

Abstract
Background
Cutaneous leishmaniasis, caused by a parasitic infection, is considered one of the most serious skin diseases in many low‐ and middle‐income countries. Old World cutaneous leishmaniasis (OWCL) is caused by species found in Africa, Asia, the Middle East, the Mediterranean, and India. The most commonly prescribed treatments are antimonials, but other drugs have been used with varying success. As OWCL tends to heal spontaneously, it is necessary to justify the use of systemic and topical treatments. This is an update of a Cochrane Review first published in 2008.
Objectives
To assess the effects of therapeutic interventions for the localised form of Old World cutaneous leishmaniasis.
Search methods
We updated our searches of the following databases to November 2016: the Cochrane Skin Specialised Register, CENTRAL, MEDLINE, Embase, and LILACS. We also searched five trials registers and checked the reference lists of included studies for further references to relevant randomised controlled trials (RCTs). We wrote to national programme managers, general co‐ordinators, directors, clinicians, WHO‐EMRO regional officers of endemic countries, pharmaceutical companies, tropical medicine centres, and authors of relevant papers for further information about relevant unpublished and ongoing trials. We undertook a separate search for adverse effects of interventions for Old World cutaneous leishmaniasis in September 2015 using MEDLINE.
Selection criteria
Randomised controlled trials of either single or combination treatments in immunocompetent people with OWCL confirmed by smear, histology, culture, or polymerase chain reaction. The comparators were either no treatment, placebo/vehicle, and/or another active compound.
Data collection and analysis
Two review authors independently assessed trials for inclusion and risk of bias and extracted data. We only synthesised data when we were able to identify at least two studies investigating similar treatments and reporting data amenable to pooling. We also recorded data about adverse effects from the corresponding search.
Main results
We included 89 studies (of which 40 were new to this update) in 10,583 people with OWCL. The studies included were conducted mainly in the Far or Middle East at regional hospitals, local healthcare clinics, and skin disease research centres. Women accounted for 41.5% of the participants (range: 23% to 80%). The overall mean age of participants was 25 years (range 12 to 56). Most studies lasted between two to six months, with the longest lasting two years; average duration was four months. Most studies were at unclear or high risk for most bias domains. A lack of blinding and reporting bias were present in almost 40% of studies. Two trials were at low risk of bias for all domains. Trials reported the causative species poorly.
Here we provide results for the two main comparisons identified: itraconazole (200 mg for six to eight weeks) versus placebo; and paromomycin ointment (15% plus 10% urea, twice daily for 14 days) versus vehicle.
In the comparison of oral itraconazole versus placebo, at 2.5 months' follow up, 85/125 participants in the itraconazole group achieved complete cure compared to 54/119 in the placebo group (RR 3.70, 95% CI 0.35 to 38.99; 3 studies; 244 participants). In one study, microbiological or histopathological cure of skin lesions only occurred in the itraconazole group after a mean follow‐up of 2.5 months (RR 17.00, 95% CI 0.47 to 612.21; 20 participants). However, although the analyses favour oral itraconazole for these outcomes, we cannot be confident in the results due to the very low certainty evidence. More side effects of mild abdominal pain and nausea (RR 2.36, 95% CI 0.74 to 7.47; 3 studies; 204 participants) and mild abnormal liver function (RR 3.08, 95% CI 0.53 to 17.98; 3 studies; 84 participants) occurred in the itraconazole group (as well as reports of headaches and dizziness), compared with the placebo group, but again we rated the certainty of evidence as very low so are unsure of the results.
When comparing paromomycin with vehicle, there was no difference in the number of participants who achieved complete cure (RR of 1.00, 95% CI 0.86, 1.17; 383 participants, 2 studies) and microbiological or histopathological cure of skin lesions after a mean follow‐up of 2.5 months (RR 1.03, CI 0.88 to 1.20; 383 participants, 2 studies), but the paromomycin group had more skin/local reactions (such as inflammation, vesiculation, pain, redness, or itch) (RR 1.42, 95% CI 0.67 to 3.01; 4 studies; 713 participants). For all of these outcomes, the certainty of evidence was very low, meaning we are unsure about these results.
Trial authors did not report the percentage of lesions cured after the end of treatment or speed of healing for either of these key comparisons.
Authors' conclusions
There was very low‐certainty evidence to support the effectiveness of itraconazole and paromomycin ointment for OWCL in terms of cure (i.e. microbiological or histopathological cure and percentage of participants completely cured). Both of these interventions incited more adverse effects, which were mild in nature, than their comparisons, but we could draw no conclusions regarding safety due to the very low certainty of the evidence for this outcome.
We downgraded the key outcomes in these two comparisons due to high risk of bias, inconsistency between the results, and imprecision. There is a need for large, well‐designed international studies that evaluate long‐term effects of current therapies and enable a reliable conclusion about treatments. Future trials should specify the species of leishmaniasis; trials on types caused by Leishmania infantum, L aethiopica, andL donovani are lacking. Research into the effects of treating women of childbearing age, children, people with comorbid conditions, and those who are immunocompromised would also be helpful.
It was difficult to evaluate the overall efficacy of any of the numerous treatments due to the variable treatment regimens examined and because RCTs evaluated different Leishmania species and took place in different geographical areas. Some outcomes we looked for but did not find were degree of functional and aesthetic impairment, change in ability to detect Leishmania, quality of life, and emergence of resistance. There were only limited data on prevention of scarring.
Keywords: Animals; Humans; Anti‐Infective Agents; Anti‐Infective Agents/therapeutic use; Antiprotozoal Agents; Antiprotozoal Agents/therapeutic use; Complementary Therapies; Cryotherapy; Hot Temperature; Hot Temperature/therapeutic use; Laser Therapy; Leishmania major; Leishmania tropica; Leishmaniasis, Cutaneous; Leishmaniasis, Cutaneous/therapy; Photochemotherapy; Randomized Controlled Trials as Topic
Treatments for Old World cutaneous leishmaniasis
Background
Old World cutaneus leishmaniasis (OWCL) is an infection caused by the Leishmania parasite, which is passed onto humans by the bite of sandflies. It is a serious skin disease associated with a broad range of signs, symptoms, and degrees of severity. We wanted to assess the competence and safety of all available treatments for OWCL.
Review question
We assessed participants with a healthy immune response who had OWCL diagnosed by laboratory methods. Treatments had to be given alone or in combination with another treatment, and they were compared against no treatment, placebo (an inactive substance) only, or another active treatment. Some of the main outcomes we were interested in included the percentage of wounds cured after the end of treatment, the number of participants completely cured after the end of treatment, speed of healing, side‐effects of treatment, and clearance of parasites (i.e. infection).
Study characteristics
We reviewed 89 clinical trials, which included 10,583 people, in total, with OWCL. We included participants of both sexes and all ages (mean 24.5 years); most participants were over 18 years of age. Most studies were carried out in single centres in different countries, mainly in the Far or Middle East, and lasted between two to six months. We included a variety of treatments, such as antimonials, antifungals, and antibiotics, which were administered either directly onto the skin or into a wound, taken by mouth, or physically applied (e.g. laser treatment, heat therapy, etc.). Most of the included studies assessed OWCL caused by two species of parasites known as Leishmania major (L. major) andLeishmania tropica (L. tropica).
Key results
The evidence is current to November 2016.
Two of the most important treatments that we assessed in this review were itraconazole, an antifungal drug taken by mouth, and paromomycin, an antibiotic applied as an ointment. Trials compared both to a placebo tablet or inactive cream (vehicle).
Participants received 200 mg itraconazole for six to eight weeks or paromomycin ointment at a concentration of 15% plus 10% urea, twice daily for 14 days.
When assessed on average 2.5 months after treatment, more participants were completely cured and cleared of the infection‐causing parasites with itraconazole than placebo, but they also had more side effects (mild stomach pain, sickness, and abnormal liver function, as well as headaches and dizziness).
When paromomycin ointment was compared with placebo, there was no difference in the number of completely cured participants or the number who were found to be cleared of parasites when assessed on average 2.5 months after treatment, but those in the paromomycin treatment group had more contained skin reactions (such as swelling, blistering, pain, redness, or itch).
However, as the certainty of the evidence for these outcomes for these particular comparisons was very low, we are not sure of the accuracy of these results.
Neither of our key treatment comparisons assessed the percentage of wounds cured after the end of treatment and speed of healing (i.e. time taken to be cured).
Quality of the evidence
The overall certainty of the evidence for the different outcomes in the two main comparisons was very low. Important reasons for this were that studies were not blinded, or had a small sample size, making the results less precise. Some of the evidence only focused on young people, and the results greatly varied between each study.
We need more research to fill in the following research gaps: 1) trials of OWCL caused by other types of infection such as L. infantum, L. aethiopica,or L. donovani; 2) involving specific subgroups of people such as children; 3) assessing effectiveness and safety of different anti‐Leishmania drugs compared with placebo in self‐healing forms of leishmaniasis or with traditional first‐choice antimonial treatment in complicated form (defined as more than four lesions over 4 cm in size, located close to an opening or small joints, for which previous treatment has failed); and 4) assessing areas such as wound healing and patient‐reported outcomes, such as quality of life. In addition, few studies assessed relevant issues such as drug resistance. International collaboration is required to improve the quality and standardisation of future trials in order to develop a better evidence‐based approach.
Summary of findings
Summary of findings for the main comparison.
Itraconazole (200 mg for 6 to 8 weeks) versus placebo for Old World cutaneous leishmaniasis
| Itraconazole (200 mg for 6‐8 weeks) versus placebo for Old World cutaneous leishmaniasis | ||||||
| Patient or population: patients with Old World cutaneous leishmaniasis Settings: Kuwait, India, and Iran Intervention: itraconazole (200 mg for 6‐8 weeks) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Itraconazole (200 mg for 6‐8 weeks) | |||||
| Percentage of lesions cured after the end of treatment | Not measured in this comparison | |||||
| Percentage of participants with complete cure Follow‐up: mean 2.5 months | Study population | RR 3.70 (0.35 to 38.99) | 244 (3 studies) | ⊕⊝⊝⊝ Very lowa | — | |
| 454 per 1000 | 1000 per 1000 (159 to 1000) | |||||
| Moderate | ||||||
| 100 per 1000 | 370 per 1000 (35 to 1000) | |||||
|
Adverse effects Mild abdominal pain and nausea Adverse effects Mild abnormal liver function |
40 per 1000 0 per 1000 |
95 per 1000 (30 to 302) 0 per 1000 (0 to 0) |
RR 2.36 (0.74 to 7.47) RR 3.08 (0.53 to 17.98) |
204 (3 studies) 84 (3 studies) |
⊕⊝⊝⊝ Very lowb ⊕⊝⊝⊝ Very lowc |
— |
| Speed of healing (time taken to be 'cured') | Neither of the studies reported speed of healing (time taken to be 'cured') in this comparison. | |||||
| Microbiological or histopathological cure of skin lesions Follow‐up: mean 2.5 months | Not estimable | Not estimable | RR 17.00 (0.47 to 612.21) | 20 (1 study) | ⊕⊝⊝⊝ Very lowd | There were zero events in the placebo group |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality/certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality/certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality/certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality/certainty: we are very uncertain about the estimate. | ||||||
aDowngraded by 4 levels due to: risk of bias (2 RCTs have many uncertain items), inconsistency (there is considerable heterogeneity ‐ I² = 73%), and imprecision (2 levels due to wide 95% confidence intervals, crossing the line of no effect). bDowngraded by 3 levels due to: risk of bias (many uncertain items in the risk of bias judgment), and imprecision (2 levels due to wide 95% confidence intervals, crossing the line of no effect). cDowngraded by 3 levels due to: risk of bias (many uncertain items in the risk of bias judgment), and imprecision (2 levels due to wide 95% confidence intervals, crossing the line of no effect). dDowngraded by 3 levels due to: risk of bias (many uncertain items in the risk of bias judgment), and imprecision (2 levels due to wide 95% confidence intervals; this outcome is only reported for one study involving 20 participants).
Summary of findings 2.
Paromomycin ointment versus vehicle for Old World cutaneous leishmaniasis
| Paromomycin ointment versus matched vehicle for Old World cutaneous leishmaniasis | |||||
| Patient or population: patients with Old World cutaneous leishmaniasis Settings: primary health centres, Iran and Tunisia Intervention: paromomycin ointment (15% + 10% urea) twice daily for 14 days Comparison: vehicle | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Vehicle | Paromomycin ointment (15% + 10% urea) twice daily for 14 days | ||||
| Percentage of lesions cured after the end of treatment | Not measured in this comparison | ||||
| Percentage of participants with complete cure Follow‐up: mean 2.5 months | Study population | RR 1.00 (0.86 to 1.17) | 383 (2 studies) | ⊕⊝⊝⊝ Very lowa | |
| 623 per 1000 | 623 per 1000 (536 to 729) | ||||
| Moderate | |||||
| 619 per 1000 | 619 per 1000 (532 to 724) | ||||
| Adverse effects Skin/local reactions | Study population | RR 1.42 (0.67 to 3.01) | 713 (4 studies) | ⊕⊝⊝⊝ Very lowb | |
| 96 per 1000 | 136 per 1000 (64 to 287) | ||||
| Moderate | |||||
| 90 per 1000 | 128 per 1000 (60 to 271) | ||||
| Speed of healing (time taken to be 'cured') | Not measured in this comparison | ||||
| Microbiological or histopathological cure of skin lesions Follow‐up: mean 2.5 months | Study population | RR 1.03 (0.88 to 1.2) | 383 (2 studies) | ⊕⊝⊝⊝ Very lowc | |
| 859 per 1000 | 884 per 1000 (756 to 1000) | ||||
| Moderate | |||||
| 792 per 1000 | 816 per 1000 (697 to 950) | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; N/A: not applicable. | |||||
| GRADE Working Group grades of evidence High quality/certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality/certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality/certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality/certainty: we are very uncertain about the estimate. | |||||
aDowngraded by 4 levels due to risk of bias (1 RCT has many uncertain risks), indirectness (2 levels because one of the studies focused on young people), and imprecision (the confidence interval around the estimate risk ratio ranges from a 14% reduction to a 17% increase in the risk ratio for healing with paromomycin). bDowngraded by 4 levels due to risk of bias (2 RCTs have many uncertain risks), indirectness (2 levels because one of the studies focused on young people), and imprecision (the confidence interval crosses the line of no effect). cDowngraded by 5 levels due to risk of bias (1 RCT has many uncertain risks), inconsistency (there is considerable heterogeneity ‐ I² = 84%), indirectness (2 levels because one of the studies focused on young people), and imprecision (the confidence interval around the estimate risk ratio ranges from a 12% reduction to a 20% increase in the risk ratio for healing with paromomycin).
Background
Please see Table 117 for a glossary of terms used.
Table 1.
Glossary
| Term | Definition |
| Antimonials | Pharmaceutical agents containing antimony. Antimony‐containing compounds (meglumine antimoniate and sodium stibogluconate) are the principal medications used to treat leishmaniases, an infection caused by a protozoan parasite. |
| Arthralgia | Pain in the joints. The causes of arthralgia are varied and range, from a joints perspective, from degenerative and destructive processes such as osteoarthritis and sports injuries to inflammation of tissues surrounding the joints, such as bursitis. |
| Cardiac arrhythmia | An arrhythmia is an abnormal heart rhythm. Many types of arrhythmia have no symptoms. When symptoms are present these may include palpitations or feeling a pause between heartbeats. More seriously there may be lightheadedness, passing out, shortness of breath, or chest pain. |
| Cutaneous necrosis | The death of living tissues in response to disease or injury. |
| Cytolysis | The degeneration or dissolution of cell caused by the disruption of cell membrane. |
| Exudate | A fluid with a high content of protein and cellular debris that has escaped from blood vessels and has been deposited in tissues or on tissue surfaces, usually as a result of inflammation. |
| Human monocytes | Monocytes are the biggest type of white blood cell in the immune system. Originally formed in the bone marrow, they are released into our blood and migrate into the connective tissue where they differentiate into macrophages. When certain germs enter the body, they quickly rush to the site of attack. |
| Hypotension | A systolic blood pressure reading (the top number) of 90 millimetres of mercury (mmHg) or less a diastolic blood pressure reading (the bottom number) of 60 mmHg or less is generally considered low blood pressure. The causes of low blood pressure can range from dehydration to serious medical or surgical disorders. |
| Immune response modifier | Any of a broad family of biomolecules that up‐ or down‐regulate, or restore immune responsiveness, which are generated after T cells recognise an antigen present on the surface of a self‐antigen‐presenting cell, which, once activated, produce multiple cytokines. |
| Immunolabeling | A biochemical process that enables the detection and localisation of an antigen to a particular site within a cell, tissue, or organ. Antigens are organic molecules, usually proteins, capable of binding to an antibody. These antigens can be visualised using a combination of antigen‐specific antibodies as well as a means of detection, called a tag, that is covalently linked to the antibody. If the immunolabeling process is meant to reveal information about a cell or its substructures, the process is called immunocytochemistry. Immunolabeling of larger structures is called immunohistochemistry. |
| In vitro | Biological processes or reactions made to occur outside the living organism in an artificial environment, such as a culture medium. |
| Intralesional meglumine antimoniate | Meglumine antimoniate (or Glucantime) is a medicine used for treating leishmaniasis. It belongs to a group of compounds known as the pentavalent antimonials. |
| Lymphadenopathies | Lymph nodes that have an abnormal in size, number or consistency; often used as a synonym for swollen or enlarged lymph nodes. Common causes of lymphadenopathy are infection, autoimmune disease, or malignancy. |
| Lymphatic channels | The vessels that transport lymph throughout the body. Lymph is a clear fluid that contains cells important for forming antibodies that fight infection. |
| Lymphokine | Any of various soluble protein mediators released by sensitised lymphocytes on contact with antigen, and believed to play a role in macrophage activation, lymphocyte transformation, and cell‐mediated immunity. They regulate immune responses through differentiation, amplification, and inhibition of cell functions. Lymphokines may also have a cytotoxic effector function. Used as biologic response modifiers in the treatment of cancer. |
| Macrophages | White blood cells (activated monocytes) that protect the body against infection and foreign substances by breaking them down into antigenic peptides recognised by circulating T cells. |
| Miltefosine | An oral alkyl phosphocholine analogue used to treat cutaneous and visceral leishmaniasis. Interacts with lipids and sterols in the Leishmania membrane resulting in inhibition of mitochondria and apoptotic cell death. |
| Mucous membranes | The mucous membranes (or mucosae or mucosas; singular mucosa) are linings of mostly endodermal origin, covered in epithelium, which are involved in absorption and secretion. They line cavities that are exposed to the external environment and internal organs. |
| Myalgia | Myalgia, or muscle pain, is a symptom of many diseases and disorders. The most common causes are the overuse or over‐stretching of a muscle or group of muscles. Myalgia without a traumatic history is often due to viral infections. Long‐term myalgias may be indicative of a metabolic myopathy, some nutritional deficiencies or chronic fatigue syndrome. |
| Nodular lymphangitis | Nodular lymphangitis is a distinct clinical entity, separate from lymphangitis. This disorder is characterised by inflammatory nodules along the lymphatics draining a primary skin infection |
| Papule | A solid, rounded growth that is elevated from the skin, usually inflammatory but nonsuppurative. A papule is usually less than 1 cm across. |
| Parenteral | Administration of a medicinal or therapeutic substance, other than through the gastrointestinal or respiratory tracts, e.g. by intravenous, intramuscular or subcuticular injection. |
| Pentamidine | Pentamidine (e.g. isethionate) is an antiprotozoal and antifungal agent of the class of aromatic diamidines, administered intravenously or intramuscularly in treatment of early African trypanosomiasis and leishmaniasis, and intravenously, intramuscularly, or by oral inhalation in treatment and prophylaxis of Pneumocystis carinii pneumonia. |
| Pentavalent antimony | Pentavalent antimonials are a group of compounds used for the treatment of leishmaniasis. The first pentavalent antimonial used was urea stibamine: first introduced in the 1930s, it fell out of favour in the 1950s due to higher toxicity compared to sodium stibogluconate. The compounds currently available for clinical use are: sodium stibogluconate (Pentostam; manufactured by GlaxoSmithKline; available in the USA and UK), which is administered by slow intravenous injection, intralesional or intramuscular injection, and meglumine antimoniate (Glucantime; manufactured by Aventis; available in Brazil, France and Italy), which is administered by intramuscular, intralesional, or intravenous injection. |
| Promastigotes | Term now generally used instead of 'leptomonad' or 'leptomonad stage' to avoid confusion with the flagellate genus Leptomonas. It denotes the flagellate stage of a trypanosomatid protozoan in which the flagellum arises from a kinetoplast in front of the nucleus and emerges from the anterior end of the organism; usually an extracellular phase, as in the insect intermediate host (or in culture) of Leishmania parasites. |
| Protozoan | Any of a group of single‐celled, usually microscopic, eukaryotic organisms, such as amoebas, ciliates, flagellates, and sporozoans. |
| ThermoMed device | The ThermoMed is a battery‐operated device that delivers precisely controlled localised current field radiofrequency heat to selectively destroy certain diseased tissue and is recommended by the World Health Organization as an alternative therapy for cutaneus leishmaniasis. |
| Thermotherapy | The treatment of disease by the application of heat. Thermotherapy may be administered as dry heat with heat lamps, diathermy machines, electric pads, or hot water bottles or as moist heat with warm compresses or immersion in warm water. Warm soaks or compresses may be used to treat local infections, relax muscles and relieve pain in patients with motor problems, and promote circulation in peripheral vascular disorders such as thrombophlebitis. |
Description of the condition
Definition
Desjeux 1996 describes leishmaniasis as "a group of diseases caused by infection with protozoan parasites of the genus Leishmania", transmitted by bites from sandflies infected with the parasite. Leishmaniasis has two main clinical forms of presentation (cutaneous and visceral), which are associated with a broad range of signs, symptoms, and degrees of severity (Herwaldt 1999; Reithinger 2007). Depending on its geographical distribution, cutaneous leishmaniasis (CL) is classified as New World cutaneous leishmaniasis (NWCL) or Old World cutaneous leishmaniasis (OWCL). The latter commonly – though not exclusively – presents as chronic, painless ulcers or nodules. Treatment of cutaneous leishmaniasis is complex and should depend on the Leishmania species involved, the area of acquisition of the infection, and the clinical form (Monge‐Maillo 2013). Management may vary from local or systemic treatment to no treatment.
Epidemiology and impact
In many tropical and subtropical low‐ and middle‐income countries (LMICs), protozoan parasites are amongst the most common infectious agents and have serious consequences for socioeconomic development (Alvar 2006; WHO 2002). The World Health Organization (WHO) considers leishmaniasis to be one of the most serious parasitic diseases, and the World Health Assembly has advocated for prioritising their control (WHO 2007).
An estimated 700,000 to 1.2 million new CL cases occur each year (Alvar 2012). CL is widely distributed, with 70% to 75% of the global incidence located mainly in 10 countries: Afghanistan, Algeria, Colombia, Brazil, Iran, Syria, Ethiopia, North Sudan, Costa Rica, and Peru (Alvar 2012). Global incidence of OWCL is estimated at more than 900,000 cases per year, distributed from the Mediterranean basin across the Near East to Northwest India, with a few foci in Central China as well as a thin band across West Africa and in the Horn of Africa (Alvar 2012; Pigott 2014). OWCL is also increasingly present in immigrants, military personnel, humanitarian aid workers, tourists, and travellers from endemic areas (Reithinger 2007).
Immunosuppression is also a factor that can increase the prevalence of CL, altering its clinical presentation and treatment response (van Griensven 2014). This factor is most closely associated with HIV, but recent years have seen a worldwide increase in other immunosuppressive conditions, mainly because of better medical care for chronic illnesses and the use of immunosuppressive drugs like tumour necrosis factor (TNF) inhibitors (van Griensven 2014). In any case, the reported leishmaniasis case figures seem to be only a fraction of the true burden, and the disease appears to be underestimated and on the rise in several countries (Alvar 2012). Leishmaniasis, as with many other neglected tropical diseases (NTDs), occurs mostly in LMICs, affecting rural and remote locations that may escape official data sources (WHO 2010).
Aetiology and transmission
Five Leishmania species cause OWCL: L major, L tropica, L infantum, L donovani, andL aethiopica (Pace 2014). Most cases are due to L major, with around 500,000 cases per year; L tropica, with 400,000 cases per year; followed by L aethiopica, with 50,000 cases per year (den Boer 2011). Transmission of leishmaniasis is through a complex life cycle involving sandflies belonging to the Phlebotomus species (Pace 2014). Non‐vector transmission (e.g. by accidental laboratory infection, blood transfusion, or organ transplantation) is possible but rare (Cardo 2006). Transmission of leishmaniasis can be either anthroponotic or zoonotic (Pace 2014).
Zoonotic cutaneous leishmaniasis (ZCL), occurring mostly in rural areas, is where the parasite is transmitted from a range of animals to humans (WHO 2010). It is geographically distributed in the Middle East, North‐western China, and North Africa, where it may be caused by L major, and in the Mediterranean basin where it is mostly caused by L infantum (Pace 2014). The Old World ZCL often heals spontaneously after two to four months, although in some cases it may persist for as long as five years (WHO 2010).
Anthroponotic cutaneous leishmaniasis (ACL) is transmitted from person to person, mainly in urban areas; it is geographically distributed in the Middle East, the Indian subcontinent, and western Asia (WHO 2010). ACL is mostly caused by L tropica and also by L donovani, which may cause post‐kala‐azar dermal leishmaniasis (PKDL) in Asia (India, Nepal, and Bangladesh) and in East Africa (Ethiopia, Kenya, and Sudan) (Alvar 2012).
Clinical manifestations
CL can affect the skin and mucous membranes and has been categorised into five different clinical forms: localised, recidivans, diffuse, mucosal, and PKDL.
Localised leishmaniasis
In the localised form, the parasite is confined to the skin (Gonzalez 2008). After an incubation period of 1 to 12 weeks, a papule or bump develops at the site of the insect bite, and the papule grows and turns into an ulcer (González 2009). A typical lesion of the localised form of CL is a painless papule or ulcer covered with an adherent crust of dried exudate and located on exposed parts of the body such as the face, arms, or legs (Gonzalez 2008; González 2009). CL due to L infantum usually causes single lesions. By contrast, L tropica frequently causes multiple lesions, as does L major, in this case often severely inflamed and ulcerated lesions that heal slowly and cause large disfiguring or disabling scars. Other variations exist as well: some people may have as many as 200 simple skin lesions, with some growing but not ulcerating (sporotrichoid), while certain Leishmania species also infect the lymphatic system, producing lesions along the lymphatic channels (nodular lymphangitis) (Gonzalez 2008). Secondary bacterial infection is common, causing pain and serious disability (Blum 2014). Most lesions heal spontaneously over months or years, leaving permanent scarring with skin thinning. Scarring of leishmaniasis typically displays a depigmented centre and a pigmented border (Reithinger 2007).
Leishmaniasis recidivans
This form appears in around five per cent of people with CL by L tropica and is characterised by microsatellite and confluent lesions that relapse and finally ulcerate on the border of previous scars (Sharifi 2010; WHO 2010).
Diffuse leishmaniasis
This form affects only the skin but with generalised skin lesions; it is seen mainly in Africa and transmitted by L aethiopica (Alrajhi 2003).
Mucosal leishmaniasis
In mucosal leishmaniasis, the parasite may spread to the mucous membranes, especially those of the nose, mouth, and throat, causing extensive damage and disfiguration. It mainly occurs in South America, but it can also be caused by species from Old World countries including L tropica, L major, and L infantum (WHO 2010).
Post‐kala‐azar dermal leishmaniasis
PKDL is a form of diffuse cutaneous leishmaniasis and a sequel of visceral leishmaniasis (VL) that may appear in affected individuals up to 20 years after being partially treated or untreated or even in those who supposedly received adequate treatment (Rathi 2005). PKDL is mostly seen in areas where L donovani is endemic, such as in Asia (India, Nepal, and Bangladesh) and East Africa (Ethiopia, Kenya, and Sudan) (Alvar 2012).
Diagnosis
Clinically diagnosed OWCL should be confirmed using the traditional diagnostic techniques of smear, parasite culture, and histological analysis of skin by aspiration, scrapings, or biopsies (Saab 2015). Circulating antibodies in the bloodstream are generally low or undetectable in cases of OWCL (Masmoudi 2013). Modern molecular diagnostic techniques, mainly the polymerase chain reaction test (PCR), appear to be the most sensitive single diagnostic test for species identification in skin samples (Faber 2003; Schallig 2002). Identification of the Leishmania species involved is essential for selecting the most appropriate treatment.
Description of the intervention
Issues of treatment in CL are difficult to deal with because there are many factors that can influence the efficacy of drugs: the size, the number, and the appearance of the lesions; the duration of the disease prior to treatment; the frequency and time to self‐healing; the frequency of relapse and re‐infection; the frequency and severity of either mucosal or diffuse involvement; immunosuppression; co‐infections; prior anti‐Leishmania treatment; and the knowledge of resistance to anti‐Leishmania drugs (Gonzalez 2008). Laboratory studies have described acquired resistance to anti‐Leishmania drugs for decades, but only recently has clinical resistance been described. Monitoring resistance is currently controversial due to an inadequate correlation between clinical and in vitro resistance and a need for knowledge about the biochemical and molecular mechanisms of resistance (Croft 2006).
The location of the lesion (e.g. face or joints) and the patient's sex and age often determine the choice of treatment (González 2009). Other factors are intrinsic and related to the different Leishmania species (Safi 2012). An effective treatment in one geographical area for a given organism may not work in a different geographical area or for a different organism in the same location. In these cases, efficacy depends not only on the Leishmania species but also on the response of the person to the parasite and factors such as immunity, variable clinical response to treatments, drug toxicity, drug resistance, HIV co‐infection and adherence (Blum 2014).
Different authors have described many treatments for OWCL (Modabber 2007; WHO 2008; WHO 2010), of which we summarise the most relevant in Table 118. Nonetheless, several authors have pointed out the lack of properly controlled clinical trials (Hepburn 2001, Herwaldt 1999; Moskowitz 1999; Monge‐Maillo 2013). Another disadvantage and paradox is the lack of availability of most of these drugs in rural and poorer areas where leishmanias appears most frequently (Gonzalez 2008). Systemic treatments are generally given to those with CL who present with big (≥ 5 cm), multiple (> 5), or disseminated lesions; in those who have simple lesions involving cosmetically sensitive areas or joints, with mucosal reaction, or with the presence of nodular lymphangitis or lymphadenopathies; or for whom local therapy has failed (Blum 2014). For people with immunosuppression, there is controversy. Some experts consider that acquired or induced immunosuppression is a risk factor for developing mucosal leishmaniasis, so they recommend systemic treatment. Meanwhile, other experts have considered different treatment for the same type of people (Blum 2012).
Table 2.
Interventions for Old World cutaneous leishmaniasis
| Drug | Doses |
| Systemic antimonials | |
| Sodium stibogluconate (Pentostam, Stibanate) Meglumine antimonate (Glucantime) Combined with pentoxifylline |
20 mgSb v+/kg/d intramuscularly or intravenously for 20‐30 days 400 mg orally 3 times a day for 10–20 days |
| Intralesional antimonials | |
| Sodium stibogluconate (Pentostam, Stibanate) Meglumine antimonate (Glucantime) |
1–5 mL per session every 3–7 days. Up to 10 sessions depending on the clinical response, but most patients require ≤ 5 sessions |
| Non‐antimonial systemic treatments | |
| Fluconazole | 200 mg orally daily for 6 weeks |
| Miltefosine | 50 mg orally three times daily for 28 days |
| Liposomal amphotericin B | 3 mg/kg/d IV on days 1‐5 and 10 (18 mg/kg total dose) |
| Non‐antimonial topical or intralesional therapies | |
| 15% paromomycin/12% methylbenzethonium chloride | Ointment twice daily for 10‐20 days |
| 15% paromomycin/0.5% gentamicin sulphate | Twice a day for 20 days |
| Physical therapies | |
| Cryotherapy with liquid nitrogen | Frozen for 10‐30 s and thaw applied locally 2‐3 times in each session, repeated every 1‐4 weeks to complete healing (usually 2‐4 sessions) |
| Local heat therapy | 50°‐55ºC for 30 s by: Infrared light Direct current electrical stimulation Ultrasound Laser Radiofrequency waves ThermoMed device |
Systemic and intralesional antimonials
Meglumine antimoniate and sodium stibogluconate
The current mainstays of systemic treatment for OWCL are the pentavalent antimony (Sbv+) compounds sodium stibogluconate (SSG) (Pentostam, Stibanate) and meglumine antimoniate (MA) (Glucantime) (Asilian 2004a; WHO 2010); they often constitute the control condition in trials of new treatments. The recommended dosage is 20 mgSbv+/kg/d intramuscularly (IM) or intravenously (IV) for 20 to 30 days (WHO 2010); oral administration is not an option. SSG and MA can also be administered intralesionally (IL) with the recommended dosage of 1 mL to 5 mL per session every 3 to 7 days. Up to 10 sessions are needed depending on the clinical response, but most people require less than five sessions.
Despite the widespread use of SSG and MA, there are concerns about their cost, toxicity, and the development of drug resistance. Parenteral antimonial drugs are associated with severe adverse and often dose‐dependent effects, including nausea, vomiting, diarrhoea, skin eruptions, dizziness, cardiac arrhythmia, hypotension, arthralgia, myalgia, abdominal discomfort, headache, reversible elevation of hepatocellular enzymes, occasional anaemia, and thrombocytopaenia (Aronson 2010; Ejaz 2014; Esfandiarpour 2002; Farajzadeh 2015; Mohebali 2007; Momeni 2002). Pain at the site of the injection is greater in intralesional administration than in the intravenous/intramuscular route (Iraji 2005; Momeni 2002; Salmanpour 2006). Whilst there is no general consensus on optimum treatment, there are active lines of research to identify alternatives to systemic antimonials (Jowkar 2012). Momeni 2002 has investigated combination therapies with allopurinol, and Sadeghian 2006a with pentoxifylline, as treatments that may help to reduce drug resistance by increasing the efficacy of the antimonials, reducing their doses, or both.
Infiltration of skin lesions (injecting a substance directly into the infected lesion) can be very painful. Adverse effects of IL treatments are burning at the site of injection, itching, inflammation, and vasovagal shock due to severe pain (Layegh 2009; Mapar 2010). A safe and efficient therapeutic method for IL injection is the use of a Dermojet device (Bogenrieder 2003). Preventing blood‐borne transmission of other infectious diseases in LMICs entails reducing the use of injections, implementing blood safety practices, and providing sterile injection equipment in healthcare centres (Kermode 2004; Simonsen 1999). Several studies provide evidence for the efficacy of intralesional pentavalent antimonials for OWCL, mainly in Asia and the Mediterranean basin (Alkhawajah 1997; Uzun 2004). Moreover, local administration reduces the systemic toxicity of antimonials; people may receive anywhere from a few injections or – for those with multiple or complicated lesions – daily injections for up to 40 days (Mujtaba 1999). (Complicated CL is defined as more than 4 lesions over 4 cm in size, which are periorificial or located close to small joints, for which previous treatment has failed. Clinically important lymphatic dissemination is present, as well as underlying immunosuppressive conditions, and "there is significant comorbidity for which systemic treatments could be contraindicated in a benefit–risk assessment (e.g. unbalanced diabetes, malnutrition)" (Gradoni 2017).)
Self‐limiting lesions are normally amenable to weekly or alternate day IL injections of SSG or MA (Alkhawajah 1997; Mujtaba 1999). According to some authors, deficient infiltration of the lesions is one of the most common and important causes of treatment failure with IL antimonials (Faghihi 2003). There are also other studies that have evaluated the association of intralesional pentavalent antimonials as a way to increase the efficacy (Asilian 2003; Munir 2008).
Non‐antimonial systemic treatments
Oral antifungal
Oral azole antifungal drugs (fluconazole, ketoconazole, itraconazole) are potential therapeutic agents in CL. The first reports of oral ketoconazole for the treatment of CL in both the New and the Old World came out in the early 1980s (Urcayo 1982; Weinrauch 1983a; Weinrauch 1983b). However, reports of liver toxicity and low cure rates for certain species led to ketoconazole being withdrawn from the market, making it necessary to search for other azoles (Alrajhi 2002). In the mid‐ to late 1980s, another azole called itraconazole was touted as a treatment for CL (Borelli 1987; Cauwenberg 1986), with different cure rates depending on the species involved (Momeni 1996; Nassiri‐Kashani 2005). Fluconazole, another antifungal azole, has also been used as an alternative therapy for CL, with good cure rates at different doses (Alrajhi 2002; Emad 2011).
Oral dapsone
Some studies have proposed the antibiotic/antileprotic drug dapsone as an inexpensive oral alternative to the treatment currently used for CL (Dogra 1986; Dogra 1991), although the main side effect of dapsone is blood cell destruction and anaemia (Dogra 1991).
Oral allopurinol
Allopurinol (a medicine used to treat gout) alters protein synthesis and inhibits the growth of Leishmania in vitro (Momeni 2002); different authors have assessed its use as a potential therapeutic agent for the treatment of both CL and VL, mainly in combination with pentavalent antimonials (Chunge 1985; Jha 1983; Kager 1981; Momeni 2002).
Oral antibiotics
Researchers have also reported several other oral antibiotics like metronidazole and cotrimoxazole as possibly promising anti‐Leishmania agents in the treatment of VL (Rodriguez‐Cuartero 1990), while others have looked into the efficacy of a short‐term course of antibiotic rifampicin for CL (Bygbjerg 1980). Oral azithromycin is another antibiotic that is effective in vitro and in mice (Minodier 2007), but research has not established it as superior to antimonials for OWCL in humans (Layegh 2007), even when combined with allopurinol (Dastgheib 2012). Thus, azithromycin needs further investigation for human leishmaniasis.
Oral pentoxifylline
Oral pentoxifylline, used in people with vascular diseases, also has anti‐Leishmania effects (Lessa 2001), decreasing the inflammatory reaction and the resulting tissue damage (Sadeghian 2006a). Pentoxifylline has a good safety profile, although nausea, arthralgias, dizziness, abdominal pain, and diarrhoea can occur (Lessa 2001).
Oral miltefosine
Oral miltefosine, which was originally developed as an anticancer drug, is active against the Leishmania membrane (Croft 2006). Miltefosine was included in the WHO essential medicines list as an anti‐leishmaniasis medicine in March 2011 (WHO 2011), and in March 2014, the US Food and Drug Administration (FDA) approved oral miltefosine for visceral, cutaneous, and mucosal leishmaniasis (FDA 2014). Miltefosine seems active against most Leishmania species, but with variable efficacy depending on geographical regions, even for the same species (Monge‐Maillo 2015; Soto 2004; Stojkovic 2007). Limited experience with miltefosine for OWCL shows efficacy, mostly against L major (Dorlo 2011; Mohebali 2007; Stojkovic 2007). The most commonly reported adverse drug reactions associated with miltefosine are transient gastrointestinal discomfort, nausea, vomiting, abdominal pain, and mild elevation of liver enzymes and serum creatinine (Mohebali 2007). Women of childbearing age require contraception beyond the end of treatment because this drug is contraindicated during pregnancy (Sindermann 2006).
Oral zinc sulphate
Early studies on oral zinc sulphate reported promising results for the treatment of CL (Sharquie 1996; Sharquie 1997; Sharquie 2001), although more recent studies have not reported good cure rates (Yazdanpanah 2011).
Oral artesunate
Artemisinin is effective against promastigotes in vitro, and artemisinin and artemether are leishmanicidal for amastigotes in infected murine macrophages (Adam 2008; Keiser J 2007). However, an RCT performed in Sudan did not show that artesunate combined with sulphamethoxypyrazine/pyrimethamine was better than placebo for OWCL (Adam 2009).
Other oral drugs
Jiang 2002 was the first to describe using omeprazole, a common treatment for peptic ulcer diseases, as a potential antiparasitic drug for the growth of L donovani in a laboratory setting, and Nilforoushzadeh 2008 concluded it was a good alternative to antimonials when these have to be given in a lower dose.
Parenteral liposomal amphotericin B
Amphotericin B, an antifungal drug used since 1960 (Sampaio 1960), is commonly used for treating American mucocutaneous leishmaniasis, HIV co‐infection, and visceral leishmaniasis (VL) in areas where Leishmania is resistant to antimonial and pentamidine drugs (Karamian 2007; Laguna 1999; Musa 2005; Sampaio 1997; Sundar 2007a; Thakur 1996). There has been little experience with this drug for OWCL due to pentavalent antimonials' dominance as the most commonly systemic regimens until now. However, the toxicity of pentavalent antimonials has prompted an increase in the administration of amphotericin B (mostly the lipid formulations), with promising results mainly with L major and L tropica (Solomon 2011; Wortmann 2010). Zanger 2011 also reported a case of OWCL due to L aethiopica in a immunosuppressed person with response to liposomal amphotericin B.
Non‐antimonial topical or intralesional therapies
Mild disease caused by L major is often self‐healing (Alkhawajah 1997; Nilforoushzadeh 2006). OWCL can be managed with local care alone and may not require other specific therapies when it is a simple CL (Shazad 2005), defined as CL: not caused by Leishmania species with common mucosal dissemination; not a diffuse, recurrent or post‐kala‐azar CL; with no lymphatic nodes affected; with small lesions; with fewer than five lesions; with lesions that are not localised in joints or aesthetic areas; or in people who are not immunocompromised (Nilforoushzadeh 2006). Topical and local therapies are attractive options that are appropriate for early self‐limiting lesions, offering reduced systemic toxicity and possibilities for outpatient treatment (Iraji 2004).
Topical antifungals
Early research on topical antifungals was based on clotrimazole, miconazole, and ketoconazole. The only RCT performed was in Saudi Arabia; investigators compared clotrimazole and miconazole and concluded that clotrimazole was more effective (Larbi 1995). Topical ketoconazole was tested in Afghanistan, but it did not significantly change the course of the lesions (Storer 2005).
Topical paromomycin (aminosidine)
Topical formulations often offer easier administration, fewer adverse effects, and sometimes cost‐effectiveness, although there may be difficulties in getting enough of the active drug absorbed through the skin (Jowkar 2012). Paromomycin is an antibiotic in the aminoglycoside family, originally identified as an anti‐Leishmania drug in the 1960s. Parenteral formulations have been used for VL (Sundar 2007b), and topical preparations for CL since 1987 (Asilian 1995). The literature uses the names paromomycin, aminosidine, monomycin, and neomycin E interchangeably, although the active principle is the same (Bryceson 1994). Two main topical preparations are available for CL: 15% paromomycin sulphate dissolved in a soft white paraffin base, either with 12% methyl benzethonium chloride (MBCL) or with 10% urea (Faghihi 2003; Shazad 2005). Paromomycin ointment combined with MBCL has been shown to be more efficacious than with urea (Iraji 2005). The original paromomycin formulation is no longer used because of its toxicity, and newer penetration‐enhancing formulations have been subjected to clinical evaluation (Davis 2003). More recently, studies have also evaluated new combinations of paromomycin plus gentamycin (Ben Salah 2009). Of the topical preparations, paromomycin ointment is commonly the first‐line treatment in uncomplicated CL (Asilian 2006). Adverse effects encountered were redness, pruritus, burning, oedema, local pain, inflammation, contact dermatitis, urticaria, or lymphadenitis with pain (Ben Salah 2013).
Intralesional zinc sulphate
Intralesional zinc sulphate had a direct anti‐Leishmania effect against L major and L tropica species in both an in vitro and in vivo study (Firooz 2005; Najim 1998).
Topical imiquimod
Topical imiquimod is an immune response modifier used for treating genital warts and premalignant skin cancer conditions, first used in combination with antimony for American CL (Arevalo 2007).
Intralesional hypertonic sodium chloride
Intralesional hypertonic sodium chloride solution (HSCS) can act by its osmotic effect to destroy the parasite as well as the surrounding tissue of the granuloma (Sharquie 1995; Sharquie 1997). It appears to be a cheap, safe, and effective local method for treating CL.
Intralesional interferon‐gamma (IFN‐γ)
Intralesional interferon (IFN‐ɣ) is a lymphokine originally used for the treatment of leprosy, cancer, HIV, and chronic granulomatous disease, and it has been shown to enhance the leishmanicidal capacity of human monocytes in vitro (Badaro 1990; Passwell 1986).
Topical aminoglycoside ointment (WR279,396)
The topical aminoglycoside ointment (WR279,396) is a hydrophilic formulation of paromomycin 15% plus a second aminoglycoside (gentamicin 0.5%) that was developed for topical administration, avoiding the potential skin irritation of other combinations performed with MBCL. Researchers in Tunisia have studied this combination of ointment (WR279,396) for L major, with better results than vehicle (Ben Salah 2009).
Intralesional metronidazole
Metronidazole was initially the first line treatment for trichomoniasis and later for amoebiasis and giardiasis. Its effectiveness for cutaneous leishmaniasis is controversial. Several case reports and a clinical trial performed in Iraq showed good cure rates for OWCL with intralesional metronidazole (Al‐Waiz 2004). However, a more recent clinical trial performed in Iran showed that metronidazole is ineffective (Mapar 2010). Moreover intralesional metronidazole injection was very painful.
Topical miltefosine
Pre‐clinical animal studies initially evaluated topical miltefosine, showing a potential benefit for NWCL and OWCL (Schmidt‐Ott 1999). A trial in Syria did not demonstrate efficacy of topical miltefosine for OWCL (Garnier 2002). However, a recent RCT in Iran showed that topical miltefosine was significantly more efficacious than meglumine antimoniate (Asilian 2014).
Topical dapsone
Dapsone is a synthetic sulfone employed orally for infectious diseases such as leprosy and for certain cutaneous disorders such as nodulocystic acne. However, the potential for systemic toxicity has limited its use in many cases. Topical formulations have also been developed especially for acne, thus reducing the possible systemic secondary effects (Stotland 2009). For OWCL an RCT in Iran compared dapsone 5% gel mask plus intralesional meglumine antimoniate (ILMA) versus cryotherapy plus ILMA, but investigators did not find any statistically significant difference in cure rates (Fekri 2015).
Topical 0.045% pharmaceutical chlorite (DAC N‐055)
DAC N‐055 seems to promote tissue regeneration, which may be of benefit for cutaneous lesions due to OWCL (Migdal 2011). A clinical trial in Afghanistan compared this treatment, both alone and in combination with bipolar high frequency electrocauterisation, versus intralesional antimonials (Stahl 2014). Use of electrocauterisation is based on the fact that physical wound debridement practised with bipolar high frequency electrosurgical cauterisation (HF‐EC) seems to speed up wound healing (Jebran 2014). The results of the RCT showed that DAC N‐055 alone was significantly more efficacious than ILMA (Stahl 2014).
Topical Thio‐Ben
An Iranian study reported promising results in OWCL using topical thioxolone plus benzoxonium chloride, or Thio‐Ben (Daie Parizi 1992; Daie Parizi 1996). Thioxolone, which has long been topically administered for the treatment of acne and psoriasis, is recognised as a safe drug. A later RCT from Iran compared topical Thio‐Ben plus cryotherapy versus meglumine antimoniate (Glucantime) plus cryotherapy (Daie Parizi 2015), showing that topical Thio‐Ben plus cryotherapy had good efficacy for OWCL, with fewer side affects than ILMA.
Physical therapies
People with OWCL may receive a range of physical treatments, including vaporisation, cauterisation, freezing, surgical excision, and the application of local heat.
Laser
Carbon dioxide (CO₂) lasers have been used to vaporise CL lesions, thereby destroying affected tissue without significant side effects in normal tissue (Asilian 2004b). Studies have shown that a single session is enough, with lesions healing within three to four weeks, with quite admissible cosmetic results, although the procedure is painful and requires local anaesthetic (Shamsi Meymandi 2011).
Trichloroacetic acid (TCA)
The efficacy of TCA for treating CL could be due to the destruction of infected tissue and skin regeneration (Nilforoushzadeh 2006).
Cryotherapy
Cryotherapy with liquid nitrogen has been used to treat individual lesions, destroying infected tissue, but it is labour intensive (Layegh 2009). As an effective, painless, low‐cost technique with few side effects, it is an especially good option for children (Layegh 2009; Salmanpour 2006). However, it is not suitable for multiple or complicated lesions (Alrajhi 2003; Bassiouny 1982; Layegh 2009; Leibovici 1986; Minodier 2007; Mosleh 2008; Ranawaka 2011).
Thermotherapy
Laboratory studies have reported that Leishmania parasites cannot multiply in macrophages when temperatures are greater than 39ºC (Berman 1981; Sacks 1983). These findings have stimulated investigations into the efficacy of thermotherapy for CL with direct‐current electrical stimulation (Sharquie 1998), ultrasound (Aram 1987), infrared light (Junaid 1986), hot‐water baths (Neva 1984), laser (Asilian 2004b; Babajev 1991; Meawad 1997; Rodriguez 1990), radiofrequency waves, and ThermoMed device (Reithinger 2005; Sadeghian 2007; Aronson 2010). The procedure is painful and may require local anaesthetic (Sadeghian 2007).
Topical photodynamic therapy
Photodynamic therapy is a light‐mediated technique that causes cytolysis of Leishmania parasites, resulting in an effective and safe therapeutic option for OWCL (Asilian 2006).
Mesotherapy
Mesotherapy is based on the use of a specific amount of variable substance (hormones, nutrients, enzymes, pharmaceuticals, and detergents, among others) and is not an invasive technique. People have received it for treating cellulite and acne scars, reducing aging skin, and rejuvenating the hands and neck (Amin 2006; Rohrich 2003). One RCT performed in Iran compared mesotherapy with ILMA, finding no difference in cure rates between the two therapeutic options (Kashani 2010).
Methods for promoting healing
Methods used in wound healing, including dressing and antiseptics, are often employed in ulcerative lesions of CL to accelerate cure, normalise epithelialisation, and reduce scarring, especially at cosmetic sites (Stahl 2014). Compromised wound healing due to repetitive trauma, contamination, and infection are major problems encountered in people with OWCL, and it is important to improve scar formation or at least not interfere with the natural healing process (Gonzalez 2008). A recent consensus panel on recommendations for chronic and acute wound dressings reported that "hydrocolloid (polymer dressings with medium absorption properties and containing carboxymethylcellulose) and low‐adherent dressings seem to be the most suitable dressings for the epithelialisation stage of chronic and acute wounds" (Vaneau 2007). The highest impact of scarring and ulcerative lesions is on the faces of young women, which exposes them to stigma and which may affect their marriage prospects (Weigel 2001; Reithinger 2005b). To assess the cosmetic impact, clinicians often use the Burn Scar Index (also known as the Vancouver Scar Scale) to document change in scar appearance (Baryza 1995), which should be ideally measured six months after completion of treatment (Modabber 2007).
Alternative therapies
Increasing treatment failure with antimonial drugs has accelerated the search for alternative therapies (Nilforoushzadeh 2007). Up to 80% of the world's population may depend on medicinal plants as the only source of remedies for this disease; modern drugs may be either too expensive or wholly inaccessible (Fatima 2005). Practitioners of both traditional and modern medicine in Iran have long used herbal remedies and honey (Nilforoushzadeh 2007; Zerehsaz 1999). Different plants with medicinal value (Azadirachta indica, Acacia nilotica, and Allium sativa), traditionally used in the west and central parts of Sudan, have proven to have active anti‐Leishmania activity on L major promastigotes in vitro (Fatima 2005; Khalid 2005). Honey is effective for wound healing through enhancement of granulation and epithelialisation stages, enhancement of debridement, and downsizing of wound malodour (Moore 2001; Pieper 2003). Studies have described that honey from flowers in Australia and New Zealand has antibacterial effects (Pieper 2003).
How the intervention might work
OWCL is a heterogeneous group of diseases that differ in their clinical presentation, prognosis, and response to different the therapeutic interventions. There are two main factors affecting response to treatment: the species of Leishmania and the clinical form of the disease.
The mechanisms of action of the different therapeutic options for OWCL are diverse and may act over different targets, which can result in a complementary effect when working in combination (González 2009). There are two main types of therapeutic options: local physical therapies and local or systemic pharmacological therapies (Gonzalez 2008). Physical therapies such as HSCS, CO₂ lasers, TCA, cryotherapy, thermotherapy, and photodynamic therapy can destroy the parasite, the infected tissues, and the locally formed granulomas (González 2009). Pharmacological therapies, such as pentavalent antimonials, amphotericin B and its lipid formulations, miltefosine, paromomycin, and pentamidine, seem to enter the host cells and act against amastigotes, compromising the parasite metabolism, altering the parasite membrane fluidity or damaging the parasite mitochondria, among other mechanisms of action (Gonzalez 2008).
Therapeutic interventions could work in several ways: healing the forms of disease that would run a chronic course without treatment (L aethiopica); avoiding the progression of the disease to more complex clinical forms (mucocutaneous leishmaniasis, diffuse cutaneous leishmaniasis, leishmaniasis recidivans, etc.); speeding the healing of those forms that would naturally cure (L major); avoiding sequelae and disfiguring residual lesions (L major, L tropica, L infantum); and decreasing the human reservoir in anthroponotic forms (L tropica).
As a wide range of interventions are available, from physical or pharmacological local therapy to parenteral treatment, and response to treatment varies depending on the Leishmania species, knowing the optimal therapeutic approach in each situation is of great interest. This review aims to analyse the different interventions in order to provide the best treatment options for OWCL.
Why it is important to do this review
Controlling CL currently depends on early detection and rapid treatment (González 2009). The mainstays of treatment have been pentavalent antimonials, but other oral and topical treatment alternatives have become available in recent years (Gonzalez 2008). Global health development policies have mainly focused on innovative research to develop effective and affordable tools to tackle neglected tropical diseases (NTDs) and provide necessary new knowledge (WHO 2010). WHO is now prioritising the delivery of drugs that are currently available and using existing resources to reduce mortality, morbidity, and disability as a result of NTDs in low‐income countries (Savioli 2006). However, evidence for the comparative effectiveness, cost‐effectiveness, and safety of different treatment strategies is needed to improve disease control.
This systematic review focused on addressing the effects of treatments for the localised form of CL due to L tropica and L major, which account for more than 90% of CL in the Old World (WHO 2010). Since the overwhelming majority of OWCL cases heal spontaneously within 3 to 18 months (Blum 2014), the rationale for using systemic and topical treatments needs to be well established and preferably stratified for different geographic regions and Leishmania species. Separate Cochrane Reviews have addressed treatments for American CL and prevention measures for all types of cutaneous and mucosal leishmaniasis (González 2009; González 2015).
The protocol of this review was first entitled 'Interventions for solitary or limited cutaneous leishmaniasis'. However, we split the clinical subject into two reviews. We amended the title of the present review, first published in 2008 (Gonzalez 2008), to 'Interventions for Old World cutaneous leishmaniasis', and we also published a separate Cochrane Review, entitled 'Interventions for American cutaneous and mucocutaneous leishmaniasis' (González 2009). Thus, some parts of the Background and Methods sections are common to both reviews.
Objectives
To assess the effects of therapeutic interventions for the localised form of Old World cutaneous leishmaniasis.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials.
Types of participants
All immunocompetent people who had localised OWCL confirmed by parasitological diagnostic methods (i.e. tissue smears, histology, culture or PCR).
Types of interventions
The interventions were either single therapy or combination therapy. The comparators were either no treatment, placebo/vehicle only, or another active compound.
1. Systemic and intralesional antimonials
1.1 Meglumine antimoniate (Glucantime) 1.2 Sodium stibogluconate (Pentostam)
2. Non‐antimonial systemic treatments
2.1 Oral antifungals 2.2 Oral dapsone 2.3 Oral allopurinol 2.4 Oral antibiotics 2.5 Oral pentoxifylline 2.6 Oral miltefosine 2.7 Oral zinc sulphate 2.8 Oral artesunate
3. Non‐antimonial topical or intralesional therapies
3.1 Topical antifungals 3.2 Topical paromomycin (aminosidine) 3.3 Intralesional zinc sulphate 3.4 Topical imiquimod 3.5 Intralesional hypertonic sodium chloride (HSCS) 3.6 Intralesional interferon‐gamma (IFN‐γ) 3.7 Topical aminoglycoside ointment (WR279,396) 3.8 Intralesional metronidazole 3.9 Topical miltefosine 3.10 Topical dapsone 3.11 Topical 0.045% pharmaceutical chlorite (DAC N‐055) 3.12 Topical Thio‐Ben
4. Physical therapies
4.1 Laser 4.2 Trichloroacetic acid 4.3 Cryotherapy 4.4 Thermotherapy 4.5. Topical photodynamic therapy 4.6. Mesotherapy
5. Measures for promoting healing
6. Alternative therapies
Types of outcome measures
We did not limit the measurement of any outcomes based on length of follow‐up.
Primary outcomes
Percentage of lesions cured after the end of treatment
Percentage of participants with a complete cure after the end of treatment
By cured, we meant that all inflammatory signs disappeared (either skin oedema, hardening, or both) and that complete scarring or healthy repair occurred in ulcerative lesions. We did not consider lesions to be healed if there was no re‐epithelialised skin, or if inflammatory signs remained after follow‐up.
Secondary outcomes
Speed of healing (time taken to be 'cured')
Duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
Degree of functional and aesthetic impairment
Prevention of scarring
Quality of life
Adverse effects
Tertiary outcomes
Change in ability to detect Leishmania by parasitological diagnostic methods (e.g. smear, PCR, or culture)
"Emergence of treatment failures (defined as a decline in the efficacy of a drug against a population of parasites previously susceptible to that compound. The definition assumes that the original susceptibility of the population is known, which is not always the case for Leishmania)" (Ponte‐Sucre 2003)
Microbiological or histopathological cure of skin lesions
Development of cell‐mediated immunity (i.e. positive leishmanin skin test)
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
For this update, we revised all our search strategies in line with current Cochrane Skin practices. Details of the previous search strategies are available in Gonzalez 2008.
We searched the following databases up to 17 November 2016:
Cochrane Skin Specialised Register, using the search strategy in Appendix 1.
Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 10) in the Cochrane Library, using the strategy in Appendix 2.
MEDLINE via Ovid (from 1946), using the strategy in Appendix 3
Embase via Ovid (from 1974) using the strategy in Appendix 4.
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy in Appendix 5.
Trials registers
We searched the following trials registers in October 2015 using the term 'cutaneous leishmaniasis'.
ISRCTN registry (www.isrctn.com).
ClinicalTrials.gov (www.clinicaltrials.gov).
Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
EU Clinical Trials Register (www.clinicaltrialsregister.eu).
Searching other resources
References from unpublished studies
We checked the bibliographies of included studies and key papers identified by our searches for further references to relevant trials.
Unpublished literature
We wrote to national programme managers, general coordinators, directors, clinicians, regional officers from endemic countries in the WHO Eastern Mediterranean Region, pharmaceutical companies, and relevant authors for further information about unpublished and ongoing trials. We also contacted the following tropical medicine centres.
Department of Infectious Diseases and Tropical Medicine at the University of Munich, Germany.
Swiss Tropical Institute, Switzerland; Prince Leopold Institute of Tropical Medicine, Belgium.
McGill Centre for Tropical Disease, Canada.
Tulane University School of Public Health & Tropical Medicine, USA.
London School of Hygiene & Tropical Medicine, UK.
Tropical Medicine at the Liverpool School of Tropical Medicine, UK.
Department of Public Health and Tropical Medicine James Cook, University of North Queensland, Australia.
Institut Pasteur, France.
Bernhard Nocht Institute, Germany.
TropEdEurop, Spain.
Adverse effects
We searched MEDLINE (Ovid) from 1946 to 30 September 2015 for adverse or side effects of interventions for Old World cutaneous leishmaniasis using the search strategy in Appendix 6.
Data collection and analysis
Some parts of this review use text that was originally published in other Cochrane Reviews (predominantly van Zuuren 2015, Ingram 2015, and Delamere 2008; the latter was an exemplar Cochrane Review at the time the original review was written).
Selection of studies
We checked the titles and abstracts identified from the searches by at least two authors (JHM, PC, PLP, BMM, EGM). If it was unclear, then two authors obtained the full text study for independent assessment (BMM, EGM). The authors decided which trials met the inclusion criteria. The authors resolved any disagreements by discussion, with referral to a third author (AR) if necessary. We describe excluded studies and reasons for exclusion in the Characteristics of excluded studies table.
Data extraction and management
At least two independent authors (MP, LR, JHM, PLP) carried out data extraction using a pre‐designed data extraction form. We extracted data for all outcomes for all relevant drugs, paying attention particularly to the doses and therapy frequencies. We resolved disagreements by discussion. We obtained the missing data from trial authors when possible.
Assessment of risk of bias in included studies
The quality assessment included an evaluation of the following components for each included study, since there is some evidence that these are associated with biased estimates of treatment effect (Higgins 2011).
The method for generating the randomisation sequence.
The method for concealing allocation – it was considered 'adequate' if personnel and participants could not have foreseen assignment.
Blinding (participants, clinicians, outcome assessors).
Loss to follow‐up in each arm (split into postrandomisation exclusions and later losses if possible), and whether participants were analysed in the groups to which they were originally randomised (intention‐to‐treat).
In addition, the quality assessment also included the following.
Declaration of sample size calculation.
Definition of inclusion and exclusion criteria.
Reporting of Leishmania species involved.
Time of follow‐up.
Baseline comparability of severity of infection, age, sex, and duration of complaint.
Conflict of interest.
We assessed these other risks of bias. When trials reported two or more incorrectly, we judged them to be at high risk. When trials reported only one incorrectly, we judged them to be at unclear risk. We recorded the information in 'Risk of bias' tables (Characteristics of included studies) and described the quality of each study based on these components.
Measures of treatment effect
We reported all outcome data with their associated 95% confidence interval (CI). We expressed results as risk ratios (RR) and 95% CIs for dichotomous outcomes. We presented continuous outcomes with the same scale as reported in each trial, with a mean change from baseline with its associated standard deviation (SD), or as weighted mean difference (MD) or standardised mean difference (SMD) if more than one study was available.
If enough information was available in the study reports, we decided to describe hazard ratios (HR) for time‐to‐event outcome data.
Unit of analysis issues
We found that most RCTs included in this review assessed participants instead of lesions as the unit of analysis. When lesions were used as unit of analysis, comparisons were performed with lesions cured over lesions at start, not taking into account the correlation of multiple lesions per participant. We only pooled together studies which reported number of participants cured (not number of lesions), due to unit of analysis issues.
The approach followed to 3‐arm trials, was comparing the arms in pairs (A vs B, B vs C, and A vs C).
We only considered parallel group designs for all clinical trials. We did not consider cross‐over trials in this review because they are an inappropriate design for treatments that can potentially cure an infectious disease. We did not find any particular additional important information in the specific search for adverse effects for every particular treatment. Authors described these qualitatively in the Results and Discussion sections.
Dealing with missing data
For all missing data from trials that were less than 10 years old, we tried to contact the authors. Only six of the trials explicitly stated intention‐to‐treat (ITT) analysis. Where an ITT was not stated, we used the numbers originally randomised to the groups in order to calculate effect estimates.
For each study, we took all participants that were randomised into account when introducing the data in our tables. We sent emails to study authors asking for more information, and we recorded these emails and their responses. When we had no response, we assumed that missing data were treatment failures. Concerning the losses to follow‐up, it was not always possible to determine the arm in which the losses occurred, making ITT analyses impossible. In that case, we introduced only available data into the tables for analysis.
Assessment of heterogeneity
To assess the consistency of the study results, we obtained the I² statistic, which measures the proportion of total variation across studies that is due to heterogeneity rather than chance. I² lies between 0% and 100%. A value of 0% indicates no observed heterogeneity, and larger values show increasing heterogeneity. We analysed statistical heterogeneity using a Chi² test (on 1 degree of freedom, with a significance level of 0.05) (Higgins 2003).
Assessment of reporting biases
In this review, the low number of studies evaluating similar interventions and comparisons did not permit an assessment of publication bias. In future updates, if a sufficient number of trials assessing similar effects are identified for inclusion in this review, publication bias will be assessed according to the recommendations on testing for funnel plot asymmetry (Egger 1997) as described in section 10.4.3.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). If asymmetry is identified we will try to assess other possible causes and these will be explored in the discussion if appropriate
Data synthesis
We performed data synthesis only when we were able to identify at least two studies investigating similar treatments and reporting data amenable to pooling. Although there is inevitably a degree of heterogeneity between the studies included in a review, we entered these into a meta‐analysis if clinical reasoning could explain the heterogeneity and if there was a coherent argument for combining the studies. We used the random‐effects model, as this is more conservative in the presence of heterogeneity.
The percentage of lesions cured after the end of treatment was the primary outcome measure if available. If this were not available, we used secondary and tertiary outcomes. To estimate differences between treatments, we pooled trials that evaluated similar interventions and controls and calculated a weighted treatment effect across trials, using a random‐effects model. Where it was not possible to perform a meta‐analysis, we summarised the data for each trial.
Subgroup analysis and investigation of heterogeneity
In view of the limited number of included studies covering any one specific intervention, we did not conduct any of the subgroup analyses that we originally planned: Leishmania species, location and severity of infection, geographical setting, diagnostic techniques, type of treatment (topical, systemic, or combination), and relapse or re‐infection.
We assessed clinical heterogeneity by examining the characteristics of the studies, the similarity between the types of participants, the interventions, the comparisons, and the outcomes as specified in the criteria for included studies.
Sensitivity analysis
We plan to carry out a sensitivity analysis by excluding studies at high risk of bias.
Summary of findings tables
We used the GRADE approach to rate certainty of the evidence (for each outcomes) and strength of recommendations (Guyatt 2008). For better understanding of the review, we highlighted the GRADE assessments in 'Summary of findings' tables of key comparisons and outcomes.
Depending on the study design, risk of bias, consistency of the results across studies, and precision of the overall estimated across studies, certainty of evidence can be high, moderate, low, or very low.
Results
Description of studies
Results of the search
The previous version of this review identified 49 trials with 5559 randomised participants (Gonzalez 2008).
The update searches of the five databases (see Electronic searches) retrieved 517 records. Our searches of other resources identified six ongoing studies. We obtained no response from the institutions we contacted to try to identify further trials.
Once we removed duplicates, we had a total of 521 records. We excluded 452 records based on titles and abstracts and obtained the full text of the remaining 69 records. We excluded 15 studies (see Characteristics of excluded studies) and included 40, taking the total number of included studies to 89 (see Characteristics of included studies).
There are eight studies awaiting assessment: (Farajzadeh 2016a; Farajzadeh 2016b; Hanif 2016; Jaffary 2016; Na‐Bangchang 2016; Rajabi 2016; Refai 2016; Sattar 2012) (see Characteristics of studies awaiting classification). We found six ongoing trials (see Characteristics of ongoing studies).
Two studies were reported in two references: Nilforoushzadeh 2006 and Shanehsaz 2015.
For a further description of our screening process, see the study flow diagram Figure 1.
Figure 1.
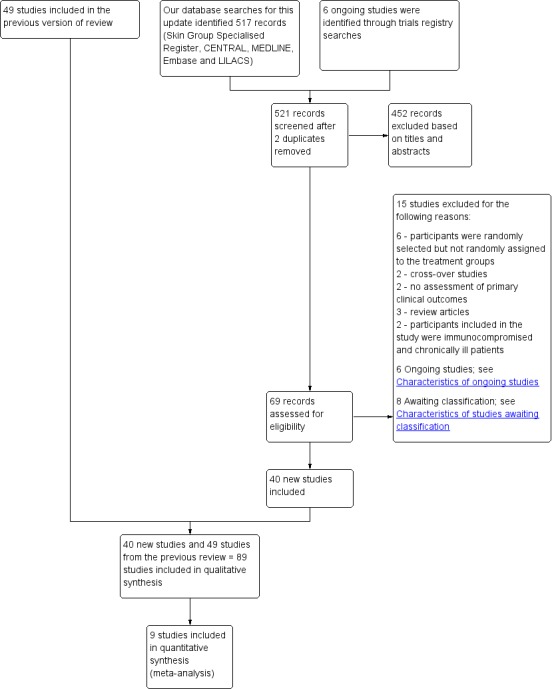
Study flow diagram.
Included studies
We included 89 studies (including 40 new to this update) involving 10,583 randomised participants with Old World cutaneous leishmaniasis (see Characteristics of included studies table).
Design
All of the studies were RCTs: 30 were placebo‐controlled, and 59 had an active treatment comparator. Seventy‐two took place after the year 2000; 85 were single‐centre studies, and the other 4 were multicentre studies (Asilian 1995; Asilian 2003; Ben Salah 2009; Ejaz 2014).
Sample sizes
The number of participants included in the individual studies ranged from 18 to 444; the most common sample size was 50 to 120 participants (interquartile range).
Setting
The studies included in this review took place in different countries, mainly in the Far or Middle East. Fifty‐three took place in Iran; seven in India; five in Africa (Sudan and Tunisia), four each in Saudi Arabia and Afghanistan; three each in Pakistan, Iraq, and Syria; two each in Kuwait and Sri Lanka; and one each in Yemen, Turkey, and the USA (recruiting Department of Defense healthcare beneficiaries who were likely to have been infected in Iraq or in Kuwait). Most studies took place in regional hospitals, teaching hospitals, local healthcare clinics, and skin disease research centres. A few studies were in military hospitals or army medical centres.
Participants
The studies had a mean of 41.5% of women participants, but this ranged from 23% to 80%. Four RCTs conducted by military institutions only included men (Aronson 2010; Ejaz 2014; Mashood 2001; Shazad 2005). Mean age in the individual studies ranged from 12 years to 56 years, with an overall mean age of 24.5 years. There were three RCTs including participants exclusively under 14 years (Asilian 1995; Asilian 2003; Lynen 1992). This review focused on L major andL tropica infections because of the paucity of studies involving L infantum,L aethiopica, orL donovani.
Interventions
Trials evaluated a wide range of interventions, which we categorised into six types.
Systemic and intralesional antimonials (8 studies).
Non‐antimonial systemic treatments (28 studies).
Non‐antimonial topical or intralesional therapies (28 studies).
Physical therapies (28 studies).
Measures for promoting healing (1 study).
Alternative therapies (5 studies).
The 89 studies covered 68 comparisons, and 59 included an active control arm. Duration of the intervention in most studies ranged between two and eight weeks (mean six weeks).
We describe examples of the first four categories in the Methods section under Types of interventions. The study on promotion of healing compared two topical interventions: diminazene aceturate (Berenil) versus cetrimide plus chlorhexidine (Savlon). Examples of alternative therapies were topical interventions based on substances like garlic, herbal extracts, or honey.
Systemic and intralesional antimonials included meglumine antimoniate (MA) and sodium stibogluconate (SSG) administered intramuscularly (IM) or intralesionally (IL). The standard dosage was 20 mg/kg/d, though some studies used higher doses such as 30 mg/kg/d or 60 mg/kg/d. The standard schedule frequency was 20 days (three weeks), but in some studies the treatment was extended until achieving cure.
Non‐antimonial systemic treatments included oral antifungals such as ketoconazole, itraconazole, and fluconazole. Trials compared itraconazole only against placebo or no treatment, whereas the trials on ketoconazole and fluconazole compared increasing dosages. Some trials compared ketoconazole versus intramuscular or intralesional SSG and intralesional meglumine antimoniate (ILMA). This treatment category also included oral dapsone versus placebo, oral allopurinol versus intramuscular meglumine antimoniate (IMMA) or intravenous sodium stibogluconate (IVSSG), oral antibiotics alone or combined versus placebo or IMMA, oral pentoxifylline alone or combined versus IMMA, oral zinc sulphate versus IMMA and oral artesunate versus placebo.
Non‐antimonial topical or intralesional therapies comprised topical antifungals like miconazole, ketoconazole, amphotericin B, and clotrimazole as well as topical paromomycin alone or in combination with oral ketoconazole, photodynamic therapy, intralesional zinc sulphate, hypertonic sodium chloride (HSCS), INF‐ɣ, metronidazole, topical imiquimod, miltefosine, dapsone, 0.045% pharmaceutical chlorite (DAC N‐055), and Thio‐Ben. The vast majority were tested against ILMA or ILSSG, with some therapies also compared to placebo/vehicle (topical paromomycin) or different regimens of paromomycin (photodynamic therapy).
Physical therapies included ablative or fractional CO₂ laser, compared against each other, IMMA, or cryotherapy. Trichloroacetic acid (TCA), alone or combined with non‐ablative CO₂ laser, was compared with ILMA. Cryotherapy, alone or combined with 15% paromomycin and/or ILMA, was compared with ILMA alone or combined. Cryotherapy was also combined with topical treatments such as 3% salicylic acid cream and 3% sodium nitrite cream and compared against cryotherapy plus 3% salicylic acid cream plus a vehicle. Thermotherapy using radiofrequency waves was compared with ILMA, ILSSG, and IMSSG. Another type of thermotherapy included electrocauterisation, tested with or without DAC N‐055.
Measures for promoting healing included topical diminazene aceturate (Berenil) versus topical cetrimide and chlorhexidine (Savlon), and trials of alternative therapies included topical interventions alone or combined compared with ILMA (topical honey plus ILMA; topical Cassia fistula alone or with fruit gel plus ILMA; and topical Achilles millefolium cream plus ILMA) or IMMA (topical herbal extract).
Heterogeneity in study design, medium to high levels of data bias, missing standard deviations, and a mix of different comparators, dosing regimens, and scheduled frequency did not, in general, permit pooling of the data or allow us to make accurate and direct comparisons of a substantial number of the interventions.
Outcomes
Three RCTs did not clearly report the time points when the primary outcome was assessed (Dandashli 2005; Mapar 2010; Salmanpour 2006), and the remaining studies reported a time ranging from the end of treatment to six months after treatment.
Percentage of lesions cured after the end of treatment. Only 18 RCTs reported the primary outcome as percentage of lesions cured at three months after the end of treatment (Al Hamdi 2010; Alkhawajah 1997; Aronson 2010; Asilian 2004a; Asilian 2006; Dandashli 2005; El‐Sayed 2010; Firooz 2005; Harms 1991; Jowkar 2012; Khatami 2013; Larbi 1995; Maleki 2012; Mujtaba 1999; Ranawaka 2015; Shamsi Meymandi 2011; Shanehsaz 2015; Sharquie 2001).
Percentage of participants with a complete cure after the end of treatment. The rest of the studies reported the percentages in terms of participants cured.
Varying numbers of studies reported the following secondary outcome measures.
Speed of healing (time taken to be 'cured') in 16 studies (Alrajhi 2002; Aronson 2010; Asilian 2004b; Ben Salah 2009; Farajzadeh 2015; Jaffary 2014b; Jebran 2014; Layegh 2009; Mujtaba 1999; Nilforoushzadeh 2007; Nilforoushzadeh 2013; Ranawaka 2015; Reithinger 2005; Sharquie 1997; Sharquie 2001; Stahl 2014).
Duration of remission and percentage of people with treated lesions that recur within more than 6 months in nine studies (Al‐Fouzan 1991; Bumb 2013; Ejaz 2014; Faghihi 2003; Fekri 2015; Mujtaba 1999; Ranawaka 2010; Ranawaka 2015; Sadeghian 2006b).
Degree of functional or aesthetic impairment (no studies measured this outcome).
Prevention of scarring in eight studies (Alkhawajah 1997; Asilian 2004b; Asilian 2006; Faghihi 2003; Mujtaba 1999; Sadeghian 2007; Sharquie 1997; Sharquie 2001).
Quality of life (no studies measured this outcome).
Adverse effects in all but eight studies (Al Hamdi 2010; Asilian 2014; Farajzadeh 2015; Kochar 2006; Nilforoushzadeh 2004; Nilforoushzadeh 2008; Nilforoushzadeh 2012; Nilforoushzadeh 2013).
Three studies did not report secondary outcomes (Jaffar 2006; Kochar 2006; Nilforoushzadeh 2004).
A number of studies also reported tertiary outcome measures.
Change in ability to detect Leishmania by parasitological diagnostic methods (e.g. smear, polymerase chain reaction (PCR), or culture). Two RCTs reported results where Leishmania was detected by parasitological diagnostic methods (e.g. PCR or culture, positive smears) (Jaffary 2014A; Jebran 2014). No studies reported emergence of resistance.
Emergence of treatment failures (defined as a decline in the efficacy of a drug against a population of parasites previously susceptible to that compound (Ponte‐Sucre 2003). The definition assumes that the original susceptibility of the population is known, which is not always the case for Leishmania). No studies reported this outcome.
Microbiological or histopathological cure of skin lesions, reported in 15 studies (Aronson 2010; Asilian 1995; Asilian 2003; Asilian 2004a; Ben Salah 1995; Dogra 1990; Dogra 1991; Dogra 1996; Harms 1991; Jebran 2014; Kashani 2010; Kochar 2000; Momeni 2002; Nilforoushzadeh 2006; Shamsi Meymandi 2011).
Development of cell‐mediated immunity (i.e. positive leishmanin skin test). One study reported the percentage of participants who developed Leishman bodies three months after treatment (Kashani 2010).
Excluded studies
We excluded 15 RCTs for the reasons described in the Characteristics of excluded studies tables and below.
Participants were randomly selected but not randomly assigned to the treatment groups.
Cross‐over studies.
No assessment of clinical primary outcomes.
Review and meta‐analysis.
Participants included in the study were immunocompromised and chronically ill patients.
Studies awaiting assessment
We identified seven studies from full‐text screening (Farajzadeh 2016a; Farajzadeh 2016b; Hanif 2016; Jaffary 2016; Na‐Bangchang 2016; Rajabi 2016; Refai 2016), and one by assessing an abstract (Sattar 2012). These studies evaluated the following.
Intralesional injection of zinc sulphate versus ILMA (Glucantime)e.
IMMA plus vehicle versus IMMA plus topical terbinafine.
Intralesional versus oral chloroquine administration.
ILMA (Glucantime) plus topical trichloroacetic acid (TCA) 50% versus ILMA alone versus fractional carbon dioxide laser.
Shiunko ointment versus vehicle.
Topical liposomal form of azithromycin versus ILMA (Glucantime).
Radiofrequency‐induced heat therapy versus ILMA (Glucantime).
Topical ointment prepared from the stem extract of Morinda citrifolia.
We will evaluate them in a future update of this review.
Ongoing studies
We identified six ongoing studies (Characteristics of ongoing studies).
Risk of bias in included studies
Please see Figure 2 for the 'Risk of bias' summary: review authors' judgments about each risk of bias item for each included study, and see Figure 3 for the 'Risk of bias' graph: review authors' judgments about each risk of bias item presented as percentages across all included studies.
Figure 2.
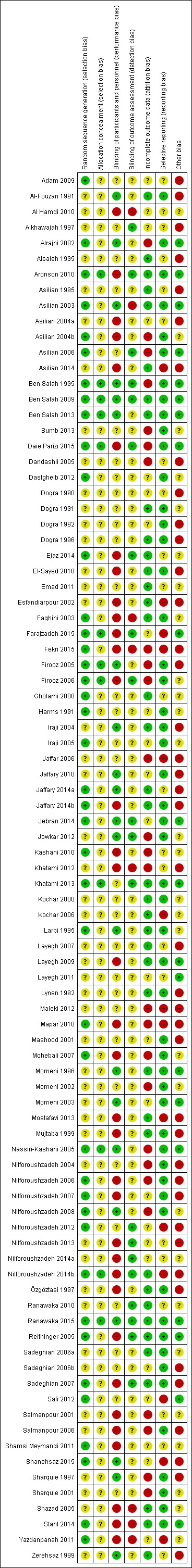
Risk of bias summary: review authors' judgments about each risk of bias item for each included study.
Figure 3.
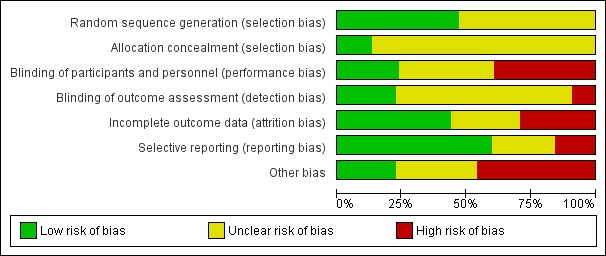
Risk of bias graph: review authors' judgments about each risk of bias item presented as percentages across all included studies.
Our assessment of the risk of bias in the included studies has broadly followed the criteria set in the protocol. We thought the quality of the RCTs was generally poor for the following reasons.
Allocation
Forty‐two (47.2%) studies clearly described an adequate method of random sequence generation, meriting a judgment of low risk of bias, whereas the risk of bias in the rest of the studies (47/89, 52.8%) was unclear (Characteristics of included studies; Figure 2; Figure 3).
Of the 42 studies that were at low risk for sequence generation, only 12 (13.5%) also used an adequate method of allocation concealment (Aronson 2010; Ben Salah 1995; Ben Salah 2009; Ben Salah 2013; Farajzadeh 2015; Firooz 2005; Firooz 2006; Khatami 2013; Nassiri‐Kashani 2005; Nilforoushzadeh 2014b; Daie Parizi 2015; Ranawaka 2015; see Characteristics of included studies for details). However, in most studies (77, 86.5%), the method used to conceal the allocation prior to assignment was unclear.
Blinding
Only 21 studies were at low risk of performance bias because they clearly reported blinding participants or used other methods that we judged as unlikely to add risk of bias. Thirty‐three studies did not provide enough information about the blinding of the participants. Among the 35 studies that did not blind the intervention and were at high risk of bias, there were some interventions that were impossible to blind due to the different administration routes (Esfandiarpour 2002; Farajzadeh 2015; Jaffary 2014b; Nilforoushzadeh 2013; Daie Parizi 2015; Sadeghian 2007; Salmanpour 2001; Salmanpour 2006).
With regard to the blinding of the outcome assessment, most studies 62/89 (69.7%) did not provide enough information about the blinding. Authors described 20 studies as blinded, and we therefore judged them to be at low risk of bias, whereas we judged 7 studies to be at high risk. See Characteristics of included studies for further details on who was blinded.
Incomplete outcome data
Of the 89 included studies, we considered the risk of bias for 39 (43.8%) to be low; for 24 (27.0%), unclear; and for 26 (29.2%), high.
Dropouts
The overall number of participants lost to follow‐up was 1439, or 13.6% of the total number of study participants included in the review. We have categorised the dropouts into groups according to the percentage of evaluable participants (Characteristics of included studies).
Intention‐to‐treat analyses
Sixty‐four studies accounted for losses to follow‐up, while the other 25 did not report dropouts. However, 55 out of the 64 studies did not carry out intention‐to‐treat (ITT) analyses, or rather they just assessed participants who completed treatment. For each study, we took all randomised participants into account when introducing the data in our tables. We assumed that missing data were treatment failures. Concerning the loss to follow‐up, it was not always possible to determine the arm in which the losses occurred, complicating ITT analyses. In that case, we introduced only available data into the tables for analysis.
Selective reporting
Of the 89 included studies, 53 (59.55%) reported all expected outcomes, meriting a judgment of low risk of bias. We considered 22 studies (25.72%) to be at unclear risk of bias. Fourteen studies (15.73%) failed to report, or reported only incompletely (making it impossible to enter the data into meta‐analysis), results for a key outcome that would be expected to have been reported, and we therefore considered these studies to be at high risk of bias.
Other potential sources of bias
We assessed other potential sources of bias such as sample size calculation, the reporting of Leishmania species and baseline comparability among intervention groups. If a study reported all three items correctly, we classified it as being at low risk of bias. If at least one of the items were not reported correctly, we classified it as being at high risk of bias, and if the study did not report enough information to assess if there were other biases present, we classified it as being at unclear risk of bias. Of the 89 included studies, we considered the risk of bias to be high in 41 (46.07%), unclear in 28 (31.46%), and low in 20 (22.47%).
Effects of interventions
Please see the 'Summary of findings' tables, where we have summarised the certainty of the body of evidence for two of the most clinically important comparisons (Table 1; Table 2). We have produced two forest plots for our primary outcome, with subgroups for each comparison (see Figure 4; Figure 5), and we have summarised adverse effects in seven tables (see Table 119; Table 120; Table 121; Table 122; Table 123; Table 124; Table 125).
Table 3.
Adverse effects of oral antibiotics
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Kochar 2000 | Quote: "Biochemical tests were done to detect any toxic effects of the drug." | Biochemical tests were done at the end of 1 week, 2 weeks and 4 weeks post‐ treatment. | I1: oral rifampicin 1200 mg/d I2: placebo |
Intervention 23 participants evaluated for AEs. Quote: "The drug was well‐tolerated and no side‐effects were seen in any participant." Placebo 23 participants evaluated for AEs. Not reported |
| Jaffar 2006 | Quote: "clinical examination, liver function tests, renal function tests" | Quote: "Before, during, and after completion of treatment" | I1: oral rifampicin 10 mg/kg/d I2: placebo |
Intervention 46 participants evaluated for AEs. Elevation liver enzymes: 1 (2%) Placebo 16 participants evaluated for AEs. Not reported |
| Kochar 2006 | Haemoglobin, leukocyte count, and liver test | Biochemical tests were done at the end of 2 week, 4 weeks and 6 weeks post‐treatment | I1: oral rifampicin + omeprazole I2: placebo |
Intervention 1 23 participants evaluated for AEs. Intervention 2 21 participants evaluated for AEs. Quote: "All participants tolerated the drug and placebo very well and no side effect was reported." |
| Layegh 2007 | Not described | Azythromycin group: monthly up to 4 months | I1: azithromycin 500 mg/d I2: IMMA 60 mg/kg/d |
Intervention 1 22 participants evaluated for AEs (35 lesions). Nausea and vomiting: 2 (9%) Intervention 2 27 participants evaluated for AEs (58 lesions). Myalgia: 3 (11%); Erythema: 1 (3.7%). |
| Adam 2009 | Quote: "Complete haemogram, including haemoglobin and liver and renal function tests Participants were questioned about expected adverse effects for 3 days (Days 5–7) following administration of the doses." | Haemogram: 1 and 2 months Clinical AEs: days 5‐7; 19‐21; 33‐35; 47‐19. |
I1: artesunate I2: placebo |
Intervention 1 20 participants evaluated for AEs. Placebo 21 participants evaluated for AEs. Skin rash with itching: 1 (4.7%) Quote: "There was no significant difference in biological tests (liver and renal function tests) in all the participants before and after treatment." |
| Ben Salah 2009 | Interview, physical examination, laboratory test, evaluation for pain, standardised questionnaire for the occurrence of systemic side effects (e.g. vertigo, tinnitus). Diminished hearing was verified with audiometer. Laboratory test. |
Quote: "Investigators observed each participant each day that the topical creams were administered and at follow‐up study visits (days 50‐100‐180). Clinical and laboratory evidence of side effects was determined on D10 and D20." | I1: WR 279,396 I2: placebo |
Intervention 50 participants evaluated for AEs. Erythema at the site of application: 15 (30%); mild pain within 30 minutes of application: 7 (14 %); mild increases and decreases in hearing acuity from baseline: 14 (28%); change hearing acuity: 14 (28%); vertigo: 0 (0%); Increase serum creatinine: 0 (0%); Death: 0 (0%) Placebo 42 participants evaluated for AEs. Erythema at the site of application: 10 (24%); mild pain within 30 minutes of application: 6 (14 %); mild increases and decreases in hearing acuity from baseline: 9 (21%); change hearing acuity: 9 (21%); vertigo: 0 (0%); increase serum creatinine: 0 (0%); death: 0 (0%) |
| Dastgheib 2012 | Quote: "Participants were interviewed and underwent laboratory tests three times" | Quote: "In Allopurinol group, the participants which were visited and received medication underwent laboratory tests three times (before, one month a2fter, and end of the treatment) | I1: azithromycin + allopurinol I2: IMMA |
Intervention 1 36 participants evaluated for AEs. Gastrointestinal complaints and headache severe: 1 (2.7%); slight gastrointestinal complications (nausea, heartburn, and epigastric pain): 3 (8.3%) Intervention 2 35 participants evaluated for AEs. Myalgia: 2 (5.7%) |
AE: adverse effect; IMMA: intramuscular meglumine antimoniate.
Table 4.
Adverse effects of topical paromomycin
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Asilian 1995 | Clinical evaluation and laboratory tests | Days 15, 45 and 105 | I1: paromomycin I2: placebo |
126 participants evaluated for AEs. During treatment: oedema, local pain, vesiculation: 1 (0.7%) After treatment: redness, pain, vesiculation, and inflammation: 8 (6.3%) Quote: "There were no significant differences in four laboratory test results of safety (SGOT, BUN, Hb, and WBC) between the groups either before or after treatment." |
| Ben Salah 1995 | Clinical evaluation, physical examination, advice to participants, laboratory test: liver function, haemoglobin and white blood cell count | Days 15, 45 and 105 | I1: paromomycin I2: placebo |
57 participants evaluated for AEs. Quote: "A local reaction (inflammation, vesication, pain and/or red ness) was recorded for 12 participants, with no significant difference between the 2 groups." Laboratory test changes: 0 (0%) |
| Özgöztasi 1997 | Not described | At the end of treatment (day 30) and 1 month post‐treatment | I1: paromomycin + MBCL I2: oral ketoconazole |
40 participants (62 lesions) evaluated for AEs Quote: "Treatment‐related adverse effects were only observed in the paromomycin group. The most common side‐effect was the development of irritant contact dermatitis. No subjects withdrew because of this adverse effect." |
| Asilian 2003 | Clinical evaluation | Days 15, 29, 45 and 105 | I1: paromomycin I2: placebo |
108 participants evaluated for AEs. Quote: "Treatment was well tolerated, and no adverse reactions to the ointment were observed or reported in either group." |
| Faghihi 2003 | Not described | Quote "Clinical evaluation and follow‐up were performed fortnightly until 1 month post treatment and then monthly until 3 months post treatment, and finally every 3 months until 1 year post treatment" | I1: paromomycin + urea I2: ILMA weekly |
Not described |
| Shazad 2005 | Not described | Week 1 and week 6 post‐treatment and at 6 months after treatment was completed | I1: paromomycin + urea I2: ILMA weekly |
30 participants evaluated for AEs. Cutaneous reactions (erythematosus, urticaria or lymphadenitis with pain): 1 (3%) Quote: "No systemic toxic reaction attributable to the drug was observed." |
| Iraji 2005 | Clinical evaluation | Days 7, 14, 21 and 30 | I1: paromomycin I2: placebo |
30 participants evaluated for AEs. Mild contact dermatitis: 3 (10%) |
| Asilian 2006 | Not described | Weekly during treatment and monthly for up 2 months | I1: photodynamic therapy I2: paromomycin I3: placebo |
19 participants (34 lesions) evaluated for AEs. Quote: "Adverse side‐effects seen in some participants in all groups were pruritus, burning, redness, discharge, oedema, and pain, but all were generally mild and tolerable." |
| Ben Salah 2009 | Quote: "Renal toxic effects and ototoxic effects from aminoglycoside exposure were ascertained by means of serum creatinine measurements at the end of therapy (at 20 days) and participants' daily reports of tinnitus and vertigo." | Quote: "Safety end points were assessed daily during therapy (20 days)." |
I1:Paromomycin ‐Gentamicin I2: paromomycin Alone I3: vehicle Control |
Intervention 1: 125 participants evaluated for AEs. Erythema: 6 (5%); local infection: 0 (0%); inflammation: 0 (0%); vesicles mild‐moderate: 31 (25%); mild oedema: 2 (2%); pain: 2 (2%); mild bronchitis: 5 (4%); paronychia: 2 (2%); superinfection: 3 (2%); upper respiratory tract infection: 0 (0%); oropharyngeal pain: 4 (3%); skin irritation: 3 (2%); tinnitus: 0 (0%); vertigo: 0 (0%); creatinine serum changes: 0 (0%) Intervention 2: 125 participants evaluated for AEs. Erythema: 7 (6%); local infection: 0 (0%); inflammation: 0 (0%); vesicles mild‐moderate: 32 (26%); mild‐moderate oedema: 3 (3%); pain: 2 (2%); bronchitis mild‐moderate: 3 (3%); paronychia: 0 (0%); superinfection: 0 (0%); upper respiratory tract infection: 2 (2%); oropharyngeal pain: 3 (2%); skin irritation: 9 (7%); tinnitus: 0 (0%); vertigo: 0 (0%); creatinine serum changes: 0 (0%) |
AE: adverse effect; BUN: blood urea nitrogen; Hb: haemoglobin; ILMA: intralesional meglumine antimoniate; SGOT: serum glutamic‐oxaloacetic transaminase; WBC: white blood cells.
Table 5.
Adverse effects of intralesional zinc sulphate
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Sharquie 1997 | Not described | Quote: "Participants were seen at 10‐15 day intervals after injection, and at 6 weeks post treatment" | I1: IL 2% zinc sulphate I2: IL 7% sodium chloride I3: ILSSG 2‐5 mL per lesion |
19 participants evaluated for AEs. Quote: "Apart from pain at the time of injection, no appreciable side‐effect was noted." |
| Iraji 2004 | Not described | Not described | I1: IL zinc sulphate I2: ILMA weekly |
31 participants evaluated for AEs. Severe pain caused vasovagal shock: 2 (6.4%) |
| Firooz 2005 | Not described | Not described | I1: IL zinc sulphate I2: ILMA weekly |
36 participants evaluated for AEs. Pain: 13 (36.1%); burning at site injection: 3 (8.4%); itching: 3 (8.4%); inflammation: 7 (19.4%) |
| Maleki 2012 | Not described | 14, 28, 42, and 56 days after starting the treatment | I1: IL 2% zinc sulphate I2: ILMA weekly |
24 participants evaluated for AEs. Quote: "The side effects seen in both groups were pain after injection and hyperpigmentation." Burning after injection and necrosis of the lesions: 24 (100%); inflammation and swelling: 3 (12.5%) |
AE: adverse effect; IL: intralesional; ILMA: intralesional meglumine antimoniate; ILSSG: intralesional sodium stibogluconate.
Table 6.
Adverse effects of intralesional hypertonic sodium chloride solution
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Sharquie 1997 | Not described | Quote: "Participants were seen at 10‐15 day intervals after injection, and at 6 week post treatment" | I1: IL 2% zinc sulphate I2: IL 7% HSCS I3: ILSSG 2‐5 mL per lesion |
17 participants evaluated for AEs. Quote: "No side‐effect other than pain at the time of injection was noted." |
| Sadeghian 2006b | Not described | Not described | I1: IL 5% HSCS I2: ILMA 0.5‐1 mL/week |
36 participants evaluated for AEs. Allergic reaction (erythema, oedema, and pruritus): 0 (0%); sporotrichoid dissemination: 3 (8.3%) |
| Ranawaka 2010 | Not described | Quote: "Participants were seen weekly for the first three injections; fortnightly for the fourth and fifth injections; then monthly until cure. Participants were followed‐up every 3 months after cure for 18 months to assess recurrences and evidence of visceralization." | I1: ILSSG I2: IL 7% HSCS |
67 participants evaluated for AEs. Leishmaniasis recidivans: 0 (0%) Quote: "There were no systemic side effects with SSG or HS. Pain during injection was the only local side effect noted with both therapies. After healing, scarring was minimal, but postinflammatory hyperpigmentation was observed in all participants for both treatments, which faded out over 6–8 months." |
AE: adverse effect; IL: intralesional; ILSSG: intralesional sodium stibogluconate; HSCS: hypertonic sodium chloride solution.
Table 7.
Adverse effects of laser
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Asilian 2004b | Not described | 1, 3, 4, 8, 12 and 24 weeks after treatment | I1: CO₂ I2: IMMA 50 mg/kg/d |
123 participants evaluated for AEs. Quote: "Complications were seen in (4) 4.5% of participants and included hyperpigmentation, persistent redness." Hypertonic scars: 5 (4%) |
| Shamsi Meymandi 2011 | Quote: "Follow‐up was performed and any side‐effects were recorded." | Quote: "Follow‐up evaluation was performed by clinical assessment of treated lesions at weeks 2, 6, 12 and 16." | I1: CO₂ I2: cryotherapy + MA |
80 participants (95 lesions) evaluated for AEs. Hyperpigmentation + trivial scar: 20 (25%); atrophic scar: 7 (8.75%); hypertrophic scar: 1 (1.25%); sporotrichoid: 1 (1.25%); raised papular lesions: 1 (1.25%); persistent erythema: 3 (3.75%); hypopigmentation + trivial scar: 4 (5%) |
| Nilforoushzadeh 2014a | Not described | Quote: "Participants were followed in the first, third, and sixth months after treatment with the final evaluation in the sixth month." | I1: ablative CO₂ laser I2: fractional CO₂ laser |
Intervention 1: 30 participants evaluated for AEs. Erythema: 2 (6.7%) Intervention 2: 30 participants evaluated for AEs. Erythema: 4 (13.3%) |
AE: adverse effect; IMMA: intramuscular meglumine antimoniate; MA: meglumine antimoniate.
Table 8.
Adverse effects of cryotherapy
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Asilian 2004a | Not described | Fortnightly until 6 months post‐treatment and 2 weeks and 4 weeks post‐treatment | I1: cryotherapy + ILMA I2: cryotherapy alone I3: ILMA alone |
Intervention 1: 100 participants evaluated for AEs. Postinflammatory hypopigmentation: 5 (5%) Intervention 2: 200 participants evaluated for AEs. Postinflammatory hypopigmentation: 10 (5%) |
| Salmanpour 2006 | Not described | Not described | I1: ILMA alone I2: cryotherapy alone I3: cryotherapy + ILMA |
Intervention 2: 20 participants evaluated for AEs. Erythema and oedema of the lesions and perilesional area: (28%) Intervention 3: 20 participants evaluated for AEs. Erythema and oedema of the lesions and perilesional area: (33%) Quote: "There were no serious side‐effects in any of the treatment groups" |
| Layegh 2009 | Not described | Quote: "Weekly for up to six weeks of treatment and six months after." | I1: cryotherapy I2: ILMA |
36 participants evaluated for AEs. Hypopigmentation: 2 (5.5%); hyperpigmentation: 7 (19.4%). Quote: "the most common adverse reactions were erythema and oedema of the treated site, which appeared during the initial hours of treatment, and blistering of the treatment site, which became evident 1–2 days after treatment and responded well to local treatment." |
| Shamsi Meymandi 2011 | Quote: "Follow‐up was performed and any side‐effects were recorded." | Quote: "Follow‐up evaluation was performed by clinical assessment of treated lesions at weeks 2, 6, 12 and 16." | I1: CO₂ I2: cryotherapy + MA |
80 participants (95 lesions) evaluated for AEs. Hyperpigmentation + trivial scar: 15 (18.7%); atrophic scar: 6 (7.5%); hypertrophic scar: 0 (0%); sporotrichoid: 0 (0%); raised papular lesions: 0 (0%); persistent erythema: 0 (0%); hypopigmentation + trivial scar: 15 (18.8%) |
| Jowkar 2012 | Quote: "During these visits the healing process of the ulcer, change of diameter and induration of lesions and complications were assessed." | Quote: "The participants were evaluated every 2 weeks up to 12 weeks." | I1: cryotherapy + 3% salicylic + 3% sodium nitrite I2: cryotherapy + 3% salicylic + placebo |
Intervention 1: 36 participants evaluated for AEs. Erythema, a burning sensation and skin irritation: 7 (19.4%) Intervention 2: 27 participants evaluated for AEs. Erythema, a burning sensation and skin irritation: 1 (3.7%) |
AE: adverse effect; ILMA: intralesional meglumine antimoniate; MA: meglumine antimoniate.
Table 9.
Adverse effects of thermotherapy
| Study | Method of assessment | Timing | Interventions | Adverse effects |
| Reithinger 2005 | Quote: "The occurrence of adverse effects was evaluated blindly by means of participant interviews and physical examinations." | Quote: "The occurrence of adverse effects was evaluated … during follow‐up visits." | I1: ILSSG 2‐5 mL per lesion I2: IMSSG 20 mg/kg I3: thermotherapy |
138‐108 participants evaluated for AEs. Secondary infections: 8 (5.7%). Quote: "The original CL ulcer often increased in size immediately after and up to 2 weeks after treatment." |
| Sadeghian 2007 | Quote: "Appearance of lesions at subsequent follow‐up visits and occurrence of unwanted side‐effects were also recorded on the form." | Weekly 4 weeks and monthly up to 6 months | I1: thermotherapy I2: ILMA weekly |
57 participants (83 lesions) evaluated for AEs. Satellite lesions: 1 (1.7%) |
| Aronson 2010 | Quote: "Interview, physical examination, laboratory testing (complete blood count, creatine phosphokinase, amylase, lipase, complete metabolic profile), and electrocardiograms." | Quote: "Daily for the first 10 days and follow‐up at 2, 6, and 12–24 months post treatment" | I1: thermotherapy I2: ILSSG |
27 participants evaluated for AEs. Serious AE: 4 (15%); ECG changes: 10 (37%); abdominal discomfort: 1 (4%); wound infection: 5 (19%); musculoskeletal: 5 (19%); headache: 3 (11%); fatigue: 5 (19%); rash: 1 (4%); blister reaction: 25 (93%); erythema: 7 (26%); oozing: 21 (78%) |
| Safi 2012 | Quote: "The occurrences of adverse effects were evaluated by means of participant interviews and physical examinations during follow‐up visits." | Quote: "During treatment, all participants were then followed for four visits at weekly intervals … After initial treatment, all participants were scheduled for four subsequent follow‐up visits: 10 days after baseline and 1‐month, 2 months and 6 months after treatment." | I1: thermotherapy I2: ILMA weekly |
189 participants evaluated for AEs Not reported |
| Bumb 2013 | Not described | Not described | I1: radiofrequency heat treatment I2: ILSSG |
Quote: "RFHT was cosmetically acceptable because it was associated with less scarring and hyperpigmentation compared with intralesional SSG injections." |
| Jebran 2014 | Quote: "In case of clinical signs for a superinfection, a smear was taken, Gram stained and microscopically evaluated for the presence of bacteria and/or fungi." | Quote: "Adverse events such as bacterial or fungal superinfections of the wounds, the formation or scars and the rate of re‐ulcerations were monitored during the treatment and follow‐up period." | I1: electrocauterisation + DAC N‐055. I2: electrocauterisation + placebo. |
Intervention 1: 38 participants evaluated for AEs. Bacterial and fungal superinfections: 3 (8.0%); Keloïd formation: 2 (5%) Intervention 2: 32 participants evaluated for AEs. Bacterial and fungal superinfections: 3 (9.0%); Keloïd formation: 2 (6%) |
AE: adverse effect; CL: cutaneous leishmaniasis; ILMA: intralesional meglumine antimoniate; ILSSG: intralesional sodium stibogluconate; IMSSG: intramuscular sodium stibogluconate.
Figure 4.

Forest Plot of Primary Outcome: Lesions Cured (Various comparisons)
Figure 5.

Forest plot of primary outcome: 1.2 Participants cured (Various comparisons)
We describe only those outcomes with data below. If a particular primary, secondary, or tertiary outcome is missing, then this is because we did not find suitable data. Pooling of outcome data across studies to provide a summary estimate of effect was only possible for five interventions and comparisons; these investigated the effects of topical paromomycin, itraconazole, radiofrequency waves, oral dapsone, and intramuscular meglumine antimoniate (IMMA). We categorised a substantial number of the studies included in this review as being at unclear or high risk of bias (see Figure 2 and Figure 3), so readers should interpret the results with caution.
We have addressed our pre‐specified outcomes under the following intervention headings.
Systemic and intralesional antimonials.
Non‐antimonial systemic treatments.
Non‐antimonial topical or intralesional therapies.
Physical therapies.
Measures for promoting healing.
Alternative therapies.
We have also referred in the text to tables where we have summarised reports of adverse effects of the different interventions.
1. Systemic and intralesional antimonials
1.1 Meglumine antimoniate
1.1.1 Different doses of intralesional meglumine antimoniate
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Pakistan compared intralesional meglumine antimoniate (ILMA) weekly versus fortnightly, until complete cure or up to eight weeks (Mujtaba 1999). Two months after treatment, 92% (102/111) of the lesions were cured in the weekly, and 85.6% (89/104) in the fortnightly group. There was no statistical difference between weekly and fortnightly administration (risk ratio (RR) 1.07, 95% confidence interval (CI) 0.98 to 1.18; N = 215; 1 study, Analysis 1.1).
Analysis 1.1.
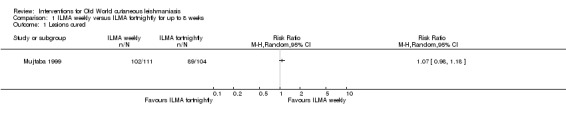
Comparison 1 ILMA weekly versus ILMA fortnightly for up to 8 weeks, Outcome 1 Lesions cured.
Secondary outcome: speed of healing (time taken to be 'cured')
Most lesions had healed at six weeks in both groups with minimal or absent scarring.
Secondary outcome: adverse effects
Authors did not report adverse effects in any of the participants, except transient pain at the site of the injections in both groups.
1.1.2 Intralesional versus intramuscular meglumine antimoniate
Primary outcome: percentage of lesions cured after the end of treatment
One RCT from Saudi Arabia compared ILMA every other day versus IMMA 6 days a week, over a 30‐day period until the lesions had blanched (Alkhawajah 1997). The authors reported a comparable cure rate: 68.6% (48/70) and 59.7% (46/77) of the of lesions in the respective groups at the end of the treatment period (RR 1.15, 95% CI 0.90 to 1.46; N = 147; 1 study, Analysis 2.1).
Analysis 2.1.
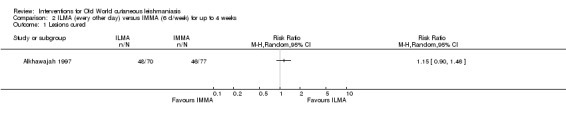
Comparison 2 ILMA (every other day) versus IMMA (6 d/week) for up to 4 weeks, Outcome 1 Lesions cured.
Secondary outcome: adverse effects
Regarding adverse effects, both groups reported pain at the site of injection, but it was greater in the ILMA group.
1.1.3 Different doses of IMMA
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Syria compared IMMA 60 mg/kg/d plus placebo versus IMMA 30 mg/kg/d plus placebo or cimetidine for three weeks (Shanehsaz 2015). Three months after treatment, 91.1% (82/90) of the lesions were cured in the IMMA 60 mg plus placebo group, 84.6% (127/150) in the IMMA 30 mg plus cimetidine group, and 78.3% (94/120) in IMMA 30 mg plus placebo group. We did not find any significant differences in cure rates between the IMMA 30 mg plus cimetidine group versus the IMMA 30 mg plus placebo group (RR 1.08, 95% CI 0.96 to 1.21; N = 270; 1 study, Analysis 3.1) or between the IMMA 30 mg plus cimetidine group versus the IMMA 60 mg group (RR 0.93, 95% CI 0.85 to 1.02; N = 240; 1 study, Analysis 4.1). Cure rates were significantly higher in the IMMA 60 mg group compared with the IMMA 30 mg plus placebo group (RR 1.16 95% CI 1.04 to 1.30; N = 210; 1 study, Analysis 5.1).
Analysis 3.1.
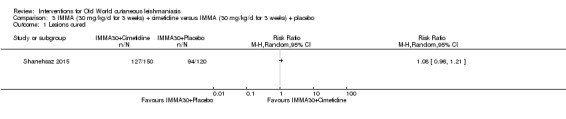
Comparison 3 IMMA (30 mg/kg/d for 3 weeks) + cimetidine versus IMMA (30 mg/kg/d for 3 weeks) + placebo, Outcome 1 Lesions cured.
Analysis 4.1.
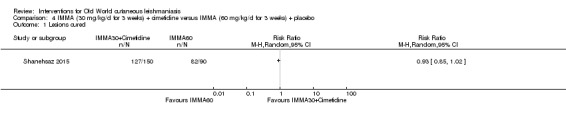
Comparison 4 IMMA (30 mg/kg/d for 3 weeks) + cimetidine versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 1 Lesions cured.
Analysis 5.1.
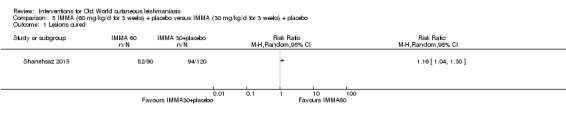
Comparison 5 IMMA (60 mg/kg/d for 3 weeks) + placebo versus IMMA (30 mg/kg/d for 3 weeks) + placebo, Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
A pooled analysis of two trials showed that cure rates in the IMMA 60 mg group were better than in the IMMA 30 mg plus placebo group (RR 0.83, 95% CI 0.71 to 0.96; N = 148; Nilforoushzadeh 2008; Shanehsaz 2015, Analysis 6.1). We assessed the certainty of evidence as low, which means we are uncertain whether there is much difference in cure rate between these two concentrations of IMMA.
Analysis 6.1.
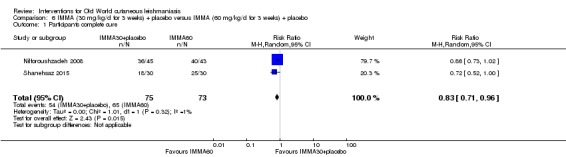
Comparison 6 IMMA (30 mg/kg/d for 3 weeks) + placebo versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 1 Participants complete cure.
An RCT from Iran compared IMMA 60 mg/kg/d plus placebo versus IMMA 30 mg/kg/d plus placebo or omeprazole for three weeks (Nilforoushzadeh 2008). Three months after treatment, there was complete cure in 93% (40/43) of participants in the IMMA 60 mg plus placebo group, 89% (32/36) in the IMMA 30 mg plus omeprazole group, and 80% (36/45) in the IMMA 30 mg plus placebo group. There were no significant differences in cure rates in any comparisons: IMMA 30 mg plus omeprazole versus IMMA 60 mg (RR 0.96, 95% CI 0.83 to 1.10; N = 79; 1 study, Analysis 7.1), IMMA 30 mg plus placebo versus IMMA 60 mg plus placebo (RR 0.86, 95% CI 0.73 to 1.02; N = 88; 1 study, Analysis 8.1), or IMMA 30 mg plus omeprazole versus IMMA 30 mg plus placebo (RR 1.11, 95% CI 0.92 to 1.34; N = 81; 1 study, Analysis 9.1).
Analysis 7.1.
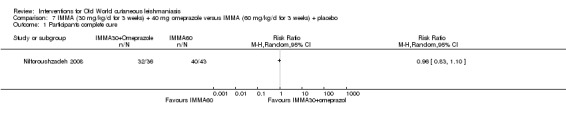
Comparison 7 IMMA (30 mg/kg/d for 3 weeks) + 40 mg omeprazole versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 1 Participants complete cure.
Analysis 8.1.

Comparison 8 IMMA (30 mg/kg/d for 3 weeks) + placebo versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 1 Participants complete cure.
Analysis 9.1.
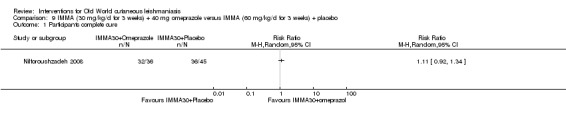
Comparison 9 IMMA (30 mg/kg/d for 3 weeks) + 40 mg omeprazole versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, Shanehsaz 2015 reported serious adverse effects in 83.3% (n = 25) participants in the IMMA 60 mg dose group and 60% (n = 18) participants in the IMMA 30 mg group; 40% (n = 12) of participants in the IMMA 60 mg dose group and 23.3% (n = 7) in the 30 mg group experienced skin hypersensitivity, while there were five and three cases, respectively, of cardiac toxicity (QT prolongation) (Analysis 6.2). Hence, there were fewer serious adverse effects (RR 0.72, 95% CI 0.52 to 1.00; N = 60, 1 study), fewer skin reactions (RR 0.58, 95% CI 0.27 to 1.28; N = 60, 1 study), and less QT prolongation (RR 0.60, 95% CI 0.16 to 2.29; N = 60, 1 study) in the IMMA 30 mg plus placebo group, but none of the results were significant.
Analysis 6.2.
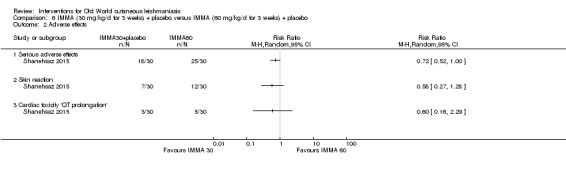
Comparison 6 IMMA (30 mg/kg/d for 3 weeks) + placebo versus IMMA (60 mg/kg/d for 3 weeks) + placebo, Outcome 2 Adverse effects.
In Nilforoushzadeh 2008, the authors reported one case of anaphylactic shock to meglumine antimoniate (not described).
1.1.4 ILMA alone versus ILMA plus silver or non‐silver dressing
Primary outcome: percentage of lesions cured after the end of treatment
One RCT from Iran compared weekly injections of ILMA for 6 weeks alone versus ILMA at the same dose plus silver or non‐silver dressing (Khatami 2013). We performed an ITT analysis considering the lesions allocated in each arm at the beginning of the study: 26 participants with 45 lesions in the ILMA‐alone group, 31 participants with 60 lesions in the ILMA plus silver dressing group, and 26 participants with 53 lesions in the ILMA plus non‐silver dressing group. At one month after treatment, 35.5% (16/45) of lesions were cured in the ILMA‐alone group, 33.3% (20/60) in the ILMA plus silver dressing group, and 35.8% (19/53) in the ILMA plus non‐silver dressing group. There were no significant differences in cure rates between groups: ILMA plus non‐silver dressing versus ILMA alone (RR 1.01, 95% CI 0.59 to 1.72; N = 98, Analysis 10.1); ILMA plus silver dressing versus ILMA alone (RR 0.94, 95% CI 0.55 to 1.60; N = 105, Analysis 11.1); or ILMA plus silver dressing versus ILMA plus non‐silver dressing (RR 0.93, 95% CI 0.56 to 1.54; N = 113, Analysis 12.1).
Analysis 10.1.
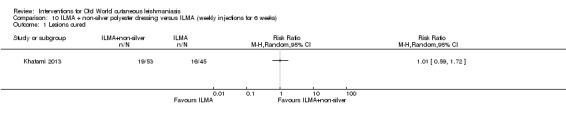
Comparison 10 ILMA + non‐silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 1 Lesions cured.
Analysis 11.1.

Comparison 11 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 1 Lesions cured.
Analysis 12.1.
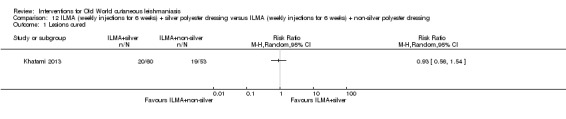
Comparison 12 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks) + non‐silver polyester dressing, Outcome 1 Lesions cured.
Secondary outcome: adverse effects
Regarding adverse effects, participants reported itching and burning in 7.5% (n = 3) of lesions in ILMA alone group, 10.9% (n = 6) of lesions in the ILMA plus silver group, and 17.7% (n = 8) lesions in the ILMA plus non‐silver group; there was also oedema in 12.5% (n = 5), 7.2% (n = 4), and 6.6% (n = 3) of the lesions, respectively. One lesion in each group showed exudation, and another lesion was accompanied by dermatitis in the ILMA plus non‐silver group. There were no significant differences in any adverse effects rates in any comparison (Analysis 10.2; Analysis 10.3; Analysis 11.2; Analysis 11.3; Analysis 12.2; Analysis 12.3).
Analysis 10.2.
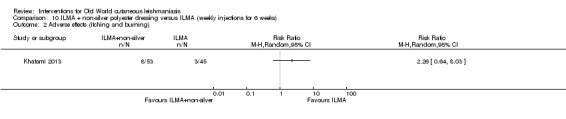
Comparison 10 ILMA + non‐silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 2 Adverse effects (itching and burning).
Analysis 10.3.
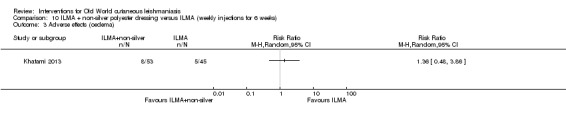
Comparison 10 ILMA + non‐silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 3 Adverse effects (oedema).
Analysis 11.2.
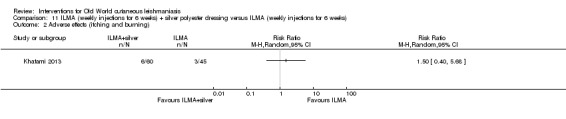
Comparison 11 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 2 Adverse effects (itching and burning).
Analysis 11.3.
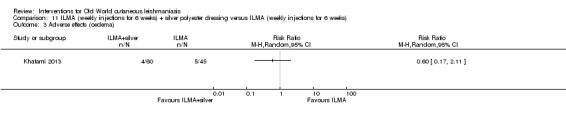
Comparison 11 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks), Outcome 3 Adverse effects (oedema).
Analysis 12.2.

Comparison 12 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks) + non‐silver polyester dressing, Outcome 2 Adverse effects (itching and burning).
Analysis 12.3.

Comparison 12 ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks) + non‐silver polyester dressing, Outcome 3 Adverse effects (oedema).
1.1.5 ILMA plus gel mask versus ILMA plus vehicle
Primary outcome: percentage of participants with a complete cure after the end of treatment
One RCT from Iran compared ILMA (weekly injections for six weeks) plus gel mask twice a day with ILMA (weekly injections for six weeks) plus placebo (Mostafavi 2013). At the end of treatment, 75% (15/20) and 65% (13/20) of participants, respectively, had complete cure. There were no significant differences in cure rates between groups (RR 1.15, 95% CI 0.77 to 1.74; N = 40; 1 study, Analysis 13.1).
Analysis 13.1.

Comparison 13 ILMA (weekly injections for 6 weeks) + gel mask twice a day versus ILMA (weekly injections for 6 weeks) + vehicle, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, none of the participants from either group suffered severe adverse effects such as local irritation or pruritus.
1.2 Sodium stibogluconate
1.2.1 Intralesional versus intramuscular sodium stibogluconate (SSG)
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared ketoconazole 200 mg three times daily for four weeks plus intralesional sodium stibogluconate (ILSSG) (100 mg/mL on days 1, 3, and 5) versus intralesional plus intramuscular sodium stibogluconate (20 mg/kg/d intralesionally on days 1, 3, and 5, and 80 mg/kg/d intramuscularly simultaneously) versus ILSSG alone (100 mg/mL on days 1, 3, and 5) (El‐Sayed 2010). The authors reported that two months after treatment, 92.3% (12/13), 93.3% (14/15), and 58.3% (7/12) of lesions were cured in the respective groups. There were no significant differences between intralesional plus intramuscular SSG versus ILSSG alone (RR 1.60, 95% CI 0.97 to 2.63; N = 27; 1 study, Analysis 14.1).
Analysis 14.1.

Comparison 14 ILSSG (20 mg/kg/d) + IMSSG (remaining total dose days 1, 3, 5) versus ILSSG (1000 mg/mL days 1, 3, 5), Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Afghanistan compared ILSSG (five injections of 2 mL to 5 mL every 5 to 7 days, depending on lesion size) for up to 29 days versus intramuscular sodium stibogluconate (IMSSG) (20 mg/kg/d for 21 days) (Reithinger 2005). Two months after treatment, 47.3% (70/148) and 18% (26/144) of participants had complete cure in the ILSSG and IMSSG groups, respectively (RR 2.62, 95% CI 1.78 to 3.86; N = 292; 1 study, Analysis 15.1).
Analysis 15.1.

Comparison 15 ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 d for 29 days) versus IMSSG (20 mg/kg/d for 3 weeks), Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
In Reithinger 2005, the speed of healing took a median of 75 days in the ILSSG group and 100 days or more for the IMSSG group (the original paper reported that the time to cure was significantly shorter for participants treated with thermotherapy; P = 0.003, by the log‐rank test).
Secondary outcome: adverse effects
Regarding adverse effects, Reithinger 2005 reported that one participant experienced bradycardia and one an undefined local reaction in the ILSSG group. In the IMSSG group, one participant reported bradycardia, one tachycardia, and one palpitation. There were no significant differences in adverse effects rates between groups (RR 0.32, 95% CI 0.03 to 3.08; N = 292; 1 study, Analysis 15.2). In El‐Sayed 2010, all participants from ILSSG group suffered pain and swelling at the intralesional site, which calmed on its own within two days. IMSSG was associated with pain at the injection site.
Analysis 15.2.
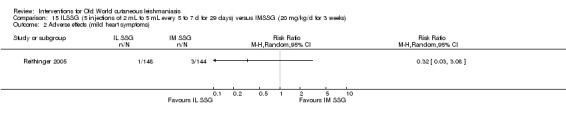
Comparison 15 ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 d for 29 days) versus IMSSG (20 mg/kg/d for 3 weeks), Outcome 2 Adverse effects (mild heart symptoms).
2. Non‐antimonial systemic treatments
2.1 Oral antifungals
2.1.1 Different doses of ketoconazole
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Kuwait compared oral ketoconazole at 600 mg/d versus 800 mg/d for six weeks or until the participant was cured, whichever occurred earlier (Alsaleh 1995). The authors reported that at the end of treatment, 66.7% (12/18) and 60% (9/15) of participants in the respective groups had a complete cure (RR 1.11, 95% CI 0.66 to 1.88; N = 33; 1 study Analysis 16.1). Thereafter, investigators followed up participants every one to two months for a period of six months, with no change.
Analysis 16.1.
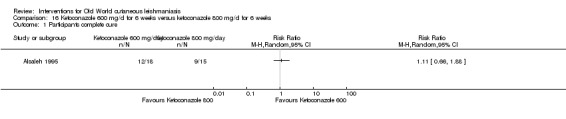
Comparison 16 Ketoconazole 600 mg/d for 6 weeks versus ketoconazole 800 mg/d for 6 weeks, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
None of the participants from either group relapsed during a six‐month follow‐up. With regard to adverse effects, one participant had nausea and vomiting in the ketoconazole 800 mg/d group. There were no significant differences in adverse effects rates between groups (RR 0.28, 95% CI 0.01 to 6.43; N = 33; 1 study, Analysis 16.2).
Analysis 16.2.
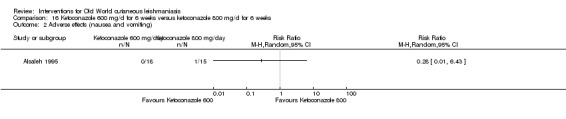
Comparison 16 Ketoconazole 600 mg/d for 6 weeks versus ketoconazole 800 mg/d for 6 weeks, Outcome 2 Adverse effects (nausea and vomiting).
2.1.2 Ketoconazole versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared oral ketoconazole 600 mg/d for 30 days versus six to eight injections of ILMA, biweekly (Salmanpour 2001). The authors reported that six weeks after treatment, 89% (57/64) and 72% (23/32) of participants in the respective groups had a complete cure. There were no significant differences between groups (RR 1.24, 95% CI 0.98 to 1.56; N = 96; 1 study, Analysis 17.1).
Analysis 17.1.
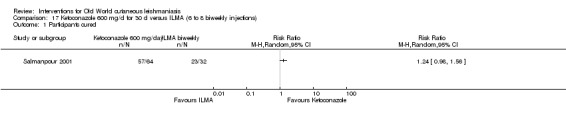
Comparison 17 Ketoconazole 600 mg/d for 30 d versus ILMA (6 to 8 biweekly injections), Outcome 1 Participants cured.
Secondary outcome: adverse effects
In the ketoconazole group the most common side effect cited was nausea and abdominal pain, and liver enzymes doubled in two participants, returning to normal two weeks after the end of treatment. There were no significant differences in adverse effects rates between groups (RR 2.54, 95% CI 0.13 to 51.36; N = 96; 1 study, Analysis 17.2). In the ILMA group, the most common side effect was redness and swelling after the injection. None of the side effects were significant enough to discontinue treatment.
Analysis 17.2.
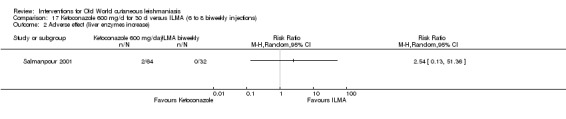
Comparison 17 Ketoconazole 600 mg/d for 30 d versus ILMA (6 to 8 biweekly injections), Outcome 2 Adverse effect (liver enzymes increase).
2.1.3 Ketoconazole plus ILSSG versus ILSSG alone versus ILSSG plus IMSSG
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared oral ketoconazole plus ILSSG for four weeks versus ILSSG plus IMSSG versus ILSSG alone (El‐Sayed 2010). The authors reported that two months after treatment, 92.3% (12/13), 93.3% (14/15), and 58.3% (7/12) of the lesions were cured in the respective groups. There were no significant differences between comparisons of either ketoconazole plus ILSSG versus ILSSG alone (RR 1.58, 95% CI 0.96 to 2.62; N = 25; 1 study, Analysis 18.1) nor between ketoconazole plus ILSSG versus ILSSG plus IMSSG (RR 0.99, 95% CI 0.80 to 1.22; N = 28; 1 study, Analysis 19.1).
Analysis 18.1.

Comparison 18 ILSSG (100 mg/mL days 1, 3, 5) + oral ketoconazole (600 mg/d for 4 weeks) versus ILSSG (100 mg/mL days 1, 3, 5), Outcome 1 Lesions cured.
Analysis 19.1.
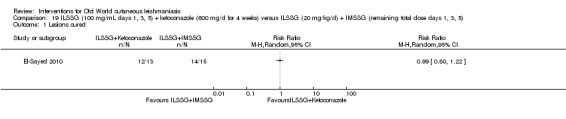
Comparison 19 ILSSG (100 mg/mL days 1, 3, 5) + ketoconazole (600 mg/d for 4 weeks) versus ILSSG (20 mg/kg/d) + IMSSG (remaining total dose days 1, 3, 5), Outcome 1 Lesions cured.
Secondary outcome: adverse effects
Regarding adverse effects, in one trial all participants complained of pain and swelling at the intralesional injection site that subsided on its own within days; no participants in the oral ketoconazole group reported adverse effects (El‐Sayed 2010).
We also found one trial comparing oral ketoconazole with topical paromomycin/MBCL (Özgöztasi 1997); we assess this comparison in the topical paromomycin section (section 3.2 below).
2.1.4 Itraconazole versus placebo
Primary outcome: percentage of participants with a complete cure after the end of treatment
Three RCTs compared itraconazole versus placebo and reported complete cure three months after the end of treatment as a primary outcome (Al‐Fouzan 1991; Dogra 1996; Nassiri‐Kashani 2005). Although heterogeneity was high (P = 0.02; I² = 73%), the three studies obtained results favouring itraconazole, so we decided to maintain the pool of results and the meta‐analysis (RR 3.70, 95% CI 0.35 to 38.99; N = 244; 3 studies, Analysis 22.1).
Analysis 22.1.
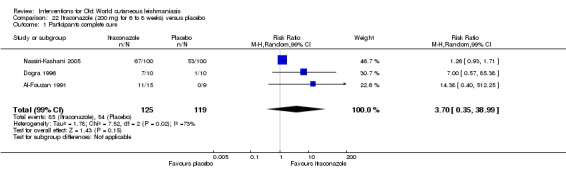
Comparison 22 Itraconazole (200 mg for 6 to 8 weeks) versus placebo, Outcome 1 Participants complete cure.
Please see Table 1, where we assessed the certainty of evidence as very low, which means we are very uncertain about the difference in cure with itraconazole compared to placebo.
Two RCTs evaluated complete cure of participants at other time points. An RCT from Iran compared oral itraconazole for three weeks versus placebo (Momeni 1996). Fifty‐one days after treatment, results showed complete cure of participants in 51.4% (36/70) and 38.6% (27/70) of the participants, respectively. There were no significant differences in cure rates between groups (RR 1.33, 99% CI 0.82 to 2.18; N = 140; 1 study, Analysis 21.1). An RCT from India compared oral itraconazole for six weeks with placebo (Dogra 1992). At the end of the treatment period, 75% (15/20) of participants in the oral itraconazole group and 0% (0/20) of the participants in the placebo groups achieved a complete cure. There were no statistically significant differences in cure rates between groups (RR 31.00, 99% CI 0.83 to 1151.33; N = 40; 1 study, Analysis 20.1).
Analysis 21.1.
Comparison 21 Itraconazole (200 mg for 3 weeks) versus placebo, Outcome 1 Participants complete cure.
Analysis 20.1.
Comparison 20 Itraconazole (200 mg for 6 weeks) versus placebo, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
In Al‐Fouzan 1991, two participants from the itraconazole group reported nausea and headache during the course of treatment, and one participant had elevated liver enzymes that returned back to normal upon discontinuation of the drug. Dogra 1992 reported nausea in 12 cases (30%) between the itraconazole and dapsone groups. In the itraconazole group, two participants (10%) also had mildly abnormal liver function that reverted after completion of therapy. In Momeni 1996, six participants in the itraconazole group and four participants in the placebo group complained of mild abdominal pain and nausea. None of the laboratory values were outside normal limits. In Dogra 1996, one participant showed abnormal liver function and one nausea in the itraconazole group. Participants in the placebo group did not report adverse effects. In Nassiri‐Kashani 2005, the most common adverse effects were gastrointestinal complaints and headache – reported only in the itraconazole group during the follow‐up period. Participants in the placebo group reported adverse effects only during the treatment period.
Participants of itraconazole group had more side effects, including mild abdominal pain and nausea (RR 2.36, 95% CI 0.74 to 7.47; 3 studies, N = 204, Analysis 22.2) as well as mild abnormal liver function (RR 3.08, 95% CI 0.53 to 17.98; N = 84, 3 studies, Analysis 22.2). However, the very low certainty of the evidence means we are uncertain about these results (Table 1).
Analysis 22.2.
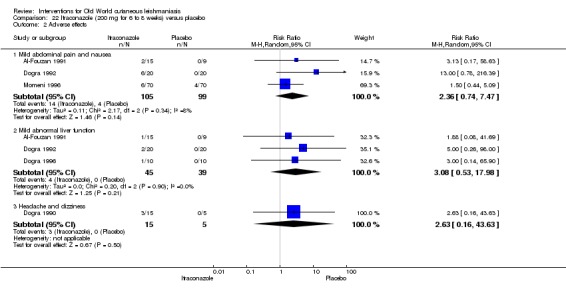
Comparison 22 Itraconazole (200 mg for 6 to 8 weeks) versus placebo, Outcome 2 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
In Dogra 1996, parasitological cure was significantly higher (Fisher's exact test P = 0.048) in participants receiving itraconazole compared to placebo. Parasitological cure occurred in 80% (8/10) of itraconazole‐treated participants but in none of the participants in the placebo group at six weeks follow‐up (RR 17.0, 95% CI 0.47 to 612.21; N = 20; 1 study, Analysis 22.3).
Analysis 22.3.
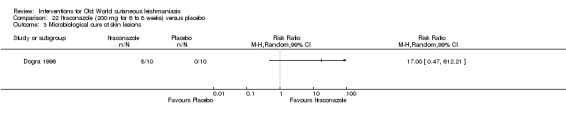
Comparison 22 Itraconazole (200 mg for 6 to 8 weeks) versus placebo, Outcome 3 Microbiological cure of skin lesions.
Please see Table 1 where we assessed the certainty of evidence.
2.1.5 Itraconazole versus no treatment
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from India compared oral itraconazole 200 mg/d for six weeks versus no treatment (Dogra 1990). The authors reported complete cure of participants in 66.7% (10/15) of the treatment group but in none of the no treatment group. Low statistical power meant that these differences could have been due to chance (RR 7.88, 99% CI 0.23 to 265.70; N = 20; 1 study, Analysis 23.1).
Analysis 23.1.
Comparison 23 Itraconazole (200 mg for 6 to 8 weeks) versus no treatment, Outcome 1 Participants complete cure.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
Participants responding to therapy had no relapses at three‐month follow‐up.
Secondary outcome: adverse effects
Regarding adverse effects, the drug was generally well tolerated, although 20% (3/15) of participants in the itraconazole group reported mild headache and dizziness. There were no significant differences in adverse effects rates between groups (RR 2.63, 95% CI 0.16 to 43.63; N = 20; 1 study, Analysis 23.2).
Analysis 23.2.

Comparison 23 Itraconazole (200 mg for 6 to 8 weeks) versus no treatment, Outcome 2 Adverse effects (headache and dizziness).
Tertiary outcomes: microbiological or histopathological cure of skin lesions
At six weeks, 66.7% (10/15) of the participants in the itraconazole group were free of parasites. Untreated participants were still parasitologically active. Low statistical power meant that these differences could have been due to chance (RR 7.88, 95% CI 0.54 to 114.56; N = 20; 1 study, Analysis 23.3).
Analysis 23.3.
Comparison 23 Itraconazole (200 mg for 6 to 8 weeks) versus no treatment, Outcome 3 Microbiological cure of skin lesions.
2.1.6 Fluconazole versus placebo
Primary outcome: percentage of lesions cured after the end of treatment
One RCT from Aleppo, Syria compared fluconazole 200 mg/d for six weeks versus placebo (Dandashli 2005). The authors reported that at the end of treatment, 28.4% (75/264) of the lesions in the fluconazole group and 9.8% (10/102) in the placebo group had completely resolved. Cure rates were higher in the oral fluconazole group compared with placebo (RR 2.90, 95% CI 1.56 to 5.38, Analysis 24.1).
Analysis 24.1.
Comparison 24 Fluconazole (200 mg for 6 weeks) versus placebo, Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
One RCT from Saudi Arabia compared oral fluconazole 200 mg/d versus placebo for six weeks (Alrajhi 2002). Three months after treatment, 59% (63/106) participants in the oral fluconazole and 21% (22/103) in the placebo group had achieved complete cure (RR 2.78, 95% CI 1.86 to 4.16; N = 209; 1 study, Analysis 24.2).
Analysis 24.2.
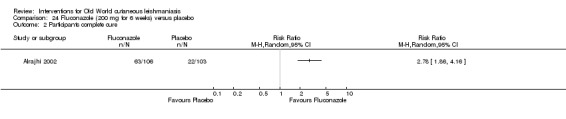
Comparison 24 Fluconazole (200 mg for 6 weeks) versus placebo, Outcome 2 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
Only Alrajhi 2002 reported the speed of healing, finding that this took a median of 8.5 weeks in the fluconazole group and 11.2 weeks in the placebo group (the original paper reported that the time to cure was significantly shorter for participants treated with thermotherapy; P < 0.001, by the log‐rank test). Also, none of the participants with complete healing had a relapse during a mean follow‐up of seven months.
Secondary outcome: adverse effects
In both studies participants from reported mild and similar side effects, although the authors did not describe them (Alrajhi 2002; Dandashli 2005).
2.1.7 Different doses of fluconazole
Primary outcome: percentage of participants with a complete cure after the end of treatment
One RCT from Iran compared oral fluconazole 400 mg/d versus 200 mg/d for six weeks (Emad 2011). The authors reported that at the end of treatment, 48.3% (29/60) of participants in the 200 mg group and 81% (47/58) in the 400 mg group achieved complete cure (RR 1.68, 95% CI 1.25 to 2.24; N = 118; 1 study, Analysis 25.1).
Analysis 25.1.

Comparison 25 Fluconazole (400 mg/d for 6 weeks) versus fluconazole (200 mg/d for 6 weeks), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, two participants in the 400 mg group discontinued treatment because of a rise of serum creatinine and liver enzymes. Participants in the 400 mg group reported 45 (75%) cases of cheilitis and 10 (16.6%) cases of nausea, compared to none in the 200 mg group (cheilitis: RR 94.08, 95% CI 5.93 to 1492.36; nausea: RR 21.71, 95% CI 1.30 to 362.21; N = 118; 1 study, Analysis 25.2).
Analysis 25.2.
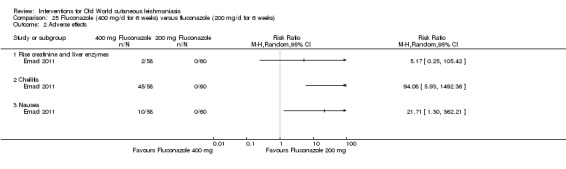
Comparison 25 Fluconazole (400 mg/d for 6 weeks) versus fluconazole (200 mg/d for 6 weeks), Outcome 2 Adverse effects.
2.2 Oral dapsone
2.2.1 Dapsone versus placebo
Primary outcome: percentage of participants with a complete cure after the end of treatment
Two RCTs comparing oral dapsone versus placebo reported complete cure after the end of treatment as a primary outcome (Dogra 1991; Dogra 1992). Although heterogeneity was high (P = 0.02; I² = 73%), both studies obtained results favouring dapsone, so we decided to maintain the pool of results and meta‐analysis (RR 24.08, 95% CI 1.44 to 403.43; N = 160; 2 studies; I² = 71%, Analysis 26.1).
Analysis 26.1.
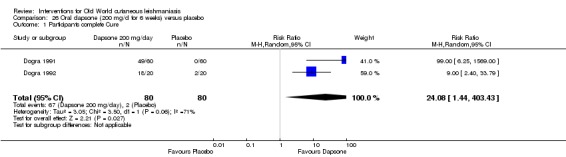
Comparison 26 Oral dapsone (200 mg/d for 6 weeks) versus placebo, Outcome 1 Participants complete Cure.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
In Dogra 1992, participants initially responding to therapy did not relapse at follow‐up, while two cases with single lesions (10%) demonstrated spontaneous healing in the placebo group at three months' follow‐up.
Secondary outcome: adverse effects
Regarding adverse effects, 30% (n = 12) of participants in the dapsone group experienced nausea. In Dogra 1991, 5% (3/60) of participants in the dapsone group developed anaemia and 15% (9/60), nausea. Nausea rates were higher in the dapsone group compared with placebo (RR 21.86, 95% CI 3.04 to 157.29; N = 160; 2 studies; I² = 0%, Analysis 26.2).
Analysis 26.2.
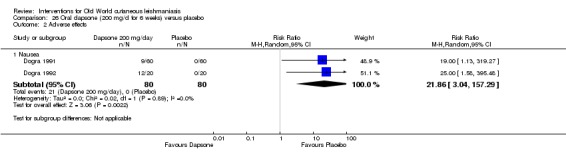
Comparison 26 Oral dapsone (200 mg/d for 6 weeks) versus placebo, Outcome 2 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Regarding the parasitological cure, 5% (3/60) of participants in the placebo group showed healing with negative smears one month after treatment (Dogra 1991).
2.3 Oral allopurinol
2.3.1 Allopurinol versus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared oral allopurinol 15 mg/kg/d for three weeks versus IMMA 30 mg/kg/d for two weeks versus oral allopurinol 15 mg/kg/d plus IMMA 30 mg/kg/d simultaneously, using the same dosage schedule (Esfandiarpour 2002). The authors reported that 18% (9/50) of participants receiving allopurinol alone achieved complete cure, compared to 24% (12/50) in the IMMA group and 46% (23/50) in the allopurinol plus IMMA group at the end of the treatment period. The group receiving allopurinol plus IMMA had better cure rates than those receiving allopurinol alone (RR 3.83, 95% CI 1.71 to 8.60; N = 100; 1 study, Analysis 27.1) or IMMA alone (RR 1.92, 95% CI 1.08 to 3.41; N = 100; 1 study, Analysis 28.1). There was no difference regarding cure rates between the groups receiving allopurinol alone versus IMMA alone (RR 0.75, 95% CI 0.35 to 1.62; N = 100; 1 study, Analysis 29.1).
Analysis 27.1.

Comparison 27 Allopurinol (15 mg/kg/d for 3 weeks) + IMMA (20 mg/kg/d for 2 weeks) versus allopurinol (15 mg/kg/d for 3 weeks), Outcome 1 Lesions cured.
Analysis 28.1.
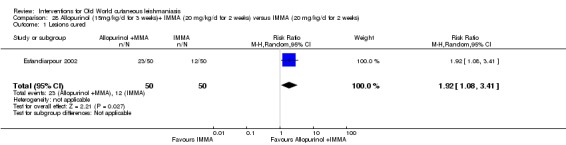
Comparison 28 Allopurinol (15mg/kg/d for 3 weeks)+ IMMA (20 mg/kg/d for 2 weeks) versus IMMA (20 mg/kg/d for 2 weeks), Outcome 1 Lesions cured.
Analysis 29.1.

Comparison 29 Allopurinol (15 mg/kg/d for 3 weeks) versus IMMA (20 mg/kg/d for 2 weeks), Outcome 1 Lesions Cured.
An RCT from Iran compared oral allopurinol 20 mg/kg/d plus IMMA 10 mg/kg/d versus IMMA 20 mg/kg/d alone for up to 28 days (Ejaz 2014). There was no data to address the primary outcome of percentage of participants with a complete cure.
Another RCT in an Iranian army camp compared oral allopurinol 20 mg/kg/d plus low‐dose IMMA 30 mg/kg/d for 20 days versus IMMA 60 mg/kg/d for 20 days (Momeni 2002). In this trial the authors reported that 51 days after treatment, 69% (25/36) and 72% (26/36) of participants, respectively, achieved a complete cure (RR 0.96, 95% CI 0.71 to 1.29; N = 72; 1 study, Analysis 30.1).
Analysis 30.1.

Comparison 30 Allopurinol (20 mg/kg/d for 3 weeks) + IMMA (30 mg/kg/d for 20 days) versus IMMA (60 mg/kg/d for 20 d), Outcome 1 Lesions cured.
Secondary outcome: adverse effects
Esfandiarpour 2002 observed a few adverse effects in the allopurinol group: three participants experienced nausea and heartburn, and two saw a mild increase in serum glutamic‐oxaloacetic transaminase and serum glutamic pyruvic transaminase levels. In Momeni 2002 participants tolerated the drugs well, and only 17% (6/36) in the combined treatment group complained of mild abdominal pain and nausea; however, 3% (1/36) of participants in the IMMA group developed skin eruption, and 11% (4/36) suffered generalised muscle pain and weakness. There was no significant difference between groups in total adverse effects (Analysis 30.2). Ejaz 2014 reported secondary infection in 12.1% (n = 21) of participants in the allopurinol plus IMMA group and in 12.5% (n = 19) of participants in the IMMA group, myalgia in 5.7% (n = 10) and 7.9% (n = 12), anorexia in 7.5% (n = 13) and 5.9% (n = 9), and ECG changes in 0.6% (n = 1) and 1.98% (n = 3), respectively. In the IMMA alone group one participant suffered chest pain; two, pain at the injection site; and five, abscess. There was no significant difference regarding adverse effects rates between the two groups (Analysis 31.1).
Analysis 30.2.
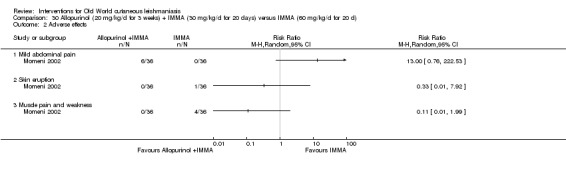
Comparison 30 Allopurinol (20 mg/kg/d for 3 weeks) + IMMA (30 mg/kg/d for 20 days) versus IMMA (60 mg/kg/d for 20 d), Outcome 2 Adverse effects.
Analysis 31.1.

Comparison 31 Allopurinol (20 mg/kg/d for 3 weeks)+ IMMA (10 mg/kg/d for 20 d) versus IMMA (20 mg/kg/d for 28 d), Outcome 1 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Only one study reported that 83.3% (30/36) of participants in the allopurinol plus IMMA group and 75% (27/36) in the IMMA alone group had parasitologically free lesions one month after the end of treatment (Momeni 2002). There was no difference regarding cure rates between groups ((RR 1.11, 95% CI 0.88 to 1.41; participants = 72; studies = 1) Analysis 30.3).
Analysis 30.3.
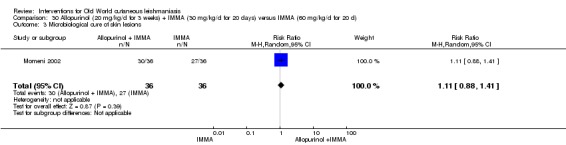
Comparison 30 Allopurinol (20 mg/kg/d for 3 weeks) + IMMA (30 mg/kg/d for 20 days) versus IMMA (60 mg/kg/d for 20 d), Outcome 3 Microbiological cure of skin lesions.
2.3.2 Oral allopurinol versus intravenous sodium stibogluconate
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT with participants from the Pakistani army compared oral allopurinol 20 mg/kg/d versus intravenous sodium stibogluconate (IVSSG) for 15 days (Mashood 2001). The authors reported at the end of the treatment period, 85% (17/20) of participants in the allopurinol group and 70% (14/20) in the IVSSG group achieved a complete cure. There was no significant difference regarding cure rates between groups (RR 1.21, 95% CI 0.86 to 1.71; N = 40; 1 study, Analysis 32.1).
Analysis 32.1.
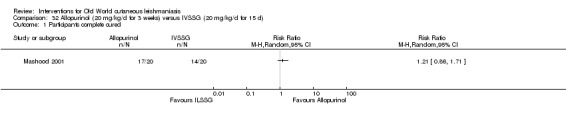
Comparison 32 Allopurinol (20 mg/kg/d for 3 weeks) versus IVSSG (20 mg/kg/d for 15 d), Outcome 1 Participants complete cured.
Secondary outcome: adverse effects
Mashood 2001 observed adverse effects that included nausea, vomiting, anorexia, and diarrhoea in 5% (1/20) of participants in the allopurinol group and 20% (4/20) of participants in the IVSSG group. In the SSG group, 10% (2/20) of participants experienced liver abnormalities and 15% (3/20), myalgia and body aches. In the allopurinol group, 5% (1/20) had elevated liver enzymes, and 10% (2/20), macular rash, but there was no myalgia or body aches. There was no significant difference in adverse effects rates between the allopurinol group and IVSSG alone group (Analysis 32.2).
Analysis 32.2.
Comparison 32 Allopurinol (20 mg/kg/d for 3 weeks) versus IVSSG (20 mg/kg/d for 15 d), Outcome 2 Adverse effects.
2.4 Oral antibiotics
For a summary of adverse effects of oral antibiotics please see Table 119.
2.4.1 Oral rifampicin versus placebo
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Saudi Arabia compared oral rifampicin 10 mg/kg/d for four to six weeks versus placebo (Jaffar 2006). Three months after treatment, 45.7% (21/46) of participants in the oral rifampicin group and 18.8% (3/16) of the placebo group achieved complete cure. There was no difference in cure rates between oral rifampicin and placebo (RR 2.43, 95% CI 0.84 to 7.08; N = 62; 1 study, Analysis 33.1).
Analysis 33.1.
Comparison 33 Oral rifampicin (10 mg/kg/d for 4 to 6 weeks) versus placebo, Outcome 1 Participants complete cure.
Two RCTs from India address the primary outcome of percentage of participants with a complete cure at the end of treatment. Kochar 2000 compared 10 mg/kg/d oral rifampicin for four weeks versus placebo, reporting that 68% (17/25) and 4% (1/25) of participants, respectively, achieved complete cure by the end of the treatment period (RR 17.00, 95% CI 2.45 to 118.19; N = 50; 1 study, Analysis 33.1). Kochar 2006 compared 10 mg/kg/d oral rifampicin plus 20 mg/d omeprazole for six weeks versus placebo, reporting that 64% (16/25) participants in the rifampicin group and 12% (3/25) in the placebo group had achieved a complete cure at the end of the treatment period (RR 5.33, 95% CI 1.77 to 16.05; N = 50; 1 study, Analysis 34.1).
Analysis 34.1.
Comparison 34 Oral rifampicin (10 mg/kg/d) + omeprazole (20 mg/d) for 6 weeks versus placebo, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Jaffar 2006 reported that one participant had elevated liver enzymes that returned back to normal upon discontinuation of the drug. Kochar 2000 reported that the drug was very well tolerated, and there were no adverse effects during therapy.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Kochar 2000 reported an absence of parasites in 8% (2/25) and 32% (8/25) of partially healed lesions in the rifampicin and placebo groups, respectively, at the end of treatment (RR 0.25, 95% CI 0.06 to 1.06; N = 50; 1 study, Analysis 33.2).
Analysis 33.2.
Comparison 33 Oral rifampicin (10 mg/kg/d for 4 to 6 weeks) versus placebo, Outcome 2 Microbiological cure of skin lesions.
2.4.2 Oral terbinafine plus cryotherapy versus IMMA plus cryotherapy
Secondary outcome: speed of healing (time taken to be 'cured')
An RCT from Iran compared cryotherapy (liquid nitrogen via a cotton swab for 10 s to 25 s every two weeks for four weeks) plus either oral terbinafine 500 mg/d for four weeks or plus IMMA 15 mg/kg/d (Farajzadeh 2015). After three months' follow‐up, the crude hazard ratio was 0.53 (95% CI 0.30 to 0.98) and after adjustment for sex, it was 0.35 (95% CI 0.19 to 0.69). A Kaplan‐Meier analysis indicated that the difference in complete treatment between the groups was not significant.
2.4.3 Azithromycin with or without allopurinol versus IMMA
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Iran compared azithromycin 500 mg for five days/month up to a maximum of four months with IMMA 60 mg/kg/d for 20 days (Layegh 2007). After 16 weeks of follow‐up, 10.3% (13/29) and 34.4% (20/58) of the lesions were cured in the respective groups (RR 0.60, 95% CI 0.18 to 2.01; N = 87; 1 study, Analysis 35.1).
Analysis 35.1.

Comparison 35 Azythromicin (500 mg/d for 5 d/month up to 4 months) versus IMMA (60 mg/kg/d for 20 d), Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared azithromycin 10 mg/kg/d (maximum 500 mg/d) plus allopurinol 10 mg/kg for one month versus IMMA 20 mg/kg/d for 20 days (Dastgheib 2012); 38.9% (14/36) and 40% (14/35) of participants, respectively, had achieved complete cure at two months after treatment (RR 0.97, 95% CI 0.55 to 1.73; N = 71, Analysis 36.1). There was no statistical difference in cure rates between azithromycin plus allopurinol versus IMMA in ulcerated (P = 0.40) and non‐ulcerated (P = 0.37) lesions or with regard to the site of lesions (P = 0.30).
Analysis 36.1.
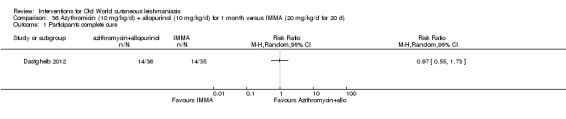
Comparison 36 Azythromicin (10 mg/kg/d) + allopurinol (10 mg/kg/d) for 1 month versus IMMA (20 mg/kg/d for 20 d), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, Dastgheib 2012 reported that one (2.7%) participant in the azithromycin plus allopurinol group had gastrointestinal complaints and severe headache, and three (8.3%) experienced slight gastrointestinal complications (nausea, heartburn, and epigastric pain). Layegh 2007 described only nausea and vomiting in two participants (9%) in the azithromycin alone group. Only Dastgheib 2012 reported myalgia in two (5.7%) participants in the IMMA group. There were no significant differences in adverse effects rates between groups (Analysis 35.2; Analysis 36.2).
Analysis 35.2.
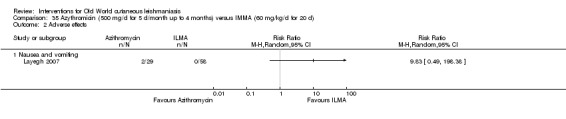
Comparison 35 Azythromicin (500 mg/d for 5 d/month up to 4 months) versus IMMA (60 mg/kg/d for 20 d), Outcome 2 Adverse effects.
Analysis 36.2.
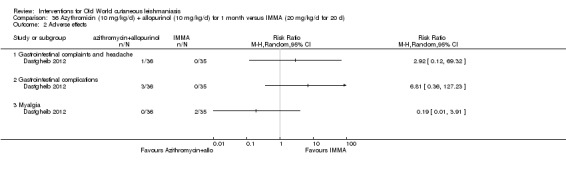
Comparison 36 Azythromicin (10 mg/kg/d) + allopurinol (10 mg/kg/d) for 1 month versus IMMA (20 mg/kg/d for 20 d), Outcome 2 Adverse effects.
2.5 Oral pentoxifylline
2.5.1 Oral pentoxifylline plus IMMA versus placebo plus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared oral pentoxifylline 400 mg three times daily plus IMMA 20 mg/kg/d versus IMMA 20 mg/kg/d plus placebo, for 20 days (Sadeghian 2006a). Three months after treatment, 81.3% (26/32) and 50% (16/32) of the participants in the respective groups had achieved complete cure (RR 1.63; 95% CI 1.11, 2.39, Analysis 37.1).
Analysis 37.1.

Comparison 37 Oral pentoxifylline (400 mg 3 times daily) + IMMA (20 mg/kg/d) for 20 d versus placebo + IMMA (20 mg/kg/d) for 20 d, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, one participant in the IMMA plus placebo group had an allergic macule‐papular itchy rash. There were no significant differences in adverse effects rates between groups (RR 0.33, 95% CI 0.01 to 7.89; N = 64; 1 study, Analysis 37.2).
Analysis 37.2.
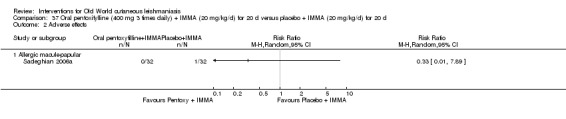
Comparison 37 Oral pentoxifylline (400 mg 3 times daily) + IMMA (20 mg/kg/d) for 20 d versus placebo + IMMA (20 mg/kg/d) for 20 d, Outcome 2 Adverse effects.
2.6 Oral miltefosine
2.6.1 Oral miltefosine versus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT in Iran compared oral miltefosine 2.5 mg/kg/d for four weeks versus IMMA 20 mg/kg/d for two weeks (Mohebali 2007). Three months after treatment, 81.3% (26/32) and 80.6% (25/31) of participants in the respective groups had achieved complete cure (RR 1.01; 95% CI 0.79, 1.28, Analysis 38.1).
Analysis 38.1.
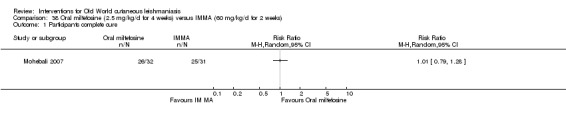
Comparison 38 Oral miltefosine (2.5 mg/kg/d for 4 weeks) versus IMMA (60 mg/kg/d for 2 weeks), Outcome 1 Participants complete cure.
An RCT from Iran compared oral miltefosine 2.5 mg/kg/d for four weeks versus IMMA 60 mg/kg/d for two weeks (Khatami 2012). At one month after treatment, 32% (10/31) and 35.7% (15/42) of the participants in the respective groups had achieved complete cure (RR 0.90, 95% CI 0.47 to 1.73; N = 73; 1 study, Analysis 39.1).
Analysis 39.1.
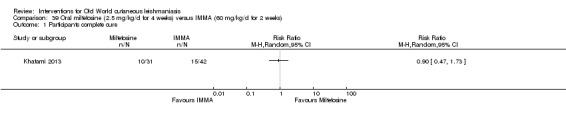
Comparison 39 Oral miltefosine (2.5 mg/kg/d for 4 weeks) versus IMMA (60 mg/kg/d for 2 weeks), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Mohebali 2007 did not report any relapse at six months post‐treatment. With regard to adverse effects, during the first week participants from the miltefosine group suffered from nausea, vomiting, diarrhoea, abdominal pain, and cough. Participants from the IMMA group had only diarrhoea and local pain. During the second week, participants receiving miltefosine suffered from nausea, vomiting, abdominal pain, headache, itch, and fever. Participants receiving IMMA developed those adverse effects as well as local pain, chest pain, and cough. Khatami 2012 only reported that adverse effects were higher in the miltefosine arm (nine cases versus one case).
2.7 Oral zinc sulphate
2.7.1 Different doses of oral zinc sulphate
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iraq compared oral zinc sulphate at different doses: 2.5 mg/kg/d, 5 mg/kg/d, or 10 mg/kg/d, versus no treatment (Sharquie 2001). The authors reported that 45 days after treatment, 66.7% (26/39) in the 2.5 mg/kg/d group, 73% (27/37) in the 5 mg/kg/d group, and 79% (31/39) in the 10 mg/kg/d group had achieved complete cure. In the control group, all lesions were still active at day 45. There were no significant differences in cure rates between oral zinc sulphate 2.5 mg/kg/d versus 5 mg/kg/d (RR 0.91, 95% CI 0.68 to 1.23; N = 76; 1 study, Analysis 40.1); 2.5 mg/kg/d versus 10 mg/kg/d (RR 0.84, 95% CI 0.64 to 1.10; N = 78; 1 study, Analysis 41.1); or 5 mg/kg/d versus 10 mg/kg/d (RR 0.92, 95% CI 0.71 to 1.18; N = 76; 1 study, Analysis 42.1).
Analysis 40.1.
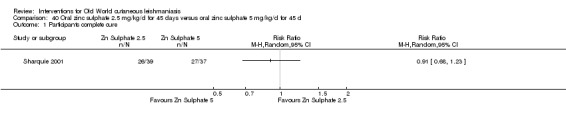
Comparison 40 Oral zinc sulphate 2.5 mg/kg/d for 45 days versus oral zinc sulphate 5 mg/kg/d for 45 d, Outcome 1 Participants complete cure.
Analysis 41.1.
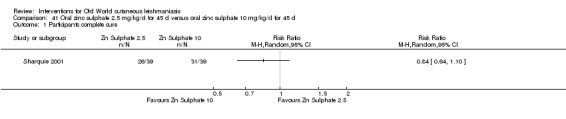
Comparison 41 Oral zinc sulphate 2.5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d, Outcome 1 Participants complete cure.
Analysis 42.1.
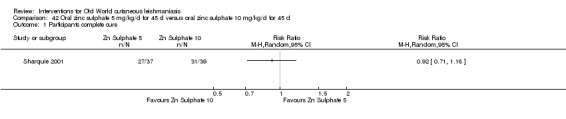
Comparison 42 Oral zinc sulphate 5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
In the zinc sulphate 2.5 mg/kg/d group, the mean time taken to cure was 30.8 days (range 21 to 45): 29.96 days (range 15 to 45) in the 10 mg/kg/d group and 28.32 days (range 15 to 45) in the 10 mg/kg/d (the original paper reported that the time to cure was not statistically significant between the different treatment groups). None of the participants recovered in the control group.
Secondary outcome: prevention of scarring
In all treatment groups there was minimal or no scarring at the site of lesions.
Secondary outcome: adverse effects
Regarding adverse effects, one participant each in the 2.5 mg/kg/d, 5 mg/kg/d, and 10 mg/kg/d group had nausea and vomiting. Only one participant from the 5 mg/kg/d group had a leishmanid reaction, while two participants in the 2.5 mg/kg/d group and two from the 5 mg/kg/d group, plus one participant in the 10 mg/kg/d group, had oedema. There were no significant differences in adverse effects rates between groups (Analysis 40.2; Analysis 41.2; Analysis 42.2).
Analysis 40.2.
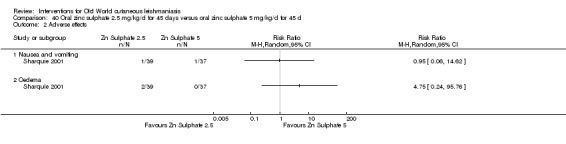
Comparison 40 Oral zinc sulphate 2.5 mg/kg/d for 45 days versus oral zinc sulphate 5 mg/kg/d for 45 d, Outcome 2 Adverse effects.
Analysis 41.2.
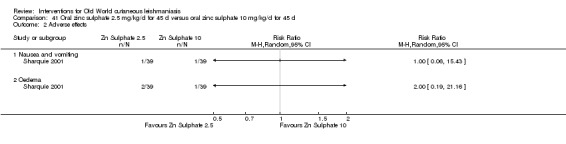
Comparison 41 Oral zinc sulphate 2.5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d, Outcome 2 Adverse effects.
Analysis 42.2.
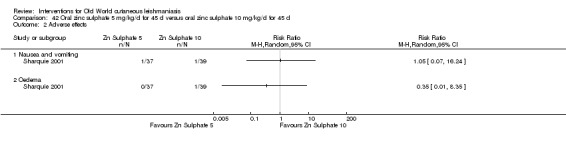
Comparison 42 Oral zinc sulphate 5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d, Outcome 2 Adverse effects.
2.7.2 Oral zinc sulphate versus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared oral zinc sulphate (ZS) in a dose of 10 mg/kg/d for 45 days versus IMMA 20 mg/kg/d for up to 20 days (Yazdanpanah 2011). The authors reported that 45 days after treatment, 30.2% (8/26) and 35.5% (26/74) of the respective groups achieved complete cure (RR 0.88, 95% CI 0.46 to 1.68; N = 100; 1 study, Analysis 43.1).
Analysis 43.1.
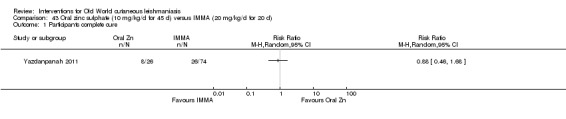
Comparison 43 Oral zinc sulphate (10 mg/kg/d for 45 d) versus IMMA (20 mg/kg/d for 20 d), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, the authors did not report any adverse effects in the oral zinc sulphate group, but six participants in the IMMA group dropped out because of adverse effects, including severe erythema and pruritus at inoculation site and severe muscular pain.
2.8 Oral artesunate
2.8.1 Oral artesunate plus sulphamethoxypyridazine/pyrimethamine versus placebo
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Sudan compared oral artesunate plus sulphamethoxypyrazine/pyrimethamine (400 mg artesunate plus 1000 mg/50 mg sulphamethoxypyrazine/pyrimethamine for four days, four times each two weeks) versus placebo (Adam 2009). The authors reported that 72 days after treatment, 90% (18/20) and 85.7% (18/21) of the respective groups had a complete cure (RR 1.05, 95% CI 0.84 to 1.32; N = 41; 1 study, Analysis 44.1).
Analysis 44.1.
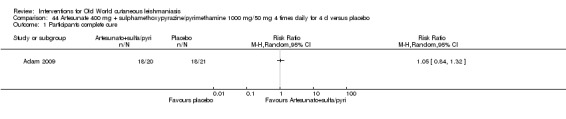
Comparison 44 Artesunate 400 mg + sulphamethoxypyrazine/pyrimethamine 1000 mg/50 mg 4 times daily for 4 d versus placebo, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
One participant in the placebo arm developed a mild skin rash with itching. There was no significant difference in biological tests (liver and renal function tests) in any of the participants before or after treatment.
3. Non‐antimonial topical or intralesional therapies
3.1 Topical antifungals
3.1.1 Topical 2% miconazole versus topical 1% clotrimazole
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Saudi Arabia compared 2% miconazole cream twice a day with 1% clotrimazole cream twice a day for 30 days (Larbi 1995). The authors reported there was no lesions healed in the miconazole group and 15% (14/89) of the lesions healed completely in the clotrimazole group at the end of treatment (RR 0.05, 95% CI 0.00 to 0.81; N = 151; 1 study, Analysis 45.1).
Analysis 45.1.
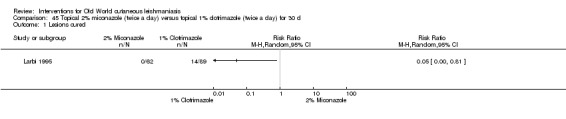
Comparison 45 Topical 2% miconazole (twice a day) versus topical 1% clotrimazole (twice a day) for 30 d, Outcome 1 Lesions cured.
Secondary outcome: adverse effects
In both treatment groups, the medication was well tolerated, with no reported adverse effects.
3.1.2 Ketoconazole cream versus vehicle
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical ketoconazole versus vehicle, twice daily for 21 days (Momeni 2003). The authors reported that 51 days after treatment, 24% (11/45) and 13% (6/45) of the participants in the respective groups achieved complete cure. There were no significant differences in cure rates between topical ketoconazole and vehicle (RR 1.83, 95% CI 0.74 to 4.53; N = 90; 1 study, Analysis 46.1).
Analysis 46.1.
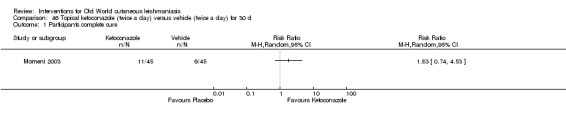
Comparison 46 Topical ketoconazole (twice a day) versus vehicle (twice a day) for 30 d, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, the drugs were well tolerated, and only two participants (group unknown) complained of mild pruritus at the site of the lesions.
3.1.3 Amphotericin B versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical liposomal amphotericin B (3 to 7 drops twice daily) versus ILMA (maximum 2 mL) for eight weeks (Layegh 2011). We performed an ITT analysis considering the participants allocated in each arm at the beginning of the study. After six months of follow‐up, 44% (22/50) and 48.3% (29/60) of the participants in the amphotericin B and ILMA groups, respectively, achieved complete cure (RR: 0.91, 95% CI 0.61 to 1.37; N = 110; 1 study, Analysis 47.1).
Analysis 47.1.

Comparison 47 Topical amphotericin B (3 to 7 drops twice daily for 8 weeks) versus ILMA (max 2 mL) once a week for 8 weeks, Outcome 1 Participants complete cure (ITT).
Secondary outcome: adverse effects
One (1.7%) participant in amphotericin B group presented hypersensitivity, and five (10%) mild pruritus around the lesions. Seven (11.7%) participants in the ILMA group suffered erythema and oedema at the injection site. There were no significant differences in any hypersensitivity rates (RR 3.59, 95% CI 0.15 to 86.19), mild pruritus rates (RR 13.16, 95% CI 0.75 to 232.30), or erythema and oedema rates (RR 0.08, 95% CI 0.00 to 1.36) between groups (N = 110; 1 study, Analysis 47.2).
Analysis 47.2.
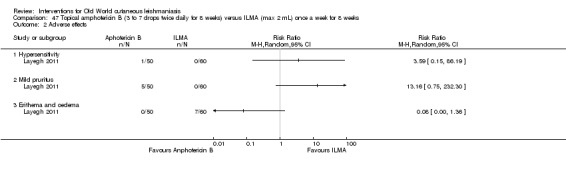
Comparison 47 Topical amphotericin B (3 to 7 drops twice daily for 8 weeks) versus ILMA (max 2 mL) once a week for 8 weeks, Outcome 2 Adverse effects.
3.2 Topical paromomycin (aminosidine)
For a summary of adverse effects of topical paromomycin please see Table 120.
3.2.1 Paromomycin versus vehicle
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared topical 15% paromomycin sulphate in 12% MBCL (PR‐MBCL) versus vehicle, twice daily for 30 days (Asilian 2006). Two months after treatment, 41.2% (14/34) and 13.3% (4/30) of the lesions in the PR‐MBCL and placebo groups, respectively, had completely healed (RR 3.09, 95% CI 1.14 to 8.37; N = 64; 1 study Analysis 48.1).
Analysis 48.1.
Comparison 48 Paromomycin 15% + 12% MBCL (twice daily for 28 d) versus vehicle (twice daily for 28 d), Outcome 1 Lesions cured.
An RCT from Iran compared topical 15% paromomycin in 10% urea (PR‐U) versus vehicle, twice daily for 30 days (Iraji 2005); 12.5% (5/40) and 17.5% (7/40) of the lesions in the respective groups had healed completely one month after treatment (RR 0.71, 95% CI 0.25 to 2.06; N = 80; 1 study, Analysis 49.1).
Analysis 49.1.
Comparison 49 Paromomycin (twice daily for 30 d) versus vehicle (twice daily for 30 d), Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
A pooled analysis of two trials showed no significant differences in cure rates between topical PR‐U and vehicle (RR 1.00; 95% CI 0.86, 1.17; Asilian 1995; Ben Salah 1995; Analysis 50.1). Please see Table 2 where we assessed the certainty of evidence as very low, meaning that we are uncertain whether there is much difference in cure rate between paromomycin and vehicle.
Analysis 50.1.
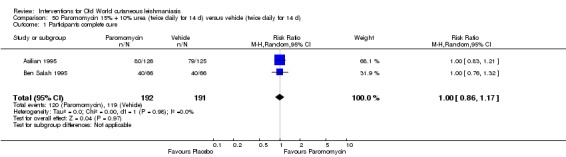
Comparison 50 Paromomycin 15% + 10% urea (twice daily for 14 d) versus vehicle (twice daily for 14 d), Outcome 1 Participants complete cure.
An RCT from Iran compared PR‐U versus vehicle, twice daily for 14 days (Asilian 1995). Two and a half months after treatment, 63.5% (80/126) and 63.2% (79/125) of those receiving PR‐U and vehicle, respectively, achieved a complete cure.
An RCT from Tunisia compared PR‐U in soft white paraffin versus vehicle, twice daily for 14 days (Ben Salah 1995). Two and a half months after treatment (day 105), 60.6% (40/66) in each group achieved complete cure.
An RCT from Tunisia compared topical 15% paromomycin plus gentamicin 0.5% versus 15% paromomycin alone versus vehicle, daily for 20 days (Ben Salah 2013). Five and a half months after treatment (day 168), 80% (101/125), 82% (102/125) and 58% (73/125) of participants, respectively, had achieved a complete cure. Paromomycin alone (RR 1.40, 95% CI 1.18 to 1.66, Analysis 51.1) and paromomycin plus gentamicin (RR 1.38, 95% CI 1.17 to 1.64, Analysis 52.1) were more efficacious than vehicle. There was no significant difference between topical 15% paromomycin plus gentamicin 0.5% and 15% paromomycin alone (RR 0.99, 95% CI 0.88 to 1.12; N = 250; 1 study, Analysis 53.1).
Analysis 51.1.
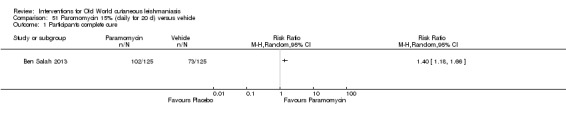
Comparison 51 Paromomycin 15% (daily for 20 d) versus vehicle, Outcome 1 Participants complete cure.
Analysis 52.1.
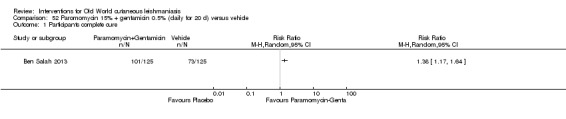
Comparison 52 Paromomycin 15% + gentamicin 0.5% (daily for 20 d) versus vehicle, Outcome 1 Participants complete cure.
Analysis 53.1.

Comparison 53 Paromomycin 15% + gentamicin 0.5% (daily for 20 d) versus paromomycin 15% alone (daily for 20 d), Outcome 1 Participants complete cure.
Secondary outcome: prevention of scarring
Only Asilian 2006 reported that at the end of the study there was no significant difference in number of deep or disfiguring scars between the PR‐MBCL group (8/34 lesions; 23.5%) and vehicle group (3/10; 10%) (RR 2.35; 95% CI 0.69 to 8.07; N = 64; 1 study, Analysis 48.2). However, we have used the number of lesions originally randomised in presenting the data in this analysis, whereas in the original paper the authors assessed scarring only in lesions that were completely healed at the end of the study (which perhaps makes more sense as a denominator). Both methods of calculating the degree of scarring failed to show any significant differences.
Analysis 48.2.
Comparison 48 Paromomycin 15% + 12% MBCL (twice daily for 28 d) versus vehicle (twice daily for 28 d), Outcome 2 Scarring.
Secondary outcome: adverse effects
Twelve participants between the PR‐U and vehicle groups in Ben Salah 1995 reported a local reaction (inflammation, vesiculation, pain and/or redness); 8 participants from the PR‐U group and 11 from the vehicle group in Asilian 1995 complained about redness, local pain, vesiculation, and inflammation; 3 participants in the PR‐U group in Iraji 2005 reported mild contact dermatitis; and participants in all three groups in Asilian 2006 reported mild and tolerable itch, burning, redness, discharge, oedema, and pain. In Ben Salah 2013, all adverse effects deemed by the investigators to be at least possibly related to a study treatment were reactions of mild or moderate severity at the application site. The paromomycin group reported a little more skin/local reaction (RR 1.42, 95% CI 0.67 to 3.01; N = 713, Analysis 50.2); however, the very low certainty of the evidence means we are not confident about these results (Table 2).
Analysis 50.2.
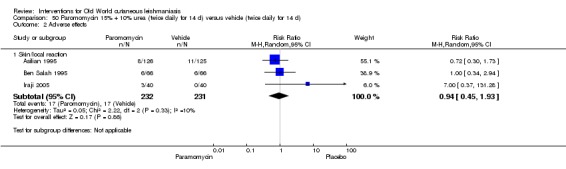
Comparison 50 Paromomycin 15% + 10% urea (twice daily for 14 d) versus vehicle (twice daily for 14 d), Outcome 2 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
In a pooled analysis of two studies comparing PR‐U versus placebo, there was no significant difference in the number of negative parasitologic smears at a mean follow‐up of 2.5 months (RR 1.03; 95% CI 0.88 to 1.20; Asilian 1995; Ben Salah 1995; Analysis 50.3). The certainty of evidence was very low, meaning we are uncertain whether there was any difference in parasitological cure between the two groups. In another study (Asilian 2006), 64.7% (22/34) in the PR‐MBCL group and 20% (6/30) in the vehicle group were parasitologically free two months after the end of treatment (RR 3.24, 95% CI 1.52 to 6.90; N = 64; 1 study, Analysis 48.3).
Analysis 50.3.
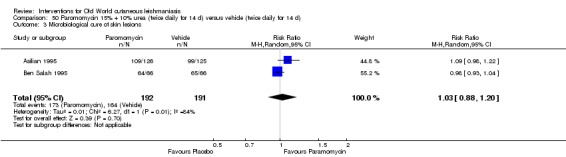
Comparison 50 Paromomycin 15% + 10% urea (twice daily for 14 d) versus vehicle (twice daily for 14 d), Outcome 3 Microbiological cure of skin lesions.
Analysis 48.3.
Comparison 48 Paromomycin 15% + 12% MBCL (twice daily for 28 d) versus vehicle (twice daily for 28 d), Outcome 3 Microbiological cure of skin lesions.
3.2.2 PR‐U versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared PR‐U ointment, twice per day for a mean duration of 45 days and a maximum duration of 3 months versus ILMA weekly (Faghihi 2003). At less than two months after treatment, the authors reported complete cure of participants in 16.6% (8/48) and 41.7% (20/48) of the PR‐U and ILMA groups, respectively (RR 0.40, 95% CI 0.20 to 0.82; N = 96; 1 study, Analysis 54.1).
Analysis 54.1.
Comparison 54 Paromomycin 15% + 10% urea (twice daily for 45 d) versus ILMA (weekly for up to 3 months), Outcome 1 Participants complete cure.
Another RCT from Iran compared PR‐U ointment twice a day for 20 days versus ILMA (dose unspecified) every other day for 20 days (Shazad 2005). One week after the end of treatment, 66.7% (20/30) and 60% (18/30) participants of the respective groups achieved complete cure (RR 1.11, 95% CI 0.75 to 1.64; N = 60; 1 study, Analysis 55.1).
Analysis 55.1.
Comparison 55 Paromomycin 15% + 10% urea (twice daily for 20 d) versus ILMA (weekly for up to 20 d), Outcome 1 Participants complete cure.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
Faghihi 2003 reported a reactivation in 6.3% (3/48) of the participants in both groups at one year after complete recovery, due to lymphatic spread in the PR‐U group and non‐lymphatic spread in the ILMA group (RR 1.00, 95% CI 0.21 to 4.71; N = 96; 1 study, Analysis 54.2). Scarring occurred in 4.2% (2/48) of participants in the PR‐U group and for 8.3% (4/48) in the ILMA group (RR 0.50, 95% CI 0.10 to 2.60; N = 96; 1 study, Analysis 54.3).
Analysis 54.2.
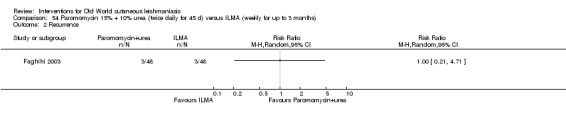
Comparison 54 Paromomycin 15% + 10% urea (twice daily for 45 d) versus ILMA (weekly for up to 3 months), Outcome 2 Recurrence.
Analysis 54.3.
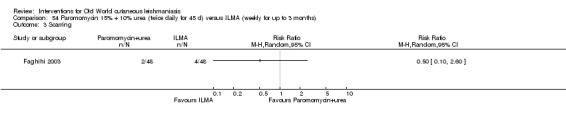
Comparison 54 Paromomycin 15% + 10% urea (twice daily for 45 d) versus ILMA (weekly for up to 3 months), Outcome 3 Scarring.
Secondary outcome: adverse effects
Regarding adverse effects, Shazad 2005 reported that 1/30 in the PR‐U group and 3/30 participants in the ILMA treatment group withdrew from the study because of cutaneous reactions like erythematosus, urticaria, or lymphadenitis with pain. They were all put on systemic MA and cured thereafter. They did not observe any systemic toxic reaction attributable to the drug. There were no significant differences in adverse effects rates between groups (RR 0.33, 95% CI 0.04 to 3.03; N = 60; 1 study Analysis 55.2).
Analysis 55.2.
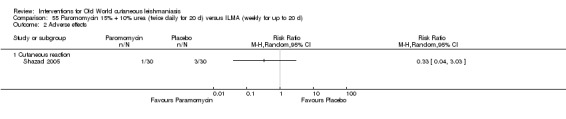
Comparison 55 Paromomycin 15% + 10% urea (twice daily for 20 d) versus ILMA (weekly for up to 20 d), Outcome 2 Adverse effects.
3.2.3 PR‐MBCL versus oral ketoconazole
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Turkey compared topical PR‐MBCL twice per day for 15 days versus oral ketoconazole 600 mg/d for 30 days (Özgöztasi 1997). One month after the end of treatment the authors reported complete cure of participants in 37.5% (15/40) of the paromomycin group and none in the ketoconazole group (RR 24.95, 95% CI 1.55 to 401.63; N = 72; 1 study, Analysis 56.1).
Analysis 56.1.

Comparison 56 Paromomycin + MBCL (twice daily for 15 d) versus ketoconazole (weekly for up to 30 d), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Irritant contact dermatitis was the most common adverse effect described in the PR‐MBCL group. In contrast, the ketoconazole‐treated participants reported no adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
The PR‐MBCL and the ketoconazole groups showed incomplete improvement (absence of parasites on culture or smear): just 20% (8/40) and 21.9% (7/32) of participants, respectively, had achieved a microbiological cure at four weeks post‐treatment (RR 0.91, 95% CI 0.37 to 2.25; N = 72; 1 study, Analysis 56.2).
Analysis 56.2.

Comparison 56 Paromomycin + MBCL (twice daily for 15 d) versus ketoconazole (weekly for up to 30 d), Outcome 2 Microbiological cure of skin lesions.
3.2.4 PR‐MBCL versus topical photodynamic therapy (PDT)
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared topical PR‐MBCL twice daily for 28 days versus topical PDT every week for four weeks (Asilian 2006). Two months after treatment, results showed complete cure of lesions in 41.2% (14/34) and 93.5% (29/31) of the PR‐MBCL and PDT groups, respectively (RR 0.44, 95% CI 0.29 to 0.66; N = 65; 1 study, Analysis 57.1).
Analysis 57.1.

Comparison 57 Paromomycin (15% + 12% MBCL twice daily for 28 days) versus PDT (weekly for 4 weeks), Outcome 1 Lesions cured.
Secondary outcome: prevention of scarring
At the end of the study, there was no statistical difference (Fisher's exact test P = 0.0558) in the number of deep or disfiguring scars between paromomycin (8/24) and PDT groups (0/31) (RR 15.54, 95% CI 0.39 to 625.57; N = 65; 1 study, Analysis 57.2).
Analysis 57.2.
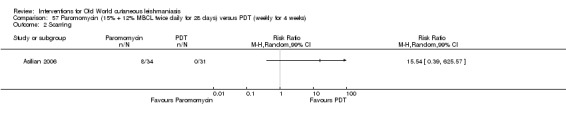
Comparison 57 Paromomycin (15% + 12% MBCL twice daily for 28 days) versus PDT (weekly for 4 weeks), Outcome 2 Scarring.
Secondary outcome: adverse effects
Both groups experienced mild and tolerable itch, burning, redness, discharge, oedema, and pain.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Two months after the end of treatment, 64.7% (22/34) of the lesions in the PR‐MBCL group were parasitologically free, as were all 31 lesions in the PDT group (RR 0.65, 95% CI 0.51 to 0.84; N = 65; 1 study, Analysis 57.3).
Analysis 57.3.
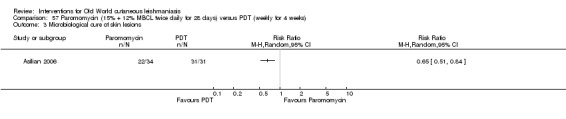
Comparison 57 Paromomycin (15% + 12% MBCL twice daily for 28 days) versus PDT (weekly for 4 weeks), Outcome 3 Microbiological cure of skin lesions.
3.2.5 Different regimens of topical paromomycin
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared two tubes of 15 g of paromomycin ointment (each tube was enough for two applications per day for 14 days) for a four‐week treatment versus one tube of 15 g of paromomycin ointment for two weeks plus vehicle for two additional weeks (Asilian 2003). Two and a half months after treatment, results showed complete cure of participants in 50% (58/117) and 37% (43/116) of the four‐week and two‐week regimens, respectively. There was no significant difference in cure rates between the four‐week and the two‐week treatment (RR 1.34, 95% CI 0.99 to 1.80; N = 233; 1 study, Analysis 58.1).
Analysis 58.1.
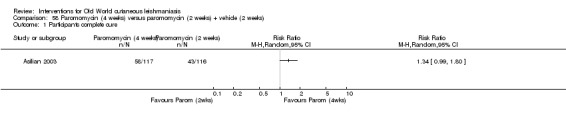
Comparison 58 Paromomycin (4 weeks) versus paromomycin (2 weeks) + vehicle (2 weeks), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Participants tolerated the treatment well, and investigators did not observe any adverse effect or adverse reactions to the ointment in either group.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Three months (105 days) after treatment initiation, there was a small difference in parasitological cure in favour of the four‐week regimen 49/117 (41%) versus 33/116 (28.4%) respectively (RR 1.47, 95% CI 1.03 to 2.11; N = 233; 1 study, Analysis 58.2).
Analysis 58.2.
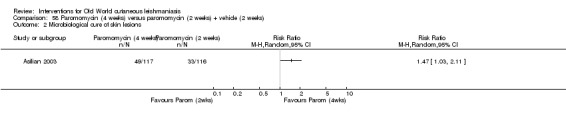
Comparison 58 Paromomycin (4 weeks) versus paromomycin (2 weeks) + vehicle (2 weeks), Outcome 2 Microbiological cure of skin lesions.
3.3 Intralesional zinc sulphate
For a summary of adverse effects of intralesional zinc sulphate please see Table 121.
3.3.1 Intralesional (IL) zinc sulphate versus ILSSG versus IL hypertonic sodium chloride solution (HSCS)
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Iraq compared mono doses of IL 2% zinc sulphate solution versus ILSSG 100 mg/mL versus IL 7% HSCS (Sharquie 1997). Investigators left a few lesions on unimportant and unexposed parts of the body as controls. At six weeks after treatment, 94.8% (36/38), 88.5% (31/35), and 85% (34/40) of the lesions, respectively, had healed completely. There were no statistical differences between any comparison: 2% zinc sulphate solution versus ILSSG (RR 1.07, 95% CI 0.93 to 1.23; N = 73; 1 study, Analysis 59.1); 2% zinc sulphate solution versus IL 7% HSCS (RR 1.11, 95% CI 0.96 to 1.30; N = 78; 1 study, Analysis 60.1), or ILSSG versus IL 7% HSCS (RR 1.04, 95% CI 0.87 to 1.24; N = 75; 1 study, Analysis 61.1). In the untreated group, nine participants with 38 lesions were followed up for six weeks, showing no decrease in the size of the lesions and no disappearance of parasites.
Analysis 59.1.
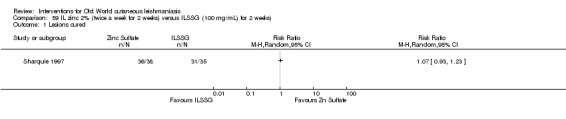
Comparison 59 IL zinc 2% (twice a week for 2 weeks) versus ILSSG (100 mg/mL) for 2 weeks), Outcome 1 Lesions cured.
Analysis 60.1.
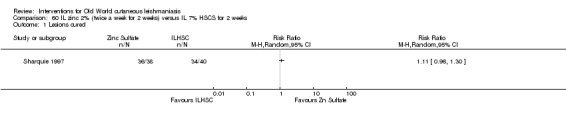
Comparison 60 IL zinc 2% (twice a week for 2 weeks) versus IL 7% HSCS for 2 weeks, Outcome 1 Lesions cured.
Analysis 61.1.
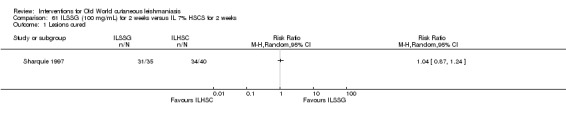
Comparison 61 ILSSG (100 mg/mL) for 2 weeks versus IL 7% HSCS for 2 weeks, Outcome 1 Lesions cured.
Secondary outcome: speed of healing (time taken to be 'cured')
Sharquie 1997 reported that in the zinc sulphate group most lesions were cured by 30 days. In the HSCS and in the ILSSG group, lesions were cured after 30 days.
Secondary outcome: prevention of scarring
In all three groups the scar was minimal or absent after healing, but all participants developed postinflammatory hyperpigmentation. In the control group, some lesions (mainly on the lower limbs) showed signs of infection.
Secondary outcome: adverse effects
Regarding adverse effects, all participants from both groups in the Sharquie 1997 study had pain at the time of the injection.
3.3.2 IL zinc sulphate versus ILMA
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Iran compared IL zinc sulphate up to six times weekly versus ILMA up to six times weekly (maximum 2 mL) (Firooz 2005). At five weeks after treatment, 22.6% (12/53) and 50.9% (27/53) of the lesions in the respective groups had healed completely (RR 0.44, 95% CI 0.25 to 0.78; N = 106; 1 study, Analysis 62.1). However, baseline characteristics were unbalanced: both mean diameter induration and mean diameter ulceration were higher in the zinc sulphate group (induration: 10.0 mm (standard deviation (SD) 11.4)), and ulceration: 2.7 mm (SD 4.6)) than in the MA group (induration: 2.4 mm (SD 8.7) and ulceration 0.6 mm (SD 3.6)). However, the authors claimed that there was no significant difference between the two groups (P > 0.05; exact p value not reported).
Analysis 62.1.

Comparison 62 IL zinc 2% (weekly for up to 6 weeks) versus ILMA (max 2 mL weekly for up to 6 weeks), Outcome 1 Lesions cured.
An RCT from Iran compared two double bouts of IL zinc sulphate, delivered at a two‐week interval, versus six weekly bouts of ILMA 60 mg/kg/d (Maleki 2012). At eight weeks after starting treatment, 33.3% (8/24) and 80% (8/10) of participants achieved complete cure in the respective groups (RR 0.42, 95% CI 0.22 to 0.79; N = 34; 1 study, Analysis 63.1).
Analysis 63.1.
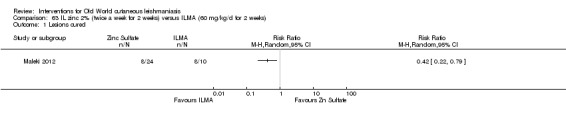
Comparison 63 IL zinc 2% (twice a week for 2 weeks) versus ILMA (60 mg/kg/d for 2 weeks), Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
One RCT from Iran compared IL zinc sulphate with ILMA at a maximum dose of 2 mL (Iraji 2004). In cases with slight to mild improvement, the authors gave another injection after two weeks. The authors reported complete cure in 53% (26/49) and 38% (21/55) of participants in the respective groups at the end of the treatment period. There were no significant differences in cure rates between groups (RR 1.39, 95% CI 0.91 to 2.13; N = 104; 1 study, Analysis 62.2).
Analysis 62.2.
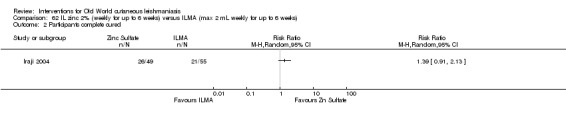
Comparison 62 IL zinc 2% (weekly for up to 6 weeks) versus ILMA (max 2 mL weekly for up to 6 weeks), Outcome 2 Participants complete cured.
Secondary outcome: adverse effects
In Firooz 2005, the most commonly observed adverse effect was pain, found in 25% (18/72) of participants: in 36.1% (13/36) of participants in the zinc sulphate group and 13.9% (5/36) in the ILMA group (RR 2.60, 95% CI 1.03 to 6.54, Analysis 62.3). In the zinc sulphate group, 8.4% (3/36) of participants complained about burning at the injection site. Itching occurred in 8.4% (3/36) and in 25% (9/36), and inflammation in 19.4% (7/36) and 22.2% (8/36) of the cases in the zinc sulphate and ILMA group, respectively. There were no significant differences in other adverse effects rates between groups (Analysis 62.3). Iraji 2004 reported pruritus, erythema, and scaling in the periphery of the injection site in three cases in the ILMA group; IL zinc sulphate was painful, and the severity caused vasovagal shock in two cases. There were no significant differences in skin reactions (RR 7.84, 95% CI 0.42 to 148.08; N = 104; 1 study, Analysis 62.3) or severe pain rates (RR 5.60, 95% CI 0.28 to 113.87; N = 104; 1 study, Analysis 62.3) between groups. In another study the side effects seen in both groups were pain after injection and hyperpigmentation (Maleki 2012). All participants in the zinc sulphate group experienced burning after injection and necrosis of the lesions, and three of these participants also had inflammation and swelling. There were no significant differences in inflammation and swelling rates between groups (RR 3.08, 95% CI 0.17 to 54.71; N = 34; 1 study, Analysis 63.2).
Analysis 62.3.
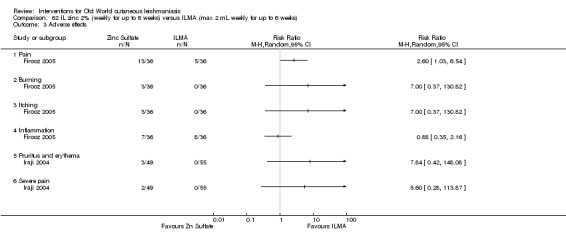
Comparison 62 IL zinc 2% (weekly for up to 6 weeks) versus ILMA (max 2 mL weekly for up to 6 weeks), Outcome 3 Adverse effects.
Analysis 63.2.
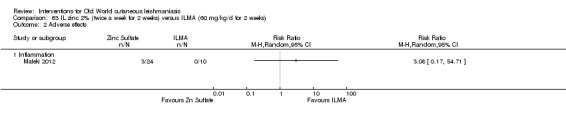
Comparison 63 IL zinc 2% (twice a week for 2 weeks) versus ILMA (60 mg/kg/d for 2 weeks), Outcome 2 Adverse effects.
3.4 Topical imiquimod
3.4.1 Imiquimod cream versus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared 5% imiquimod cream three times per week for 28 days plus IMMA 20 mg/kg/d for 14 days versus vehicle plus IMMA at the same dose (Firooz 2006). At 3.5 months after treatment, 44.1% (26/59) and 40% (24/60) of participants in the treatment and control groups, respectively, achieved complete cure (RR 1.10, 95% CI 0.72 to 1.68; N = 119; 1 study, Analysis 64.1).
Analysis 64.1.
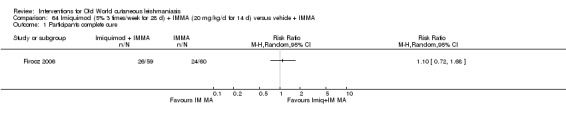
Comparison 64 Imiquimod (5% 3 times/week for 28 d) + IMMA (20 mg/kg/d for 14 d) versus vehicle + IMMA, Outcome 1 Participants complete cure.
Secondary outcome: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
At four months after treatment, relapse occurred in 1/32 participants available for assessment and treated with imiquimod plus IMMA, and in 3/37 control participants (RR 0.39, 95% CI 0.04 to 3.52; N = 69; 1 study, Analysis 64.2).
Analysis 64.2.
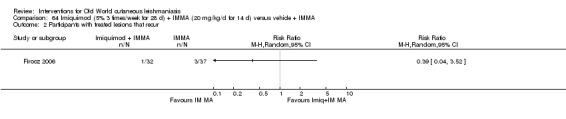
Comparison 64 Imiquimod (5% 3 times/week for 28 d) + IMMA (20 mg/kg/d for 14 d) versus vehicle + IMMA, Outcome 2 Participants with treated lesions that recur.
Secondary outcome: adverse effects
The only adverse effects related to topical treatment were moderate itch and a burning sensation in three imiquimod‐treated participants. There were no significant differences between groups (RR 7.12, 95% CI 0.38 to 134.84; N = 119; 1 study, Analysis 64.3).
Analysis 64.3.
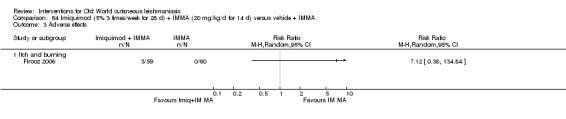
Comparison 64 Imiquimod (5% 3 times/week for 28 d) + IMMA (20 mg/kg/d for 14 d) versus vehicle + IMMA, Outcome 3 Adverse effects.
3.5 Intralesional hypertonic sodium chloride solution
For details about the adverse effects of intralesional hypertonic sodium chloride solution (HSCS) please see Table 122.
3.5.1 IL 7% HSCS versus ILSSG
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Sri Lanka compared IL HSCS (0.2 mL to 4 mL per lesion and maximum 5 injections) versus ILSSG (maximum 2 mL and 5 injections) (Ranawaka 2010). Average duration of treatment was 8.78 weeks and 5.11 weeks, respectively. At 18 months of follow‐up, 92.2% (86/96) and 100% (136/136) lesions had healed completely in the respective groups (RR 0.87, 95% CI 0.79 to 0.96; N = 147; 1 study, Analysis 65.1). There was no difference in treatment response between therapies with regard to the size, duration, or location (head, trunk, upper, and lower extremities) of the lesions.
Analysis 65.1.
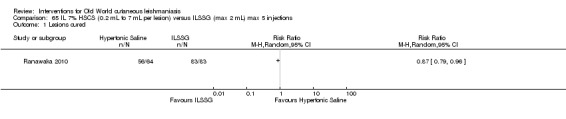
Comparison 65 IL 7% HSCS (0.2 mL to 7 mL per lesion) versus ILSSG (max 2 mL) max 5 injections, Outcome 1 Lesions cured.
Secondary outcome: prevention of scarring
In both groups the scar was minimal or absent after healing, but all participants developed postinflammatory hyperpigmentation.
Secondary outcome: adverse effects
There were no systemic adverse effects with ILSSG or HSCS in Ranawaka 2010. Pain during injection was the only local side effect noted with both therapies. After healing, scarring was minimal, but all participants receiving both treatments had postinflammatory hyperpigmentation, which faded over six to eight months.
3.5.2 IL 5% HSCS versus ILMA
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared IL HSCS (0.5 mL to 1 mL) with ILMA (0.5 mL to 1 mL) weekly for 6 to 10 weeks (Sadeghian 2006b). At the end of treatment, 25% (9/36) and 33.3% (12/36) lesions had healed in the respective groups (RR 0.75, 95% CI 0.36 to 1.56; N = 72; 1 study, Analysis 66.1).
Analysis 66.1.
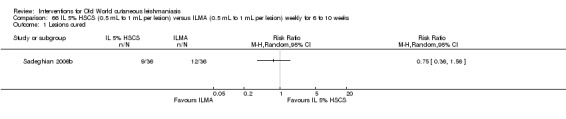
Comparison 66 IL 5% HSCS (0.5 mL to 1 mL per lesion) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 6 to 10 weeks, Outcome 1 Lesions cured.
Secondary outcome: adverse effects
In the HSCS group, there were three cases of sporotrichotic dissemination but with no allergic reactions. In the ILMA group, there were three cases of sporotrichotic dissemination, two satellite lesions, and two allergic reactions including redness, oedema, and severe itch around the lesions. There were no significant differences in sporotrichotic dissemination rates (RR 1.00, 95% CI 0.22 to 4.63; N = 72; 1 study) or allergic reactions rates (RR 0.20, 95% CI 0.01 to 4.03; N = 72; 1 study) between groups (Analysis 66.2).
Analysis 66.2.
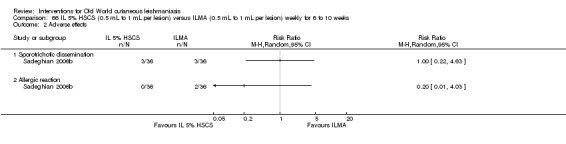
Comparison 66 IL 5% HSCS (0.5 mL to 1 mL per lesion) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 6 to 10 weeks, Outcome 2 Adverse effects.
3.5.3 IL 7% HSCS versus IL ciprofloxacin solution
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iraq compared IL 7% HSCS (0.1 mL to 0.5 mL per lesion) with IL ciprofloxacin solution 2 mg/mL (0.1 mL to 0.5 mL per lesion) (Al Hamdi 2010). At two months after treatment, 76.2% (16/21) and 81.5% (22/27) of the lesions had healed in the respective groups. There was no difference in cure rates between IL HSCS and IL ciprofloxacin (RR 1.07, 95% CI 0.79 to 1.44; N = 48; 1 study, Analysis 67.1).
Analysis 67.1.
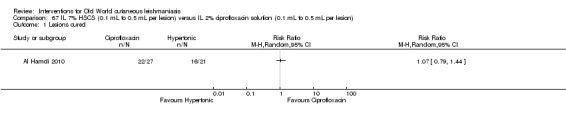
Comparison 67 IL 7% HSCS (0.1 mL to 0.5 mL per lesion) versus IL 2% ciprofloxacin solution (0.1 mL to 0.5 mL per lesion), Outcome 1 Lesions cured.
3.5.4 IL 10% HSCS versus IL 15% HSCS versus ILSSG
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Sri Lanka compared IL 10% HSCS versus IL 15% HSCS versus ILSSG (0.2 mL to 4 mL per lesion and maximum five injections) (Ranawaka 2015). The authors reported that three months after treatment, 93% (268/290), 93.6% (92/98), and 96.3% (226/235) of the lesions in the respective groups had completely healed. There was no difference in cure rates between any comparison: IL 15% HSCS versus IL 10 % HSCS (RR 1.02, 95% CI 0.96 to 1.08; N = 388, Analysis 68.1); ILSSG versus IL 10% HSCS (RR 1.04, 95% CI 1.00 to 1.08; N = 525, Analysis 69.1); and ILSSG versus IL 15% HSCS (RR 1.02, 95% CI 0.97 to 1.08; N = 333; 1 study, Analysis 70.1).
Analysis 68.1.
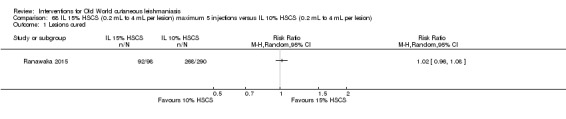
Comparison 68 IL 15% HSCS (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 1 Lesions cured.
Analysis 69.1.
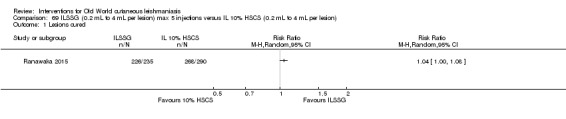
Comparison 69 ILSSG (0.2 mL to 4 mL per lesion) max 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 1 Lesions cured.
Analysis 70.1.
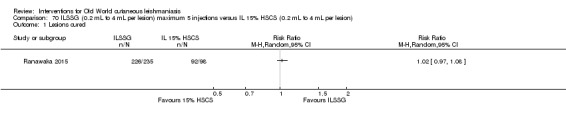
Comparison 70 ILSSG (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 15% HSCS (0.2 mL to 4 mL per lesion), Outcome 1 Lesions cured.
Secondary outcome: speed of healing (time taken to be 'cured')
There was no difference in speed of healing between IL 15% HSCS versus IL 10% HSCS (MD −1.45, 95% CI −4.34 to 1.44 weeks; N = 388; 1 study, Analysis 68.3). Lesions in ILSSG group cured significantly faster than in 10% HSCS group (MD −5.20, 95% CI −6.31 to −4.09 weeks; N = 525, Analysis 69.3) and in IL 15% HSCS group (MD −6.65, 95% CI −9.41 to −3.89 weeks; N = 333, Analysis 70.3).
Analysis 68.3.
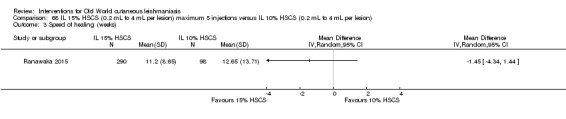
Comparison 68 IL 15% HSCS (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 3 Speed of healing (weeks).
Analysis 69.3.
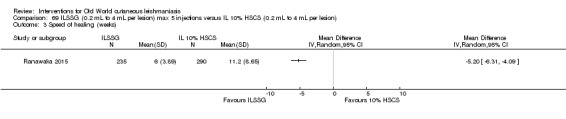
Comparison 69 ILSSG (0.2 mL to 4 mL per lesion) max 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 3 Speed of healing (weeks).
Analysis 70.3.
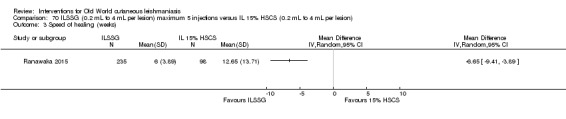
Comparison 70 ILSSG (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 15% HSCS (0.2 mL to 4 mL per lesion), Outcome 3 Speed of healing (weeks).
Secondary outcome: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
After six months of follow‐up, the authors described four cases of relapse in 10% HSCS group, four cases in 15% HSCS group and three in the ILSSG group (Ranawaka 2015). There was no significant difference in this outcome in any comparisons: 15% HSCS versus 10% HSCS (RR 2.93, 95% CI 0.75 to 11.50; N = 362; 1 study, Analysis 68.2), ILSSG versus 10% HSCS (RR 0.90 95% CI 0.20 to 3.96, Analysis 69.2), and ILSSG versus 15% HSCS (RR 1.02, 95% CI 0.97 to 1.08, Analysis 70.2).
Analysis 68.2.

Comparison 68 IL 15% HSCS (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 2 Recurrence.
Analysis 69.2.
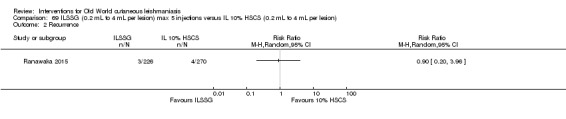
Comparison 69 ILSSG (0.2 mL to 4 mL per lesion) max 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 2 Recurrence.
Analysis 70.2.
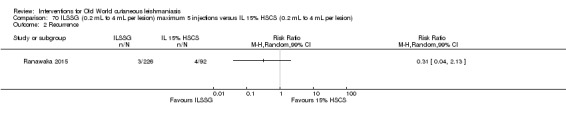
Comparison 70 ILSSG (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 15% HSCS (0.2 mL to 4 mL per lesion), Outcome 2 Recurrence.
Secondary outcome: adverse effects
All participants experienced pain during injections, particularly in the 15% HSCS group. All lesions presented postinflammatory hyperpigmentation, which faded over six to eight months. Ulceration and necrosis occurred in 30% (30/98) of the lesions treated with 15% HSCS and in 3.1% (9/290) of the lesions in the 10% HSCS group. There were statistically significant differences in ulceration and necrosis rates between the IL 15% HCS group and the 10% HSCS group (RR 9.86, 95% CI 4.86 to 20.04; N = 388; 1 study, Analysis 68.4) and between the IL 15% HCS group and ILSSG group (RR 145.41, 95% CI 8.98 to 2354.65; N = 333; 1 study, Analysis 70.4), with more events in the IL 15% HCS group.
Analysis 68.4.
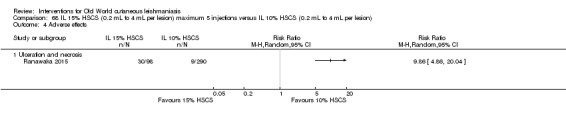
Comparison 68 IL 15% HSCS (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 4 Adverse effects.
Analysis 70.4.
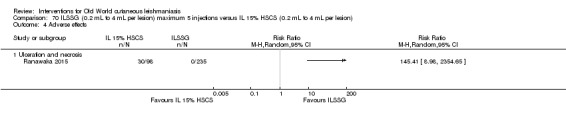
Comparison 70 ILSSG (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 15% HSCS (0.2 mL to 4 mL per lesion), Outcome 4 Adverse effects.
3.6 IL interferon‐gamma (IFN‐γ)
3.6.1 IL IFN‐ γ versus ILMA
Primary outcome: percentage of lesions with a complete cure after the end of treatment
An RCT from Syria compared IL IFN‐γ versus ILMA once weekly for five weeks (Harms 1991). At one month after treatment, 3% (1/37) and 76% (29/38) of the lesions had healed completely in the respective groups. The cure rates in the IL IFN‐γ group were significantly lower than in the ILMA group (RR 0.04, 95% CI 0.01 to 0.28; N = 76; 1 study, Analysis 71.1).
Analysis 71.1.
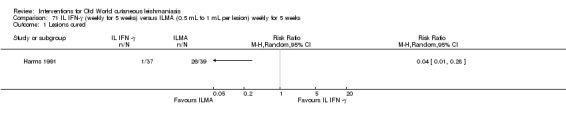
Comparison 71 IL IFN‐γ (weekly for 5 weeks) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 5 weeks, Outcome 1 Lesions cured.
Secondary outcome: adverse effects
In the IFN‐γ arm, pain at the injection site of was mild on 68 occasions, moderate on 47, and severe on 38. In the ILMA group it was mild on 55 occasions, moderate on 51, and severe on 40. Other adverse effects included local maculopapular erythema in the ILMA group and headache in the IFN‐γ group.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Parasitological assessment one month after the end of treatment showed 65% (24/37) and 100% (38/38) of the lesions were parasite‐free in the IFN‐γ and ILMA groups, respectively (RR 0.65, 95% CI 0.51 to 0.83; N = 75; 1 study, Analysis 71.2).
Analysis 71.2.
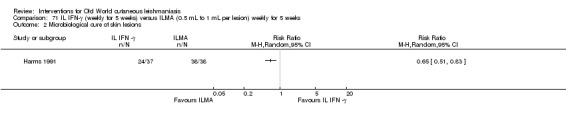
Comparison 71 IL IFN‐γ (weekly for 5 weeks) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 5 weeks, Outcome 2 Microbiological cure of skin lesions.
3.7 Topical aminoglycoside ointment (WR279,396)
3.7.1 WR279,396 ointment versus placebo
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Tunisia compared WR279,396 (a third generation aminoglycoside ointment) versus vehicle twice daily for 20 days (Ben Salah 2009). After a six‐month follow‐up, 86% (43/50) and 64% (27/42) of the participants in the respective groups achieved complete cure (RR 1.34, 95% CI 1.04 to 1.72; N = 92, Analysis 72.1).
Analysis 72.1.
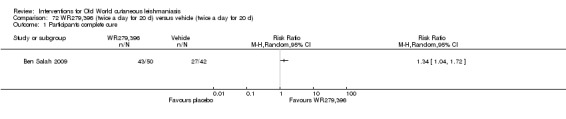
Comparison 72 WR279,396 (twice a day for 20 d) versus vehicle (twice a day for 20 d), Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Regarding adverse effects, 30% (n = 15) of participants from the WR279,396 and 24% (n = 10) from the vehicle groups suffered from erythema at the site of application (RR 1.26, 95% CI 0.63 to 2.50; N = 92; 1 study); 14% from both groups (n = 7 and n = 6, respectively) had mild pain within 30 minutes of application (RR 0.98, 95% CI 0.36 to 2.69; N = 92; 1 study); 28% (n = 14) and 21% (n = 9) presented mild increases and decreases in hearing acuity from baseline; 28% (n = 14) and 21% (n = 9) had changed hearing acuity (RR 1.31, 95% CI 0.63 to 2.71; N = 92; 1 study), and there were no deaths reported in either group. See Analysis 72.2.
Analysis 72.2.

Comparison 72 WR279,396 (twice a day for 20 d) versus vehicle (twice a day for 20 d), Outcome 2 Adverse effects.
3.8 Intralesional metronidazole
3.8.1 IL metronidazole versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT compared weekly intralesional metronidazole (2.5 mg to 10 mg = 0.5 mL to 2 mL for each lesion) with weekly ILMA (150 mg to 600 mg = 0.5 mL to 2 mL for each lesion) for up to eight weeks (Mapar 2010). At the end of treatment, 16.6% (3/18) and 72.2% (13/18) of participants in the respective groups achieved complete cure (RR 0.23, 95% CI 0.08 to 0.67; N = 36, Analysis 73.1).
Analysis 73.1.
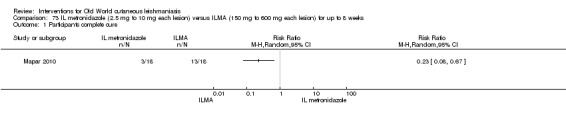
Comparison 73 IL metronidazole (2.5 mg to 10 mg each lesion) versus ILMA (150 mg to 600 mg each lesion) for up to 8 weeks, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Authors did not describe any systemic or local adverse effects with metronidazole, but two participants in the ILMA group suffered local inflammatory reactions with oedema and induration that diminished in about 10 days. There were no significant differences in local inflammatory reactions rates between groups (RR 0.20, 95% CI 0.01 to 3.89; N = 36; 1 study, Analysis 73.2). In both groups the participants reported severe pain at the site of injections, but in the ILMA group it was intolerable.
Analysis 73.2.
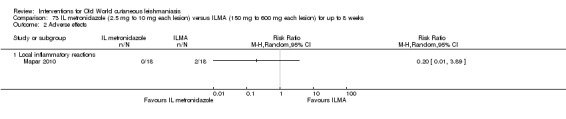
Comparison 73 IL metronidazole (2.5 mg to 10 mg each lesion) versus ILMA (150 mg to 600 mg each lesion) for up to 8 weeks, Outcome 2 Adverse effects.
3.9 Topical miltefosine
3.9.1 Topical miltefosine versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical miltefosine ointment 6% once daily versus ILMA twice a week for up to 28 days (Asilian 2014). At one month after treatment, 81.3% (26/32) and 50.0% (16/32) of the participants in the respective groups achieved complete cure (RR 1.63, 95% CI 1.11 to 2.39; N = 64; 1 study, Analysis 74.1).
Analysis 74.1.
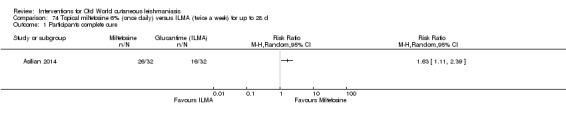
Comparison 74 Topical miltefosine 6% (once daily) versus ILMA (twice a week) for up to 28 d, Outcome 1 Participants complete cure.
3.10 Topical dapsone
3.10.1 Niosomal dapsone 5% gel mask plus ILMA versus cryotherapy plus ILMA
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared niosomal dapsone 5% gel mask (twice a day) plus ILMA (weekly) versus cryotherapy (every two weeks) plus ILMA (weekly) for up to 16 weeks (Fekri 2015). At 16‐week follow‐up, 82% (29/35) and 86% (33/38) of the lesions had healed in the respective groups (RR 0.95, 95% CI 0.79 to 1.16; N = 73; 1 study, Analysis 75.1).
Analysis 75.1.
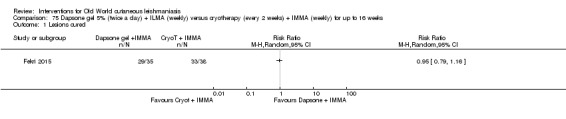
Comparison 75 Dapsone gel 5% (twice a day) + ILMA (weekly) versus cryotherapy (every 2 weeks) + IMMA (weekly) for up to 16 weeks, Outcome 1 Lesions cured.
3.11 Topical 0.045% pharmaceutical chlorite (DAC N‐055)
3.11.1 Topical 0.045% pharmaceutical chlorite (DAC N‐055) with and without bipolar high frequency electrocauterisation versus ILSSG
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Afghanistan compared 0.045% pharmaceutical chlorite (DAC N‐055) plus bipolar high frequency electrocauterisation for 15 minutes versus 0.045% DAC N‐055 for 15 minutes alone versus ILSSG 0.6 mL for up to 75 days (Stahl 2014). After a 75‐day follow‐up, the authors reported a complete cure in 100% (23/23), 87% (20/23), and 65% (15/23) of the participants in the respective groups. There was no difference between DAC N‐055 plus electrocauterisation versus DAC N‐055 alone (RR 1.15, 95% CI 0.96 to 1.37; N = 46; 1 study, Analysis 76.1). DAC N‐055 plus electrocauterisation was more efficacious than ILSSG (RR 1.52, 95% CI 1.12 to 2.05; N = 46; 1 study, Analysis 77.1), and DAC N‐055 alone was not more efficacious than ILSSG (RR 1.33, 95% CI 0.95 to 1.87; N = 46; 1 study, Analysis 78.1).
Analysis 76.1.

Comparison 76 DAC‐055 + MWT (for 15 min) versus DAC‐055 alone for up to 75 d, Outcome 1 Participants complete cure.
Analysis 77.1.
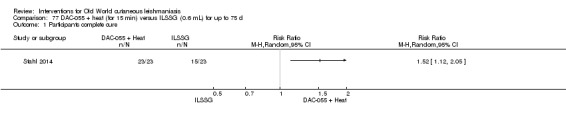
Comparison 77 DAC‐055 + heat (for 15 min) versus ILSSG (0.6 mL) for up to 75 d, Outcome 1 Participants complete cure.
Analysis 78.1.
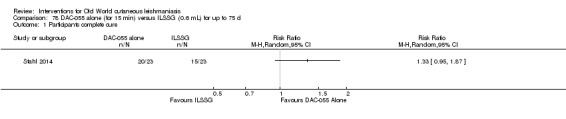
Comparison 78 DAC‐055 alone (for 15 min) versus ILSSG (0.6 mL) for up to 75 d, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
The hazard ratio for DAC N‐055 versus ILSSG was 4.4 (95% CI 2.2 to 8.7) in the per protocol analysis (P < 0.001) and 3.2 (95% CI 1.6 to 6.3) in the intention‐to‐treat analysis (P < 0.001). The lesion closed three to four times faster in the DAC N‐055 group than in the ILSSG group.
Secondary outcome: adverse effects
Regarding adverse effects, four participants developed reulceration of lesions in the DAC N‐055 plus electrocauterisation group, three in the DAC N‐055 alone group, and seven in the ILSSG group. There were no significant differences in reulceration rates between DAC N‐055 plus electrocauterisation versus DAC N‐055 alone (RR 1.33, 95% CI 0.34 to 5.30; N = 46; 1 study, Analysis 76.2). Two participants (8.7%) developed keloids and keloid scars in the DAC N‐055 plus electrocauterisation group.
Analysis 76.2.
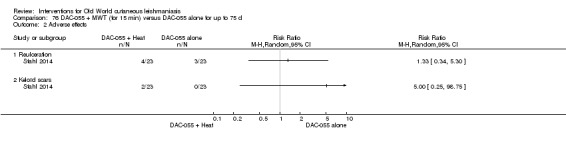
Comparison 76 DAC‐055 + MWT (for 15 min) versus DAC‐055 alone for up to 75 d, Outcome 2 Adverse effects.
3.12 Topical Thio‐Ben
3.12.1 Topical Thio‐Ben plus cryotherapy versus ILMA plus cryotherapy
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared 1 mL to 2 mL of Thio‐Ben tincture daily plus cryotherapy (liquid nitrogen (−195°C) fortnightly versus ILMA weekly (dose of 0.5 mL to 2 mL per lesion) plus cryotherapy (Daie Parizi 2015). After three months' follow‐up 91% (20/22) and 92% (23/25) of the lesions had healed completely in the respective groups. There was no difference between Thio‐Ben plus cryotherapy and ILMA plus cryotherapy (RR 0.99, 95% CI 0.83 to 1.18; N = 47; 1 study, Analysis 79.1).
Analysis 79.1.
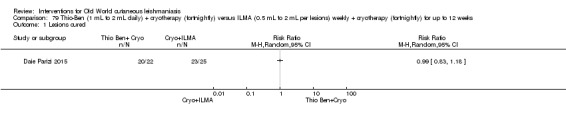
Comparison 79 Thio‐Ben (1 mL to 2 mL daily) + cryotherapy (fortnightly) versus ILMA (0.5 mL to 2 mL per lesions) weekly + cryotherapy (fortnightly) for up to 12 weeks, Outcome 1 Lesions cured.
Secondary outcome: speed of healing (time taken to be 'cured')
On average, lesions required the same amount of time to achieve a complete cure: 2.45 months in the Thio‐Ben plus cryotherapy group and 2.41 months in the ILMA plus cryotherapy group (Daie Parizi 2015).
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
The rate of relapse was slightly, but not significantly, higher in the Thio‐Ben group 20% (4/22) than in the ILMA group 13% (3/25) (RR 1.52, 95% CI 0.38 to 6.04; N = 47; 1 study, Analysis 79.2).
Analysis 79.2.
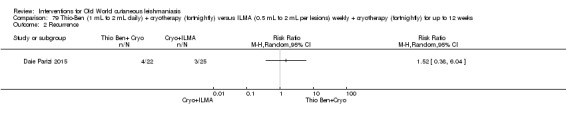
Comparison 79 Thio‐Ben (1 mL to 2 mL daily) + cryotherapy (fortnightly) versus ILMA (0.5 mL to 2 mL per lesions) weekly + cryotherapy (fortnightly) for up to 12 weeks, Outcome 2 Recurrence.
Secondary outcome: adverse effects
Regarding adverse effects, investigators observed itching and mild erythema in almost all cases with different degrees of severity, and oedema especially after cryotherapy in almost all lesions. Participants from the ILMA plus cryotherapy group experienced pain at the injection site in almost all cases, while three participants reported dizziness and nausea (RR 0.16, 95% CI 0.01 to 2.96; N = 47; 1 study), and one had a hypersensitive reaction (RR 0.38, 95% CI 0.02 to 8.80; N = 47; 1 study, Analysis 79.3). There was poor adherence to treatment particularly among children.
Analysis 79.3.
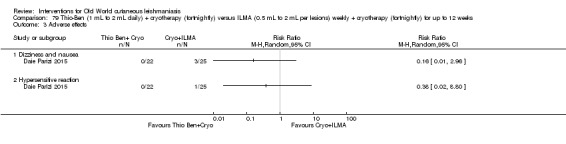
Comparison 79 Thio‐Ben (1 mL to 2 mL daily) + cryotherapy (fortnightly) versus ILMA (0.5 mL to 2 mL per lesions) weekly + cryotherapy (fortnightly) for up to 12 weeks, Outcome 3 Adverse effects.
4. Physical therapies
4.1 Laser
See Table 123 for a summary of adverse effects of lasers and Table 124 for a summary of adverse effects of cryotherapy.
4.1.1 CO₂ laser versus IMMA
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared CO₂ laser applied to the lesion (30 W, continuous) versus IMMA (50 mg/kg/d) for 15 days (this treatment was repeated after 15 days of rest) (Asilian 2004b). At six weeks after treatment, 11% (20/183) and 12% (30/250) of the lesions had healed completely in the respective groups (RR 0.91, 95% CI 0.53 to 1.55; N = 433; 1 study, Analysis 80.1).
Analysis 80.1.
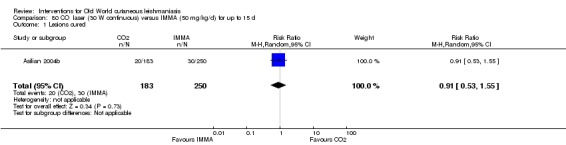
Comparison 80 CO₂ laser (30 W continuous) versus IMMA (50 mg/kg/d) for up to 15 d, Outcome 1 Lesions cured.
Secondary outcomes: speed of healing
The time taken to be cured was one month for the CO₂ laser group and three months for the IMMA group.
Secondary outcome: prevention of scarring
In the laser group, study authors considered most scars to be 'acceptable', except in five cases (4%) in which the lesions were located on the joints or neck, and the resulting scars were raised and hypertrophic.
Secondary outcome: adverse effects
In the laser group, 3% (4/123) of participants experienced hyperpigmentation and persistent redness (RR 12.28, 95% CI 0.67 to 226.62; N = 433; 1 study, Analysis 80.2), and five participants had hypertrophic scarring (RR 0.16, 95% CI 0.01 to 2.96; N = 47; 1 study, Analysis 80.2). In the IMMA group, 9% (22/250) of participants reported myalgia, sensitivity, headache, urticaria, and nausea; these were higher in the IMMA group than in the laser group (RR 0.03, 95% CI 0.00 to 0.50; N = 433; 1 study, Analysis 80.2).
Analysis 80.2.
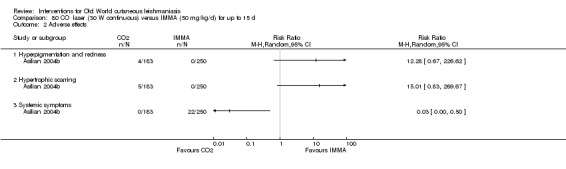
Comparison 80 CO₂ laser (30 W continuous) versus IMMA (50 mg/kg/d) for up to 15 d, Outcome 2 Adverse effects.
4.1.2 CO₂ laser versus cryotherapy plus ILMA
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared CO₂ laser (30 W, continuous) applied to the lesion (repeated maximum 3 to 5 times) versus combined cryotherapy (liquid nitrogen 10 s to 25 s) biweekly plus ILMA (0.5 mL to 2 mL per lesion) weekly until complete cure or up to 12 weeks (Shamsi Meymandi 2011). At 12 weeks, 94.7% (90/95) and 77.9% (74/95) of the lesions had healed in the respective groups. CO₂ laser was more efficacious than cryotherapy plus ILMA (RR 1.22, 95% CI 1.08 to 1.37; N = 190; 1 study, Analysis 81.1).
Analysis 81.1.
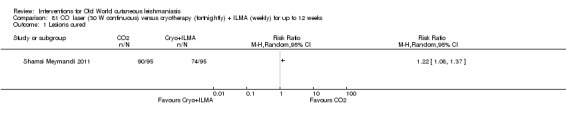
Comparison 81 CO₂ laser (30 W continuous) versus cryotherapy (fortnightly) + ILMA (weekly) for up to 12 weeks, Outcome 1 Lesions cured.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
There was no worsening (increase in size of the lesions) or relapse after the two types of treatment.
Secondary outcome: adverse effects
The authors reported adverse effects to participants (80 participants per group). In the laser group, 25% (n = 20) of participants had hyperpigmentation and trivial scarring, compared to 18.7% (n = 15) in the cryotherapy plus ILMA group (RR 1.33, 95% CI 0.73 to 2.44; N = 190; 1 study); 8.75% (n = 7) and 18.8% (n = 15), respectively, had atrophic scars (RR 0.47, 95% CI 0.20 to 1.09; N = 190; 1 study), and 5% (n = 4) in the laser group only had hypopigmentation plus trivial scarring (RR 9.00, 95% CI 0.49 to 164.88; N = 190; 1 study; Analysis 81.2).
Analysis 81.2.
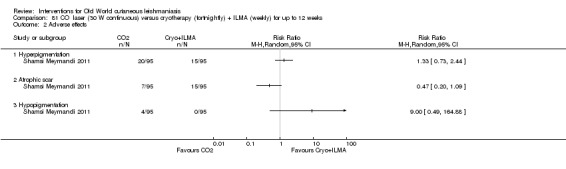
Comparison 81 CO₂ laser (30 W continuous) versus cryotherapy (fortnightly) + ILMA (weekly) for up to 12 weeks, Outcome 2 Adverse effects.
4.1.3 Ablative CO₂ laser versus fractional CO₂ laser
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared ablative CO₂ laser (10 ns, frequency: use of 20 kHz, and power: 25 Kw for one session) versus fractional CO₂ laser (energy: 25, one pulse, pass: 1, dot cycle: 6) until complete cure or up to 12 weeks (Nilforoushzadeh 2014a). At six months' follow‐up, 46.7% (28/60) and 76.7% (46/60) of participants in the respective groups achieved a complete cure (RR 0.61, 95% CI 0.45 to 0.83; N = 120; 1 study, Analysis 82.1).
Analysis 82.1.

Comparison 82 Ablative CO₂ laser (25 kW for 1 session) versus 3 weeks fractional CO₂ laser, Outcome 1 Partcipants complete cure.
Secondary outcome: adverse effects
Nilforoushzadeh 2014a reported erythema in two participants (3.3%) in the ablative CO₂ laser group and four (6.7%) in the fractional CO₂ laser group (RR 0.50, 95% CI 0.10 to 2.63; N = 120; 1 study, Analysis 82.2).
Analysis 82.2.
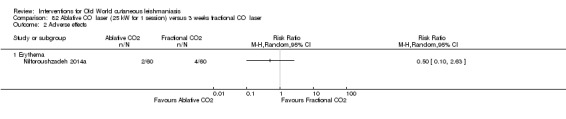
Comparison 82 Ablative CO₂ laser (25 kW for 1 session) versus 3 weeks fractional CO₂ laser, Outcome 2 Adverse effects.
4.2 Trichloroacetic acid
4.2.1 Trichloroacetic acid versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared trichloroacetic acid (TCA) 50% (wt/vol) applied fortnightly until complete re‐epithelialisation or up to three times versus ILMA (0.5 mL to 2 mL per lesion) weekly until complete re‐epithelialisation of the lesions or up to six weeks (Nilforoushzadeh 2006). At the end of the treatment period, 65% (26/40) and 57.5% (23/40) of participants in the respective groups achieved a complete cure (RR 1.13, 95% CI 0.80 to 1.60; N = 80; 1 study, Analysis 83.1).
Analysis 83.1.
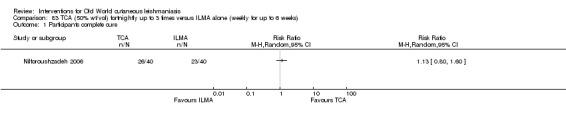
Comparison 83 TCA (50% wt/vol) fortnightly up to 3 times versus ILMA alone (weekly for up to 6 weeks), Outcome 1 Participants complete cure.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
Recurrence occurred in 12.5% (5/40) of the cases in the TCA group and 10% (4/40) in the ILMA group after three months' follow‐up (RR 1.25, 95% CI 0.36 to 4.32; N = 80; 1 study, Analysis 83.2).
Analysis 83.2.
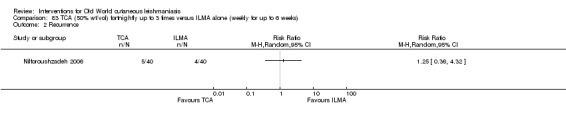
Comparison 83 TCA (50% wt/vol) fortnightly up to 3 times versus ILMA alone (weekly for up to 6 weeks), Outcome 2 Recurrence.
Secondary outcome: adverse effects
The observed adverse effects included mild erythema and itch (two cases in the ILMA group), with no significant differences between groups (RR 0.20, 95% CI 0.01 to 4.04; N = 80; 1 study, Analysis 83.3).
Analysis 83.3.
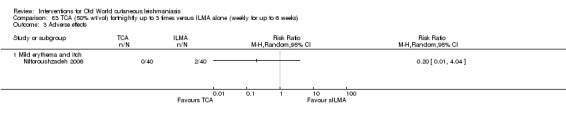
Comparison 83 TCA (50% wt/vol) fortnightly up to 3 times versus ILMA alone (weekly for up to 6 weeks), Outcome 3 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Parasitological assessment at week 6 showed 72.5% (29/40) and 75% (30/40) of the lesions were parasite‐free in the TCA and ILMA groups, respectively. Parasitologic tests were positive at week 6 in 27.5% (11/40) and 25% (10/40) of the cases in the TCA and ILMA groups, respectively. There were no significant differences in parasitologic test rates between groups ((RR 0.97, 95% CI 0.74 to 1.26; participants = 80; studies = 1 Analysis 83.4).
Analysis 83.4.
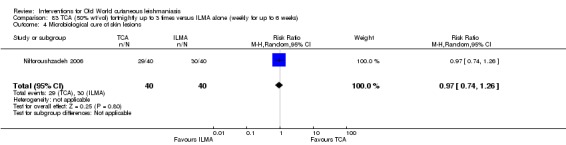
Comparison 83 TCA (50% wt/vol) fortnightly up to 3 times versus ILMA alone (weekly for up to 6 weeks), Outcome 4 Microbiological cure of skin lesions.
4.2.2 Non‐ablative laser plus TCA versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared non‐ablative laser (weekly for four consecutive weeks) plus TCA (applied weekly for up to two weeks) versus ILMA (twice a week up to eight weeks) alone (Nilforoushzadeh 2012). At six months' follow‐up, 42.1% (16/30) and 63.2% (24/30) of the participants in the respective groups achieved a complete cure (RR 0.67, 95% CI 0.46 to 0.97; N = 60, Analysis 84.1).
Analysis 84.1.
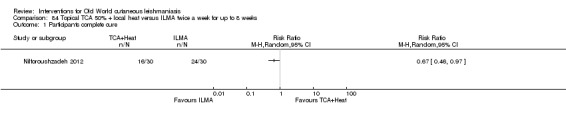
Comparison 84 Topical TCA 50% + local heat versus ILMA twice a week for up to 8 weeks, Outcome 1 Participants complete cure.
Primary outcome: percentage of lesions cured after the end of treatment
The authors performed a subgroup analysis according to sex. In the male group, 36.3% (8/22) of the lesions treated with laser plus TCA, and 53.3% (8/15) receiving ILMA, had healed completely at six months (RR 0.68, 99% CI 0.26 to 1.77; N = 37). In females, cure rates in the respective groups were 50% (8/16) and 69.6% (16/23) (RR 0.72, 99% CI 0.34 to 1.50; N = 39; Analysis 84.2).
Analysis 84.2.
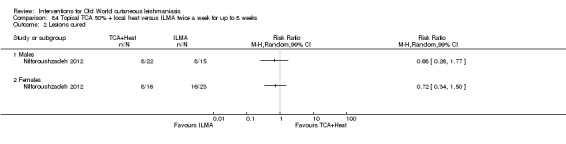
Comparison 84 Topical TCA 50% + local heat versus ILMA twice a week for up to 8 weeks, Outcome 2 Lesions cured.
4.2.3 TCA plus ILMA versus factional laser plus ILMA versus ILMA alone
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared TCA (applied weekly for up to eight consecutive weeks until the lesion was frosted) plus ILMA versus fractional laser (fortnightly two sessions) plus ILMA versus ILMA alone twice a week until complete re‐epithelialisation of the lesions or up to eight weeks (Nilforoushzadeh 2013). At six months' follow‐up, 90% (27/30) of the participants in the TCA plus ILMA group, 87% (20/23) in the fractional laser plus ILMA group, and 38.5% (10/26) in the ILMA alone group achieved complete cure. Cure rates were significantly higher in the TCA plus ILMA group compared with ILMA alone (RR 2.34, 95% CI 1.42 to 3.86; N = 56; 1 study, Analysis 85.1). ILMA alone was less efficacious than fractional laser and ILMA (RR 2.26, 95% CI 1.36 to 3.77; N = 49; 1 study, Analysis 86.1). There was no difference in complete cure rates between TCA plus ILMA versus fractional laser plus ILMA (RR 1.03, 95% CI 0.85 to 1.26; N = 53; 1 study, Analysis 87.1).
Analysis 85.1.
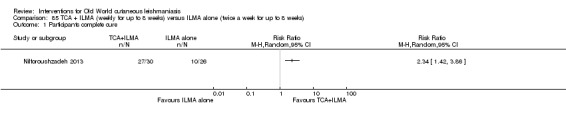
Comparison 85 TCA + ILMA (weekly for up to 8 weeks) versus ILMA alone (twice a week for up to 8 weeks), Outcome 1 Participants complete cure.
Analysis 86.1.
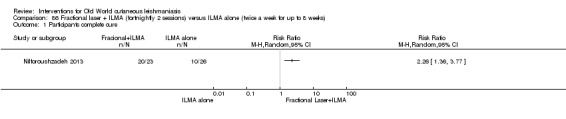
Comparison 86 Fractional laser + ILMA (fortnightly 2 sessions) versus ILMA alone (twice a week for up to 8 weeks), Outcome 1 Participants complete cure.
Analysis 87.1.
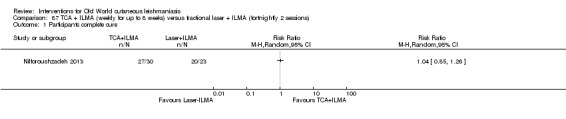
Comparison 87 TCA + ILMA (weekly for up to 8 weeks) versus fractional laser + ILMA (fortnightly 2 sessions), Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
Analysis of time to heal data in the Nilforoushzadeh 2013 study only showed significant differences in mean time to heal between TCA plus ILMA versus ILMA alone groups (MD −1.60, 95% CI −2.35 to −0.85 weeks, Analysis 85.2).
Analysis 85.2.
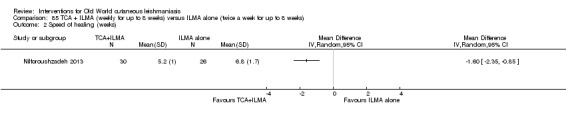
Comparison 85 TCA + ILMA (weekly for up to 8 weeks) versus ILMA alone (twice a week for up to 8 weeks), Outcome 2 Speed of healing (weeks).
Secondary outcome: adverse effects
Nilforoushzadeh 2013 reported adverse effects in 80% (n = 72) of participants. There were five (6.9%) cases of local irritation, two (2.8%) cases of infection, one (1.4%) case of enlargement of lesion, and three (4.2%) cases of satellite lesions. The study did not describe the adverse effects by intervention.
4.2.4 TCA plus ILMA versus ILMA alone
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared TCA 50% (applied fortnightly up to frosting the lesion) plus ILMA (twice a week until complete resolution of the lesions or up to eight weeks) versus ILMA alone (twice a week until complete resolution of the lesions or up to eight weeks) (Nilforoushzadeh 2014b). At the end of the treatment period, 85.7% (78/91) and 80% (76/95) of the participants in the respective groups achieved a complete cure (RR 1.07, 95% CI 0.94 to 1.22; N = 186; 1 study Analysis 88.1). The authors also performed an analysis by the type of lesions (papule, nodule, plaque or ulcerative nodule), but they did not observe any difference between groups.
Analysis 88.1.

Comparison 88 TCA fortnightly up to 8 weeks + ILMA (twice a week) versus ILMA alone (weekly for up to 8 weeks), Outcome 1 Participants complete cure.
4.3 Cryotherapy
4.3.1 Cryotherapy alone versus ILMA plus cryotherapy versus ILMA monotherapy
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared a combination of cryotherapy (freezing time was 10 s to 30 s with a thawing interval of 20 s) plus ILMA (after 5 to 10 min of cryotherapy) versus cryotherapy alone versus ILMA monotherapy (0.2 mL to 1.5 mL per session per week) (Salmanpour 2006). Ninety per cent (18/20), 70% (14/20), and 75% (15/20) of the participants in the respective groups had achieved a complete cure; however, authors did not report the time of this assessment. There were no statistical difference in cure rates between any comparisons: cryotherapy plus ILMA versus cryotherapy alone (RR 1.29, 95% CI 0.93 to 1.77; N = 40; 1 study, Analysis 89.1); cryotherapy plus ILMA versus ILMA alone (RR 1.20, 95% CI 0.90 to 1.61; N = 40; 1 study, Analysis 90.1), or cryotherapy alone versus ILMA alone (RR 0.93, 95% CI 0.64 to 1.37; N = 40; 1 study, Analysis 91.1).
Analysis 89.1.
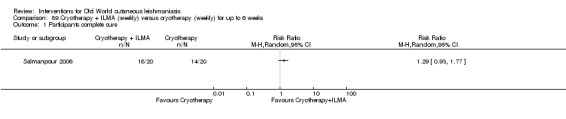
Comparison 89 Cryotherapy + ILMA (weekly) versus cryotherapy (weekly) for up to 6 weeks, Outcome 1 Participants complete cure.
Analysis 90.1.
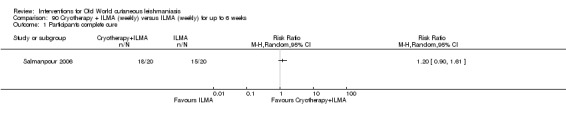
Comparison 90 Cryotherapy + ILMA (weekly) versus ILMA (weekly) for up to 6 weeks, Outcome 1 Participants complete cure.
Analysis 91.1.
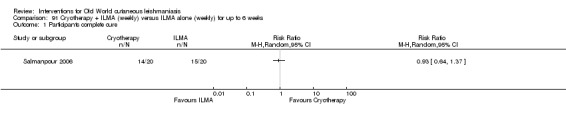
Comparison 91 Cryotherapy + ILMA (weekly) versus ILMA alone (weekly) for up to 6 weeks, Outcome 1 Participants complete cure.
An RCT from Iran compared cryotherapy (liquid nitrogen (−195°C) twice to the lesion for 10 s to 15 s, with a thawing interval of 20 s) versus ILMA monotherapy (0.2 mL to 1.5 mL per lesion) weekly for up to six weeks (Layegh 2009). At six months' follow‐up, 58.3% (21/36) and 27.8% (10/36) of the cryotherapy and ILMA group, respectively, achieved a complete cure (RR 2.10, 95% CI 1.16 to 3.81; N = 72; 1 study, Analysis 92.1).
Analysis 92.1.
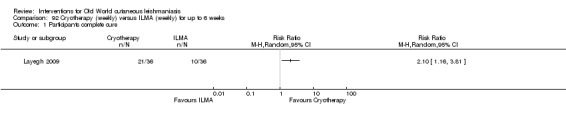
Comparison 92 Cryotherapy (weekly) versus ILMA (weekly) for up to 6 weeks, Outcome 1 Participants complete cure.
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared cryotherapy (liquid nitrogen (−195°C) 10 s to 25 s with a thawing interval of 20 s) plus ILMA (after thawing, 0.5 mL to 2 mL per lesion) versus cryotherapy alone versus ILMA alone (0.5 mL to 2 mL per lesion) fortnightly until complete cure or for up to six weeks (Asilian 2004a). At the end of the treatment period, 80.5% (120/149), 52.2% (120/230) and 52.5% (84/160) of the lesions in the respective groups had healed completely. Cure rates were significantly higher in the cryotherapy plus ILMA group compared with cryotherapy alone (RR 1.54, 95% CI 1.33 to 1.79; N = 379; 1 study, Analysis 93.1) and ILMA alone (RR 1.53, 95% CI 1.30 to 1.81; N = 309; 1 study Analysis 94.1). There were no significant differences in cure rates between ILMA alone and cryotherapy alone groups (RR 0.99, 95% CI 0.82 to 1.20; N = 390; 1 study, Analysis 95.1).
Analysis 93.1.
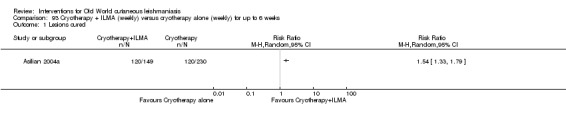
Comparison 93 Cryotherapy + ILMA (weekly) versus cryotherapy alone (weekly) for up to 6 weeks, Outcome 1 Lesions cured.
Analysis 94.1.
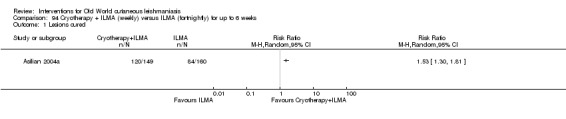
Comparison 94 Cryotherapy + ILMA (weekly) versus ILMA (fortnightly) for up to 6 weeks, Outcome 1 Lesions cured.
Analysis 95.1.
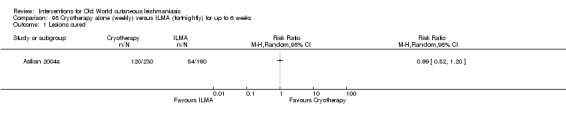
Comparison 95 Cryotherapy alone (weekly) versus ILMA (fortnightly) for up to 6 weeks, Outcome 1 Lesions cured.
Secondary outcome: speed of healing (time taken to be 'cured')
Only one study reported that none of the cured lesions recurred during the six‐month follow‐up period (Asilian 2004a).
Secondary outcome: adverse effects
Asilian 2004a reported mild adverse side effects, such as postinflammatory hypopigmentation: 5/100 of cases in the combined cryotherapy and ILMA group and 10/230 in the cryotherapy group. There were no significant differences in adverse effects rates between groups (Analysis 93.2; Analysis 94.2; Analysis 95.2). In Salmanpour 2006 there were no serious side effects in any of the treatment groups. However, 35% of the cases in the cryotherapy plus ILMA group, 60% in the cryotherapy alone group, and 20% in the ILMA alone group showed erythema and oedema of the lesions and perilesional area, but these adverse effects rates were not statistically different (Analysis 89.2; Analysis 90.2; Analysis 91.2). The most common adverse effects reported by Layegh 2009 were erythema and oedema at the treated site; these adverse reactions appeared during the initial hours after treatment. Layegh 2009 also described blistering at the treatment site, which became evident one or two days after treatment with a good response to local treatment. Other minor adverse effects are shown in Table 124.
Analysis 93.2.
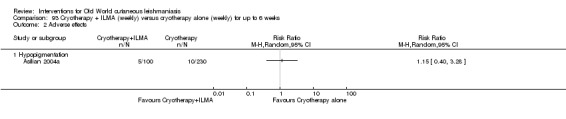
Comparison 93 Cryotherapy + ILMA (weekly) versus cryotherapy alone (weekly) for up to 6 weeks, Outcome 2 Adverse effects.
Analysis 94.2.
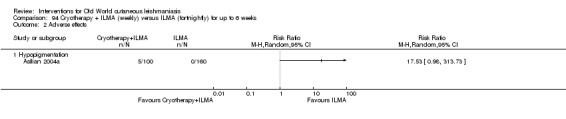
Comparison 94 Cryotherapy + ILMA (weekly) versus ILMA (fortnightly) for up to 6 weeks, Outcome 2 Adverse effects.
Analysis 95.2.
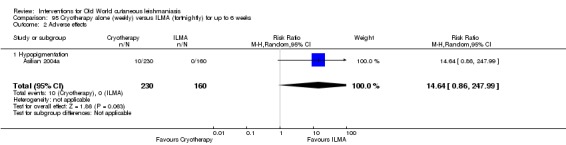
Comparison 95 Cryotherapy alone (weekly) versus ILMA (fortnightly) for up to 6 weeks, Outcome 2 Adverse effects.
Analysis 89.2.
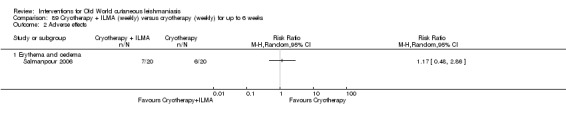
Comparison 89 Cryotherapy + ILMA (weekly) versus cryotherapy (weekly) for up to 6 weeks, Outcome 2 Adverse effects.
Analysis 90.2.
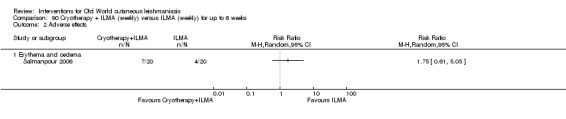
Comparison 90 Cryotherapy + ILMA (weekly) versus ILMA (weekly) for up to 6 weeks, Outcome 2 Adverse effects.
Analysis 91.2.
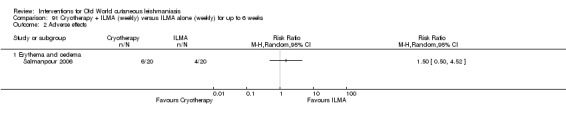
Comparison 91 Cryotherapy + ILMA (weekly) versus ILMA alone (weekly) for up to 6 weeks, Outcome 2 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Only Asilian 2004a reported that all cured lesions showed negative direct smears at the end of treatment and six weeks after treatment.
4.3.2 Triple combination of cryotherapy plus 15% paromomycin plus ILMA versus ILMA monotherapy
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared a combined triple therapy of cryotherapy (fortnightly for up to 3 sessions) plus 15% paromomycin plus 10% urea cream applied twice a day for 4 weeks plus ILMA (twice a day for four weeks after cryotherapy) versus ILMA monotherapy twice every week until complete healing or for a maximum of six weeks (Nilforoushzadeh 2004). At six weeks after treatment, 84% (68/81) and 75% (57/76) of the participants in the respective groups had achieved complete cure. There were no significant differences in cure rates between groups (RR 1.12, 95% CI 0.95 to 1.31; N = 157; 1 study, Analysis 96.1).
Analysis 96.1.

Comparison 96 Cryotherapy (fortnightly) + 15% paromomycin + 10% urea cream (twice a day) + ILMA (twice a day for 4 weeks) versus ILMA (twice a week) for up to 6 weeks, Outcome 1 Participants complete cure.
4.3.3 Triple combination cryotherapy plus 3% salicylic acid cream plus 3% sodium nitrite cream versus cryotherapy plus 3% salicylic acid cream plus vehicle
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared cryotherapy (once a week) plus 3% salicylic acid cream (twice a day) plus 3% sodium nitrite cream (twice a day) versus cryotherapy plus 3% salicylic acid cream for up to 12 weeks (Jowkar 2012). At the end of the treatment period, 83.3% (30/36) and 74.1% (20/27) of the lesions in the respective groups had healed completely (RR 1.13, 95% CI 0.86 to 1.47; N = 63; 1 study, Analysis 97.1).
Analysis 97.1.

Comparison 97 Cryotherapy (weekly) + 3% salicylic + 3% sodium nitrite cream (twice a day) for up to 12 weeks versus cryotherapy (weekly) + 3% salicylic cream (twice a day), Outcome 1 Lesions cured.
Secondary outcome: adverse effects
Jowkar 2012 reported erythema, a burning sensation, and skin irritation in seven participants (19.4%) in the cryotherapy plus salicylic acid plus sodium nitrate group, and in one participant (3.7%) in the cryotherapy plus salicylic acid plus vehicle group. Statistically, this was not a significant difference (RR 5.25, 95% CI 0.69 to 40.17; N = 63; 1 study, Analysis 97.2). See Table 124.
Analysis 97.2.

Comparison 97 Cryotherapy (weekly) + 3% salicylic + 3% sodium nitrite cream (twice a day) for up to 12 weeks versus cryotherapy (weekly) + 3% salicylic cream (twice a day), Outcome 2 Adverse effects.
4.4 Thermotherapy
For a summary of adverse effects of thermotherapy please see Table 125.
4.4.1 Thermotherapy using radiofrequency waves versus ILMA
Primary outcome: percentage of lesions cured after the end of treatment
One RCT from Iran compared controlled localised heating using a radiofrequency heat generator (4 MHz, maximum output 90 W weekly) versus ILMA (0.1 mL to 4 mL per lesion) for four consecutive weeks (Sadeghian 2007). Six months after treatment, 80.7% (67/83) and 55.3% (52/94) of the lesions in the respective groups had healed completely. Cure rates were significantly higher in the thermotherapy group compared with ILMA (RR 1.46, 95% CI 1.18 to 1.80; N = 177; 1 study, Analysis 98.1).
Analysis 98.1.

Comparison 98 Radiofrequency waves versus ILMA (1 mL to 7 mL per lesion) weekly for 4 weeks, Outcome 1 Lesions cured.
We assessed the certainty of evidence as very low.
Primary outcome: percentage of participants with a complete cure after the end of treatment
There were two studies assessing complete cure after the end of treatment (Sadeghian 2007; Safi 2012). Heterogeneity was substantial (P = 0.08; I² = 68%), although both effects are in the same direction. The pooled analysis showed no significant difference in cure rates between thermotherapy and ILMA groups (RR 1.23, 95% CI 0.97 to 1.55; N = 499; 2 studies, Analysis 98.2).
Analysis 98.2.
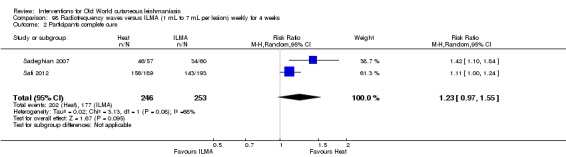
Comparison 98 Radiofrequency waves versus ILMA (1 mL to 7 mL per lesion) weekly for 4 weeks, Outcome 2 Participants complete cure.
We assessed the certainty of evidence as very low.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
Only the study from Iran reported not finding relapse from lesions with complete response in either group after six months of follow‐up (Sadeghian 2007).
Secondary outcome: prevention of scarring
Sadeghian 2007 reported that although all participants with complete response in both groups had scars, the size of the scars was smaller in the heat‐treated group (15.9 mm before and 11.2 mm after treatment) than in the ILMA‐treated group (15.0 mm before and 14.8 mm after treatment).
Secondary outcome: adverse effects
In Sadeghian 2007, allergic reactions such as erythema, oedema, and pruritus occurred in the ILMA group in four participants with 10 lesions, and there were also four cases of post‐treatment sporotrichotic lesions and three cases of satellite lesions. There was only one case with satellite lesions after treatment in the ILMA group. Safi 2012 reported mild and minimal adverse effects only in the ILMA group.
Participants treated with controlled localised heating reported fewer allergic reactions (RR 0.05, 95% CI 0.00 to 0.76; N = 117, 1 study, Analysis 98.3) but with very low‐certainty evidence.
Analysis 98.3.
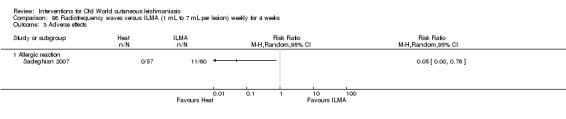
Comparison 98 Radiofrequency waves versus ILMA (1 mL to 7 mL per lesion) weekly for 4 weeks, Outcome 3 Adverse effects.
4.4.2 Thermotherapy using radiofrequency waves versus intralesional or intravenous sodium stibogluconate (SSG)
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iraq and Kuwait compared IVSSG (20 mg/kg/d) for 10 doses versus one session of thermotherapy using radiofrequency waves (50 uCTM applied for 30 s) (Aronson 2010). Two months after treatment, 73.3% (63/86) and 59.5% (50/84) of lesions had healed completely in the respective groups. There was no difference in cure rates between groups ((RR 1.23, 95% CI 0.99 to 1.53; participants = 170; studies = 1); Analysis 99.1).
Analysis 99.1.

Comparison 99 Radiofrequency waves (50 uCTM applied for 30 s) versus ILSSG (10 days of 20 mg/kg/d), Outcome 1 Lesions cured.
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Afghanistan compared ILSSG (five injections every five to seven days depending on the lesion size) for up to 29 days versus IMSSG (20 mg/kg/d) daily for 21 days versus thermotherapy using radiofrequency waves (one session of > 1 consecutive application at 50ºC for 30 s depending on lesion size) (Reithinger 2005). Two months after treatment, 47.3% (70/148), 18% (26/144) and 54% (75/139) of the participants in the respective group had achieved a complete cure. Cure rates were significantly higher in the thermotherapy group compared with IMSSG (RR 2.99, 95% CI 2.04 to 4.37; N = 283; 1 study, Analysis 100.1), but there was no difference compared with ILSSG (RR 1.14, 95% CI 0.91 to 1.43; N = 287; 1 study, Analysis 101.1).
Analysis 100.1.

Comparison 100 Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus IMSSG (20 mg/kg/d for 3 weeks), Outcome 1 Participants complete cure.
Analysis 101.1.

Comparison 101 Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 days), Outcome 1 Participants complete cure.
An RCT from India compared ILSSG (2 mL to 5 mL per lesion twice a week, for seven injections over 29 days versus thermotherapy using radiofrequency waves (one treatment of two or more consecutive applications at 50ºC for 30 s depending on lesion size) (Bumb 2013). Six months after treatment, 98% and 94% of participants in the respective groups had achieved a complete cure (RR 1.04, 95% CI 0.96 to 1.13; N = 100; 1 study, Analysis 102.1).
Analysis 102.1.
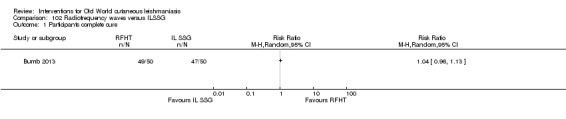
Comparison 102 Radiofrequency waves versus ILSSG, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
In the Reithinger 2005 study, the speed of healing took a median of 75 days for the ILSSG group, 100 days or more for the IMSSG group, and 53 days for the thermotherapy group (the original paper reported that the time to cure was significantly shorter for participants treated with thermotherapy; P = 0.003, by the log‐rank test). Bumb 2013 described a median time to heal of 11 weeks (95% CI 8 to 12) in the thermotherapy group and 10 weeks (95% CI 8 to 12) in the ILSSG group. In Aronson 2010, time to heal was similar (P = 0.24) in both groups in the analysis using blinded reading of photographs.
Secondary outcome: adverse effects
Reithinger 2005 described secondary infection in 3.7% (5/148) of participants in the ILSSG group, 1.4% (2/144) in the IMSSG group, and 5.7% (8/139) in the thermotherapy group, and there were no statistical differences in either comparison: thermotherapy versus ILSSG (RR 4.14, 99% CI 0.55 to 31.03; N = 283; 1 study, Analysis 100.2) or thermotherapy versus IMSSG (RR 1.70, 99% CI 0.41 to 7.17; N = 287; 1 study, Analysis 101.2). The authors also reported one participant with bradycardia and one an undefined local reaction in the ILSSG group. In the IMSSG group, one participant reported bradycardia, one tachycardia, and one palpitation. In the thermotherapy group, some participants experienced superficial second degree burns. In Bumb 2013, radiofrequency heat treatment (RFHT) was associated with less scarring and hyperpigmentation compared with ILSSG. Aronson 2010 reported four participants (15%) in the thermotherapy group and three (11%) in the ILSSG group suffered serious adverse effects (RR 1.30, 95% CI 0.30 to 5.64; N = 170; 1 study, Analysis 99.2). See Table 125.
Analysis 100.2.

Comparison 100 Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus IMSSG (20 mg/kg/d for 3 weeks), Outcome 2 Adverse event (secondary infection).
Analysis 101.2.

Comparison 101 Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 days), Outcome 2 Adverse event (secondary infection).
Analysis 99.2.

Comparison 99 Radiofrequency waves (50 uCTM applied for 30 s) versus ILSSG (10 days of 20 mg/kg/d), Outcome 2 Adverse effects (serious).
4.4.3 Electrocauterisation plus DAC n‐055 or electrocauterisation
Secondary outcome: speed of healing (time taken to be 'cured')
An RCT from Afghanistan compared bipolar high‐frequency electrocauterisation plus daily moist‐wound‐treatment (MWT) with polyacrylate hydrogel including 0.045% DAC N‐055 versus electrocauterisation plus vehicle (Jebran 2014). The speed of healing took a median of 43.15 days for the electrocauterisation plus DAC N‐055 group and 42 days for the electrocauterisation group.
Secondary outcome: adverse effects
Six participants in each group had bacterial and fungal superinfections (8% in the electrocauterisation plus DAC n‐055 group and 9% in the electrocauterisation group; RR 0.85, 95% CI 0.29 to 2.50; N = 135; 1 study, Analysis 103.1), and two participants in electrocauterisation plus DAC n‐055 group and four participants in the electrocauterisation group reported Keloïd formation (5% and 6%, respectively; RR 0.42, 95% CI 0.08 to 2.24; N = 135; 1 study, Analysis 103.1). Other minor adverse effects are shown in Table 125.
Analysis 103.1.
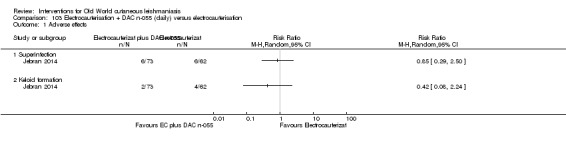
Comparison 103 Electrocauterisation + DAC n‐055 (daily) versus electrocauterisation, Outcome 1 Adverse effects.
4.5 Topical photodynamic therapy (PDT)
4.5.1 Topical PDT versus vehicle
Primary outcome: percentage of lesions cured after the end of treatment
An RCT from Iran compared topical PDT every week for four weeks (0% 5‐aminolevulinic acid (5‐ALA) hydrochloride in a water‐in‐oil cream) (Asilian 2006). Lesions irradiated using visible red light at 100 J/cm² per treatment session) with vehicle twice daily also for 28 days. Two months after treatment, 93.5% (29/31) of the lesions in the PDT group and 13.3% (4/30) in the vehicle groups healed completely. Cure rates were significantly higher in the PDT group compared with vehicle (RR 7.02, 95% CI 2.80 to 17.55; N = 61; 1 study, Analysis 104.1).
Analysis 104.1.
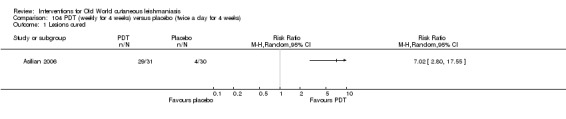
Comparison 104 PDT (weekly for 4 weeks) versus placebo (twice a day for 4 weeks), Outcome 1 Lesions cured.
Secondary outcome: prevention of scarring
At the end of the study, none of the lesions had deep or disfiguring scars in the PDT group. However, 20% (3/30) of the lesions in the vehicle group had deep or disfiguring scars (RR 0.14, 95% CI 0.01 to 2.57; N = 61; 1 study, Analysis 104.2). Both groups had mild and tolerable itch, burning, redness, discharge, oedema, and pain as adverse effects.
Analysis 104.2.
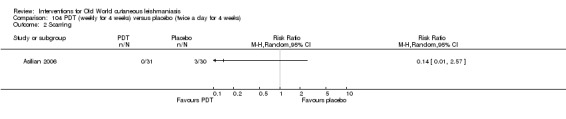
Comparison 104 PDT (weekly for 4 weeks) versus placebo (twice a day for 4 weeks), Outcome 2 Scarring.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Two months after treatment, parasitological cure rates were significantly higher in the PDT group (100%, 31/31 lesions) compared to placebo (20%, 6/30) (RR 4.69, 95% CI 2.37 to 9.31; N = 61; 1 study, Analysis 104.3).
Analysis 104.3.
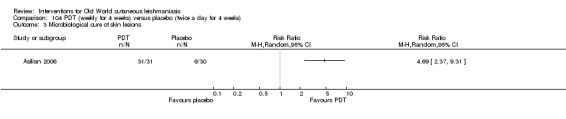
Comparison 104 PDT (weekly for 4 weeks) versus placebo (twice a day for 4 weeks), Outcome 3 Microbiological cure of skin lesions.
The same author also compared PDT with topical paromomycin plus methyl benzethonium chloride, which we analyse in section 3.2, 'Topical paromomycin (aminosidine)'.
4.6 Mesotherapy
4.6.1 Mesotherapy versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
One RCT from Iran compared mesotherapy (0.05 mL injection of MA in each shot in the depth of 2 mm) versus ILMA (0.1 mL of MA) for up six weeks (Kashani 2010). Three months after treatment, 83.3% (25/30) and 86.7% (26/30) of participants in the respective groups had achieved complete cure (RR 0.96, 95% CI 0.78 to 1.19; N = 60; 1 study, Analysis 105.1).
Analysis 105.1.

Comparison 105 Mesotherapy gun (0.5 mL of MA weekly) versus ILMA (0.1 mL weekly) for up to 6 weeks, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
The authors did not report significant improvement in ulcer, induration, erythema, or scar size between the two groups. However, the study described faster improvement in participants who received mesotherapy compared to ILMA: ulcerated lesions improved at the rate of 0.27 cm per week (SD 0.14) and 0.15 cm per week (SD 0.72), respectively, with an improvement for erythema of 0.31 cm per week (SD 0.15) and 0.22 cm per week (SD 0.07).
Secondary outcome: adverse effects
Pain severity evaluated by VAS was significantly higher in the ILMA group than in the mesotherapy group (7.67 cm versus 6.06 cm; P = 0.005). Two participants in the mesotherapy group and one in the ILMA group suffered an allergic reaction. There were no significant differences in allergic reaction rates between groups (RR 2.00, 95% CI 0.19 to 20.90; N = 60; 1 study; Analysis 105.2).
Analysis 105.2.
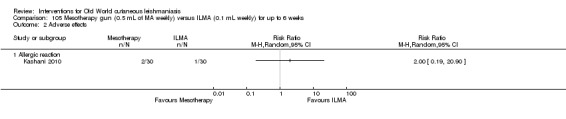
Comparison 105 Mesotherapy gun (0.5 mL of MA weekly) versus ILMA (0.1 mL weekly) for up to 6 weeks, Outcome 2 Adverse effects.
Tertiary outcomes: development of cell‐mediated immunity (i.e. positive leishmanin skin test)
Three months after treatment, there were positive Leishmania antibodies in three participants in the mesotherapy group and two participants in the ILMA group. There were no significant differences in adverse effects rates between groups (RR 1.50, 95% CI 0.27 to 8.34; N = 60; 1 study) (Analysis 105.3).
Analysis 105.3.
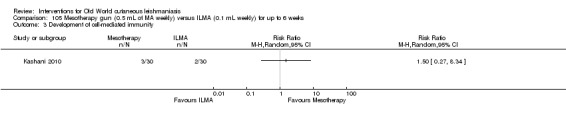
Comparison 105 Mesotherapy gun (0.5 mL of MA weekly) versus ILMA (0.1 mL weekly) for up to 6 weeks, Outcome 3 Development of cell‐mediated immunity.
5. Measures for promoting healing
5.1 Topical diminazene aceturate (Berenil) versus topical cetrimide plus chlorhexidine (Savlon)
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Sudan compared diminazene aceturate solution versus cetrimide plus chlorhexidine solution for 50 days (Lynen 1992). At the end of the treatment period, 80% (28/35) and 57% (20/35) of participants of in the respective groups achieved a complete cure, with a statistically significant difference in between groups and in favour of the diminazene aceturate solution (RR 1.40, 95% CI 1.01 to 1.95; N = 70; 1 study, Analysis 106.1).
Analysis 106.1.
Comparison 106 Diminazene aceturate solution (weekly) versus cetrimide + chlorhexidine solution for 50 d, Outcome 1 Participants complete cure.
Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within six months, one year, two years, and three years
In the diminazene aceturate group, three re‐ulcerations occurred (two after 20 days and one after 25 days of cure). In the chlorhexidine group, two re‐ulcerations occurred after 35 days of cure.
Secondary outcome: adverse effects
Extreme drying of ulcers and surrounding skin occurred in participants in the diminazene aceturate solution (Berenil) group. Participants from the cetrimide plus chlorhexidine solution (Savlon) group experienced a slight burning sensation and drying of the skin at the site of treatment.
6. Alternative therapies
6.1 Garlic cream
6.1 Topical garlic cream versus vehicle
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared 5% garlic cream treatment versus vehicle twice a day under occlusion with sterile gauze for three hours, for three weeks (Gholami 2000). At 40 days after treatment, 18.75% (18/96) and 20% (15/75) of the participants in the respective groups had achieved a complete cure. There were no significant differences in cure rates between groups (RR 0.94, 95% CI 0.51 to 1.73; N = 171; 1 study, Analysis 107.1).
Analysis 107.1.
Comparison 107 Topical garlic (twice a day) versus vehicle for 3 weeks, Outcome 1 Participants complete cure.
6.2 Herbal extract
6.2.1 Topical herbal extract versus IMMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical herbal extract Z‐HE, covered with a dressing for five consecutive days plus placebo injection (0.5 mL saline) versus IMMA (15 mg/kg/d to 20 mg/kg/d) plus vehicle (petrolatum and charcoal powder) for 20 consecutive days (Zerehsaz 1999). Six weeks after treatment, 74.4% (64/86) and 27.1% (23/85) of participants in the respective groups had achieved complete cure. There was a statistically significant difference in cure rates between topical herbal extract Z‐HE compared with IMMA (RR 2.75, 95% CI 1.90 to 3.98; N = 171; 1 study, Analysis 108.1).
Analysis 108.1.

Comparison 108 Topical herbal extract + placebo (5 d) versus IMMA (15‐20/mg/kg/d) + vehicle for 20 d, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Participants in the IMMA reported urticaria and generalised itch.
6.3 Honey
6.3.1 Topical honey plus ILMA versus ILMA monotherapy
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical honey soaked gauze twice a day plus ILMA (weekly) versus ILMA (weekly) for four weeks (Nilforoushzadeh 2007). Results showed that two and a half to three months after treatment, 46% (23/50) and 64% (32/50) of the participants in the respective groups had achieved a complete cure, but with no significant difference between them (RR 0.72, 95% CI 0.50 to 1.04; N = 100; 1 study, Analysis 109.1).
Analysis 109.1.
Comparison 109 Topical honey (twice a day) + ILMA (weekly) versus ILMA (weekly) for 4 weeks, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
The mean time taken to be cured after omitting dropouts was 7.04 weeks and 6.3 weeks in the honey plus ILMA and the ILMA groups, respectively.
6.4 Cassia fistula
6.4.1 Topical Cassia fistula fruit gel plus ILMA versus ILMA plus vehicle
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical Cassia fistula fruit gel plus ILMA (0.5 mL to 2 mL) twice a week versus ILMA (0.5 mL to 2 mL) twice a week plus vehicle (Jaffary 2010). Results showed that three months after treatment, compete healing occurred in 67.1% and 41.4% of the participants in the respective groups. There was a statistically significant difference in cure rates in favour of Cassia fistula fruit gel as an adjuvant to ILMA compared with ILMA monotherapy (RR 1.62, 95% CI 1.17 to 2.24; N = 140; 1 study, Analysis 110.1).
Analysis 110.1.

Comparison 110 Cassia fistula (topical gel) + ILMA (0.5 mL to 2 mL), twice a week versus ILMA (0.5 mL to 2 mL), twice a week + vehicle, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Nine (12.9%) participants in each group suffered from adverse effects such as itching and erythema (RR 1.00, 95% CI 0.42 to 2.37; N = 140; 1 study, Analysis 110.2).
Analysis 110.2.
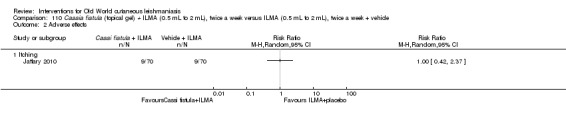
Comparison 110 Cassia fistula (topical gel) + ILMA (0.5 mL to 2 mL), twice a week versus ILMA (0.5 mL to 2 mL), twice a week + vehicle, Outcome 2 Adverse effects.
6.4.2 Topical concentrated boiled Cassia fistula versus topical Cassia fistula hydroalcoholic versus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical concentrated boiled Cassia fistula versus topical Cassia fistula hydroalcoholic versus ILMA (0.5 mL to 2 mL), twice a week for four weeks (Jaffary 2014b). Results showed that four months after treatment, 40% (22/55), 36% (20/55), and 65% (36/55) of the participants in the respective groups had achieved complete cure. Efficacy of ILMA was higher than concentrated boiled extract (RR 0.61, 95% CI 0.42 to 0.89; N = 110; 1 study, Analysis 111.1) and hydroalcoholic extract of Cassia fistula (RR 0.56, 95% CI 0.37 to 0.83; N = 110; 1 study, Analysis 112.1), but there was no significant difference between application of concentrated boiled extract and hydroalcoholic extract of Cassia fistula (RR 1.10, 95% CI 0.68 to 1.77; N = 110; 1 study, Analysis 113.1).
Analysis 111.1.

Comparison 111 Cassia fistula boiled (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 1 Participants complete cure.
Analysis 112.1.
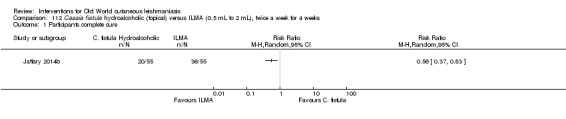
Comparison 112 Cassia fistula hydroalcoholic (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 1 Participants complete cure.
Analysis 113.1.

Comparison 113 Cassia fistula boiled (topical) versusC fistula hydroalcoholic (topical) for 4 weeks, Outcome 1 Participants complete cure.
Secondary outcome: speed of healing (time taken to be 'cured')
Participants receiving ILMA healed more quickly than those treated with concentrated boiled extract of Cassia fistula (MD −1.80 weeks, 95% CI −3.49 to −0.11; N = 110; 1 study, Analysis 111.2), but there was no difference between concentrated boiled extract of Cassia fistula and hydroalcoholic extract (MD −0.30 weeks, 95% CI −1.70 to 1.10; N = 110; 1 study, Analysis 113.2). There was also no difference between hydroalcoholic extract of Cassia fistula and ILMA (MD −1.50 weeks, 95% CI −3.20 to 0.20; N = 110; 1 study, Analysis 112.2).
Analysis 111.2.
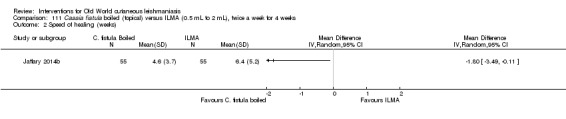
Comparison 111 Cassia fistula boiled (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 2 Speed of healing (weeks).
Analysis 113.2.
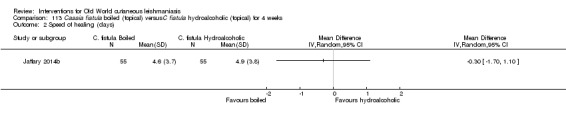
Comparison 113 Cassia fistula boiled (topical) versusC fistula hydroalcoholic (topical) for 4 weeks, Outcome 2 Speed of healing (days).
Analysis 112.2.
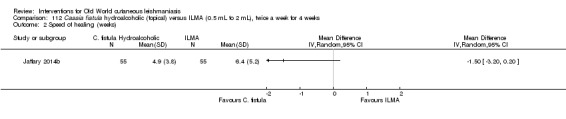
Comparison 112 Cassia fistula hydroalcoholic (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 2 Speed of healing (weeks).
Secondary outcome: adverse effects
Three (5.5%) participants in the concentrated boiled extract group and two (3.6%) in the other groups withdrew from the study due to an allergic reaction to the medications. There was no significant difference among these three groups in this regard: boiled extract of Cassia fistula versus ILMA (RR 1.50, 95% CI 0.26 to 8.63; N = 110; 1 study, Analysis 111.3), hydroalcoholic extract of Cassia fistula versus ILMA (RR 1.00, 95% CI 0.15 to 6.85; N = 110; 1 study, Analysis 112.3), or boiled extract of Cassia fistula versus hydroalcoholic extract of Cassia fistula (RR 1.50, 95% CI 0.26 to 8.63; N = 110; 1 study, Analysis 113.3).
Analysis 111.3.
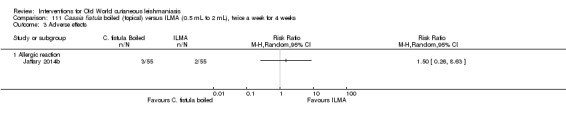
Comparison 111 Cassia fistula boiled (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 3 Adverse effects.
Analysis 112.3.
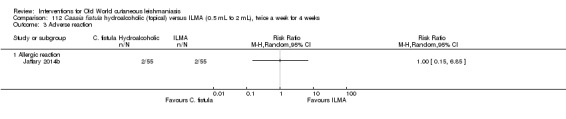
Comparison 112 Cassia fistula hydroalcoholic (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks, Outcome 3 Adverse reaction.
Analysis 113.3.

Comparison 113 Cassia fistula boiled (topical) versusC fistula hydroalcoholic (topical) for 4 weeks, Outcome 3 Adverse effects.
6.5Achilles millefolium
6.5.1 TopicalAchilles millefolium cream plus ILMA versus vehicle plus ILMA
Primary outcome: percentage of participants with a complete cure after the end of treatment
An RCT from Iran compared topical gel of 5% Achilles millefolium (twice daily) plus ILMA (weekly injections at 20 mg/kg/d) versus ILMA (weekly injections of 20 mg/kg/d) plus vehicle (twice daily) for four weeks (Jaffary 2014a). Two months after treatment, 53% (16/30) and 40% (12/30) of the participants in the respective groups had achieved a complete cure. There was no difference in cure rates between Achilles millefolium as an adjuvant to ILMA compared with ILMA monotherapy (RR 1.33, 95% CI 0.77 to 2.31; N = 60; 1 study, Analysis 114.1).
Analysis 114.1.
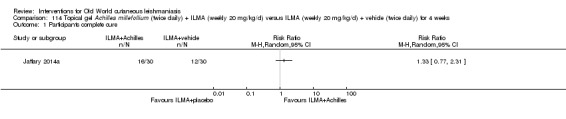
Comparison 114 Topical gel Achilles millefollium (twice daily) + ILMA (weekly 20 mg/kg/d) versus ILMA (weekly 20 mg/kg/d) + vehicle (twice daily) for 4 weeks, Outcome 1 Participants complete cure.
Secondary outcome: adverse effects
Eight (26.6%) participants in the Achilles millefolium group reported mild or moderate severe itching and redness, and one experienced severe itching and increasing wound discharge. In the control group, two participants reported mild itching (6.6%) and one, severe itching (3.3%). There were no significant differences in itching rates between groups (RR 3.00, 95% CI 0.90 to 10.01; N = 60; 1 study, Analysis 114.2)
Analysis 114.2.
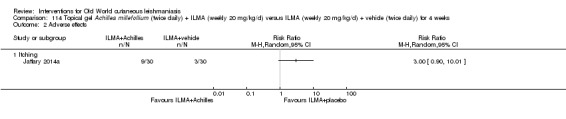
Comparison 114 Topical gel Achilles millefollium (twice daily) + ILMA (weekly 20 mg/kg/d) versus ILMA (weekly 20 mg/kg/d) + vehicle (twice daily) for 4 weeks, Outcome 2 Adverse effects.
Tertiary outcomes: microbiological or histopathological cure of skin lesions
Six weeks after treatment, 33% of the participants in the Achilles millefolium group and 40% in the control group had parasitological cure (RR 0.83, 95% CI 0.43 to 1.63; N = 60; 1 study, Analysis 114.3).
Analysis 114.3.
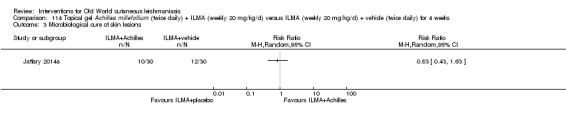
Comparison 114 Topical gel Achilles millefollium (twice daily) + ILMA (weekly 20 mg/kg/d) versus ILMA (weekly 20 mg/kg/d) + vehicle (twice daily) for 4 weeks, Outcome 3 Microbiological cure of skin lesions.
Results from the MEDLINE search for adverse effects
We performed a MEDLINE search for adverse or side effects combined with therapeutic terms. However, we could only find general papers reporting known adverse effects derived from the evaluated drugs that are already mentioned in the Background under the Description of the intervention section.
Discussion
Summary of main results
The randomised controlled trials (RCTs) included in this review assessed a broad range of treatments and many different clinical questions. Yet together, there were few opportunities to describe and pool useful data. We have some concerns regarding the precision of data reported in several studies. Furthermore, because most RCTs were at high risk of bias, it was difficult to conclude whether one treatment was more beneficial than the comparator much of the time. Many interventions ruled ineffective in an essentially inconclusive study could still prove to have some benefit if evaluated with greater statistical power. Nonetheless, this review accurately documents the existing RCT evidence on the usefulness of treatments, and that relevant information can be extracted for practice and future research.
We found 89 RCTs that covered more than 20 different interventions, broadly categorised into six main groups: antimonial drugs, non‐antimonial systemic treatments, non‐antimonial topical and intralesional treatments, physical therapies, methods for promoting healing, and alternative therapies. Given that sample size per se is not a source of potential bias but a source of potential imprecision that may lead to bias, care has to be exercised in not concluding that the evaluation of the efficacy of interventions of small‐sized RCTs was untrustworthy.
It was difficult to evaluate the efficacy of any of the multiple treatments due to the variable treatment regimens examined and because the studies evaluated different Leishmania species and took place in different geographical areas. Moreover, most of the studies compared different regimens, and only a few RCTs compared treatments with sham therapies.
We created 'Summary of findings' tables for two comparisons (Table 1; Table 2); however, where measured, we rated all of the key outcomes included as very low‐certainty evidence, so we are not confident that the results found are conclusive.
-
A pooled analysis of three RCTs comparing itraconazole versus placebo favoured itraconazole with very low certainty of evidence, in terms of the following outcomes.
Percentage of participants with complete cure (N = 244).
Adverse effects (mild abdominal pain and nausea (N = 204) and mild abnormal liver function (N = 84).
Microbiological or histopathological cure of skin lesions (N = 20, only in one RCT).
-
A pooled analysis of two RCTs comparing paromomycin ointment plus 10% urea versus vehicle showed no difference in cure rate between interventions with a very low certainty of evidence, in terms the following outcomes.
Percentage of participants with complete cure (N = 383).
Microbiological or histopathological cure of skin lesions (N = 383).
However, the paromomycin group had more skin/local reactions, as assessed in four studies (N = 713).
Neither of the key comparisons offer data on the percentage of lesions cured after the end of treatment and speed of healing (i.e. time taken to be cured).
Overall completeness and applicability of evidence
Not all of the trials assessed the primary, secondary, and tertiary outcomes that we hoped to evaluate in this review. Our two main comparisons did not report our primary outcome of the percentage of lesions cured, and the comparisons that did measure this outcome (along with percentage of participants cured) lacked data on long‐term effects. Assessing the two primary outcomes, we established that dealing with participants rather than with numbers of lesions proved to be a better cure criteria: lesions cured did not provide a realistic reflection of the number of participants that achieved complete cure or rather, whether all their lesions had healed. In addition, from a clinical perspective, it may be more relevant to know whether a person is completely or partially cured irrespective of the number of lesions fully healed by the tested drug. In fact, only 18 RCTs reported the primary outcome as percentage of lesions cured; the rest reported the percentages in terms of participants cured.
The main secondary outcome reported in our studies was the description of adverse effects, which is key when deciding a specific therapeutic option. Several RCTs also reported other secondary outcomes, but none reported the degree of functional and aesthetic impairment or quality of life. Only two tertiary outcomes were reported ('Microbiological or histopathological cure of skin lesions' in 15 trials and 'Development of cell‐mediated immunity (i.e. positive leishmanin skin test)' in just one trial.
Many of the included studies did not specify the infecting species of leishmaniasis or made assumptions regarding the disease‐causing species. We did not find any RCTs related to L aethiopica infections. There is also a lack of evidence for L infantum andL donovani: of the 41 new studies included in this updated systematic review, only two studies assessed L donovani‐infected participants, and another one assessed the efficacy of a treatment in one L infantum‐infected participant together with 66L major‐ and oneL tropica‐infected participants. Twenty studies reported L major, L tropica, or both.
Most included participants were adults, and all were immunocompetent; thus, our findings are not necessarily applicable to subsets of people such as women of childbearing age, children, people with co‐morbid conditions, and immunocompromised individuals with no drug interactions.
The difficulty in determining the actual time point for cure in clinically significant terms in these studies is due to a lack of any universal measure for successful cure in OWCL. In fact, we found that most of the included RCTs did not explore cure rates past three months after treatment cessation. On the contrary, in most cases they assessed the primary outcome at the end of treatment or before three months.
The applicability of the results of this review to clinical practice is limited. The most important limitation in order to determine a specific conclusion for clinical practice is the short follow‐up period reported in most studies. This impedes finding the real cure rate for OWCL because it is not possible to clearly confirm healing without long‐term assessment (e.g. until at least six months after treatment has finished). There are no studies evaluating liposomal amphotericin B as a systemic treatment. Currently, because of the toxicity of pentavalent antimonials in most developed countries when systemic treatment is needed for OWCL, liposomal amphotericin B could be a good option, but at the time of writing there was no published information regarding its effectiveness.
Quality of the evidence
Most included trials were poorly designed and reported. Poor reporting is a major issue, and most studies were at unclear risk for one or more important bias domains in the 'Risk of bias' assessment. Risk of bias was generally moderate due to performance, attrition, or reporting bias. Most studies did not guard properly against performance bias (by blinding participants and healthcare professionals). Blinded outcome assessment was unclear. Adequate randomisation was reported in 47.2% (42/89) of the included studies. Only 13.5% (12/89) reported adequate allocation concealment. Double‐blinding was found in 44.9% (40/89) of the studies. There was inadequate description of baseline characteristics in 10.1% (9/89) of the studies. Thirty point three per cent (27/89) of the studies reported sample size calculation. Only two studies assessed compliance (Firooz 2006; Lynen 1992). The causative parasite was not mentioned in 32.6% (29/89) of the studies, and 34.8% (31/89) of the trials mentioned the endemic nature of the parasite in the area, assuming that was the species causing the disease. The timing for outcome assessment was not reported in 2.2% (2/89) of the trials.
Because resources for clinical research into neglected diseases are limited, there is a need to prioritise and carry out properly designed clinical trials. We found many mistakes in the write‐up of published manuscripts. Thus, it is essential that submitted journal manuscripts undergo rigorous peer review.
For the two main comparisons, we created 'Summary of findings' tables and conducted GRADE assessments on the certainty of evidence for the key outcomes, which we found to be very low. Risk of bias, as highlighted above, was one of the reasons for downgrading the evidence. We also downgraded evidence for imprecision, as many of the confidence intervals were very wide, likely due to the small sample size and low number of events in many of the studies assessed. Furthermore, in two cases (for 'participants complete cure' in the comparison itraconazole versus placebo, and 'microbiological/histopathological cure of skin lesions' in the comparison paromomycin ointment versus vehicle), we downgraded evidence for inconsistency due to the high I² values and the inconsistent direction or size of effect. In addition, we also observed indirectness in the outcomes measured in the comparison paromomycin ointment versus vehicle, as the evidence assessed only focused on young people.
Overall, considerable numbers of participants withdrew or were lost to follow‐up. Most trial authors stated that they performed an adherence assessment, but results were seldom shown in the assessed studies.
Potential biases in the review process
Studies with more positive effects are more likely to be published than those with less conclusive results (Chalmers 2009), or those written in languages other than English (Bigby 2003). To tackle the problem of publication bias, we wrote to authors from endemic countries and the WHO asking for information. Besides, we searched databases of ongoing trials and others as well. However, the fact that eight studies have not yet been incorporated and are awaiting classification may be a source of potential bias.
Agreements and disagreements with other studies or reviews
Khatami 2007 performed a non‐Cochrane systematic review on acute Old World cutaneous leishmaniasis. They included 50 studies with a total of 5515 participants. However, we detected quite a few inconsistent results. Firstly, review authors cited 51 RCTs instead of the 50 reported in the abstract and the Materials and methods, and we excluded three of these in our review for several reasons (Dogra 1986; El On 1992; Trau 1987): one was a cross‐over trial, which is an inappropriate design for evaluating potential curative treatments in an infection (El On 1992), and we considered the generation of the randomisation sequence to be inadequate in the other two. We considered another four trials to be non‐randomised in our review (Crawford 2005; el‐Safi 1990; Mapar 2001; Vardy 2001). One RCT that Khatami 2007 included dealt with two case reports (el‐On 1985). Secondly, if clinical trials reported in the paper's tables III to VII are included, review authors mentioned only 47 studies, while omitting 4. However, we do agree with Khatami 2007 in terms of the variable quality and methods of RCTs, providing weak evidence for treatment of OWCL, and regarding the importance of properly designing RCTs to improve their quality and to provide better evidence for the treatment of OWCL.
Two non‐systematic reviews gathered evidence on Old World cutaneous leishmaniasis treatments (Blum 2014;Monge‐Maillo 2013). The fact that these reviews did not use a systematic approach confers many biases and does not assure inclusion of all published studies. One study was more restrictive on selecting the studies and included only controlled clinical trials (Blum 2014), whereas the other was more permissive and also included other published data based on retrospective studies and large case series. However, review authors analysed the methodology of each of the studies, considering that only clinical trials were able to give a high quality of evidence (Monge‐Maillo 2013). Moreover, these studies were not so restrictive regarding the period of follow‐up needed and considered all the end points reported (percentages of lesions and of participants cured). In this way review authors considered certain therapeutic regimens such as dapsone for L tropica or itraconazole for L major to have a a higher quality of evidence than in the Cochrane Review. Even though these reviews (Blum 2014, Monge‐Maillo 2013) do not have the methodological rigour of a Cochrane Review, they are relevant in clinical practice because they give therapeutic recommendations stratified according to their level of evidence and Leishmania species implicated.
Authors' conclusions
We have updated information from randomised controlled trials (RCTs) of treatments for Old World cutaneous leishmaniasis (OWCL) and summarised the best available evidence using quantitative and qualitative methods. We have endeavoured to provide information to help clinicians choose the most appropriate treatment. We have been careful not to be too prescriptive because the purpose of this systematic review is to present information rather than offer advice.
There are few treatments for CL with robust evidence from multiple randomised trials. This may be because OWCL can heal spontaneously – in many cases within a couple of months – without any need for therapeutic intervention. L major‐caused CL can heal spontaneously in 40% to 70% of cases at 3 months and close to 100% at 12 months. Spontaneous cure rates for L tropica‐caused CL are 1% at 3 months, 68% at 12 months, and usually close to 100% in three years (Asilian 1995; Ben Salah 1995; Zakraoui 1995). Therefore, in many cases people with CL do not receive any drug. When clinicians do prescribe treatment, they usually prefer less toxic local treatments, reserving systemic treatments (azole drugs, miltefosine, antimonials, amphotericin B formulations) for complex cases.
We cannot make firm recommendations about the use of our two key comparisons.
Itraconazole versus placebo.
Paromomycin ointment (15% plus 10% urea) versus vehicle.
Due to the very low certainty of the evidence, and even though our analyses favoured itraconazole in terms of complete cure and lesions cured, we cannot be sure our results are conclusive. Similarly, although we found no difference between paromomycin ointment and vehicle in terms of these same outcomes, the very low certainty of the evidence casts doubt on our results, as it also does we do when we report more mild adverse effects with itraconazole and paromomycin ointment.
Our other key outcomes of microbiological or histopathological cure of skin lesions and speed of healing (i.e. time taken to be cured) were not measured for these key comparisons. None of the included studies assessed participant‐focused measures of success, such as quality of life and degree of functional and aesthetic impairment.
We have identified gaps in knowledge that imply difficulties and limits in terms of clinical practice. RCTs that compare local versus systemic treatment are scarce, and the results discordant.
Also, there is in many cases a lack of knowledge on when treatment is really needed, since OWCL can also heal spontaneously.
Although we included 89 studies in this review, most of them explore different therapeutic regimens: not only different drugs, but even the same drug administered locally or systemically or with different doses or regimens. This means that only one RCT evaluated most regimens with a specific Leishmania species and with a different clinical presentation, which may reduce the strength of the recommendation and limit the possibility to extrapolate the results to other Leishmania species.
Twenty‐nine studies did not isolate the Leishmania species. This can make it difficult to apply the result to clinical practice because a particular therapeutic regimen may have good results among a specific Leishmania species but not with another OWCL species. In addition, the same Leishmania species may vary its response to a certain treatment when the only difference is the geographical area where the infection was acquired (Monge‐Maillo 2013). Therefore, it may be difficult for physicians to extrapolate the published results to their daily clinical practice.
Before starting treatment for localised CL, and especially in the zoonotic form, people with OWCL need to be informed of the possibility of spontaneous healing and the lack of evidence for some treatments. Healthcare practitioners can still play an important role in providing information and wound healing management even if there is no good evidence for any special regimen of healing support.
The eight studies in Studies awaiting classification may alter the conclusions of the review once assessed.
This updated systematic review has identified the need for large, well‐conducted RCTs to assess the benefits and harms of interventions for OWCL.
Design
We considered only one RCT to be at low risk of bias in all domains (Ranawaka 2015). To encourage the implementation of well‐designed clinical trials specifically aimed at developing effective treatments (both primary and adjuvant), González 2010 developed guidelines for clinical trials of cutaneous leishmaniasis. However, it is evident from the newly included RCTs that improvement of study quality and standardisation of outcomes is still needed. The COMET (Core Outcome Measures in Effectiveness Trials) Initiative (www.cometinitiative.org) is working to improve the standardisation of study outcomes by raising awareness of the need for systematised methodologies that acknowledge the complexity of CL and define reproducible, measurable, and clinically meaningful outcomes (Olliaro 2013). Future trials need to ensure they follow this guidance and also adhere to the CONSORT Statement (Schulz 2010), especially to improve the reporting of important bias domains, the causative parasite, and timing of outcome assessment. Future studies should also be sufficiently powered.
Resources are particularly limited for research into neglected diseases in LMICs, even those that present major public health problems. Cost and licensing entanglements (freedom to operate) need to be considered before investing money in conducting new trials. Prioritisation for clinical research in OWCL is a necessity.
Participants
A number of interventions are currently used in women of childbearing age and children, in those with comorbidities, and in immunocompromised individuals with no drug interactions. Future trials should assess safety and efficacy in those subgroups.
Leishmania species
The current evidence for different types of clinical management of OWCL and particularly for species such as L infantum,L aethiopica,L major,L tropica, andL donovani is either lacking or of very low quality. Eighteen studies did not even report the species of leishmaniasis, which can theoretically be easily determined with the use of new DNA techniques. However, these techniques require an investment in infrastructures that is unaffordable for the resource‐restricted laboratories located in disease‐endemic countries. Of the 23 studies that reported the Leishmania species, only 11 (48%) confirmed which caused the development of the disease, and the other 12 mentioned the endemic nature of a specific parasite strain and assumed it was the disease‐causing species. Since treatment sensitivity is species‐dependent, species identification is critical in future trials, for the choice of the best treatment outcome, with the fewest side effects and late complications (de Vries 2015).
Outcomes
None of the studies addressed measurements of quality of life, degree of functional and aesthetic impairment, or relevant issues such as drug resistance or change in ability to detect Leishmania. Outcomes evaluating participants' values and preferences are needed to ensure greater responsiveness of practice guidelines and support shared decision‐making.
With only limited data on prevention of scarring, the development of successful approaches to enhance wound healing or diminish scar formation within targeted areas, or both, will lead to a lower risk of developing scars in these sites. These issues are also possible future trial priorities.
Reducing adverse effects derived from treatment should also be a priority in research. Intramuscular or intravenous drugs are associated with more severe adverse effects. There is a need for less painful and better‐tolerated novel treatment modalities, particularly for children. Alternatives to intramuscular or intravenous treatments should be a research priority, as well as efficacious, well‐tolerated and inexpensive oral agents with enough sensitivity to treat all target species, with few serious adverse effects.
Future studies should assess the long‐term effects of treatment, as healing cannot be clearly confirmed without a long‐term assessment (e.g. at least six months after treatment has finished). For this, it is important to make an a priori decision about the postintervention time frame, based on the maximum length of time after the event during which improvement is attributed to the intervention (Goodman 2007).
Interventions
Future trials should aim at assessing the efficacy of treatments, ideally administered with a single dose of drug or with a short regimen, as this improves adherence. Other lines of research on interventions are for oral or self‐administered treatments requiring minimal supervision (the route of administration can be topical but oral is preferred).
Although frequently used and recommended for the treatment of localised OWCL, we found limited evidence on the use of wound healing to treat OWCL, so this is an important area to test.
Furthermore, as highlighted in the previous review, there is a need for more evidence of the effectiveness and safety of different anti‐Leishmania drugs compared with placebo in self‐healing forms of leishmaniasis or with traditional first‐line antimonials in complicated form, as the basis to recommend alternative safe, efficacious, and affordable treatments. The number of new studies using an active drug as a comparator has doubled, contrasting with fewer placebo‐controlled studies.
Acknowledgements
The authors wish to acknowledge Alireza Firooz for translating and extracting data from Iranian papers, Nieves Plana for her help in developing this review, and Ludovic Reveiz for his help in data extracting for the review.
The Cochrane Skin editorial base wishes to thank Sam Gibbs, Cochrane Dermatology Editor for this review; Matthew Grainge, Statistical Editor; Ching‐Chi Chi, Methods Editor; the clinical referees, Sabha Mushtaq and Iraj Sharifi; and Meggan Harris, who copy‐edited the review.
Appendices
Appendix 1. Cochrane Skin Specialised Register/CRS
(solitary or limited or "old world" or localised or diffuse or cutaneous) AND (leishmania*))
Appendix 2. CENTRAL (Cochrane Library) search strategy
#1 MeSH descriptor: [Leishmaniasis, Cutaneous] this term only #2 (solitary or limited or localised or old world or diffuse or cutaneous):ti,ab,kw #3 leishmania*:ti,ab,kw #4 #2 and #3 #5 #1 or #4
Appendix 3. MEDLINE (Ovid) search strategy
1. leishmania$.mp. 2. (solitary or limited or old world or localised or diffuse or cutaneous).mp. 3. 1 and 2 4. Leishmaniasis, Cutaneous/ 5. 3 or 4 6. randomised controlled trial.pt. 7. controlled clinical trial.pt. 8. randomized.ab. 9. placebo.ab. 10. clinical trials as topic.sh. 11. randomly.ab. 12. trial.ti. 13. 6 or 7 or 8 or 9 or 10 or 11 or 12 14. exp animals/ not humans.sh. 15. 13 not 14 16. 5 and 15
[Lines 6‐15: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision)]
Appendix 4. Embase (Ovid) search strategy
1. skin leishmaniasis/ 2. leishmania$.mp. 3. (solitary or limited or old world or localised or diffuse or cutaneous).mp. 4. 2 and 3 5. 1 or 4 6. crossover procedure.sh. 7. double‐blind procedure.sh. 8. single‐blind procedure.sh. 9. (crossover$ or cross over$).tw. 10. placebo$.tw. 11. (doubl$ adj blind$).tw. 12. allocat$.tw. 13. trial.ti. 14. randomised controlled trial.sh. 15. random$.tw. 16. or/6‐15 17. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 18. human/ or normal human/ 19. 17 and 18 20. 17 not 19 21. 16 not 20 22. 5 and 21
Appendix 5. LILACS search strategy
(cutaneous and leishmaniasis) or (cutanea and leishmaniosis) or (old and world and leishman$) or ((solitar$ or locali$ or limited) and leishman$)
In LILACS we searched using the above terms and the Controlled clinical trials topic‐specific query filter.
Appendix 6. Adverse effects search strategy in MEDLINE (Ovid)
1. exp product surveillance, postmarketing/ or exp adverse drug reaction reporting systems/ or exp clinical trials, phase iv/ 2. adverse event$.mp. 3. adverse effect$.mp. 4. exp hypersensitivity/ or exp drug hypersensitivity/ or exp drug eruptions/ or exp hypersensitivity, delayed/ or exp hypersensitivity, immediate/ 5. exp hypersensitivity, immediate/ or exp anaphylaxis/ or exp conjunctivitis, allergic/ or exp dermatitis, atopic/ or exp food hypersensitivity/ or exp respiratory hypersensitivity/ or exp urticaria/ 6. side effect$.mp. 7. exp Poisoning/ 8. exp Substance‐Related Disorders/ 9. exp Drug Toxicity/ 10. exp Abnormalities, Drug‐Induced/ 11. exp Teratogens/ 12. exp Mutagens/ 13. exp Carcinogens/ 14. exp dermatitis, contact/ or exp dermatitis, allergic contact/ or exp dermatitis, irritant/ or exp dermatitis, phototoxic/ 15. photoallergic reaction$.mp. 16. exp dermatitis, allergic contact/ or exp dermatitis, photoallergic/ 17. sensiti?ation.mp. 18. fetal abnormalit$.mp. 19. exp Drug Monitoring/ 20. harm$ effect$.mp. 21. (toxic effect$ or drug effect$).mp. 22. undesirable effect$.mp. 23. (safe or safety).mp. 24. toxicity.mp. 25. noxious.mp. 26. serious reaction$.mp. 27. complication$.mp. 28. tolerability.mp. 29. (adverse adj3 (effect$ or reaction$ or event$ or outcome$)).mp. 30. Tachyphylaxis/ci, de [Chemically Induced, Drug Effects] 31. *Itraconazole/ 32. *Ketoconazole/ 33. *Paromomycin/ 34. *Allopurinol/ 35. *Amphotericin B/ 36. aminosidine sulphate.mp. 37. pentamidine isethionate.mp. or *Pentamidine/ 38. *Aminoglycosides/ 39. miltefosine.mp. 40. thermotherapy.mp. 41. *Granulocyte‐Macrophage Colony‐Stimulating Factor/ 42. *Mefloquine/ 43. *Immunotherapy/ 44. *BCG Vaccine/ or bacillus calmette guerin.mp. 45. *Meglumine/ 46. sodium stibogluconate.mp. 47. or/1‐30 48. or/31‐46 49. 47 and 48 50. (solitary or limited or old world or localised or diffuse or cutaneous).mp. 51. leishmania$.mp. 52. 50 and 51 53. exp Leishmaniasis, Cutaneous/ 54. 52 or 53 55. 49 and 54
Data and analyses
Comparison 1.
ILMA weekly versus ILMA fortnightly for up to 8 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2.
ILMA (every other day) versus IMMA (6 d/week) for up to 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 3.
IMMA (30 mg/kg/d for 3 weeks) + cimetidine versus IMMA (30 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4.
IMMA (30 mg/kg/d for 3 weeks) + cimetidine versus IMMA (60 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 5.
IMMA (60 mg/kg/d for 3 weeks) + placebo versus IMMA (30 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 6.
IMMA (30 mg/kg/d for 3 weeks) + placebo versus IMMA (60 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 2 | 148 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.71, 0.96] |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Serious adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Skin reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Cardiac toxicity 'QT prolongation' | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 7.
IMMA (30 mg/kg/d for 3 weeks) + 40 mg omeprazole versus IMMA (60 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 8.
IMMA (30 mg/kg/d for 3 weeks) + placebo versus IMMA (60 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 9.
IMMA (30 mg/kg/d for 3 weeks) + 40 mg omeprazole versus IMMA (60 mg/kg/d for 3 weeks) + placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 10.
ILMA + non‐silver polyester dressing versus ILMA (weekly injections for 6 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (itching and burning) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects (oedema) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 11.
ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (itching and burning) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects (oedema) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 12.
ILMA (weekly injections for 6 weeks) + silver polyester dressing versus ILMA (weekly injections for 6 weeks) + non‐silver polyester dressing
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (itching and burning) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects (oedema) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 13.
ILMA (weekly injections for 6 weeks) + gel mask twice a day versus ILMA (weekly injections for 6 weeks) + vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 14.
ILSSG (20 mg/kg/d) + IMSSG (remaining total dose days 1, 3, 5) versus ILSSG (1000 mg/mL days 1, 3, 5)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 15.
ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 d for 29 days) versus IMSSG (20 mg/kg/d for 3 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (mild heart symptoms) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 16.
Ketoconazole 600 mg/d for 6 weeks versus ketoconazole 800 mg/d for 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (nausea and vomiting) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 17.
Ketoconazole 600 mg/d for 30 d versus ILMA (6 to 8 biweekly injections)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effect (liver enzymes increase) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 18.
ILSSG (100 mg/mL days 1, 3, 5) + oral ketoconazole (600 mg/d for 4 weeks) versus ILSSG (100 mg/mL days 1, 3, 5)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 19.
ILSSG (100 mg/mL days 1, 3, 5) + ketoconazole (600 mg/d for 4 weeks) versus ILSSG (20 mg/kg/d) + IMSSG (remaining total dose days 1, 3, 5)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 20.
Itraconazole (200 mg for 6 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected |
Comparison 21.
Itraconazole (200 mg for 3 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected |
Comparison 22.
Itraconazole (200 mg for 6 to 8 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 3 | 244 | Risk Ratio (M‐H, Random, 99% CI) | 3.70 [0.35, 38.99] |
| 2 Adverse effects | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Mild abdominal pain and nausea | 3 | 204 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [0.74, 7.47] |
| 2.2 Mild abnormal liver function | 3 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 3.08 [0.53, 17.98] |
| 2.3 Headache and dizziness | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 2.63 [0.16, 43.63] |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected |
Comparison 23.
Itraconazole (200 mg for 6 to 8 weeks) versus no treatment
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected | |
| 2 Adverse effects (headache and dizziness) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 24.
Fluconazole (200 mg for 6 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 25.
Fluconazole (400 mg/d for 6 weeks) versus fluconazole (200 mg/d for 6 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Rise creatinine and liver enzymes | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Cheilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 26.
Oral dapsone (200 mg/d for 6 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete Cure | 2 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 24.08 [1.44, 403.43] |
| 2 Adverse effects | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Nausea | 2 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 21.86 [3.04, 157.29] |
Comparison 27.
Allopurinol (15 mg/kg/d for 3 weeks) + IMMA (20 mg/kg/d for 2 weeks) versus allopurinol (15 mg/kg/d for 3 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 3.83 [1.71, 8.60] |
Comparison 28.
Allopurinol (15mg/kg/d for 3 weeks)+ IMMA (20 mg/kg/d for 2 weeks) versus IMMA (20 mg/kg/d for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [1.08, 3.41] |
Comparison 29.
Allopurinol (15 mg/kg/d for 3 weeks) versus IMMA (20 mg/kg/d for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions Cured | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.35, 1.62] |
Comparison 30.
Allopurinol (20 mg/kg/d for 3 weeks) + IMMA (30 mg/kg/d for 20 days) versus IMMA (60 mg/kg/d for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Mild abdominal pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Skin eruption | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Muscle pain and weakness | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Microbiological cure of skin lesions | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.88, 1.41] |
Comparison 31.
Allopurinol (20 mg/kg/d for 3 weeks)+ IMMA (10 mg/kg/d for 20 d) versus IMMA (20 mg/kg/d for 28 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Secondary infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 ECG changes | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Chest pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Pain injection site | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Abscess | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 32.
Allopurinol (20 mg/kg/d for 3 weeks) versus IVSSG (20 mg/kg/d for 15 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Abdominal symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Liver abnormalities | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Rash | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 33.
Oral rifampicin (10 mg/kg/d for 4 to 6 weeks) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Evaluated 3 months after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Evaluated after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 34.
Oral rifampicin (10 mg/kg/d) + omeprazole (20 mg/d) for 6 weeks versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 35.
Azythromicin (500 mg/d for 5 d/month up to 4 months) versus IMMA (60 mg/kg/d for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Nausea and vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 36.
Azythromicin (10 mg/kg/d) + allopurinol (10 mg/kg/d) for 1 month versus IMMA (20 mg/kg/d for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Gastrointestinal complaints and headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Gastrointestinal complications | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 37.
Oral pentoxifylline (400 mg 3 times daily) + IMMA (20 mg/kg/d) for 20 d versus placebo + IMMA (20 mg/kg/d) for 20 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Allergic macule‐papular | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 38.
Oral miltefosine (2.5 mg/kg/d for 4 weeks) versus IMMA (60 mg/kg/d for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 39.
Oral miltefosine (2.5 mg/kg/d for 4 weeks) versus IMMA (60 mg/kg/d for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 40.
Oral zinc sulphate 2.5 mg/kg/d for 45 days versus oral zinc sulphate 5 mg/kg/d for 45 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Nausea and vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 41.
Oral zinc sulphate 2.5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Nausea and vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 42.
Oral zinc sulphate 5 mg/kg/d for 45 d versus oral zinc sulphate 10 mg/kg/d for 45 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Nausea and vomiting | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 43.
Oral zinc sulphate (10 mg/kg/d for 45 d) versus IMMA (20 mg/kg/d for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 44.
Artesunate 400 mg + sulphamethoxypyrazine/pyrimethamine 1000 mg/50 mg 4 times daily for 4 d versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 45.
Topical 2% miconazole (twice a day) versus topical 1% clotrimazole (twice a day) for 30 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 46.
Topical ketoconazole (twice a day) versus vehicle (twice a day) for 30 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 47.
Topical amphotericin B (3 to 7 drops twice daily for 8 weeks) versus ILMA (max 2 mL) once a week for 8 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Hypersensitivity | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Mild pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Erithema and oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 48.
Paromomycin 15% + 12% MBCL (twice daily for 28 d) versus vehicle (twice daily for 28 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Scarring | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 49.
Paromomycin (twice daily for 30 d) versus vehicle (twice daily for 30 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.25, 2.06] |
Comparison 50.
Paromomycin 15% + 10% urea (twice daily for 14 d) versus vehicle (twice daily for 14 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 2 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.86, 1.17] |
| 2 Adverse effects | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Skin/local reaction | 3 | 463 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.45, 1.93] |
| 3 Microbiological cure of skin lesions | 2 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.88, 1.20] |
Comparison 51.
Paromomycin 15% (daily for 20 d) versus vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 52.
Paromomycin 15% + gentamicin 0.5% (daily for 20 d) versus vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 53.
Paromomycin 15% + gentamicin 0.5% (daily for 20 d) versus paromomycin 15% alone (daily for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 54.
Paromomycin 15% + 10% urea (twice daily for 45 d) versus ILMA (weekly for up to 3 months)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Scarring | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 55.
Paromomycin 15% + 10% urea (twice daily for 20 d) versus ILMA (weekly for up to 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Cutaneous reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 56.
Paromomycin + MBCL (twice daily for 15 d) versus ketoconazole (weekly for up to 30 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 57.
Paromomycin (15% + 12% MBCL twice daily for 28 days) versus PDT (weekly for 4 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Scarring | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected | |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 58.
Paromomycin (4 weeks) versus paromomycin (2 weeks) + vehicle (2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 59.
IL zinc 2% (twice a week for 2 weeks) versus ILSSG (100 mg/mL) for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 60.
IL zinc 2% (twice a week for 2 weeks) versus IL 7% HSCS for 2 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 61.
ILSSG (100 mg/mL) for 2 weeks versus IL 7% HSCS for 2 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 62.
IL zinc 2% (weekly for up to 6 weeks) versus ILMA (max 2 mL weekly for up to 6 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participants complete cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Burning | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Itching | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Inflammation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Pruritus and erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Severe pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 63.
IL zinc 2% (twice a week for 2 weeks) versus ILMA (60 mg/kg/d for 2 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Inflammation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 64.
Imiquimod (5% 3 times/week for 28 d) + IMMA (20 mg/kg/d for 14 d) versus vehicle + IMMA
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participants with treated lesions that recur | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Itch and burning | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 65.
IL 7% HSCS (0.2 mL to 7 mL per lesion) versus ILSSG (max 2 mL) max 5 injections
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 66.
IL 5% HSCS (0.5 mL to 1 mL per lesion) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 6 to 10 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Sporotrichotic dissemination | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 67.
IL 7% HSCS (0.1 mL to 0.5 mL per lesion) versus IL 2% ciprofloxacin solution (0.1 mL to 0.5 mL per lesion)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 68.
IL 15% HSCS (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Ulceration and necrosis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 69.
ILSSG (0.2 mL to 4 mL per lesion) max 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Ulceration and necrosis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Analysis 69.4.

Comparison 69 ILSSG (0.2 mL to 4 mL per lesion) max 5 injections versus IL 10% HSCS (0.2 mL to 4 mL per lesion), Outcome 4 Adverse effects.
Comparison 70.
ILSSG (0.2 mL to 4 mL per lesion) maximum 5 injections versus IL 15% HSCS (0.2 mL to 4 mL per lesion)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected | |
| 3 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Ulceration and necrosis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 71.
IL IFN‐γ (weekly for 5 weeks) versus ILMA (0.5 mL to 1 mL per lesion) weekly for 5 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 72.
WR279,396 (twice a day for 20 d) versus vehicle (twice a day for 20 d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Mild pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Hearing acuity problems | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 73.
IL metronidazole (2.5 mg to 10 mg each lesion) versus ILMA (150 mg to 600 mg each lesion) for up to 8 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Local inflammatory reactions | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 74.
Topical miltefosine 6% (once daily) versus ILMA (twice a week) for up to 28 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 75.
Dapsone gel 5% (twice a day) + ILMA (weekly) versus cryotherapy (every 2 weeks) + IMMA (weekly) for up to 16 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 76.
DAC‐055 + MWT (for 15 min) versus DAC‐055 alone for up to 75 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Reulceration | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Keloïd scars | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 77.
DAC‐055 + heat (for 15 min) versus ILSSG (0.6 mL) for up to 75 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Reulceration | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Keloïd scars | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Analysis 77.2.
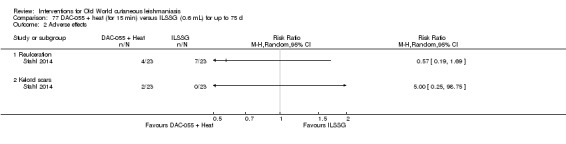
Comparison 77 DAC‐055 + heat (for 15 min) versus ILSSG (0.6 mL) for up to 75 d, Outcome 2 Adverse effects.
Comparison 78.
DAC‐055 alone (for 15 min) versus ILSSG (0.6 mL) for up to 75 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Reulceration | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Keloïd scars | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Analysis 78.2.
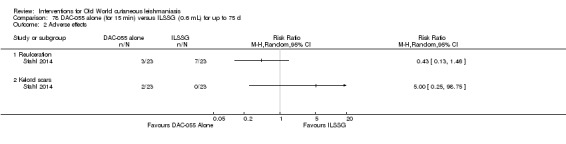
Comparison 78 DAC‐055 alone (for 15 min) versus ILSSG (0.6 mL) for up to 75 d, Outcome 2 Adverse effects.
Comparison 79.
Thio‐Ben (1 mL to 2 mL daily) + cryotherapy (fortnightly) versus ILMA (0.5 mL to 2 mL per lesions) weekly + cryotherapy (fortnightly) for up to 12 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Dizziness and nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Hypersensitive reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 80.
CO₂ laser (30 W continuous) versus IMMA (50 mg/kg/d) for up to 15 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | 433 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.53, 1.55] |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Hyperpigmentation and redness | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Hypertrophic scarring | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Systemic symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 81.
CO₂ laser (30 W continuous) versus cryotherapy (fortnightly) + ILMA (weekly) for up to 12 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Hyperpigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Atrophic scar | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Hypopigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 82.
Ablative CO₂ laser (25 kW for 1 session) versus 3 weeks fractional CO₂ laser
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Partcipants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 83.
TCA (50% wt/vol) fortnightly up to 3 times versus ILMA alone (weekly for up to 6 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Recurrence | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Mild erythema and itch | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Microbiological cure of skin lesions | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.74, 1.26] |
Comparison 84.
Topical TCA 50% + local heat versus ILMA twice a week for up to 8 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Lesions cured | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected | |
| 2.1 Males | 1 | Risk Ratio (M‐H, Random, 99% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Females | 1 | Risk Ratio (M‐H, Random, 99% CI) | 0.0 [0.0, 0.0] |
Comparison 85.
TCA + ILMA (weekly for up to 8 weeks) versus ILMA alone (twice a week for up to 8 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 86.
Fractional laser + ILMA (fortnightly 2 sessions) versus ILMA alone (twice a week for up to 8 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 87.
TCA + ILMA (weekly for up to 8 weeks) versus fractional laser + ILMA (fortnightly 2 sessions)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Analysis 87.2.
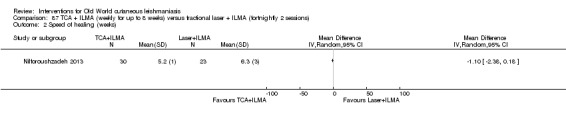
Comparison 87 TCA + ILMA (weekly for up to 8 weeks) versus fractional laser + ILMA (fortnightly 2 sessions), Outcome 2 Speed of healing (weeks).
Comparison 88.
TCA fortnightly up to 8 weeks + ILMA (twice a week) versus ILMA alone (weekly for up to 8 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 89.
Cryotherapy + ILMA (weekly) versus cryotherapy (weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Erythema and oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 90.
Cryotherapy + ILMA (weekly) versus ILMA (weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Erythema and oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 91.
Cryotherapy + ILMA (weekly) versus ILMA alone (weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Erythema and oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 92.
Cryotherapy (weekly) versus ILMA (weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 93.
Cryotherapy + ILMA (weekly) versus cryotherapy alone (weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Hypopigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 94.
Cryotherapy + ILMA (weekly) versus ILMA (fortnightly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Hypopigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 95.
Cryotherapy alone (weekly) versus ILMA (fortnightly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | 390 | Risk Ratio (M‐H, Random, 95% CI) | 14.64 [0.86, 247.99] |
| 2.1 Hypopigmentation | 1 | 390 | Risk Ratio (M‐H, Random, 95% CI) | 14.64 [0.86, 247.99] |
Comparison 96.
Cryotherapy (fortnightly) + 15% paromomycin + 10% urea cream (twice a day) + ILMA (twice a day for 4 weeks) versus ILMA (twice a week) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 97.
Cryotherapy (weekly) + 3% salicylic + 3% sodium nitrite cream (twice a day) for up to 12 weeks versus cryotherapy (weekly) + 3% salicylic cream (twice a day)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Mild skin symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 98.
Radiofrequency waves versus ILMA (1 mL to 7 mL per lesion) weekly for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participants complete cure | 2 | 499 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.97, 1.55] |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 99.
Radiofrequency waves (50 uCTM applied for 30 s) versus ILSSG (10 days of 20 mg/kg/d)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects (serious) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 100.
Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus IMSSG (20 mg/kg/d for 3 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse event (secondary infection) | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected |
Comparison 101.
Radiofrequency waves (1 treatment of > 1 consecutive application at 50ºC for 30 s) versus ILSSG (5 injections of 2 mL to 5 mL every 5 to 7 days)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse event (secondary infection) | 1 | Risk Ratio (M‐H, Random, 99% CI) | Totals not selected |
Comparison 102.
Radiofrequency waves versus ILSSG
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 103.
Electrocauterisation + DAC n‐055 (daily) versus electrocauterisation
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Superinfection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Keloid formation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 104.
PDT (weekly for 4 weeks) versus placebo (twice a day for 4 weeks)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Lesions cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Scarring | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 105.
Mesotherapy gun (0.5 mL of MA weekly) versus ILMA (0.1 mL weekly) for up to 6 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Development of cell‐mediated immunity | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 106.
Diminazene aceturate solution (weekly) versus cetrimide + chlorhexidine solution for 50 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 107.
Topical garlic (twice a day) versus vehicle for 3 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 108.
Topical herbal extract + placebo (5 d) versus IMMA (15‐20/mg/kg/d) + vehicle for 20 d
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 109.
Topical honey (twice a day) + ILMA (weekly) versus ILMA (weekly) for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 110.
Cassia fistula (topical gel) + ILMA (0.5 mL to 2 mL), twice a week versus ILMA (0.5 mL to 2 mL), twice a week + vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Itching | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 111.
Cassia fistula boiled (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 112.
Cassia fistula hydroalcoholic (topical) versus ILMA (0.5 mL to 2 mL), twice a week for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Speed of healing (weeks) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Adverse reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 113.
Cassia fistula boiled (topical) versusC fistula hydroalcoholic (topical) for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Speed of healing (days) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 114.
Topical gel Achilles millefollium (twice daily) + ILMA (weekly 20 mg/kg/d) versus ILMA (weekly 20 mg/kg/d) + vehicle (twice daily) for 4 weeks
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants complete cure | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Itching | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Microbiological cure of skin lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
What's new
Last assessed as up‐to‐date: 17 November 2016.
| Date | Event | Description |
|---|---|---|
| 15 November 2017 | New citation required and conclusions have changed | The incorporation of GRADE into this updated review means that compared with the previous version of the review, we are now less certain of the evidence on which we base our conclusions. |
| 15 November 2017 | New search has been performed | The corresponding author is now Julio Heras‐Mosteiro. Of the authors from the first review, only Mariona Pinart and Ludovic Reitez have participated in this update. There are seven new authors: Begoña Monge‐Maillo, Patricia Lopez‐Pereira, Emely Garcia‐Carrasco, Pedro Campuzano Cuadrado, Rogelio López‐Vélez, Irene Mendez Roman, and Ana Royuela. We have not searched CINAHL for this update. |
History
Protocol first published: Issue 1, 2005 Review first published: Issue 4, 2008
| Date | Event | Description |
|---|---|---|
| 17 May 2012 | Amended | The lead author's contact details have been edited. |
| 18 April 2008 | Amended | Converted to new review format. |
| 21 December 2007 | New citation required and conclusions have changed | Substantive amendment |
Differences between protocol and review
Differences between the protocol and the current update
For differences between other published versions, please see the 'Differences between protocol and review' section within the original publications.
The protocol of this review was first entitled 'Interventions for solitary or limited cutaneous leishmaniasis'. However, the clinical subject was split into two reviews. The title of this review was amended to 'Interventions for Old World cutaneous leishmaniasis' (Gonzalez 2008). A Cochrane Review entitled 'Interventions for American cutaneous and mucocutaneous leishmaniasis' was also published (González 2009). This decision stemmed from the fact that the Leishmania species in the geographical areas involving the Old World differ from the ones affecting the New World. Due to the title change and also in response to referee comments, we modified the Background considerably. Also, our Objectives are now focused on the localised form of cutaneous leishmaniasis (CL) in the Old World rather than the solitary or limited form of CL.
Compared with the published protocol, there were some alterations in the tasks completed by review authors: JHM, PC, PLP, BMM, or EGM checked the titles and abstracts identified from the searches rather than UG and LR; BMM and EGM obtained the full text study for independent assessment when it was unclear if a study was relevant rather than NH and WF. Any disagreements were discussed with AR rather than UG, and MP, LR, JHM, or PLP carried out data extraction rather than MC, NH, or UG. This is because they are no longer authors of the updated version of the systematic review.
Types of participants: we modified this to 'immunocompetent people who have localised OWCL' in accordance with the aforementioned changes.
Types of interventions: we added a list of interventions in response to past referees' comments and to ease readability. We also changed the scope of the interventions from 'all doses and regimens of therapeutic interventions (including topical, systemic, and non‐pharmacological treatments) for solitary or localised cutaneous leishmaniasis' to interventions for Old World cutaneous leishmaniasis, in accordance with the aforementioned changes. We also replaced placebo as a comparison with vehicle because a topical comparison in an RCT should correctly be termed a vehicle rather than a placebo, as the vehicle in a dermatologic drug product enhances delivery and efficacy of the active compound.
Types of outcome measures: also following advice from past referees, we clarified the primary outcomes, and we added a phrase to define emergence of resistance to the tertiary outcomes. We also added a second primary outcome following advice from past referees: 'Percentage of participants with a complete cure after the end of treatment'. We omitted the term "around 3 months" in our primary outcome because it was not precise. We decided not to limit both primary outcomes by length of follow‐up since the studies reporting these outcomes prior to 3 months of follow‐up may report interesting clinical information.
Electronic searches: for the update of this review, we did not search the CINAHL database. This database focuses on nursing and allied health literature, content not particularly relevant to our review topic. We did not search the Cochrane Database of Abstracts of Reviews of Effectiveness (DARE) or MedCarib. DARE contains information on systematic reviews, and the MedCarib database is focused on the Caribbean region. We did not update our search of the American College of Physicians (ACP) journal club. We considered these databases unlikely to yield further references to relevant trials, based on our search results from the previous edition of this review. For the update of this review, we searched the following databases, which we considered relevant for the identification of ongoing trials.
The US National Institutes of Health Ongoing Trials Register (www.clinicaltrials.gov).
The Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
The World Health Organization International Clinical Trials Registry platform (www.who.int/trialsearch).
The EU Clinical Trials Register (www.clinicaltrialsregister.eu/).
Searching other resources > Unpublished literature: although planned in the protocol, we did not contact Centro Dermatológico Federico Lleras Acosta, Colombia, because the Leishmania species in Colombia differ from the ones affecting the Old World. Instead we wrote to National Programme Managers, General Coordinators, Directors, Clinicians, and WHO‐EMRO Regional Officers of endemic countries in order to find relevant studies. Although planned in the protocol, we did not update our search of the conference proceedings. Conference proceedings were considered unlikely to yield further references to relevant trials, based on our search results from the previous edition of this review.
Searching other resources >Adverse effects: although planned in the protocol, we did not contact adverse reaction‐reporting bodies. The review team considered it preferable and more efficient to search MEDLINE for adverse or side effects using the search strategy in Appendix 6.
Data collection and analysis: we updated some of the methods from what we had planned in the protocol, which was published many years ago.
Measures of treatment effect: we planned in the protocol to express results as number needed to treat where appropriate, for a range of plausible control event rates. We did not do this because the great variety found among different participant populations made it impossible to obtain a range of plausible control event rates. As the Cochrane Handbook for Systematic Reviews of Interventions says, "Risk ratios and relative risk reductions remain crucial because relative effect tends to be substantially more stable across risk groups than does absolute benefit" (Higgins 2011). We decided to describe hazard ratios (HR) for time‐to‐event outcomes data when the studies did. We have followed the recommendation, "Conducting a meta‐analysis using summary information from published papers or trial reports is often problematic as the most appropriate summary statistics are typically not presented", and we have not calculated them because we did not have enough information from studies (Higgins 2011).
Assessment of heterogeneity: In the protocol, we had not planned how to assess clinical heterogeneity, but we recorded this in the update. Also, we had planned to explore reasons for heterogeneity using sensitivity and/or subgroup analyses, but we did not do this because there were too few studies to perform a sensitivity and/or subgroup analyses.
Data synthesis: Although not planned in the protocol, we decided to only undertake data synthesis if we were able to identify two or more studies investigating similar treatments and reporting data that could be pooled. We did this because the previous systematic review chose this approach, and we consider that defining a minimum number of studies is necessary to be informative in the data synthesis phase. Where it was not possible to perform a meta‐analysis, we summarised the data for each trial.
We decided not to meta‐analyse studies when I² was above 75% and effect estimates crossed the no‐effect line. However, we did meta‐analyse studies with a high I² if none of the confidence intervals crossed the line of no effect, and we discuss the reasons for such significant heterogeneity.
Although not planned in the protocol, where an ITT was not stated, we used the numbers originally randomised to the groups in order to calculate effect estimates. We did this to avoid overestimating the effect of the intervention (to reduce attrition bias). Concerning the losses to follow‐up, it was not always possible to determine within which arm the losses occurred, and therefore perform ITT analyses.
In the protocol, we had planned that for each trial, we would report other commonly reported outcomes in a table with the cure rates at follow‐up, based on the reported clinical, microbiological, histopathological, or polymerase chain reaction (PCR) results. We did not do this because there were few studies assessing all outcomes. In our manuscript we stated that: "Two RCTs reported results where Leishmania was detected by parasitological diagnostic methods (e.g. PCR or culture, positive smears) (Jaffary 2014A; Jebran 2014). No studies reported emergence of resistance.."
Unit of analysis issues: in the protocol we planned to list quasi‐randomised and non‐randomised controlled studies but not discuss them further; however, in the review, we decided to focus only on RCTs, as in our inclusion criteria.
Dealing with missing data: in the protocol, we did not specify how to deal with missing data; therefore, in this review, we specified that we would treat missing data as treatment failures.
Reporting bias: in the protocol we did not specify if we would investigate reporting bias. In the review we planned to investigate it but was unable to due to low number of studies included in the meta‐analyses.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Omdurman Hospital for Tropical Diseases, Sudan Study period: August 2007 to March 2008 (7 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: people with cutaneous leishmaniasis, confirmed microscopically by finding amastigotes in slit skin smears, if they had not received any previous treatment (Antimonial) Exclusion criteria: pregnant women, people weighing < 10 kg, malnourished people and those with a history of allergy to sulphonamides or artemisinins N randomised: 41 (group 1: n = 20, group 2: n = 21) Withdrawals: 0 N assessed: 41 (100%) (group 1: n = 20, group 2: n = 21) Mean age (SD): group 1: 30.6 years (18.0), group 2: 28.5 years (15.3) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 8 weeks Co‐interventions: pentosan was administered to participants who failed treatment with the study drug as well as to participants who had received placebo tablets |
|
| Outcomes |
Healing rates: disappearance and/or shrinkage of the lesions. Lesions were identified, measured and numbered by their specific location on the participant's body before treatment and after 36 days and 72 days. Reported at the end of treatment. Adverse effects: participants were questioned about expected adverse effects for 3 days (days 5–7) following administration of the doses. These were considered drug‐related if they were not reported at presentation. |
|
| Notes |
Study funding sources: Dafra Pharma NV/SA, Turnhout, Belgium Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated block‐randomisation, blinded to the treating physician, was used to allocate patients to the two treatment arms" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double‐blind, placebo controlled clinical trial"; "blinded to the treating physician…" Comment: not fully reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts are unclear |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available; not registered in a prospective clinical trial registry |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective clinical trial Setting/location: outpatient clinic of Basra teaching hospital, south Iraq Study period: April 2004 to March 2005 (11 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania major and L tropica Inclusion criteria: all patients with cutaneous leishmaniasis who were diagnosed clinically by the same dermatologist Exclusion criteria: ≥ 20 lesions; pregnancy, lactation; hypersensitivity to pentavalent antimonials or local anaesthetic; serious medical illness, lesion in proximity to mucous membranes, face, or cartilage; implanted metallic devices; unwillingness to avoid procreation for at least 2 months N randomised: 38 participants, 70 lesions: group 1 = 35, group 2 = 35 Withdrawals: group 1: 14 lesions; group 2: 8 lesions N assessed: lesions assessed: group 1: 21 (60%); group 2: 27 (77%) Age range: 1.5 to 64 years (1.5–45 years in group 1 and 3–64 years in group 2) with a mean of 21.1 years Sex: 52.5% males, 47.5% females Baseline data:
|
|
| Interventions |
Type of interventions:
Both drugs were injected into the lesions in amounts of 0.1–0.5 mL according to the size of the lesion Duration of intervention: 8 weeks |
|
| Outcomes |
Clinical cure: resolution of active lesion with or without scarring. A scoring system was specially designed as follows. The diameter of lesions was recorded in mm using a ruler and scored as: 0 (total healing); 1 (0 cm to < 0.5 cm); 2 (0.5 cm to < 1 cm); 3 (1 cm to < 1.5 cm); 4 (1.5 cm to < 2 cm); 5 (2 cm to < 2.5 cm); or 6 (≥ 2.5 cm). The degree of induration was assessed by palpation in comparison with the participant's normal skin and given the following scores: 0, 0.5, 1, 1.5, 2, or 3. The degree of erythema was assessed visually and scored as: 0, 0.5, 1, 1.5, 2, or 3. Ulceration was scored as: 1 (present) or 0 (absent). The scores of these 4 parameters were added to give a total score for each lesion. Time points reported: the changes in total score between weeks: lesions were assessed at the start and again at 2‐week intervals after treatment for 8 weeks: 0, 2, 4, 6, and 8 weeks of treatment. Follow‐up continued for 8 weeks until complete healing took place |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not reported; probably an open trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not reported; probably an open trial. No information on how lesions were assessed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | After excluding participants who defaulted on treatment, 21/35 lesions were analysed in group 1 (HSCS) and 27/35 in group 2 (ciprofloxacin) |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. Not registered in a clinical trial registry. Pre‐specified outcomes of the review were reported. Tables were not available in the links of the journal, and .pdf does not work. No adverse effects reported in the text |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Kuwait Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica or L major in the area Inclusion criteria: positive for leishmanial parasites (amastigotes) on microscopic examination. Women of childbearing age were instructed to use potent and adequate contraceptive measures before the initiation of treatment. Exclusion criteria: not described N randomised: 24. Oral itraconazole: 15; placebo: 9 Withdrawals: 0 N assessed: N = 24. Oral itraconazole: 15; placebo: 9 Age range: 12‐52 years Sex: male/female: 13/11 Baseline data: single or multiple lesions, active being nodule, nodule‐ulcerative, or ulcerative. The site of lesions including both groups was 75% on upper limbs; 46% on lower limbs; 25% on the face, and 4% on the trunk. The duration of the lesion varied between 1 and 14 months. |
|
| Interventions |
Type of interventions:
Duration of intervention: 6‐8 weeks Duration of follow‐up: 12 weeks post‐treatment |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' 2 months after treatment. The response to treatment was graded as excellent (reduction in size of lesion by 80% up to complete clearance); good (reduction in size of lesion by 50%) and poor when there is minimal or no change of lesion. Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within 6 months and 1, 2, and 3 years (for a period up to 3 months after suspension of the drug) Adverse effects Time points reported: 8 weeks and 12 weeks post‐treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The participants were randomly divided into two groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two groups" No further information about allocation concealment was provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The drug and the placebo were supplied in capsules with the same shape. No information about blinding of personnel was provided but it is not likely to add risk of bias being oral administration |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding of outcome assessment was described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Al‐Ahssa, Saudi Arabia Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major in the area Inclusion criteria: only a few (1‐3) simple lesions, on a non‐facial site; clinically confirmed CL by direct slit smears and/or in skin‐punch biopsies of the active, infiltrated edge of a representative lesion. Exclusion criteria: multiple or disseminated lesions, lesions aged > 6 months, pregnancy, chronic illness, immunologically compromised condition, hyperallergic reaction to the trial drugs, treatment with regular medications which may affect specific therapy, treatment with antileishmanial drugs within the previous 6 months, the presence of scars of previously healed lesions N participants (lesions) randomised: 80. IMMA group: 40 (77); ILMA group: 40 (70) Withdrawals: 13 (13). IMMA group: 9 (9); ILMA group: 4 (4) N assessed: 67. IMMA group: 31 (68); ILMA group: 36 (66) Mean age (range): range 13‐42 years. IMMA group: 29.8 years (15‐41); ILMA group: 31.5 years (13‐42) Sex: male: 48, female: 19 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention:
Duration of follow‐up: 1 month post‐treatment |
|
| Outcomes |
Primary outcome: percentage of lesions 'cured' at the end of treatment Secondary outcomes: prevention of scarring Adverse effects Time points reported: days 15, 30 |
|
| Notes |
Study funding sources: none described Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "enrolled patients were randomly assigned to one of two treatment groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Quote: "enrolled patients were randomly assigned to one of two treatment groups" Comment: no further information about allocation concealment was provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about blinding of participants and personnel was provided but the different administration via of the drug is impossible to blind, although it is unlikely to add risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Lesions were assessed, whenever a patient came for his or her injection (s), by an observer who was unaware of the treatment the patient was receiving." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about dropouts |
| Selective reporting (reporting bias) | Unclear risk | No information about adverse effects was provided |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Al‐Ahsaa and Ryyadh, Saudi Arabia Study period: 15 months Sample size calculation: to detect a difference of 22% in the rate of healing between the placebo group and the treatment group, assuming a healing rate of 45% in the placebo group, with a power of 90% and a two‐sided type I error of 5%, 101 subjects were needed in each group. To compensate for loss to follow‐up, 25% more participants were to be enrolled in each group. |
|
| Participants |
Type of Leishmania:L major 56 participants (27%) Inclusion criteria: age > 12 years, presence of lesions parasitologically confirmed leishmaniasis, non‐use of antileishmanial therapy during previous 2 months Exclusion criteria: pregnancy, potential for pregnancy, breastfeeding; presence of lesions on the face or ears; presence of more than 10 lesions; history of liver disease; elevated serum, creatinine concentration, abnormal results on liver‐function tests; allergy to fluconazole N randomised: 209. 106 were assigned to the fluconazole group, and 103 were assigned to the placebo group Withdrawals: 63 received the container of capsules at the first visit and never returned for follow‐up: 37 participants assigned to receive placebo and 26 participants of fluconazole group One participant in the placebo group, who was therefore excluded from the analyses. N assessed: 145. Fluconazole group: 80, placebo group: 65 Age and sex: not described Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks Co‐interventions: SSG was offered during follow‐up if oral therapy was considered to have failed (14 participants in the fluconazole group and 33 in the placebo group) Duration of follow‐up: 1 year post‐treatment |
|
| Outcomes |
Healing rates:
Adverse effects Time points reported: 6 weeks, 3 months of follow‐up, 1 year post‐treatment |
|
| Notes |
Baseline imbalances: because of the criteria for inclusion and the limited number of women at risk for Leishmania in the study areas, there was only one female participant. Most of the participants were foreign construction workers or farmers originally from countries where CL is not endemic. One of 5 participants was a local national. Study funding sources: supported in part by a grant (no. 146‐1414) from Pfizer and the Ministry of Health of Saudi Arabia Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the randomisation sequence was generated from a random‐number table" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The randomisation sequence was generated from a random‐number table" Comment: no further information about allocation concealment was provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "assigned to receive either fluconazole (Diflucan, Pfizer, New York) in the form of a 200‐mg capsule once daily for six weeks or a matching placebo" "An independent observer evaluated the rates of compliance and side effects by interviewing patients and counting their remaining capsules." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further information about blinding of outcome assessment was provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High rate of drops out: 63/209 (30.14%). Missing outcome data imbalanced in numbers across groups. Fluconazole orally 200 mg: 26. Placebo group: 37. An ITT analysis was performed |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Kuwait Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp not specified Inclusion criteria: only the smear‐positive cases included in the study Exclusion criteria: participants younger than 14 years and pregnant nursing women N randomised: 33. Group 1, ketoconazole 600 mg: 18; Group 2, ketoconazole 800 mg: 15 Withdrawals: 7. Group 1: 3, Group 2: 4 N assessed: 26. Group 1: 15, Group 2: 11 Age range: 14‐66 years Sex (male/female): 26/7 Severity of illness: 1‐8 lesions. Site of the lesions in the ketoconazole 600 mg group: 46% on the upper extremities and 24/% on the lower extremities. In the ketoconazole 800 mg: 58% on the upper extremities; 21% on the lower extremities and 21% on head and neck Ketoconazole 600 mg: MNL: 3.56 (range 2‐8). MDLBT: 3.4 months (range 1.5‐7) Ketoconazole 800 mg: MNL: 3.27 (range 1‐6). MDLBT: 4.5 months (range 1‐12) |
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks or until the participant was cured (whatever occurred early) Duration of follow‐up: 6 months |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment ( If there was more than 90% improvement of these parameters: re‐epithelisation and decrease in the size and inflammation of the lesions, with a negative smear for Leishmania parasites) Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within 6 months Adverse effects Time points reported: 1, 2, 4, 6, 8 weeks post‐treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | No information about allocation concealment was provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about blinding was provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about blinding was provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to judge. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Walter Reed Army Medical Center (WRAMC) in Washington Study period: 24 months (2004‐6) Sample size calculation: a sample size of 27 participants per treatment group was planned, assuming a 73% cure rate for ThermoMed (TM), 99% for SSG, controlling for a probability of a type I error at alpha = 0.05 and was predicted to have 80% power to determine a 26% difference in outcome. |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: eligible participants were Department of Defense healthcare beneficiaries with parasitologically confirmed cutaneous leishmaniasis. At Walter Reed Army Medical Center (WRAMC) in Washington. All participants were likely infected in Iraq or Kuwait. All were treatment naive. Exclusion criteria: 20 lesions; pregnancy, lactation; hypersensitivity to pentavalent antimonials or local anaesthetic; serious medical illness; lesion in proximity to mucous membranes, face, or cartilage; implanted metallic devices; unwillingness to avoid procreation for at least 2 months N randomised: 56, IVSSG: 28. Localised TM device heat treatment: 28 Participants with clinical failure at 2 months were offered cross‐over treatment. Afer 2 months: SSG: 29 (−1, +3), TM: 25 (−3, +1). Withdrawals: 2. TM: 1 (lesion not amenable to heat), SSG: 1 (not confirmed L major) N assessed: 54 (96.43%) completed treatment: TM: 27 (96.43%), SSG: 27 (96.43%). 53 (94.64%) completed 12 months follow‐up: TM: 27 (96.43%), SSG: 26 (92.86%) Median age (range): TM: 25 years (20‐53); SSG: 24 years (18‐57) Sex: TM: males: 28 (100%), SSG: males: 27 (96%), females: 1 (4%) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: TM: 1 session. SSG: 10 days. Co‐interventions: participants randomised to thermotherapy received oral antibiotics for secondary bacterial infections of the leishmaniasis lesion(s) prior to treatment. SSG arm participants were treated concurrently with antibiotics. Follow‐up: 2, 6, and 12–24 months post‐treatment. |
|
| Outcomes |
Clinical cure of the lesions: clinical cure was defined as complete epithelialisation or visually healed at 2 ± 1 month after completion of therapy and no reactivation in 12 months after the start of treatment. Clinical failure was less than complete epithelialisation or visually not healed at 2 ± 1 month after treatment completion. Relapse failure was defined as skin lesion persistence at the treatment site or elsewhere in the period up to 12 months after start of therapy, regardless of appearance at 2 ± 1 month after treatment completion Laboratory cure of the lesions: microbiological cure: they looked for an eradication of the infection Time to healing: survival analysis of time to healing for the 2 treatment arms Adverse effects: toxicity profile Time points reported: 2, 6, 12 months. Toxicity profile: daily physician evaluations |
|
| Notes |
Study funding sources: this trial was supported by Walter Reed Army Medical Center, The North Atlantic Regional Medical Command, and the US Army Medical and Materiel Development Agency. Possible conflicts of interest: the authors have declared that no competing interests exist |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The statistician (RH) generated the randomisation plan in blocks of 4 subjects using www.randomization.com. The research pharmacist made assignments using the randomisation plan in sequential order. |
| Allocation concealment (selection bias) | Low risk | The allocation sequence was unavailable to investigators until completion of the trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Sequential photographs were independently assessed by blinded leishmaniasis experts, who were clinicians experienced in the treatment of CL, with a tiebreaker assessment when needed. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2/56 withdrawals. An ITT analysis was performed |
| Selective reporting (reporting bias) | Low risk | Clinical Trial Registration: ClinicalTrials.gov NCT 00884377; all prespecified outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: 8 primary health centres around Borkhar, north of Isfahan, Iran Study period: 14 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: infections here were thought to be caused entirely by L major parasites, although there is probably some L tropica infection within the city of Isfahan Inclusion criteria: 2 years or older, single lesion that was parasitologically positive, < 5 cm in diameter, at least 3 cm from the eyes, lesion present < 4 months Exclusion criteria: pregnant or nursing mothers, previously treated for leishmaniasis, intercurrent illness or a history of allergy to aminoglycoside N randomised: 251, aminosidine group: 126 (134 lesions); placebo group: 125 (134 lesions) Withdrawals: not described N assessed: aminosidine group: 123 lesions; placebo group: 123 lesions Age (years): aminosidine group: < 15 years: 114, >15 years: 12; placebo group: < 15 years: 105, > 15 years: 20 Sex (male/female): aminosidine group: 64/62; placebo group: 67/58 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 14 days Co‐interventions: additional treatment, usually parenteral antimony, was given if lesions were judged to have worsened (25 participants in the PR‐treated group and 28 in the placebo group) Duration of follow‐up: 105 days after starting treatment |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 2.5 months after treatment. Definite cure was defined as complete epithelialisation on days 45 or 105 Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesion Time points reported: 15, 45, 105 days |
|
| Notes |
Study funding sources: this work was supported in part by he UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases (TDR) and Isfahan University of Medical Sciences Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation of treatment was carried out in Geneva (Switzerland) but did not state how that was done. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Identical ointment tubes were numbered and allocated to consecutive eligible participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups (16 drops out of 251, 10%) |
| Selective reporting (reporting bias) | Unclear risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: 3 primary health centres around Borkhar district north of Isfahan, Iran Study period: not described Sample size calculation: determined on the basis of a 2‐week cure rate of 50% at day 45 and unexpected 4‐week cure rate of 70% |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: participants with a single parasitologically confirmed lesion, < 5 cm in diameter Exclusion criteria: lesions duration > 4 months and < 2 years, pregnant or nursing mothers, treated previously, any intercurrent illness or history of allergy to aminoglycoside N randomised: 233. 117 were allocated to receive 4 weeks of active treatment and 116 to receive 2 weeks of active treatment Withdrawals: 17, 9 in 4 weeks of active treatment and 8 in 2 weeks of active treatment N assessed: 216, 108 in each group. Mean age (SD): 4 weeks of active treatment: 9.2 years (8.9); 2 weeks of active treatment: 9.0 years (8.8) Sex ratio (male/female): 4 weeks of active treatment: 47/53; 2 weeks of active treatment: 44/56 Baseline data: ulcerated lesion (SD): 4 weeks of active treatment 85 (79); 2 weeks of active treatment 95 (88) |
|
| Interventions |
Type of interventions:
Co‐interventions: if lesions were bad enough, they were treated with antimonate Duration of follow‐up: 105 days after starting treatment |
|
| Outcomes |
Healing rates: 'Clinical cure' was defined as > 50% re‐epithelialisation of the original lesion, and 'clinical and parasitological cure' as either complete re‐epithelialisation or clinical cure plus a parasitologically negative smear. The primary study endpoints were clinical cure and clinical and parasitological cure at day 29, when the 4 weeks of active treatment ended Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: days 29, 45, 105 |
|
| Notes |
Study funding sources: this investigation was supported by the UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list after participants returned their first used tube after 2 weeks |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The vehicle and active ointments looked and smelled identical. Outcome is not likely to be influenced by the lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The clinical and parasitological evaluators were blinded to each other's assessment. All efforts were made to reduce the introduction of bias into this study; however, duration of the lesion, which was self‐reported by the participants or their guardians, could introduce bias if the durations were significantly different in the 2 arms by chance. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Diseases and Leishmania Research Center of Isfahan (Iran) Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania but spp not declared Inclusion criteria: confirmed diagnosis parasitologically and clinically, lesions with duration of < 8 weeks Exclusion criteria: participants with a history of > 8 weeks, those with allergy to antimonials, lactating or pregnant women N randomised participants (lesions): 400 (539). Combined cryotherapy and ILMA: 100 (149), cryotherapy alone: 200 (230), ILMA alone: 100 (160) Withdrawals: 30 participants (46 lesions) N assessed (lesions): 370 (493). Group 1: 93 (132); group 2: 185 (210); group 3: 92 (151) Age: range 2‐65 years. Mean: group 1, 32 years; group 2, 27 years; group 3, 25 years Sex (male/female): 186/184. Group 1, 46/47; group 2, 95/90; group 3, 45/47 Baseline data: not described |
|
| Interventions |
Type of interventions:
Different modalities of treatment were not given to the same participant in different lesions (i.e. each participant received ILMA alone, cryotherapy alone, or combined cryotherapy and ILMA). Duration of intervention: fortnightly until complete cure or for up to 6 weeks Duration of follow‐up: 6 months |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 2.5 months after treatment Adverse effects: mild adverse side effects, such as postinflammatory hypopigmentation and hyperpigmentation Tertiary outcomes: microbiological or histopathological cure of skin lesions |
|
| Notes |
Baseline imbalances: in number of participants and lesions among the groups Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The 400 patients were randomly divided into three groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The 400 patients were randomly divided into three groups" Comment: no further information was provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatment doesn't appear as blinded and the different treatments are difficult to blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No good information about adverse effects. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin and Leishmaniasis Research Center of Isfahan (Iran) Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not reported Inclusion criteria: age 7‐70 years, disease confirmed clinically and by laboratory methods, lesions present, surface area of lesions ≤ 5 cm² Exclusion criteria: disease duration > 4 months; pregnant or breastfeeding; chronic disease; immune suppression; and sporotrichoid forms N participants randomised (lesions): 233 (433). 123 (183) were included in the CO₂ laser group and 110 (250) in the IMMA group Withdrawals: 59 (112). CO₂ laser group: 40 (72), IMMA group: 19 (40) N assessed (lesions): 174 (321). CO₂ laser group: 83 (111), IMMA group: 91 (210) Age: range 12‐60 years Sex (male/female): 55/68 included in the CO₂ laser group; 40/70 in the IMMA group (control group) Baseline imbalances: no Severity Illness: in 47% of cases, participants had one lesion and in 53% of cases they had 2‐5 lesions. There were more lesions on the upper limbs (43%), and lesions were < 5 cm². In the remaining, the lesion duration was 2‐4 months. Mean number of lesions: CO₂ laser group: 1.49; IMMA group: 2.27 |
|
| Interventions |
Type of interventions:
Co‐interventions: in the first group, lesions were locally anaesthetised by injection of 1%–2% lidocaine Duration of follow‐up: 24 weeks |
|
| Outcomes |
Healing rates:
Adverse effects |
|
| Notes |
Study funding sources: none declared Possible conflicts of interest: none described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: the author was contacted. The randomisation was through coin flip method. |
| Allocation concealment (selection bias) | Unclear risk | Comment: the author was contacted. The randomisation was through coin flip method, but the method to conceal the allocation was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No data provided, but the treatment is unlikely to be blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided about blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imbalance in the missing data between the groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Department of Dermatology, Isfahan University of Medical Sciences (Iran) Study period: September 2004 to May 2005 (8 months) Sample size calculation: after following a formula, sample size and number of lesions would be a maximum of 20 participants and 40 lesions, respectively, for each group |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: confirmation of typical CL by positive Giemsa‐stained direct smear for Leishman‐Donovan bodies Exclusion criteria: > 2 lesions, lesions with > 20 mm induration diameter, duration of the disease > 2 months, previous use of any anti‐leishmanial treatments, pregnant or nursing women, children < 5 years of age, serious concomitant medical problems, history of seizure, photosensitivity N participants randomised (lesions): 60 (99). Photodynamic therapy (PDT): 20 (31); topical paromomycin: 20 (35); vehicle: 20 (33) Withdrawals (n = 3): 1 participant with one lesion in topical paromomycin group and 2 participants with 3 lesions (one participant had 2 lesions and the other had one) in vehicle group did not complete the study because they used the ointment irregularly N assessed (lesions): 57 (95). Photodynamic therapy (PDT): 20 (31); topical paromomycin: 19 (34); placebo: 18 (30) Age: range 5‐59 years. Mean (SD): PDT 22.2 years (15); topical paromomycin: 24.2 years (17); vehicle 22.3 years (15) Sex (n, (%)): PDT: female: 12 (60)/male: 8 (40); topical paromomycin: female: 8 (42.1)/male: 11 (57.9); vehicle: female: 11 (61.1)/male: 7 (38.9) Severity of illness: the most common sites of lesions (< 10) were on the extremities. Total number of lesions: PDT 31; topical paromomycin 34; vehicle 30 MNL: PDT 1.55; topical paromomycin 1.75, vehicle 1.65 Mean duration of lesions (SD): PDT 38 days (11); topical paromomycin 35 days (11); vehicle 36 days (11.6) |
|
| Interventions |
Type of interventions:
Duration of intervention: 4 weeks Duration of follow‐up: these groups were followed for 2 months after the end of treatment |
|
| Outcomes |
Healing rates: percentage of lesions 'cured' 2 months after treatment; prevention of scarring Scale: 'complete improvement' was defined as loss of induration and other signs of inflammation, complete re‐epithelialisation and return to normal skin texture as well as 'parasitological cure', i.e. a negative Giemsa‐stained direct smear. 'Partial improvement' was considered as flattening, reduction in size and induration without complete re‐epithelialisation. All lesions showing no decrease in size or induration were regarded as 'treatment failures'. Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: days 7, 14, 21, 28, 90 |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The participants were randomly divided into three groups of 20 subjects each, using computer‐based randomisation." |
| Allocation concealment (selection bias) | Unclear risk | No information about allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Subjects in both the paromomycin and placebo groups and the clinician treating them were blinded with respect to the topical treatment received. However, it was not possible to blind subjects in the PDT group." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The dermatologist was not aware of the type of the treatment that the patient was receiving." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No defaults were included in the analyses. No good information about adverse effects. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised, prospective, clinical trial Setting/location: Skin and Leishmaniasis Research Center of Isfahan, Barkhar Health Center and Department of Dermatology, Isfahan University of Medical Sciences (Iran) Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: fewer than 4 lesions of < 1 month old and < 3 cm, lesions not located on the face, > 15 years of age, lesions located neither on muscles nor were sporotrichoid lesions; receiving no medication for leishmaniasis Exclusion criteria: history of susceptibility to MA or miltefosine; pregnant or breastfeeding; immunosuppressed; had taken systemic medication interfering with p‐450 enzyme; or history of kidney, liver, and heart failure N randomised: 64. Topical miltefosine: 32, ILMA: 32 Withdrawals: 0 N assessed: 64 (100%). Topical miltefosine: 32, ILMA: 32 Mean age (SD): 23.12 years (13.30) Sex: topical miltefosine: 17 males (53%) and 15 females (47%). ILMA: 16 males (50%) and 16 females (50%) Baseline data: mean (SD) size of the lesions before treatment in the group treated with miltefosine and ILMA was 4.4 cm (3.1) and 2.3 cm (2.2), respectively. |
|
| Interventions |
Type of interventions:
Duration of intervention: 28 days |
|
| Outcomes |
Clinical cure: cure of cutaneous leishmaniasis was defined as follows:
Adverse effects: participants were questioned about expected adverse effects for 3 days (days 5–7) following administration of the doses. These were considered drug‐related if they were not reported at presentation. Time points reported: at the end of treatment, 1 month after treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Open trials. Photography was done from all the lesions both at the first visit and all the follow‐up visits |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | High risk | Protocol not available; not registered; in clinical trial registry; adverse effects not reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Sidi‐ Bouzid. Tunisia Study period: 9 months Sample size calculation: the ideal sample size was estimated to be 120 based on a rate of success of treatment of 80%, 30%‐45% self‐healing in the vehicle group (type I error = 0.01and type II error = 0.10), and 10%‐20% loss to follow‐up |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: aged 2‐60 years, single lesion diagnosed by the presence of parasite in stained dermal smears, no previous anti‐leishmanial treatment Exclusion criteria: known allergy, adverse reactions to aminoglycoside antibiotics, multiple lesions, an active lesion measuring > 5 cm in diameter, if their ulcerated lesion had already persisted for more than 4 months, lesions < 3 cm from the eye, who by the physician's judgment required systemic antimonial treatment: participants with serious concomitant diseases, under medication for other illnesses likely to interfere with this study; pregnant women or nursing mothers. N randomised: 132. Paromomycin group: 66; vehicle: 66 Withdrawals: 17. Paromomycin group: 9; vehicle: 8 N assessed: 115. Paromomycin group: 57; vehicle: 58 Mean age (SD): paromomycin group: 19.2 years ( 2.31); vehicle: 18.2 years (1.65) Sex (ratio M:F): paromomycin group: 1.04; vehicle: 1.07 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: twice daily for 14 days Duration of follow‐up: 105 days |
|
| Outcomes |
Healing rates: percentage of participants with complete re‐epithelisation of the lesion, 2.5 months after treatment initiation (105 days) Parasitological cure: percentage of lesions with negative smear and culture, 2.5 months after treatment initiation (105 days) Adverse effects Time points reported: days 15, 45, 105 |
|
| Notes |
Study funding sources: this investigation received funding from the United Nations Development Program/World Bank/World Health Organization Special Program for Research and TraininginTropical Diseases(grant ID:TDK910677) Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The tubes containing drug or placebo were supplied by the WHO/TDR, randomly numbered, and were given in numerical order to patients as they were admitted into the study." |
| Allocation concealment (selection bias) | Low risk | Quote: "The tubes containing drug or placebo were supplied by the WHO/TDR, randomly numbered, and were given in numerical order to patients as they were admitted into the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The code remained unknown to patients and investigators until the study had been completed" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The code remained unknown to patients and investigators until the study had been completed" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No defaults were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Sidi Bouzid,Tunisia, and Paris, France Study period: 22 months Sample size calculation: the protocol calculated a sample size of 50 participants per group with 80 percent power and a Type I error rate of 5 percent to detect a 30% difference in the proportion of participants achieving CCR, assuming a CCR proportion of 35% in the vehicle group and 65 percent in WR279, 396 participants, with a 5% expected rate of loss to follow‐up |
|
| Participants |
Type of Leishmania: L major, L tropica, L infantum Inclusion criteria: aged 5‐75 years, presence of parasitologically confirmed CL, lesions that were primarily ulcerative (i.e. not purely verrucous or nodular) and measured ≥ 1 cm² and ≤ 5 cm². Exclusion criteria: history of known or suspected hypersensitivity or idiosyncratic reactions to aminoglycoside; previous use of antileishmanial drugs (within 3 months) or nephrotoxic or ototoxic drugs; prior diagnosis of leishmaniasis; more than 5 lesions, or a lesion in the face that in the opinion of the attending dermatologist could potentially cause significant disfigurement; significant medical problems as determined by history or laboratory studies; breastfeeding and pregnancy N randomised: 92. WR279,396: 50; vehicle: 42 Withdrawals (n = 2): WR279,396: 1; vehicle: 1 N assessed (%): 90 (95.7). WR279,396: 49 (94.2), vehicle: 41 (97.6) Age < 18 years ‐ n (%): WR278,396: 47 (94), vehicle: 33 (79) Sex: male: 54, female: 38 Severity of illness: N lesions: 1: 54, 2: 16, 3: 13, 4 or 5: 9 Total lesion area (median, IQR): WR279,396: 128 (85 to 223), vehicle: 154 (70 to 264) Index lesion area (median, IQR): WR279,396: 92 (55 to 141), vehicle: 115 (50 to 172) Leishmania spp ‐ n (%): L major: WR279,396: 32 (64), vehicle: 24 (57). L Infantum: WR279,396: 1 (2), placebo: 0 (0). L tropica: WR279,396: 1 (2), vehicle: 0 (0). Unidentified: WR279,396: 16 (32), placebo: 18 (43) |
|
| Interventions |
Type of interventions:
Duration of intervention: 20 days |
|
| Outcomes |
Clinical cure: defined as complete re‐epithelialisation (i.e. length 6 width of ulceration = 060) of the index lesion by day 50 or a 50% re‐epithelialisation by day 50 followed by complete re‐epithelialisation on or before day 100 with no relapse ever having occurred from day 50 through day 180. Relapse was defined as an increase in the area of ulceration relative to the previous measurement. Participants who did not complete the 180‐day period of observation were considered to have failed to achieve complete clinical response (CCR) because relapse could not be fully assessed. Complete clinical response at the index lesion according to baseline characteristics: influence of baseline factors on effect of treatment. Time to healing: time course of complete re‐epithelialisation measured at 20, 50, 100, 180 days since start of treatment. Adverse effects: immediate: observed within 30 min of application. Delayed: observed just prior to next application |
|
| Notes |
Study funding sources: the Office of the Surgeon General (OTSG), Chief, Human Subjects Protection Division, U.S. Army MRMC, Fort Detrick, MD 21702‐5012. IND50,098 HSRRB Protocol#1791. Co‐sponsor: Institute Pasteur, Rue du Dr. Roux, Paris, France Possible conflicts of interest: MG has no financial competing interests |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A sequence of genuine random numbers for the randomisation procedure was obtained from the 'fourmilab.ch/hotbits' website by a member of the Department of Chemical Information, Walter Reed Army Institute of Research, Silver Spring, Maryland and purged of duplicates. The random numbers are generated by a process which takes advantage of the inherent uncertainty in the quantum mechanical laws of nature. Specifically, they are generated by timing successive pairs of radioactive decays detected by a Geiger‐Muëller tube interfaced to a computer. This process is better than the pseudo‐random number algorithms typically used in computer programs. The randomisation of the study drugs was done by an independent group, Fischer BioServices, Rockville, Maryland a contractor to The U.S. Army Medical Research Acquisition Activity (USAMRAA), Ft. Detrick, Maryland." |
| Allocation concealment (selection bias) | Low risk | The randomisation of the study drugs was done by an independent group; however, allocation concealment is not fully described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The vehicle lacked the active components and trace amounts of colorings agents to match the appearance and maintain the blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Investigators, who were blinded to whether participants received WR279,396 or placebo‐vehicle, evaluated lesions for clinical response on D20 (i.e. the end of the treatment period), D50 (i.e., 30 days after the conclusion of treatment), D100, and D180". Investigators measured all lesions in 2 perpendicular directions and took photographs at the following time points: prior to therapy, at the end of therapy |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 49 of 50 participants randomised to WR279,396 and 41 of 42 participants randomised to placebo‐vehicle completed the study. With one exception, applications of study drugs were conducted according to the protocol |
| Selective reporting (reporting bias) | Low risk | ClinicalTrials.gov NCT00703924. Pre‐specified outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Sidi Bouzid, Tunisia Study period: January 2008 to July 2011 (42 months) Sample size calculation: the sample size of 375 participants was based on estimated rates of final clinical cure of 94% in the paromomycin – gentamicin group and 71% in the vehicle‐control group, as shown in a previous study. On the basis of these rates, a sample size of 125 participants in each of these 2 groups provided a statistical power of 99% to detect a significant difference in the rates of final clinical cure rates (94% vs 71%). |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: aged 5‐65 years; good health besides cutaneous leishmaniasis if female; absence of pregnancy and lactation; the presence of ≤ 5 lesions, with an index lesion that was ulcerative; lesions measured 1‐5 cm in diameter; lesions confirmed to contain Leishmania by means of culture or microscopical examination of lesion material Exclusion criteria: included clinically significant lymphadenopathy or mucosal involvement, against which a topical agent would not be expected to be effective N randomised: 383 (129 were assigned to paromomycin–gentamicin; 128 were assigned to paromomycin; 126 were assigned to vehicle control) Withdrawals: 8 participants (4, paromomycin–gentamicin; 3, paromomycin; 1, vehicle control) N assessed: 375 participants (125 in each of the 3 groups) Mean age (SD): 24 years (16) (paromomycin–gentamicin, 23 years (16); paromomycin, 25 years (16); vehicle 23 years (15) Sex: male 193 (51%) (paromomycin–gentamicin, 56 (45%); paromomycin, 68 (54%); vehicle control, 69 (55%)) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 20 days |
|
| Outcomes |
Final cure of index lesion Total re‐epithelialisation of ulcerated lesions at 42 days Adverse effects Time points reported: 168 days |
|
| Notes |
Study funding sources: the study was sponsored by the Office of the Surgeon General, Department of the Army Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A permuted block randomisation schema was generated using nQuery, employing random block sizes, for the first 330 randomisation numbers. As 7 subjects were randomised that did not receive treatment at the time when the new randomisation list was generated, the plan in generating the new list was to balance the assignments in the new list for these 7 subjects to achieve the 1:1:1 allocation balance overall. The second randomisation was performed using SAS Version 9.2." |
| Allocation concealment (selection bias) | Low risk | The randomisation of the study drugs was done by an independent group. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The placebo consisted of the vehicle without the active components and trace amounts of colorings agents to match the appearance and maintain the blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The modified intention‐to‐treat population consisted of patients who received at least one dose of study treatment. We tested two hypotheses using a fixed testing‐sequence procedure with an overall two‐sided alpha level of 0.05 or less." |
| Selective reporting (reporting bias) | Low risk | ClinicalTrials.gov number, NCT00606580. Pre‐specified outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised, phase I, open‐label, clinical trial Setting/location: Department of Dermatology at SP Medical College and PBM Hospital, Bikaner, India Study period: June 2009 to December 2010 (19 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: aged ≥ 4 years, 4 or fewer lesions, a parasitologically confirmed diagnosis of CL by demonstration of organisms (L tropica bodies) in the lesion smear or biopsy Exclusion criteria: included lesion size > 5 cm diameter, prior treatment failure with SSG, treatment for CL within 2 months of enrolment into the study, any chronic condition that might prevent the patient from completing the study therapy and subsequent follow‐up N randomised: 100; radiofrequency‐induced heat therapy (RFHT): 50; ILSSG: 50 Withdrawals: not described N assessed: 100 Median (range) age: RFHT: 20 years (4‐70); ILSSG: 20.5 years (4‐85) Sex: RFHT: male 27, female 23; ILSSG: male 20, female 30 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: RFHT, 5 days; ILSSG, 4 weeks Co‐interventions: all participants were prescribed oral nonsteroidal anti‐inflammatory drugs and topical antibacterial cream (fusidic acid cream) for 5 days |
|
| Outcomes |
Cure rate, assessed as follows:
Time points reported: 6, 8, 10, 12, 16, and 20 weeks after the initiation of treatment, and at 5, 6, 9, 12 and 18 months post‐treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were N randomised in a 1:1 ratio" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The clinician who recorded healing was blinded to the modality of treatment." Comment: not clear how the assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information about numbers or reasons for withdrawals during treatment. It seems that there was withdrawals only in the follow‐up: 44% at 18 months in RFHT group and 16% at 18 months in the SSG group. Losses to follow‐up were heterogeneous between groups and no ITT analyses were performed. |
| Selective reporting (reporting bias) | Low risk | Outcomes of interest were reported in results |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised single blind parallel clinical trial Setting/location: Leishmaniasis Center in Dadbin Health Care Clinic in Kerman, Iran Study period: 15 months. Participants recruitment: 8 months (December 2012 to August 2013) Sample size calculation: based on the data from previous study, and by considering the confidence interval of 95%, sample size was determined as 23 lesions per group. Regarding to a possible loss in follow‐up period, finally 32 lesions were allocated in each group |
|
| Participants |
Type of Leishmania: L amastigote (Leishman‐Donovan bodies) Inclusion criteria: positive direct smear or documented skin biopsy for L amastigote (Leishman‐Donovan bodies), > 2 years of age, no previous therapy for leishmaniasis, ulcerated lesions for those lesions that were included in Thio‐Ben + cryotherapy (TC) group Exclusion criteria: pregnancy, lesions on the eyelids and lips, having more than 5 lesions, positive history of confirmed immunodeficiency disorders or immunosuppression, disease duration for more than a year, positive history of allergy or hypersensitivity to thioxolone, benzoxonium chloride, or pentavalent antimony compounds. N randomised: 64 lesions of 47 participants, Thio‐Ben + cryotherapy (TC) group (32 lesions, 22 participants), ILMA (32 lesions, 25 participants) Withdrawals: 16 lesions of 9 participants (10 lesions from TC and 6 from ILMA group) were removed from the study arms because of their poor adherence to the trial protocol and lost to follow‐up. Additionally, one participant (with one lesion) was excluded because of developing a hypersensitive reaction to MA in the course of the treatment. Lesions assessed: 48 lesions in 47 participants. TC: 22, ILMA: 25 Mean age (SD): TC 23.5 years (16.4), ILMA 25.4 years (19.4) Sex (n (%)): TC: male 6 (27.2%), female: 16 (72.7%); ILMA: male 19 (76%), female: 6 (24%) Baseline data:
|
|
| Interventions |
Type of interventions:
Co‐intervention: in addition, at the beginning, and then every 2 weeks, cryotherapy with liquid nitrogen (−195°C) was performed for all lesions in both groups Duration of intervention: 3 months or until the lesion was cured, whichever came first Duration of follow‐up: 6 months after the termination of the treatment to evaluate the incidence of relapse (reported at 1 month, 2 months, 5 months) |
|
| Outcomes |
Clinical cure of the lesions:
Adverse effects Relapse: participants that developed with relapse of the lesions at the previously involved area Time points reported: clinical cure was reported when it occurs or for a maximum of 3 months. Adverse effects were assessed at follow‐up during therapy; they were the safety endpoints of the treatments, and 6 months after the termination of the treatment |
|
| Notes |
Study funding sources: this study was supported and funded by the Vice Chancellor for Research, Kerman University of Medical Sciences. The founder had no financial or proprietary interest in any material or method used in this study and had no role in study design, data collection and analysis, or preparation of the manuscript. Possible conflicts of interest: no conflict of interest |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised allocation was performed by blocked randomisation method with block size of 2 |
| Allocation concealment (selection bias) | Low risk | Randomised allocation was performed by blocked randomisation method and was prepared by the analyst of the research team who had no clinical involvement in the trial. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding was impossible |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessing the outcomes and data analyst were blinded to group assignment. The photographs were reviewed by the dermatologist of the research team who was blinded to study groups. The photographs did not contain any indicator that could help differentiate the group of the lesion. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The numbers of withdrawals were high in both groups: 31.25% (10/32) in TC group and 18.75 % (6/32) in the ILMA group. Participants were removed from the study arms because of their poor adherence to the trial protocol and lost to follow‐up. No intention‐to‐treat analysis was done. |
| Selective reporting (reporting bias) | Low risk | The study published reported our outcomes |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Aleppo, Syria Study period: June 2009 to December 2010 (19 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: not described Exclusion criteria: not described N randomised: 79 Withdrawals: 14 N assessed: 65. 46 participants (264 lesions) in the fluconazole group and 19 participants (102 lesions) in the placebo group Age (years): not described Sex: not described Baseline data: not described |
|
| Interventions |
Type of interventions:
Duration of follow‐up: not reported |
|
| Outcomes |
Healing rates: percentage of lesions 'cured' (follow‐up not reported) Adverse effects Time points reported: not described |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared This is an abstract |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned to treatment" Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Quote: "randomly assigned to treatment" Comment: no further information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about blinding was provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about blinding was provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Side effects were mild and similar in both groups" Comment: no further information on adverse effects was provided. Withdrawals: 14/79 (17.7%) and no ITT analyses were performed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Not possible to allocate to high or low risk. |
| Other bias | High risk | Sample size calculation and baseline comparability were not correctly reported |
| Methods |
Study design: prospective randomised controlled trial Setting/location: dermatology clinic in Saadi Hospital, an academic center in Fars Province, Iran Study period: December 2008 to March 2010 (15 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: participants that presented with skin lesions suspicious of CL, not receiving any previous treatment, positive for leishmaniasis with direct smears Exclusion criteria: pregnant and nursing women; children < 12 years old; lesions on the face; disease for more than 3 months; more than 5 active lesions; participants with any serious systemic disease or previous history of sensitivity to MA, allopurinol, or azithromycin; any difficulty in laboratory results before initial treatment (CBC diff, LFT, BUN, Cr); those who refused to sign the written informed consent form N randomised: 86 participants Withdrawals: 14 participants (6 in azithromycin + allopurinol group and 8 IMMA group) one lost follow‐up because of adverse effect N assessed: 71 participants (36 azithromycin + allopurinol group and 35 IMMA group) Mean age (SD; range): 38.2 years (12.6); range 16‐64. Azithromycin + allopurinol group: 39.7 years (12.6); IMMA: 36.8 years (12.8) Sex: 28 females and 53 males (azithromycin + allopurinol group: 13 females and 23 males; IMMA group: 15 females and 20 males Baseline data: most participants in azithromycin + allopurinol group had more than 3 lesions (72.3%) and in the IMMA group most participants also had more than 3 lesions (65.8%) |
|
| Interventions |
Type of interventions:
Duration of intervention: group 1, 2 months; group 2, 20 days. Co‐interventions: in case of any secondary bacterial infection, participants were with oral cephalexin for 10 days after which the antileishmanial was administered |
|
| Outcomes |
Cure rate, assessed as follows:
Adverse effects Time points reported: 2 months after completing treatment |
|
| Notes |
Study funding sources: funded by deputy of research, Shiraz University of Medical Sciences Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A total of 71 participants who met the inclusion criteria in the trial were randomly divided into two treatment groups according to simple even and odd number allocation" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Fourteen of 86 patients dropped out due to poor compliance (six patients in the combination therapy and eight in the Glucantime group). The number and reason for their withdrawal were almost the same in both treatment groups. One patient also developed GI complications and headache while taking the combination therapy of azithromycin and allopurinol; therefore, overall 71 subjects completed the study." Comment: no ITT analyses were performed. However, withdrawals accounted for <20% and were homogeneous among the treatment groups. |
| Selective reporting (reporting bias) | Low risk | Our primary outcomes (cure and adverse effects) were described in Methods and reported in Results |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: India Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica in the area Inclusion criteria: participants with cutaneous Leishmania confirmed by the presence of L tropica bodies in the slit skin smear stained with Leishman stain. Exclusion criteria: women of child‐bearing age N randomised: 20 Withdrawals: 0 N assessed: 20 (100%). Intervention group: 15, control group: 5 Age: range 14‐56 years. Baseline data: single or multiple lesions, the duration of the lesions varied from 4 to 16 weeks |
|
| Interventions |
Type of interventions:
Duration of follow‐up: 6 weeks, but 3 months for relapses assessment |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' at the end of treatment. The essential criteria for declaring the participant was cured was complete disappearance of the induration or redness in the nodular form and complete healing in the ulcerative form, accompanied by smear positivity conversion Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within 3 months Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: clinical cure: 4 weeks, clinical and parasitological cure: 6 weeks |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Proportion of 2 to 4:1 Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Selective reporting (reporting bias) | Unclear risk | All of the study's pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: India Study period: not described Sample size calculation: the number of subjects was determined by the standard method of Schork 1967 |
|
| Participants |
Type of Leishmania: L tropica in the area Inclusion criteria: demonstration of Leishmania from skin lesion by the slit smear technique. Exclusion criteria: pregnant women and children < 12 years old, suffering from any chronic illness, immunocompromised, allergic to sulphones, prior therapy for cutaneous Leishmania in any form, patients with scars of healed leishmanial lesions, lesions of > 4 months duration N randomised: 120. 60 in each group Withdrawals: 0 N assessed: 120 (100%), 60 in each group Age: range 15‐56 years Sex: 52 males/68 females Baseline data: the duration of the lesions ranged from 3 weeks to 3 months. Lesions were situated mainly on the exposed parts of the body (face, arms and feet). 46 participants (24 in dapsone group and 22 in placebo group) had a single lesion while 74 participants had multiple lesions (maximum 13). |
|
| Interventions |
Type of interventions:
Duration of intervention: every 12 h for 6 weeks Duration of follow‐up: 6 weeks |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' at the end of treatment Secondary outcome: adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: clinical response: days 15 and 45. Clinical and parasitological response: 6 weeks |
|
| Notes |
Informed consent obtained: yes Study funding sources: — Possible conflicts of interest: — |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "... and was randomly allocated to receive either tablets of dapsone (100 mg) or placebo tablets which were identical in appearance" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "... and was randomly allocated to receive either tablets of dapsone (100 mg) or placebo tablets which were identical in appearance" Comment: no further information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind therapeutic trial" Comment: participants looks like blinded but no description about personnel blinding were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind therapeutic trial" Comment: no further information about blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All relevant outcome data were provided |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: India Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica in the area Inclusion criteria: participants with localised CL and only smear positive cases Exclusion criteria: not being smear positive N randomised: 60, 20 in each group Withdrawals: 0 N assessed: 60 (100%), 20 in each group Age (years): ≥ 15 years Sex: not described Severity of illness: 8, 11, and 12 participants had multiple lesions in the itraconazole, dapsone and placebo groups respectively. |
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks Duration of follow‐up: 6 weeks, but 3 months for assessment of relapses |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment. Strict clinical and parasitological criteria were followed to asses cure. Adverse effects Time points reported: healing rates at the end of treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: India Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania:L major and tropica in the area Inclusion criteria: participants with localised CL. Demonstration of parasites from skin lesions by slit smear examination Exclusion criteria: women of child‐bearing age, children < 18 years old, participants suffering from any chronic illness, immunocompromised, prior therapy for CLs in any form, scars of healed leishmanial lesions, lesions of 4 months or more duration, participants showing abnormality in liver function tests N randomised: 20 Withdrawals: 0 N assessed: 20 (100%) Age: range 19‐62 years Sex: 15 males/5 females Baseline data: the duration of the lesions ranged from 2 weeks to 16 weeks, they were mainly seen on exposed parts of the body. 9 had a single lesion and 11 participants had a multiple lesion. |
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks Duration of follow‐up: 3 months |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' 3 months after treatment Secondary outcome: adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: clinical response: day 15. Clinical and parasitological response: 6 weeks |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were allocated randomly to receive capsules of itraconazole 100 mg or identical placebo". Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were allocated randomly to receive capsules of itraconazole 100 mg or identical placebo". Comment: no further information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind therapeutic trial" Comment: participants looks like blinded but no description about personnel blinding was provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind therapeutic trial" Comment: participants looks like blinded but no description about assessment blinding was provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes were reported |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Pakistan at dermatology departments of Combined Military Hospital, Kharian Camtonment, Combines Military Hospital, Quetta and Combined Military Hospital, Muzaffarabad Study period: January 2008 to December 2010 (35 months) Sample size calculation: assuming equal‐sized groups, keeping the equivalence trial design, standardised difference of 0.05 and power at 90%, sample size was calculated as 150 participants in each arm using Altman nomogram. A total of 300 participants were needed for the study and 24 extra participants were recruited to cater for dropouts. |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: men and women over 18 years of age having parasitologically proven CL requiring systemic therapy, willing for admission to hospital for the study and regular follow‐up visits, and consenting not to use any other treatment for CL during trial Exclusion criteria: pregnant, compromised immune system (i.e. diabetics or cancer patients), diffuse cutaneous or visceral leishmaniasis, complete or incomplete treatment with antimony compounds in the last 3 months, history of hepatic, renal, or cardiovascular disease N randomised: 324. Group 1: 151; group 2: 173 Withdrawals: not described N assessed: 324 (100%) Age: mean 27.9 years (SD 6.5), range 17‐48 years Sex: all were male soldiers Baseline data: most participants (39.2%) had a single lesion, but one participant had 15 lesions. Maximum lesions were found on exposed parts of body, arms and legs accounted for 42.3% and 47.2% of lesions respectively. Plaques were the most common morphological pattern seen (82.7%). Mean (SD) size of lesions at baseline was 28.8 mm (16.1). Group 1: 29.7 mm (16.4); group 2: 28 mm (15.8). Mean induration at baseline was 17.5 mm (11.6). Group 1: 17.7 mm (11.5); group 2: 18 mm (12.8) |
|
| Interventions |
Type of interventions:
Duration of intervention: 28 days maximum |
|
| Outcomes |
Successful treatment: was defined as complete re‐epithelialisation of the ulcer and disappearance of the induration, or reduction of more than 50% of the ulcer and the indurations areas in relation to the last clinical evaluation Adverse effects: serious adverse event was defined as life‐threatening, or prolongation of existing hospitalisation, or causing persistent or significant disability. Non‐serious adverse effects, not qualifying the above criteria were clinically judged by the investigator to be definitely related, probably related, possibly related, or not related to the trial medication. Time points reported: follow‐up phase lasted 6 months |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation through a random number table and allocation ratio being 1:1. |
| Allocation concealment (selection bias) | Unclear risk | "Randomisation sequence generation and allocation of medicine was done by lead investigators at the three research set ups" Comment: no information on the method of allocation concealment was provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded from the treatment groups; however, it is not clear how this was done. No placebo was provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "There were 18 dropouts due to various complications, 9 belonging to each group" Comment: ITT analyses were performed. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. The trial was registered with www.anzctr.org.au and the registration number is ACTRN12607000295448. Some outcomes registered in the protocol were not reported: successful treatment; therapeutic failure; reactivation |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: the Saudi Hospital in Sanaa, Yemen Study period: 21 months; participants recruited from June 2006 to June 2007 Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp Inclusion criteria: all had the clinical signs of cutaneous leishmaniasis and a majority of their cutaneous lesions were found smear positive for mastigotes. 4 participants with a negative smear preparation were diagnosed by tissue culture using NNN (Novey, McNeal and Nicolle) medium Exclusion criteria: skin lesions of more than 8 weeks duration, allergic to antimonial drugs, lactating or pregnant N participants randomised (lesions): N = 30: ILSSG, n = 10 participants (12 lesions), ILSSG + IMSSG, n = 10 (15), ILSSG + oral ketoconazole: n = 10 (13) Withdrawals: no N assessed: 30 (100%) Age: range 12‐50 years (mean 23.5, SD 14). Group 1: 12‐50 (24.3, SD 15); group 2: 13‐48 years (22.5, SD 14); group 3: 12‐49 years (25.1, SD 12) Sex (male/female): group 1, 5/5; group 2, 7/3; group 3, 4/6 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: in all groups, treatment was continued until clearance or for a maximum of 3 treatment cycles at 4 week intervals (12 weeks) Follow‐up: performed monthly for 6 months after the last treatment |
|
| Outcomes |
Clinical cure: the cure was indicated by complete re‐epithelialisation, the disappearance of oedema, induration and other signs of inflammation and a negative Giemsa‐stained direct smear of a scraping of the skin at the lesion site. In the 4 participants diagnosed only by a positive culture before enrolment, the cure was indicated by a negative culture at the end of the 12 weeks. Adverse effects Time points reported: 4, 8, 13 weeks. Adverse effects: at the end of treatment |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "A follow‐up assessment was performed by the treating clinician, the patient and by comparing the serial photographs" Comment: open trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes were reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: PCR‐proven L major cutaneous infection, age > 12 years, not using anti‐Leishmania therapies during the past 2 months, duration of lesions < 4 moths Exclusion criteria: pregnancy, breastfeeding, presence of lesions on ears and/or face, number of lesions >10, history of liver or kidney disease N randomised: 120 participants (fluconazole 200 mg: 60; fluconazole 400 mg: 60) Withdrawals: 2 in the 400 mg group N assessed: 118 participants Mean (SD) age: fluconazole 200 mg, 36.45 years (15.34); fluconazole 400 mg, 35.38 years (13.81) Sex (male/female): 65/55 (fluconazole 200 mg, 30/30; fluconazole 400 mg, 35/25) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks |
|
| Outcomes |
Complete healing: defined as complete re‐epithelialisation of the lesions at intervals of 2, 4 and 6 weeks Adverse effects Time points reported: 6 weeks |
|
| Notes |
Study funding sources: grant from Shiraz University of Medical Sciences, Shiraz, Iran Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were then randomized into two equal groups to receive either fluconazole 100 mg or 200 mg twice daily." Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 dropouts were reported in the fluconazole 400 mg group. ITT analysis were performed. |
| Selective reporting (reporting bias) | Unclear risk | Our primary outcomes (Complete healing and adverse effects) were described in Methods and reported in Results. |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica in the area Inclusion criteria: positive direct smear or documented skin biopsy, not pregnant or nursing at the time of therapy, no serious systemic illness, no sensitivity to antimonial drugs or AL Exclusion criteria: not having received treatment for at least 2 months before entry into the study. N randomised: 150. 50 in each group Withdrawals: 0 N assessed: 150 (100%). 50 in each group Age: mean age 14 years. Group 1 (AL): 14, group 2 (IMMA): 14.5, group 3 (AL + IMMA): 14 Sex: male/female: 69/81. 23/27 in each group Baseline data: the common sites of involvement were the face and extremities. By groups were as follows: oral AL: face: 62%; upper limbs: 34%; lower limbs: 4%, and trunk: nil. IMMA: face: 48%; upper limbs: 38%; lower limbs: 14%, and trunk: nil. AL + IMMA: face: 78%; upper limbs: 15%; lower limbs: 6%, and trunk: 2%. Most participants had plaque‐type, papular, and nodular lesions. By groups: AL: plaque: 64%, papule: 29%, and nodule: 7%. IMMA: plaque: 71%, papule: 18%, and nodule: 11%. AL + IMMA: plaque: 77%, papule: 19%, and nodule: 4%. Mean number of lesions: group 1, 1.5; group 2, 1.8; group 3, 1.4. Most had multiple lesions (≤ 7 lesions) Mean duration of lesions: group 1, 8 months; group 2, 7.4 months; group 3, 8 months |
|
| Interventions |
Type of interventions:
Duration of follow‐up: one month |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' at the end of treatment. The response to treatment was graded as excellent (reduction in the size of the lesion by at least 80% or complete clearance), good (reduction in the size of the lesion by 50%), and poor (minimal or no change in the lesion) Secondary outcome: adverse effects Time points reported: end of treatment and 2 and 4 weeks later |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients were randomly divided into three groups." Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "the patients were randomly divided into three groups." Comment: no further information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: the treatment it is not possible to blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no reference to the outcome assessment blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no missing outcome data |
| Selective reporting (reporting bias) | High risk | Comment: no reference to adverse effects |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Isfahan, Iran Study period: 15 months Sample size calculation: yes |
|
| Participants |
Type of Leishmania: L major is endemic in Isfahan Inclusion criteria: clinical and parasitological diagnosis of CL Exclusion criteria: pregnant or had > 3 lesions, ulcerative lesions, lesions with cartilage or lymphatic involvement or hypersensitivity to the drug N randomised: 96 participants, 48 in each group Withdrawals: 0 N assessed (lesions): 96 (190). 48 (95) in each group Age (years): age range 1‐48 years (mean age of 16 years old and a median of 14.5 years) Sex: male/female: 40/56 Baseline data: all lesions treated were papules or early nodules with mean diameter of about 4 mm. Mean number of lesions per person in each group: 1 |
|
| Interventions |
Type of interventions:
Duration of intervention: treatment was continued in both groups for 3 months or until complete recovery (return to the normal tissue texture without any atrophic changes or scar formation) Duration of follow‐up: one year |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' within 2 months after treatment. Complete recovery or cure was defined as re‐epithelialisation and return to normal tissue texture in less than 2 months, with no residual scar or relapse after follow‐up of up to 1 year Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within one year; prevention of scarring Time points reported: completely recovered (cured) (< 2 months). Healed (in 2–3 months) |
|
| Notes |
Study funding sources: Isfahan University of Medical Sciences (Pharmacy College and its Research Laboratory) and the Amin Leishmaniasis Research Group in Isfahan City Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "In the period of recruitment, […] they were randomised by fixed block random allocation". |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatment description do not appear as blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | In statistical methods section, quote "There was no blinding method used." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Afzalipour Hospital, Kerman University of Medical Sciences, Iran Study period: 1 year (2011‐12) Sample size calculation: a sample size of 40 participants per treatment group was planned, a probability of a type I error at alpha = 0.05 and beta = 0.1 to determine a 20% difference between topical terbinafine compared with control group |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: participants between 2‐60 years and without serious medical illness were included, not have received any other leishmanicidal treatment during last 3 months, duration of their lesions should be less than 6 months Exclusion criteria: cardiac, hepatic, renal and other systemic diseases; pregnant and nursing women; hypersensitivity to trial drugs N randomised: 80, 40 in each group Withdrawals: 0 N assessed: 80, 40 in each group Mean (SD) age: terbinafine: 16.3 years (14.8), IMMA: 20.74 years (19.4) Sex (n, %): terbinafine: male 18 (45); female 18 (45); the sex of 4 (10) participants was unknown. IMMA: male: 16 (40); female: 18 (45); the sex of 6 (15) participants was unknown Baseline data:
|
|
| Interventions |
Type of interventions:
Co‐interventions: Both groups received cryotherapy every 2 weeks for 4 weeks Duration of intervention: group 1: 4 weeks, group 2: 3 weeks Duration of follow up: patients were followed monthly for 3 months after the treatment. |
|
| Outcomes |
Clinical cure: clinical response was determined based on the following criteria:
Time to clinical cure: development of oartial and complete response to treatment in IMMA and terbinafine groups. Time points reported: the improving rate determined by measuring indurations at baseline, in the middle (day 10 for IMMA group and day14th for terbinafine group), and at the end of the study (day 21 for IMMA group and day 28 for terbinafine group) |
|
| Notes |
Study funding sources: Kerman University of Medical Sciences accepted the financial support of this study Possible conflicts of interest: the authors declare that there is no conflict of interest. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation sequence was obtained by the use of a randomisation table. |
| Allocation concealment (selection bias) | Low risk | A simple block randomisation list with a block size of 4 was accumulated by a team member who was not involved in the enlistment and follow‐up of the participants. The randomisation allocation concealment was carried out by sending the randomisation numbers in envelopes to a dermatologist who was responsible for giving the assigned treatment after each participant was en‐rolled. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Different administration of treatments: oral terbinafine and IMMA. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was an assessor blind trial in which the outcome assessor was unaware of the drugs used by the participants. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | High risk | The study published does not report 2 primary outcomes: adverse effects and recurrence |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Afzalipour hospital affiliated to Kerman University of Medical Sciences and Dadbin Health Center, Kerman/Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: — Inclusion criteria: age over 7 years, positive smear or biopsy, providing informed consent Exclusion criteria: pregnant or lactating, lesions on face, more than 5 lesions, duration longer than one year, size larger than 3 cm, any treatment in past month, history of allergy to MA or dapsone, systemic diseases, taking immunosuppressive drugs in past 6 months, lupoid or sporotrichoid forms N randomised: 73 Withdrawals: 5 N assessed (lesions): 68 (73). Group 1: 33 (35); group 2: 35 (38) Mean (SD) age: 29.6 years (15.4) in group 1, 31.6 years (20.4) in group 2 Sex (male/female): intervention group: 17/16, group 2: 14/17 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: until complete healing or max 16 weeks |
|
| Outcomes |
Healing response: complete healing (100% epithelialisation and loss of induration), moderate healing (50‐99% epithelialisation and loss of induration), no response (less than 50% epithelialisation and loss of induration) Time points reported: 16 weeks after beginning intervention, 1 year later for recurrence |
|
| Notes |
Study funding sources: Kerman University of Medical Sciences Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open study |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No reasons for missing outcome data |
| Selective reporting (reporting bias) | High risk | One outcome of interest in the review is not reported: adverse effects, so that it cannot be entered in a meta‐analysis |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Isfahan, Iran Study period: 6 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major endemic in the area Inclusion criteria: the presence of parasitologically confirmed lesion(s) of CL, aged 12‐60 years, and otherwise healthy on the basis of medical history and physical examination Exclusion criteria: women of childbearing age without adequate effective contraception; pregnant or breastfeeding; duration of lesions > 8 weeks; the presence of lesions on face, joints or near the mucous membranes; the presence of > 5 lesions or any lesion with a diameter > 5 cm; history of any antileishmanial therapy in the past 4 weeks N participants randomised (lesions): 72 (106). 36 (53) treated with zinc sulphate (ZS) and 36 (53) treated with ILMA Withdrawals participants (lesions): 37 (56). ZS group 23 (34): unresponsive: 12, lost to follow‐up: 5, non‐medical events: 2, protocol deviation: 2, complications: 1, patient request: 1. ILMA 14 (22): unresponsive: 2, lost to follow‐up: 10, non‐medical events: 1, patient request: 1 N assessed (lesions): 35 (50). 13 (19) treated with ZS and 22 (31) treated with ILMA Mean (SD) age: 20.2. years (9.6). ZS group: 18.1 years (6.1); ILMA: 22.3 years (11.2) Sex (male/female): 33/39. ZS group: 19/17; ILMA group: 14/22. Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: up to 6 weeks Duration of follow‐up: 5 weeks |
|
| Outcomes |
Healing rates: percentage of lesions 'cured' 5 weeks after treatment. Complete re‐epithelialisation of each ulcer with marked reduction in induration with or without scarring was considered as the main efficacy parameter. Amount of injection into each lesion Adverse effects: the pain experienced by the participant (using a verbal analogue scale on a 0‐10 scale) was recorded Time points reported: 6 weeks |
|
| Notes |
Study funding sources: this study was supported by a grant from Chancellery of Research, Tehran University of Medical Sciences. Possible conflicts of interest: none declared The plot and the figures were not matched accordingly |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Table of random numbers |
| Allocation concealment (selection bias) | Low risk | The randomisation sequence was concealed from the investigators until the data entry was completed and the data bank was locked. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Comment: the preparation and coding of drugs were done by a pharmacist outside of the research team and investigators were blinded to them |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High rate of dropouts (37/72, 54%). Missing outcome data imbalanced in numbers and reasons across intervention groups. ZS group: 23. ILMA group: 14. 'As‐treated' analysis done with substantial departure of the intervention received from that assigned at randomisation |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Mashad, Iran Study period: 18 months Sample size calculation: 45 participants per treatment group were needed to have 80% power to detect a significant difference in the expected cure rate of 50% in the vehicle group and the desired cure rate of 80% in the imiquimod‐treated group at week 8 with a type Ierror level of 0.05. To compensate for 20% estimated dropout, 54 participants would be required in each group. |
|
| Participants |
Type of Leishmania: L tropica endemic in the area Inclusion criteria: parasitologically proven cases of CL based on positive smear or culture, otherwise healthy participants Exclusion criteria: pregnant or lactating women, duration of the lesions > 6 months, number of lesions > 5, any lesions > 5 cm, history of any standard course of treatment with antimonials, history of allergy to antimonials, serious systemic illnesses, participation in any drug trials in the last 60 days N participants randomised (lesions): 119 (252). 59 (128) participants in the imiquimod group and 60 (124) in the vehicle group Withdrawals: 30 participants. Imiquimod group (n = 17): 9 lost to follow‐up, 4 inadequate efficacy, 3 protocol deviation, 1 consent withdrawal. Vehicle group (n = 13): 9 lost to follow‐up, 2 inadequate efficacy, 2 protocol deviation N assessed: 89. 42 participants in the imiquimod group and 47 in the vehicle group Mean (SD) age: 27.0 years (SD 1, range 12‐60). Imiquimod group: 7.4 years (13); vehicle group: 6.5 years (12) Sex: approximately 50%‐55% of the participants were female (35/59 in the imiquimod group, and 31/60 in the vehicle group) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: imiquimod and vehicle 3 times per week for 28 days, IMMA 20 mg/kg/d for 14 days Duration of follow‐up: 20 weeks after initiation of treatment |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' 3.5 months after treatment. Clinical cure of the participants, defined as more than 75% reduction in the size of lesions compared with baseline Secondary outcomes: the relapse rate (defined as a reappearance of lesions at the site or periphery of previously healed lesions or an increase in the size of lesions after initial improvement) was assessed 16 weeks after the end of treatment Adverse effects Time points reported: at the end of the treatment period (week 4) and 4 weeks later Intention‐to‐treat analysis of rates of complete re‐epithelialisation, clinical cure, and clinical improvement at weeks 4, 8,and 20 after initiation of treatment |
|
| Notes |
Study funding sources: this study was supported by the Small Grants Scheme for Operational Research in Tropical and Other Communicable Diseases from the Joint World Health Organization Eastern Mediterranean Region Division of Communicable Diseases and the Special Program for Research and Training in Tropical Diseases. Possible conflicts of interest: the funding source was involved in the study design, in the writing of the manuscript, and in the decision to submit the manuscript for publication, but not in the collection, analysis, or interpretation of data |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly divided into 2 groups according to a list made by a simple randomisation block design" |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomisation allocation concealment was performed by sending the randomisation numbers in envelopes to a pharmacist who was responsible for giving the assigned treatment after each eligible patient was enrolled." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatment unlikely to be blinded. This study was a single‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "To keep the trial blinded, the physicians who were responsible for evaluation of patients were uninvolved in the process of allocation and drug dispensing and were unaware of the drug used by the patients." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals: 30/119 (25.2%); no ITT analyses performed |
| Selective reporting (reporting bias) | Low risk | All relevant outcomes reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: 24 months Sample size calculation: yes |
|
| Participants |
Type of Leishmania:L major Inclusion criteria: CL confirmed with direct smear, age 5‐50 years, maximum number of lesions 3, duration of disease < 100 days Exclusion criteria: previous treatment for leishmaniasis, use of immunosuppressives, history of chronic systemic disease, lesions on face, pregnancy or lactating, age < 5 years, duration of disease > 100 days N randomised: 197 Withdrawals: 26 N assessed: 171. 96 were treated with garlic 5% cream and 75 with vehicle Mean age: garlic, 18.5 years; vehicle, 23.7 years Sex (male/female): garlic, 51/45; placebo, 38/37 Baseline data: not reported |
|
| Interventions |
Type of interventions:
Duration of intervention: both applied twice daily under occlusion with sterile gauze for 3 hours, for 20 days Duration of follow‐up: 60 days (after the 3 week treatment, participants were followed for another period of 40 days) |
|
| Outcomes |
Healing rates: percentage of participants 'cured' one month (40 days) after treatment Time points reported: yes |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared We only have the abstract. The author (A Khamesipour) was contacted and kindly agreed to extract the data from the original paper written in Persian. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | List generated by a computer |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low rates of dropouts (26/197; < 25%) |
| Selective reporting (reporting bias) | Unclear risk | All relevant outcomes reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Aleppo, Syria Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: up to 3 lesions diagnosed clinically and parasitologically as CL; absence of chronic or intercurrent systemic disease Exclusion criteria: pregnancy and no prior antimonial medication N randomised and withdrawals: not reported N participants assessed (lesions): 40 (75) ILMA: 20 (38); IFN‐γ: 20 (37) Mean age (range): ILMA: 21 years (6‐60); IFN‐γ: 27 years (12‐57) Sex (male/female): 17/23. ILMA: 9/11; IFN‐γ: 2/12 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: once weekly for 5 weeks Duration of follow‐up: 10 weeks |
|
| Outcomes |
Primary outcome: percentage of lesions 'cured' one month after treatment Secondary outcome: adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: weeks 3, 6, 10 |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers table method used |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information about allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information about blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no information about blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No losses to follow‐up were reported. |
| Selective reporting (reporting bias) | Low risk | Comment: all relevant outcomes provided |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Department of Dermatology, Isfahan Medical University, Iran Study period: 10 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp: due to a previous study in the area, it was likely that participants were infected with L major Inclusion criteria: proven leishmaniasis based on typical lesions of ACL and a positive direct smear. Number of lesions < 4 and duration of lesion < 12 weeks Exclusion criteria: cases of reinfection, pregnant or nursing women, those who had lesion on the face or joints and participants with sporotrichoid or erysipeloid lesions N randomised: 104. ILMA: 55, zinc sulphate (ZS): 49 Withdrawals: 38. ILMA: 20, ZS: 18. 13 due to occurrence of new lesions: 7 from ILMA group and 6 from ZS group; 6 participants with sporotrichoid spread: 4 from the ILMA group and 2 from the ZS group; 19 participants lost to follow‐up: 9 from the ILMA group and 10 from the ZS group N assessed: 66. ILMA: 35, ZS: 31 Mean age (range): ILMA: 11 years (2‐67); ZS: 12 years (3‐64) Sex (male/female): 50/54. ILMA: 14/21; ZS: 17/14 Baseline imbalances: sex distribution in each group Severity of illness: Mean duration of lesions (SD) (weeks): ILMA: 6.73 ±0.53 ;ZS: 7.64±0.12 . |
|
| Interventions |
Type of interventions:
In cases where there was a slight to mild improvement, another injection was given after 2 weeks Duration of intervention: 2‐6 weeks Duration of follow‐up: 6 weeks |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment. The results of treatment were graded according to the system of Sharquie 1997 slight = 1, mild = 2, moderate = 3, marked = 4, 5 = total clearance of the lesion and parasite not detected in the affected area by smear Adverse effects Time points reported: 2, 4, 6 weeks |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were treated randomly with intralesional injection of any of 2 similar 50‐mL vials marked A (MA) or B (ZS) by an independent physician. ZS solution was prepared by dissolving 2 g ZS (ZNSO4W 7H2O) per 100 mL of bidistilled deionised water. The blinding was unlikely to have been broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | The study's pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: not described Sample size calculation: the required sample size, 40 in each treatment group, was estimated, assuming one in every 4 cases would not provide full data, using the formula n = [Z1 − (a/2) + Z1 − b] 2 × [P1(1 − P1) + P2(1 − P2)]/(P1 − P2)2, and setting P1 (the paromomycin efficacy) to 0.7, P2 (the vehicle efficacy) to 0.3, a to 0.05, b to 0.1, Z1 − (a/2) to 1.96, and Z(1 − b) to 1.64 |
|
| Participants |
Type of Leishmania: L tropica and L major are responsible for hyperendemic CL in rural areas and endemic CL in parts of many cities in Iran Inclusion criteria: had the clinical signs of CL and all of their cutaneous lesions were found smear‐positive for amastigotes Exclusion criteria: cases who had first noticed a skin lesion > 3 months previously, had lesions on their face, had lesions with diameter of > 3 cm, had received previous treatment, or who were lactating or pregnant N randomised: 80, 40 in each group Withdrawals: 15. Intervention group: 10; control group: 5 N assessed: 65. Intervention group: 30; control group: 35 Age: range 8‐55 years. Mean intervention group: 21.4 years; control group: 21.5 years Sex (male/female): 33/32. Intervention group: 19/11; control group: 14/21 Baseline data: mean duration of lesions (years): intervention group: 1.65; control group: 1.75 |
|
| Interventions |
Type of interventions:
Duration of intervention: 30 days Duration of follow‐up: for clinical and parasitological follow‐up on day 30 and for only parasitological follow‐up on day 60 |
|
| Outcomes |
Healing rates: percentage of participants 'cured' one month after treatment, clinical and parasitological cure
Adverse effects Time points reported: days 30 (clinical and parasitological follow‐up) and 60 (parasitological follow‐up) |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The cases enrolled were assigned to two treatment groups, placebo or paromomycin, using computer‐based randomisation." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study was described as "double blind" but no description about allocation method was given. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding of outcome assessment described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No data about dropouts were given |
| Selective reporting (reporting bias) | Low risk | Relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Saudi Arabia (Bahrain) Study period: 12 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: proved positive for Leishmania parasites (amastigotes) on microscopic examination or biopsy Exclusion criteria: not described N randomised: 62. Rifampicin: 46; placebo: 16. Rifampicin: 2 groups were analysed; group 1a (children aged 3‐11 years ‐ mean age, 7.5 years), and group 1b (adults aged 12‐65 years ‐ mean age, 33 years). 32 participants enrolled in group 1a and 30 in group 1b. Out of these, 8 participants in each group (16 in total) served as control to receive the placebo. Withdrawals: 21 participants lost to follow‐up. Rifampicin: 12; placebo: 9 N assessed: 41. Rifampicin: 34; placebo: 7. Number participants imbalanced between groups Age: range 3‐65 years; mean 20 years Sex (male/female): 43/19 Baseline data: the duration of the lesions varied between 1 and 12 months (mean 2.6 months). The lesions of CL were single or multiple and were mostly over the extremities (upper limbs 51% and lower limbs 38%) and to a lesser extent on the face 30%. Most of the lesions were active being nodular, nodule‐ulcerative, or ulcerative. 2 of the participants were members of the same family. However, the rest of the participants had a negative family history. |
|
| Interventions |
Type of interventions:
Duration of intervention: 2 equally divided doses during meals for 4‐6 weeks Duration of follow‐up: 3 months |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' 3 months after treatment Secondary outcomes: duration of remission and percentage of people with treated lesions that recur up to 3 months of follow‐up Adverse effects |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Comment: "Randomized" but no further information was provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Each subject in the control group was given placebo, which was supplied, in capsules/suspension identical in shape and colour to that given in the rifampicin group." Participants look blinded but no information about personnel were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: "Double‐blinded" but no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There is a 38.87% of follow‐up, the percentage is very different between the groups, and it isn't explained. |
| Selective reporting (reporting bias) | High risk | No adverse effects data available |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Disease and Leishmaniasis Research Center of Isfahan (SDLRC), Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: not described Exclusion criteria: not described N randomised: 140 participants Withdrawals: 64 N assessed: 76 participants (47 with ILMA and topical Cassia fistula fruit gel and 29 participants in vehicle group) Age: not described Sex: not described Baseline data: not described |
|
| Interventions |
Type of interventions:
Duration of intervention: not described |
|
| Outcomes |
Cure: complete cure, partial cure and treatment failure Adverse effects (itching and erythema) Time points reported: 12 weeks |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared We only have the abstract |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "140 patients with cutaneous leishmaniasis referring to Skin Diseases and Leishmaniasis Research Center of Isfahan (SDLRC) were randomly allocated in two groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: the vehicle came exactly in the same colour and shape as the drug and the application procedure was also the same. We think the outcome is unlikely to be influenced by lack of blinding of personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There is no information about withdrawals. |
| Selective reporting (reporting bias) | Low risk | Our primary outcomes (cured and adverse effects) were described in the Methods and reported in the text. |
| Other bias | High risk | Sample size calculation, inclusion criteria, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: double‐blind, randomised controlled study Setting/location: Isfahan, Iran Study period: January 2009 to February 2010 (13 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: met the histological criteria for presence of parasite, age > 5 years, acute leishmanial lesions smaller than 5 cm², fewer than 5 lesions, more than 3 months of disease duration Exclusion criteria: pregnancy, women of childbearing age, history of administration of immune suppressive drugs in the last 6 months or anti‐leishmanial drugs in the last month N randomised: 60 participants, 30 in each group Withdrawals: 0 N assessed: 60, 30 in each group Mean (SD) age: 25.29 years (4.01). Achillea millefolium 5%: 25.8 years (3.7); vehicle: 24.6 years (4.2). Sex: 15 (25%) female; 45 (75%) male. A millefolium: 7 (23.3%) female, 23 (76.6%) male; vehicle: 8 (26.6%) female, 22 (73.3%) male) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 4 weeks Co‐interventions: all participants received 4 weekly an injections of MA (Glucantime, Paris, France) at a dose of 20 mg/kg |
|
| Outcomes |
Definition: complete or partial cure of lesions Time points reported: week 12 (visits months ‐ 3) |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "During this double‐blind randomised study, 60 patients were randomised into two treatment groups by using random allocation computer software." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "During this double‐blind randomised study ..." Quote: "Both gels were identical in terms of the colour and consistency." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up were reported, therefore ITT analyses performed |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to judge |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised parallel clinical trial Setting/location: Skin Diseases and Leishmaniasis Research Center (SDLRC), Isfahan, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: 6‐60 years old, with positive results of leishmaniasis smear test Exclusion criteria: size of lesion > 3 cm, > 5 lesions, lesion duration > 12 weeks, lesion located on the eyelid or < 2 cm away from the eye, pregnant or lactating N randomised: 165, 55 in each of 3 treatment groups Withdrawals: 7; 3 (5.5%), participants were in the concentrated boiled extract group and 2 (3.6%) participants in the other groups withdrew from the study due to the allergic reaction to the medications N assessed: 158. Concentrated boiled extract of Cassia fistula: 52; hydroalcoholic extract of C fistula: 53; ILMA: 53 Mean (SD) age: boiled extract of C fistula: 20.6 years (12.4); hydroalcoholic extract of C fistula: 19.8 years (11.5); ILMA: 22.9 years (13.6) Sex (n,(%)): boiled extract of C fistula: male: 35 (63.6), female: 20 (36.4); hydroalcoholic extract of C fistula: male: 29 (52.7), female: 26 (47.3); ILMA: male: 25 (45.5), female: 30 (54.5) Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: until complete resolution of the lesion (complete epithelialisation) or for a maximum duration of 4 weeks. Follow‐up: 3 months after completing the project, they were examined every month to assess the recurrence of the lesions |
|
| Outcomes |
Clinical and parasitological cure: lesions were considered completely cured (improvement), if the participants achieved both clinical and parasitological resolutions (negative results of direct smear). Participants were considered resistant to the treatment, if no clinical changes were observed in the lesion or it was worsened. Time to healing: comparison of complete cure time of the cutaneous Leishmaniasis lesions between 3 groups Adverse effects: allergic reaction to the medications Time points reported: week 1, 2, 3, 4, 16, complete cure |
|
| Notes |
Study funding sources: the study was self‐funded Possible conflicts of interest: there was no conflict of interest |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The participants were randomly allocated to 3 groups using table of random numbers. |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Different administration of treatments: 2 groups: apply the extract‐soaked gauze; third group: ILMA |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The numbers and reasons (allergic reaction to the medications) of withdrawals were similar and small in 2 of the groups 3.5% (3/28) in concentrated boiled group vs 3.6% (2/28) in the other groups. |
| Selective reporting (reporting bias) | Low risk | The study published reports our primary outcomes: cured and adverse effects |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Leishmania Clinic of the German Medical Service (GMS), Darwaze‐e‐Lahory, 1st district, Kabul, Afghanistan Study period: 36 months (2004‐7) Sample size calculation: sample size was calculated based on the primary outcome (i.e. complete wound healing). A number of 40 participants each was calculated to be necessary to obtain a power of 80% (significance level 5%). A presumed dropout rate of 20% led to a minimum total enrolment of 100 participants |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: male or female participants of the GMS Clinic; 5 years of age or older; with non‐ulcerated (papule/nodule) or ulcerated lesions (ulcer with or without crust); presence of at least one parasitologically confirmed CL lesion and no prior history of leishmaniasis and/or anti‐parasitic treatment Exclusion criteria: participants below age 5; with the presence of CL lesion(s) on or immediately adjacent to the nose, lips or eyes; pregnancy, or serious co morbidities N randomised: 135. Group 1: 73; group 2: 62 Withdrawals: due to protocol breach: group 1, n = 14; group 2, n = 8. Lost to follow‐up: group 1, n = 21; group 2, n = 22 N assessed: 113 (83.7%): group 1: 59 (80.82%); group 2: 54 (87.09%). Mean age (range): group 1: 18.62 years (5‐66); group 2: 18.16 years (6‐63) Sex (male/female): 93/42; group 1: n = 51/22; group 2: n = 42/20. Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: time to wound closure Co‐interventions: systemic antibiotics were only given if bacteria were detected and the wound infection was severe. Neither local nor systemic antifungal therapy was allowed to exclude anti‐protozoal effects on the Leishmania infection |
|
| Outcomes |
Primary outcome ‐ time to wound closure (days): the time needed for complete epithelialisation of the lesion wound was defined and photodocumented. Secondary outcome ‐ microbiological cure: the parasite load (Leishmania/g) of the lesions was taken prior to electrocauterisation (1st time), after wound closure (2nd) and after 6 months (3rd) Adverse effects: such as bacterial or fungal superinfections of the wounds, the formation or scars and the rate of re‐ulcerations were monitored during the treatment and follow‐up period |
|
| Notes |
Possible conflicts of interest: not described Study funding sources: this study was supported by the German‐Academic Exchange Service (travel grants to KWS, AFJ and FM), the Senior Expert Service (travel grants to KWS) and the Interdisciplinary Center of Clinical Research at the Universitätsklinikum Erlangen (IZKF, project A49 to US and CB). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were randomly assigned with the help of a GMS computer at a ratio of 1:1" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "RS and KWS were responsible for the enrolment of the patients and their assignment to the treatment groups." Comment: not further information was provided regarding the method for allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The participants, the medical personnel treating and evaluating the patients, the investigators analysing the clinical course of treated lesions, as well as the lab members performing the parasitological analyses were blinded. The preparation of the two different gels (marked as jelly A and B), which could not be visually distinguished, was performed by a technician who was not involved in the treatment of patients". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The participants, the medical personnel treating and evaluating the patients, the investigators analysing the clinical course of treated lesions, as well as the lab members performing the parasitological analyses were blinded. The preparation of the two different gels (marked as jelly A and B), which could not be visually distinguished, was performed by a technician who was not involved in the treatment of patients". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 14 participants of group 1 and 8 participants of group 2 were retrospectively excluded due to deviations from the study protocol. For the follow‐up analysis, 6 months after wound healing, only 70 participants could be included, as 21 participants of group I and 22 participants of group II did not observe their appointments. |
| Selective reporting (reporting bias) | Low risk | Main outcomes were included. Trial was registered at clinicaltrial.gov: NCT00947362 |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: double‐blind, randomised placebo‐controlled clinical trial Setting/location: south of Iran Study period: October 2007 to January 2009 (15 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major and L tropica Inclusion criteria: men and women aged at least 12 years old, positive smear and/or polymerase chain reaction (PCR) for CL, cutaneous lesions of less than 4 months' duration Exclusion criteria: pregnancy and lactation, facial lesions, lesions of more than 4 months' duration, complete or incomplete treatment with a systemic or topical medication within the previous month, drug sensitivity N randomised: 100 participants Withdrawals: 37 participants N assessed: 63 participants (36 in treatment and 27 in control group) Age: range 12‐62 years (mean 32.6 (SD 12) in the treatment group and 33.1 (14) in the vehicle group) Sex (male/female): 35/28 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 12 weeks Co‐interventions: simultaneously, cryotherapy once a week and aqueous cream containing 3% salicylic acid (emulsifying ointment 30 g, phenoxyethanol 1 g and freshly boiled and cooled water 69 g) |
|
| Outcomes |
Clinical response:
Adverse effects Time points reported: 12 weeks |
|
| Notes |
Study funding sources: not described Possible conflicts of interest: no conflicts of interest |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "This investigation was designed as a 12‐week, double blind, N randomised, placebo‐controlled study and its protocol was approved by the ethics committee of the vice chancellor of research affairs of Shiraz University." Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The identically coded and packaged creams were applied two times daily." Quote: "The study was blinded to both the patients and the assessing physician." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study was blinded to both the patients and the assessing physician." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 100 participants were enrolled but only 63 completed the study. Reasons for withdrawal were not reported. |
| Selective reporting (reporting bias) | Low risk | Our primary outcomes (cure and adverse effects) were described in Methods and reported in Results |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Center for Research & Training in Skin Diseases & Leprosy, Tehran University of Medical Sciences, Tehran, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica and L major Inclusion criteria: the participants with CL who attended to Center for Research & Training in Skin Diseases & Leprosy, Tehran University of Medical Sciences, Tehran, Iran. The diagnosis was based on clinical findings and physical examination and confirmed by Giemsa‐stained direct smear of the lesion Exclusion criteria: ore than 5 lesions; lesions greater than 5 cm in diameter; received anti‐CL treatment during the previous month; lesions on the face and around vital organs; diabetic individuals; pregnant, lactating female participants; subjects with active infectious diseases N randomised: 85. Group 1: mesotherapy administration of meglumine antimonate: 41. Group 2: ILMA injection: 44 Withdrawals: 25. Group 1: 11 (7 lost to follow‐up, 3 inadequate efficacy, 1 allergic reaction). Group 2: 14 (10 lost to follow‐up, 2 inadequate efficacy, 2 allergic reaction) N assessed: 60 (70.59%). Group 1: 30 (73.17%), group 2: 30 (68.18%) Mean (SD) age: Group 1: 25.24 years (16), group 2: 24.32 years (15.26) Sex (male/female): Group 1: 19/11, group 2: 17/13 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks or until complete cure |
|
| Outcomes |
Clinical cure: 3 categories were defined for ulcer healing: complete cure: complete re‐epithelialisation of the ulcer with loss of induration; partial cure: 25 to 99% re‐epithelialisation of lesions; failure: less than 25% re‐epithelialisation of lesions. Improvement of the lesions: ulcer improvement (cm), induration improvement (cm), induration improvement (cm), speed of improvement (cm/week), speed of erythema improvement (cm/week), scare size (cm) Microbiological cure: Leismanian body: negative, positive. Pain severity during injection: visual analogue scale (VAS; 0 to 10). Amount of drug used (mL/cm²) and number of treatment sessions Adverse effects Time points reported: 3 months after treatment. Adverse effects: after the each treatment session. |
|
| Notes |
Study funding sources: this study was supported by a research grant from Center for Research & Training in Skin Diseases & Leprosy, Tehran University of Medical Sciences, Tehran, Iran Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were randomised according to a computer generated simple randomisation list to be treated with intralesional injection of MA using either the classic method or mesotherapy gun." |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 14/44 participants with the conventional technique and 11/41 with mesotherapy gun withdrawn the study (29.4%) |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available; not registered in a trial registry; SD not provided for pain severity (VAS) Insufficient information to permit judgment |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Bam and Mashhad in Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: parasitologically proven cases of CL; otherwise healthy subjects on the basis of medical history, physical examination and results of blood tests; age 12–50 years; body weight >40 kg; willingness to participate in the trial and signing the informed consent Exclusion criteria: not described N randomised: 138 participants (75 in IMMA and 63 in miltefosine) Withdrawals: 33 in IMMA and 32 in miltefosine N assessed: 42 in IMMA arm and 31 in miltefosine Age (years): not described Sex: not described Baseline data: not described |
|
| Interventions |
Type of interventions: Group 1: oral miltefosine 2.5/kg/d for 28 days Group 2: IMMA 60 mg/kg/d for 14 days Duration of intervention: not described Co‐interventions: not described |
|
| Outcomes |
Healing of their lesions and occurrence Adverse effects Time points reported: participants were evaluated weekly for 28 days and then 1 month later |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote:"This open N randomised controlled trial was carried out in Bam and Mashhad in Iran …" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote:"This open N randomised controlled trial was carried out in Bam and Mashhad in Iran …" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote:"This open N randomised controlled trial was carried out in Bam and Mashhad in Iran…" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Losses to follow‐up quite high (around 50%) Intervention group: 32/63= 50.8% Control group: 33/75= 44% |
| Selective reporting (reporting bias) | Unclear risk | It is an abstract. At least they reported complete cure of cases and adverse effects. |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: randomised, assessor‐blind, controlled clinical trial Setting/location: Kashan, Tehran, Iran Study period: September 2008 to April 2010 (19 months) Sample size calculation: 56 lesions per treatment group were needed to have 80% power to detect a significant difference in the expected cure rate of 60% in the ILMA alone group and the desired cure rate of 85% in the ILMA and either silver or non‐silver dressing‐treated group at week 10 with a type I error level of 0.05. Compensating for a 20% loss‐to‐follow‐up, recruiting 68 (round up to 70) lesions per treatment group looked reasonable. |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: parasitologically confirmed cases of CL based on positive smear and/or culture; otherwise healthy subjects on the basis of medical history; age of 12‐60 years; willingness to participate in the study and signing the informed consent form (by the participant or his/her parent/guardian in cases younger than 18 years). Exclusion criteria: pregnant or lactating women; duration of lesion more than 3 months; number of lesions more than 5; ulcer size greater than 5 cm in largest diameter; history of receiving full course standard treatment (antimonials); history of allergy to MA or silver; serious systemic illnesses (as judged by the physician); participation in any drug trials in the last 60 days; indication for systemic treatment with MA; presence of secondary bacterial infection of the lesion according to clinical appearance N randomised: 83 participants (158 lesions) ILMA: 26 participants (45 lesions); ILMA + non‐silver dressing: 26 participants (53 lesions); ILMA + silver dressing: 31 participants (60 lesions) Withdrawals: 10 participants (18 lesions) N assessed: 73 (140 lesions) Mean (SD) age: 28.81 years (14.45). ILMA: 32.88 years (12.92); ILMA + non‐silver dressing: 30.31 years (15.41); ILMA + silver dressing: 24.13 years (13.97) Sex: male 39; ILMA: male 8; ILMA + non‐silver dressing: male 11; ILMA + silver dressing: male 20 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks |
|
| Outcomes |
Clinical cure (complete healing defined as more than 75% reduction, clinical improvement defined as 50%–75% reduction, and no response to treatment defined as less than 50% reduction in the size of the lesion compared with baseline) Relapse: defined as a reappearance of lesions at the site or periphery of previously healed lesions or an increase in the size of lesions after initial improvement was assessed 5 months after the termination of treatment Adverse effects: itching and burning, exudation, oedema, and dermatitis Time points reported: oucomes were assessed at the end of the treatment period (end of week 6), then at 4 weeks and 5 months after the last treatment session |
|
| Notes |
Study funding sources: the budget of the research project has been provided by the Vice‐Chancellery of Research of Tehran University of Medical Sciences. Possible conflicts of interest: the authors have declared that no competing interests exist. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A random sequence generated by using the online software Random Sequence Generator, which is available at URL: www.random.org [22]. It was done by an investigator with no clinical involvement in the trial. R. Talaee was responsible for enrolment of the patients." |
| Allocation concealment (selection bias) | Low risk | Quote: "The method for randomisation concealment was to use sequentially numbered, opaque, sealed envelopes (SNOSE). The envelopes were kept in a safe box, which was only accessible to A. Khatami who was responsible for assigning the patients to the interventions." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An assessor who was blinded to the type of treatment visited the patients at weekly intervals during the treatment period and 1‐ month and 5 months after the last treatment session" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An intent‐to‐treat analysis was performed at 2 time points (end of the treatment period (day 42) and one month later (day 72)). The numbers of and the reasons for withdrawals were not significantly different between the 3 groups: 11% ILMA group; 19% ILMA + non‐silver and 6.45% ILMA + silver |
| Selective reporting (reporting bias) | Low risk | Comment: all relevant outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: India Study period: 5 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: participants with a confirmed diagnosis of L tropica. The diagnosis was confirmed by demonstrations of L tropica bodies (amastigote stage of L tropica under oil immersion). Exclusion criteria: no mention about inclusion or exclusion criteria N randomised: 50. Rifampicin 1200 mg/d orally: 25; placebo: 25 Withdrawals: 4. Rifampicin 1: 2 (lost to follow‐up); placebo: 2 (withdrew due to exacerbation of lesions) N assessed: 46. Rifampicin: 23; placebo: 23 Mean (SD) age: rifampicin 29.54 years (17.34); placebo: 26.96 years (10.96) Sex (male/female): rifampicin: 10/13; placebo: 9/14 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 2 divided doses for 4 weeks Duration of follow‐up: 4 weeks Co‐interventions: the 2 withdrew due to exacerbation of lesions in placebo group and were treated by another therapeutic regimen |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment: a cure was defined as complete healing and disappearance of the lesion or reversible hypopigmentation at the site of lesion. Incomplete or partial healing was defined as a reduction in the size of a lesion and the absence of parasites on smear. A treatment failure or non‐healing was defined as the absence of any change in the lesion and persistence of parasites on smear Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: at the end of the treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Comment: "Randomized" but no further information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: "Double‐blinded" but no further information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: "Double‐blinded" but no further information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups |
| Selective reporting (reporting bias) | Low risk | Comment: all relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: hospital, Bikaner, India Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: participants with a confirmed diagnosis of anthroponotic CL. Exclusion criteria: — N randomised: 50. Intervention group: 25, placebo group: 25 Withdrawals: 6 lost to follow‐up. Intervention group: 2, placebo group: 4 N assessed: 44. Intervention group: 23, placebo group: 21 Age: intervention group: n = 12, 16‐20 years; n = 5, 21‐30 years; n = 3, 31‐40 years; n = 2, 41‐50 years; n = 2, 51‐60 years; n = 0, 61‐70 years. Placebo group: n = 10, 16‐20 years; n = 4, 21‐30 years; n = 5, 31‐40 years; n = 3, 41‐50 years; n = 2, 51‐60 years; n = 1, 61‐70 years Sex (male/female): 34/16. Intervention group: 17/8; placebo group: 17/8 Male predominance was probably due to the habit of sleeping in open spaces outside the house without mosquito nets and improper clothing during night when the sandflies are active. Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks Duration of follow‐up: 6 weeks |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment: complete healing; partial response; no response Time points reported: at the end of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized" Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Comment: "Randomized" but no further information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: "Double‐blinded" but no further information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: "Double‐blinded" but no further information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across group |
| Selective reporting (reporting bias) | High risk | Comment: no adverse effects information reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Saudi Arabia Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania:L major Inclusion criteria: only participants in whom parasites (amastigotes) were demonstrated were enrolled Exclusion criteria: pregnancy, chronic illness, immunocompromised or hyper allergic reaction to the trial drugs, treatment with regular medications such as antituberculous agents and steroids, which might have affected specific therapy, treatment with anti‐leishmanial drugs within the previous months, the presence of scars of previously treated lesions N randomised: 58. 2% miconazole cream: 27; 1% clotrimazole cream: 31 Withdrawals: 4: 2% miconazole cream: 4 (lost to follow‐up); 1% clotrimazole cream: 0 N participants assessed (lesions): 54 (151). 2% miconazole cream:n23 (62); 1% clotrimazole cream: 31 (89) Mean (SD) age: 2% miconazole cream: 19.5 years (15.4); 1% clotrimazole cream: 21.5 years (12.8) Sex (male/female): 42/12. 2% miconazole cream: 17/6; 1% clotrimazole cream: 25/6 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: twice daily for 30 consecutive days Duration of follow‐up: 30 days |
|
| Outcomes |
Healing rates: percentage of lesions 'cured' at the end of treatment. Response to treatment was based on results of repeated parasitologic evaluation and/or clinical changes using the same parameters as in the follow‐up. On this basis, response to treatment was graded into 4 categories:
Adverse effects Time points reported: at the end of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was carried out by using a table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients admitted into the trial were given unlabeled tubes containing either 1% clotrimazole cream or 2% miconazole" Comment: we think that the outcome is unlikely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Of 27 participants treated with 2% miconazole, 4 defaulted and were lost to follow‐up; they were excluded from analysis" (4/27 = 14.28%) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but he published reports probably include all expected outcomes. |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised, prospective Setting/location: dermatology department of Qaem University Hospital, Mashhad, Iran Study period: from October 2004 through November 2005 (13 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not reported (although they reported that CLL is endemic and L tropica and, less commonly, L major are the most prevalent species) Inclusion criteria: aged > 2 years with CL of < 6‐months' duration; no treatment of the current infection during the last 3 months; and no history of a renal or liver disease Exclusion criteria: pregnant women; history of intolerance or allergy to Glucantime or macrolides; simultaneous use of antacids containing Al and Mg, theophyline, phenytoin, barbiturates, carbamazepine, and cyclosporin N randomised: 49: azithromycin: 22 (35 lesions), IMMA: 27 (58 lesions). Withdrawals: 2 (azithromycin group) N assessed: 47 participants; 20 (29 lesions) in the azithomicin group and 27 (58 lesions) in the IMMA group. Mean (SD) age: range 4‐70 years; azithromycin: 21.77 years (17.13) and IMMA: 22.78 years (13.17) Sex (male/female): 24/25; azithromycin: 8/14; IMMA: 16/11 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: up to 4 months (azithromicin) and 20 days (IMMA) Co‐interventions: participants showing no favourable response after 4 months of azithromycin therapy would receive IMMA at the same dose as the other group, at the end of the study. |
|
| Outcomes |
Clinical cure: clinical response was determined on the basis of the lesions' size, border, induration, and infiltration of papulonodular lesions; ulcerative lesions were evaluated on the basis of the ulcer depth, diameter, crusting, and the extent of re‐epithelialisation
Recurrence Adverse effects Time points reported: participants in the azithromycin group were evaluated monthly up to a maximum of 4 months, but participants in the IMMA group were evaluated both at the end of the treatment period (day 20) and 45 days later. Recurrence was reported after the 16‐week follow‐up period. |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The 49 patients who met our inclusion criteria were randomly divided into two groups." Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 48 of 50 participants randomised completed the study. 2/20 participants of the azithromycin group withdrew because of adverse effects. Applications of study drugs were conducted according to the protocol |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Dermatology Clinic of Qaem Hospital in Mashad, Iran Study period: 10 months Sample size calculation: to detect a 22% difference in the cure rate between cryotherapy and ILMA groups and assuming a 57% cure rate in the cryotherapy group, with a 65% power and a 10% 2‐sided type I error, 40 persons were needed in each group. The 79 participants who met our criteria were randomly divided into 2 groups: 40 participants received cryotherapy and 39 participants received ILMA |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: positive direct skin smears for CL, were ≤ 13 years of age, had visited the dermatology clinic from September 2006 through June 2007, and had lesions with a duration of < 12 weeks to exclude any natural self‐healing during follow‐up Exclusion criteria: > 13 years of age, had a lesion history > 3 months, allergic to antimonial drugs, simultaneously using any other therapeutic method N randomised: 79. Cryotherapy: 40, ILMA: 39 Withdrawals: 7: cryotherapy group: 4, ILMA: 3 N assessed: 72 (91.14%): cryotherapy group: 36 (90%), ILMA: 36 (92.31%) Mean (SD) age: 6.8 years (3.4) in cryotherapy group and 6.2 years (3.4) in ILMA group Sex (male/female): cryotherapy: 18/22; ILMA: 20/19 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks |
|
| Outcomes |
Clinical cure: complete cure was defined as full re‐epithelialisation; disappearance of oedema, induration, and other signs of inflammation; and a negative direct skin smear result. Score:
Time to healing Adverse effects Time points reported: at the end of treatment and 6 months (clinical cure); at 4, 5 and 6 weeks (time to healing); at the end of treatment, after 6 weeks (adverse effects) |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Open trial; lesions of all participants were photographed. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4 participants in the cryotherapy group and 3 participants in the ILMA group were excluded because they received 2 medications simultaneously, did not complete the treatment course, did not visit the clinic for follow‐up 6 months later, or changed their address and were lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | Protocol not available; not registered; however, pre‐specified outcomes of the review were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Dermatology Clinic of Ghaem Hospital, Mashhad, Iran Study period: March 2008 to September 2010 (30 months) Sample size calculation: to detect a 35% difference in the cure rate between the 2 groups of liposomal AmB and ILMA, assuming a 90% cure rate in liposomal AmB group with a 95% power and a 5% 2‐sided type I error, 36 subjects were required in each group. Also regarding a loss to follow‐up of 20%, this number was raised to 50 participants for each group |
|
| Participants |
Type of Leishmania: L tropica, L major Inclusion criteria: cutaneous leishmaniasis approved by either a direct smear stained with Giemsa or a positive skin biopsy of lesions with less than 6‐month duration, in cases with a previous history of anti‐leishmaniasis therapy, a 3‐month treatment‐free interval from the last treatment course Exclusion criteria: pregnancy, breastfeeding, taking any other specific treatment while participating in the study, past medical history of any local or systemic disease during the last 2 months, a significant underlying disease such as cardiac, renal, or liver dysfunction N randomised: 110. Group 1: 50; group 2: 60 Withdrawals: 34 did not adhere. Group 1: 11; group 2: 23 N assessed: group 1: 39 (78%); group 2: 37 (61.7%) Mean (SD) age: group 1: 20.54 years (18.72); group 2: 25.30 years (15.70) Sex (male/female): group 1: 23/27; group 2: 21/39 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 8 weeks |
|
| Outcomes |
Clinical response: determined on the basis of the lesion induration size of and the extent of re‐epithelialisation in ulcerative ones. Scales: clinical response:
Adverse effects Time points reported: at the end of treatment, and 6 months after termination. Adverse effects were reported at the end of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The 110 patients who met our inclusion criteria were randomly divided into two groups." Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT analyses performed Losses to follow‐up per group: Intervention group: 11/50 (22%) Control group: 23/60 (38.3%) |
| Selective reporting (reporting bias) | Unclear risk | Number of lesions higher in the liposomal ampB group (P < 0.05) whereas number of papule and plaque lesions where higher in the ILMA group (P = 0.011) Outcomes described in the methods were reported in the results section. However, only one time point was reported Protocol not available; not registered in a clinical trial registry |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: school children, Sudan Study period: 80 days Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp not specified but L major was presumed to be the causative parasite. Inclusion criteria: all positive smears Exclusion criteria: children who had received diminazene aceturate or previous treatment in hospital N randomised: 70. 35 in each group Withdrawals: 8. Berelin: 3; Savlon: 5. 2 children in Berelin group were not enrolled because they received diminazene aceturate before. 2 in Savlon group were excluded because they were treated for CL in the hospital N assessed: 62. Berelin: 32; Savlon: 30 Mean (SD) age: Berelin: 9.3 years (4.6); Savlon: 10.4 years (2.8) Sex (male/female): Berelin: 18/15; Savlon: 20/13 Baseline imbalances: comparability with regard to sex, age, number, size, duration and location of ulcer, and previous treatment taken
|
|
| Interventions |
Type of interventions:
Duration of treatment: 50 days Duration of follow‐up: one month |
|
| Outcomes |
Primary outcome: percentage of participants 'cured' at the end of treatment. Cure was defined when the skin lesion was completely closed and covered with scar tissue, without the possibility to evoke secretions on pressure and if this apparent cure was sustained for at least 2 weeks Secondary outcomes: duration of remission and percentage of people with treated lesions that recur 20 to 35 days after cure Adverse effects Time points reported: at the end of treatment and at the end of follow‐up. |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | A double‐blind study design was attempted, but could not be sustained as Savlon is a well‐known product and soapy on application |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low rates of dropouts (8/70, < 2.5%) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is probably that the published reports include all expected outcomes |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Imam Reza Hospital in Mashhad, Iran Study period: March to September 2004 (7 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: confirmation of cutaneous leishmaniasis based on direct smear, ≤ 3 lesions, duration of the disease < 12 weeks, aged 7‐60 years, completion of informed consent form by participant or parents of minor participants, dry cutaneous leishmaniasis Exclusion criteria: pregnant or breastfeeding women; children < 7 years, lesions on ear, nose, joints, and near the eye; > 3 lesions; application of any kind of treatment for cutaneous leishmaniasis, duration of the disease longer than 12 weeks (to omit spontaneous healing cases during the follow‐up period), recurrent infection, wet cutaneous leishmaniasis N randomised: 45 participants Withdrawals: 11 N assessed: 34 participants (24, group 1; 10, group 2) Age: not described Sex: not described Baseline data: mean size of lesions (SD): group 1, 1.27 cm (0.81); group 2, 0.98 cm (0.61) |
|
| Interventions |
Type of interventions:
Duration of intervention: group 1, 2 weeks; group 2, 6 weeks |
|
| Outcomes |
Clinical response
Time points reported: 8 weeks |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "This N randomised controlled trial was performed from March to September 2004 on patients with cutaneous leishmaniasis admitted to Imam Reza Hospital in Mashhad" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 45 participants were randomised into 2 groups. 11 were lost to follow‐up (24%). No ITT analyses were performed High imbalance between groups |
| Selective reporting (reporting bias) | High risk | No baseline data. Side effects not clearly reported (no statistical analyses performed either) |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Ahvaz city, southern Iran Study period: 12 months (2002) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: clinically and parasitologically positive (Leishman bodies in Giemsa‐stained direct smear of the lesion scrapings), duration of lesions < 3 months, aged 15‐40 years, no evidence of secondary bacterial infections, no mucosal involvement, no history of previous treatment of cutaneous leishmaniasis, or of allergic reactions to MA or metronidazole Exclusion criteria: history of underlying disease (such as cardiac, renal, or pulmonary); pregnant or breastfeeding; cutaneous leishmaniasis localised on or near the joints N randomised: 36. Group 1: 18, group 2: 18 Withdrawals: 8. Group 1: 2, group 2: 6 N assessed: 28 (77.78%). Group 1: 16, group 2: 12 Age: mean 28.8 years (range 15‐40) Sex (male/female): 21/15 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 8 weeks |
|
| Outcomes |
Time points reported: clinical cure: after 8 injections. Adverse effects: after the treatment. |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The method of randomisation was selecting a card among 36 cards with odd or even numbers |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 28/36 participants completed the study (77.8%) |
| Selective reporting (reporting bias) | High risk | No protocol available. Adverse effects not reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: skin department of PNS Shifa, a naval hospital in Karachi. Endemic areas of Balochistan province, Pakistan Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp not mentioned Inclusion criteria: never received any treatment for their skin disease Exclusion criteria: very young and very old people, those who had received some treatment for the disease, people suffering from diffuse CL or leishmaniasis recidivans, people with some known cardiac, renal or hepatic disease N randomised: 40. 20 in each group Withdrawals: 0 Participants assessed: 40 (100%). 20 in each group Age: range 20‐40 Sex: 100% male Baseline data. Nodular, ulcerative and crusted forms were the most common morphological patterns seen. Most lesions were on hands and feet.
|
|
| Interventions |
Type of interventions:
Duration of intervention: 15 days Duration of follow‐up: 3 months |
|
| Outcomes |
Healing rates: percentage of participants 'cured' at the end of treatment Adverse effects Time points reported: at the end of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is probably that the published reports include all expected outcomes |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Golestan province, in northeastern Iran Study period: 20 November 2005 to 20 July 2006 (8 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: observation of Leishman bodies (amastigotes) in dermal lesions, no previous use of anti‐leishmanial drugs, no previously confirmed leishmaniasis (by scar or clinically compatible history), no pregnant or lactating women, no acute or chronic medical condition and no history of allergy, female participants of childbearing age were included after giving consent for effective contraception during therapy and until 3 months thereafter Exclusion criteria: not explicitly reported, but can be inferred from the above inclusion criteria N randomised: 63. Group 1: 32; group 2: 31 Withdrawals: 5. Group 1: 4 (lost after first week because lack of gastrointestinal tolerance); group 2: 1 (lost to follow‐up after 3 months) N assessed: 63 (100%). Group 1: 28; group 2: 30 Mean age: group 1: 20.2 years; group 2: 16.8 years Sex (male/female): 35/28. Group 1: 19/13; group 2: 16/15 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: miltefosine: 28 days. IMMA: 14 days Duration of follow‐up: 6 months |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 3 months after treatment Secondary outcomes: duration of remission and percentage of people with treated lesions that recur within 6 months Adverse effects Time points reported: 2 weeks, 3 months, 6 months |
|
| Notes |
Study funding sources: this study was financially supported by Medical Sciences/University of Tehran, Iran (from grant for full professor) Possible conflicts of interest: not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "For randomisation of patients into two groups, we used balanced block method" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "This study was an open‐label, N randomised comparison of miltefosine to meglumine antimonate in 63 patients" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imbalance in numbers and reasons for missing data across intervention groups (4/32 = 12.5% miltefosine group; 1/31=3.22% IMMA group) Comment: the results for the dropouts were not excluded from the statistical analysis. We think missing outcome data likely to be related to true outcome. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is probably that the published reports include all expected outcomes |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: not described Sample size calculation: assuming a 30% spontaneous cure rate with placebo and the minimum usefulness of a 60% cure rate with itraconazole, and a 10% dropout rate, the sample size was calculated as 70 individuals in each group |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: age > 12 years, diagnosis of CL confirmed by laboratory test results, no previous treatment for leishmaniasis, no serious concomitant medical problems Exclusion criteria: age < 12 years, pregnant and nursing women, patients who had lesions on the face, lesions lasting > 4 months N randomised: 140. Itraconazole: 70; placebo: 70 Withdrawals: itraconazole: 5 N assessed (lesions): 131. Itraconazole: 65 (219); placebo: 66 (262) Age: mean 26.3 years (range 12‐46) Sex (male/female): 90/41. Itraconazole: 44/21; placebo: 46/20 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 3 weeks Duration of follow‐up: 51 days |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 51 days after treatment Adverse effects Time points reported: at the end of treatment (day 21); on day 51 (1 month after the end of treatment) |
|
| Notes |
Study funding sources: this work was supported by the World Health Organization Special Program for Research and Training in Tropical Diseases and the research programme of Isfahan University of the Medical Sciences. Possible conflicts of interest: not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Carried out by the WHO Special Programme for Research and Training in Tropical Diseases group in Geneva, Switzerland" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical capsules containing 100 mg of itraconazole or placebo were used in the study. The 2 treatment groups received either itraconazole as coded capsules for 21 days or placebo as coded capsules with the same dosages. Review authors judge that the outcome is not likely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing outcome data unlikely to be related to true outcome |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. The study published reports our primary outcomes: 'cured' and adverse effects |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Research Centre in Isfahan city. Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: age > 5 years, diagnosis of CL confirmed by laboratory test results, no previous treatment for leishmaniasis, no serious concomitant medical problems, signed written informed consent Exclusion criteria: pregnant and nursing women, age < 5 years, lesions lasting for > 4 months N randomised: 72. Group 1: 36; group 2: 36 Withdrawals: 6, lost to follow‐up. Group 1: 5; group 2: 1 N participants assessed (lesions): 66 (196). Group 1: 31 (91); group 2: 35 (105) Mean age (male/female): 21.9 years/17.6 years (range 5‐48 years). Group 1: 23.6 years/17.4 years; group 2: 20.2 years /18.1 years Sex (male/female): 50 (75.7%)/16 (24.3%). Group 1: 26/5; group 2: 24/11 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 20 days Duration of follow‐up: 51 days (1 month after the end of treatment) |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 51 days after treatment. On day 21 (end of therapy), the clinical response to treatment was coded subjectively as: apparent cure, partial response, and failure. After 1 month of follow‐up (day 51), the participants were described as showing definitive cure if all the lesions had healed and direct smears of the lesions were negative Adverse effects Tertiary outcomes: microbiological or histopathological cure of skin lesions Time points reported: day 21, and 1 month after the end of treatment: day 51 |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Seventy‐two patients were assigned randomly to two groups." Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imbalance in numbers across intervention groups: 5/36 (13.89%), AL + IMMA group; 1/36 (2.78%), IMMAgroup. We think missing outcome data likely to be related to true outcome. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: endemic areas in the north of Isfahan city, Iran Study period: not described Sample size calculation: sample size calculation was based on 90% significance and 90% power to detect a difference between vehicle and ketoconazole. Assuming a 15% spontaneous cure with placebo, a minimum usefulness of a 60% cure with ketoconazole cream and about a 10% dropout, the sample size was calculated as 45 individuals in each group. |
|
| Participants |
Type of Leishmania: Leishmania spp: L major and L tropica Inclusion criteria: age > 5 years, laboratory‐confirmed diagnosis of CL, no previous treatment for leishmaniasis, no serious concomitant medical problems Exclusion criteria: pregnant and nursing women, lesions on the face, lesions of > 4 months duration N randomised: 90 Withdrawals: 17 N assessed: 73 Age: mean 19.9 years (range 6‐60) Sex (male/female): 45/28 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: twice daily for 21 days. |
|
| Outcomes |
Healing rates: clinical response to treatment was coded subjectively as: apparent cure, partial response or failure. Definitive cure was defined if at day 51, the lesions were healed and direct smears of the lesions were negative. Adverse effects Time points reported: the clinical response of the lesions was reported on days 21 (end of therapy) and 51 |
|
| Notes |
Study funding sources: supported by a research programme of Isfahan University of Medical Sciences Possible conflicts of interest: not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The study was designed as a double‐blind, N randomised, placebo‐controlled trial" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Placebo and ketoconazole creams were identical cream containing either 25 ketoconazole 2% or placebo were used in the study. The two treatment groups received either the ketoconazole cream or placebo twice daily for 21 days as coded tubes with the same dosage" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: skin and CL clinic in Gaz in Isfahan, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: size of ulcer less than 5 cm in diameter, ≤ 5 lesions, duration of disease < 3 months Exclusion criteria: patients who had lesions on the face near the eyes or on the nose or ears, pregnancy, lactation, lupoid or sporotrichoid lesions, use of immunosuppressor drugs in the past 6 months and severe adverse effects, exacerbation of the disease N randomised: 40 participants (19 in prepared gel group and 21 vehicle gel mask) Withdrawals: 1 participant in prepared gel group N assessed: 39. 21 (100%) in vehicle gel group, and 18 in prepared gel group Mean age: 28.2 years Sex (male/female): 25/15 Baseline data: not described |
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks Co‐interventions: all of the participants received the standard treatment including ILMA weekly and cryotherapy every 2 weeks for a maximum of 6 weeks |
|
| Outcomes |
Treatment outcome, defined as:
Time points reported: 2 months after the end of treatment for relapse |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Open trials. Photography was done from all the lesions both at the first visit and all the follow‐up visits |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | High risk | Protocol not available; not registered; in clinical trial registry; adverse effects not reported |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Pakistan Study period: around 4‐5 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp not mentioned Inclusion criteria: clinical diagnosis of CL and positive Giemsa‐stained smear of parasite Exclusion criteria: > 5 lesions and pregnant women N participants randomised (lesions): 104 (215). Group 1: n = 49 (111); group 2: n = 55 (104) Withdrawals: 8 N assessed: 96. group 1: n = 49; group 2, n = 47 Age:
Sex (male/female): 58/38 Baseline imbalances: comparability regarding age, sex, number, and site of lesions and duration of the disease
|
|
| Interventions |
Type of interventions:
Duration of intervention: until complete cure or up to 8 weeks |
|
| Outcomes |
Healing: complete cure (100%); partial (> 50%; < 50%); failure (nil) Speed of healing (time taken to be 'cured') Duration of remission and percentage of people with treated lesions that recur within 6 months and 1, 2, and3 years:time assessment not specified Prevention of scarring Adverse effects Time points reported: at 2, 4, 6 and 8 weeks of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two treatment groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Patients in group A were treated with weekly and those in group B with fortnightly intralesional injections" Comment: we think the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts: 8/104: 7.6% lost to follow‐up Comment: the results for the defaulters were excluded from the statistical analysis. We think missing outcome data unlikely to be related to true outcome. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: October 2000 to February 2001 (5 months) Sample size calculation: to detect a clinically relevant difference of at least 25% between placebo (35% estimated response rate) and itraconazole (60% estimated response rate) in the primary outcome parameter with a power of 90% and a 2‐sided type I error of 5%, a total of 164 subjects (82 in each group) were required. To compensate for dropouts, 20% more participants were enrolled in each group (in total 200 participants). |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: aged 12‐60 years; presence of parasitologically confirmed lesion(s) of CL; healthiness on the basis of physical examination, medical history, and the results of blood biochemistry and haematology, carried out < 2 weeks before the start of the trial Exclusion criteria: women of child‐bearing age without adequate effective contraception, pregnant or breastfeeding, duration of lesions > 45 days, presence of lesions on the face or near the mucous membranes, > 5 lesions, any lesion with a diameter > 3 cm, history of any systemic antileishmanial therapy, known hypersensitivity/allergy to itraconazole, receiving any drug with known interaction with itraconazole N randomised: 200 Withdrawals: 42 in total. 17 in the Itraconozale group and 25 in the placebo group N assessed: 158 Age: not described Baseline imbalances: comparable with regard to age, sex, and duration of the lesions
|
|
| Interventions |
Type of interventions:
Duration of intervention: 8 weeks |
|
| Outcomes |
Healing rates: for clinical assessment a 5‐point scoring system was employed: cure (complete re‐epithelialisation of all of the lesions), moderate improvement (more than 50% reduction in the size of the lesions), mild improvement (less than 50% reduction in the size of the lesions), no change, and worsening Adverse effects: at each visit, all of the participants were asked about any adverse effects. Time points reported: after completion of treatment (8 weeks), and at the end of the 3 months follow‐up (healing rates) |
|
| Notes |
Study funding sources: supported by a grant from Janssen‐Cilag Pharmaceuticals Inc. Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation sequence was generated from a random number table" |
| Allocation concealment (selection bias) | Low risk | Quote: "Concealed from the investigator until the data entry was completed and the data bank was locked" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients were randomly allocated to receive either itraconazole in the form of two 100‐mg capsules or identical placebo capsules once daily for 8 weeks." Comment: we think the outcome is unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imbalance in numbers and reasons for missing data across intervention groups (17% itraconozale group; 25% placebo group). Comment: the results for the defaulters were not excluded from the statistical analysis. We think missing outcome data likely to be related to true outcome. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not mentioned Inclusion criteria: CL confirmed with direct smear, aged 1‐20 years, maximum number of lesions 3, maximum size of lesion 5 cm, duration of disease less than 8 weeks Exclusion criteria: history of allergy to MA, pregnancy or lactating N randomised: 210 Withdrawals: 53: 29/105 in group 1 and 24/105 in group 2 N assessed: 157. Group 1: 76, group 2: 81 Age: not described Baseline data: not described |
|
| Interventions |
Type of interventions:
Duration of intervention: 6 weeks |
|
| Outcomes |
Healing rates: percentage of participants 'cured' 2 weeks after treatment Adverse effects Time points reported: not described |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High rates of dropouts: 53/210 (25.2%) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability were not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Disease and Leishmaniasis Research Center (SDLRC), Isfahan, Iran Study period: around 5 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not mentioned Inclusion criteria: CL confirmed with direct smear Exclusion criteria: pregnant or nursing mothers, treated previously for CL, any known intercurrent illness or a history of allergy to MA, palpebral lesions, having > 5 lesions, lesion > 3 cm, duration of lesions was > 12 weeks N randomised: 80: 40 in each group Withdrawals: 7 N assessed: 73: 38 in group 1 and 35 in group 2 Age: range 5‐75 years Baseline data: the most common clinical type of the lesions in both groups was papule (50% in TCA and 44% in the ILMA group) and nodule (25% in TCA and 24% in the ILMA group). Mean number of lesions: TCA: 1.2; ILMA: 1.4 |
|
| Interventions |
Type of interventions:
Duration of intervention: up to 6 weeks |
|
| Outcomes |
Healing: complete cure was defined as complete healing and re‐epithelialisation of the lesion(s); partial care as partial clinical improvement with reduction in erythema, induration and size of the lesions; treatment failure as the absence of any change or worsening of the lesion(s) Duration of remission and percentage of people with treated lesions that recur after 3 months of follow‐up Adverse effects Microbiological or histopathological cure of skin lesions Time points reported: after the last treatment session, 1 and 3 months post‐treatment. Complete cure was reported at 4 and 6 weeks of the treatment period. Recurrence was assessed at 3 months follow‐up |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random digit table |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Unblindness is a limitation of this study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "For 80 patients, which participated in the study, 73 cases completed the study" Comment: the results for the dropouts were excluded from the statistical analysis. We think missing outcome data likely to be related to true outcome. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Disease and Leishmaniasis Research Center (SDLRC), Isfahan, Iran Study period: 4 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not mentioned Inclusion criteria: CL confirmed with direct smear, no history of systemic or topical therapy for CL, absence of the malnutrition or severe predisposing disease such as cardiac, renal or hepatic disease and other contraindication for MA Exclusion criteria: pregnant or lactating mothers, lesions > 3 months old and treated with the drugs that had interaction with MA N randomised: 100, 50 in each group Withdrawals: 23 (13 in the honey group and 10 in the ILMA group). However, in the abstract authors stated that 10 participants left the study. N assessed: 90 (45 in each group) Age: range 7‐70 years Baseline data: the most common clinical type of the lesions in both groups was plaque (60% in honey + ILMA and 55.6% in the ILMA‐alone group). Mean number of lesions: honey + ILMA: 1.3; ILMA: 1.7 |
|
| Interventions |
Type of interventions:
Duration of intervention: once weekly until complete healing of the ulcer or for maximum of 6 weeks |
|
| Outcomes |
Healing: complete healing was defined as disappearance of the induration and complete re‐epithelialisation of the ulcer. Complete healing of the lesions was defined as complete clinical and parasitological healing (negative direct smear); partial healing of the lesions was defined as the decrease of the size and indurations of the lesions; non‐responsive was defined as no clinical change or progression of the lesions Speed of healing (time taken to be 'cured'): expressed as mean healing time Adverse effects Time points reported: assessed weekly for 6 consecutive weeks and at the end of the 2nd, 3rd, and 4th month. Cure was assessed at the end of treatment and follow‐up |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: no competing interests |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random allocation software (ver 1.0, May 2004; Saghaei)" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | We think the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Balance in numbers and reasons for missing data across intervention groups (13/45 (28.8%) in honey + ILMA group; 10/45 (22.2%) in ILMA‐alone group) Comment: the results for the defaulters were excluded from the Kaplan‐Meyer analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: relevant outcomes were reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Skin Diseases and Leishmaniasis Research Center. Isfahan, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica and L major Inclusion criteria: all the participants had positive smear for Leishmania body and had not received any topical or systemic therapy for leishmaniasis, aged 7‐70 years, lesion not more than 3 months Exclusion criteria: pregnant or lactating; history of cardiac, renal, hepatic diseases; any contraindication for the treatment N randomised: 150. Group 1: 50; group 2: 50; group 3: 50 Withdrawals: 26. Group 1: 7; group 2: 14; group 3: 5 N assessed: 124 (82.66%). Group 1: 43 (86%); group 2: 36 (72%); group 3: 45 (90%) Mean (SD) age: group 1: 33.1 years (17.5); group 2: 27.8 years (9.8); group 3: 29.1 years (14.7) Sex (male/female): 88 (70.97%)/36 (29.03%)
Baseline data:
|
|
| Interventions |
Type of interventions:
The oral placebo was identical in appearance to omeprazole capsules and was administered in the similar way Duration of intervention: 3 weeks |
|
| Outcomes |
Clinical cure: rate of complete response, 3 months (12 weeks) after starting treatment. Scale: complete healing of the lesions was regarded as complete clinical and parasitological healing (negative direct smear). Partial healing of the lesions was regarded as decrease of the size and indurations of the lesions, and no response was regarded as no clinical change or progression of the lesions. Time to healing: proportion of complete improvement in the 3 groups during the course of treatment Adverse effects: at the end of treatment Time points reported: clinical cure: 12 weeks. Time to healing: 2, 4, 6, 8, 12 weeks. Adverse effects: at the end of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: they declared no competing interests. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were selected and randomised by random allocation software into 3 groups. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The oral placebo was completely similar to omeprazole capsules and was administered in the similar way; low dose of IMMA was prepared by addition of normal saline to IMMA Both the investigating physicians and the participants were blinded to the type of treatment and drug codes were revealed only at the end of the study. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Isfahan University of Medical Science, Isfahan, Iran Study period: March 2008 to March 2009 (12 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: unclear Exclusion criteria: pregnant, children < 5 years, palpebral lesions (less than 20 mm from palpebral margin), > 5 lesions, > 12 weeks duration of leishmaniasis, history of any specific anti‐leishmaniasis therapy, significant underlying diseases N randomised: 80 lesions Withdrawals: 4 lesions N assessed: 76 lesions (38 lesions in each group) and 60 patients (30 patients in each group) Mean (SD) age: 5.11 years (13.3) (range 5‐50) Sex: unclear Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: non‐ablative radiofrequency + topical TCA 50%: 4 weeks; ILMA: 8 weeks |
|
| Outcomes |
Treatment responses:
Time points reported: 6 months of treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "This computer‐based randomised controlled trial was conducted from March 2008 to March 2009" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The examinations and measurements were performed by the investigators who were blinded to the type of treatment." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Originally 80 lesions were studied and randomly assigned to the 2 groups. However, due to missing data, only 76 were finally included in the analysis. No ITT analysis |
| Selective reporting (reporting bias) | High risk | Complete cure was described in Methods and Results, but partial cure and treatment failure was not reported in the Results section. No adverse effects reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Cutaneous Leishmaniasis and Skin Diseases Research Center, Isfahan, Iran Study period: 2010 to 2011 (12 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: age older than 5 years, diameter less than 3 cm, the duration of disease less than 12 weeks, lesions with less than 2 cm distance from eyelid, < 5 lesions Exclusion criteria: pregnancy; lactation; colloid scar; lesions on cartilage, eyelids or joints; history of using other drugs against CL, treatment with immune system inhibitors N randomised: 90 (30 people in each group) Withdrawals: not described N assessed: 90 (30 people in each group) Mean (SD) age: 24.5years (14) Sex (male/female):21/9, 23/7 and 21/9 Baseline data: Number of lesions:single lesion in 53 (58.9%), 2 lesions in 29 (32.2%) and 3 lesions in 8 patients (8.9%). The type of lesion: plaque in 32 patients (35.6%), nodule in 44 (48.4%), plaque and nodule in 8 (8.9%), and papule in 6 patients (6.7%) Distribution of lesions: hand 43, foot 15, face 8, trunk 5, hand and trunk 4, hand and neck 4, neck 4, hand and foot 4, hand and face 2, ear 1 |
|
| Interventions |
Type of interventions:
All patients in three groups were followed weekly for 3 months and then after 4 and 6 months to assess improvement of lesions and size of scar. |
|
| Outcomes |
Clinical response, according to standard quartile grading:
The quantitative 4‐point scale used included:
Time points reported: 6 months |
|
| Notes |
Study funding sources: Isfahan University of Medical Sciences Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Impossible for blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote "Evaluation of patients were done by two physicians who were not aware of the treatment group. These two physicians did the evaluations separately. The reduction in this size of ulcer and the scar was compared with the same lesion" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: prospective randomised controlled trial Setting/location: Central Research Center of Skin Diseases and Leishmaniasis and Novin Laser Center; Isfahan, Iran Study period: June 2010 to September 2011 (15 months) Sample size calculation: sample size was calculated as 60 cases in each group with further rate of 10% dropouts and exclusions |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: leishmaniasis scar diagnosed clinically on participants by a dermatologist Exclusion criteria: pregnancy, lactation, colloid scar, treatment with immune system inhibitors, use of isotretinoin or fillers for the past 6 months, use of dermabrasion or skin resurfacing for the past 12 months, skin types of IV to V1 N randomised: 120 (60 people in case group and 60 in control group) Withdrawals: not described N assessed: not described Mean (SD) age: 27.21 years (11.52) (range 6‐45) Sex (male/female): 51%/49% Baseline imbalances: none Severity of illness: mean (SD) scar size: laser CO₂ group: 2.3 cm (0.3) and fractional CO₂ laser: 3.4 cm (0.4) |
|
| Interventions |
Type of interventions:
|
|
| Outcomes |
Clinical response, according to standard quartile grading:
The quantitative 4‐point scale used included:
Time points reported: 6 months |
|
| Notes |
Study funding sources: Isfahan University of Medical Sciences Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quotes: "A photo was taken from patients before and after treatment with a digital camera ... Evaluation of participants were done by 2 physicians who were not aware of the treatment group. These 2 physicians did the evaluations separately. The reduction in this size of ulcer and the scar was compared with the same lesion" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Disease and Leishmaniasis Research Center, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: participants with positive direct smear for CL; 6‐60 years of age; without any previous history of systemic or topical therapy for CL; lesions < 3 cm, < 12 week duration, and not located within 2 cm distance of palpebral margin Exclusion criteria: pregnant and lactating women N randomised: 200 participants Withdrawals: not described N assessed: 187; group 1: 91; group 2: 96 Mean (SD) age: 10.7 years (22.04); group 1: 10 years (20.6); group 2: 11.3 years (23.3) Sex (male/female): 101/86 Baseline data: mean (SD) lesion area was 329 mm² (118.7) in group 1 and 359 mm² (55.6) in group 2 |
|
| Interventions |
Type of interventions:
Duration of intervention: complete resolution of the lesions or end of 8 weeks |
|
| Outcomes |
Clinical cure: complete re‐epithelialisation of the lesion and lack of induration. Clinical response was measured at the end of treatment and was defined as complete cure (negative direct smear and clinical healing), partial cure (partial clinical improvement with decreasing erythema, induration, and lesion size), non‐cure or treatment failure (no clinical change or worsening of the lesions) Time points reported: 8 weeks |
|
| Notes |
Study funding sources: the study is self‐funded Possible conflicts of interest: there is no conflict of interest |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random allocation software (version 1.0, May 2004, Saghaei) was used for randomisation |
| Allocation concealment (selection bias) | Low risk | Participants with confirmed cutaneous leishmaniasis referred to the Skin Diseases and Leishmaniasis Research Center were selected and randomised by random allocation software into 3 groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The measurements were done before treatment and at the end of eighth week by the investigators who were blinded to the type of treatment." Comment: Lesions were photographed before and after completion of the treatment course. Not clear how blinding was preserved |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | From a total of 200 randomised participants, 96 participants in ILMA group and 91 participants in the combination group completed the study Losses to follow‐up were below 25% and homogeneous between the groups |
| Selective reporting (reporting bias) | High risk | Protocol not available, not registered. Adverse effects not reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Anuradhapura Teaching Hospital, Anuradhapura, Sri Lanka Study period: 32 months. Participant selection: April 2006 to June 2007 (15 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L donovani Inclusion criteria: participants with CL who attended to Anuradhapura Teaching Hospital, Anuradhapura, Sri Lanka from April 2006 to June 2007 Exclusion criteria: not described N randomised: a total of 154 participants with 229 lesions were included in the study. IL 7% HSCS: 67; ILSSG: 87 Withdrawals: (4.5%: 7/154). HSCS: 3, ILSSG: 4 N assessed: 147 (95.45%) CL participants with 222 lesions completed treatments. HSCS: 64 (95.52%); ILSSG: 83 (95.40%). Age: mean 32 years (range 16 months to 74 years) Sex (male/female): 99/55, M:F ratio of 1.8:1 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: HSCS: 1‐29 weeks, depending on the size and the duration of the lesions (mean 8.78 weeks). SSG: 1‐13 weeks, depending on the size and the duration of the lesions (mean 5.11 weeks) Participants were seen weekly for the first 3 injections; fortnightly for the fourth and fifth injections; then monthly until cure. |
|
| Outcomes |
Clinical cure: the percentage of lesions that were cured with SSG and HSCS Number of injections required per each lesion in total for cure (scales: 1‐10 injections) Adverse effects: systemic side effects with SSG or HSCS. Local side effects: pain during injection. Scarring after healing, postinflammatory hyperpigmentation Time points reported: at the end of treatment. Participants were followed‐up every 3 months after cure for 18 months to assess recurrences and evidence of visceralisation |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind" Insufficient information to permit judgment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double blind", "At each visit the patients were examined by the primary investigator who was blind to the therapy" Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4.5% (7/154) did not complete the study |
| Selective reporting (reporting bias) | Unclear risk | No protocol was available; not registered. Insufficient information to permit judgment SD not provided for duration of treatment and number of injections |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised double‐blind clinical trial Setting/location: Skin Clinic of Anuradhapura Teaching Hospital, Sri Lanka Study period: January 2012 to February 2013 (14 months) Sample size calculation: approximately 473 participants per group were required to enable the detection of a treatment difference of 5% with a lowest cure rate of one group of 90% |
|
| Participants |
Type of Leishmania: L donovani Inclusion criteria: the study recruited participants with suspected CL who consented to participation. Exclusion criteria: pregnant or lactating women, subjects with a history of cardiac, renal, or hepatic disease N participants randomised (lesions): 444 (643). 10% HSCS: 192 (297); 15% HSCS: 82 (101); ILSSG: 170 (245) Withdrawals (lesions): 17 (20) lost to follow‐up. 10% HSCS: 8 (7); 15% HSCS: 3 (3); ILSSG: 6 (10) N assessed (lesions): 427 (623); 10% HSCS: 79 (98); 15% HSCS: 184 (290); ILSSG: 164 (235) Age: mean 32.7 years (range 14 months to 88 years). Most (60.8%) were aged 16–45 years Sex (male/female): 286/158 Baseline data: the average delay in presentation was 8 months (range: 4 weeks to 5 years). 91% of lesions were located on exposed areas of the body. Most (69.8%) participants had a single lesion. |
|
| Interventions |
Type of interventions:
Duration of intervention: participants were seen weekly for the first 3 injections, fortnightly for the 4th and 5th injections, and then monthly until a cure was achieved. |
|
| Outcomes |
Clinical and parasitological cure: responses were graded as slight (10% improvement from the initial status of the lesion), mild (20%–30% improvement), moderate (50% improvement), marked (80%–90% improvement), or as total if the lesion had cleared and parasites were not detected in the affected area by smear or culture. Both marked improvements and total clearance were considered to represent a cure. Reported at the end of treatment. Duration of treatment: in weeks, mean (range) Time required to achieve the cure of the lesion: mean and median times taken for cure for the different treatments groups. Every 5 weeks until achieve 30 weeks. Injections per lesion, mean (range): number of injections for cure per lesion Relapse: L recidivans Adverse effects: local side effects: at the site of the lesion caused by the injections: pain during injections, postinflammatory hyperpigmentation, scarring, postinflammatory depigmentation, ulceration and necrosis, L recidivans. Systemic side effects: no systemic or significant local side effects with either SSG or 10% HSCS. Time points reported: participants were followed up every 3 months after cure for more than 6 months to assess recurrences and evidence of visceralisation |
|
| Notes |
Study funding sources: none reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The treatment options were documented separately and packed in opaque envelopes numbered consecutively according to the randomisation schedule at a ratio of 2: 2: 1 |
| Allocation concealment (selection bias) | Low risk | The allocation sequence was concealed from the researcher who enrolled and assessed the participants by the use of sequentially numbered, opaque, sealed, and stapled envelopes that were impermeable even to intense light. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participants and the investigator (who assessed the participants for their clinical response) were blind to the type of therapy allocated. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | At each visit, the participant was examined by the primary investigator, who was blinded to the therapy. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The numbers of withdrawals were similar and small in the 3 groups: 4.1% (8/192) in group 1, 3.6% (3/82) in group 2, and 3.5% (6/170) in group 3. |
| Selective reporting (reporting bias) | Low risk | The study published reports our primary outcome and adverse effects. |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Khair Khana Clinic; Kabul, Afghanistan Study period: 100 days Sample size calculation: to detect a 20% difference in the cure rate between the SSG and thermotherapy groups, assuming an 80% cure rate in the SSG groups, with a 90% power and a 5% 2‐sided type I error, 98 subjects were needed in each group. To compensate for anticipated loss to follow‐up, 40% more participants were enrolled in each group. |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: age > 5 years; the presence of a single, parasitologically confirmed CL lesion; no prior history of disease and/or antimonial treatment Exclusion criteria: the presence of a CL lesion located on or immediately adjacent to the nose, lips, or eyes; pregnancy; breastfeeding; major surgery in the previous 3 months; presence of any uncontrolled medical condition; anticipated unavailability for follow‐up N randomised: 431. Group 1: 148, group 2: 144, group 3: 139 Withdrawals: 172 in total: 30 decided to withdraw after allocation and 142 did not complete the 4 visits or the follow‐up N assessed: 259. Group 1: 93, group 2: 58, group 3: 108 Age: range 10‐20 years Sex (male/female): 200/201 Baseline data: the lesions were primarily located on the face (43.4% of participants), on the hands (38.2%), legs (15.9%) and arms (2.4%)
|
|
| Interventions |
Type of interventions:
Duration of intervention: not described |
|
| Outcomes |
Percentage of participants 'cured' at 100 days after treatment initiation and by time to cure: cure was defined as the complete re‐epithelialisation of the CL lesion, with no evidence of papules, inflammation, or induration Speed of healing (time taken to be 'cured') Adverse effects: the occurrence of adverse effects was evaluated blindly by means of participant interviews and physical examinations during follow‐up visits Time points reported: trial endpoint was 100 days after start of therapy |
|
| Notes |
Study funding sources: the HNI Malaria and Leishmaniasis Control Program is funded by the European Union. This study was supported by the United Nation Children's Fund/United Nation Development Program/WorldBank/World Health Organization Special Program for Research and Training in Tropical Diseases, the Afghan Research Evaluation Unit, and the Leveen Family Fund. Possible conflicts of interest: since March 2004, R Reithinger has been a part‐time employee of Thermosurgery Technologies. All other authors: no conflicts |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients then proceeded to pick 1 of 3 identical cardboard pieces out of a hat (the cardboard had been labelled with different treatment codes on one of its sides, the codes being non‐visible to the patient). After patients were randomly assigned to receive a treatment, the cardboard piece picked was returned to the hat." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: no blinding of participants and personnel seemed to be performed. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The occurrence of adverse effects was evaluated blindly by means of patient interviews and physical examinations during follow‐up visits", "Microscopic examination was performed blindly." Comment: microscopic examination and adverse effects were performed blindly but no description about clinical examination was given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 172/431 (39.9%) withdrew from the study. However, ITT analyses were performed. Quote: "An intention‐to‐treat analysis of the data (... including the patients lost to follow‐up, who were considered to have had treatment failure) yielded similar results for the comparison of the odds of cure for the different treatments" |
| Selective reporting (reporting bias) | Low risk | Comment: relevant outcomes were reported |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: 3 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: CL in whom the diagnosis was confirmed by laboratory demonstration of the parasite in the lesions by direct smear Exclusion criteria: allergic to antimonial drugs or pentoxifylline, lactating or pregnant, history of systemic illness N randomised: 64, 32 in each group Withdrawals: 1 participant withdrew from the MA + placebo group N participants assessed (lesions): 63. Group 1: 32 (143), group 2: 31 (164) Mean age: group 1: 27 years, group 2: 31 years Sex (male/female): 29/34 Baseline data: duration of the disease approximately 1.3 months.
|
|
| Interventions |
Type of interventions:
Duration of intervention: 20 days Duration of follow‐up: 3 months |
|
| Outcomes |
Healing rates:
Adverse effects Time points reported: after end of treatment and 3 months' follow‐up |
|
| Notes |
Study funding sources: not described Possible conflicts of interest: not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into two groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Study was described as "double blind" but has no description about allocation method. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study was described as "double blind" but has no description about the blinding method. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One patient withdrew in the placebo group. Reasons for withdrawal were reported. However, no ITT analyses were performed. |
| Selective reporting (reporting bias) | Low risk | Comment: relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Diseases and Leishmaniasis Research Center in Isfahan, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania:L major endemic in the area. Inclusion criteria: participant with positive smear for Leishman bodies, of both sexes and > 5 years old who did not have indication for systemic therapy Exclusion criteria: facial lesions or lesions on joints, sporotrichoid type, lupoid leishmaniasis, erysipeloid type and other atypical forms of CL, pregnant women, history of cardiovascular and renal diseases N randomised: 72, 36 in each group Withdrawals: 0 N assessed: 72 Mean age: HSCS, 20.52 years; ILMA group, 18.67 years Baseline data: the most common site of lesions was on the extremities and the least common site was the trunk in both groups. The size range of lesions were from 0.5‐4 cm² (IL HSCL group) and from 0.5‐4 cm² (ILMA group) |
|
| Interventions |
Type of interventions:
Duration of intervention: weekly 6‐10 weeks Duration of follow‐up: 6 months |
|
| Outcomes |
Healing: complete improvement is defined as completely clinical re‐epithelialisation with no signs of induration and inflammation with negative smear for Leishman bodies at the end of the treatment period. Partial improvement is defined as the re‐epithelialising lesion has become smaller but not cured. If the lesion has become larger or has not been differed it will be called as no response to treatment. Time points reported: at 6 and 10 weeks of treatment and 6 months follow‐up |
|
| Notes |
Study funding sources: not described Possible conflicts of interest: not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "This N randomised controlled clinical trail study with simple sampling." "72 patients randomly divided in two equal groups". Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | Comment: relevant outcomes were reported |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Skin Diseases and Leishmaniasis Research Center, Iran Study period: 7 months Sample size calculation: not reported |
|
| Participants |
Type of Leishmania: not reported Inclusion criteria: smear from a suspected CL lesion was confirmed positive for Leishmania Exclusion criteria: pregnant women, children < 5 years of age, patients with facial lesions, those who had already received or were under other specific antileishmanial therapy, significant underlying diseases N randomised: 117 Withdrawals: 0 N participants assessed (lesions): 117: 57 (83) lesions in group 1 and 60 (94) in group 2 Mean age: group 1, 25.12 years; group 2, 22.6 years Sex (male/female): group 1, 37/20; group 2, 29/31 Baseline data: the shape of lesions was papule, nodule and plaque‐like. Lesions were located in the trunk and upper and lower limbs.
|
|
| Interventions |
Type of interventions:
Duration of intervention: once weekly for 4 consecutive weeks Duration of follow‐up: 6 months |
|
| Outcomes |
Percentage of participants 'cured':
Duration of remission and percentage of people with treated lesions that recur within 6 months Prevention of scarring Adverse effects Time points reported: at the end of treatment, and 6 months after |
|
| Notes |
Study funding sources: technical and financial support from the joint WHO Eastern Mediterranean Region (EMRO), Division of Communicable Diseases (DCD) and the WHO Special Program for Research and Training in Tropical Diseases (TDR): the EMRO/TDR Small Grant Scheme for Operational Research in Tropical and other Communicable Disease. Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly allocated to one of two treatment groups using a random number list generated by Epi Info (a software package designed to provide easy form and database construction, data entry, and analysis with epidemiologist statistics, map sand graphs)" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Group 1 was treated with controlled localised heating using an RF heat generator. Group 2 was treated with ILMA |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Follow‐up evaluation was made by clinical assessment of treated lesions by a second dermatologist who was blinded to the method of treatment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Leishmaniasis clinic of the National Malaria and Leishmaniasis Control Program in Darul‐Aman District, Kabul City, Afghanistan Study period: not described Sample size calculation: to detect an 8% difference in the cure rate between the current standard therapy (ILMA) and thermotherapy by radio frequency groups, assuming 90% cure rate in the conventional therapy with an 80% power and a 5% type I error, 174 subjects were needed in each group. To compensate for anticipated loss to follow‐up, sample size inflated by 10%. Hence, the final sample size, figuring research design, rounded to nearest 10 turned out to be 390 subjects, with 195 assigned to each treatment group. |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: aged > 5 years; presence of a single, parasitologically confirmed CL lesion Exclusion criteria: history of previous infections from anthroponotic CL that was treated with antimonial medications, lesions located within 2 cm of the eyelids or on the lips or nose N randomised: 390 (195 assigned to each treatment group) Withdrawals: 8 participants (6 in group 1; 2 in group 2). N assessed: 382 (189 in group 1; 193 in group 2). Median (interval) age: group 1, 14 years (5‐70); group 2, 13 years (5‐75) Sex (male/female): group 1, 85/104; group 2, 92/101 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 5 weeks in ILMA group |
|
| Outcomes |
Cure was defined as complete re‐epithelialisation of the lesion with no inflammation and resolution of the papule and/or nodule. Failure was defined as no improvement, with the lesion unchanged or worse compared with its status at the start of treatment. Lesions that showed some improvement but did not meet the criteria for cure were considered treatment failures Adverse effects Time points reported: 6 months after treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were asked to pick one of two identical cardboard pieces out of a box (the cardboard had been labelled with different treatment codes on one of its sides) for their group assignment. After patients were assigned to a treatment group, the cardboard piece picked was returned to the box." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were losses to follow‐up and no ITT analyses were performed. Also, the distribution of the lesions and the size was uneven between the 2 arms. Quote: "More papules (57.7%) were treated by thermotherapy compared to (42.3%) those which were treated by Glucantime. Nodules were mostly treated by Glucantime (58.9%), whereas all ulcers were treated by Glucantime. The lesion size interval was [1–4 cm²] with median of 1 cm² for thermotherapy by radio frequency and 2 cm² for Glucantime." This is important because the authors found out the following significant associations: Quote: "Type and size of lesion, were significantly associated with the outcome variable (cure/failure). Type of lesion was significantly associated with treatment outcome in this sample, taking papules as reference (Table V). Those who had nodules were 41% less likely and those with ulcers were 80% less likely to be cured compared with those with papules" |
| Selective reporting (reporting bias) | High risk | Adverse effects were described in Methods but not in Results. They mention them vaguely in the Discussion section. |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Iran Study period: 18 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania:Leishmania spp: L tropica and L major Inclusion criteria: patient with CL confirmed parasitologically by direct skin smears or skin biopsies. Exclusion criteria: children < 3 years of age, pregnant and lactating women, cases with concomitant renal, liver or heart disease, and any event of any laboratory abnormality prior to initiation of treatment N randomised: 96 Withdrawals: 0 N assessed: 96 (100%): 64 in the ketoconazole group and 32 in the ILMA group Age: range 3‐64 years Sex (male/female): 44/52 Baseline data: In the ketoconazole group:
In the MA group:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 30 days in the ketoconazole group. There is no mention about frequency of injections in MA group. |
|
| Outcomes |
Healing rates:
Adverse effects Time points reported: participants were followed every 2 weeks for the duration of the treatment and every 6 weeks after cessation of treatment up to 24 weeks |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly allocated to two treatment groups using a simple randomisation method" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Group 1, which included 64 participants, was designated as the group treated with oral ketoconazole. Group 2, which included 32 participants, was designated as the group that received ILMA. The outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imbalance in numbers and reasons for missing data across intervention groups: 7/57, ketoconazole; 9/23, ILMA |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgment |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Dermatology Clinic of Faghihi Hospital, Shiraz University of Medical Sciences, Shiraz, Iran Study period: unclear Sample size calculation: not reported |
|
| Participants |
Type of Leishmania: not reported Inclusion criteria: participant with CL confirmed Exclusion criteria: not described N randomised: 60 Withdrawals: 0 N assessed: 60 participants distributed into the 3 groups of equal number (n = 20) Age: not reported Sex: not reported Baseline data: lesions were located on the head, neck and lower and upper extremities. Lesion duration was divided in 2 groups: a duration of < 3 months and > 3 months. Regarding lesion size, the authors divided the sizes in 3 groups: < 1 cm, 1‐3 cm and > 3 cm. |
|
| Interventions |
Type of interventions:
Duration of intervention: weekly for a total of 6‐8 times for each case |
|
| Outcomes |
Healing rates: percentage of participants 'cured': timing not reported Adverse effects Time points reported: not reported |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No explanation was provided for missing outcome data. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports included all expected outcomes, including those that were pre‐specified. |
| Other bias | High risk | Sample size calculation, reporting of Leishmania spp involved and baseline comparability were not correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: dermatology clinic and leishmaniasis research centre of Afzalipour hospital of Kerman, province of Iran Study period: 21 months Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: aged 7–60 years, confirmed CL Exclusion criteria:chronic systemic disease (such as renal failure, myocarditis, hepatitis and pancreatitis), immune suppression, breastfeeding, pregnancy, sporotrichoid and lupoid forms, maximum diameter of lesions > 3 cm, disease duration more than 1 year, more than 2 lesions present, history of sensitisation to MA,facial and joint lesions, receiving other specific anti Leishmania therapies N randomised: 191. Group 1: 96, group 2: 95 Withdrawals: group 1: 16 (14: no follow‐up, 2 enrolled in other treatment), group 2: 15 (8 no follow‐up, 4 enrolled in other treatment, 3 did not adhere to treatment schedule) N assessed: 160 (83.77%). Group 1: participants: 80 (83.33%), group 2: participants: 80 (84.21%) Age: range 7‐60 years Sex (male/female): Randomised patients: group 1: 50(52%)/46 (48%), group 2 39 (41%)/56 (59%). Assessed patients: group 1: 42 (50%)/38 (46%); group 2: 31 (39%)/49 (61%) Baseline data (N lesions): group 1: 80, GB: 80 Locationof the target lesion:
Type of lesions:
Case type: New, group 1: 67 (83.3%); group 2: 69 (86.3%) Recurrence, group 1: 0 (0%); group 2: 1 (1.3%) Failure of previous treatment, group 1: 8 (10%); group 2: 0 (0%) Missed previous treatment, group 1: 5 (6.3%); group 2: 10 (12.5%) |
|
| Interventions |
Type of interventions:
Duration of intervention: 16 weeks |
|
| Outcomes |
Clinical cure of the lesions: defined as complete re‐epithelialisation of 100% (± scar), complete flattening of induration Laboratory cure of the lesions: negative smear of the lesions compared with baseline Cure rate based on weeks of follow‐up Adverse effects of 2 types of treatments Time points reported: weeks 2, 6, 12 and 16 |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation sequence was generated by the use of a randomisation Table. A simple block randomisation list with block size of 4 was prepared by a member of the study. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The randomisation allocation concealment was performed by sending the randomisation number in envelopes to a member who was responsible for giving the assigned treatment after each eligible patient was enrolled. The recruited patients were referred to this member to receive their assigned treatments." Comment: unclear because no mention of opaque and numbered envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 31 withdrawals (16/96 (16.7%) in the ILMA + cryotherapy and 15/95 (15.8%) in the CO₂ laser group) Comment: no ITT analyses were performed. However, withdrawals accounted for < 20% and were homogeneous among the treatment groups. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. Not registered. Some pre‐specified outcomes were reported. Data on individuals not presented. Conflicting data is presented between the abstract and the tables |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised double‐blind placebo‐controlled clinical trial Setting/location: Aleppo University Hospital Clinic, Aleppo, Syria Study period: 12 months (from July 2009 to July 2010) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: not described Inclusion criteria: parasitological confirmation; aged 5–65 years; normal values of liver, kidney, and pancreas function tests and electrocardiogram before treatment Exclusion criteria: pregnant and lactating women; history of cardiac, renal, and hepatic diseases; treatment with other drugs during the month before commencement of the study N randomised: 90, 30 in each group Withdrawals: not described, but it seems that there were no dropouts N assessed: 90, 30 in each group Sex (male/female): group 1, 21 (70%)/9 (30%); group 2, 17 (56.7%)/13 (43.4%); group 3, 20 (66.7%)/10 (33.3%) Mean (SD) age: group 1, 26.3 years (11.2); group 2, 24.2 years (12.5); group 3, 25.1 years (12.4) Baseline data: Mean number of lesions (SD): group 1, 3.0 (1.92); group 2, 3.0 (1.92); group 3, 3.0 (1.92) |
|
| Interventions |
Type of interventions:
Duration of intervention: 3 weeks for 3 groups |
|
| Outcomes |
Clinical and parasitological cure: the effectiveness of the treatment was classified in 3 levels: complete response of the lesions was regarded as complete clinical and parasitological healing (negative direct smear). Partial response of the lesions was regarded as decrease of the size and indurations of the lesions, and no response was regarded as no clinical change or progression of the lesions Time to healing: proportion of complete response in 3 groups during the course of treatment Adverse effects: complications Time points reported: clinical and parasitological cure: 12 weeks. Adverse effects: 6, 8, 12 weeks |
|
| Notes |
Study funding sources: this project was funded by the University of Aleppo, Aleppo, Syria Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were recruited and randomised by random allocation software into 3 treatment groups of 30 subjects each. |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding; the oral placebo was completely identical in appearance to the cimetidine tablet, they were the same colour, and drug codes were revealed only at the end of the study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study. The adverse effects are not properly described, although questionnaires regarding response to treatment and drug side effects were completed and in Table 2 there are data, numbers (%). |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Baghdad, Iraq Study period: October 1994 to November 1995 (8 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp: L major and L tropica endemic in the area Inclusion criteria: confirmed cases of CL by smear or culture, or both; acute CL with a history of 12 weeks or less Exclusion criteria: cases of reinfection N randomised: 85 Withdrawals: 22 participants (participants who did not show up after the first or second injection were excluded). Losses were not reported by group N assessed: 63. Group 1, 19; group 2, 17; group 3, 18; group 4, 9 Age: range 3 months to 65 years Sex (male/female): 28/37 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: not described |
|
| Outcomes |
Healing: responses were graded according to scale (Sharquie 1988):
Speed of healing (time taken to be 'cured'); expressed in days Prevention of scarring Adverse effects Time points reported: participants were followed up every 10‐15 days for a period of 45 days. At the end of the 6 weeks follow‐up period, the lesions were reassessed and parasitological proof of cure or otherwise was sought by smear and/or culture |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Of the 85 participants initially recruited, 63 participants were followed‐up for the full observation period. Participants who did not show up after the first or second injection were excluded. We do not know numbers of missing data across intervention groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported. |
| Methods |
Study design: randomised controlled trial Setting/location: Iraq (outpatient department) Study period: October 1994 to May 1995 and November 1995 to May 1996 (15 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major and L tropica Inclusion criteria: CL confirmed by smear/culture; acute CL of ≤ 12 weeks duration (to exclude the possibility of self healing); those for whom systemic treatment was indicated including those for whom having multiple lesions ( > 5) or large lesions ( > 4 cm²), that could not be injected, or who had lesions near to critical areas such as the eye; very young children for whom no local injection was attempted; participants who refused local treatment (2%) Exclusion criteria: received antileishmanial treatment, either local or systemic, cases of reinfection N randomised: 130. Group 1, 39; group 2, 37; group 3, 39; group 4, 15 Withdrawals: 27. Group 1, 8; group 2, 8; group 3, 7; group 4, 3 N assessed: 104. Group 1, 31; group 2, 29; group 3, 32; group 4, 12 Mean (SEM) age: group 1, 20.57 years (3.20); group 2, 22.55 years (2.72); group 3, 13.83 years (2.45); group 4, 25.74 years (4.45) Sex (male/female): 62/68. Group 1, 13/18; group 2, 19/10; group 3, 11/21; group 4, 6/6 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: ZS groups one capsule every 8 hours Duration of follow‐up: 6 weeks |
|
| Outcomes |
Healing: responses were graded according to scale (Sharquie 1988):
Speed of healing (time taken to be 'cured'); expressed in days Prevention of scarring Adverse effects Time points reported: participants were followed up every 10‐15 days for a period of 45 days. At the end of the 6 weeks follow‐up period, the lesions were reassessed and parasitological proof of cure or otherwise was sought by smear and/or culture |
|
| Notes |
Study funding sources: none declared Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropouts: 26/130 (25%) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Military Base Clinic, Iran Study period: January to October 2001 (10 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L major Inclusion criteria: proven cases of CL, healthy apart from CL, lesions not in close proximity to a vital organ or joint, number of lesions 1 to 3, ulcer size < 5 cm in diameter, onset of the lesions < 3 months, no previous standard anti‐Leishmania treatment, no history of allergy to the paromomycin family Exclusion criteria: not reported N randomised: 60, 30 in each group Withdrawals: 4. Group 1: 1; group 2: 3, N assessed: 36. Group 1: 29; group 2: 27 Age: Group 1: approx 20.6 years; group 2: approx 21.7 years Sex: all men Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: 20 days |
|
| Outcomes |
Healing rates:
Adverse effects Time points reported: follow‐up and clinical evaluation were performed at week 1 and 6 after completion of treatment. Participants were visited once at 6 months after treatment was completed. |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study was described as open and no blinding was done |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Study was described as open and no blinding was done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: relevant outcomes were reported |
| Selective reporting (reporting bias) | Low risk | Comment: relevant outcomes were reported |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: single‐centre, 3‐armed, open label, randomised, controlled, phase IIb trial Setting/location: Leishmania and Malaria Centre (LMC) of the Provincial Balkh Civil Hospital Mazar‐e‐Sharif (Afghanistan) Study period: November 2010 to March 2011 (15 months) Sample size calculation: the sample size calculation was based on the per protocol (PP) analysis, defined as all participants evaluable with respect to the primary endpoint |
|
| Participants |
Type of Leishmania: in 44/69 participants analysed, 28 (64%) lesions were infected with L tropica and 16 (36%) with L major Inclusion criteria: CL lesions with Leishmania‐positive Giemsa smears without prior CL treatment Exclusion criteria: age < 12 years; more than one skin lesion (to exclude intra‐individual variations in this phase IIb analysis); lesion age > 3 months; lesions located on eye lids, lips or nose; drug addiction; coinfection with Mycobacterium tuberculosis, HIV, or diabetes N randomised: 87 participants: group 1, 24 (27.5%), group 2, 32 (36.8%), and group 3, 31 (35.6%) Withdrawals: lost after registration: group 2, 3; group 3, 3. Not followed up for the full treatment period: SSG, 1; group 2, 6; group 3, 3. Mixed treatment: group 3, 2 N assessed: 81 (93.10%). group 1: 24; group 2: 29; group 3: 28 Mean age: 29 years (95% CI 25–33) Sex (male/female): 32 (46%)/37 (54%) Baseline data: comparable in all 3 regimens |
|
| Interventions |
Type of interventions:
Duration of intervention: for all groups: daily treatments (with the exception of Fridays) during the first week, followed by topical treatments at the centre twice a week until the end of week 4 and thereafter once a week until wound closure In group 1, the SSG treatment was discontinued after week 4. In groups 2 and 3, participants dressed their lesion themselves after week 4 with the topical NaClO₂‐basic‐crème between their visits at the centre until lesion closure |
|
| Outcomes |
Primary outcome was the ratio of closed versus open wounds at day 75 (D75) in the PP analysis for each regimen Speed of healing: not reported in Methods Adverse effects: not reported in Methods 6 visits were scheduled in the first week, 2 visits per week from weeks 2 to 4, and 1 visit per week thereafter until complete wound closure. Follow‐up visits were required once a month until day 180 after treatment start |
|
| Notes | The funders had no role in study design, data collection, data analysis, and interpretation, decision to publish, or preparation of the manuscript. KWS and HCS are members of the Board and CB is a member of the non‐profit NGO Waisenmedizin PACEM e.V. promoting access to essential medicine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Each patient was randomly assigned to one of the 3 regimens by the random allocation generator in the computer‐based Leishmedoc system." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study was described as open and no blinding was done |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "the present trial could not be conducted as either a double or a single blinded trial due to the physical nature of the applied interventions" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The intention‐to‐treat analysis (ITT) is solely added as additional information. Patients that could not be evaluated were patients that were lost immediately after registration before treatment started. 81 out of 87 patients (93.1%) were suitable for the ITT and 69 (79.3%) for the PP analysis" |
| Selective reporting (reporting bias) | Low risk | Clinicaltrials.gov ID: NCT00996463. Registered: 15 October 2009 |
| Other bias | Low risk | Other items assessed correctly reported |
| Methods |
Study design: randomised, prospective, double‐blind trial Setting/location: Dermatology Department, Ghaem Hospital, Mashhad University of Medical Sciences, Iran Study period: October 2006 to May 2008 (19 months) Sample size calculation: not described |
|
| Participants |
Type of Leishmania: L tropica Inclusion criteria: proven acute CL by a positive direct smear were selected for this study. Duration of lesions less than 6 months and no anti‐leishmaniasis treatment received during 2 months ago Exclusion criteria: pregnant or nursing women, participants with hepatic, renal, and heart diseases N randomised: 115. Group 1: 30, group 2: 85 Withdrawals: excluded because of side effects: group 2, 6. Not followed up for the full treatment period: group 1, 4; group 2, 5 N assessed: 100 (86.96%). Group 1: 26 (86.67%); group 2: 74 (87.06%) Mean (SD) age: group 1, 32 years (23), group 2: 25 years (19) Sex (male/female): group 1, 11/15, group 2: 39/35 Baseline data:
|
|
| Interventions |
Type of interventions:
Duration of intervention: group 1: 45 days, group 2: 20 days |
|
| Outcomes |
Decreased indurations of lesion (lesions were measured by palpation and ruler): in aspect of response to treatment in comparison with first visit participants graded as 4 improvement rate groups:
Decreased indurations of lesion after 45 days from completing treatment period: the same scale Adverse effects: severe muscular pain and topical reaction of inoculation site (severe erythema and pruritus) Time points reported: at the end of clinical treatment. After 45 days from completing treatment period. |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using simple randomisation". |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not done. At each visit all lesions were reexamined by the same dermatologist. The size and induration of lesions were measured by palpation and ruler. No photographs were taken |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 100 out of 115 participants completed the study. 6 participants who received systemic MA were excluded because of side effects that included severe muscular pain and topical reaction of inoculation site (severe erythema and pruritus) |
| Selective reporting (reporting bias) | High risk | Protocol not available. Not registered. Alhtough most pre‐specified outcomes were reported, adverse effects were not clearly reported (only for those excluded from the analysis) |
| Other bias | Unclear risk | There was not enough information in the publication to assess if there were other biases present. |
| Methods |
Study design: randomised controlled trial Setting/location: Fars province, Iran Study period: not described Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp: L major and L tropica in the area Inclusion criteria: diagnosed as having CL based on positive smears from lesions, and in some cases, cultures and histopathologic studies were also performed. Exclusion criteria: pregnant, nursing, or serious concomitant diseases. N randomised: 171 Withdrawals: 0 N assessed: 171 participants: 86 participants in the herbal extract Z‐HE group and 85 participants in the IMMA (15 to 20 mg/kg/d) Age: range 10 months to 69 years Sex (male/female): 84/87 Baseline data: the duration of the disease was < 4 months. Most participants had papular and papulonodular lesion(s), although other clinical forms including ulcerative, eczematoid, hyperkeratotic, and erysipeloid types were also present. Most participants had multiple lesions, and the most common sites were the face and the extremities. |
|
| Interventions |
Type of interventions:
Duration of intervention: 20 consecutive days |
|
| Outcomes |
Healing rates: complete cure was defined as clinical improvement with complete healing and re‐epithelialisation of the lesion(s), partial cure as partial clinical improvement with reduction in infiltration erythema, and size of the lesion(s), and failure as the absence of any changes in the lesion(s) or progression and worsening of the lesion(s) Adverse effects Time points reported: 6 weeks post‐treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgment |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
| Methods |
Study design: randomised controlled trial Setting/location: Turkey Study period: around 8 weeks Sample size calculation: not described |
|
| Participants |
Type of Leishmania: Leishmania spp: L tropica in the area Inclusion criteria: confirmed diagnosis of CL Exclusion criteria: pregnant, nursing, or serious concomitant diseases N randomised: 72 Withdrawals: 0 N assessed: 72 (100%): 40 participants in group 1 and 32 participants in group 2 Age: most were children aged 10 years or younger Sex: both sexes Baseline data: the most common site of the lesion was the face and most of the participants had one papulonodular lesion, with duration of the lesions varying from 1 to 12 months
|
|
| Interventions |
Type of interventions:
Duration of intervention: 4 weeks |
|
| Outcomes |
Healing rates: percentage of participants 'cured' one month after treatment Adverse effects Time points reported: at the end of treatment and 4 weeks post‐treatment |
|
| Notes |
Study funding sources: not reported Possible conflicts of interest: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were divided randomly into two treatment groups" Comment: insufficient detail was reported about the method used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "An open‐labeled, N randomised controlled trial to evaluate the efficacy of paromomycin ointment as compared with ketoconazole" Comment: we think the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "No subjects withdrew because of this adverse effect" Comment: no dropouts, so ITT analyses were performed. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that the published reports include all expected outcomes. |
| Other bias | High risk | Sample size calculation and reporting of Leishmania spp involved was not correctly reported |
ACL: anthroponotic cutaneous leishmaniasis; AL: allopurinol; CCR: complete clinical response; CL: cutaneous leishmaniasis; HF‐EC: high frequency electrocauterisation; HSCS: hypertonic sodium chloride solution; IL: intralesional; ILMA: intralesional meglumine antimoniate; ILSSG: intralesional sodium stibogluconate; IM: intramuscular; IMMA: intramuscular meglumine antimoniate; IQR: interquartile range; IV: intravenous; MA: meglumine antimoniate; MBCL: methyl benzethonium chloride; MDLBT: median duration of lesions before therapy; MNL: median number of lesions; MSL: median size of lesions; MWT: moist wound treatment; PDT: topical photodynamic therapy; PR: paromomycin; RF: radiofrequency; RFHT: radiofrequency heat therapy; SD: standard deviation, SEM: standard error of the mean; SSG: sodium stibogluconate; TCA: trichloroacetic acid; TM: ThermoMed; ZS: zinc sulphate;
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alavi‐Naini 2012 | Review |
| Banihashemi 2015 | Inclusion criteria of the patients: patients with lupoid leishmaniasis, chronic recurrent leishmaniasis, that typically follows an acute form of cutaneus leishmaniasis |
| Bumb 2010 | Not an RCT. "Selected patients were categorised into two groups (groups A and B) of 110 persons each. Alternate patients were included in group A and B, respectively" |
| Dogra 1986 | They stated that participants were randomly selected for the study, but not randomly assigned to the treatment groups. |
| Dogra 1994 | They stated that participants were randomly selected for the study, but not randomly assigned to the treatment groups. |
| Dorlo 2012 | Review |
| El On 1992 | Cross‐over study |
| Frankenburg 1993 | It measured immunity parameters but did not show any clinical results. |
| Kim 2009 | This was a meta‐analysis. |
| Moosavi 2005 | The method of generation of randomisation was inappropriate. The author was contacted and stated that after enrolling the participants, they did consecutively allocate ILMA for odd number participants and used topical paromomycin for even number participants. |
| Nilforoushzadeh 2010 | They stated that participants were randomly selected for the study, but not randomly assigned to the treatment groups. |
| Nilforoushzadeh 2011 | Included immunocompromised and chronically ill patients |
| Siavash 2013 | Only studied electrocardiogram and biochemical adverse effects of meglumine antimoniate |
| Singh 1995 | They stated that participants in both groups were randomly selected. However, they did not have an initial population eligible for the study that was randomly divided in the 2 treatment groups. But rather one group of people that were randomly selected for one group and another group of participants that were also randomly selected to form part of the other group. Besides, the duration of treatments was not the same and the follow‐up was also different for both treatment groups. |
| Trau 1987 | No clear data is available for multiple lesions. Only lesions from participants with multiple lesions were randomised. The lesions from participants with single lesions a cross‐over study was performed. |
RCT: randomised controlled trial.
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Double‐blind randomised controlled trial This study was carried out in some clinics of Kerman University of Medical Sciences (Iran), from October 2008 to December 2010. Sample size calculation: a sample size of 40 participants per treatment group was planned, a probability of a type I error at alpha = 0.05 and beta = 0.1 to determine a 20% difference between intralesional injection of ZS 2 % solution with ILMA (Glucantime) |
| Participants |
Type of Leishmania: L tropica Inclusion criteria: the presence of parasitological confirmed lesion(s) of CL and aged 5–60 years Exclusion criteria: disease duration more than 6 months, those with a history of hypersensitivity to MA (Glucantime) or ZS, pregnant or nursing women, those with more than 5 lesions, those with lesions with size of 5 cm or more, and a history of any anti‐leishmanial therapy during last 4 weeks N randomised: 80, 40 in each group Withdrawals: 34, 18 in the ZS‐treated group and 16 patients in the MA (Glucantime)‐treated group (lost to follow‐up in all of the participants except in one in the ZS‐treated group, who had severe necrosis) N assessed: 46; 22 patients in the ZS‐treated group and 24 patients in the MA (Glucantime)‐treated group There was no significant difference between compared groups' baseline characteristics |
| Interventions |
Group 1: intralesional injection of ZS 2% vials once a week for 10 weeks or sooner in case of complete resolution of the lesions Group 2: ILMA once a week for 10 weeks or sooner in case of complete resolution of the lesions Co‐intervention: in both groups cryotherapy was performed once every other week for 10 weeks |
| Outcomes |
Clinical response: no response to treatment was defined if the area of a lesion decreased less than 75%, partial treatment if decreased 75% to 99%, and complete treatment if decreased 100% compared to its baseline area Time to healing (partial/complete treatment) Adverse effects: there were no major side effects in either group. Pain was observed in all participants of both groups. In ZS group 4 participants developed necrosis of the site of the injection while this was not observed in MA (Glucantime) group at all Time points reported: on average, each participant was followed for 4.1 weeks (SD 2.6). |
| Notes |
Study funding sources: none declared Possible conflicts of interest: there is no conflict of interest |
| Methods | Triple‐blind randomised controlled trial Performed in Shahid Dadbin Clinic of Leishmaniasis Research Center (Iran), at Kerman University of Medical Sciences, between 2008 and 2010 Sample size calculation: a sample size of 44 participants per treatment group was planned with a probability of a type I error at alpha = 0.05 and beta = 0.1 to determine a 20% difference between topical terbinafine and vehicle in outcome. |
| Participants |
Type of Leishmania: L major and L tropica in the area Inclusion criteria: < 2 years of age; parasitological diagnosis of cutaneous leishmaniasis from all lesions with direct smear; ulcerative lesions with duration of less than one month that were not on exposed areas or joints; the size of lesions has to be < 3 cm; ≤ 3 lesions; no anti‐leishmanial therapy during last 2 weeks, and with no history of hypersensitivity to MA Exclusion criteria: pregnant or nursing women, patients with hepatic, renal, or heart diseases N randomised: 88, 44 in each group Withdrawals: 36 in topical terbinafine + MA; 29 in vehicle + MA N assessed (3 months): 9 in topical terbinafine + MA; 15 in vehicle + MA There are significant differences between participants' sex in intervention and control groups |
| Interventions |
Group 1: IMMA 20 mg/kg/d + topical terbinafine for 20 days 3 months follow‐up after completing of the treatment phase Group 2: IMMA 20 mg/kg/d + vehicle (Mahan Vaseline) for 20 days Visited at days: 0, 14, 30 and 44 |
| Outcomes |
Clinical cure, defined as:
Time to healing (partial/complete treatment) |
| Notes |
Study funding sources: none declared Possible conflicts of interest: there is no conflict of interest. |
| Methods | Randomised controlled study Department of Dermatology, Sheikh Zayed Medical College/Hospital, Rahim Yar Khan, Pakistan, from November 2013 to June 2014 Sample size: was calculated by using a formula for 2 proportions; expected proportion for duration of treatment in one group was taken as 55, and in second group 51 days; power of study was taken as 80, and standard error of 5% |
| Participants |
Type of Leishmania: not reported Inclusion criteria: patients of all age groups except neonates, with single or multiple parasitologically confirmed lesions of cutaneous leishmaniasis with no past history of disease and not previously treated with SSG or MA Exclusion criteria: retinal abnormality or visual field defects, pregnancy, hepatic dysfunction, known hypersensitivity to chloroquine and those having visceral leishmaniasis N randomised: 86; 48 intralesional chloroquine, 38 oral chloroquine Withdrawals: 0 N assessed: 86; 48 intralesional chloroquine, 38 oral chloroquine |
| Interventions | Intralesional and oral chloroquine administration Co‐interventions: lesions with secondary bacterial infection before, during, or after treatment were treated with topical antibiotics. If such infections required systemic treatment, an antibiotic that has no activity against Leishmania (e.g. erythromycin) was given. |
| Outcomes |
Cure was defined as the complete re‐epithelialisation of the ulcerated lesions, with no evidence of papules, inflammation, or induration Quantitative variables like, duration, cost and total dose of treatment were calculated Adverse effects Time points reported: < 4 weeks, 5‐8 weeks, ≥ 9 weeks |
| Notes |
Study funding sources: none declared Possible conflicts of interest: none declared |
| Methods | Interventional randomised controlled study was conducted in the Skin Disease and Leishmaniasis Research Center of Isfahan University of Medical Sciences, Isfahan, Iran, between April 2010 and May 2011 Sample size calculation: not described |
| Participants |
Leishmania type: not described Inclusion criteria: patients older than 5 years with biopsy‐confirmed cutaneous leishmaniasis were eligible if they had the following criteria: lesion diameter < 3 cm, disease duration < 12 weeks, lesion‐to‐eyelid distance > 2 cm, and no history of systemic or topical therapy for cutaneous leishmaniasis Exclusion criteria: pregnancy, lactation, immunosuppressive therapy, and serious side effects of medication N randomised: 90. 30 in each group (ILMA (Glucantime) alone, ILMA + TCA 50%, or fractional CO₂ laser) Withdrawals: 14 N assessed: 76 participants (84.5%) completed treatment and were followed up for 60 days |
| Interventions | ILMA alone, ILMA + TCA 50% or fractional CO₂ laser for up to 8 weeks Participants in all 3 groups were followed up weekly for the next 6 months. |
| Outcomes | The clinical response was rated as complete improvement (complete re‐epithelialisation of the lesion with negative direct skin smear result), partial improvement (50%–75% improvement of the lesion size), and no change in the lesion appearance. Time to healing Adverse effects The improvement of scar was also evaluated according to the participant's satisfaction, the morphology of the lesion, the level of induration, and the level of atrophy and scored as follows: score of 1 (less than 5% improvement), score of 2 (25% to 50% improvement), score of 3 (51% to 75% improvement), and score of 4 (76% to 100% improvement) |
| Notes |
Study funding sources: none declared Possible conflicts of interest: there is no conflict of interest. |
| Methods | Exploratory phase II, randomised, vehicle‐controlled, single‐centre (Ankober Health Center, North Shewa zone of Amhara region, Ethiopia) study Sample size calculation: not described |
| Participants |
Leishamania type: not described Inclusion criteria: patients with localised cutaneous leishmaniasis (LCL) aged 18–65 years with a new, uncomplicated, localised, single lesion on the face or arms and positive parasite smear by microscopy Exclusion criteria: cutaneous leishmaniasis with secondary infection, concomitant diseases (mucocutaneous or visceral leishmaniasis), history of anti‐leishmanial treatment within the past 6 months, abnormality of biochemical and/or haematological laboratory tests, known hypersensitivity to any of the Shiunko components, or pregnancy (positive urine HCG test), breastfeeding, or possibility of becoming pregnant during the study N randomised: 40 were randomised to receive vehicle and Shiunko ointment (20 participants for each group) Withdrawals: 2 participants did not receive complete treatment, and 9 were not completely followed up. In the vehicle group, 2 and 5 cases did not have complete treatment and follow‐up, respectively. N assessed: 38 participants had complete treatment and 31, complete follow‐up. In the vehicle group, 18 and 15 cases had complete treatment and follow‐up, respectively. |
| Interventions |
Group 1: the composition of Shiunko was as follows: 2.04 g shikon, 1.02 g tohki, 16.92 g sesame oil, and 6.76 g honeycomb wax Group 2: the vehicle contained 20 g wax Co‐interventions: cryotherapy was to be offered to all participants who were withdrawn or discontinued from the study for safety reasons Vehicle and Shiunko ointment applied on the lesion twice a day for 4 weeks Follow‐up: 16 weeks |
| Outcomes |
Cure: complete cure was defined as complete wound closure and re‐epithelialisation without inflammation or infiltration and absence of parasite (amastigotes) within 12 weeks after the end of treatment. Partial response was defined as improvement of Leishmania signs (skin oedema, erythema, and/or hardening) and/or reduction in size but not total disappearance of the lesion and absence of parasite (amastigotes) within 12 weeks after the end of treatment. Treatment failure was defined as failure of the lesion size to decrease and/or lack of lesion sign improvement or re‐epithelialisation and/or presence of leishmanial amastigotes in the lesion 12 weeks after end of treatment (week 16) Adverse effects Clinical and parasitological assessments were performed before treatment, weekly for 4 weeks, and then 4, 8, and 12 weeks after the end of treatment |
| Notes |
Study funding sources: the study was supported by the Institute of Tropical Medicine, Nagasaki University (Japan), ArmauerHansen Research Institute (Ethiopia), and Okusa Co., Ltd. (Japan). Possible conflicts of interest: there is no conflict of interest. |
| Methods | Clinical randomised trial conducted from March 2008 through 2009 in the dermatology department of Quaem University Hospital, Mashhad, Iran Sample size calculation: not described |
| Participants |
Leishamania type: not described Inclusion criteria: positive smear for leishmaniasis with disease duration of less than 12 weeks, no treatment for the current condition before and volunteer to participate in the study Exclusion criteria: pregnancy, breastfeeding, history of simultaneous treatment with other methods before or during the trial, history of local cutaneous or systemic diseases in the past 2 months, history of intolerance or allergy to macrolides, severe underlying diseases such as cardiovascular, renal, or hepatic diseases N randomised: 96; 26 (43 lesions) azithromycin, 40 (54 lesions) MA (Glucantime) Withdrawals: 2 azithromycin group N assessed: 94; 24 (40 lesions) azithromycin, 40 (54 lesions) MA |
| Interventions |
Group 1: topical liposomal form of azithromycin was administered for the first group twice daily. Group 2: the other group was treated by weekly ILMA with a volume of 0.5–2 cm³ into each lesion till complete blanching of the lesion occurred Clinical evaluations were performed every week during the treatment course (8 weeks) by a single dermatologist in both groups. The participants were followed up for recurrence or complications 6 and 12 months after the end of the treatment course. |
| Outcomes |
Complete cure was defined as full re‐epithelialisation, disappearance of oedema, induration, and other signs of inflammation, and a negative direct skin smear result Improvement rate (%): complete improvement (full re‐epithelialisation of the lesions for ulcerative ones or disappearance of induration and erythema; significant improvement (decrease in induration size > 60%); moderate improvement (decrease in induration size between 30% and 60%; slight improvement (decrease in induration size of < 30%). Adverse effects |
| Notes |
Study funding sources: none declared Possible conflicts of interest: none declared |
| Methods | Interventional randomised study Setting: Sri Lanka Sample size calculation: not described |
| Participants |
Leishmania type:L donovani Inclusion criteria: Laboratory‐confirmed CL patients with single lesions Exclusion criteria: — N randomised: test group (n = 98); control group (n = 115) Withdrawals: — N assessed: — |
| Interventions |
Group 1: single session of radiofrequency induced heat therapy (RFHT) at 50ºC for 30 s Group 2: received weekly ILSSG until cure or 10 doses Participants were followed up fortnightly for 12 weeks to assess clinical response and adverse effects. |
| Outcomes |
Cure rate Adverse effects Time points reported: 8, 10, and 12 weeks |
| Notes | Poster |
| Methods | Randomised controlled trial |
| Participants | 40 participants (no more data described in abstract) |
| Interventions | Topical ointment prepared from the stem extract of Morinda citrifolia (no more data described in abstract) |
| Outcomes | Improvement (no more data described in abstract) |
| Notes | Article request to authors |
CL: cutaneous leishmaniasis; HCG: human chorionic gonadotropin; ILMA: intralesional meglumine antimoniate; IM: intramuscular; MA: meglumine antimoniate; SSG: sodium stibogluconate; ZS: zinc sulphate.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | A clinical trial to assess the safety and effect of heat therapy in comparison to standard intra‐lesional sodium stibogluconate for cutaneous leishmaniasis |
| Methods | Randomised controlled trial |
| Participants |
Inclusion criteria: suggestive skin lesions (papules, nodules, plaques, ulcers and nodule‐ulcers) who were clinically diagnosed by a consultant dermatologist and parasitologically confirmed as CL Exclusion criteria: non‐localised leishmaniasis (VL, MCL, leishmaniasis recidivans, diffuse cutaneous leishmaniasis and post‐kala‐azar dermal leishmaniasis); use of prior concomitant treatment for leishmaniasis including any traditional medicines; lesions close to nasal, oral, urogenital, anal areas (mucosae) and the eyes; multiple lesions; any chronic or concomitant illnesses and people on pace makers and any metallic devices; pregnancy and breastfeeding mothers; children < 12 years of age; immunocompromised states including HIV/AIDS and use of immunosuppressants like steroids; alcohol abuse; not capable of understanding and complying with the study protocol; known hypersensitivity or allergy to treatment Age: 12‐90 years |
| Interventions | The device used was the ThermoMed 1.8, ThermoSurgery Technologies portable battery operated device. The generator has received clearance by the US Food and Drug Administration for the treatment of CL. It delivers precisely controlled localised radiofrequency waves to selectively destroy diseased tissue. Heat therapy was administered as a single session. The lesion and surrounding normal skin was cleaned with normal saline. Then the area was anaesthetised with 2% lignocaine using a 1 cc syringe. The device was used in according to manufacturers instructions (ThermoMed 1.8; Thermosurgery). It generates a 6.78 mHz frequency applied with a hand set that is attached to 2 applicator electrodes which were placed on the diseased skin. A temperature of 50ºC was applied for 30 s to cover an area about 49‐73 mm², according to the size of electrodes (3 sizes are provided). Then the applicator is moved to another area of the lesion until the entire area is treated. Once treatment begins the temperature is measured by a thermistor embedded in the applicator which ensures that the applied temperature remains constant. After treatment the lesions were covered with gauze to prevent secondary infections and participants were given an antibiotic cream (soframycin 1% per 30 g depending on the size of lesion to cover the lesion) to be applied twice a day for 3 days. |
| Outcomes |
Recurrence Treatment efficacy was measured by the percentage of participants clinically cured by 8 weeks, 10 weeks, 12 weeks and 16 weeks after initiation of treatment in both groups.
Any adverse effects like secondary bacterial infections, allergy to treatment or hypersensitivity and worsening of the lesion clinically was recorded during or after treatment with either therapy |
| Starting date | December 2014 |
| Contact information | Dr Fathima Wardha Refa PGIM address: No. 160, Professor Nandadasa Kodagoda Mawatha (Norris Canal Road), 00700, Colombo 7. 2.Department of Parasitology, Faculty of Medicine Colombo University of Colombo address: No 25, Kynsey Road, Colombo 8 0080, Sri Lanka. 7 MACLEOD ROAD COLOMBO 4, Sri Lanka +94 727800003 or +94 11 2580093 or+94 11 2699284 wardharefai@yahoo.com |
| Notes | — |
| Trial name or title | Comparison between the efficacy of intralesional placebo and nitric oxide releasing patch versus placebo patch and Glucantime in the treatment of cutaneous leishmaniasis |
| Methods | Randomised, placebo‐controlled trial |
| Participants |
Inclusion criteria: diagnosis of acute zoonotic cutaneous leishmaniasis, proven parasitology by direct smear; aged 18‐50 years; lesion diameter ≤ 3 cm; 1 lesion, not located on the face near the eyes, joints, cartilage, on the nose and ears; < 3 months passed from diseases times Exclusion criteria: pregnancy and lactation; risk of presence or incidence of sporotrichoid or satellite lesions; history of hepatitis, heart, or kidney disorder; taking immunosuppressive medications (over the last 6 months); local or systemic treatment against leishmaniasis in the last 3 months; history of heart attack or high blood pressure; liver disease; anaemia; history of surgery for head and brain haemorrhage; taking blood pressure medications, antidepressants, or drugs used for angina |
| Interventions |
Group 1: nitroglycerin patch (the pharmacy of Nour and Ali Asghar hospital in Isfahan) once a day covering 5.08 to 7.62 cm of lesion + intralesional injection of placebo (injection of distilled water) once a week (15‐30 mg) until whitening of base of the ulcer for 8 weeks or until complete cure Group 2: ILMA (Meglusan) from Avenue de Scheut company in Belium, once a week (15‐30 mg) until whitening of base of the ulcer + placebo patch (the pharmacy of Nour and Ali Asghar hospital in Isfahan) once a day covering 5.08 to 7.62 cm of lesion for 8 weeks or until complete cure |
| Outcomes |
Improvement rate. Timepoint: 1, 2, 3, 4, 7, 8 weeks during treatment and 12, 16, 20 weeks after end of treatment. Method of measurement: observation and examination Induration. Timepoint: 1, 2, 3, 4, 7, 8, 12, 16, 20 weeks after end of treatment. Method of measurement: physical examination Lesion area. Timepoint: 8, 12, 20 weeks after end of treatment |
| Starting date | August 2013 |
| Contact information | Fariba Jaffary Skin Diseases and Leishmaniasis Research Center, Isfahan University of Medical Sciences, Sedigheh Tahereh (AS) Research Centers Complex, Khorram Ave., Isfahan, Iran Isfahan Iran, Islamic Republic Of Iran. 00983113373736 jaffary@pharm.mui.ac.ir |
| Notes | — |
| Trial name or title | The effect of MJ1 (topical dairy extract) versus routine care for treatment of cutaneous leishmaniasis (rural) in Isfahan Iran:a randomised controlled clinical trial (RCTs) |
| Methods | Randomised controlled trial |
| Participants |
Inclusion criteria: cutaneous leishmaniasis of parasitology confirmed that they have not been previously treated; any other medical problem (such as heart disease, renal, hepatic, or haematological); available for follow‐up; informed consent to participate in the study Exclusion criteria: secondary infections, history of allergic reactions |
| Interventions |
Group 1: in this group patients apply MJ1 skin cream 3 times a day, they are trained to use it for a month with a diameter of 3 mm on the rub of the lesion without dressing Group 2: skin cream MJ1 ‐ 3 times a day ‐ for a month |
| Outcomes |
Lesion size, lesion diameters measured Timepoint: 1, 2, 3, 4 and 8 weeks after starting treatment, participants are evaluated. Method of measurement: at each visit, the 2 dimensions are measured and the area calculated |
| Starting date | December 2013 |
| Contact information | Dr Mohsen Janghorbani Hezar jarib Street, School of Public Health, Isfahan University of Medical Sciences Isfahan Iran, Islamic Republic Of 00983117922774 janghorbani@hlth.mui.ac.ir |
| Notes | — |
| Trial name or title | Phase 2 study of the efficacy of daylight activated photodynamic therapy in the treatment of cutaneous leishmaniasis |
| Methods | Randomised, parallel assignment, open label |
| Participants | Age: ≥ 18 years |
| Interventions |
Group 1: photodynamic therapy Application of Metvix 16% cream followed by exposure to daylight for 2.5 h Group 2: cryotherapy for 2 times 20 s |
| Outcomes | Eradiation of amastigotes |
| Starting date | September 2009 |
| Contact information | Prof Claes D. Enk, Hadassah Medical Organization |
| Notes | — |
| Trial name or title | Pilot study of efficacy of topical nano‐liposomal meglumine antimoniate (Glucantime) or paromomycin in combination with systemic Glucantime for the treatment of anthroponotic cutaneous leishmaniasis (ACL) caused by Leishmania tropica |
| Methods | Randomised, parallel assignment, double‐blind |
| Participants |
Inclusion criteria: aged 12‐60 years; parasitologically proven CL due toL tropica; history of failure to at least one full course of systemic meglumine antimoniate (Glucantime); general good health based on history and physical examination; ≤ 4 lesions; lesion size < 3 cm; signed informed consent voluntarily and knowingly; guardian's signature for volunteer less than 18 years old Exclusion criteria: pregnant or lactating women and those who are planning to be pregnant in next 60 days; use of other types of treatment for CL; involvement in any other drug or vaccine trial during the study period; known heart, kidney, liver diseases based on history and physical exam; abnormal ECG Age: 12‐60 years |
| Interventions |
Group 1: liposomal paromomycin (liposomes containing 10% paromomycin
) + liposomal MA Group 2: liposomal MA (Glucantime) Group 3: placebo + liposomal MA |
| Outcomes | Complete cure equal to complete re‐epithelialisation of all lesions |
| Starting date | March 2011 |
| Contact information | Ali Khamesipour, Tehran University of Medical Sciences |
| Notes | — |
| Trial name or title | Randomised, double‐blind, controlled study on efficacy and safety of intralesional metronidazole vs intralesional sodium stibogluconate in L donovani cutaneous leishmaniasis |
| Methods | Randomised controlled trial |
| Participants |
Inclusion criteria: > 12 years of age, positive slit skin smear and/or skin biopsy for Leishmania parasites Exclusion criteria: pregnancy, breastfeeding, known renal impairment, known liver impairment, congestive cardiac failure |
| Interventions |
Group 1: intralesional metronidazole 0.2‐4 mL per lesion depending on the size of the lesion, weekly until cure or maximum of 10 injections Group 2: intralesional stibogluconate 0.2‐4 mL per lesion depending on the size of the lesion, weekly until cure or maximum of 10 injections |
| Outcomes |
Rate of clinical cure Adverse effects – anticipated local side effects are pain, ulceration, scarring, postinflammatory hyperpigmentation or depigmentation. Systemic side effects are not anticipated as the drug is given intralesionally. |
| Starting date | November 2014 |
| Contact information | Ranthilaka R Ranawaka 0112855200 ranthilaka37@yahoo.com |
| Notes | — |
CL: cutaneous leishmaniasis; ECG: electrocardiogram; ILMA: intralesional meglumine antimoniate.
Contributions of authors
JHM was the contact person with the editorial base; he coordinated contributions from the co‐authors and wrote the final draft of the review. PLP, EGC, BMM, and JHM screened papers against eligibility criteria. PLP obtained data on ongoing and unpublished studies. MP, JHM, and PLP appraised the quality of papers. MP, PLP, EGC, PC, and JHM extracted data for the review and sought additional information about papers. JHM, PLP, EGC, BMM, MP, RLV, and PC entered data into RevMan. AR, JHM, BMM, and RLV analysed and interpreted data. AR and JHM worked on the Methods sections. BMM and RLV drafted the clinical sections of the Background and responded to the clinical comments of the referees. AR and JHM responded to the methodology and statistics comments of the referees. IMR was the consumer co‐author and checked the review for readability and clarity, as well as ensuring outcomes were relevant to consumers. JHM Is the guarantor of the update.
Disclaimer
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Sources of support
Internal sources
-
Cochrane Madrid. Hospital Universitario Ramón y Cajal. Carretera de Colmenar Km 3.1 Madrid 28034, Spain.
Methodology support
External sources
Office of Control of Neglected Tropical Diseases (WHO/CDS/NTD/IDM), Communicable Disease Cluster, World Health Organization, Switzerland.
-
The National Institute for Health Research (NIHR), UK.
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Julio Heras‐Mosteiro: none known. Begoña Monge‐Maillo: none known. Mariona Pinart: none known. Patricia Lopez Pereira: none known. Emely Garcia‐Carrasco: none known. Pedro Campuzano Cuadrado: none known. Ana Royuela: none known. Irene Mendez Roman: none known. Rogelio López‐Vélez: none known.
Iraj Sharifi, one of the clinical referees, was a co‐author on the included studies Khatami 2012 and Daie Parizi 2015.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
- Adam I, Hagelnur A. Artesunate plus sulfamethoxypyrazine/pyrimethamine for the treatment of cutaneous leishmaniasis: a double‐blind, placebo‐controlled clinical trial. International Journal of Antimicrobial Agents 2009;34(4):380‐1. [PUBMED: 19409761 ] [DOI] [PubMed] [Google Scholar]
- al‐Fouzan AS, al‐Saleh QA, Najem NM, Rostom AI. Cutaneous leishmaniasis in Kuwait. Clinical experience with itraconazole. International Journal of Dermatology 1991;30(7):519‐21. [PUBMED: 1663089] [DOI] [PubMed] [Google Scholar]
- Al Hamdi K, Awad AH, Moker HM. Evaluation of intralesional 0.2% ciprofloxacin as a treatment for cutaneous leishmaniasis. Eastern Mediterranean Health Journal 2010;16(1):89‐93. [PUBMED: 20214164] [PubMed] [Google Scholar]
- Alkhawajah AM, Larbi E, al‐Gindan Y, Abahussein A, Jain S. Treatment of cutaneous leishmaniasis with antimony: intramuscular versus intralesional administration. Annals of Tropical Medicine and Parasitology 1997;91(8):899‐905. [PUBMED: 9579209] [DOI] [PubMed] [Google Scholar]
- Alrajhi AA, Ibrahim EA, Vol EB, Khairat M, Faris RM, Maguire JH. Fluconazole for the treatment of cutaneous leishmaniasis caused by leishmania major. New England Journal of Medicine 2002;346(12):891‐5. [PUBMED: 11907288] [DOI] [PubMed] [Google Scholar]
- Alsaleh QA, Dvorak R, Nanda A. Ketoconazole in the treatment of cutaneous leishmaniasis in Kuwait. International Journal of Dermatology 1995;34(7):495‐7. [PUBMED: 7591417] [DOI] [PubMed] [Google Scholar]
- Aronson NE, Wortmann GW, Byrne WR, Howard RS, Bernstein WB, Marovich MA, et al. A randomized controlled trial of local heat therapy versus intravenous sodium stibogluconate for the treatment of cutaneous Leishmania major infection. PLoS Neglected Tropical Diseases 2010;4(3):e628. [PUBMED: 20231896] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asilian A, Jalayer T, Whitworth JA, Ghasemi RL, Nilforooshzadeh M, Olliaro P. A randomized, placebo‐controlled trial of a two‐week regimen of aminosidine (paramomycin) ointment for treatment of cutaneous leishmaniasis in Iran. American Journal of Tropical Medicine and Hygiene 1995;53(6):648‐51. [PUBMED: 8561269] [DOI] [PubMed] [Google Scholar]
- Asilian A, Jalayer T, Nilforoosshzadeh M, Ghassemi RL, Peto R, Wayling S, et al. Treatment of cutaneous leishmaniasis with aminosidine (paromomycin) ointment: double‐blind, randomized trial in the Islamic Republic of Iran. Bulletin of the World Health Organization 2003;81(5):353‐9. [PUBMED: 12856053] [PMC free article] [PubMed] [Google Scholar]
- Asilian A, Sadeghinia A, Faghihi G, Momeni A. Comparative study of the efficacy of combined cryotherapy and intralesional meglumine antimoniate (Glucantime) vs. cryothrapy and intralesional meglumine antimoniate (Glucantime) alone for the treatment of cutaneous leishmaniasis. International Journal of Dermatology 2004;43(4):281‐3. [PUBMED: 15090013] [DOI] [PubMed] [Google Scholar]
- Asilian A, Sharif A, Faghihi G, Enshaeieh Sh, Shariati F, Siadat AH. Evaluation of CO laser efficacy in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 2004;43(10):736‐8. [PUBMED: 15485530] [DOI] [PubMed] [Google Scholar]
- Asilian A, Davami M. Comparison between the efficacy of photodynamic therapy and topical paromomycin in the treatment of Old World cutaneous leishmaniasis: a placebo‐controlled, randomized clinical trial. Clinical & Experimental Dermatology 2006;31(5):634‐7. [PUBMED: 16780497] [DOI] [PubMed] [Google Scholar]
- Asilian A, Omrani Sh, Nilforoushzadeh MA. Comparing the effects of topical miltefosine and glucantime in treatment of cutaneous leishmaniasis. Journal of Isfahan Medical School 2014;31(269):2257‐63. [Google Scholar]
- Ben Salah A, Zakraoui H, Zaatour A, Ftaiti A, Zaafouri B, Garraoui A, et al. A randomized, placebo‐controlled trial in Tunisia treating cutaneous leishmaniasis with paromomycin ointment. American Journal of Tropical Medicine and Hygiene 1995;53(2):162‐6. [PUBMED: 7677218] [DOI] [PubMed] [Google Scholar]
- Ben Salah A, Buffet PA, Morizot G, Ben Massoud N, Zâatour A, Ben Alaya N, et al. WR279,396, a third generation aminoglycoside ointment for the treatment of Leishmania major cutaneous leishmaniasis: a phase 2, randomized, double blind, placebo controlled study. PLoS Neglected Tropical Diseases 2009;3(5):e432. [PUBMED: 19415122] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Salah A, Ben Messaoud N, Guedri E, Zaatour A, Ben Alaya N, Bettaieb J, et al. Topical paromomycin with or without gentamicin for cutaneous leishmaniasis. New England Journal of Medicine 2013;368(6):524‐32. [PUBMED: 23388004 ] [DOI] [PubMed] [Google Scholar]
- Bumb RA, Prasad N, Khandelwal K, Aara N, Mehta RD, Ghiya BC, et al. Long‐term efficacy of single‐dose radiofrequency‐induced heat therapy vs. intralesional antimonials for cutaneous leishmaniasis in India. British Journal of Dermatology 2013;168(5):1114‐9. [PUBMED: 23298394] [DOI] [PubMed] [Google Scholar]
- Daie Parizi MH, Karvar M, Sharifi I, Bahrampour A, Heshmat Khah A, Rahnama Z, et al. The topical treatment of anthroponotic cutaneous leishmaniasis with the tincture of thioxolone plus benzoxonium chloride (Thio‐Ben) along with cryotherapy: A single‐blind randomized clinical trial. Dermatologic Therapy 2015;28(3):140‐6. [PUBMED: 25847678] [DOI] [PubMed] [Google Scholar]
- Dandashi A. Treatment of cutaneous leishmaniasis with fluconazole: a randomized double‐blind, placebo‐controlled trial. Journal of the European Academy of Dermatology and Venereology : JEADV 2005;19(Suppl 2):43. [DOI] [PubMed] [Google Scholar]
- Dastgheib L, Naseri M, Mirashe Z. Both combined oral azithromycin plus allopurinol and intramuscular Glucantime yield low efficacy in the treatment of Old World cutaneous leishmaniasis: a randomized controlled clinical trial. International Journal of Dermatology 2012;51(12):1508‐11. [PUBMED: 23171020] [DOI] [PubMed] [Google Scholar]
- Dogra J, Aneja N, Lal BB, Mishra SN. Cutaneous leishmaniasis in India: Clinical experience with itraconazole (R51 211 Janssen). International Journal of Dermatology 1990;29(9):661‐2. [PUBMED: 2177041] [DOI] [PubMed] [Google Scholar]
- Dogra J. A double‐blind study on the efficacy of oral dapsone in cutaneous leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1991;85(2):212‐3. [PUBMED: 1887473] [DOI] [PubMed] [Google Scholar]
- Dogra J. Cutaneous leishmaniasis in India: evaluation of oral drugs (dapsone versus itraconazole). European Journal of Dermatology 1992;2:568‐9. [Google Scholar]
- Dogra J, Saxena VN. Itraconazole and leishmaniasis: a randomised double‐blind trial in cutaneous disease. International Journal for Parasitology 1996;26(12):1413‐5. [PUBMED: 9024895] [DOI] [PubMed] [Google Scholar]
- Ejaz A, Qadir SNUR, Malik N, Bari AU. Comparison of low‐dose meglumine antimoniate/ allopurinol combination therapy with full dose meglumine antimoniate alone in the treatment of cutaneous leishmaniasis ‐ A randomized controlled trial. Journal of Pakistan Association of Dermatologists 2014;24(2):108‐114. [EMBASE: 373774478] [Google Scholar]
- El‐Sayed M, Anwar AE. Intralesional sodium stibogluconate alone or its combination with either intramuscular sodium stibogluconate or oral ketoconazole in the treatment of localized cutaneous leishmaniasis: a comparative study. Journal of the European Academy of Dermatology and Venereology : JEADV 2010;24(3):335‐40. [PUBMED: 19744259] [DOI] [PubMed] [Google Scholar]
- Emad M, Hayati F, Fallahzadeh MK, Namazi MR. Superior efficacy of oral fluconazole 400 mg daily versus oral fluconazole 200 mg daily in the treatment of cutaneous leishmania major infection: a randomized clinical trial. Journal of the American Academy of Dermatology 2011;64(3):606‐8. [PUBMED: 21315963] [DOI] [PubMed] [Google Scholar]
- Esfandiarpour I, Alavi A. Evaluating the efficacy of allopurinol and meglumine antimoniate (Glucantime) in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 2002;41(8):521‐4. [PUBMED: 12207774] [DOI] [PubMed] [Google Scholar]
- Faghihi G, Tavakoli‐kia R. Topical paromomycin vs intralesional meglumine antimoniate in cutaneous leishmaniasis. Clinical and Experimental Dermatology 2003;28(1):13‐6. [PUBMED: 12558620] [DOI] [PubMed] [Google Scholar]
- Farajzadeh S, Esfandiarpour I, Haghdoost AA, Mohammadi S, Mohebbi A, Mohebbi E, Mostafavi M. Comparison between combination therapy of oral terbinafine and cryotherapy versus systemic meglumine antimoniate and cryotherapy in cutaneous leishmaniasis: a randomized clinical trial. Iranian Journal of Parasitology 2015;10(1):1‐8. [PUBMED: 25904940] [PMC free article] [PubMed] [Google Scholar]
- Fekri A, Rahnama Z, Khalili M, Dookhani AP, Khazaeli P, Beigi KB. The efficacy of co‐administration of topical niosomal dapsone gel and intralesional injection of glucantime in cutaneous leishmaniasis in comparison with cryotherapy plus intralesional injection of glucantime. Journal of Kerman University of Medical Sciences 2015;22(2):117‐32. [PUBMED: 603502875] [Google Scholar]
- Firooz A, Khatami A, Khamesipour A, Nassiri‐Kashani M, Behnia F, Nilforoushzadeh M, et al. Intralesional injection of 2% zinc sulphate solution in the treatment of acute Old World cutaneous leishmaniasis: a randomized, double‐blind, controlled clinical trial. Journal of Drugs in Dermatology 2005;4(1):73‐9. [PUBMED: 15696988] [PubMed] [Google Scholar]
- Firooz A, Khamesipour A, Ghoorchi MH, Nassiri‐Kashani M, Eskandari SE, Khatami A, et al. Imiquimod in combination with meglumine antimoniate for cutaneous leishmaniasis: a randomized assessor‐blind controlled trial. Archives of Dermatology 2006;142(12):1575‐9. [PUBMED: 17178983 ] [DOI] [PubMed] [Google Scholar]
- Gholami A, Khamesipour A, Momeni A, Ghazanfari T, Nilforoushzadeh MA, Darajeh Z, et al. Treatment of cutaneous leishmaniasis with 5% garlic cream: a randomized, double‐blind study. Iranian Journal of Dermatology 2000;3(3):2‐6. [CENTRAL: CN‐00454251] [Google Scholar]
- Harms G, Chehade AK, Douba M, Roepke M, Mouakeh A, Rosenkaimer F, et al. A randomized trial comparing a pentavalent antimonial drug and recombinant interferon‐gamma in the local treatment of cutaneous leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1991;85(2):214‐6. [PUBMED: 1909469] [DOI] [PubMed] [Google Scholar]
- Iraji F, Vali A, Asilian A, Shahtalebi M, Momeni AZ. Comparison of intralesionally injected zinc sulphate with meglumine antimoniate in the treatment of acute cutaneous leishmaniasis. Dermatology 2004;209(1):46‐9. [PUBMED: 15237267] [DOI] [PubMed] [Google Scholar]
- Iraji F, Sadeghinia A. Efficacy of paromomycin ointment in the treatment of cutaneous leishmaniasis: results of a double‐blind, randomized trial in Isfahan, Iran. Annals of Tropical Medicine and Parasitology 2005;99(1):3‐9. [PUBMED: 15701249] [DOI] [PubMed] [Google Scholar]
- Jaffar H. Rifampicin in cutaneous leishmaniasis ‐ a therapeutic trial in Saudi Arabia. Journal of Pakistan Association of Dermatologists 2006;16(1):4‐9. [EMBASE: 44265597] [Google Scholar]
- Jaffary F, Nilforoushzadeh MA, Ansari N, Rahimi M. Treatment of cutaneous leishmaniasis: cassia fistula fruit gel‐intralesional glucantime Vs. placebo gel‐ intralesional glucantime combination. Tehran University Medical Journal 2010;67(10):705‐11. [Google Scholar]
- Jaffary F, Nilforoushzadeh MA, Tavakoli N, Zolfaghari B, Shahbazi F. The efficacy of Achilles millefolium topical gel along with intralesional injection of glucantime in the treatment of acute cutaneous leishmaniasis major. Advanced Biomedical Research 2014;3:111. [PUBMED: 24804185 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffary F, Nilforoushzadeh MA, Moradi S, Derakhshan R, Ansari N. Concentrated extracts of Cassia fistula versus intralesional injection of meglumine antimoniate in treatment of acute cutaneous leishmaniasis. Journal of Skin and Stem Cell 2014;1(1):e16631. [EMBASE: 604236618] [Google Scholar]
- Jebran AF, Schleicher U, Steiner R, Wentker P, Mahfuz F, Stahl HC, et al. Rapid healing of cutaneous leishmaniasis by high‐frequency electrocauterization and hydrogel wound care with or without DAC N‐055: a randomized controlled phase IIa trial in Kabul. PLoS Neglected Tropical Diseases 2014;8(2):e2694. [PUBMED: 24551257 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowkar F, Dehghani F, Jamshidzadeh A. Is topical nitric oxide and cryotherapy more effective than cryotherapy in the treatment of old world cutaneous leishmaniasis?. Journal of Dermatological Treatment 2012;23(2):131‐5. [PUBMED: 20964568 ] [DOI] [PubMed] [Google Scholar]
- Kashani MN, Sadr B, Nilforoushzadeh MA, Arasteh M, Babakoohi S, Firooz A. Treatment of acute cutaneous leishmaniasis with intralesional injection of meglumine antimoniate: comparison of conventional technique with mesotherapy gun. International Journal of Dermatology 2010;49(9):1034‐7. [PUBMED: 20883265 ] [DOI] [PubMed] [Google Scholar]
- Khatami A, Rahshenas M, Bahrami M, Ghoorchi MH, Eskandari SE, Sharifi I, et al. Miltefosine in treatment of cutaneous leishmaniasis: a randomized controlled trial (Poster P‐31). 6th International Dermato‐Epidemiology Association Congress, Malmo, Sweden, 26‐28 August 2012. British Journal of Dermatology 2012;167(2):e23. [EMBASE: 71050777] [Google Scholar]
- Khatami A, Talaee R, Rahshenas M, Khamesipour A, Mehryan P, Tehrani S, et al. Dressings combined with injection of meglumine antimoniate in the treatment of cutaneous leishmaniasis: a randomized controlled clinical trial. PloS One 2013;8(6):e66123. [PUBMED: 23826087] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochar DK, Aseri S, Sharma BV, Bumb RA, Mehta RD, Purohit SK. The role of rifampicin in the management of cutaneous leishmaniasis. QJM : Monthly Journal of the Association of Physicians 2000;93(11):733‐7. [PUBMED: 11077029] [DOI] [PubMed] [Google Scholar]
- Kochar DK, Saini G, Kochar SK, Sirohi P, Bumb RA, Mehta RD, et al. A double blind, randomised placebo controlled trial of rifampicin with omeprazole in the treatment of human cutaneous leishmaniasis. Journal of Vector Borne Diseases 2006;43(4):161‐7. [PUBMED: 17175700] [PubMed] [Google Scholar]
- Larbi EB, al‐Khawajah A, al‐gindan Y, Jain S, Abahusain A, al‐Zayer A. A randomized, double‐blind, clinical trial of topical clotrimazole versus mizonazole for treatment of cutaneous leishmaniasis in the eastern province of Saudi Arabia. American Journal of Tropical Medicine and Hygiene 1995;52(2):166‐8. [PUBMED: 7872446] [DOI] [PubMed] [Google Scholar]
- Layegh P, Yazdanpanah MJ, Vosugh EM, Pezeshkpoor F, Shakeri MT, Moghiman T. Efficacy of azitromycin versus systemic meglumine antimoniate (Glucantime) in the treatment of cutaneous leishmaniasis. American Journal of Tropical Medicine and Hygiene 2007;77(1):99‐101. [PUBMED: 17620637] [PubMed] [Google Scholar]
- Layegh P, Pezeshkpoor F, Soruri AH, Naviafar P, Moghiman T. Efficacy of cryotherapy versus intralesional meglumine antimoniate (Glucantime) for treatment of cutaneous leishmaniasis in children. American Journal of Tropical Medicine and Hygiene 2010;80(2):172‐5. [PUBMED: 19190206] [PubMed] [Google Scholar]
- Layegh P, Rajabi O, Jafari MR, Emamgholi Tabar Malekshah P, Moghiman T, Ashraf H, et al. Efficacy of topical liposomal amphotericin B versus intralesional meglumine antimoniate (Glucantime) in the treatment of cutaneous leishmaniasis. Journal of Parasitology Research 2011;2011:656523. [DOI: 10.1155/2011/656523; PUBMED: 22174993 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynen L, Damme W. Local application of diminazene aceturate: an effective treatment for cutaneous leishmaniasis?. Annales de la Société Belge de Médecine Tropicale 1992;72(1):13‐9. [PUBMED: 1567264] [PubMed] [Google Scholar]
- Maleki M, Karimi G, Tafaghodi M, Raftari S, Nahidi Y. Comparison of intralesional two percent zinc sulfate and glucantime injection in treatment of acute cutaneous leishmaniasis. Indian Journal of Dermatology 2012;57(2):118‐22. [PUBMED: 22615508] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapar MA, Omidian M. Intralesional injections of metronidazole versus meglumine antimoniate for the treatment of cutaneous leishmaniasis. Jundishapur Journal of Microbiology 2010;3(2):79‐83. [EMBASE: 2010406291] [Google Scholar]
- Mashhood AA, Hussain K. Efficacy of allopurinol compared with pentostam in the treatment of old world cutaneous leishmaniasis. Journal of the College of Physicians and Surgeons‐‐Pakistan : JCPSP 2001;11(6):367‐70. [EMBASE: 2001264054] [Google Scholar]
- Mohebali M, Fotouhi A, Hooshmand B, Zarei Z, Akhoundi B, Rahnema A, et al. Comparison of miltefosine and meglumine antimoniate for the treatment of zoonotic cutaneous leishmaniasis (ZCL) by a randomized clinical trial in Iran. Acta Tropica 2007;103(1):33‐40. [PUBMED: 17586452] [DOI] [PubMed] [Google Scholar]
- Momeni AZ, Jalayer T, Emamjomeh M, Bashardost N, Ghassemi RL, Meghdadi M, et al. Treatment of cutaneous leishmaniasis with itraconazole. Randomized doubl‐blind study. Archives of Dermatology 1996;132(7):784‐6. [PUBMED: 8678570] [PubMed] [Google Scholar]
- Momeni AZ, Reiszadae MR, Aminjavaheri M. Treatment of cutaneous leishmaniasis with a combination of allopurinol and low‐dose meglumine antimoniate. International Journal of Dermatology 2002;41(7):441‐3. [PUBMED: 12121563] [DOI] [PubMed] [Google Scholar]
- Momeni AZ, Aminjavaheri M, Omidghaemi MR. Treatment of cutaneous leishmaniasis with ketoconazole cream. Journal of Dermatological Treatment 2003;14(1):26‐9. [PUBMED: 12745852] [DOI] [PubMed] [Google Scholar]
- Mostafavi SA, Shatalebi MA, Attar AM, Hejazi H, Mottaghinejad A, Fadaei R, et al. Preparation and evaluation of clinical effects of glucantime gel mask. Journal of Isfahan Medical School 2013;31(231):389‐99. [EMBASE: 2014593999] [Google Scholar]
- Mujtaba G, Khalid M. Weekly vs. fortnightly intralesional meglumine antimoniate in cutaneous leishmaniasis. International Journal of Dermatology 1999;38(8):607‐9. [PUBMED: 10487452] [DOI] [PubMed] [Google Scholar]
- Nassiri‐Kashani M, Firooz A, Khamesipour A, Mojtahed F, Nilforoushzadeh M, Hejazi H, et al. A randomized, double‐blind, placebo‐controlled clinical trial of itraconazole in the treatment of cutaneous leishmaniasis. Journal of the European Academy of Dermatology and Venereology: JEADV 2005;19(1):80‐3. [PUBMED: 15649196] [DOI] [PubMed] [Google Scholar]
- Nilforoushzadeh MA. Efficacy of combined triple therapy (paromomycin ointment, cryotherapy and intralesional Glucantime) in comparison with intralesional Glucantime for treatment of acute cutaneous leishmaniasis. Iranian Journal of Dermatology 2004;3(7):136‐9. [Google Scholar]
- Nilforoushzadeh MA, Jaffary F, Reiszadeh MR. Comparative effect of topical trichloroacetic acid and intralesional meglumine antimoniate in the treatment of acute cutaneous leishmaniasis. International Journal of Pharmacology 2006;2(6):633‐6. [EMBASE: 2007084731] [Google Scholar]; Nilforoushzadeh MA, Reiszadeh MR, Jafari F. Topical trichloroacetic acid compared with intralesional glucantime injection in the treatment of acute wet cutaneous leishmaniasis: An open clinical trial. Iranian Journal of Dermatology 2003;2(6):34‐9. [Google Scholar]
- Nilforoushzadeh MA, Jaffary F, Moradi S, Derakhshan R, Haftbaradaran E. Effect of topical honey application along with intralesional injection of glucantime in the treatment of cutanneous leishmaniasis. BMC Complementary and Alternative Medicine 2007;7:13. [PUBMED: 17466071] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilforoushzadeh MA, Jaffary F, Ansari N, Siadat AH, Nilforoushan Z, Firouz A. A comparative study between the efficacy of systemic meglumine antimoniate therapy with standard or low dose plus oral omeprazole in the treatment of cutaneous leishmaniasis. Journal of Vector Borne Diseases 2008;45(4):287‐91. [PUBMED: 19248655] [PubMed] [Google Scholar]
- Nilforoushzadeh MA, Naeeni FF, Sattar N, Haftbaradaran E, Jaffary F, Askari G. The effect of intralesional meglumine antimoniate (Glucantime) versus a combination of topical trichloroacetic acid 50% and local heat therapy by non‐ablative radiofrequency on cutaneous leishmaniasis lesions. Journal of Research in Medical Sciences 2012;17(1 Suppl 1):S97‐S102. [EMBASE: 2013471627] [Google Scholar]
- Nilforoushzadeh MA, Siadat AH, Ansari N, Haftbaradaran E, Ahmadi E. A comparison between the effects of glucantime, topical trichloroacetic acid 50% plus glucantime, and fractional laser plus glucantime on cutaneous leishmaniasis lesions. Journal of Isfahan Medical School 2013;30(221):2450‐9. [EMBASE: 2013245548] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilforoushzadeh MA, Siadat AH, Haftbaradaran E, Minaravesh M. Comparative study of the efficacy of CO2 and fraxel lasers in treatment of cutaneous leishmaniasis scars. Journal of Isfahan Medical School 2014;31(269 SPEC.ISSUE):2277‐84. [EMBASE: 2014574591] [Google Scholar]
- Nilforoushzadeh MA, Jaffary F, Derakhsahan R, Haftbaradaran E. Comparison between intralesional meglumine antimoniate and combination of trichloroacetic acid 50% and intralesional meglumine antimoniate in the treatment of acute cutaneous leishmaniasis: a randomized clinical trial. Journal of Stem Cells 2014;1(1):e16633. [DOI: 10.5812/jssc.16633; EMBASE: 2015013267] [DOI] [Google Scholar]
- Özgöztasi O, Baydar I. A randomized clinical trial of topical paromomycin versus oral ketoconazole for treating cutaneous leishmaniasis in Turkey. International Journal of Dermatology 1997;36(1):61‐3. [EMBASE: 1997097334] [DOI] [PubMed] [Google Scholar]
- Ranawaka RR, Weerakoon HS. Randomized, double‐blind, comparative clinical trial on the efficacy and safety of intralesional sodium stibogluconate and intralesional 7% hypertonic sodium chloride against cutaneous leishmaniasis caused by L. donovani. Journal of Dermatological Treatment 2010;21(5):286‐93. [PUBMED: 20438389] [DOI] [PubMed] [Google Scholar]
- Ranawaka RR, Weerakoon HS, Silva SH. Randomized, double‐blind, controlled, comparative study on intralesional 10% and 15% hypertonic saline versus intralesional sodium stibogluconate in Leishmania donovani cutaneous leishmaniasis. International Journal of Dermatology 2015;54(5):555‐63. [PUBMED: 25600472] [DOI] [PubMed] [Google Scholar]
- Reithinger R, Mohsen M, Wahid M, Bismullah M, Quinnell RJ, Davies CR, et al. Efficacy of thermotherapy to treat cutaneous leishmaniasis caused by Leishmania tropica in Kabul, Afghanistan: a randomized, controlled trial. Clinical Infectious Diseases 2005;40(8):1148‐55. [PUBMED: 15791515] [DOI] [PubMed] [Google Scholar]
- Sadeghian G, Nilforoushzadeh MA. Effect of combination therapy with systemic glucantime and pentoxifylline in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 2006;45(7):819‐21. [PUBMED: 16863518] [DOI] [PubMed] [Google Scholar]
- Sadeghian G, Nilfroushzadeh MA, Siadat AH. A comparison between intralesional hypertonic sodium chloride solution and meglumine antimoniate in the treatment of cutaneous leishmaniasis. Egyptian Dermatology Online Journal 2;1:8. [Google Scholar]
- Sadeghian G, Nilfroushzadeh MA, Iraji F. Efficacy of local heat therapy by radiofrequency in the treatment of cutaneous leishmaniasis, compared with intralesional injection of meglumine antimoniate. Clinical and Experimental Dermatology 2007;32(4):371‐4. [PUBMED: 17376205] [DOI] [PubMed] [Google Scholar]
- Safi N, Davis GD, Nadir M, Hamid H, Robert LL, Case AJ. Evaluation of thermotherapy for the treatment of cutaneous leishmaniasis in Kabul, Afghanistan: a randomized controlled trial. Military Medicine 2012;177(3):345‐351. [PUBMED: 22479925] [DOI] [PubMed] [Google Scholar]
- Salmanpour R, Handjani F, Nouhpisheh MK. Comparative study of the efficacy of oral ketoconazole with intra‐lesional meglumine antimoniate (Glucantime) for the treatment of cutaneous leishmaniasis. Journal of Dermatological Treatment 2001;12:159‐62. [PUBMED: 12243707 ] [DOI] [PubMed] [Google Scholar]
- Salmanpour R, Razmavar MR, Abtahi N. Comparison of intralesional meglumine antimoniate, cryotherapy and their combination in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 2006;45(9):1115‐6. [PUBMED: 16961529] [DOI] [PubMed] [Google Scholar]
- Shamsi Meymandi S, Zandi S, Aghaie H, Heshmatkhah A. Efficacy of CO(2) laser for treatment of anthroponotic cutaneous leishmaniasis, compared with combination of cryotherapy and intralesional meglumine antimoniate. Journal of the European Academy of Dermatology and Venereology : JEADV 2011;25(5):587‐91. [PUBMED: 20666876] [DOI] [PubMed] [Google Scholar]
- Shanehsaz SM, Ishkhanian S. A comparative study between the efficacy of oral cimetidine and low‐dose systemic meglumine antimoniate (MA) with a standard dose of systemic MA in the treatment of cutaneous leishmaniasis. International Jornal of Dermatology 2015;54(7):834‐8. [PUBMED: 26108265] [DOI] [PubMed] [Google Scholar]; Shanehsaz SM, Ishkhanian S. Therapeutic and adverse effects of standarddose and low‐dose meglumine antimoniate during systemic treatment of Syrian cutaneous leishmaniasis patients. Journal of Pakistan Association of Dermatologists 2014;24(2):115‐121. [EMBASE: 2014549292] [Google Scholar]
- Sharquie KE, Najim RA, Farjou IB. A compararive controlled trial of intralesionally‐ administered zinc sulphate, hypertonic sodium chloride and pentavalent antimony compound against cutaneous leishmaniasis. Clinical and Experimental Dermatology 1997;22(4):169‐73. [PUBMED: 9499605] [PubMed] [Google Scholar]
- Sharquie KE, Najim RA, Farjou IB, Al‐timimit DJ. Oral zinc sulphate in the treatment of acute cutaneous leishmaniasis. Clinical and Experimental Dermatology 2001;26(1):21‐6. [PUBMED: 11260171] [DOI] [PubMed] [Google Scholar]
- Shazad B, Abbaszadeh B, Khamesipour A. Comparison of topical paromomycin sulfate (twice/day) with intralesional meglumine antimoniate for the treatment of cutaneous leishmaniasis caused by L. major. European Journal of Dermatology 2005;15(2):85‐7. [PUBMED: 15757817] [PubMed] [Google Scholar]
- Stahl HC, Ahmadi F, Schleicher U, Sauerborn R, Bermejo JL, Amirih ML, et al. A randomized controlled phase IIb wound healing trial of cutaneous leishmaniasis ulcers with 0.045% pharmaceutical chlorite (DAC N‐055) with and without bipolar high frequency electro‐cauterization versus intralesional antimony in Afghanistan. BMC Infectious Diseases 2014;14:619. [PUBMED: 25420793] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanpanah MJ, Banihashemi M, Pezeshkpoor F, Khajedaluee M, Famili S, Tavakoli Rodi I, et al. Comparison of oral zinc sulfate with systemic meglumine antimoniate in the treatment of cutaneous leishmaniasis. Dermatology Research & Practice 2011;269515:1‐4. [DOI: 10.1155/2011/269515; PUBMED: 21747837] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerehsaz F, Salmanpour R, Farhad H, Ardehali S, Panjehshahin MR, Tabei SZ, et al. A double‐blind randomized clinical trial of a topical herbal extract (Z‐HE) vs. systemic meglumine antimoniate for the treatment of cutaneous leishmaniasis in Iran. International Journal of Dermatology 1999;38(8):610‐2. [PUBMED: 10487453] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
- Alavi‐Naini R, Fazaeli A, O'Dempsey T. Topical treatment modalities for old world cutaneous leishmaniasis: a review. Prague Medical Report 2012;113(2):105‐18. [PUBMED: 22691282] [DOI] [PubMed] [Google Scholar]
- Banihashemi M, Yazdanpanah MJ, Amirsolymani H, Yousefzadeh H. Comparison of lesion improvement in lupoid leishmaniasis patients with two treatment approaches: trichloroacetic Acid and intralesional meglumine antimoniate. Journal of Cutaneous Medicine and Surgery 2015;19(1):35‐9. [PUBMED: 25775661] [DOI] [PubMed] [Google Scholar]
- Bumb RA, Mehta RD, Ghiya BC, Jakhar R, Prasad N, Soni P, et al. Efficacy of short‐duration (twice weekly) intralesional sodium stibogluconate in treatment of cutaneous Leishmaniasis in India. British Journal of Dermatology 2010;163(4):854‐8. [PUBMED: 20500797] [DOI] [PubMed] [Google Scholar]
- Dogra J, Lal BB, Misra SN. Dapsone in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 1986;25(6):398‐400. [PUBMED: 3531044] [DOI] [PubMed] [Google Scholar]
- Dogra J, Aneja N. Leishmaniasis and itraconazole: a controlled clinical trial on cutaneous subtypes. International Journal of Antimicrobial Agents 1994;4(4):309‐11. [PUBMED: 18611622] [DOI] [PubMed] [Google Scholar]
- Dorlo TP, Balasegaram M, Beijnen JH, Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. Journal of Antimicrobial Chemotherapy 2012;67(11):2576‐97. [PUBMED: 22833634 ] [DOI] [PubMed] [Google Scholar]
- el‐On J, Halevy S, Grunwald MH, Weinrauch L. Topical treatment of Old World cutaneous leishmaniasis caused by Leishmania major: a double blind control study. Journal of the American Academy of Dermatology 1992;27(2 Pt 1):227‐31. [PUBMED: 1430361] [DOI] [PubMed] [Google Scholar]
- Frankenburg S, Gross A, Jonas F, Klaus S. Effect of topical paromomycin on cell‐mediated immunity during cutaneous leishmaniasis. International Journal of Dermatology 1993;32(1):68‐70. [PUBMED: 8425810] [DOI] [PubMed] [Google Scholar]
- Kim DH, Chung HJ, Bleys J, Ghohestani RF. Is paromomycin an effective and safe treatment against cutaneous leishmaniasis? A meta‐analysis of 14 randomized controlled trials. PLoS Neglected Tropical Diseases 2009;3(2):e381. [PUBMED: 19221595] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi Z, Nakhli A, Rassaii S. Comparing the efficiency of topical paromomycin with intralesional meglumine antimoniate for cutaneous leishmaniasis. International Journal of Dermatology 2005;44(12):1064‐5. [PUBMED: 16409282] [DOI] [PubMed] [Google Scholar]
- Ali NM, Fariba J, Elaheh H, Ali N. The efficacy of 5% trichoroacetic acid cream in the treatment of cutaneous leishmaniasis lesions. Journal of Dermatological Treatment 2012;23(2):136‐9. [PUBMED: 21034291] [DOI] [PubMed] [Google Scholar]
- Nilforoushzadeh MA, Jaffary F, Ansari N, Moradi S, Siadat AH. The comparison between trichloroacetic acid 50% and CO2 laser in the treatment of cutaneous leishmaniasis scar. Indian Journal of Dermatology 2011;56(2):171‐3. [PUBMED: 21716542] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanehsaz SM, Ishkhanian S. Electrocardiographic and biochemical adverse effects of meglumine antimoniate during the treatment of Syrian cutaneous leishmaniasis. Egyptian Dermatology Journal 2013;9(1):5. [Google Scholar]
- Singh S, Singh R, Sundar S. Failure of ketaconazole in oriental sore in India. Journal of Chemotherapy 1995;7(4):202‐3. [PUBMED: 8904156] [PubMed] [Google Scholar]
- Trau H, Schewach‐Millet M, Shoham J, Doerner T, Shor R, Passwell JH. Topical application of human fibroblast interferon (IFN) in cutaneous leishmaniasis. Israel Journal of Medical Sciences 1987;23(11):1125‐7. [PUBMED: 3325468] [PubMed] [Google Scholar]
References to studies awaiting assessment
- Farajzadeh S, Hakimi Parizi M, Haghdoost AA, Mohebbi A, Mohammadi S, Pardakhty A, et al. Comparison between intralesional injection of zinc sulfate 2 % solution and intralesional meglumine antimoniate in the treatment of acute old world dry type cutaneous leishmaniasis: a randomized double‐blind clinical trial. Journal of Parasitic Diseases 2016;40(3):935‐9. [PUBMED: 27605813] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajzadeh S, Heshmatkhah A, Vares B, Mohebbi E, Mohebbi A, Aflatoonian M, et al. Topical terbinafine in the treatment of cutaneous leishmaniasis: triple blind randomized clinical trial. Journal of Parasitic Diseases 2016;40(4):1159‐64. [DOI: 10.1007/s12639-014-0641-1; PUBMED: 27876906] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanif MM, Akram K, Mustafa G. Intralesional versus oral chloroquine in cutaneous leishmaniasis: comparison of outcome, duration of treatment and total dose of drug. Journal of the College of Physicians and Surgeons Pakistan 2016;26(4):260‐2. [PUBMED: 27097693] [PubMed] [Google Scholar]
- Jaffary F, Nilforoushzadeh MA, Siadat A, Haftbaradaran E, Ansari N, Ahmadi E. A comparison between the effects of Glucantime, topical trichloroacetic acid 50% plus Glucantime, and fractional carbon dioxide laser plus Glucantime on cutaneous leishmaniasis lesions. Dermatology Research and Practice 2016;2016:6462804. [PUBMED: 27148363] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na‐Bangchang K, Ahmed O, Hussein J, Hirayama K, Kongjam P, Aseffa A, et al. Exploratory, phase II controlled trial of shiunko ointment local application twice a day for 4 weeks in Ethiopian patients with localized cutaneous leishmaniasis. Evidence‐Based Complementary and Alternative Medicine 2016;2016:5984709. [PUBMED: 27195014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi O, Layegh P, Hashemzadeh S, Khoddami M. Topical liposomal azithromycin in the treatment of acute cutaneous leishmaniasis. Dermatologic Therapy 2016;29(5):358‐63. [DOI: 10.1111/dth.12357; PUBMED: 27073044] [DOI] [PubMed] [Google Scholar]
- Refai W, Madarasingha N, Weerasingha S, Senarath U, Silva A, Fernandopulle R, et al. Efficacy, safety and cost‐effectiveness of thermotherapy for L. donovani‐induced cutaneous leishmaniasis: a randomized controlled clinical trial. International Journal of Infectious Diseases 2016;45:74. [CENTRAL: CN‐01142616] [Google Scholar]
- Sattar FA, Ahmed F, Ahmed N, Sattar SA, Malghani MA, Choudhary MI. A double‐blind, randomized, clinical trial on the antileishmanial activity of a Morinda citrifolia (Noni) stem extract and its major constituents. Natural Product Communications 2012;7(2):195‐6. [PUBMED: 22474954] [PubMed] [Google Scholar]
References to ongoing studies
- ACTRN12614001288617. A clinical trial to assess the safety and effect of heat therapy in comparison to standard intra‐lesional sodium stibogluconate for cutaneous leishmaniasis [In patients with cutaneous leishmaniasis, thermotherapy was compared with standard intra‐lesional therapy with regard to efficacy and safety]. anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12614001288617 Date first received: 10 December 2014.
- IRCT138904091159N7. Comparison between two groups in the treatment of cutaneous leishmaniasis [Comparison between the efficacy of intralesional placebo and nitric oxide releasing patch versus placebo patch and glucantime in the treatment of cutaneous leishmaniasis]. www.irct.ir/searchresult.php?id=1159&number=7 Date first received: 2 September 2013.
- IRCT2013092414746N1. Treatment of patients with ZCL [The effect of MJ1 (Topical dairy extract) versus routine care for treatment of cutaneous leishmaniasis (rural) in Isfahan Iran :a randomized controled clinical trial (RCTs) ‐]. apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT2013092414746N1 Date first received: 20 February 2014.
- NCT00840359. Study of the Efficacy of Daylight Activated Photodynamic Therapy in the Treatment of Cutaneous Leishmaniasis [Phase 2 Study of the Efficacy of Daylight Activated Photodynamic Therapy in the Treatment of Cutaneous Leishmaniasis]. clinicaltrials.gov/ct2/show/NCT00840359 Date first received: 8 February 2009.
- NCT01050777. Efficacy of Topical Liposomal Form of Drugs in Cutaneous Leishmaniasis [Pilot Study of Efficacy of Topical Nano‐liposomal Meglumine Antimoniate (Glucantime) or Paromomycin in Combination With Systemic Glucantime for the Treatment of Anthroponotic Cutaneous Leishmaniasis (ACL) Caused by Leishmania Tropica]. clinicaltrials.gov/ct2/show/NCT01050777 Date first received: 13 January 2010.
- SLCTR/2014/028. Treatment for leishmaniasis [Randomized, double blind, controlled study on efficacy and safety of intralesional metronidazole vs intralesional sodium stibogluconate in L. donovani cutaneous leishmaniasis]. slctr.lk/trials/276 Date first received: 18 October 2014.
Additional references
- Adam I, Elhardello OA, Elhadi MO, Abdalla E, Elmardi KA, Jansen FH. The antischistosomal efficacies of artesunate–sulfamethoxypyrazine–pyrimethamine and artemether–lumefantrine administered as treatment for uncomplicated Plasmodium falciparum malaria. Annals of Tropical Medicine and Parasitology 2008;102(1):39‐44. [PUBMED: 18186976] [DOI] [PubMed] [Google Scholar]
- Al‐Waiz M, Sharquie KE, Al‐Assir M. Treatment of cutaneous leishmaniasis by intralesional metronidazole. Saudi Medical Journal 2004;25(10):1512‐3. [PUBMED: 15494841] [PubMed] [Google Scholar]
- Alrajhi AA. Cutaneous leishmaniasis of the Old World. Skin Therapy Letter 2003;8(2):1‐4. [PUBMED: 12728282] [PubMed] [Google Scholar]
- Alvar J, Yactayo S, Bern C. Leishmaniasis and poverty. Trends in Parasitology 2006;22(12):552‐7. [PUBMED: 17023215] [DOI] [PubMed] [Google Scholar]
- Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 2012;7(5):e35671. [PUBMED: 22693548] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SP, Phelps RG, Goldberg DJ. Mesotherapy for facial skin rejuvenation: a clinical, histologic, and electron microscopic evaluation. Dermatologic Surgery 2006;32(12):1467‐1472. [PUBMED: 17199654] [DOI] [PubMed] [Google Scholar]
- Aram H, Leibovici V. Ultrasound‐induced hyperthermia in the treatment of cutaneous leishmaniasis. Cutis 1987;40(4):350‐3. [PUBMED: 3677796] [PubMed] [Google Scholar]
- Arevalo I, Tulliano G, Quispe A, Spaeth G, Mathlashewski, Llanos‐cuantas A, et al. Role of imiquimod and parenteral meglumine antimoniate in the intitial treatment of cutaneous lesihmaniasis. Clinical Infectious Diseases 2007;44(12):1549‐54. [PUBMED: 17516397] [DOI] [PubMed] [Google Scholar]
- Babajev KB, Babajev OG, Korepanov VI. Treatment of cutaneous leishmaniasis using a carbon dioxide laser. Bulletin of the World Health Organization 1991;69(1):103‐6. [PUBMED: 1905204] [PMC free article] [PubMed] [Google Scholar]
- Badaro R, Falcoff E, Badaro FS, Carvalho EM, Pedral‐Sampaio D, Barral A, et al. Treatment of visceral leishmaniasis with pentavalent antimony and interferon gamma. New England Journal of Medicine 1990;322(1):16‐21. [PUBMED: 2104665] [DOI] [PubMed] [Google Scholar]
- Baryza MJ, Baryza GA. The Vancouver Scar Scale: an administration tool and its interrater reliability. Journal of Burn Care Rehabilitation 1995;16(5):535‐8. [PUBMED: 8537427] [DOI] [PubMed] [Google Scholar]
- Bassiouny A, Meshad M, Talaat M, Kutty K, Metawaa B. Cryosurgery in cutaneous leishmaniasis. British Journal of Dermatology 1982;107(4):467‐74. [PUBMED: 7126453] [DOI] [PubMed] [Google Scholar]
- Berman JD, Neva FA. Effect of temperature on multiplication of Leishmania amastigotes within monocyte‐derived macrophages in vitro. American Journal of Tropical Medicine and Hygiene 1981;30(2):318‐21. [PUBMED: 7235124] [DOI] [PubMed] [Google Scholar]
- Bigby M, Williams H. Appraising Systematic Reviews and Meta‐analyses. Archives of Dermatology 2003;139(6):795‐798. [PUBMED: 12810513] [DOI] [PubMed] [Google Scholar]
- Blum J, Lockwood DN, Visser L, Harms G, Bailey MS, Caumes E, et al. Local or systemic treatment for New World cutaneous leishmaniasis? Re‐evaluating the evidence for the risk of mucosal leishmaniasis. International Health 2012;4(3):153‐63. [PUBMED: 24029394] [DOI] [PubMed] [Google Scholar]
- Blum J, Buffet P, Visser L, Harms G, Bailey MS, Caumes E, et al. LeishMan recommendations for treatment of cutaneous and mucosal leishmaniasis in travelers, 2014. Journal of Travel Medicine 2014;21(2):116‐29. [PUBMED: 24745041] [DOI] [PubMed] [Google Scholar]
- Bogenrieder T, Lehn N, Landthaler M, Stolz W. Treatment of Old World cutaneous leishmaniasis with intralesionally injected meglumine antimoniate using a Dermojet device. Dermatology 2003;206(3):269‐72. [PUBMED: 12673089] [DOI] [PubMed] [Google Scholar]
- Borelli D. A clinical trial of itraconazole in the treatment of deep mycoses and leishmaniasis. Reviews of Infectious Diseases 1987;9(Suppl 1):S57‐63. [PUBMED: 3027848] [DOI] [PubMed] [Google Scholar]
- Bryceson AD, Murphy A, Moody AH. Treatment of 'Old World' cutaneous leishmaniasis with aminosidine ointment: results of an open study in London. Transactions of the Royal Society of Tropical Medicine and Hygiene 1994;88(2):226‐8. [PUBMED: 8036683] [DOI] [PubMed] [Google Scholar]
- Bygbjerg IC, Knudsen L, Kieffer M. Failure of rifampicin therapy to cure cutaneous leishmaniasis. Archives of Dermatology 1980;116(9):988. [PUBMED: 7416770] [PubMed] [Google Scholar]
- Cardo LJ. Leishmania: risk to the blood supply. Transfusion 2006;46(9):1641‐45. [PUBMED: 16965594] [DOI] [PubMed] [Google Scholar]
- Cauwenberg G, Doncker P. Itraconazole (R 51 211): a clinical review of its anti mycotic activity in dermatology, gynecology, and internal medicine. Drug Development Research 1986;8:317‐23. [DOI: 10.1002/ddr.430080136] [DOI] [Google Scholar]
- Chalmers I, Glasziou P. Avoidable waste in the production and reporting of research evidence. Lancet 2009;374(9683):86‐9. [PUBMED: 19525005] [DOI] [PubMed] [Google Scholar]
- Chunge CN, Gachihi G, Muigai R, Wasunna K, Rashid JR, Chulay JD, et al. Visceral leishmaniasis unresponsive to antimonial drugs. III. Successful treatment using a combination of sodium stibogluconate plus allopurinol. Transactions of the Royal Society of Tropical Medicine and Hygiene 1985;79(5):715‐8. [PUBMED: 3006296] [DOI] [PubMed] [Google Scholar]
- Crawford R, Holmes D, Meymandi S. Comparative study of the efficacy of combined imiquimod 5% cream and intralesional meglumine antimoniate versus imiquimod 5% cream and intralesional meglumine antimoniate alone for the treatment of cutaneous leishmaniasis. Journal of the American Academy of Dermatology 2005;52:S118. 15858507 [Google Scholar]
- Croft SL, Seifert K, Yardley V. Current scenario of drug development for Leishmaniasis. Indian Journal of Medical Research 2006;123(3):399‐410. [PUBMED: 16778319] [PubMed] [Google Scholar]
- Daie Parizi MH. Treatment of cutaneous leishmaniasis with local application of the tincture of Thioxolone and benzoxonium chloride (first research in the world) at annual congress of Iranian society of pediatrics, Tehran, Iran, Oct 1992. The Annual Book of Congress 1992;1:121‐5. [Google Scholar]
- Daie Parizi MH, Shamsaddini S. Comparison between topical treatment with Thioxolone, Benzoxonium Chloride tincture and intralesional injection of Meglumine Antimoniate on cutaneous Leishmaniasis. Journal of Kerman University of Medical Sciences 1996;3(1):7‐14. [Google Scholar]
- Davies CR, Kaye P, Croft SL, Sundar S. Leishmaniasis: new approaches to disease control. BMJ 2003;326(7385):377‐82. [PUBMED: 12586674 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vries HJ, Reedijk SH, Schallig HD. Cutaneous leishmaniasis: recent developments in diagnosis and management. American Journal of Clinical Dermatology 2015;16(2):99‐109. [PUBMED: 25687688] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamere FM, Sladden MJ, Dobbins HM, Leonardi‐Bee J. Interventions for alopecia areata. Cochrane Database of Systematic Reviews 2008;2:CD004413. [DOI: 10.1002/14651858.CD004413.pub2] [DOI] [PubMed] [Google Scholar]
- Boer M, Argaw D, Jannin J, Alvar J. Leishmaniasis impact and treatment access. Clinical Microbiology and Infection 2011;17(10):1471–7. [PUBMED: 21933305] [DOI] [PubMed] [Google Scholar]
- Desjeux P. Leishmaniasis. Public health aspects and control. Clinical Dermatology 1996;14(5):417‐23. [PUBMED: 8889319] [DOI] [PubMed] [Google Scholar]
- Dorlo TP, Thiel PP, Schoone GJ, Stienstra Y, Vugt M, Beijnen JH, et al. Dynamics of parasite clearance in cutaneous leishmaniasis patients treated with miltefosine. PLoS Neglected Tropical Diseases 2011;5(12):e1436. [PUBMED: 22180803] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger M, Davey Smith G, Schneider M, Minder C. Biasin meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629–34. [PUBMED: 9310563] [DOI] [PMC free article] [PubMed] [Google Scholar]
- el‐On J, Weinrauch L, Livshin R, Even‐Paz Z, Jacobs GP. Topical treatment of recurrent cutaneous leishmaniasis with ointment containing paromomycin and methylbenzethonium chloride. British Medical Journal (Clinical Research Ed.) 1985;291(6497):704‐5. [PUBMED: 3929905] [DOI] [PMC free article] [PubMed] [Google Scholar]
- el‐Safi SH, Murphy AG, Bryceson AD, Neal RA. A double‐blind clinical trial of the treatment of cutaneous leishmaniasis with paromomycin ointment. Transactions of the Royal Society of Tropical Medicine and Hygiene 1990;84(5):690‐1. [PUBMED: 2278070] [DOI] [PubMed] [Google Scholar]
- Faber WR, Oskam L, Gool T, Kroon NC, Knegt‐Junk KJ, Hofwegen H, et al. Value of diagnostic techniques for cutaneous leishmaniasis. Journal of the American Academy of Dermatology 2003;49(1):70‐4. [PUBMED: 12833011] [DOI] [PubMed] [Google Scholar]
- Fatima F, Khalid A, Nazar N, Abdalla M, Mohomed H, Toum AM, et al. In vitro assessment of anti ‐ cutaneous leishmaniasis activity of some Sudanese plants. Turkiye Parazitoloji Dergisi 2005;29(1):3‐6. [PUBMED: 17167733] [PubMed] [Google Scholar]
- US Food, Drugs Administration. News and Events ‐ FDA News Release 2014. www.fda.gov/NewsEvents/Newsroom/PrressAnnouncements/ucm389671.htm (acessed prior to 10 October 2016).
- Garnier T, Croft SL. Topical treatment for cutaneous leishmaniasis. Current Opinion in Investigational Drugs (London, England : 2000) 2002;3(4):538‐44. [PUBMED: 12090720] [PubMed] [Google Scholar]
- González U, Pinart M, Rengifo‐Pardo M, Macaya A, Alvar J, Tweed JA. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD004834.pub2] [DOI] [PubMed] [Google Scholar]
- González U, Pinart M, Reveiz L, Rengifo‐Pardo M, Tweed J, Macaya A, et al. Designing and reporting clinical trials on treatments for cutaneous leishmaniasis. Clinical Infectious Diseases 2010;51(4):409‐19. [PUBMED: 20624067] [DOI] [PubMed] [Google Scholar]
- González U, Pinart M, Sinclair D, Firooz A, Enk C, Vélez ID, et al. Vector and reservoir control for preventing leishmaniasis. Cochrane Database of Systematic Reviews 2015, Issue 8. [DOI: 10.1002/14651858.CD008736.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AC. Beware the "Texas sharp shooter" in rate ratios of progression. BMJ 2007;334(7591):440. [PUBMED: 17332546] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradoni L, López‐Vélez R, Mokni M. World Health Organization. Manual on case management and surveillance of the leishmaniasis in the WHO European Region. www.who.int/leishmaniasis/resources/EURO_WHO_Leish_manual_on_case_management_and_surveillance_9789289052511_2017.pdf?ua=1 (accessed prior to 15 November 2017).
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [PUBMED: 18436948] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn NC. Management of cutaneous leishmaniasis. Current Opinion in Infectious Diseases 2001;14(2):151‐4. [PUBMED: 11979125] [DOI] [PubMed] [Google Scholar]
- Herwaldt BL. Leishmaniasis. Lancet 1999;354(9185):1191‐9. [PUBMED: 10513726] [DOI] [PubMed] [Google Scholar]
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [PUBMED: 12958120] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JP, Altman DG, Gotzsche PC, Juni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928. [PUBMED: 22008217] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram JR, Woo PN, Chua SL, Ormerod AD, Desai N, Kai AC, et al. Interventions for hidradenitis suppurativa. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD010081.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha TK. Evaluation of allopurinol in the treatment of kala‐azar occurring in North Bihar, India. Transactions of the Royal Society of Tropical Medicine and Hygiene 1983;77(2):204‐7. [PUBMED: 6868102] [DOI] [PubMed] [Google Scholar]
- Jiang S, Meadows J, Anderson SA, Mukkada AJ. Antileishmanial activity of the antiulcer agent omeprazole. Antimicrobial agents and chemotherapy 2002;46(8):2569‐74. [PUBMED: 12121934] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junaid AJ. Treatment of cutaneous leishmaniasis with infrared heat. International Journal of Dermatology 1986;25(7):470‐2. [PUBMED: 3771049] [DOI] [PubMed] [Google Scholar]
- Kager PA, Rees PH, Wellde BT, Hockmeyer WT, Lyerly WH. Allopurinol in the treatment of visceral leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1981;75(4):556‐9. [PUBMED: 6275579] [DOI] [PubMed] [Google Scholar]
- Karamian M, Motazedian MH, Mehrabani D, Gholami K. Leishmania major infection in a patient with visceral leishmaniasis: treatment with Amphotericin B. Parasitology Research 2007;101(5):1431‐4. [PUBMED: 17659388] [DOI] [PubMed] [Google Scholar]
- Keiser J, Utzinger J. Artemisinins and synthetic trioxolanes in the treatment of helminth infections. Current Opinion in Infectious Diseases 2007;20(6):605–12. [PUBMED: 17975411] [DOI] [PubMed] [Google Scholar]
- Kermode M. Unsafe injections in low‐income country health settings: need for injection safety promotion to prevent the spread of blood‐borne viruses. Health Promotion International 2004;19(1):95‐103. [PUBMED: 14976177] [DOI] [PubMed] [Google Scholar]
- Fatima F, Khalid A, Nazar N, Abdalla M, Mohomed H, Toum AM, et al. In vitro assessment of anti ‐cutaneous leishmaniasis activity of some Sudanese plants. Türkiye Parazitoloji Dergisi 2005;29(1):3‐6. [PUBMED: 17167733] [PubMed] [Google Scholar]
- Khatami A, Firooz A, Gorouhi F, Dowlati Y. Treatment of acute Old World cutaneous leishmaniasis: a systematic review of the randomized controlled trials. Journal of the American Academy of Dermatology 2007;57(2):335.e1‐29. [PUBMED: 17337090] [DOI] [PubMed] [Google Scholar]
- Laguna F, López‐Vélez R, Pulido F, Salas A, Torre‐Cisneros J, Torres E, et al. Treatment of visceral leishmaniasis in HIV‐infected patients: a randomized trial comparing meglumine antimoniate with amphotericin B. Spanish HIV‐Leishmania Study Group. AIDS 1999;13(9):1063‐9. [PUBMED: 10397536] [DOI] [PubMed] [Google Scholar]
- Leibovici V, Aram H. Cryotherapy in acute cutaneous leishmaniasis. International Journal of Dermatology 1986;25(7):473‐5. [PUBMED: 3533799] [DOI] [PubMed] [Google Scholar]
- Lessa HA, Machado P, Lima F, Cruz AA, Bacellar O, Guerreiro J, et al. Successful treatment of refractory mucosal leishmaniasis with pentoxifylline plus antimony. American Journal of Tropical Medicine and Hygiene 2001;65(2):87‐9. [PUBMED: 11508396] [DOI] [PubMed] [Google Scholar]
- Mapar MA, Kavoosi H, Dabbagh MA. Assessment of the effect of topical opium in treatment of cutaneous leishmaniasis. Iranian Journal of Dermatology 2001;4(16):23‐8. [Google Scholar]
- Masmoudi A, Hariz W, Marrekchi S, Amouri M, Turki H. Old World cutaneous leishmaniasis: diagnosis and treatment. Journal of Dermatological Case Reports 2013;7(2):31‐41. [PUBMED: 23858338] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meawad OB. Selective heat therapy in cutaneous leishmaniasis: a preliminary experience using the 585 nm pulsed dye laser. Journal of the European Academy of Dermatology and Venereology: JEADV 1997;8(3):241‐4. [EMBASE: 27286057] [Google Scholar]
- Migdal C, Serres M. Reactive oxygen species and oxidative stress. Medical Science 2011;27(4):405‐412. [PUBMED: 21524406] [DOI] [PubMed] [Google Scholar]
- Minodier P, Parola P. Cutaneous leishmaniasis treatment. Travel Medicine and Infectious Disease 2007;5(3):150‐8. [PUBMED: 17448941] [DOI] [PubMed] [Google Scholar]
- Modabber F, Buffet PA, Torreele E, Milon G, Croft SL. Consultative meeting to develop a strategy for treatment of cutaneous leishmaniasis. Institute Pasteur, Paris. 13‐15 June, 2006. Kinetoplastid Biology and Disease 2007;6:3. [PUBMED: 17456237] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monge‐Maillo B, López‐Vélez R. Therapeutic options for old world cutaneous leishmaniasis and new world cutaneous and mucocutaneous leishmaniasis. Drugs 2013;73(17):1889‐920. [PUBMED: 24170665] [DOI] [PubMed] [Google Scholar]
- Monge‐Maillo B, López‐Vélez R. Miltefosine for visceral and cutaneous leishmaniasis: drug characteristics and evidence‐based treatment recommendations. Clinical Infectious Diseases 2015;60(9):1398‐404. [PUBMED: 25601455] [DOI] [PubMed] [Google Scholar]
- Moore OA, Smith LA, Campbell F, Seers K, McQuay KJ, Moore RA. Systematic review of the use of honey as a wound dressing. BMC Complementary and Alternative Medicine 2001;1:2. [PUBMED: 11405898] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz PF, Kurban AK. Treatment of cutaneous leishmaniasis: Retrospective and advances for the 21st century. Clinical Dermatology 1999;17(3):305‐15. [PUBMED: 10384870] [DOI] [PubMed] [Google Scholar]
- Mosleh IM, Geith E, Natsheh L, Schönian G, Abotteen N, Kharabsheh S. Efficacy of a weekly cryotherapy regimen to treat Leishmania major cutaneous leishmaniasis. Journal of the American Academy of Dermatology 2008;58(4):617‐24. [PUBMED: 18249466] [DOI] [PubMed] [Google Scholar]
- Munir A, Janjua SA, Hussain I. Clinical efficacy of intramuscular meglumine antimoniate alone and in combination with intralesional meglumine antimoniate in the treatment of old world cutaneous leishmaniasis. Acta Dermatovenerologica Croatica: ADC 2008;16(2):60‐4. [PUBMED: 18541100] [PubMed] [Google Scholar]
- Musa AM, Khalil EA, Mahgoub FA, Hamad S, Elkadaru AM, Hassan AM. Efficacy of liposomal amphotericin B (AmBisome) in the treatment of persistent post‐kala‐azar dermal leishmaniasis (PKDL). Annals of Tropical Medicine and Parasitology 2005;99(6):563‐9. [PUBMED: 16156969] [DOI] [PubMed] [Google Scholar]
- Najim RA, Sharquie KE, Farjou IB. Zinc sulphate in the treatment of cutaneous leishmaniasis: an in vitro and animal study. Memórias do Instituto Oswaldo Cruz 1998;93(6):831‐7. [PUBMED: 9921312] [DOI] [PubMed] [Google Scholar]
- Neva FA, Petersen EA, Corsey R, Bogaert H, Martinez D. Observations on local heat treatment for cutaneous leishmaniasis. American Journal of Tropical Medicine and Hygiene 1984;33(5):800‐4. [PUBMED: 6091468] [DOI] [PubMed] [Google Scholar]
- Olliaro P, Vaillant M, Arana B, Grogl M, Modabber F, Magill A, et al. Methodology of clinical trials aimed at assessing interventions for cutaneous leishmaniasis. PLoS Neglected Tropical Diseases 2013;7(3):e2130. [PUBMED: 23556016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace D. Leishmaniasis. Journal of Infection 2014;69(Suppl 1):S10‐S18. [PUBMED: 25238669] [DOI] [PubMed] [Google Scholar]
- Passwell JH, Shor R, Shoham J. The enhancing effect of interferon‐beta and ‐gamma on the killing of Leishmania tropica major in human mononuclear phagocytes in vitro. Journal of Immunology 1986;136(8):3062‐6. [PUBMED: 3082979] [PubMed] [Google Scholar]
- Pieper B, Caliri MH. Nontraditional wound care: A review of the evidence for the use of sugar, papaya/papain, and fatty acids. Journal of Wound, Ostomy, and Continence Nursing 2003;30(4):175–83. [PUBMED: 12851592] [DOI] [PubMed] [Google Scholar]
- Pigott DM, Golding N, Messina JP, Battle KE, Duda KA, Balard Y, et al. Global database of leishmaniasis occurrence locations, 1960‐2012. Scientific Data 2014;30(1):140036. [PUBMED: 25984344] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte‐Sucre A. Physiological consequences of drug resistance in Leishmania and their relevance for chemotherapy. Kinetoplastid Biology and Disease 2003;2(1):14. [PUBMED: 14613496] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranawaka RR, Weerakoon HS, Opathella N. Liquid nitrogen cryotherapy on Leishmania donovani cutaneous leishmaniasis. Journal of Dermatological Treatment 2011;22(4):241‐5. [PUBMED: 20818996] [DOI] [PubMed] [Google Scholar]
- Rathi SK, Pandhi RK, Chopra P, Khanna N. Post‐kala‐azar dermal leishmaniasis: a histopathological study. Indian Journal of Dermatology, Venereology and Leprology 2005;71(4):250‐53. [PUBMED: 16394433] [DOI] [PubMed] [Google Scholar]
- Reithinger R, Aadil K, Kolaczinski J, Mohsen M, Hami S. Social impact of leishmaniasis, Afghanistan. Emerging Infectious Diseases 2005;11(4):634‐6. [PUBMED: 15834984] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infectious Diseases 2007;7(9):581‐96. [PUBMED: 17714672] [DOI] [PubMed] [Google Scholar]
- Rodriguez ME, Inguanzo P, Ramos A, Perez J. Treatment of cutaneous leishmaniasis with CO2 laser radiation. Revista Cubana de Medicina Tropical 1990;42(2):197‐202. [PUBMED: 2128547] [PubMed] [Google Scholar]
- Rodriguez‐Cuartero A, Pérez‐Blanco FJ, López‐Fernández A. Co‐trimoxazole for visceral leishmaniasis. Infection 1990;18(1):40. [PUBMED: 2312176] [DOI] [PubMed] [Google Scholar]
- Rohrich RJ, Janis EJ, Reisman NR. Use of off‐label and non‐approved drugs and devices in plastic surgery. Plastic and Reconstructive Surgery 2003;112(1):241‐243. [PUBMED: 12832901] [DOI] [PubMed] [Google Scholar]
- Saab M, Hage H, Charafeddine K, Habib RH, Khalifeh I. Diagnosis of cutaneous leishmaniasis: why punch when you can scrape?. American Journal of Tropical Medicine and Hygiene 2015;92(3):518‐22. [PUBMED: 25561563] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks DL, Barral A, Neva F. Thermosensitivity patterns of Old vs. New World cutaneous strains of Leishmania growing within mouse peritoneal macrophages in vitro. American Journal of Tropical Medicine and Hygiene 1983;32(2):300‐4. [PUBMED: 6837841] [DOI] [PubMed] [Google Scholar]
- Sampaio SA, Godoy JT, Paiva L, Dillon NL, Lacaz CS. The treatment of American (mucocutaneous) leishmaniasis with amphotericin B. Archives of Dermatology 1960;82:627‐35. [PUBMED: 13745957] [DOI] [PubMed] [Google Scholar]
- Sampaio RN, Marsden PD. Treatment of the mucosal form of leishmaniasis without response to glucantime, with liposomal amphotericin B. Revista da Sociedade Brasileira de Medicina Tropical 1997;30(2):125‐8. [PUBMED: 9148335] [DOI] [PubMed] [Google Scholar]
- Savioli L, Engels D, Daumerie D, Jannin J, Alvar J, Asiedu K, et al. Response from Savioli and colleagues from the Department of Neglected Tropical Diseases, World Health Organization. PLoS medicine 2006;3(6):e283. [PUBMED: 16789805] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallig HD, Oskam L. Molecular biological applications in the diagnosis and control of leishmaniasis and parasite identification. Tropical Medicine and International Health 2002;7(8):641‐51. [PUBMED: 12167091] [DOI] [PubMed] [Google Scholar]
- Schmidt‐Ott R, Klenner T, Overath P, Aebischer T. Topical treatment with hexadecylphosphocholine (Miltex) efficiently reduces parasite burden in experimental cutaneous leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1999;93(1):85‐90. [PUBMED: 10492799] [DOI] [PubMed] [Google Scholar]
- Schork MA, Ramington RD. The determination of sample size in disease studies in which drop out or non‐adherence is a problem. Journal of Chronic Diseases 1967;20(4):233‐9. [PUBMED: 6023231] [DOI] [PubMed] [Google Scholar]
- Schulz KF, Altman DG, Moher D, for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomized trials. Annals of Internal Medicine 2010;152(11):726‐32. [DOI: 10.7326/0003-4819-152-11-201006010-00232] [DOI] [PubMed] [Google Scholar]
- Sharifi I, Fekri AR, Aflatoonian MR, Khamesipour A, Mahboudi F, Dowlati Y, et al. Leishmaniasis recidivans among school children in Bam, South‐east Iran, 1994‐2006. International Journal of Dermatology 2010;49(5):557‐561. [PUBMED: 20534092] [DOI] [PubMed] [Google Scholar]
- Sharquie KE, Al‐Talib K, Chu AC. Intralesional therapy of cutaneous leishmaniasis with sodium stibogluconate antimony. British Journal of Dermatology 1988;119(1):53‐7. [PUBMED: 2841964] [DOI] [PubMed] [Google Scholar]
- Sharquie KE. A new intralesional therapy for cutaneousd leishmaniasis with hypertonic sodium chloride solution. Journal of Dermatology 1995;22(10):732‐7. [PUBMED: 8586751] [DOI] [PubMed] [Google Scholar]
- Sharquie KE, Al‐Azzawi KE. Intralesional therapy for cutaneous leishmaniasis with 2% zinc sulfate solution. Journal of Pan‐Arab League of Dermatologists 1996;7:41‐6. [Google Scholar]
- Sharquie KE, Al‐Hamamy H, El‐Yassin D. Treatment of cutaneous leishmaniasis by direct current electrotherapy: the Baghdadin device. Journal of Dermatology 1998;25(4):234‐7. [PUBMED: 9609980] [DOI] [PubMed] [Google Scholar]
- Simonsen L, Kane A, Lloyd J, Zaffran M, Kane M. Unsafe injections in the developing world and transmission of bloodborne pathogens: a review. Bulletin of the World Health Organization 1999;77(10):789‐800. [PUBMED: 10593026] [PMC free article] [PubMed] [Google Scholar]
- Sindermann H, Engel J. Development of miltefosine as an oral treatment for leishmaniasis. Transactions of the Royal Society of Tropical Medicine and Hygiene 2006;100(Suppl 1):S17‐20. [PUBMED: 16730362] [DOI] [PubMed] [Google Scholar]
- Solomon M, Pavlotsky F, Leshem E, Ephros M, Trau H, Schwartz E. Liposomal amphotericin B treatment of cutaneous leishmaniasis due to Leishmania tropica. Journal of the European Academy of Dermatology and Venereology 2011;25(8):973‐7. [PUBMED: 21129042] [DOI] [PubMed] [Google Scholar]
- Soto J, Arana BA, Toledo J, Rizzo N, Vega JC, Diaz A, et al. Miltefosine for new world cutaneous leishmaniasis. Clinical Infectious Diseases 2004;38(9):1266‐72. [PUBMED: 15127339] [DOI] [PubMed] [Google Scholar]
- Stojkovic M, Junghanss T, Krause E, Davidson RN. First case of typical Old World cutaneous leishmaniasis treated with miltefosine. International Journal of Dermatology 2007;46(4):385‐7. [PUBMED: 17442078] [DOI] [PubMed] [Google Scholar]
- Storer E, Wayte J. Cutaneous leishmaniasis in Afghani refugees. Australasian Journal of Dermatology 2005;46(2):80–3. [PUBMED: 15842398] [DOI] [PubMed] [Google Scholar]
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. American Journal of Clinical Dermatology 2009;10(4):221‐7. [PUBMED: 19489655] [DOI] [PubMed] [Google Scholar]
- Sundar S, Chakravarty J, Rai VK, Agrawal N, Singh SP, Chauhan V, et al. Amphotericin B treatment for Indian visceral leishmaniasis: response to 15 daily versus alternate‐day infusions. Clinical infectious Diseases 2007;45(5):556‐61. [PUBMED: 17682988] [DOI] [PubMed] [Google Scholar]
- Sundar S, Jha TK, Thakur CP, Sinha PK, Bhattacharya SK. Injectable paromomycin for Visceral leishmaniasis in India. New England Journal of Medicine 2007;356(25):2571‐81. [PUBMED: 17582067] [DOI] [PubMed] [Google Scholar]
- Thakur CP, Pandey AK, Sinha GP, Roy S, Behbehani K, Olliaro P. Comparison of three treatment regimens with liposomal amphotericin B (AmBisome) for visceral leishmaniasis in India: a randomized dose‐finding study. Transactions of the Royal Society of Tropical Medicine and Hygiene 1996;90(3):319‐22. [PUBMED: 8758093] [DOI] [PubMed] [Google Scholar]
- Urcayo FG, Zaias N. Oral ketoconazole in the treatment of cutaneous leishmaniasis. International Journal of Dermatology 1982;21(7):414‐6. [PUBMED: 6290403] [DOI] [PubMed] [Google Scholar]
- Uzun S, Durdu M, Culha G, Allahverdiyev AM, Memisoglu HR. Clinical features, epidemiology, and efficacy and safety of intralesional antimony treatment of cutaneous leishmaniasis: recent experience in Turkey. Journal of Parasitology 2004;90(4):853‐9. [PUBMED: 15357081] [DOI] [PubMed] [Google Scholar]
- Griensven J, Carrillo E, López‐Vélez R, Lynen L, Moreno J. Leishmaniasis in immunosuppressed individuals. Clinical Microbiology and Infection 2014;20(4):286‐299. [PUBMED: 24450618] [DOI] [PubMed] [Google Scholar]
- Zuuren EJ, Fedorowicz Z, Carter B, Linden MMD, Charland L. Interventions for rosacea. Cochrane Database of Systematic Reviews 2015, Issue 4. [DOI: 10.1002/14651858.CD003262.pub5] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaneau M, Chaby G, Guillot B, Martel P, Senet P, Téot L, et al. Consensus panel recommendations for chronic and acute wound dressings. Archives of Dermatology 2007;143(10):1291‐4. [PUBMED: 17938343] [DOI] [PubMed] [Google Scholar]
- Vardy D, Barenholz Y, Naftoliev N, Klaus S, Gilead L, Frankenburg S. Efficacious topical treatment for human cutaneous leishmaniasis with ethanolic lipid amphotericin B. Transactions of the Royal Society of Tropical Medicine and Hygiene 2001;95(2):184‐6. [PUBMED: 11355557] [DOI] [PubMed] [Google Scholar]
- Weigel MM, Armijos RX. The traditional and conventional medical treatment of cutaneous leishmaniasis in rural Ecuador. Revista Panamericana de Salud Publica [Pan American Journal of Public Health] 2001;10(6):395‐404. [PUBMED: 11820108] [DOI] [PubMed] [Google Scholar]
- Weinrauch L, Livshin R, El‐On J. Cutaneous leishmaniasis treatment with ketoconazole. Cutis 1983;32(3):288‐90, 294. [PUBMED: 6313298] [PubMed] [Google Scholar]
- Weinrauch L, Livshin R, Even‐Paz Z, El‐On J. Efficacy of ketoconazole in cutaneous leishmaniasis. Archives of Dermatology 1983;275(5):353‐4. [PUBMED: 6318670] [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO). Leishmaniasis. World Health Report 2002. www.who.int/whr/2002/annex/en/ (accessed 2 April 2004).
- World Health Organization (WHO). Control of leishmaniasis. Report by the Secretariat 22 March 2007. apps.who.int/gb/archive/pdf_files/WHA60/A60_10‐en.pdf (accessed before 11 October 2016).
- World Health Organization (WHO). Report of the Fifth Consultative Meeting on Leishmania/HIV Co‐infection. Addis Ababa, Ethiopia, 20‐22 March 2007. www.who.int/leishmaniasis/resources/Leishmaniasis_hiv_coinfection5.pdf (accessed before 11 October 2016). [WHO/CDS/NTD/IDM/2007.5]
- World Health Organization (WHO). Control of the leishmaniases: report of a meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva 22‐26 March 2010. WHO technical report series 949. apps.who.int/iris/bitstream/10665/44412/1/WHO_TRS_949_eng.pdf (accessed before 11 October 2016). [ISBN 9789241209496 ]
- World Health Organization (WHO). Application for Inclusion of Miltefosina on WHO Model List of Essential Medicines. Submitted to the EML Secretariat for consideration. November 2010. www.who.int/selection_medicines/committees/expert/18/applications/Miltefosine_application.pdf (accessed 11 October 2016).
- Wortmann G, Zapor M, Ressner R, Fraser S, Hartzell J, Pierson J, et al. Lipsosomal amphotericin B for treatment of cutaneous leishmaniasis. American Journal of Tropical Medicine and Hygiene 2010;83(5):1028‐33. [PUBMED: 21036832] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakraoui H, Ben Salah A, Ftaiti A, Marrakchi H, Zaatour A, Zaafouri B, et al. Spontaneous course of lesions of Leishmania major cutaneous leishmaniasis in Tunisia. Annales de Dermatologie et Venereogie 1995;122(6‐7):405‐7. [PUBMED: 8526421] [PubMed] [Google Scholar]
- Zanger P, Kotter I, Raible A, Gelanew T, Schonian G, Kremsner PG. Case report: Successful treatment of cutaneous leishmaniasis caused by Leishmania aethiopica with liposomal amphothericin B in an immunocompromised traveler returning from Eritrea. American Journal of Tropical Medicine and Hygiene 2011;84(5):692‐4. [PUBMED: 21540377] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeglin O. Infectiology and Tropical Dermatology Part 17: cutaneous leishmaniasis ‐ a casuistry with after assessment [Infektiologie und Tropendermatologie ‐ Teil 17: kutaneleishmaniasis ‐ eine kasuistik mit nachbegutachtung]. Dermatology 2009;15(4):246ff. [Google Scholar]
References to other published versions of this review
- González U, Pinart M, Reveiz L, Alvar J. Interventions for Old World cutaneous leishmaniasis. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD005067.pub3] [DOI] [PubMed] [Google Scholar]