

Abstract
Here, we present the genome of the industrial ethanol production strain Brettanomyces bruxellensis CBS 11270. The nuclear genome was found to be diploid, containing four chromosomes with sizes of ranging from 2.2 to 4.0 Mbp. A 75 Kbp mitochondrial genome was also identified. Comparing the homologous chromosomes, we detected that 0.32% of nucleotides were polymorphic, i.e. formed single nucleotide polymorphisms (SNPs), 40.6% of them were found in coding regions (i.e. 0.13% of all nucleotides formed SNPs and were in coding regions). In addition, 8,538 indels were found. The total number of protein coding genes was 4897, of them, 4,284 were annotated on chromosomes; and the mitochondrial genome contained 18 protein coding genes. Additionally, 595 genes, which were annotated, were on contigs not associated with chromosomes. A number of genes was duplicated, most of them as tandem repeats, including a six-gene cluster located on chromosome 3. There were also examples of interchromosomal gene duplications, including a duplication of a six-gene cluster, which was found on both chromosomes 1 and 4. Gene copy number analysis suggested loss of heterozygosity for 372 genes. This may reflect adaptation to relatively harsh but constant conditions of continuous fermentation. Analysis of gene topology showed that most of these losses occurred in clusters of more than one gene, the largest cluster comprising 33 genes. Comparative analysis against the wine isolate CBS 2499 revealed 88,534 SNPs and 8,133 indels. Moreover, when the scaffolds of the CBS 2499 genome assembly were aligned against the chromosomes of CBS 11270, many of them aligned completely, some have chunks aligned to different chromosomes, and some were in fact rearranged. Our findings indicate a highly dynamic genome within the species B. bruxellensis and a tendency towards reduction of gene number in long-term continuous cultivation.
Introduction
The yeast, Brettanomyces bruxellensis (syn. Dekkera bruxellensis- the last issue of the taxonomic monography of the yeasts [1] mentioned D. bruxellensis as the valid name of this species, however, according to the recently introduced principle “one species, one name” [2] we use the older name B. bruxellensis in this study), is regarded as a major contaminant in wine [3, 4] and bioethanol production [5, 6]. However, it is also involved in certain economically relevant, spontaneous fermentations, such as the production of Belgian Lambic beer [7–9]. It has also been found to be the production yeast in a continuous ethanol production process with cell recirculation, after outcompeting the initially inoculated Saccharomyces cerevisiae [10]. B. bruxellensis has an ethanol tolerance similar to S. cerevisiae, and has the ability to grow at low sugar concentrations. This explains why it usually becomes important in the later stages of wine or beer production, or in sugar limited continuous fermentations [11]. The mechanism of outcompeting S. cerevisiae is not completely known at present. It has been speculated that the ability of B. bruxellensis to assimilate nitrate may play a role, such as in some Brazilian ethanol production plants, where nitrate can come into the fermentation with the substrate, sucrose from sugarcane [12]. However, outcompetition of S. cerevisiae by B. bruxellensis has been observed in nitrate-free, glucose-limited fermentations, and thus, the competitiveness of the yeast could rather be due to a higher affinity for the substrate and/or a more efficient energy metabolism [13].
B. bruxellensis has several interesting metabolic capabilities, such as the (strain dependent) ability to ferment cellobiose to ethanol [14, 15], to assimilate nitrate [12] and even xylose [16]. Due to its robustness and its ability to assimilate the above-mentioned sugars, it has been regarded as a potential candidate to convert lignocellulose-hydrolysate to ethanol, and after some adaptation to the substrate, it performed as well as S. cerevisiae [17, 18].
Apart from being a biotechnologically important organism, B. bruxellensis can also serve as a model for yeast evolution. It separated from the S. cerevisiae lineage prior to the lineage-specific whole genome duplication. Interestingly, similar to S. cerevisiae, it developed a fermentative, Crabtree-positive life-style in a case of parallel evolution, possibly through the loss of a regulatory element affecting expression of genes associated with respiration [19, 20]. Those losses might have been facilitated by partial amplifications of the genome, which relaxed the selective pressure for ordered expression from the amplified genes [21]. Extensive chromosome polymorphisms and–rearrangements in different B. bruxellensis strains have been demonstrated by pulsed field electrophoresis. Such rearrangements are common in non-sexual species, and therefore, the description of B. bruxellensis as a sexual species has been called into question [22].
Due to the emergence of next generation sequencing (NGS) methods, a variety of genomes of B. bruxellensis wine- and beer strains has been sequenced to date [21, 23–29]; however, annotated genomes of isolates from industrial ethanol plants are yet to be reported. The majority of the sequenced genomes seems to be diploid [30]; yet some allotriploid wine strains, containing a third set of chromosomes with a sequence slightly different from the other two chromosomes, have been identified [31]. Chromosome polymorphism has been demonstrated on the level of complete genome sequences in D. bruxellensis UMY321 wine isolate generated by Nanopore MinION Sequencing [29]. Genome assemblies from short sequencing reads usually produce short scaffolds, making it difficult to follow events of rearrangements, amplifications or deletions of large chromosomal fragments [23–28]. In a recent study, we presented a method that enabled assembly of scaffolds representing chromosomes, using a combination of two complementary sequencing platforms (Illumina, PacBio) and structural mapping provided by the OpGen method [32]. We now annotated the genome of the industrial isolate CBS 11270, enabling genetic analysis to determine ploidy, to understand the distribution of genes over the four identified chromosomes, to identify gene content and possible amplifications and–losses on the chromosomes, and to determine polymorphisms within our strain of interest and when compared to another strain of the same species.
Materials and methods
Assembly
The genome assembly was described earlier [32]. However, for the present study, the genome assembly was additionally subjected to manual curation, which is depicted in the Results section.
Annotation
The annotation of the B. bruxellensis CBS 11270 genome assembly was performed using the reference annotation of the existing assembly of B. bruxellensis CBS 2499 and matching annotation, version 2.0, available from the JGI website (http://genome.jgi.doe.gov/vista_embed/?organism=Dekbr2). Gene models were computed using the Maker package (version 2.31.8, PMID: 22192575) based on protein sequences from the reference assembly in combination with a fungi specific repeat library. Rather than simply projecting the existing annotation through syntenic mapping of the scaffolds, this approach re-built the reference annotation on top of our assembly, thus more effectively taking into account any difference in sequence or structure. While we also tried different permutations of RNA-sequences-based annotations, detailed manual inspection indicated that the protein-guided annotation best met our needs with respect to the comparative analyses we wished to perform. Further and more densely sampled transcriptome data may change this view in the future. We used EMBLmyGFF3 tool to deposit genome annotation at European Nucleotide Archive (ENA) [33].
Repeat analysis
Repeat Masker (http://www.repeatmasker.org) was used with default settings to mask known repeats present in the CBS 11270 genome.
SNP analysis
Genome sequences’ dictionaries were created using Picard tools version1.107 (http://broadinstitute.github.io/picard/). The Illumina reads [32] were mapped to the reference (genome of B. bruxellensis CBS 2249) and the new assembly dictionaries by using BWA version 0.7.4 [34]. The files resulting from mapping were, in the case of SAM files, indexed and sorted using samtools version 1.2 [35] and the read coverage was counted for both the reference and the new assembly.
The mapped Illumina reads were run through the GATK HaplotypeCaller version 2.8–1 [36] pipeline, using default settings, to identify the various variants (SNP and indels), and their location and frequency (including allele frequency) present in the reference and the new assembly. We used FreeBayes (1.1.0) for haplotype sampling analysis.
Gene copy number analysis
The software CNVnator version 0.3 was used to identify copy number variations (CNV) [37].
We set a window size of one hundred for all steps of CNV analysis: generation of histograms of the read depth, calculation of statistical significances for the fragments with unusual read depth, partitioning of the chromosome into regions with similar read depth and CNVs identification.
Comparative analysis of genome assemblies
Comparative analysis of the genome assemblies of B. bruxellensis CBS 11270 and CBS 2499 was performed using the Multiple genome alignment tool Mauve version 2.3.1 under default settings of the “Progressive Mauve” function [38].
Comparative analysis of gene content
Comparison of the gene content between the two genomes was done using program BLASTN 2.2.29+ with a culling limit of one, in order to collect only the best hit, since the objective was to determine presence/absence of homologues [39]. The cut off E value for genes to be considered homologous was 1e-10 [24]. The search for strain specific gene duplications was performed without constraining a culling limit.
BLASTP 2.2.29+ was used for comparison of proteins to identify substitutions of amino acids.
Results
Genome structure
In a previous study [32], the genome assembly of CBS 11270 was demonstrated to be organized in four large chromosomes. Analysis of the B. bruxellensis genome assembly [32] using BLASTN showed that a 1 Megabase pairs (Mbp) fragment from nucleotide 2,619,547 to 3,634,467 of chromosome 1 was duplicated. Based on coverage information, we verified that this was an assembly artefact and adjusted the assembly by removal of this fragment using a custom R script. This reduced the number of regions in the genome assembly with a lower than average depth of aligned Illumina reads (Fig 1) [40]. The finalized assembly of the CBS 11270 genome consists of four chromosomes, spanning 4 Mbp (chromosome 1), 3.3 Mbp (chromosome 2), 3.7 Mbp (chromosome 3), and 2.2 Mbp (chromosome 4) respectively. The determined chromosome sizes are in line with results from pulsed field electrophoresis [16]. Additionally, 394 contigs of totally 2.1 Mbp (13.7% of the total genome size) were assembled but could not be associated with chromosomes (See Data availability section for accession numbers) [32]. These sequences are, (i) Illumina contigs with no alignments to the optical map assembly (mostly contigs shorter than 40 Kbp), or (ii) unaligned flanks of optical-map-aligned contigs or (iii) flanks or contigs with ambiguous alignments or iv) unique PacBio contigs [41]. The total size of the nuclear genome was thus determined to be 15.3 Mbp, which is comparable to other B. bruxellensis strains that have been sequenced [21, 23–28]. 97.3% of Illumina reads mapped to the genome assembly draft, of them 88.8% aligned to chromosome sequences and 11.2% to contigs that could not be associated with chromosomes.
Fig 1.
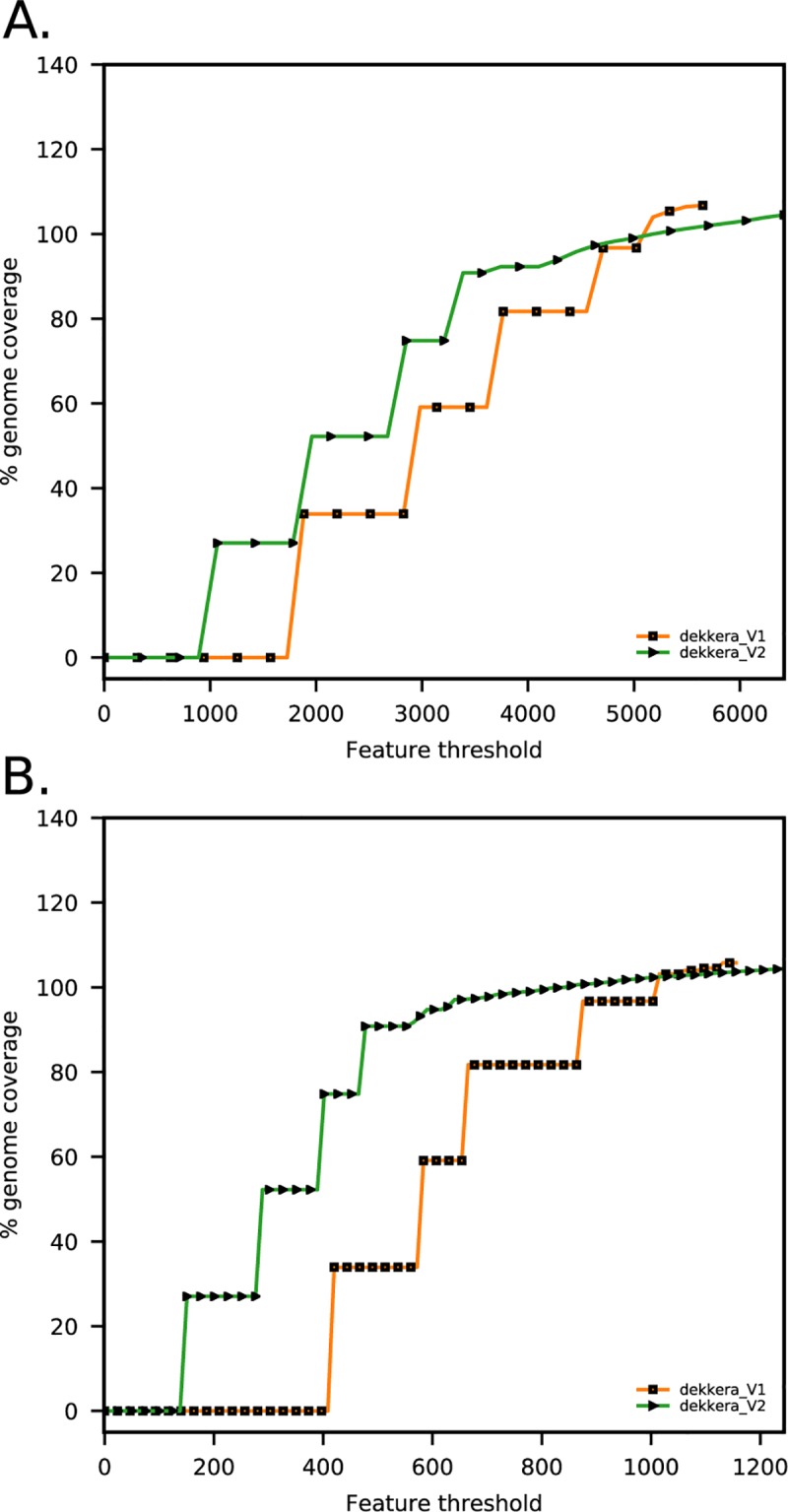
Feature response curves (FRC) computed for all features (A) and low coverage features (B) (adapted from [32]). FRC are shown for HGAP, allpaths, dekkera_V1 (final assembly presented in [32]) and dekkera_V2 (assembly presented in this work). The decreased amount of features when removing the duplicated fragment of chromosome 1 in dekkera_V1 assembly is mostly attributed to the loss of regions below normal read coverage (B). Such regions are often indicative of incorrect repeat expansions made by the assembly program [32].
A contig of 75 Kbp (scaffold 39309 produced by ABySS), representing mitochondrial DNA was also assembled.
An investigation of heterozygous sites by SNP-analysis showed that the ploidy of CBS 11270 is more than haploid. Average frequency of a particular allele at a heterozygous site in diploid genome is expected to be about 0.5. In a triploid genome partial heterozygous site would have allele frequency of 0.33 or 0.66. The average allele frequency at heterozygous sites was determined to be 0.5 (S1 File), suggesting that the genome of B. bruxellensis CBS 11270 is diploid. This conclusion is corroborated by results of haplotype sampling analysis (S1 Fig). In contrast to two highly abundant Australian wine strains, AWRI1499 and AWRI1608, additional chromosomes forming an allotriploid hybrid genome [23] were not observed in CBS 11270.
Genome annotation
The genome was annotated by using the annotation of B. bruxellensis CBS 2499 [21] as reference (see Methods). We identified 4897 protein encoding genes (Table 1), which was fewer than in other B. bruxellensis strains (see below). Chromosome 1 contained 1433 genes; chromosome 2, 1052; chromosome 3, 1191; and chromosome 4, 608 genes. The location of some genes that were discussed in our earlier study [42] is illustrated in Fig 2. Additionally, 595 genes were annotated on contigs not associated with chromosomes, 18 protein encoding genes were detected on the mitochondrial contig.
Table 1. Annotation details of the B. bruxellensis CBS 11270 nuclear genome.
| Annotation feature | Counts |
|---|---|
| Genes | 4879 |
| mRNAS | 4881 |
| Exons | 6342 |
| Introns | 1461 |
| Mean introns per mRNA | 0.30 |
| Mean intron length | 217 bp |
| Mean CDS length | 1358 bp |
| Mean mRNA length | 1423 bp |
Fig 2.
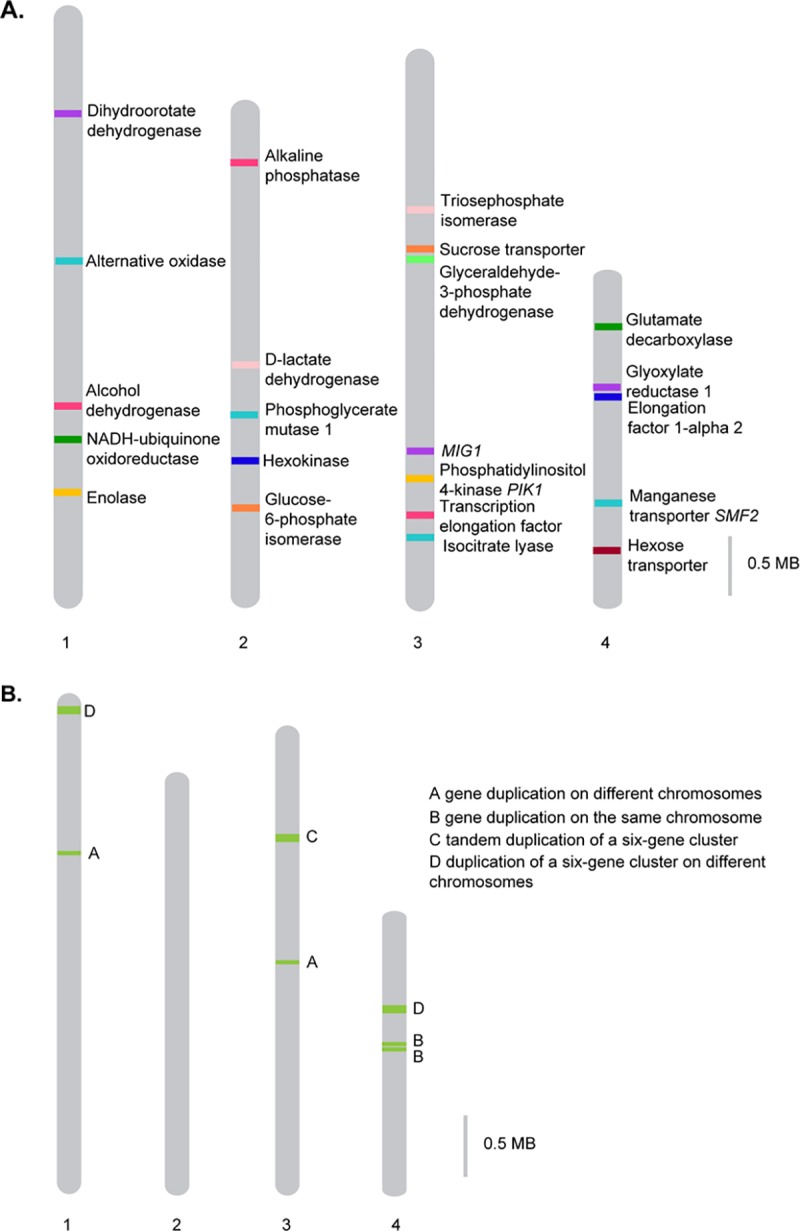
Location of certain genes (A) and duplicated genes (B) on the chromosomes of CBS 11270.
Heterozygosity
Analysis of polymorphisms between the homologous chromosomes was performed by mapping the CBS 11270 reads to the de novo assembly of the CBS 11270 genome. We detected 49,890 single nucleotide polymorphisms (SNPs) (Table 2), constituting 0.32% of the genome size (see S1 and S2 Files). The majority of observed nucleotide variation is due to transitions (i.e. purine-purine or pyrimidine-pyrimidine exchanges), which were observed three times more frequently than transversions (Table 3). Variants were identified in almost all parts of the genome, but with different frequencies at different chromosomal sites (Fig 3). 28,806 variants were detected in non-coding regions (S1 and S2 Files). 21,084 variants occurred in coding sequences, and in total 2668 genes with SNPs were identified (S3 File). 17,423 variants caused amino acid substitutions. The number of variants per gene was highly variable, more than 2,000 genes did not show any SNP (S4 File), 1016 had only 1–3 SNPs. On the other hand, 592 genes had 10 and more SNPs per gene, and 19 of them even had 35 to 75 variants per gene (S1 Table). Some of these genes are shown in Table 4. The normalised by gene length distribution of SNPs per gene Kbp is shown in S2 Fig.
Table 2. Statistics of variant analysis in heterozygous sites in genome of CBS 11270 and between genomes of CBS 11270 and CBS 2499.
| Variant type | Variant counts in heterozygous sites in genome of CBS 11270 | Variant counts between genomes of CBS 11270 and CBS 2499 |
|---|---|---|
| SNP counts | 49890 | 88534 |
| Indel counts | 8538 | 8133 |
| Total variant counts | 58230 | 96421 |
Table 3. Counts of different types of nucleotide transversions and transitions in heterozygous sites in the genome of CBS 11270 and between the genomes of CBS 11270 and CBS 2499.
| SNP type | Counts in the genome of CBS 11270 | Counts in the genome of CBS 2499 |
|---|---|---|
| A/C | 3459 | 5934 |
| A/G | 17729 | 32159 |
| T/G | 3547 | 5845 |
| T/A | 4351 | 7602 |
| C/T | 17919 | 31975 |
| C/G | 2890 | 5036 |
Fig 3. Number and location of variants on chromosomes of B. bruxellensis CBS 11270.

Y-axes represent the number of variants per 10,000 bp. Black bars show occurrence of variants. Red color denotes chromosome margins.
Table 4. Survey of the genes with the highest numbers of SNPs in CBS 11270 and CBS 2499.
| Gene ID and position in CBS 11270 | Name in CBS 2499 | Variants in CBS 11270 | Variants in CBS 2499 |
|---|---|---|---|
| General negative regulator of transcription subunit 1; BRETBRUG00000001221; chr1: 3243008–3249016 |
jgi|Dekbr2|64613|fgenesh1_pm.2_#_424 | 75 | 102 |
| AP-1 accessory protein LAA1; BRETBRUG00000001265; chr1: 3343683–3350348 |
jgi|Dekbr2|5806|gm1.2215_g | 64 | 108 |
| DNA repair protein RAD50; BRETBRUG00000002017; chr2: 1651095–1655015 |
jgi|Dekbr2|23614|fgenesh1_kg.1_#_362_#_Locus3870v1rpkm14.01 | 75 | 84 |
| Protein SNQ2; BRETBRUG00000002706; chr3:961178–965875 | jgi|Dekbr2|172416|CE84544_34760 | 63 | 93 |
| General negative regulator of transcription subunit 2; BRETBRUG00000002734; chr3: 1042035–1042658 |
jgi|Dekbr2|64613|fgenesh1_pm.2_#_424 | 11 | 102 |
The results of CNV analysis are presented in S5 File and S3 Fig. CNV analysis showed that 372 genes were present on only one of the two homologous chromosomes (S6 File). Most of the genes with reduced copy number were present in clusters (S2 Table). Only 47 of these genes, 17 on chromosome 1, 9 on chromosome 2, 11 on chromosome 3 and 10 on chromosome 4, were not associated with clusters of deleted genes. There was a relatively high number of smaller clusters–two clusters contained two and four genes, and three were formed of seven genes. However, there were also bigger clusters of deleted genes; clusters of 13, 14, 23 and 33 genes with reduced copy number on chromosomes of CBS 11270 were identified. Some clusters were located in close proximity to each other and other clusters were well separated (S2 Table).
8538 indels were found in the CBS 11270 genome. We have also found micro/mini satellites in some indels (S1 File). Indels varied in size from 1 to 128 nucleotides. The size of the indels inversely correlated to the frequency: single nucleotide indels occurred 4207 times; indels with a length of 10 nucleotides, 74 times; and indels with 20 nucleotides, 36 times. The longest indel covered 128 nucleotides (Fig 4A). In total, SNPs and indels constituted 58,230 variant counts.
Fig 4.

Distribution of indels of different size in heterozygous sites in the genome of CBS 11270 (A) and between genomes of CBS 11270 and CBS 2499 (B).
Gene amplifications
Evidence for amplified genes was investigated by CNV and BLASTN analysis. Twenty genes were found to be duplicated. Six of these genes (Table 5) were found to be duplicated according to copy number analysis only, but were not found in the assembled genome using a BLASTN search. This lack of assembly of duplicated genes is a common problem in genome analysis, typically due to the collapse of repeated regions during assembly [32]. However, most of the amplified genes could be localized to the assembled chromosomes (Table 6, Fig 2). The amplified genes belong to a broad range of GO-categories, including regulation of transcription and replication (e.g. U6 snRNA-associated Sm-like protein LSm8 or histone acetyltransferase ESA1), enzymes (e.g. hexokinase-1 or indoleamine C3-dioxygenase), regulators of cellular processes (e.g. flocculation protein gene FLO5 or temperature shock-inducible protein 1), and transport (e.g. allantoin permease or GABA-specific permease, but no sugar transporters).
Table 5. Duplicated genes revealed by CNV analysis and not identified in the genome assembly.
| Gene name | Gene ID and location |
|---|---|
| F0249 protein VIBHAR | BRETBRUG00000000385 |
| chr1 1018607 1018993 | |
| Probable NADPH dehydrogenase | BRETBRUG00000002123 |
| chr2 2039150 2040358 | |
| Chromatin modification-related protein EAF1 | BRETBRUG00000002201 |
| chr2 2253321 2256215 | |
| Copia protein | BRETBRUG00000002416 |
| chr2 2815514 2816992 | |
| Copia protein | BRETBRUG00000002417 |
| chr2 2817465 2818151 | |
| Repressor of filamentous growth | BRETBRUG00000003666 |
| chr3 3312856 3314625 |
Table 6. List of duplicated genes resolved by genome assembly.
| Gene name | Gene ID and location | |||
|---|---|---|---|---|
| A | tRNA (guanine(10)-N2)-methyltransferase | BRETBRUG00000001038 | BRETBRUG00000003060 | |
| chr1 2703203 2705172 | chr3 1805200 1807169 | |||
| B | Methylthioribulose-1-phosphate dehydratase | BRETBRUG00000003998 | BRETBRUG00000004000 | |
| chr4 1127888 1128565 | chr4 1129334 1130011 | |||
| C | Hexokinase-1 | BRETBRUG00000001423 | BRETBRUG00000004091 | |
| chr1 3856343 3857854 | chr4 1381931 1383442 | |||
| Flocculation protein FLO5 | BRETBRUG00000001424 | BRETBRUG00000004090 | ||
| chr1 3864230 3868254 | chr4 1371498 1375532 | |||
| Temperature shock-inducible protein 1 | BRETBRUG00000001425 | BRETBRUG00000004089 | ||
| chr1 3870223 3870528 | chr4 1369224 1369529 | |||
| Putative transcriptional regulatory protein | BRETBRUG00000001426 | BRETBRUG00000004088 | ||
| chr1 3874728 3876680 | chr4 1363072 1365024 | |||
| Uncharacterized transcriptional regulatory protein | BRETBRUG00000001427 | BRETBRUG00000004087 | ||
| chr1 3877690 3879264 | chr4 1360488 1362062 | |||
| Allantoin permease | BRETBRUG00000001428 | BRETBRUG00000004086 | ||
| chr1 3880591 3882306 | chr4 1357446 1359161 | |||
| D | Indoleamine 2 2C3-dioxygenase | BRETBRUG00000003493 | BRETBRUG00000003500 | BRETBRUG00000003492 |
| chr3 2804460 2805389 | chr3 2823355 2824602 | chr3 2801423 2802706 | ||
| Histone acetyltransferase ESA1 | BRETBRUG00000003494 | BRETBRUG00000003501 | ||
| chr3 2807345 2808712 | chr3 2826194 2827561 | |||
| Serine/threonine-protein phosphatase 2A activator | BRETBRUG00000003495 | BRETBRUG00000003502 | ||
| chr3 2808829 2809905 | chr3 2827678 2828226 | |||
| GABA-specific permease | BRETBRUG00000003496 | BRETBRUG00000003503 | ||
| chr3 2810290 2811957 | chr3 2829138 2830805 | |||
| V-type proton ATPase subunit a 2C vacuolar isoform | BRETBRUG00000003497 | BRETBRUG00000003504 | ||
| chr3 2813515 2816040 | chr3 2832363 2834888 | |||
| Protein PNS1 | BRETBRUG00000003498 | BRETBRUG00000003505 | ||
| chr3 2816967 2818574 | chr3 2835815 2837422 | |||
Letter in first column indicates guide-mark in Fig 2B.
Amplification of genes on either the same or on different chromosomes was observed. Interchromosomal single gene duplication was observed for a gene encoding for tRNA (guanine(10)-N2)-methyltransferase (letter A in Table 6 and Fig 2). This gene has copies on chromosome 1 and chromosome 3. One intrachromosomal single gene duplication was identified on chromosome 4 (a gene coding for methylthioribulose-1-phosphate dehydratase, letter B in Table 6 and Fig 2). The methylthioribulose-1-phosphate dehydratase gene copies are separated by the gene encoding for U6 snRNA-associated Sm-like protein LSm8.
There were also two examples of amplified gene clusters. These clusters both contain six genes. One of the clusters (letter C in Table 6 and Fig 2) contains genes encoding for hexokinase-1, flocculation protein FLO5, temperature shock-inducible protein 1, putative transcriptional regulatory protein, an uncharacterized transcriptional regulatory protein and allantoin permease. One copy of this cluster is located in chr1:3,856,343–3,882,306 and the other copy in chr4:1,383,442–1,357,446. The other six-gene cluster (letter D in Table 6 and Fig 2) comprises genes encoding for indoleamine C3-dioxygenase, histone acetyltransferase ESA1, serine/threonine-protein phosphatase 2A activator, GABA-specific permease, V-type proton ATPase subunit (vacuolar isoform) and protein PNS1. This gene cluster forms a tandem copy chr3:2,804,460–2,818,574 and chr3:2,823,355–2,837,422. Interestingly, in the first copy of the gene cluster, the gene encoding for indoleamine C3-dioxygenase forms itself a tandem duplication (one more copy of this gene is present between chr3:2,801,423–2,802,706), but the gene is not duplicated in the other copy of the gene cluster.
It is notable that none of the duplicated genes in the genome of strain CBS 11270 had analogous duplications in the genome of the wine strain CBS 2499 [21].
Centromers, simple, low complexity and interspersed repeat analysis
Analysis of centromere structures on chromosomes of B. bruxellensis is presented in S3 Table. Partial sequences of B. bruxellensis CBS 2499 centromeres [43] were identified on chromosome 1 (CEN1), and on chromosomes 1, 2, and 4 (CEN2). None of the identified centromer structures were found on chromosome 3.
Repeats identified in the CBS 11270 genome are summarized in S7 File. 2% of the genome consisted of repeats. We found 53 non-long terminal repeat- (LTR) retrotransposons (48 Long Interspersed Nuclear Elements (LINE) and five Short Interspersed Nuclear Elements (SINE)), 6 DNA transposons (DNA/TcMar-Tigger, DNA/hAT-Ac, DNA/hAT-Charlie), 3228 simple repeats, 771 low complexity repeats, two snRNA, five rRNA and 55 tRNA.
Interestingly, five of the duplicated genes (Table 7) did contain mainly simple and low complexity repeats both in the upstream and downstream flanking regions. Two of these genes (both coding for GABA-specific permease) even contained the non-LTR retrotransposon AmnL2-1 LINE/L2 in the downstream regions. Three of the duplicated genes contained repeats only in downstream regions and two others had retrotransposon AmnL2-1 LINE/L2 in their upstream regions.
Table 7. Characterization of repeats in flanking regions of duplicated genes.
| Gene id and location | Gene name | Downstream repeat | Upstream repeat |
|---|---|---|---|
| BRETBRUG00000003060 | tRNA (guanine(10)-N2)-methyltransferase | (AAGATAG)n Simple_repeat 1807186 1807233 | (T)n Simple_repeat 1805044 1805068 |
| chr3 1805200 1807169 | (CTC)n Simple_repeat 1807517 1807558 | ||
| (AGTAA)n Simple_repeat 1808962 1809011 | |||
| BRETBRUG00000003500 | Indoleamine 2 2C3-dioxygenase | A-rich Low_complexity 2826505 2826555 | |
| chr3 2823355 2824602 | |||
| BRETBRUG00000003494 | Histone acetyltransferase ESA1 | A-rich Low_complexity 2807656 2807706 | |
| chr3 2807345 2808712 | |||
| BRETBRUG00000003495 | Serine/threonine-protein phosphatase 2A activator | (T)n Simple_repeat 2810049 2810080 | A-rich Low_complexity 2807656 2807706 |
| chr3 2808829 2809905 | |||
| BRETBRUG00000003496 | GABA-specific permease | AmnL2-1 LINE/L2 2812187 2812238 | (T)n Simple_repeat 2810049 2810080 |
| chr3 2810290 2811957 | |||
| BRETBRUG00000003497 | V-type proton ATPase subunit a 2C vacuolar isoform | AmnL2-1 LINE/L2 2812187 2812238 | |
| chr3 2813515 2816040 | |||
| BRETBRUG00000003501 | Histone acetyltransferase ESA1 | (T)n Simple_repeat 2828899 2828928 | |
| chr3 2826194 2827561 | |||
| BRETBRUG00000003502 | Serine/threonine-protein phosphatase 2A activator | (T)n Simple_repeat 2828899 2828928 | A-rich Low_complexity 2826505 2826555 |
| chr3 2827678 2828226 | |||
| BRETBRUG00000003503 | GABA-specific permease | AmnL2-1 LINE/L2 2831034 2831085 | (T)n Simple_repeat 2828899 2828928 |
| chr3 2829138 2830805 | |||
| BRETBRUG00000003504 | V-type proton ATPase subunit a%2C vacuolar isoform | AmnL2-1 LINE/L2 2831034 2831085 | |
| chr3 2832363 2834888 |
Comparative genome analysis of B. bruxellensis CBS 11270 and CBS 2499
To compare the genome organization of CBS 11270 with another B. bruxellensis strain, we aligned the scaffolds obtained for the wine strain CBS 2499 [21] to the chromosomes of CBS 11270 (S4 Fig) using the multiple genome alignment tool Mauve 2.3.1 (see Methods). This program recognizes regions similar to the reference as blocks. Those regions could be composed of several scaffolds when they align to a larger reference scaffold, or they can also be part of a scaffold, if the rest of the scaffold does not align at this position. Scaffolds 4, 6, 27 and a substantial part of scaffold 2 form a block with high similarity to the segment between 0.1 Mbp to 2.5 Mbp of chromosome 1. Moreover, also a part of scaffold 3 mapped to chromosome 1. Two blocks of scaffold 2 had another order as compared to the homologous regions of chromosome 1. Scaffolds 17, 1, 29, 15 and 12 almost completely covered chromosome 2. The first segment of chromosome 3 up to 1.6 Mbp was almost completely covered by scaffolds 18, 5, 8 and 14. Apart from this, parts of scaffolds 2, 3 and 13 mapped to chromosome 3. Major parts of scaffolds 20, 10, 16, 21, 7, 11, 9 and 24 were similar to parts of chromosome 4, whereas only the last third of scaffold 19 aligned to chromosome 4.
We also investigated single nucleotide polymorphisms (SNPs) and indels between the two strains, by mapping the CBS 11270 reads to the CBS 2499 genome (see Methods and S8 File). CBS 11270 differed from CBS 2499 in 96.421 variants: 88.534 SNPs and 8.133 indels. 10.626 variants are homozygous, 85.552 heterozygous with one allele common to CBS 2499 and 243 are potential heterozygous variants. with both alleles different from CBS2499.
42,064 inter-strain variants were located inside open reading frames (ORF), i.e. 47.5% of total inter-strain variants are in coding regions, which is higher than the proportion of heterozygous sites in ORFs of CBS 11270 (38%). 28,679 variants caused amino acid substitution. Almost all genes (4,410) were polymorphic between B. bruxellensis CBS 11270 and CBS 2499. 46,450 variants were in non-coding regions (see S9 File). A list of the genes containing variants and a list of genes without variants is presented in S10 and S11 Files, respectively. As observed in the intra-strain heterozygosity pattern (see above), transitions were three times more abundant than transversions, and the number of variants per gene and gene counts were in inverse relationship. 839 genes were found with one variant. In one gene, annotated as gm1.2215_g (AP-1 accessory protein) (Table 4), 108 variants were found. Other genes with a high number of variants included: fgenesh1_pm.2_#_424 b (Ccr4-Not transcription complex subunit (NOT1) 100 variants, CE91624_56964 (hypothetical protein) 97 variants, CE84544_34760 (multidrug transporter) 87 variants, fgenesh1_kg.1_#_362_#_Locus3870v1rpkm14.01 (DNA repair protein RAD50) 84 variants, e_gw1.2.1034.1 (Midasin) 76 variants, estExt_Genewise1Plus.C_5_t20257 (RNA helicase) 75 variants, gm1.2263_g (hypothetical protein) 69 variants, estExt_Genewise1Plus.C_6_t20136 (N-glycosylated protein) 68 variants, gm1.360_g (protein of unknown function) 63 variants.
The size of the indels ranged from 1 to 201 nucleotides (Fig 4B). Indels of almost all sizes were most often sequences from CBS 2499 absent in CBS 11270 rather than the opposite. 3,571 single nucleotide indels were found in CBS 11270 compared to CBS 2499. In total, 8,133 indels were observed in CBS 11270 compared to CBS 2499. These 8,133 indels had a total length of 41,233 nucleotides.
Gene content differences between CBS 11270 and CBS 2499
CBS 11270 and CBS 2499 differed in their gene contents. 19 genes were found in CBS 2499 but not in CBS 11270, by using BLASTN-search versus whole CBS 11270 genome assembly (see S4 Table). Table 8 shows some of these genes present in the genome of CBS 2499 but absent in CBS 11270. Most of these genes are hypothetical proteins. Two genes involved in transport through plasma membrane, a gene encoding for Na+/H+ antiporter involved in sodium and potassium efflux and a putative transmembrane sensor transporter were absent from the CBS 11270 genome. A gene involved in antioxidant metabolism, s-formylglutathione hydrolase was also absent in CBS 11270. A gene coding for maltase was absent in CBS 11270, which is not consistent with the ability of this strain to grow on maltose [16].
Table 8. Genes in the B. bruxellensis CBS 2499 genome absent in the CBS 11270 genome.
| Gene name | Best blast hit |
|---|---|
| jgi|Dekbr2|26744|fgenesh1_kg.19_#_10_#_Locus2611v2rpkm45.30 | Na+/H+ antiporter involved in sodium and potassium efflux through the plasma membrane [Ogataea parapolymorpha DL-1]. |
| jgi|Dekbr2|8850|gm1.5259_g | maltase [Brettanomyces bruxellensis AWRI1499]. |
| jgi|Dekbr2|8855|gm1.5264_g | putative transmembrane sensor transporter [Brettanomyces bruxellensis AWRI1499]. |
| jgi|Dekbr2|145681|CE57809_24 NA jgi|Dekbr2|51831|e_gw1.23.15.1 |
s-formylglutathione hydrolase [Brettanomyces bruxellensis AWRI1499] |
31 genes were identified in CBS 11270 that were not present in CBS 2499 (S5 Table). Further analysis would be required to verify the absence of these genes in CBS 2499.
Discussion
This study represents the first genomic investigation of a B. bruxellensis-strain that functions as an ethanol production strain [12, 16]. Using the recently developed assembly of the CBS 11270 genome to scaffolds of chromosome size [44] we could associate a major part, 86.4% of the genome sequences, to the assembled four chromosomes.
Due to the re-construction of chromosomes we could identify larger re-arrangements of the genome, and we found that the B. bruxellensis-genome is highly flexible. Scaffolds identified earlier in the wine isolate CBS 2499 [21] were split or arranged differently in CBS 11270. For instance, parts of scaffold 2 of CBS 2499 mapped to chromosomes 1 and 3 in CBS 11270, and parts of scaffold 2 were in a different order compared to CBS 2499. Alternatively, the mismatch of contigs order between two genomes could arise from assembly errors [45]. The combination of various sequencing and assembly strategies aimed to strengthen the accuracy of the CBS 11270 genome sequence [32]. The size of our identified four chromosomes was in the range from 2.2–4 Mbp, which fits to results obtained by pulsed field electrophoresis. The pulsed field investigations even indicated a potential fifth chromosome of about 500 kb [18], and it is possible that some of our non-assembled contigs belong to this chromosome. However, using our assembly approach we could not confirm its existence [32]. Large differences between different B. bruxellensis strains in chromosome size and -number have been demonstrated by pulsed field electrophoresis, with chromosome sizes ranging from below 1 Mbp up to 6 Mbp, and chromosome numbers up to nine [22]. We also found a number of deletions (in the largest case about 149099 bp were missing in one of the homologues of chromosome 1, leading to a deletion of 33 genes) in homologous chromosomes. These findings strongly indicate a very flexible genome of B. bruxellensis. Chromosome re-arrangements have mainly been observed in non-sexual species such as Candida glabrata or Candida albicans [46, 47]. Ordered meiosis seems to be difficult or impossible when there is such flexibility of chromosomes. Ascospores have been observed in B. bruxellensis [48], but no further investigation of those ascospores has been reported, and thus there is no genetic evidence for the existence of a sexual cycle in B. bruxellensis. On the other hand, the existence of allotriploid wine strains indicates mating activity even over species borders [23, 26]. Possibly, B. bruxellensis uses a similar program of genetic recombination as has been described for C. albicans, where mating is followed by a mitotic chromosome loss [49].
In general, we found a very high variability in the genome of the industrial strain. The number of SNPs when comparing the homologous chromosomes (44,022, i.e. 0.34% of the total haploid genome) was higher than the variability between distantly related S. cerevisiae strains. In S. cerevisiae the number of variants is lower and varies between strains: 39, 4894, 7955, 13,914, 25,298 between S288C and BY4716, A364A, W303, FL100, CEN.PK, S1278b, SK1 [50], YJSH1 [51], respectively. Curtins et al. reported 342,900 heterozygotic sites within the genome of the wine isolate AWRI1499 [26]. Distribution of SNPs along the chromosomes was uneven, with local maxima of the SNP-frequence (Fig 3), indicating the location of highly polymorphic sequences or highly repetitive sequences, similar to that observed for chromosomes of S. cerevisiae [50–52].
There was a considerable interstrain- variability, more than 88,000 SNPs were identified in CBS 11270 compared to the wine strain CBS 2499. In total, 96,421 variants (SNPs and indels) were found between the two strains. This was slightly higher but still in the same order of what has been found when comparing several wine strains, ST05.12/22 and AWRI 1499 (79,627 variants), and ST05.12/22 and CBS 2499 (82,676 variants) These numbers illustrate, that there is a high diversity within the species B. bruxellensis.
Among the SNPs, transitions were about three times as frequent as transversions. Although there are double as many possibilities for transversions to occur, the transition to transversion bias has been observed in almost all known biological systems. Transitions are only in half the cases resulting in amino acid exchanges compared to transversions, however, as it has recently been pointed out, the background of the transition:transversion bias is not really understood [53].
We found 18 genes with high SNP density (more than 35 SNPs per gene), suggesting that they may be under some selective pressure (Weihong Qi 2009) [54]. Indeed, Yi-Cheng Guo et al (2016) showed that the genes of the transcription system in B. bruxellensis CBS 2249 exhibited faster evolution than other genes [55]. We identified 77 SNPS in a gene coding for a general negative regulator of transcription subunit 1 (BRETBRUG00000002734) in CBS 11270 (see Table 4).
The ecosystem from which this strain has been isolated is very different from that of wine and beer strains [13, 44, 56]. The industrial conditions, consisting of year-long continuous cultivation with cell recirculation at constant low pH (3.5), considerable ethanol concentrations (about 60 g/l), and relatively high temperature (37°C) [12], provide a stressful, but relatively constant environment. Constant environments often result in reductive evolution, resulting in gene losses within the strains under these conditions [29, 56]. Frequently, loss-of-function mutations can provide a selection advantage in those environments [57]. However, although we observed a substantial loss of heterozygosity, i.e. loss of one of the homologous genes in 372 cases, we did not find a substantial loss of function within known metabolic pathways. Massive loss of heterozygosity was also shown in D. bruxellensis wine strain UMY321 [29]. There may be various challenges for the strain in the ethanol process, for instance during cell recirculation, or when interacting with the high number of lactic acid bacteria in the process [10, 58], which provide a certain selectivity for multiple metabolic pathways. Previous experiments showed that isolates from this process are able to ferment cellobiose [14], and that CBS 11270 can adapt to inhibitors of lignocellulose hydrolysate [18] and thus can cope with conditions that are quite different from a starch-based ethanol process. In diploids, events other than merely gene losses, such as mutations modifying gene expression, may provide a fitness advantage for the respective strain [49], and further investigation may be required to identify mutations that are specific for the ethanol production environment.
B. bruxellensis is a unique yeast with an amazing competitiveness in the stressful environments of wine-, beer- and bioethanol production. Many traits of its physiology are still not understood. A variety of isolates from wine and beer production have been sequenced to date. Here, we present the first genome of an ethanol production strain in chromosome-sized scaffolds which may serve as a reference to reconstruct chromosomes of strains from a variety of environments. This will help to reconstruct mutational events that are correlated to the adaptation to different environments, and thus, contribute to understanding of the unique features of B. bruxellensis physiology. Moreover, our study demonstrates the enormous flexibility of the B. bruxellensis genome. This flexibility may be utilized in artificial evolution experiments in appropriate long-term cultivations, and thus, together with the recently developed methods for genetic manipulation of this yeast [49], provide a tool for obtaining strains for future biotechnological applications [13, 44, 56].
Supporting information
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
The authors acknowledge support of NBIS (National Bioinformatics Infrastructure Sweden), Science for Life Laboratory, the National Genomics Infrastructure and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. This project was supported by a Formas Mobility Starting Grant, the Swedish Energy Authority (Energimyndigheten) and the MicroDrive-program of the Swedish University of Agricultural Sciences. We wish to thank Dr. Su-Lin Hedén for proofreading the manuscript.
Data Availability
The annotated sequence of the nuclear chromosomes, contigs not associated with chromosomes and mitochondrial DNA for B. bruxellensis strains CBS 11270 has been deposited in the European Nucleotide Archive (ENA) with the accession numbers from LS990926 to LS990930 for chromosomes and mitochondrial contigs and from UFQA01000001 to UFQA01000432 for contigs not associated with chromosomes.
Funding Statement
This work received funding from FORMAS (Mobility starting grant #2016-00767) to IAT and from Swedish Energy Authority (#34134-1) to VP. This project was supported by the Swedish University of Agricultural Sciences (MicroDrive-program). There was no additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kurtzman CP, Fell JW, Boekhout T. The yeasts, a taxonomic study. 2011. 5.ed. [Google Scholar]
- 2.Hawksworth DL, Crous PW, Redhead SA, Reynolds DR, Samson RA, Seifert KA et al. The amsterdam declaration on fungal nomenclature. 2011. IMA fungus 2: 105–112. 10.5598/imafungus.2011.02.01.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loureiro V, Malfeito-Ferreira M. Spoilage yeasts in the wine industry. Int J Food Microbiol. 2003. September 1;86(1–2):23–50. [DOI] [PubMed] [Google Scholar]
- 4.Agnolucci M, Tirelli A, Cocolin L, Toffanin A. Brettanomyces bruxellensis yeasts: impact on wine and winemaking. World J Microbiol Biotechnol. 2017;33(10):180 10.1007/s11274-017-2345-z [DOI] [PubMed] [Google Scholar]
- 5.Leite FC, Basso TO, Pita Wde B, Gombert AK, Simoes DA, de Morais MA Jr. Quantitative aerobic physiology of the yeast Dekkera bruxellensis, a major contaminant in bioethanol production plants. FEMS Yeast Res. 2013. February;13(1):34–43. 10.1111/1567-1364.12007 [DOI] [PubMed] [Google Scholar]
- 6.Liberal ATD, Basilio ACM, Resende AD, Brasileiro BTV, da Silva EA, de Morais JOF, et al. Identification of Dekkera bruxellensis as a major contaminant yeast in continuous fuel ethanol fermentation. J Appl Microbiol. 2007. February;102(2):538–47. 10.1111/j.1365-2672.2006.03082.x [DOI] [PubMed] [Google Scholar]
- 7.van Nedervelde L, Debourg A. Properties of Belgian acid beers and their microflora. II. Biochemical properties of Brettanomyces yeasts. Cerev Biotechnol. 1995;20:43–8. [Google Scholar]
- 8.van Oevelen D, Spaepen M, Timmermans P, Verachtert H. Microbiological aspects of spontaneous wort fermentation in the production of lambic and gueuze. J Inst Brew,. 1977;83:356–60. [Google Scholar]
- 9.Crauwels S, Van Opstaele F, Jaskula-Goiris B, Steensels J, Verreth C, Bosmans L, et al. Fermentation assays reveal differences in sugar and (off-) flavor metabolism across different Brettanomyces bruxellensis strains. FEMS Yeast Res. 2017;17(1). [DOI] [PubMed] [Google Scholar]
- 10.Passoth V, Blomqvist J, Schnürer J. Dekkera bruxellensis and Lactobacillus vini form a stable ethanol-producing consortium in a commercial alcohol production process. Appl Environ Microbiol. 2007. July;73(13):4354–6. 10.1128/AEM.00437-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blomqvist J, Passoth V. Dekkera bruxellensis—spoilage yeast with biotechnological potential, and a model for yeast evolution, physiology and competitiveness. FEMS Yeast Res. 2015. June;15(4):fov021 10.1093/femsyr/fov021 [DOI] [PubMed] [Google Scholar]
- 12.de Barros Pita W, Leite FC, de Souza Liberal AT, Simoes DA, de Morais MA Jr. The ability to use nitrate confers advantage to Dekkera bruxellensis over S. cerevisiae and can explain its adaptation to industrial fermentation processes. Antonie Van Leeuwenhoek. 2011. June;100(1):99–107. 10.1007/s10482-011-9568-z [DOI] [PubMed] [Google Scholar]
- 13.Blomqvist J, Nogue VS, Gorwa-Grauslund M, Passoth V. Physiological requirements for growth and competitiveness of Dekkera bruxellensis under oxygen-limited or anaerobic conditions. Yeast. 2012. July;29(7):265–74. 10.1002/yea.2904 [DOI] [PubMed] [Google Scholar]
- 14.Blomqvist J, Eberhard T, Schnürer J, Passoth V. Fermentation characteristics of Dekkera bruxellensis strains. Appl Microbiol Biot. 2010. Jul;87(4):1487–97. [DOI] [PubMed] [Google Scholar]
- 15.Spindler DD, Wyman CE, Grohmann K, Philippidis GP. Evaluation of the Cellobiose-Fermenting Yeast Brettanomyces-Custersii in the Simultaneous Saccharification and Fermentation of Cellulose. Biotechnol Lett. 1992. May;14(5):403–7. [Google Scholar]
- 16.Schifferdecker A. Development of molecular biology tools for the wine and beer yeast Dekkera bruxellensis. Lund: Lund university; 2015. [Google Scholar]
- 17.Blomqvist J, South E, Tiukova I, Momeni MH, Hansson H, Ståhlberg J, et al. Fermentation of lignocellulosic hydrolysate by the alternative industrial ethanol yeast Dekkera bruxellensis. Lett Appl Microbiol. 2011. July;53(1):73–8. 10.1111/j.1472-765X.2011.03067.x [DOI] [PubMed] [Google Scholar]
- 18.Tiukova IA, de Barros Pita W, Sundell D, Haddad Momeni M, Horn SJ, Ståhlberg J, et al. Adaptation of Dekkera bruxellensis to lignocellulose-based substrate. Biotechnol Appl Bioc. 2014. Jan-Feb;61(1):51–7. [DOI] [PubMed] [Google Scholar]
- 19.Rozpedowska E, Hellborg L, Ishchuk OP, Orhan F, Galafassi S, Merico A, et al. Parallel evolution of the make-accumulate-consume strategy in Saccharomyces and Dekkera yeasts. Nat Commun. 2011;2:302 10.1038/ncomms1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng J, Guo X, Cai P, Cheng X, Piskur J, Ma Y, et al. Parallel Evolution of Chromatin Structure Underlying Metabolic Adaptation. Mol Biol Evol. 2017;34(11):2870–8. 10.1093/molbev/msx220 [DOI] [PubMed] [Google Scholar]
- 21.Piskur J, Ling Z, Marcet-Houben M, Ishchuk OP, Aerts A, LaButti K, et al. The genome of wine yeast Dekkera bruxellensis provides a tool to explore its food-related properties. Int J Food Microbiol. 2012. July 2;157(2):202–9. 10.1016/j.ijfoodmicro.2012.05.008 [DOI] [PubMed] [Google Scholar]
- 22.Hellborg L, Piskur J. Complex nature of the genome in a wine spoilage yeast, Dekkera bruxellensis. Eukaryot Cell. 2009. November;8(11):1739–49. 10.1128/EC.00115-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borneman AR, Zeppel R, Chambers PJ, Curtin CD. Insights into the Dekkera bruxellensis genomic landscape: comparative genomics reveals variations in ploidy and nutrient utilisation potential amongst wine isolates. PLoS Genet. 2014. February;10(2):e1004161 10.1371/journal.pgen.1004161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crauwels S, Van Assche A, de Jonge R, Borneman AR, Verreth C, Troels P, et al. Comparative phenomics and targeted use of genomics reveals variation in carbon and nitrogen assimilation among different Brettanomyces bruxellensis strains. Appl Microbiol Biot. 2015. July 2. [DOI] [PubMed] [Google Scholar]
- 25.Crauwels S, Zhu B, Steensels J, Busschaert P, De Samblanx G, Marchal K, et al. Assessing genetic diversity among Brettanomyces yeasts by DNA fingerprinting and whole-genome sequencing. Appl Environ Microbiol. 2014. July;80(14):4398–413. 10.1128/AEM.00601-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curtin CD, Borneman AR, Chambers PJ, Pretorius IS. De-novo assembly and analysis of the heterozygous triploid genome of the wine spoilage yeast Dekkera bruxellensis AWRI1499. PLoS One. 2012. 7(3):e33840 10.1371/journal.pone.0033840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valdes J, Tapia P, Cepeda V, Varela J, Godoy L, Cubillos FA, et al. Draft genome sequence and transcriptome analysis of the wine spoilage yeast Dekkera bruxellensis LAMAP2480 provides insights into genetic diversity, metabolism and survival. FEMS Microbiol Lett. 2014. December;361(2):104–6. 10.1111/1574-6968.12630 [DOI] [PubMed] [Google Scholar]
- 28.Woolfit M, Rozpedowska E, Piskur J, Wolfe KH. Genome survey sequencing of the wine spoilage yeast Dekkera (Brettanomyces) bruxellensis. Eukaryot Cell. 2007. April;6(4):721–33. 10.1128/EC.00338-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fournier T, Gounot JS, Freel K, Cruaud C, Lemainque A, Aury JM, et al. High-Quality de Novo Genome Assembly of the Dekkera bruxellensis Yeast Using Nanopore MinION Sequencing. G3 (Bethesda). 2017;7(10):3243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avramova M, Cibrario A, Peltier E, Coton M, Coton E, Schacherer J, et al. Brettanomyces bruxellensis population survey reveals a diploid-triploid complex structured according to substrate of isolation and geographical distribution. Sci Rep-Uk. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curtin CD, Pretorius IS. Genomic insights into the evolution of industrial yeast species Brettanomyces bruxellensis. FEMS Yeast Res. 2014. November;14(7):997–1005. 10.1111/1567-1364.12198 [DOI] [PubMed] [Google Scholar]
- 32.Olsen R, Bunikis I, Tiukova I, Holmberg K, Lotstedt B, Pettersson O, V., et al. De novo assembly of Dekkera bruxellensis: a multi technology approach using short and long-read sequencing and optical mapping. GigaScience. 2015. November; 4(56). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norling M, Jareborg N, Dainat J. EMBLmyGFF3: a converter facilitating genome annotation submission to European Nucleotide Archive. BMC Res Notes. 2018;11(1):584 10.1186/s13104-018-3686-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009. July 15;25(14):1754–60. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009. August 15;25(16):2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011. May;43(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011. June;21(6):974–84. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004. July; 14(7): 1394–403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990. October 5;215(3):403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 40.Vezzi F, Narzisi G, Mishra B (2012) Feature-by-feature-evaluating de novo sequence assembly. PloS one 7: e31002 10.1371/journal.pone.0031002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou S, Wei F, Nguyen J, Bechner M, Potamousis K, Goldstein S, et al. (2009) A single molecule scaffold for the maize genome. PLoS genetics 5: e1000711 10.1371/journal.pgen.1000711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiukova IA, Petterson ME, Tellgren-Roth C, Bunikis I, Eberhard T, Pettersson OV, et al. Transcriptome of the alternative ethanol production strain Dekkera bruxellensis CBS 11270 in sugar limited, low oxygen cultivation. PloS one. 2013;8(3):e58455 10.1371/journal.pone.0058455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishchuk OP, Vojvoda Zeljko T, Schifferdecker AJ, Mebrahtu Wisen S, Hagstrom AK, Rozpedowska E, et al. Novel Centromeric Loci of the Wine and Beer Yeast Dekkera bruxellensis CEN1 and CEN2. PloS one 2016. 11: e0161741 10.1371/journal.pone.0161741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steensels J, Daenen L, Malcorps P, Derdelinckx G, Verachtert H, Verstrepen KJ Brettanomyces yeasts-From spoilage organisms to valuable contributors to industrial fermentations. International J of Food Microbiol. 2015. 206: 24–38. [DOI] [PubMed] [Google Scholar]
- 45.Heydari M, Miclotte G, Demeester P, Van de Peer Y, Fostier J. Evaluation of the impact of Illumina error correction tools on de novo genome assembly. Bmc Bioinformatics. 2017;18 10.1186/s12859-016-1415-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janbon G, Sherman F, Rustchenko E Appearance and properties of L-sorbose-utilizing mutants of Candida albicans obtained on a selective plate. Genetics. 1999. 153: 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polakova S, Blume C, Zarate JA, Mentel M, Jorck-Ramberg D, Stenderup J, et al. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci U S A. 2009. February 24;106(8):2688–93. 10.1073/pnas.0809793106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Walt JP. Dekkera, new genus of Saccharomycetaceae. Antonie Van Leeuwenhoek Journal of Microbiology and Serology. 1964;30(3). [DOI] [PubMed] [Google Scholar]
- 49.Bennett RJ, Johnson AD. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. Embo J. 2003. May 15;22(10):2505–15. 10.1093/emboj/cdg235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schacherer J, Ruderfer DM, Gresham D, Dolinski K, Botstein D, Kruglyak L Genome-wide analysis of nucleotide-level variation in commonly used Saccharomyces cerevisiae strains. PloS one 2007. 2: e322 10.1371/journal.pone.0000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Zhang W, Zheng D, Zhou Z, Yu W, Zhang L, et al. Genomic evolution of Saccharomyces cerevisiae under Chinese rice wine fermentation. Genome biology and evolution 2014. 6: 2516–2526. 10.1093/gbe/evu201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song W, Dominska M, Greenwell PW, Petes TD Genome-wide high-resolution mapping of chromosome fragile sites in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 2014. 111: E2210–2218. 10.1073/pnas.1406847111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stoltzfus A, Norris RW On the Causes of Evolutionary Transition:Transversion Bias. Molecular biology and evolution 2016. 33: 595–602. 10.1093/molbev/msv274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi W, Kaser M, Roltgen K, Yeboah-Manu D, Pluschke G Genomic diversity and evolution of Mycobacterium ulcerans revealed by next-generation sequencing. PLoS pathogens 2009. 5: e1000580 10.1371/journal.ppat.1000580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo YC, Zhang L, Dai SX, Li WX, Zheng JJ, Li GH, et al. Independent Evolution of Winner Traits without Whole Genome Duplication in Dekkera Yeasts. PloS one 2016. 11: e0155140 10.1371/journal.pone.0155140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wernegreen JJ For better or worse: genomic consequences of intracellular mutualism and parasitism. Current opinion in genetics & development 2005. 15: 572–583. 10.1016/j.gde.2005.09.013 [DOI] [PubMed] [Google Scholar]
- 57.Lang GI, Murray AW, Botstein D The cost of gene expression underlies a fitness trade-off in yeast. Proceedings of the National Academy of Sciences of the United States of America 2009. 106: 5755–5760. 10.1073/pnas.0901620106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tiukova I, Eberhard T, Passoth V Interaction of Lactobacillus vini with the ethanol-producing yeasts Dekkera bruxellensis and Saccharomyces cerevisiae. Biotechnology and Applied Biochemistry 2014. 61: 40–44. 10.1002/bab.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(TXT)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
The annotated sequence of the nuclear chromosomes, contigs not associated with chromosomes and mitochondrial DNA for B. bruxellensis strains CBS 11270 has been deposited in the European Nucleotide Archive (ENA) with the accession numbers from LS990926 to LS990930 for chromosomes and mitochondrial contigs and from UFQA01000001 to UFQA01000432 for contigs not associated with chromosomes.