is negative,27 as has been recently noted.3
2.3. Speciation Experiments: The Formation of Dimeric Rh2 Species
With the polymer growth kinetics in hand, we turned to identifying the species that formed during catalysis using NMR spectroscopy. The low catalyst loadings used for polymerization (0.1–1 mol %) meant that these speciation studies were performed instead at 10 mol % 2a to obtain good signal/noise (sealed NMR tube, 1,2-F2C6H4). Under these in situ conditions, 11B NMR spectroscopy showed the formation of a mixture of (H2BNMeH)n, (HBNMe)3, and (H2B)2(μ-H)(NMeH) [td, δ −22.3 ppm28], with the latter potentially signaling free BH3 by loss of amine. 31P{1H} NMR spectroscopy under these conditions showed the initial formation, after 5 min, of two new dimeric complexes: a bridging hydrido-aminoborane 3a, [Rh2(DPEphos)2(μ-H)(μ-H2B=NHMe)][BArF4], and an amidodiboryl 4a, [Rh2(κ2-P,P-DPEphos)2(σ,μ-(H2B)2NHMe)][BArF4] (Figure 2A). After 2 h 4a is dominant (80%), but the mixture slowly returns to favoring 3a after 5 h (Figure S22). Complex 3a can be prepared as the only organometallic species by addition of H2/2 equiv of H3B·NMeH2 to 1a. Boronium [BH2(NMeH2)2]+ [δ −7.1 ppm, J(BH) = 110 Hz, cf. authentic sample δ −7.4 ppm, J(BH) = 117 Hz, 1,2-F2C6H410] is also observed under these conditions,29 in line with the reported mechanism for the formation of analogous complexes with [Rh2(R2P(CH2)nPR2)2(μ-H)(μ-H2B=NR′2)]+ motifs.30,31 Here, attack of free amine (from B–N bond cleavage32) at a precursor σ-amine–borane complex generates a neutral dimeric Rh–hydride and [BH2(NMeH2)2]+, for which subsequent proton transfer and NMeH2 loss result in the bridging amino–borane motif. NMR and ESI–MS data for 3a are fully consistent with its formulation (Supporting Materials) and are very closely related to previously reported [Rh2(iPr2P(CH2)3PiPr2)2(μ-H)(μ-H2B=NH2)][BArF4].30 Attempts to characterize these products using single-crystal X-ray diffraction were frustrated by the formation of oily materials. The identity of 4 was only revealed using the [Al(OC(CF3)3)4]− anion,33 by a single-crystal study of 4b, [Rh2(κ2-P,P-DPEphos)2(σ,μ-(H2B)2NHMe)][Al(OC(CF3)3)4], which comes from a slow (days) recrystallization of 3b, formed in situ from [Rh(κ2-P,P-DPEphos)(NBD)][Al(OC(CF3)3)4] 1b /H3B·NMeH2 activated with H2 (Figure 2B). 4b is not isolated pure, formed alongside 3b (∼5% by 31P{1H} NMR spectroscopy) and (H2BNMeH)n. The NMR data for 4b, aside from the signals due to the anion, are the same as for 4a, as are the ESI–MS data.
Figure 2.
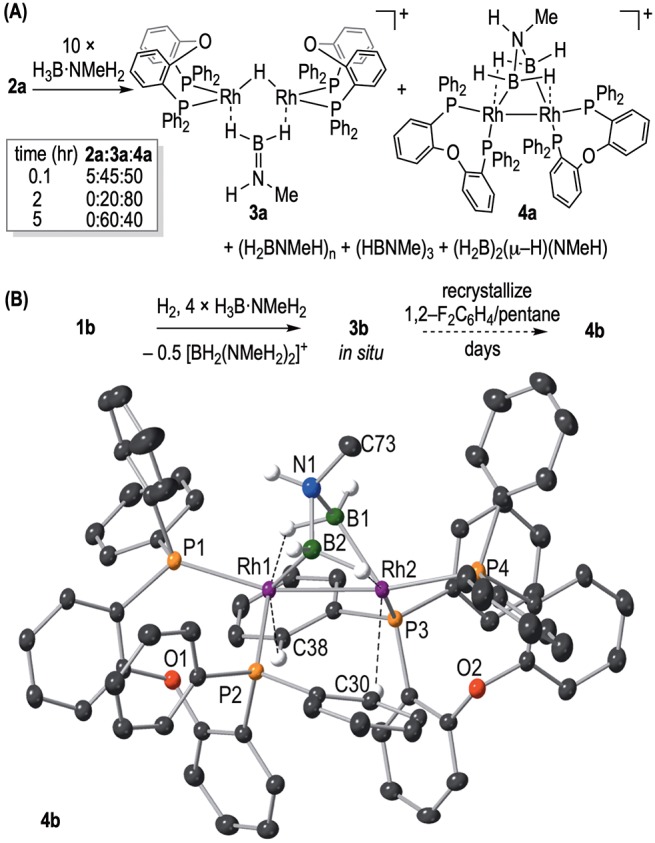
(A) Addition of H3B·NMeH2 to 2a (10 mol %) to form 3a and 4a, 1,2-F2C6H4 solvent. (B) Synthesis and solid-state structure of the cationic portion of 4. Selected bond lengths (Å) and angles (deg): Rh1–Rh2 2.6421(4); Rh1–B1 2.326(5), Rh1–B2 2.096(6); Rh2–B1 2.107(5), Rh1–B2, 2.328(5); Rh1–C38 2.997(5), B1–N1, 1.59(1), B2–N1 1.56(1); P1–Rh1–Rh2 162.59(3), P2–Rh1–Rh2, 95.31(3).
The structure of the cation in 4b has a Rh2 core [Rh–Rh 2.6421(4) Å] with a bridging amido–bisboryl ligand that has two α-BH···Rh agostic interactions with the proximal Rh centers [e.g., Rh2–B1 2.107(5), Rh1···B1 2.326(5) Å]. Such a description results in formally Rh(II) centers with a Rh–Rh bond accounting for the diamagnetism. An alternative description of the bonding in 4b is a diborylmethylammonium complex that would result in the Rh centers being formally Rh(0). The DPEphos ligand adopts a κ2-PP motif, with two of the phosphines (P2, P3) trans to the BH agostic interaction and cis to the Rh–Rh bond, while P1 and P4 lie trans to the Rh–Rh bond and couple to both Rh centers in the 31P{1H} NMR spectrum [e.g., J(RhP) = 139, 102 Hz]. The four 31P environments are chemically inequivalent. There is no evidence for a Rh–H–Rh bridging hydride (NMR, ESI–MS), and the α-BH···Rh are observed as two broad doublets at δ −8.86 and −9.44 ppm [J(PH) ≈ 70 Hz] in the 1H{11B} NMR spectrum.34 The 11B NMR spectrum shows a broad signal at δ 9.4 ppm. These data show that the solid-state structure is retained in solution. As the NMeH group forces C1 symmetry in the molecule, this also shows that the amido–bisboryl ligand is not undergoing rapid and reversible dissociation or hydride fluxionality. A Quantum Theory of Atoms in Molecules (QTAIM) study of the bonding in the cation of 4b (Figure 3) indicates a Rh–Rh interaction, with the presence of a bond path and bond critical point (BCP) between Rh1 and Rh2. BCPs are also present between Rh1–HAB1 and Rh2–HCB2, giving evidence for the α-BH···Rh agostic interactions. This is supported, for example, through examination of the BCP metrics of bridging B1–HA/B2–HC, which show a weaker (lower electron density, ρ(r), and total energy density, H(r)) B–H bond with less symmetrical bonding (larger ellipticity, ε) than for terminal B1–HB/B2–HD, as expected for B–H bonds involved in agostic interactions. Comparatively weak CH···Rh agostic interactions (ρ(r) = 0.02, H(r) = 0.00) between phenyl groups and each Rh center are also observed in the QTAIM analysis and also observed experimentally, e.g., Rh1···C38, 2.997(5) Å. Consistent with such interactions, a broad asymmetric signal is observed at δ 3.94 ppm (2 H) in the 1H NMR spectrum of 4b that is attributed to agostic Rh···HCphenyl interactions, similar to that observed in [Ru(PiPr3)2(H)(H2)(C6H5C5H4N)][BArF4] (δ 4.14 ppm).354b is a rare example of a complex with both C–H and B–H agostic interactions.36,37
Figure 3.
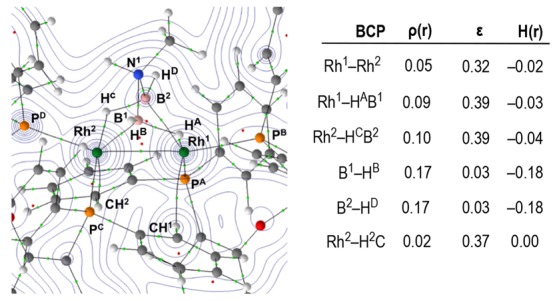
Contour plot of the electron density of the central cationic portion of 4b presented in the {Rh1N1Rh2} plane with projected stationary points, bond paths, bond critical points (BCPs; green), and ring critical points (RCPs; red). The associated table shows selected BCP metrics (a.u.; average data for indicated bonds).
Related structures to 4b that show bridging “BNB”,20,38 α-BH···Rh agostic,39 or amino–boryl motifs9,40 have been reported before. However, as far as we are aware, the amido–bisboryl structure is a new motif in metalloborane chemistry. Perhaps most closely related to 4b is a Rh-dimer with P–C activated Xantphos-Ph ligands and a bridging N,N-dimethylaminodiboranate unit ([H3BNMe2BH3]−) that is isolated at the end of dehydrocoupling of H3B·NMe2H when using catalyst A. Interestingly, this is also a competent catalyst for H3B·NMeH2 dehydropolymerization.20 While we currently can only speculate on the mechanism of formation of 4, it is connected to 3 by simple addition of BH3 and loss of H2. Under catalytic conditions 3 likely forms first, while the role of 4 is less clear. To help resolve the identity of the active species in catalysis, kinetic studies were undertaken, taking 2a, 3a, and 4b as precatalysts.
2.4. Kinetic Studies of Dehydropolymerization As Followed by H2 Evolution
The kinetics of dehydropolymerization were followed by volumetric studies of H2 generation using a eudiometer. In all cases ∼1.1 equiv of H2 was measured and very little N-trimethylborazine was observed by 11B NMR spectroscopy (<5%, Figure S23), indicating that evolved H2 is a good proxy for transient41 H2B=NMeH equivalents formed and subsequent polymer chain growth. A significant induction period was observed prior to faster turnover (e.g., ∼60 min, 0.4 mol %), that gets longer with increase in [2a]0 (Figures 4A and S24; e.g., 0.1 mol %, tind = 33 min; 1 mol %, tind = 110 min). An induction period has also been noted for catalyst A in H3B·NMeH2 dehydropolymerization9 as well as for [Rh(Ph2P(CH2)3PPh2)(FC6H5)][BArF4], C, in H3B·NMe2H dehydrocoupling (10 and 5 min, respectively, at 0.2 mol %).42 For this latter system, increased [Rh]TOTAL also led to longer induction periods, and a subsequent study showed the initial formation of an amino–borane-bridged dimer analogous to 3a.30 While the observation of an induction period might suggest a heterogeneous system here,43−45 addition of excess Hg or substoichiometric PPh3 during productive turnover did not significantly reduce reaction rate, and no darkening of the reaction was noted, pointing toward homogeneous catalysis (Figure S25). Overall, the kinetics evolve in a sinusoidal manner, with a rate maximum reached approximately at the midpoint (e.g., 0.4 mol %, νmax = 4.1(2) × 10–5 M s–1). This behavior is suggestive of a long induction period coupled to rate-attenuation as the substrate is depleted. There is a noninteger dependence of the maximum rate on the initial catalyst concentration (Figure S28), which hints at more complex kinetics. Using 0.223 M D3B·NMeH2 or H3B·NMeD2 at 0.4 mol % 2a, kinetic isotope effects (KIEs) determined from νmax were k(BH)/k(BD) = 1.1 ± 0.1 and k(NH)/k(ND) = 2.2 ± 0.1, which suggests that N–H bond cleavage is involved in the turnover-limiting step. These data are very similar to those measured for A.9 The polymerization is not living as recharging 2a gives approximately the same Mn, at a similar rate for second recharge (Figure S31). A short induction period was noted for each recharge, which reflects the reformation of 3a at the end of catalysis (vide infra).
Figure 4.

H2B=NMeH equivalents from H2 evolution (eudiometer) in the dehydropolymerization of H3B·NMeH2 (0.223 M 1,2-F2C6H4, 20 °C). Each set of comparative runs used the same batch of solvent and H3B·NMeH2. (A) [2a] = 0.4 and 1 mol % Rh and 0.4 mol % + 1 equiv of H3B·THF; (B) 2a, 3a, and 4b at 0.4 mol % [Rh]TOTAL, kobs measured for [4b]. (C) [Rh]TOTAL versus kobs using 4b as a catalyst.
Use of in situ generated dimeric 3a leads to a shorter, but still significant, induction period (∼30 min, Figure 4B) and a similar profile and rate maximum as for 2a. In contrast, reaction of crude 4b resulted in no detectable induction period. Furthermore, H2 evolution (a proxy for H2B=NMeH formation) followed a first-order profile (Figure 4B, kobs = 3.2(1) × 10–4 s–1), and this allowed for a half-order dependency on initial catalyst concentration, i.e., [Rh]TOTAL, to be estimated (Figures 4C and S30).
The polymers isolated from these H2 evolution studies using 3a and 4b are similar by GPC analysis but slightly longer compared to that from 2a at equivalent [Rh]TOTAL (Table 1, entries 7, 9, and 10). Speciation studies at 1 mol % 2a return only 3a at the end, which suggests that, if formed, 4a must be consumed under the conditions of catalysis. Overall these data show the following: a change in H2-evolution kinetics on moving from 2a (complex) to 4 (pseudo first-order), that 4 likely sits close to the actual catalyst, and that 3 still requires an induction process to bring it on-cycle. The approximately half-order dependence in [Rh]TOTAL when using 4a as a precatalyst suggests a lower-order (ligation or nuclearity) active catalyst that is in a rapid equilibrium with a higher-order inactive species, as is discussed later.
2.5. Kinetic Studies: Doping Experiments and the Promoting Effect of NMeH2
Seeking to understand the observed kinetics, and in particular the underlying reason for the induction period, the influence of various species that may be present, or formed, during catalysis was examined. Addition of 1 equiv of H3B·THF (in 50 μL of THF) to 0.4 mol % [2a]/H3B·NMeH2/1,2-F2C6H4 solvent increased the induction period significantly (Figure 4A) and gave significantly shorter polymer (Table 1, entry 11), while 10 equiv halts catalysis, possibly by the formation of inactive boron-rich species (see Supporting Information).32 Added [H2B(NMeH2)2][BArF4] (10 equiv) significantly slows catalysis, now taking 24 h for completion to produce very short polymer (Mn = 2 800 g/mol, Đ = 2.3). This argues against its role in productive catalysis, in contrast with other systems,10,29,46 in particular the [Rh(Xantphos-iPr)]+ system, where it promotes catalysis.10 At low relative concentrations, H3B·THF presumably acts to titrate out NMeH2, while we propose that excess [H2B(NMeH2)2]+ acts to poison catalysis, possibly sequestering NMeH2 via N–H···NMeH2 hydrogen bonding, as noted for related bis(phosphine)boronium salts.47 The control experiment of THF addition (50 μL) reduced the induction period to 30 min and produced polymer comparable to nondoped experiments (Table 1, entry 13). The most dramatic change came from addition of ∼2 equiv of NMeH2 (in 50 μL of THF) to 0.4 mol % [2a]/H3B·NMeH2. This resulted in a kinetic profile for H2 evolution that now showed no induction period and pseudofirst-order kinetics for hydrogen evolution (kobs = 3.7(1) × 10–4 s–1), similar to that of 4b at the same [Rh]TOTAL. Isolated polymer, however, was considerably longer (Mn = 27 400 g/mol, Đ = 1.9) than for when just 2a was used. As expected, under open conditions Mn increases (Mn = 32 100 g/mol, Đ = 1.6), albeit to a lesser extent than compared with the analogous nondoped experiments (cf. entries 14/15 and 2/7, Table 1). These observations, alongside the speciation data at 10 mol %, which demonstrate that 3a is likely the first formed species, show that free NMeH2 formed from B–N bond cleavage is key to not only bringing the catalyst on-cycle but also promoting propagation or attenuating chain-transfer/termination, leading to higher molecular weights of isolated polymer. Given these observations, the role of NMeH2 was next investigated.
2.6. Rh–Amine Adducts As Effective Precatalysts
We first sought to understand the likely species generated in situ by addition of amine to the precatalyst, 2a. Addition of ∼2 equiv of NMeH2 (in THF) to 2a gave the simple bisamine complex [Rh(κ2-P,P-DPEphos)(NMeH2)2][BArF4], 6, which reacts rapidly (on time of mixing) with H2 in situ to form the corresponding dihydride [Rh(κ2-P,P-DPEphos)(H)2(NMeH2)2][BArF4], 5 (Scheme 4). Complex 5 reversibly, but slowly, loses H2 under extended degassing to reform complex 6, and thus we suggest that, under the conditions of dehydropolymerization, 5 would be persistent. NMR spectroscopic data are fully consistent with the proposed structures (see later), but under these conditions of synthesis isolating pure samples of 5 and 6 in bulk has proved difficult; and a 1:1 mixture of 5/6 is conveniently prepared from 1a/∼2 × NMeH2/H2/degas and used directly in catalysis (see Supporting Information). Complex 5 is the sole organometallic product on addition of ∼2 equiv of NMeH2 to a 1:3 mixture of 3a/4a, alongside HB(NMeH)2 [δ(11B) 28.6 ppm, J(BH) = 127 Hz], demonstrating the role of NMeH2 in both generating 3, via boronium formation,29,30 and bringing dimeric 3 and 4 back to monometallic species. Complex 6 (and 5 on subsequent addition of H2 in solution) can be prepared as a free-flowing pure solid in bulk via an alternative route, from addition of NMeH2 to [Rh(κ2-P,P-DPEphos)(η6-o-Me2C6H4)][BArF4], 7,48 which enables definitive characterization by NMR spectroscopy. However, this involves laborious multiple triturations with cold pentane, and thus, the in situ prepared mixture is more convenient to use. Notable NMR spectroscopic data for 6 are the observation of equivalent NMeH2 groups in the 1H NMR spectrum, while for 5 addition of H2 makes these groups inequivalent and diastereotopic; two Rh–H environments are observed, one of which shows a large trans coupling to 31P [J(HP) = 182 Hz], and inequivalent phosphorus environments are observed in the 31P{1H} NMR spectrum (Supporting Information). Data from H2-evolution kinetics and isolated polymer using isolated 6 fit well with the trends apparent from using the 5/6 in situ mixture (Table 1 and Figure 5).
Scheme 4. Synthesis of Amine Adducts; [BArF4]− Anions Not Shown and DPEphos Ligand Shown in Truncated Form.

Figure 5.
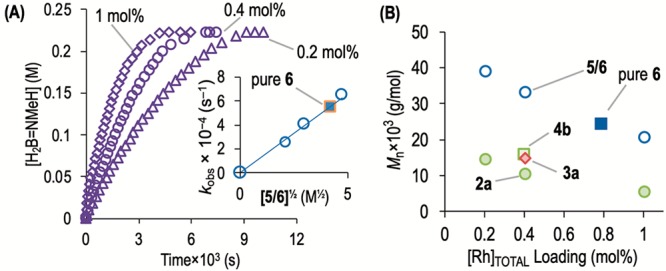
(A) H2B=NMeH equivalents from H2 evolution (eudiometer) in the dehydropolymerization of H3B·NMeH2 (0.223 M 1,2-F2C6H4, 20 °C). Each set of comparative runs used the same batch of solvent and H3B·NMeH2. 5/6 (∼50:50) at various loadings + 0.05 μL of THF, inset = kobs versus [5/6]0.5. (B) Comparison of Mn and Đ versus [5/6], pure 6, 2a, 3a, and 4b (under H2-evolution measurement conditions).
Using in situ generated 5/6 gave pseudo first-order plots for H2 evolution (e.g., 0.4 mol %, kobs = 4.1(1) × 10–4 s–1) with no induction period observed. These were also half-order in [Rh]TOTAL (Figure 5A). Half-order behavior is indicative of either a rapid equilibrium between species of different nuclearity, e.g., monomer–dimer, prior to the turnover-limiting step, in which the higher nuclearity species is inactive but dominant,49 or the rapid and reversible dissociation of a ligand that reveals a low concentration of an active species.50 Monomer/dimer equilibria have been proposed in polymerization systems previously,51−53 and in amine–borane dehydrocoupling specifically.49,54,55 While addition of 10 equiv of NMeH2 caused no significant change in rate (kobs = 4.2(1) × 10–4 s–1), suggesting that NMeH2 dissociation is not occurring, the polymer isolated from this experiment was insoluble in THF. We thus cannot rule out a change in mechanism. We discount rapid and reversible H2 loss as the reason for the observed half-order kinetics because under conditions of measurement H2 effectively becomes saturated and constant. Speciation studies with excess NMeH2 (10 equiv, [Rh]TOTAL = 5 mol %) revealed 5 to be the only observed organometallic species. No significant change in kinetics was observed on addition of excess Hg, or 0.2 equiv of PPh3, during catalysis—suggesting a homogeneous system.56 The use of these in situ prepared amine complexes 5/6 leads to polymer with greater Mn (but still inverse with regard to [Rh]TOTAL), while Đ is kept relatively low (Figure 5B, e.g., 1 mol %, Mn = 20 600 g/mol, Đ = 1.5). Thus, the added amine—whether bound or free—not only brings the catalyst onto cycle but also promotes greater apparent degrees of polymerization. Whether this is by faster propagation or attenuation of termination is not currently known.
Following catalysis by 31P{1H} NMR spectroscopy using pure 5 (1 mol %) showed that during productive catalysis a single organometallic species is observed (albeit with low signal-to-noise) as a doublet at δ 41 ppm [J(RhP) = 150 Hz], which slowly resolves to complex 3 at the end of catalysis. Importantly, the same species is observed when starting with precatalyst 4b (0.5 mol %, 1 mol % [Rh]TOTAL). This strongly suggests that both precatalysts evolve to a common species—the identity of which remains to be resolved.
Interestingly, the promoting effect of NMeH2 is not operative in the [Rh(Xantphos-iPr)(H)2]+ system,10 which is suggested to involve a different mechanism, where dehydrogenation and chain propagation occur at different metal centers in a bifunctional catalyst. Thus, independently prepared [Rh(mer-κ3-POP-Xantphos-iPr)(H)2(NMeH2)][BArF4], 8 (see Supporting Information), does not dehydropolymerize H3B·NMeH2, returning unchanged substrate after 1 h (0.2 mol %, 0.111 M H3B·NMeH2). This is probably due to the relatively strongly bound amine blocking access of H3B·NMeH2 to the metal center, at which the Xantphos-iPr is also not hemilabile (Figure S1), so that σ-complex formation by coordination of amine–borane, and subsequent dehydrogenation by BH/NH activation, does not take place. The broader promoting effects of NMeH2 are, however, evident in other cationic {Rh(chelating phosphine)}+ systems that are suggested to undergo a coordination/dehydrogenation/chain-growth mechanism. Under the specific conditions reported here, both [Rh(Xantphos-Ph)]+, A,9 and [Rh(Ph2P(CH2)3PPh2)]+, C,16,42 systems show increased Mn, slightly lower Đ, and no induction periods when ∼2 equiv of NMeH2 is added to the precatalyst, compared to the nondoped controls (Table 2).
Table 2. Effect of Added Amine in Selected Cationic Rh Catalysts, Mn (g/mol) and Đ; 0.223 M, 0.2 mol % Catalyst, H2 Measurement Conditions, 1,2-F2C6H4; [BArF4]− Anions Not Shown.
| catalyst | no added amine | ∼2 equiv of NMeH2 |
|---|---|---|
| [Rh(DPEphos)(H2B(NMe3)(CH2)2tBu)]+2a | 14500 (1.7) | 34800 (1.5) |
| [Rh(Xantphos-Ph)(H2B(NMe3)(CH2)2tBu)]+ A | 40500 (1.7) | 60900 (1.6) |
| [Rh(PH2P(CH2)3PPh2)(C6H5F)]+ C | 63100 (1.7) | 78900 (1.6) |
2.7. Discussion of Proposed Mechanistic Landscape
Bringing these observations together, we propose an overall mechanism shown in Scheme 5, in which the induction period that gets longer with increased [2a] can also now be explained. NMeH2, generated by slow B–N bond cleavage of H3B·NMeH2, at a rate that is independent of [2a], first promotes the formation of 3a and then more slowly the active precatalyst 5. In this model, higher concentrations of 2a result in more 3a needing to be first formed, via hydride abstraction and boronium formation, and then converted to the active catalyst with an unchanged amount of NMeH2, thus leading to a longer induction period. The active catalyst is closely related to both 5/6 and 4a, but we suggest both of these sit outside of the productive cycle, as their structures and reactivity are incompatible with the observed kinetics. The insensitivity in rate to added NMeH2suggests this does not reversibly dissociate, while a sensible model in which dimeric 4a, with its Rh–Rh bond and bridging amido–bisboryl ligand, undergoes rapid and reversible dissociation (vide supra) or loss of ligand is not obvious. Moreover, 4b reacts rapidly with NMeH2 to form 5, suggesting that if formed in catalysis it is not persistent. In addition, the fact that both 5 and 4b evolved to the same, currently unresolved, organometallic species under catalytic conditions suggests that both sit just outside of the productive catalytic cycle. While we cannot currently confidently comment on the nature of the actual catalyst for dehydrogenation, chain growth, or the termination process, the half-order relationship in [Rh]TOTAL and the observation of dimeric species (3 and 4) suggest that such Rh2 motifs may be intimately involved. The strong, and persistent, inverse relationship between Mn and [Rh]TOTAL, coupled with the sensitivity to H2, suggests a coordination/insertion/chain-growth mechanism for which NMeH2 also modifies chain length—possibly by attenuating chain termination. On the basis of the half-order kinetics observed from the dehydrogenation studies, we suggest three possible general motifs for the active catalyst (Scheme 6): one which invokes a monomer–dimer equilibrium in which one of the monomers is the active catalyst (A), and one in which a persistent dimer reversibly loses a bound ligand (B). Scenario A is reminiscent of the unsymmetrical Rh2 hydride dimers that can form in Rh-catalyzed alkene hydrogenations,57 while scenario B is supported by the recent report that dimeric early transition-metal complexes have been shown to act as competent catalysts for H3B·NMeH2 dehydropolymerization.7 A third possibility is that deprotonation of bound NMeH2 provides an active Rh–NMeH amido motif, similar to the bifunctional catalysts developed by Schneider and co-workers (C).11
Scheme 5. Pathways for Catalyst Activation and Catalysis in the Dehydropolymerization of H3B·NMeH2 Using [Rh] = {Rh(DPEphos)}+ Precatalysts.
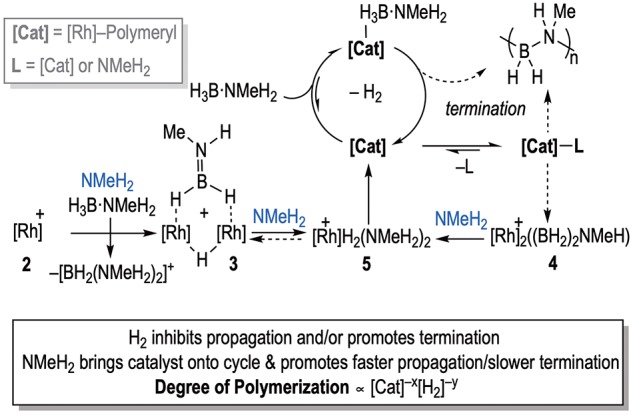
Anions are not shown. [Cat] may be mono- or bimetallic.
Scheme 6. Generalized Possible Active Species in Catalysis.

P = phosphine, L = ligand (e.g., NMeH2, or amine–borane-derived fragment). All structures shown are representative, and the actual number of hydrides/coordination geometry is undetermined.
3. Conclusions
We have shown that a combination of catalyst loading, H2, and NMeH2 can be used to control the dehydropolymerization of H3B·NMeH2 in a {Rh(DPEphos}+-based catalyst. We proposed this to be an important observation and one that may show some generality, building upon the already demonstrated improvement in catalyst lifetimes on addition of amine.8 The ability to control polymerization by catalyst loading, NMeH2 addition, and H2 in {Rh(DPEphos)}+ and {Rh(Xantphos-Ph)}+ systems is markedly different from that found for the {Rh(Xantphos-iPr)}+ catalyst and further supports that a different mechanism operates between the two sets, which may be related to the preferred coordination geometry of the ligands: DPEphos and Xantphos-Ph prefer cis-κ2-P,P while Xantphos-iPr generally adopts mer-κ2-P,O,P motifs. The amine systems we describe thus provide a tractable platform for further detailed mechanistic studies, and efforts are directed to determining the details of the propagating species and termination events so that fine control of the overall process, and thus the polymer produced, can be realized. It will be interesting to see if this effect of added amine is a more general observation across the now numerous2,3 dehydropolymerization catalysts from across the transition metals.
Acknowledgments
We are thankful for EPSRC EP/M024210 (A.S.W.) and DTP (G.M.A., N.A.B.). We thank Professor Steven Armes (Sheffield) for stimulating discussions. The research leading to these results received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013)/ERC Grant Agreement 340163.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b00081.
Author Present Address
∥ School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, United Kingdom.
The authors declare no competing financial interest.
Supplementary Material
References
- Leitao E. M.; Jurca T.; Manners I. Catalysis in Service of Main Group Chemistry Offers a Versatile Approach to P-Block Molecules and Materials. Nat. Chem. 2013, 5, 817–829. 10.1038/nchem.1749. [DOI] [PubMed] [Google Scholar]
- Colebatch A. L.; Weller A. S. Amine-Borane Dehydropolymerization: Challenges and Opportunities. Chem. - Eur. J. 2019, 25, 1379–1390. 10.1002/chem.201804592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.; Anke F.; Trose M.; Beweries T. Recent Advances in Transition Metal Catalysed Dehydropolymerisation of Amine Boranes and Phosphine Boranes. Coord. Chem. Rev. 2019, 380, 260–286. 10.1016/j.ccr.2018.09.016. [DOI] [Google Scholar]
- Staubitz A.; Robertson A. P. M.; Sloan M. E.; Manners I. Amine– and Phosphine–Borane Adducts: New Interest in Old Molecules. Chem. Rev. 2010, 110, 4023–4078. 10.1021/cr100105a. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Presa Soto A.; Manners I. Iridium-Catalyzed Dehydrocoupling of Primary Amine-Borane Adducts: A Route to High Molecular Weight Polyaminoboranes, Boron-Nitrogen Analogues of Polyolefins. Angew. Chem., Int. Ed. 2008, 47, 6212–6215. 10.1002/anie.200801197. [DOI] [PubMed] [Google Scholar]
- Dietrich B. L.; Goldberg K. I.; Heinekey D. M.; Autrey T.; Linehan J. C. Iridium-Catalyzed Dehydrogenation of Substituted Amine Boranes: Kinetics, Thermodynamics, and Implications for Hydrogen Storage. Inorg. Chem. 2008, 47, 8583–8585. 10.1021/ic801161g. [DOI] [PubMed] [Google Scholar]
- Trose M.; Reiß M.; Reiß F.; Anke F.; Spannenberg A.; Boye S.; Lederer A.; Arndt P.; Beweries T. Dehydropolymerisation of Methylamine Borane Using a Dinuclear 1,3-Allenediyl Bridged Zirconocene Complex. Dalton Trans. 2018, 47, 12858–12862. 10.1039/C8DT03311K. [DOI] [PubMed] [Google Scholar]
- Glüer A.; Förster M.; Celinski V. R.; Schmedt auf der Günne J.; Holthausen M. C.; Schneider S. A Highly Active Iron Catalyst for Ammonia Borane Dehydrocoupling at Room Temperature. ACS Catal. 2015, 5, 7214–7217. 10.1021/acscatal.5b02406. [DOI] [Google Scholar]
- Johnson H. C.; Leitao E. M.; Whittell G. R.; Manners I.; Lloyd-Jones G. C.; Weller A. S. Mechanistic Studies of the Dehydrocoupling and Dehydropolymerization of Amine-Boranes Using a [Rh(Xantphos)]+ Catalyst. J. Am. Chem. Soc. 2014, 136, 9078–9093. 10.1021/ja503335g. [DOI] [PubMed] [Google Scholar]
- Adams G. M.; Colebatch A. L.; Skornia J. T.; McKay A. I.; Johnson H. C.; Lloyd-Jones G. C.; Macgregor S. A.; Beattie N. A.; Weller A. S. Dehydropolymerization of H3B·NMeH2 to Form Polyaminoboranes Using [Rh(Xantphos-Alkyl)] Catalysts. J. Am. Chem. Soc. 2018, 140, 1481–1495. 10.1021/jacs.7b11975. [DOI] [PubMed] [Google Scholar]
- Marziale A. N.; Friedrich A.; Klopsch I.; Drees M.; Celinski V. R.; Schmedt auf der Günne J.; Schneider S. The Mechanism of Borane–Amine Dehydrocoupling with Bifunctional Ruthenium Catalysts. J. Am. Chem. Soc. 2013, 135, 13342–13355. 10.1021/ja311092c. [DOI] [PubMed] [Google Scholar]
- Staubitz A.; Sloan M. E.; Robertson A. P. M.; Friedrich A.; Schneider S.; Gates P. J.; Schmedt auf der Günne J.; Manners I. Catalytic Dehydrocoupling/Dehydrogenation of N-Methylamine-Borane and Ammonia-Borane: Synthesis and Characterization of High Molecular Weight Polyaminoboranes. J. Am. Chem. Soc. 2010, 132, 13332–13345. 10.1021/ja104607y. [DOI] [PubMed] [Google Scholar]
- Rossin A.; Peruzzini M. Ammonia–Borane and Amine–Borane Dehydrogenation Mediated by Complex Metal Hydrides. Chem. Rev. 2016, 116, 8848–8872. 10.1021/acs.chemrev.6b00043. [DOI] [PubMed] [Google Scholar]
- De Albuquerque Pinheiro C. A.; Roiland C.; Jehan P.; Alcaraz G. Solventless and Metal-Free Synthesis of High-Molecular-Mass Polyaminoboranes from Diisopropylaminoborane and Primary Amines. Angew. Chem., Int. Ed. 2018, 57, 1519–1522. 10.1002/anie.201710293. [DOI] [PubMed] [Google Scholar]
- Jurca T.; Dellermann T.; Stubbs N. E.; Resendiz-Lara D. A.; Whittell G. R.; Manners I. Step-Growth Titanium-Catalysed Dehydropolymerisation of Amine–Boranes. Chem. Sci. 2018, 9, 3360–3366. 10.1039/C7SC05395A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colebatch A. L.; Hawkey Gilder B. W.; Whittell G. R.; Oldroyd N. L.; Manners I.; Weller A. S. A General, Rhodium-Catalyzed, Synthesis of Deuterated Boranes and N-Methyl Polyaminoboranes. Chem. - Eur. J. 2018, 24, 5450–5455. 10.1002/chem.201800737. [DOI] [PubMed] [Google Scholar]
- Jenkins A. D.; Jones R. G.; Moad G. Terminology for Reversible-Deactivation Radical Polymerization Previously Called “Controlled” Radical or “Living” Radical Polymerization. Pure Appl. Chem. 2009, 82, 483–491. 10.1351/PAC-REP-08-04-03. [DOI] [Google Scholar]
- Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium-Catalyzed Hydroformylation: Effect of the Bite Angle. Organometallics 1995, 14, 3081–3089. 10.1021/om00006a057. [DOI] [Google Scholar]
- Esteruelas M. A.; Nolis P.; Oliván M.; Oñate E.; Vallribera A.; Vélez A. Ammonia Borane Dehydrogenation Promoted by a Pincer-Square-Planar Rhodium(I) Monohydride: A Stepwise Hydrogen Transfer from the Substrate to the Catalyst. Inorg. Chem. 2016, 55, 7176–7181. 10.1021/acs.inorgchem.6b01216. [DOI] [PubMed] [Google Scholar]
- Johnson H. C.; Weller A. S. P–C-Activated Bimetallic Rhodium Xantphos Complexes: Formation and Catalytic Dehydrocoupling of Amine–Boranes. Angew. Chem., Int. Ed. 2015, 54, 10173–10177. 10.1002/anie.201504073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams G. M.; Weller A. S. POP-Type Ligands: Variable Coordination and Hemilabile Behaviour. Coord. Chem. Rev. 2018, 355, 150–172. 10.1016/j.ccr.2017.08.004. [DOI] [Google Scholar]
- Johnson H. C.; McMullin C. L.; Pike S. D.; Macgregor S. A.; Weller A. S. Dehydrogenative Boron Homocoupling of an Amine–Borane. Angew. Chem., Int. Ed. 2013, 52, 9776–9780. 10.1002/anie.201304382. [DOI] [PubMed] [Google Scholar]
- Merle N.; Koicok-Köhn G.; Mahon M. F.; Frost C. G.; Ruggerio G. D.; Weller A. S.; Willis M. C. Transition Metal Complexes of the Chelating Phosphine Borane Ligand Ph2PCH2PH2P·BH3. Dalton Trans. 2004, 3883–3892. 10.1039/B413650K. [DOI] [PubMed] [Google Scholar]
- Johnson H. C.; Torry-Harris R.; Ortega L.; Theron R.; McIndoe J. S.; Weller A. S. Exploring the Mechanism of the Hydroboration of Alkenes by Amine–Boranes Catalysed by [Rh(Xantphos)]+. Catal. Sci. Technol. 2014, 4, 3486–3494. 10.1039/C4CY00597J. [DOI] [Google Scholar]
- Han D.; Joksch M.; Klahn M.; Spannenberg A.; Drexler H. J.; Baumann W.; Jiao H.; Knitsch R.; Hansen M. R.; Eckert H.; Beweries T. Iridium(III) Hydrido Complexes for the Catalytic Dehydrogenation of Hydrazine Borane. Dalton Trans. 2016, 45, 17697–17704. 10.1039/C6DT03068H. [DOI] [PubMed] [Google Scholar]
- Kim J.; Soares J.; Rempel G. Use of Hydrogen for the Tailoring of the Molecular Weight Distribution of Polyethylene in a Bimetallic Supported Metallocene Catalyst System. Macromol. Rapid Commun. 1998, 19, 197–199. 10.1002/(SICI)1521-3927(19980401)19:4<197::AID-MARC197>3.0.CO;2-4. [DOI] [Google Scholar]
- Ravve A.Principles of Polymer Chemistry, 3rd ed.; Springer: New York, 2012. [Google Scholar]
- Johnson H. C.; Robertson A. P. M.; Chaplin A. B.; Sewell L. J.; Thompson A. L.; Haddow M. F.; Manners I.; Weller A. S. Catching the First Oligomerization Event in the Catalytic Formation of Polyaminoboranes: H3B·NMeHBH2·NMeH2 bound to Iridium. J. Am. Chem. Soc. 2011, 133, 11076–11079. 10.1021/ja2040738. [DOI] [PubMed] [Google Scholar]
- Roselló-Merino M.; López-Serrano J.; Conejero S. Dehydrocoupling Reactions of Dimethylamine-Borane by Pt(II) Complexes: A New Mechanism Involving Deprotonation of Boronium Cations. J. Am. Chem. Soc. 2013, 135, 10910–10913. 10.1021/ja404655v. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Beattie N. A.; Pike S. D.; Macgregor S. A.; Weller A. S. The Simplest Amino–Borane H2B=NH2 Trapped on a Rhodium Dimer: Pre-Catalysts for Amine–Borane Dehydropolymerization. Angew. Chem., Int. Ed. 2016, 55, 6651–6656. 10.1002/anie.201600898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colebatch A. L.; McKay A. I.; Beattie N. A.; Macgregor S. A.; Weller A. S. Fluoroarene Complexes with Small Bite Angle Bisphosphines: Routes to Amine–Borane and Aminoborylene Complexes: Fluoroarene Complexes with Small Bite Angle Bisphosphines: Routes to Amine–Borane and Aminoborylene Complexes. Eur. J. Inorg. Chem. 2017, 2017, 4533–4540. 10.1002/ejic.201700600. [DOI] [Google Scholar]
- B–N bond cleavage would also generate “BH3”. At 10 mol % 2a/H3B·NMeH2, ESI–MS shows the formation of 3a, 4a, and an additional species with the empirical formula [Rh2(DPEphos)2(B2H5)]+, m/z = 1309.16. Addition of H3B·THF to 2a gives this as the dominant species. Bridging B2H5 units have been previously reported, e.g.:Jacobsen G. B.; Andersen E.; Housecroft C. E.; Hong F. E.; Buhl M. L.; Long G. J.; Fehlner T. P. Main-Group Chemistry in a Metal Framework: Preparation and Characterization of (Diborane)hexacarbonyldiiron and its Conjugate Base [B2H5Fe2(CO)6]. Inorg. Chem. 1987, 26, 4040–4046. 10.1021/ic00271a016. [DOI] [Google Scholar]
- Riddlestone I. M.; Kraft A.; Schaefer J.; Krossing I. Taming the Cationic Beast: Novel Developments in the Synthesis and Application of Weakly Coordinating Anions. Angew. Chem., Int. Ed. 2018, 57, 13982–14024. 10.1002/anie.201710782. [DOI] [PubMed] [Google Scholar]
- For {Rh(Xantphos-Ph)}+ a similar signal was observed at 5 mol %/H3B·NMe2H. We speculated upon a β-BH–agostic–amidoborane motif, but an alternative formulation could be a structure similar to 4.
- Toner A.; Matthes J.; Gründemann S.; Limbach H.-H.; Chaudret B.; Clot E.; Sabo-Etienne S. Agostic Interaction and Intramolecular Proton Transfer from the Protonation of Dihydrogen Ortho Metalated Ruthenium Complexes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6945. 10.1073/pnas.0608979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassen A.; Gloaguen Y.; Vendier L.; Duhayon C.; Poblador-Bahamonde A.; Raynaud C.; Clot E.; Alcaraz G.; Sabo-Etienne S. B–H, C–H, and B–C Bond Activation: The Role of Two Adjacent Agostic Interactions. Angew. Chem., Int. Ed. 2014, 53, 7569–7573. 10.1002/anie.201404753. [DOI] [PubMed] [Google Scholar]
- Rudolf G. C.; Hamilton A.; Orpen A. G.; Owen G. R. A ‘Sting’ on Grubbs’ Catalyst: An Insight into Hydride Migration between Boron and a Transition Metal. Chem. Commun. 2009, 553–555. 10.1039/B816036H. [DOI] [PubMed] [Google Scholar]
- Daly S. R.; Kim D. Y.; Girolami G. S. Lanthanide N,N-Dimethylaminodiboranates as a New Class of Highly Volatile Chemical Vapor Deposition Precursors. Inorg. Chem. 2012, 51, 7050–7065. 10.1021/ic201852j. [DOI] [PubMed] [Google Scholar]
- Chaplin A. B.; Weller A. S. B–H Activation at a Rhodium(I) Center: Isolation of a Bimetallic Complex Relevant to the Transition-Metal-Catalyzed Dehydrocoupling of Amine–Boranes. Angew. Chem., Int. Ed. 2010, 49, 581–584. 10.1002/anie.200905185. [DOI] [PubMed] [Google Scholar]
- Tang C. Y.; Phillips N.; Bates J. I.; Thompson A. L.; Gutmann M. J.; Aldridge S. Dimethylamine Borane Dehydrogenation Chemistry: Syntheses, X-Ray and Neutron Diffraction Studies of 18-Electron Aminoborane and 14-Electron Aminoboryl Complexes. Chem. Commun. 2012, 48, 8096–8098. 10.1039/c2cc33361a. [DOI] [PubMed] [Google Scholar]
- Metters O. J.; Chapman A. M.; Robertson A. P. M.; Woodall C. H.; Gates P. J.; Wass D. F.; Manners I. Generation of Aminoborane Monomers RR’N=BH2 from Amine–Boronium Cations [RR’NHBH2L]+: Metal Catalyst-Free Formation of Polyaminoboranes at Ambient Temperature. Chem. Commun. 2014, 50, 12146–12149. 10.1039/C4CC05145A. [DOI] [PubMed] [Google Scholar]
- Dallanegra R.; Robertson A. P. M.; Chaplin A. B.; Manners I.; Weller A. S. Tuning the [L2Rh···H3B·NR3]+ Interaction Using Phosphine Bite Angle. Demonstration by the Catalytic Formation of Polyaminoboranes. Chem. Commun. 2011, 47, 3763–3765. 10.1039/c0cc05460g. [DOI] [PubMed] [Google Scholar]
- Crooks A. B.; Yih K.-H.; Li L.; Yang J. C.; Özkar S.; Finke R. G. Unintuitive Inverse Dependence of the Apparent Turnover Frequency on Precatalyst Concentration: A Quantitative Explanation in the Case of Ziegler-Type Nanoparticle Catalysts Made from [(1,5-COD)Ir(μ-O2C8H15)]2 and AlEt3. ACS Catal. 2015, 5, 3342–3353. 10.1021/acscatal.5b00347. [DOI] [Google Scholar]
- Jaska C. A.; Manners I. Heterogeneous or Homogeneous Catalysis? Mechanistic Studies of the Rhodium-Catalyzed Dehydrocoupling of Amine–Borane and Phosphine–Borane Adducts. J. Am. Chem. Soc. 2004, 126, 9776–9785. 10.1021/ja0478431. [DOI] [PubMed] [Google Scholar]
- Sonnenberg J. F.; Morris R. H. Distinguishing Homogeneous from Nanoparticle Asymmetric Iron Catalysis. Catal. Sci. Technol. 2014, 4, 3426–3438. 10.1039/C4CY00468J. [DOI] [Google Scholar]
- Pal S.; Kusumoto S.; Nozaki K. Dehydrogenation of Dimethylamine–Borane Catalyzed by Half-Sandwich Ir and Rh Complexes: Mechanism and the Role of Cp* Noninnocence. Organometallics 2018, 37, 906–914. 10.1021/acs.organomet.7b00889. [DOI] [Google Scholar]
- Shuttleworth T. A.; Huertos M. A.; Pernik I.; Young R. D.; Weller A. S. Bis(Phosphine)Boronium Salts. Synthesis, Structures and Coordination Chemistry. Dalton Trans. 2013, 42, 12917–12925. 10.1039/c3dt50817j. [DOI] [PubMed] [Google Scholar]
- Hooper J. F.; Chaplin A. B.; González-Rodríguez C.; Thompson A. L.; Weller A. S.; Willis M. C. Aryl Methyl Sulfides as Substrates for Rhodium-Catalyzed Alkyne Carbothiolation: Arene Functionalization with Activating Group Recycling. J. Am. Chem. Soc. 2012, 134, 2906–2909. 10.1021/ja2108992. [DOI] [PubMed] [Google Scholar]
- Sewell L. J.; Huertos M. A.; Dickinson M. E.; Weller A. S.; Lloyd-Jones G. C. Dehydrocoupling of Dimethylamine Borane Catalyzed by Rh(PCy3)2H2Cl. Inorg. Chem. 2013, 52, 4509–4516. 10.1021/ic302804d. [DOI] [PubMed] [Google Scholar]
- Boller T. M.; Murphy J. M.; Hapke M.; Ishiyama T.; Miyaura N.; Hartwig J. F. Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc. 2005, 127, 14263–14278. 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- Shapiro P. J.; Schaefer W. P.; Labinger J. A.; Bercaw J. E.; Cotter W. D. Model Ziegler-Natta Α-Olefin Polymerization Catalysts Derived from [{(η5-C5Me4)SiMe2(η1-NCMe3)}(PMe3)Sc(η2-H)]2 and [{(η5-C5Me4)SiMe2(η1-NCMe3)}Sc(η2-CH2CH2CH3)]2. Synthesis, Structures, and Kinetic and Equilibrium Investigations of the Catalytically Active Species in Solution. J. Am. Chem. Soc. 1994, 116, 4623–4640. 10.1021/ja00090a011. [DOI] [Google Scholar]
- Chamberlain B. M.; Jazdzewski B. A.; Pink M.; Hillmyer M. A.; Tolman W. B. Controlled Polymerization of Dl-Lactide and Ε-Caprolactone by Structurally Well-Defined Alkoxo-Bridged Di- and Triyttrium(III) Complexes. Macromolecules 2000, 33, 3970–3977. 10.1021/ma0000834. [DOI] [Google Scholar]
- Bochmann M.; Lancaster S. J. Monomer–Dimer Equilibria in Homo- and Heterodinuclear Cationic Alkylzirconium Complexes and Their Role in Polymerization Catalysis. Angew. Chem., Int. Ed. 1994, 33, 1634–1637. 10.1002/anie.199416341. [DOI] [Google Scholar]
- Kumar A.; Ishibashi J. S. A.; Hooper T. N.; Mikulas T. C.; Dixon D. A.; Liu S.-Y.; Weller A. S. The Synthesis, Characterization and Dehydrogenation of Sigma-Complexes of BN-Cyclohexanes. Chem. - Eur. J. 2016, 22, 310–322. 10.1002/chem.201502986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Kam L.; Trerise R.; Williams T. J. Ruthenium-Catalyzed Ammonia Borane Dehydrogenation: Mechanism and Utility. Acc. Chem. Res. 2017, 50, 86–95. 10.1021/acs.accounts.6b00482. [DOI] [PubMed] [Google Scholar]
- Widegren J. A.; Finke R. G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A: Chem. 2003, 198, 317–341. 10.1016/S1381-1169(02)00728-8. [DOI] [Google Scholar]
- Duckett S. B.; Newell C. L.; Eisenberg R. Observation of New Intermediates in Hydrogenation Catalyzed by Wilkinson’s Catalyst, RhCl(PPh3)3, Using Parahydrogen-Induced Polarization. J. Am. Chem. Soc. 1994, 116, 10548–10556. 10.1021/ja00102a023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.