

Abstract
As a central hub of cellular metabolism and signaling, the mitochondrion is a crucial organelle whose dysfunction can cause disease and whose activity is intimately connected to aging. We review how the mitochondrial network maintains proteomic integrity, how mitochondrial proteotoxic stress is communicated and resolved in the context of the entire cell, and how mitochondrial systems function in the context of organismal health and aging. A deeper understanding of how mitochondrial protein quality control mechanisms are coordinated across these distinct biological levels should help explain why these mechanisms fail with age and, ultimately, how routes to intervention might be attained.
Keywords: mitochondria, proteostasis, aging, stress, unfolded protein response (UPR)
Introduction
The decline in the health of an organism over time derives from dysfunction at the cellular level, which largely arises from the progressive accumulation of damage to proteins and organelles (Fig. 1A). Work over the past decades has identified the mitochondrion as an organelle whose function imparts a significant effect on aging. Here, we review a set of underlying pathways and molecular machines that are central to the mitochondrial contribution to aging.
Figure 1.
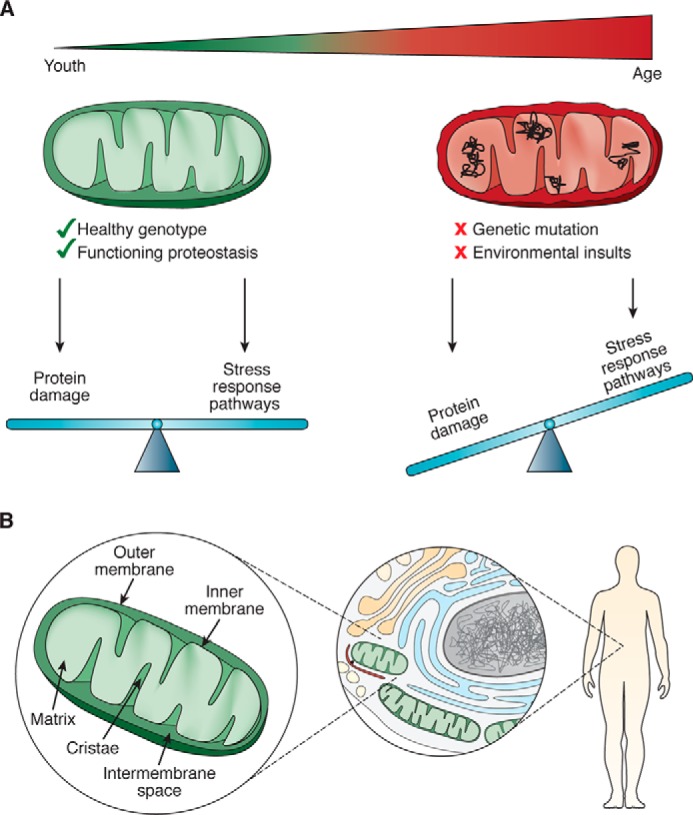
Factors that impact mitochondrial proteostasis. A, mitochondria must balance the damage that occurs to its proteins with activation of pathways evolved to counteract this damage. B, mitochondrial proteostasis occurs at multiple scales: at the level of the mitochondrion, the mitochondrion's interaction with the rest of the cell, and with the organism as it impacts metabolism, health, and life span.
The key functions mitochondria are tasked with–energy homeostasis, metabolism, and apoptosis–rely upon an elaborate network of proteins, many of them multisubunit complexes. Several challenges face the mitochondrion with respect to establishing and maintaining a functional proteome. First, having descended from an ancestral bacterium, the mitochondrial proteome and genome are separated from the rest of the cell by inner and outer membranes (Fig. 1B). Only ∼13 of its ∼1,100 resident proteins are encoded by the mitochondrial genome (mtDNA) (1); therefore, a vast majority of mitochondrial proteins have to be folded following translation in the cytoplasm and import into mitochondria. Moreover, once imported into the organelle, mitochondrial proteins are physically separated from the cytoplasmic protein-folding machinery, therefore requiring mitochondrion-localized machinery for their maintenance. The second challenge is that a number of key mitochondrial protein complexes contain subunits encoded by both the nuclear and mitochondrial genomes, and an imbalance in the expression from these two genomes can be detrimental to mitochondrial protein homeostasis. A third challenge is that the primary energy-producing process inside the mitochondrion, oxidative phosphorylation (OXPHOS),4 creates damaging reactive oxygen species (ROS) as a by-product of the electron transport chain (ETC). These ROS threaten not only the OXPHOS machinery but also other mitochondrial proteins, lipids, and the mtDNA.
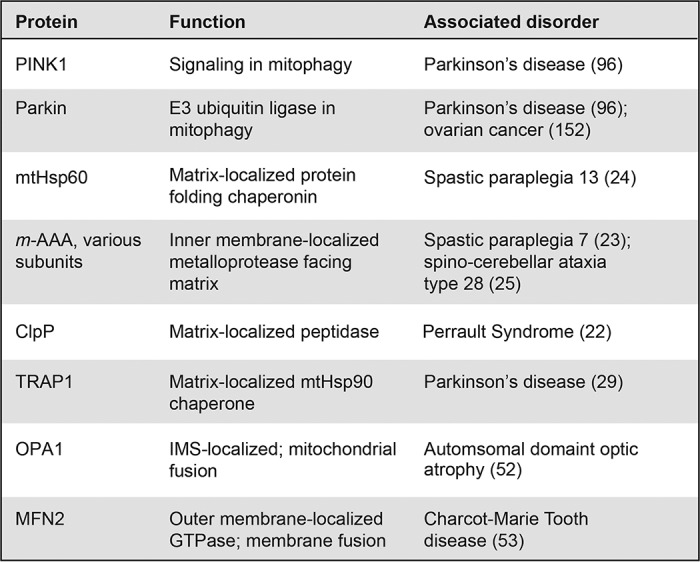
To defend against these proteomic challenges, mitochondria employ several mechanisms to maintain protein homeostasis, or “proteostasis” within the organelle. In general terms, proteostasis mechanisms exist to monitor and control all steps in the life of a protein, including biogenesis, folding, localization, and degradation. Proteostasis of mitochondrial proteins includes mitochondria-localized chaperones and proteases that re-fold or degrade individual mis-folded proteins, as well as bulk mitochondrial organelle degradation, inter-organellar communication, and trans-cellular signaling, all of which impact the quality of proteins functioning within mitochondria. Defects in these mitochondrial proteostasis defense pathways in these different layers of biological complexity can have substantial impacts on organismal health and aging. A number of mutations in genes encoding mitochondrial proteostasis machinery result in accelerated proteostatic collapse, and many eventually manifest in age-associated diseases (Table 1). In addition, acute environmental insults, such as exposure to mitochondria-targeted pesticides, herbicides, and antibiotics are thought to increase the proteostatic burden on mitochondria and are known to be pathogenic to humans (2). An increased focus on mitochondrial proteostasis as it connects to cellular and organismal health should yield a more informed perspective on the etiology and treatment of aging and aging-related disease.
Table 1.
Genetic mutations in mitochondrial protein quality control factors associated with disease and age-related disorders
Mitochondrial proteostasis at the organellar level
Types of damage that accumulate in mitochondria
To trace the connection between mitochondrial proteostasis and organismal health, we first review the types of damage to the mitochondrial proteome and the molecular machines that function within the organelle to repair this damage. Much like other cellular proteins, mitochondrial proteins misfold, mis-assemble in protein complexes, and aggregate over time, and these events all pose a threat to proteostasis if left unresolved. In some cases, this type of damage can be reversed locally through protein-folding molecular chaperones. In contrast, terminally damaged proteins, such as those containing carbonylated residues due to exposure to ROS (3), must be degraded. In addition to direct protein damage, the mitochondrial proteome can be adversely affected by mutations or deletions in the mitochondrial genome. Each mitochondrion carries multiple copies of mtDNA, and a mutation in even a fraction of these copies can result in the synthesis of misfolded proteins. Mutations and deletions in mtDNA can also perturb mitochondrial proteostasis because they affect ATP production and increase ROS production (4–8).
The accumulation of mitochondrial proteome damage directly impacts organismal health and aging. Compromised OXPHOS proteins perturb the overall structure and function of mitochondria, particularly in organisms of advanced age (9). Carbonylation of mitochondrial proteins from ROS damage has been observed to increase with age in multiple model organisms (3), and inducing excessive ROS in mice or humans causes Parkinson's disease (PD)-like symptoms (2). Furthermore, mutations in mtDNA accumulate with age, resulting in dysfunction of its encoded proteins, which can have direct physiological consequences. For example, mtDNA mutations have been observed in the substantia nigra of the midbrain and are more prevalent in PD patients compared with age-matched controls (10, 11). In addition, mutations in POLG, the gene encoding the mitochondrial DNA polymerase responsible for replication, cause deletions in, and copy number variations of, mtDNA and correlate with inheritance of parkinsonism (12), as well as a diverse panel of progressive neurological and muscular symptoms (13). The relevant mouse model of Polg mutation, termed the mutator mouse, accumulates mutations and deletions and exhibits premature aging phenotypes (7).
Mitochondria-localized mechanisms to combat damage
The canonical cytoplasmic proteostasis systems, such as the proteasome and heat-shock proteins, do not function inside mitochondria. For this reason, the mitochondrion has evolved a dedicated set of molecular machines, such as a network of resident chaperones, proteases, and other quality control factors, whose function is to protect the proteome (Fig. 2). The mitochondrion in particular bears a substantial burden with respect to protein folding, because all imported polypeptides must be folded immediately upon import and integrated into elaborate protein complexes that function inside the mitochondrion. Molecular chaperones are essential to correctly fold proteins after they are imported as nascent polypeptides into mitochondria and to re-fold any unfolded or misfolded proteins that may cause proteotoxic damage (Fig. 2A). A key chaperone, the mitochondrial Hsp70 (mtHsp70) is associated with the inner membrane import complex on the matrix side and actively mediates the import and folding of nascent proteins (14). In addition, a mitochondrial isoform of the heat-shock protein TRAP1 (mtHsp90) and the large chaperonin Hsp60/10 complex also contribute to the folding of matrix-localized polypeptides that require additional assistance (15–17). Separately, SOD2 in the matrix and SOD1 in the intermembrane space (IMS) convert damaging ROS into less toxic H2O2 and O2 before they can damage proteins (18, 19).
Figure 2.

Protein quality control machines at work within mitochondria. A, protein folding chaperones that function in the mitochondrial matrix to fold nascent polypeptides or repair mis-folded proteins. B, proteins can be damaged by the ROS generated by components of the OXPHOS machinery. Proteases in the intermembrane space and matrix degrade these damaged proteins. C, fission and fusion work dynamically to alter the shape of the mitochondrial network to dilute or segregate areas of damage.
In parallel with chaperone-mediated protein folding and repair, mitochondrial proteostasis also relies on a set of dedicated proteases to resolve irreversible protein damage through degradation (Fig. 2B). The mitochondrial proteome has a high turnover rate: measurements from yeast reveal that as much as 6–12% of the mitochondrial proteome is degraded in each generation (20). Proteins with terminal damage or orphan subunits from multimeric complexes are frequent targets for the mitochondrion's numerous proteases. In the matrix, these include LonP and ClpP, whereas the inner membrane contains two protease complexes, m-AAA (AFG3L2, AFG3L1, and SPG7) and i-AAA (YME1L), which face the matrix and IMS, respectively (21). These inner-membrane proteases are especially important for functional integrity of the OXPHOS machinery, which are particularly susceptible to oxidative damage due to their ROS-releasing activity.
Defects in these chaperone and protease machines are associated with diverse organismal pathologies (Table 1). In some cases, loss of mitochondrial quality control machinery causes severe organismal pathology starting in early or mid-life: mutations in the CLPP protease have been associated with Perrault syndrome, resulting in neonatal or early childhood sensorineural hearing loss (22). In addition, two degenerative hereditary spastic paraplegias can be caused by mutations in the mitochondrial protease SPG7 or the chaperone HSP60 (23, 24), and spinocerebellar ataxia type 28 results from mutations in the m-AAA component, AFG3L2 (25). Mutation in, or decreased expression of, mitochondrial chaperones is also associated with aging-related diseases such as PD and Alzheimer's disease (AD) (26–28). For example, loss of TRAP1 by mutation has been associated with late-onset PD, as well as decreased OXPHOS activity and loss of membrane potential in patient cells (29). In addition, SOD2 is up-regulated in both AD (30) and PD patients (31), presumably in response to oxidative stress within mitochondria from these diseases. As an organism ages, its capacity to fight proteostatic insults appears to decrease: for example, basal Lonp expression decreases with age in mice (32); similarly, the ability of mice and Caenorhabditis elegans to induce expression of mitochondrial chaperones decreases with age (33, 34). There is an unfortunate paradox in these data, since the need for such protective measures increases with age. Inherited neurological disorders are not the only pathology derived from loss of mitochondrial quality control; the mitochondrial matrix protease CLPP is highly expressed in leukemias (35) and multiple tumors (36), suggesting that cancer cells may experience a high proteotoxic burden within mitochondria.
Beyond managing their internal proteome, mitochondria continuously undergo opposing fusion and fission events, and this dynamic process is necessary for maintaining a healthy mitochondrial network (Fig. 2C). In healthy cells, mitochondria are fused into a large network, with multiple copies of the mtDNA and contiguous inner and outer membranes (37). Mitochondrial fusion, which mixes contents of different mitochondrial membranes as well as soluble components, relies on MFN1/MFN2 and OPA1 for outer and inner membrane fusion, respectively (38, 39).
In healthy cells, fusion dilutes the effects of small amounts of damage within the larger network (40). In fact, some stresses promote hyper-fusion in a process involving MFN1, OPA1, and the scaffolding inner membrane protein, SLP2 (40). Such hyper-fusion has been shown to be vital to cell survival during stresses like starvation (41) and may stave off fission-induced apoptosis. In contrast, mitochondrial fission partitions damaged mitochondria away so the health of the overall mitochondrial network can recover (42). Mitochondrial fission is driven by the dynamin-related protein DRP1/DNM1 and OMA1 (43–45). DRP1/DNM1 is recruited to mitochondrial fission sites by several identified outer mitochondrial membrane receptors, such as FIS1 and MFF (46–48). Under conditions of stress, OMA1 induces mitochondrial fragmentation by proteolytically cleaving OPA1 (49, 50). In addition to recovering the mitochondrial network, mitochondrial fission also plays a critical role in mitophagy (see below) and the initial steps of apoptosis, or programmed cell death (51).
Mutations in several components of the mitochondrial dynamics machinery also yield organismal pathologies, particularly neurological disorders. Mutation of OPA1 causes dominant optic atrophy (52), and mutations of MFN2 that disrupt the morphology and distribution of the mitochondrial network can cause Charcot-Marie-Tooth syndrome type 2A, characterized by dystrophy of peripheral muscle (53, 54). Remarkably, a knockout of Mfn2 in the mouse leads to specific loss of dopaminergic neurons, a phenotype observed in PD patients (55, 56).
A cellular perspective: mitochondrial proteostasis in the context of the cell
The mitochondrial genome has lost the vast majority of its protein-coding genes, and its proteome is therefore derived overwhelmingly from nuclear transcription, cytoplasmic translation, and polypeptide import into the mitochondrion. For this reason, whereas mitochondrial chaperones, proteases, and fusion/fission machinery act internally to maintain mitochondrial proteostasis, the overall integrity of mitochondrial function requires cooperation and communication with other cellular compartments.
Mitochondrial–nuclear communication: mtUPR
Originally identified in mammalian cells (57), the mitochondrial unfolded protein response (mtUPR) has been established as a prominent line of defense for mitochondrial proteotoxic stress in mammals, Drosophila, and C. elegans (57–60) (Fig. 3A). In a pathway named after the unfolded protein response of the endoplasmic reticulum (ER–UPR) (61), the mtUPR senses proteotoxic stress within mitochondria and enacts a gene expression program to recover organellar proteostasis. Like the ER–UPR, the mtUPR up-regulates target genes that include organelle-specific chaperones and proteases. An additional goal of the mtUPR is to alleviate the demands on stressed mitochondria by shifting metabolism away from mitochondrial-dependent OXPHOS and toward cytoplasmic glycolysis (62).
Figure 3.

Mitochondrial proteostasis mechanisms that function at the level of the cell. A, mitochondrial–nuclear communication as described in C. elegans. Mitochondria signal proteotoxic stress to the nucleus to up-regulate genes that will aid in restoring mitochondrial health as well as enact epigenetic changes across the genome. We focus on the ATFS-1 and LIN-65/MET-2/DVE-1 pathways. During basal conditions (No stress), the MLS targets ATFS-1 for mitochondrial import and subsequent degradation. During mitochondrial stress (High stress), low mitochondrial membrane potential and blocked mitochondrial import impairs mitochondrial targeting of ATFS-1, instead allowing NLS-dependent nuclear localization of ATFS-1, where it activates various gene transcription pathways. NLS, nuclear localization sequence; MLS, mitochondrial localization sequence. For a complete summary of all identified mtUPR players, we direct the reader to two recent mtUPR-focused reviews (154, 155). B, mechanisms of mitochondrial degradation via mitophagy for bulk degradation (top) or specialized, vesicle-dependent degradation (bottom). Mitophagy by either the PINK1/Parkin or NIX pathways in humans requires recognition of defective mitochondria, followed by recruitment and engulfment by an autophagosome membrane, and shuttling to the lysosome. Under basal conditions (No stress), PINK1 is targeted to the mitochondria and rapidly degraded. In one described mechanism, this occurs via mitochondrial import and N-terminal cleavage by the inner membrane protease PARL (99). N-terminally cleaved PINK1 (PINK1′) is then shuttled to the cytoplasm for proteasomal degradation. MDC (Saccharomyces cerevisiae) and MDV (humans) have both been shown to selectively target mitochondrial protein and other metabolite cargo for degradation. C, mitochondrial–cytoplasmic quality control mechanisms include the MCSR from C. elegans and proteasome-dependent degradation of mitochondrial outer membrane proteins. HSF-1, heat-shock factor 1.
Activation of the mtUPR arises from a wide range of proteotoxic stresses, including blocking mitochondrial translation (60), depletion of mtDNA (63, 64), targeted impairment of mitochondrial chaperones or proteases (65, 66), excessive ROS (67), ETC impairment (58), or expression of a misfolded protein (57). It is unclear how misfolded or damaged proteins are recognized by mtUPR machinery. One possibility is through the oligopeptides generated by the protease ClpP (66) after degrading compromised proteins. However, many of the stresses that induce the mtUPR converge on decreased mitochondrial protein import. To this end, work in C. elegans, where the mtUPR is best-studied, showed that the transcription factor ATFS-1, a primary activator of the mtUPR (68), is highly sensitive to mitochondrial import efficiency. As a primary activator of the mtUPR, ATFS-1 is uniquely suited to communicate mitochondrial stress to the nucleus as it contains both mitochondrial and nuclear localization sequences that are differentially utilized under basal and stressed conditions. Under basal conditions when mitochondrial membrane potential is high and protein import is robust, the N-terminal mitochondrial targeting sequence routes the nascent ATFS-1 protein to the mitochondrial import machinery. Immediately following import, it is rapidly degraded to undetectable levels by LonP (Fig. 3A, No stress) (68). However, during mitochondrial stress, protein import is compromised; this causes the ATFS-1 protein to accumulate in the cytoplasm, allowing its C-terminal nuclear localization sequence to access the nuclear import machinery (Fig. 3A, High stress). Once in the nucleus, ATFS-1 activates the transcription of genes to restore mitochondrial health such as chaperones, protein import machinery, ROS detoxification genes, as well as innate immunity and glycolysis factors to improve mitochondrial and cellular health (62). Consistent with ATFS-1 transcription factor activity being sensitive to import efficiency, a direct block in protein import, thereby limiting ATFS-1 import into mitochondria, leads to activation of the mtUPR (68).
Beyond ATFS-1, evidence suggests that additional methods for mtUPR activation also occur: of the 700 transcripts found to be induced during mitochondrial stress, only ∼400 required atfs-1 for induction (68). This additional regulation may come, in part, from the histone demethylases JMJD-1.2 and JMJD-3.1, which facilitate access to mtUPR response gene promoters (69), as well as the transcription factor DVE-1 and its ubiquitin-like cofactor UBL-5 (Fig. 3) (66, 70). The joint action of DVE-1 and UBL-5 is additionally driven by H3K9 dimethylation by MET-2 and its cofactor LIN-65 to up-regulate the mtHsp70 and Hsp60 chaperones upon mitochondrial stress (71). These chromatin factors promote nuclear localization of DVE-1 and enact the epigenetic changes required for mitochondrial stress signaling (71). How independent the ATFS-1 and DVE-1/LIN-65/MET-2 branches of mtUPR activation are is not fully known.
Although best-studied in C. elegans, the mtUPR was first discovered in mammalian cells (57) and remains an active area of investigation. Notably, a mammalian ortholog of ATFS-1, ATF5, has been shown to regulate the mtUPR in mammalian cells and to up-regulate transcription of mammalian orthologs of many C. elegans quality control genes in response to mitochondrial stress (72). Although such evolutionary conservation of function is noteworthy, it is clear that the mammalian mtUPR involves additional factors, such as the transcription factors CHOP and CEBPB (C/EBPβ), which have binding sites in many mtUPR-responsive genes (57, 73), including ATF5 itself (74). Furthermore, recent evidence has suggested that the mtUPR works closely alongside the integrated stress response in responding to mitochondrial stress, and that this may be a prominent mechanism for responding to stress in mammalian systems (75, 76). Finally, although the majority of mtUPR-described mechanisms focuses on proteotoxic stress in the mitochondrial matrix, an unfolded protein response specific to the IMS in human cells has also been reported (77).
An important aspect of the mtUPR is its close relationship to aging and organismal health. In C. elegans, Drosophila, and mice, mitochondrial stresses that activate the mtUPR can extend life span and improve health (58, 60, 69, 78, 79). For example, disrupted balance of expression from the mitochondrial and nuclear genomes, via altered expression of a mitochondrial ribosomal subunit, strongly correlates with increased longevity in mice (60). Furthermore, higher expression of jmjd-1.2/Phf8 or jmjd-3.1/Jmjd3 correlates with life span extension in both C. elegans and mice (69). In Drosophila, knockdown of ETC components to activate mtUPR promotes expression of genes regulated by the FoxO transcription factor to extend life span (78). In a recent study, knockout of Clpp in mice led to a compensatory mitochondrial stress response that increased insulin sensitivity and protected mice from diet-induced obesity (79). Finally, a recent study points to the mammalian mtUPR as a component of hematopoietic stem cell regeneration (80). There can, however, be a fitness compromise associated with this mtUPR-mediated extended life span. C. elegans animals often become developmentally delayed or less reproductively fit upon mtUPR activation (81, 82). In addition, ectopic activation of the mtUPR in dopaminergic neurons can cause cell death (83), suggesting that chronic activation of the mtUPR may be detrimental to cell survival. Rather than be interpreted as a longevity panacea, mtUPR-mediated effects for promoting health span may be context-dependent, highlighting the need for further investigation.
Mitophagy: removal of proteotoxic damage in bulk
During stresses such as accumulation of misfolded proteins or loss of membrane potential, the mtUPR up-regulates a host of mitochondrial proteases that degrade aberrant proteins. However, when these stresses induce too much damage within an individual mitochondrion, the entire organelle can be degraded in a process known as mitophagy (mitochondrial-specific autophagy) (Fig. 3B). Although healthy mitochondria rapidly re-fuse back into the network following fission (84, 85), unhealthy mitochondria are poor at fusion, remain separated from the network, and are recognized by the mitophagy machinery. This segregation and pruning approach allows mitochondria harboring mutant mtDNA or with a critically high burden of misfolded proteins to be degraded, thereby facilitating the recovery of the rest of the network. Many stresses that activate the mtUPR also activate mitophagy, leading to the idea that the mtUPR and mitophagy are complementary: the mtUPR may act as a first line of defense to combat insults to mitochondrial proteostasis, whereas mitophagy acts to remove the unsalvageable mitochondria (86).
The precise mechanisms by which the mitophagy machinery recognizes defective mitochondria remain incompletely understood, but they are generally thought to target mitochondria with reduced membrane potential (85), increased ROS (87), blocked mitochondrial protein import (88), or excess mis-folded proteins (89). Although the exact molecular players differ between yeast and mammalian systems, general principles of mitophagy involve recognition of a damaged mitochondrion, subsequent engulfment by the autophagosome membrane, and shuttling to the lysosome, or vacuole in yeast, for degradation.
In yeast, where mitophagy mechanisms have been best studied, autophagosome components Atg11 and Atg8 recognize the outer membrane mitochondrial protein Atg32 (90, 91). In mammalian systems, mitophagy mechanisms are more complex, involving at least two distinct pathways (Fig. 3B). Much of what is known about mitophagy in mammals focuses on BNIP3L (NIX)-driven mechanisms, in which the outer membrane protein NIX binds to the autophagosomal protein MAP1LC3A (LC3) to initiate mitophagy (92–94). Interestingly, NIX is also up-regulated during hypoxia (95), suggesting that NIX is broadly involved in restoring mitochondrial proteostasis through mitophagy during stress. An additional mechanism of mitophagy in mammalian cells is the PINK1/PRKN (Parkin) pathway. PINK1 and Parkin have gained special attention from the discovery that mutations in both proteins result in autosomal-recessive forms of PD (Table 1) (96). Similar to ATFS-1, PINK1 recognition of defective mitochondria is also regulated by mitochondrial protein import. In the absence of stress, PINK1 is imported and constitutively degraded; this occurs either by protease degradation in the mitochondrial matrix (97, 98) or by cleavage by the IMS-localized protease PARL followed by translocation to the cytoplasm and N-end rule pathway degradation by the proteasome (Fig. 3B, No stress) (99, 100). During stress, mitochondrial import efficiency decreases, and PINK1 accumulates on the mitochondrial surface (Fig. 3B, High stress) (101). This PINK1 accumulation recruits the E3 ubiquitin ligase Parkin to the mitochondrial surface, where it polyubiquitinates outer membrane proteins (102–104). It has been proposed that the autophagosome recognizes ubiquitylated mitochondria through the ubiquitin-binding protein SQSTM1 (p62), as it has been observed to accumulate on mitochondria and bind LC3 (105), but the exact mechanism of autophagic recognition remains unclear (103, 106).
NIX and PINK1/Parkin homologs have also been found to regulate mitophagy in C. elegans (107). Importantly, they have been shown to interface with other genetic pathways such as SKN-1, the transcription factor that regulates mitochondrial biogenesis, and the DAF-16 insulin/IGF-1 pathway to regulate aging (107). In mice, a PINK1 knockout exhibits impaired mitochondrial respiration and synaptic plasticity (108). Overall, mitophagy is an essential pathway that eliminates defective organelle activated under high levels of mitochondrial proteostatic stress to promote steady-state mitochondrial integrity and healthy aging.
Asymmetric cell division: controlling mitochondrial inheritance
Mitochondrial fission followed by mitophagy eliminates the damaged fraction of the mitochondrial network. However, another opportunity to eliminate damaged mitochondria occurs during the process of cell division. Typically, during mitosis, cytoskeletal components strategically organize mitochondria to equally split the mitochondrial network between the two daughter cells (109, 110). However, individual mitochondria can be actively segregated to either the mother or the daughter cell based on their quality. This active parsing of mitochondria has been demonstrated in systems as diverse as budding yeast and human mammary stem cells (111, 112) with substantial impacts on the health and life span of the daughter cells. In budding yeast, the healthiest mitochondria are passed to the daughter cell to promote longer replicative life span (111, 113, 114). The underlying mechanism involves mitochondrial interactions with the actin cytoskeleton (115) and mitochondrial fusion: only mitochondria that undergo fusion at the bud tip are retained and passed on to the daughter cell (116). In higher eukaryotes, segregation of healthy from unhealthy compartments is critical to maintain cellular and tissue homeostasis (117, 118). A recent study demonstrated that mammalian epithelial stem-like cells divide asymmetrically to retain the newest mitochondria and pass older mitochondria on to the differentiating daughter cell (112). Interestingly, only the mitochondria, and not other subcellular components such as the lysosome, ribosome, or Golgi apparatus, were asymmetrically apportioned, suggesting there is a crucial aspect of mitochondrial homeostasis required for stem cell maintenance.
Inter-organellar proteostasis
Mitophagy evolved as a mechanism to degrade an entire mitochondrion, but smaller-scale degradation mechanisms that transport damaged mitochondrial components for processing elsewhere in the cell are an emerging area of investigation. Two prominent mechanisms that have recently been described are mitochondria-derived vesicles (MDVs) and compartments (MDCs), which transport protein- and other metabolite-containing vesicular bodies to other organelles in the cell. MDVs have been reported to transport specific mitochondrial cargo to the peroxisome (119) as well as oxidized mitochondrial proteins for degradation in the lysosome (120). Although the formation and lysosomal targeting of these vesicles seem to be independent of mitochondrial fission and canonical mitophagy mechanisms, intracellular MDV trafficking relies on both Parkin and PINK1 and can be triggered by mitochondrial oxidative stress (120, 121). In yeast, a different vesicular body, the MDC, is involved in the direct removal of mitochondrial proteins for degradation in the vacuole (122). In contrast to MDVs in human cells, MDC release relies directly on the fission and autophagy machinery. Recent evidence for direct mitochondria–lysosome contacts in mammalian cells further highlights the importance of understanding this inter-organelle communication (123).
The role of these mitochondria-derived vesicular bodies in physiological contexts remains to be defined. However, growing evidence suggests that direct mitochondrial contacts with the endoplasmic reticulum (ER) impact human health: disturbance of these contact sites in the brain is observed in numerous neurodegenerative diseases (124, 125) and in primary human myotubes from patients with type 2 diabetes (126). These ER–mitochondria contact sites (also called mitochondria-associated ER membranes, or MAMs), have recently emerged as mediators of mitochondrial homeostasis. One of the primary roles of MAMs is coordinating Ca2+ signaling and lipid metabolism between the ER and mitochondria, which regulate OXPHOS and apoptosis (127–129). How MAMs as inter-organellar contact sites impact mitochondrial proteostatic health is an area of new investigation. ER–mitochondria contact sites are enriched for stress-induced cytoplasmic protein aggregates, which are later captured by mitochondria for degradation (130). In addition, MAMs have been shown to directly control mitochondrial homeostasis by regulating mitochondrial fission sites and mtDNA replication (131, 132). The functional homology of ER–mitochondria contact sites from yeast to humans (129, 133) highlights the importance of mitochondrial communication in signaling and cellular homeostasis.
Proteostasis through mitochondrial–cytoplasmic communication
A burgeoning area of research is in the coordination of proteostasis mechanisms among different cellular compartments. In one such mechanism discovered in C. elegans, termed mitochondrial-to-cytosolic stress response (MCSR), mitochondrial proteotoxic stress from pathogenic protein aggregates up-regulates not only the mtUPR but the cytosolic unfolded protein response as well, including heat-shock protein 1 (hsp-1) (Fig. 3C). This occurs via changes in lipid biosynthesis that act as the signal between the mitochondria and cytosol and serves to protect the cell as a whole from proteotoxic insults (134). Importantly, the MCSR was conserved in a cell culture model of Huntington's disease, suggesting a broad evolutionary need to coordinate intracellular stress-response pathways.
There may also exist mechanisms for coordinated protein degradation between mitochondrial and cytoplasmic compartments. The cytoplasmic proteasome has been implicated in extracting and degrading misfolded proteins from the mitochondrial outer membrane (135, 136). Additionally, it was proposed that cytosolic protein aggregates may be targeted to mitochondria for degradation by mitochondrial proteases (137). Although these mechanisms remain to be further explored, it is interesting to consider why the cell may route damaged proteins to different compartments of the cell and what the physiological consequences may be in aging.
Global effects of a local process: the whole-organism impact of mitochondrial proteostasis
Aging is a phenotype visible to the naked eye but is intricately connected to mitochondrial dysfunction at the cellular and subcellular levels. Studies of mitochondrial proteostasis offer an interesting and clear example of this connection. A remarkable set of studies established that dysfunction in the ETC (81, 138–140) or induction of ROS (141) can lead to life span extension. Interestingly, there is only a small window during development in which this effect can occur (81), providing nuance to the long-standing hypothesis that the level of ROS generated over the lifetime of an organism directly correlates with its rate of aging (142). Rather, these newer findings suggest that during development, the organism senses energy-production capacity and permanently adjusts organismal physiology to ultimately impact life span. Connection between mitochondrial output and altered life span depends on two histone demethylases (69), suggesting that mitochondrial dysfunction in early development can alter the epigenomic landscape that regulates gene expression during adulthood.
These findings prompted the question of whether the entire organism or, rather, specific tissues are responsible for sensing mitochondrial health and orchestrating the downstream effects that impact life span. Remarkably, studies in C. elegans have shown that mitochondrial dysfunction in neurons alone is sufficient to account for the life span extension (58). Furthermore, up-regulation of the mtUPR in one tissue (neurons) was sufficient to activate the mtUPR in another (the intestine). These data point to a cell-nonautonomous mechanism for communication among mitochondria in separate tissues and prompted the hypothesis that neuronal mitochondria communicate as master regulators with the rest of the organism via a currently unidentified molecule termed a “mitokine” (58). Studies in C. elegans have proposed that this mitokine may derive from secreted neuropeptides (143, 144).
In mice, two separate instances of trans-cellular mitochondrial communication by mitokine have been reported that appear to rely on a mechanism distinct from the one observed in C. elegans. Specifically, a block in mitophagy in muscle cells caused these cells to secrete an FGF21 mitokine into the serum (145). Follow-up in C2C12 cells showed that FGF21 expression is induced as part of an ATF4-dependent integrated stress response, as the FGF21 promoter contains ATF4-binding sites (120). Independently, a GDF15 mitokine was shown to be released from muscle cells upon perturbation of mitochondrial translation or mutation of POLG, and it was confirmed in a muscle cell culture model that GDF15 is up-regulated in a CHOP-dependent but ATF4-independent manner (146). It was recently reported that patients with mtDNA mutations that cause mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) exhibit abnormally high levels of GDF15, which the authors suggest could be used as a biomarker for mitochondrial disease (147). At the organismal level, both of these muscle mitokines suppress sensitivity to insulin, highlighting the powerful impact of tissue-specific stress on the metabolic state of the entire organism. In a separate study, it was shown that mitochondrial dysfunction in Drosophila, when combined with mutation in Ras, leads to cell nonautonomous progression of tumor growth via the IL6 and WNT signaling pathways (148), suggesting that the mitochondria in the tumor microenvironment can impact severity of tumorigenesis.
Although the biochemical and physiological specifics of these three examples–from C. elegans, mice, and Drosophila–are distinct, they clearly demonstrate how mitochondria in a single tissue can communicate the state of their proteome across the organism. Thus, although the mitochondrion is not typically thought of as a major component of the endocrine system, these recent data prompt a new perspective of considering mitochondria in organismal signaling.
A separate cell-nonautonomous mechanism impacting mitochondrial proteostasis is the transfer of mitochondria between cells (149), suggesting that the mitochondrial proteostatic burden can be relieved through transfer or exchange of organelles between cells. For instance, it has been reported that healthy mitochondria can be sent through tunneling nanotubes from untreated PC12 cells to UV-irradiated cells to rescue these cells and avoid apoptosis (150). However, in a process termed trans-mitophagy, depolarized mitochondria can be extruded from optical ganglion cells at the optic nerve head and subsequently engulfed and degraded by surrounding astrocytes (151). It is an interesting notion that in diseases such as PD, in which mitophagy is an important regulator of disease progression, trans-mitophagy may also play a role.
Concluding remarks
The phrase “the mitochondrion is the powerhouse of the cell” is both an educational truism and a misrepresentation of the sophistication of this organelle. In reality, the mitochondrion is not an isolated factory, tirelessly and reliably feeding energy to the rest of the cell. Instead, its function is tightly integrated with that of the rest of the cell, and a set of dedicated circuits has evolved to sense and resolve mitochondrial stress. Failure to do so, whether due to genetic or environmental insults, has profound effects on the entire organism.
At the foundation of robust cell function is a set of mechanisms for surveillance of mitochondrial proteomic integrity. These mechanisms face unique challenges because of the physical and genetic separation of the mitochondrion from the rest of the cell. As a consequence, not one but multiple mechanisms at different layers of biological complexity have evolved to regulate mitochondrial proteostasis. Initial control is imposed by intra-organellar machinery composed of a network of chaperones and proteases that fold and degrade mitochondrial proteins and complexes, as well as coordinated mitochondrial dynamics operating through fission and fusion.
Importantly, mitochondrial proteostasis is also maintained by factors beyond the organelle: coordination with other organelles is crucial in maintaining mitochondrial and cellular health. These recent discoveries point to a new perspective of mitochondria as not only a regulator of metabolism but also of cellular proteostasis. Genetic evidence connects dysfunction within every tier of mitochondrial proteostasis to aging and age-related disease, especially to those of the central nervous system. One challenge is to understand why such defects in mitochondrial proteostasis, which presumably occur systemically, can have such tissue-specific effects. Equally compelling is the emerging role for mitochondrial proteomic health in the progression of certain cancers (152, 153). In principle, these mitochondrial proteostasis pathways contain a wealth of druggable circuits. Given how integrated the mitochondrion is into overall cell and organismal function, an additional challenge is to understand how to selectively regulate mitochondrial function without unwanted cellular and organismal effects. Finally, recent evidence on the cell nonautonomous coordination of mitochondrial proteostasis suggests that assessing the role of mitochondrial dysfunction in aging requires an understanding of the mitochondrial signaling circuits that operate across different tissues. Implicit in this line of reasoning is the tantalizing notion that targeted control over mitochondrial proteostasis in one tissue may result in a systemic improvement in health.
Acknowledgments
We acknowledge Hope Henderson and Dr. Ryo Higuchi-Sanabria for their insightful comments on the manuscript and Drs. Andrew Murley, Jenni Durieux, and Joseph Daniele for helpful conversation. We apologize for the inability to cite the literature more exhaustively due to space limitations.
Note added in proof
While this manuscript was under review, Zhang et al. (156) reported that neuronal mitochondrial stress can be communicated via the canonical Wnt pathway to distal tissue.
The authors declare that they have no conflicts of interest with the contents of this article.
- OXPHOS
- oxidative phosphorylation
- ROS
- reactive oxygen species
- ETC
- electron transport chain
- PD
- Parkinson's disease
- IMS
- intermembrane space
- AD
- Alzheimer's disease
- mtUPR
- mitochondrial unfolded protein response
- ER–UPR
- unfolded protein response of the ER
- ER
- endoplasmic reticulum
- MDV
- mitochondria-derived vesicle
- MDC
- mitochondria-derived compartment
- MAM
- mitochondria-associated ER membrane
- MCSR
- mitochondrial-to-cytosolic stress response.
References
- 1. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S.-E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., and Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dick F. D. (2006) Parkinson's disease and pesticide exposures. Br. Med. Bull. 79–80, 219–231 [DOI] [PubMed] [Google Scholar]
- 3. Nyström T. (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24, 1311–1317 10.1038/sj.emboj.7600599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corral-Debrinski M., Stepien G., Shoffner J. M., Lott M. T., Kanter K., and Wallace D. C. (1991) Hypoxemia is associated with mitochondrial DNA damage and gene induction. Implications for cardiac disease. JAMA 266, 1812–1816 10.1001/jama.1991.03470130092035 [DOI] [PubMed] [Google Scholar]
- 5. Corral-Debrinski M., Horton T., Lott M. T., Shoffner J. M., McKee A. C., Beal M. F., Graham B. H., and Wallace D. C. (1994) Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 23, 471–476 10.1006/geno.1994.1525 [DOI] [PubMed] [Google Scholar]
- 6. Coskun P. E., Beal M. F., and Wallace D. C. (2004) Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. U.S.A. 101, 10726–10731 10.1073/pnas.0403649101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahlqvist K. J., Hämäläinen R. H., Yatsuga S., Uutela M., Terzioglu M., Götz A., Forsström S., Salven P., Angers-Loustau A., Kopra O. H., Tyynismaa H., Larsson N.-G., Wartiovaara K., Prolla T., Trifunovic A., and Suomalainen A. (2012) Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in polg mutator mice. Cell Metab. 15, 100–109 10.1016/j.cmet.2011.11.012 [DOI] [PubMed] [Google Scholar]
- 8. Ross J. M., Stewart J. B., Hagström E., Brené S., Mourier A., Coppotelli G., Freyer C., Lagouge M., Hoffer B. J., Olson L., and Larsson N.-G. (2013) Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415 10.1038/nature12474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brandt T., Mourier A., Tain L. S., Partridge L., Larsson N.-G., and Kühlbrandt W. (2017) Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. eLife 6, e24662 10.7554/eLife.24662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kraytsberg Y., Kudryavtseva E., McKee A. C., Geula C., Kowall N. W., and Khrapko K. (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520 10.1038/ng1778 [DOI] [PubMed] [Google Scholar]
- 11. Bender A., Krishnan K. J., Morris C. M., Taylor G. A., Reeve A. K., Perry R. H., Jaros E., Hersheson J. S., Betts J., Klopstock T., Taylor R. W., and Turnbull D. M. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517 10.1038/ng1769 [DOI] [PubMed] [Google Scholar]
- 12. Luoma P., Melberg A., Rinne J. O., Kaukonen J. A., Nupponen N. N., Chalmers R. M., Oldfors A., Rautakorpi I., Peltonen L., Majamaa K., Somer H., and Suomalainen A. (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet 364, 875–882 10.1016/S0140-6736(04)16983-3 [DOI] [PubMed] [Google Scholar]
- 13. Stumpf J. D., Saneto R. P., and Copeland W. C. (2013) Clinical and molecular features of POLG-related mitochondrial disease. Cold Spring Harb. Perspect. Biol. 5, a011395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schneider H. C., Berthold J., Bauer M. F., Dietmeier K., Guiard B., Brunner M., and Neupert W. (1994) Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 371, 768–774 10.1038/371768a0 [DOI] [PubMed] [Google Scholar]
- 15. Ostermann J., Horwich A. L., Neupert W., and Hartl F. U. (1989) Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature 341, 125–130 10.1038/341125a0 [DOI] [PubMed] [Google Scholar]
- 16. Höhfeld J., and Hartl F. U. (1994) Role of the chaperonin cofactor Hsp10 in protein folding and sorting in yeast mitochondria. J. Cell Biol. 126, 305–315 10.1083/jcb.126.2.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Felts S. J., Owen B. A., Nguyen P., Trepel J., Donner D. B., and Toft D. O. (2000) The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 275, 3305–3312 10.1074/jbc.275.5.3305 [DOI] [PubMed] [Google Scholar]
- 18. Weisiger R. A., and Fridovich I. (1973) Mitochondrial superoxide dismutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 248, 4793–4796 [PubMed] [Google Scholar]
- 19. Sturtz L. A., Diekert K., Jensen L. T., Lill R., and Culotta V. C. (2001) A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria: a physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089 [DOI] [PubMed] [Google Scholar]
- 20. Augustin S., Nolden M., Müller S., Hardt O., Arnold I., and Langer T. (2005) Characterization of peptides released from mitochondria. J. Biol. Chem. 280, 2691–2699 10.1074/jbc.M410609200 [DOI] [PubMed] [Google Scholar]
- 21. Baker M. J., Tatsuta T., and Langer T. (2011) Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 3, a007559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jenkinson E. M., Rehman A. U., Walsh T., Clayton-Smith J., Lee K., Morell R. J., Drummond M. C., Khan S. N., Naeem M. A., Rauf B., Billington N., Schultz J. M., Urquhart J. E., Lee M. K., Berry A., et al. (2013) Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 92, 605–613 10.1016/j.ajhg.2013.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R., Dürr A., Fontaine B., and Ballabio A. (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93, 973–983 10.1016/S0092-8674(00)81203-9 [DOI] [PubMed] [Google Scholar]
- 24. Hansen J. J., Dürr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M. N., Davoine C.-S., Brice A., Fontaine B., Gregersen N., and Bross P. (2002) Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 70, 1328–1332 10.1086/339935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Bella D., Lazzaro F., Brusco A., Plumari M., Battaglia G., Pastore A., Finardi A., Cagnoli C., Tempia F., Frontali M., Veneziano L., Sacco T., Boda E., Brussino A., Bonn F., et al. (2010) Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 42, 313–321 10.1038/ng.544 [DOI] [PubMed] [Google Scholar]
- 26. Jin J., Hulette C., Wang Y., Zhang T., Pan C., Wadhwa R., and Zhang J. (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics 5, 1193–1204 10.1074/mcp.M500382-MCP200 [DOI] [PubMed] [Google Scholar]
- 27. Park S. J., Shin J. H., Jeong J. I., Song J. H., Jo Y. K., Kim E. S., Lee E. H., Hwang J. J., Lee E. K., Chung S. J., Koh J.-Y., Jo D.-G., and Cho D.-H. (2014) Down-regulation of mortalin exacerbates Aβ-mediated mitochondrial fragmentation and dysfunction. J. Biol. Chem. 289, 2195–2204 10.1074/jbc.M113.492587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kong P., Lei P., Zhang S., Li D., Zhao J., and Zhang B. (2018) Integrated microarray analysis provided a new insight of the pathogenesis of Parkinson's disease. Neurosci. Lett. 662, 51–58 10.1016/j.neulet.2017.09.051 [DOI] [PubMed] [Google Scholar]
- 29. Fitzgerald J. C., Zimprich A., Carvajal Berrio D. A., Schindler K. M., Maurer B., Schulte C., Bus C., Hauser A.-K., Kübler M., Lewin R., Bobbili D. R., Schwarz L. M., Vartholomaiou E., Brockmann K., Wüst R., et al. (2017) Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson's disease. Brain 140, 2444–2459 10.1093/brain/awx202 [DOI] [PubMed] [Google Scholar]
- 30. De Leo M. E., Borrello S., Passantino M., Palazzotti B., Mordente A., Daniele A., Filippini V., Galeotti T., and Masullo C. (1998) Oxidative stress and overexpression of manganese superoxide dismutase in patients with Alzheimer's disease. Neurosci. Lett. 250, 173–176 10.1016/S0304-3940(98)00469-8 [DOI] [PubMed] [Google Scholar]
- 31. Marttila R. J., Lorentz H., and Rinne U. K. (1988) Oxygen toxicity protecting enzymes in Parkinson's disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J. Neurol. Sci. 86, 321–331 10.1016/0022-510X(88)90108-6 [DOI] [PubMed] [Google Scholar]
- 32. Bota D. A., Ngo J. K., and Davies K. J. (2005) Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 38, 665–677 10.1016/j.freeradbiomed.2004.11.017 [DOI] [PubMed] [Google Scholar]
- 33. Przybysz A. J., Choe K. P., Roberts L. J., and Strange K. (2009) Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech. Ageing Dev. 130, 357–369 10.1016/j.mad.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walther D. M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R. I., Dobson C. M., Vendruscolo M., Mann M., and Hartl F. U. (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932 10.1016/j.cell.2015.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cole A., Wang Z., Coyaud E., Voisin V., Gronda M., Jitkova Y., Mattson R., Hurren R., Babovic S., Maclean N., Restall I., Wang X., Jeyaraju D. V., Sukhai M. A., Prabha S., et al. (2015) Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27, 864–876 10.1016/j.ccell.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seo J. H., Rivadeneira D. B., Caino M. C., Chae Y. C., Speicher D. W., Tang H.-Y., Vaira V., Bosari S., Palleschi A., Rampini P., Kossenkov A. V., Languino L. R., and Altieri D. C. (2016) The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 14, e1002507 10.1371/journal.pbio.1002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meeusen S., McCaffery J. M., and Nunnari J. (2004) Mitochondrial fusion intermediates revealed in vitro. Science 305, 1747–1752 10.1126/science.1100612 [DOI] [PubMed] [Google Scholar]
- 38. Koshiba T., Detmer S. A., Kaiser J. T., Chen H., McCaffery J. M., and Chan D. C. (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862 10.1126/science.1099793 [DOI] [PubMed] [Google Scholar]
- 39. Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G. V., Rudka T., Bartoli D., Polishuck R. S., Danial N. N., De Strooper B., and Scorrano L. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 126, 177–189 10.1016/j.cell.2006.06.025 [DOI] [PubMed] [Google Scholar]
- 40. Tondera D., Grandemange S., Jourdain A., Karbowski M., Mattenberger Y., Herzig S., Da Cruz S., Clerc P., Raschke I., Merkwirth C., Ehses S., Krause F., Chan D. C., Alexander C., Bauer C., et al. (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600 10.1038/emboj.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gomes L. C., Di Benedetto G., and Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 10.1038/ncb2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tatsuta T., and Langer T. (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314 10.1038/sj.emboj.7601972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mozdy A. D., McCaffery J. M., and Shaw J. M. (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151, 367–380 10.1083/jcb.151.2.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tieu Q., and Nunnari J. (2000) Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 151, 353–366 10.1083/jcb.151.2.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Westermann B. (2010) Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- 46. Yoon Y., Krueger E. W., Oswald B. J., and McNiven M. A. (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23, 5409–5420 10.1128/MCB.23.15.5409-5420.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gandre-Babbe S., and van der Bliek A. M. (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 19, 2402–2412 10.1091/mbc.e07-12-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu R., and Chan D. C. (2015) The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 26, 4466–4477 10.1091/mbc.E15-08-0591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quirós P. M., Ramsay A. J., Sala D., Fernández-Vizarra E., Rodríguez F., Peinado J. R., Fernández-García M. S., Vega J. A., Enríquez J. A., Zorzano A., and López-Otín C. (2012) Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 31, 2117–2133 10.1038/emboj.2012.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baker M. J., Lampe P. A., Stojanovski D., Korwitz A., Anand R., Tatsuta T., and Langer T. (2014) Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 33, 578–593 10.1002/embj.201386474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jagasia R., Grote P., Westermann B., and Conradt B. (2005) DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 433, 754–760 10.1038/nature03316 [DOI] [PubMed] [Google Scholar]
- 52. Alexander C., Votruba M., Pesch U. E., Thiselton D. L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., Bhattacharya S. S., and Wissinger B. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215 10.1038/79944 [DOI] [PubMed] [Google Scholar]
- 53. Lawson V. H., Graham B. V., and Flanigan K. M. (2005) Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 65, 197–204 10.1212/01.wnl.0000168898.76071.70 [DOI] [PubMed] [Google Scholar]
- 54. Chen H., McCaffery J. M., and Chan D. C. (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562 10.1016/j.cell.2007.06.026 [DOI] [PubMed] [Google Scholar]
- 55. Lee J. S., Yoon Y. G., Yoo S. H., Jeong N. Y., Jeong S. H., Lee S. Y., Jung D.-I., Jeong S.-Y., and Yoo Y. H. (2012) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. J. Cell. Physiol. 227, 2856–2869 10.1002/jcp.23027 [DOI] [PubMed] [Google Scholar]
- 56. Pham A. H., Meng S., Chu Q. N., and Chan D. C. (2012) Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826 10.1093/hmg/dds311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao Q., Wang J., Levichkin I. V., Stasinopoulos S., Ryan M. T., and Hoogenraad N. J. (2002) A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419 10.1093/emboj/cdf445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Durieux J., Wolff S., and Dillin A. (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 10.1016/j.cell.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Owusu-Ansah E., Song W., and Perrimon N. (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712 10.1016/j.cell.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Houtkooper R. H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R. W., and Auwerx J. (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kozutsumi Y., Segal M., Normington K., Gething M. J., and Sambrook J. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- 62. Nargund A. M., Fiorese C. J., Pellegrino M. W., Deng P., and Haynes C. M. (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR. Mol. Cell 58, 123–133 10.1016/j.molcel.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martinus R. D., Garth G. P., Webster T. L., Cartwright P., Naylor D. J., Høj P. B., and Hoogenraad N. J. (1996) Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240, 98–103 10.1111/j.1432-1033.1996.0098h.x [DOI] [PubMed] [Google Scholar]
- 64. Yoneda T., Benedetti C., Urano F., Clark S. G., Harding H. P., and Ron D. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066 10.1242/jcs.01275 [DOI] [PubMed] [Google Scholar]
- 65. Münch C., and Harper J. W. (2016) Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534, 710–713 10.1038/nature18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Haynes C. M., Petrova K., Benedetti C., Yang Y., and Ron D. (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480 10.1016/j.devcel.2007.07.016 [DOI] [PubMed] [Google Scholar]
- 67. Fiorese C. J., and Haynes C. M. (2017) Integrating the UPR(mt) into the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 52, 304–313 10.1080/10409238.2017.1291577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nargund A. M., Pellegrino M. W., Fiorese C. J., Baker B. M., and Haynes C. M. (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 10.1126/science.1223560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S. D., Quiros P. M., Steffen K. K., Williams E. G., Mouchiroud L., Tronnes S. U., Murillo V., Wolff S. C., Shaw R. J., Auwerx J., and Dillin A. (2016) Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 10.1016/j.cell.2016.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Benedetti C., Haynes C. M., Yang Y., Harding H. P., and Ron D. (2006) Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239 10.1534/genetics.106.061580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tian Y., Garcia G., Bian Q., Steffen K. K., Joe L., Wolff S., Meyer B. J., and Dillin A. (2016) Mitochondrial stress induces chromatin reorganization to promote longevity and UPR. Cell 165, 1197–1208 10.1016/j.cell.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fiorese C. J., Schulz A. M., Lin Y.-F., Rosin N., Pellegrino M. W., and Haynes C. M. (2016) The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043 10.1016/j.cub.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aldridge J. E., Horibe T., and Hoogenraad N. J. (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2, e874 10.1371/journal.pone.0000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Teske B. F., Fusakio M. E., Zhou D., Shan J., McClintick J. N., Kilberg M. S., and Wek R. C. (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 24, 2477–2490 10.1091/mbc.e13-01-0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Baker B. M., Nargund A. M., Sun T., and Haynes C. M. (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8, e1002760 10.1371/journal.pgen.1002760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Quirós P. M., Prado M. A., Zamboni N., D'Amico D., Williams R. W., Finley D., Gygi S. P., and Auwerx J. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045 10.1083/jcb.201702058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Papa L., and Germain D. (2011) Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396–1402 10.1242/jcs.078220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Borch Jensen M., Qi Y., Riley R., Rabkina L., and Jasper H. (2017) PGAM5 promotes lasting FoxO activation after developmental mitochondrial stress and extends life span in Drosophila. eLife 6, e26952 10.7554/eLife.26952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bhaskaran S., Pharaoh G., Ranjit R., Murphy A., Matsuzaki S., Nair B. C., Forbes B., Gispert S., Auburger G., Humphries K. M., Kinter M., Griffin T. M., and Deepa S. S. (2018) Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 19, e45009 10.15252/embr.201745009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C. M., and Chen D. (2015) A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 10.1126/science.aaa2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dillin A., Hsu A.-L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A. G., Kamath R. S., Ahringer J., and Kenyon C. (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 10.1126/science.1077780 [DOI] [PubMed] [Google Scholar]
- 82. Lee S. S., Lee R. Y., Fraser A. G., Kamath R. S., Ahringer J., and Ruvkun G. (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48 [DOI] [PubMed] [Google Scholar]
- 83. Martinez B. A., Petersen D. A., Gaeta A. L., Stanley S. P., Caldwell G. A., and Caldwell K. A. (2017) Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson's disease. J. Neurosci. 37, 11085–11100 10.1523/JNEUROSCI.1294-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Legros F., Lombès A., Frachon P., and Rojo M. (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 13, 4343–4354 10.1091/mbc.e02-06-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., et al. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pellegrino M. W., and Haynes C. M. (2015) Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 13, 22 10.1186/s12915-015-0129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang H., Bosch-Marce M., Shimoda L. A., Tan Y. S., Baek J. H., Wesley J. B., Gonzalez F. J., and Semenza G. L. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 10.1074/jbc.M800102200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 88. Bertolin G., Ferrando-Miguel R., Jacoupy M., Traver S., Grenier K., Greene A. W., Dauphin A., Waharte F., Bayot A., Salamero J., Lombès A., Bulteau A.-L., Fon E. A., Brice A., and Corti O. (2013) The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 9, 1801–1817 [DOI] [PubMed] [Google Scholar]
- 89. Jin S. M., and Youle R. J. (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Okamoto K., Kondo-Okamoto N., and Ohsumi Y. (2009) Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 17, 87–97 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- 91. Kanki T., Wang K., Cao Y., Baba M., and Klionsky D. J. (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schweers R. L., Zhang J., Randall M. S., Loyd M. R., Li W., Dorsey F. C., Kundu M., Opferman J. T., Cleveland J. L., Miller J. L., and Ney P. A. (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505 10.1073/pnas.0708818104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sandoval H., Thiagarajan P., Dasgupta S. K., Schumacher A., Prchal J. T., Chen M., and Wang J. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235 10.1038/nature07006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Novak I., Kirkin V., McEwan D. G., Zhang J., Wild P., Rozenknop A., Rogov V., Löhr F., Popovic D., Occhipinti A., Reichert A. S., Terzic J., Dötsch V., Ney P. A., and Dikic I. (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51 10.1038/embor.2009.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bruick R. K. (2000) Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. U.S.A. 97, 9082–9087 10.1073/pnas.97.16.9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cookson M. R. (2012) Parkinsonism due to mutations in PINK1, Parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb. Perspect. Med. 2, a009415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thomas R. E., Andrews L. A., Burman J. L., Lin W.-Y., and Pallanck L. J. (2014) PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 10, e1004279 10.1371/journal.pgen.1004279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Greene A. W., Grenier K., Aguileta M. A., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., and Fon E. A. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 10.1038/embor.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., and Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yamano K., and Youle R. J. (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hasson S. A., Kane L. A., Yamano K., Huang C.-H., Sliter D. A., Buehler E., Wang C., Heman-Ackah S. M., Hessa T., Guha R., Martin S. E., and Youle R. J. (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504, 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Narendra D., Tanaka A., Suen D.-F., and Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Narendra D. P., Jin S. M., Tanaka A., Suen D.-F., Gautier C. A., Shen J., Cookson M. R., and Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298 10.1371/journal.pbio.1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y.-S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., and Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., and Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- 106. Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y.-S., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., and Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Palikaras K., Lionaki E., and Tavernarakis N. (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 10.1038/nature14300 [DOI] [PubMed] [Google Scholar]
- 108. Kitada T., Pisani A., Porter D. R., Yamaguchi H., Tscherter A., Martella G., Bonsi P., Zhang C., Pothos E. N., and Shen J. (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446 10.1073/pnas.0702717104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Warren G., and Wickner W. (1996) Organelle inheritance. Cell 84, 395–400 10.1016/S0092-8674(00)81284-2 [DOI] [PubMed] [Google Scholar]
- 110. Rohn J. L., Patel J. V., Neumann B., Bulkescher J., Mchedlishvili N., McMullan R. C., Quintero O. A., Ellenberg J., and Baum B. (2014) Myo19 ensures symmetric partitioning of mitochondria and coupling of mitochondrial segregation to cell division. Curr. Biol. 24, 2598–2605 10.1016/j.cub.2014.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McFaline-Figueroa J. R., Vevea J., Swayne T. C., Zhou C., Liu C., Leung G., Boldogh I. R., and Pon L. A. (2011) Mitochondrial quality control during inheritance is associated with life span and mother-daughter age asymmetry in budding yeast. Aging Cell 10, 885–895 10.1111/j.1474-9726.2011.00731.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Katajisto P., Döhla J., Chaffer C. L., Pentinmikko N., Marjanovic N., Iqbal S., Zoncu R., Chen W., Weinberg R. A., and Sabatini D. M. (2015) Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343 10.1126/science.1260384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lai C.-Y., Jaruga E., Borghouts C., and Jazwinski S. M. (2002) A mutation in the ATP2 gene abrogates the age asymmetry between mother and daughter cells of the yeast Saccharomyces cerevisiae. Genetics 162, 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Klinger H., Rinnerthaler M., Lam Y. T., Laun P., Heeren G., Klocker A., Simon-Nobbe B., Dickinson J. R., Dawes I. W., and Breitenbach M. (2010) Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells. Exp. Gerontol. 45, 533–542 10.1016/j.exger.2010.03.016 [DOI] [PubMed] [Google Scholar]
- 115. Higuchi R., Vevea J. D., Swayne T. C., Chojnowski R., Hill V., Boldogh I. R., and Pon L. A. (2013) Actin dynamics affect mitochondrial quality control and aging in budding yeast. Curr. Biol. 23, 2417–2422 10.1016/j.cub.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Higuchi-Sanabria R., Charalel J. K., Viana M. P., Garcia E. J., Sing C. N., Koenigsberg A., Swayne T. C., Vevea J. D., Boldogh I. R., Rafelski S. M., and Pon L. A. (2016) Mitochondrial anchorage and fusion contribute to mitochondrial inheritance and quality control in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 27, 776–787 10.1091/mbc.E15-07-0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rujano M. A., Bosveld F., Salomons F. A., Dijk F., van Waarde M. A., van der Want J. J., de Vos R. A., Brunt E. R., Sibon O. C., and Kampinga H. H. (2006) Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 4, e417 10.1371/journal.pbio.0040417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hernebring M., Fredriksson Å., Liljevald M., Cvijovic M., Norrman K., Wiseman J., Semb H., and Nyström T. (2013) Removal of damaged proteins during ES cell fate specification requires the proteasome activator PA28. Sci. Rep. 3, 1381 10.1038/srep01381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Neuspiel M., Schauss A. C., Braschi E., Zunino R., Rippstein P., Rachubinski R. A., Andrade-Navarro M. A., and McBride H. M. (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108 10.1016/j.cub.2007.12.038 [DOI] [PubMed] [Google Scholar]
- 120. Soubannier V., McLelland G.-L., Zunino R., Braschi E., Rippstein P., Fon E. A., and McBride H. M. (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141 10.1016/j.cub.2011.11.057 [DOI] [PubMed] [Google Scholar]
- 121. McLelland G.-L., Soubannier V., Chen C. X., McBride H. M., and Fon E. A. (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hughes A. L., Hughes C. E., Henderson K. A., Yazvenko N., and Gottschling D. E. (2016) Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife 5, e13943 10.7554/eLife.13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wong Y. C., Ysselstein D., and Krainc D. (2018) Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386 10.1038/nature25486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Calì T., Ottolini D., Negro A., and Brini M. (2012) α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929 10.1074/jbc.M111.302794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Area-Gomez E., and Schon E. A. (2016) Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 38, 90–96 10.1016/j.gde.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tubbs E., Theurey P., Vial G., Bendridi N., Bravard A., Chauvin M. A., Ji-Cao J., Zoulim F., Bartosch B., Ovize M., Vidal H., and Rieusset J. (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294 10.2337/db13-1751 [DOI] [PubMed] [Google Scholar]
- 127. Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., and Hajnóczky G. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 10.1083/jcb.200604016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Friedman J. R., Kannan M., Toulmay A., Jan C. H., Weissman J. S., Prinz W. A., and Nunnari J. (2018) Lipid homeostasis is maintained by dual targeting of the mitochondrial PE biosynthesis enzyme to the ER. Dev. Cell 44, 261–270.e6 10.1016/j.devcel.2017.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hirabayashi Y., Kwon S.-K., Paek H., Pernice W. M., Paul M. A., Lee J., Erfani P., Raczkowski A., Petrey D. S., Pon L. A., and Polleux F. (2017) ER–mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630 10.1126/science.aan6009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhou C., Slaughter B. D., Unruh J. R., Guo F., Yu Z., Mickey K., Narkar A., Ross R. T., McClain M., and Li R. (2014) Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159, 530–542 10.1016/j.cell.2014.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Friedman J. R., Lackner L. L., West M., DiBenedetto J. R., Nunnari J., and Voeltz G. K. (2011) ER tubules mark sites of mitochondrial division. Science 334, 358–362 10.1126/science.1207385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lewis S. C., Uchiyama L. F., and Nunnari J. (2016) ER–mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549 10.1126/science.aaf5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S., and Walter P. (2009) An ER–mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 10.1126/science.1175088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kim H.-E., Grant A. R., Simic M. S., Kohnz R. A., Nomura D. K., Durieux J., Riera C. E., Sanchez M., Kapernick E., Wolff S., and Dillin A. (2016) Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. Cell 166, 1539–1552.e16 10.1016/j.cell.2016.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Neutzner A., Youle R., and Karbowski M. (2007) Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found. Symp. 287, 4–14 [PubMed] [Google Scholar]
- 136. Taylor E. B., and Rutter J. (2011) Mitochondrial quality control by the ubiquitin–proteasome system. Biochem. Soc. Trans. 39, 1509–1513 10.1042/BST0391509 [DOI] [PubMed] [Google Scholar]
- 137. Ruan L., Zhou C., Jin E., Kucharavy A., Zhang Y., Wen Z., Florens L., and Li R. (2017) Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446 10.1038/nature21695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Feng J., Bussière F., and Hekimi S. (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644 10.1016/S1534-5807(01)00071-5 [DOI] [PubMed] [Google Scholar]
- 139. Dell'agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., Prelle A., Roubertoux P., Rizzuto R., and Zeviani M. (2007) Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444 10.1093/hmg/ddl477 [DOI] [PubMed] [Google Scholar]
- 140. Copeland J. M., Cho J., Lo T. Jr, Hur J. H., Bahadorani S., Arabyan T., Rabie J., Soh J., and Walker D. W. (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598 10.1016/j.cub.2009.08.016 [DOI] [PubMed] [Google Scholar]
- 141. Schroeder E. A., Raimundo N., and Shadel G. S. (2013) Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 17, 954–964 10.1016/j.cmet.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Harman D. (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 10.1093/geronj/11.3.298 [DOI] [PubMed] [Google Scholar]
- 143. Berendzen K. M., Durieux J., Shao L.-W., Tian Y., Kim H.-E., Wolff S., Liu Y., and Dillin A. (2016) Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166, 1553–1563.e10 10.1016/j.cell.2016.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Shao L.-W., Niu R., and Liu Y. (2016) Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 26, 1182–1196 10.1038/cr.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kim K. H., Jeong Y. T., Oh H., Kim S. H., Cho J. M., Kim Y.-N., Kim S. S., Do Hoon Kim Hur K. Y., Kim H. K., Ko T., Han J., Kim H. L., Kim J., Back S. H., Komatsu M., et al. (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19, 83–92 [DOI] [PubMed] [Google Scholar]
- 146. Chung H. K., Ryu D., Kim K. S., Chang J. Y., Kim Y. K., Yi H.-S., Kang S. G., Choi M. J., Lee S. E., Jung S.-B., Ryu M. J., Kim S. J., Kweon G. R., Kim H., Hwang J. H., et al. (2017) Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165 10.1083/jcb.201607110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Fujita Y., Ito M., Kojima T., Yatsuga S., Koga Y., and Tanaka M. (2015) GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion 20, 34–42 10.1016/j.mito.2014.10.006 [DOI] [PubMed] [Google Scholar]
- 148. Ohsawa S., Sato Y., Enomoto M., Nakamura M., Betsumiya A., and Igaki T. (2012) Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490, 547–551 [DOI] [PubMed] [Google Scholar]
- 149. Rustom A., Saffrich R., Markovic I., Walther P., and Gerdes H.-H. (2004) Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010 10.1126/science.1093133 [DOI] [PubMed] [Google Scholar]
- 150. Wang X., and Gerdes H.-H. (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 22, 1181–1191 10.1038/cdd.2014.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Davis C.-H., Kim K.-Y., Bushong E. A., Mills E. A., Boassa D., Shih T., Kinebuchi M., Phan S., Zhou Y., Bihlmeyer N. A., Nguyen J. V., Jin Y., Ellisman M. H., and Marsh-Armstrong N. (2014) Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 111, 9633–9638 10.1073/pnas.1404651111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Denison S. R., Wang F., Becker N. A., Schüle B., Kock N., Phillips L. A., Klein C., and Smith D. I. (2003) Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22, 8370–8378 10.1038/sj.onc.1207072 [DOI] [PubMed] [Google Scholar]
- 153. Kang B. H., Plescia J., Dohi T., Rosa J., Doxsey S. J., and Altieri D. C. (2007) Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 131, 257–270 10.1016/j.cell.2007.08.028 [DOI] [PubMed] [Google Scholar]
- 154. Qureshi M. A., Haynes C. M., and Pellegrino M. W. (2017) The mitochondrial unfolded protein response: signaling from the powerhouse. J. Biol. Chem. 292, 13500–13506 10.1074/jbc.R117.791061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Shpilka T., and Haynes C. M. (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120 10.1038/nrm.2017.110 [DOI] [PubMed] [Google Scholar]
- 156. Zhang Q., Wu X., Chen P., Liu L., Xin N., Tian Y., and Dillin A. (2018) The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174, 870–883.e17 10.1016/j.cell.2018.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]