

Abstract
Background
Despite the high prevalence of apathy in Alzheimer’s disease (AD), and its harmful effects, there are currently no therapies proven to treat this symptom. Recently, a number of pharmacological therapies have been investigated as potential treatments for apathy in AD.
Objectives
Objective 1: To assess the safety and efficacy of pharmacotherapies for the treatment of apathy in Alzheimer's disease (AD). Objective 2: To assess the effect on apathy of pharmacotherapies investigated for other primary outcomes in the treatment of AD.
Search methods
We searched the Specialized Register of the Cochrane Dementia and Cognitive Improvement Group (ALOIS), MEDLINE, Embase, CINAHL, PsycINFO, LILACS, ClinicalTrials.gov and the World Health Organization (WHO) portal, ICTRP on 17 May 2017.
Selection criteria
Eligible studies were double‐blind, randomized, placebo‐controlled trials (RCTs) investigating apathy as a primary or secondary outcome in people with AD.
Data collection and analysis
Three review authors extracted data. We assessed the risks of bias of included studies using Cochrane methods, and the overall quality of evidence for each outcome using GRADE methods. We calculated mean difference (MD), standardized mean difference (SMD) or risk ratio (RR) with 95% confidence intervals on an intention‐to‐treat basis for all relevant outcome measures.
Main results
We included 21 studies involving a total of 6384 participants in the quantitative analyses. Risk of bias is very low to moderate. All studies reported appropriate methods of randomization and blinding. Most studies reported appropriate methods of allocation concealment. Four studies, three with methylphenidate and one with modafinil, had a primary aim of improving apathy. In these studies, all participants had clinically significant apathy at baseline. Methylphenidate may improve apathy compared to placebo. This finding was present when apathy was assessed using the apathy evaluation scale (AES), which was used by all three studies investigating methylphenidate: MD ‐4.99, 95% CI ‐9.55 to ‐0.43, n = 145, 3 studies, low quality of evidence, but not when assessed with the neuropsychiatric inventory (NPI)‐apathy subscale, which was used by two of the three studies investigating methylphenidate: MD ‐0.08, 95% CI ‐3.85 to 3.69, n = 85, 2 studies, low quality of evidence. As well as having potential benefits for apathy, methylphenidate probably also slightly improves cognition (MD 1.98, 95% CI 1.06 to 2.91, n = 145, 3 studies, moderate quality of evidence), and probably improves instrumental activities of daily living (MD 2.30, 95% CI 0.74 to 3.86, P = 0.004, n = 60, 1 study, moderate quality of evidence), compared to placebo. There may be no difference between methylphenidate and placebo in the risk of developing an adverse event: RR 1.28, 95% CI 0.67 to 2.42, n = 145, 3 studies, low quality of evidence. There was insufficient evidence from one very small study of modafinil to determine the effect of modafinil on apathy assessed with the FrSBe‐apathy subscale: MD 0.27, 95% CI ‐3.51 to 4.05, n = 22, 1 study, low quality of evidence. In all other included studies, apathy was a secondary outcome and participants were not selected on the basis of clinically significant apathy at baseline. We considered the evidence on apathy from these studies to be indirect and associated with publication bias. There was low or very low quality of evidence on cholinesterase inhibitors (ChEIs) (six studies), ChEI discontinuation (one study), antipsychotics (two studies), antipsychotic discontinuation (one study), antidepressants (two studies), mibampator (one study), valproate (three studies) and semagacestat (one study).
Authors' conclusions
Methylphenidate may demonstrate a benefit for apathy and may have slight benefits for cognition and functional performance in people with AD, but this finding is associated with low‐quality evidence. Our meta‐analysis is limited by the small number of studies within each drug class, risk of bias, publication bias, imprecision and inconsistency between studies. Additional studies should be encouraged targeting people with AD with clinically significant apathy which investigate apathy as a primary outcome measure, and which have a longer duration and a larger sample size. This could increase the quality of evidence for methylphenidate, and may confirm whether or not it is an effective pharmacotherapy for apathy in AD.
Plain language summary
Drug treatments for apathy in Alzheimer's disease
Review question
We wanted to know whether there are any medications that are safe and effective for reducing apathy in people with Alzheimer's disease.
Background
Apathy is a state of reduced interest, lack of initiative and reduced activity. It is a very common symptom of Alzheimer's disease. It is often persistent and it is known to be linked to a lower quality of life, faster decline and more burden on caregivers. Effective treatments of apathy could improve the quality of life for people with Alzheimer's disease and their families.
What we did
We searched for randomized controlled trials (RCTs) up to May 2017 which had compared any medicine with a placebo (dummy pill) and measured the effect on apathy in people with Alzheimer's disease. We were only interested in trials in which it was decided randomly whether the people taking part got the drug of interest or the placebo; this was to make sure that the comparison was as fair as possible.
What we found
We found 21 RCTs involving more than 6300 people with Alzheimer’s disease. Four trials of two different medicines (methylphenidate and modafinil) had been done specifically to study apathy, so all the people taking part were known to be significantly apathetic before the trial started. The other 17 trials had other primary aims, but reported some data on apathy. The trials were generally well designed and conducted. From the three trials with methylphenidate, we found that it may improve apathy, although this depended on how the apathy was measured. The people taking methylphenidate also did slightly better than those taking placebo on scales measuring cognition (thinking, remembering, etc.) and some daily activities, but it was not clear that these effects were big enough to be important in practice. We found no evidence that it caused more side effects than placebo. The quality of this evidence was low or moderate, so we cannot be certain that other similar studies would not have different results. There was only one very small trial with modafinil and there was no evidence that it was effective for apathy. The other 17 trials studied a variety of medicines and included people who were not necessarily significantly apathetic to start with. We therefore thought they were only indirectly relevant to our review question. It is also highly likely that other trials of the same drugs have measured apathy but have not published the results, so we were concerned about possible publication bias (that the studies we found could have been a biased subset). We therefore thought the quality of evidence for all these other medicines was low or very low, meaning that we can have limited or little confidence in the results.
Conclusions
Current evidence suggests that methylphenidate may be useful for treating apathy in Alzheimer's disease. However, more trials should be done specifically targeting apathy in order to improve the overall quality of the evidence.
Summary of findings
Background
Description of the condition
Current evidence suggests that Alzheimer’s disease (AD), a debilitating neurodegenerative disease, is increasing in prevalence amongst the ageing population. The World Alzheimer Report estimates that by 2015 47 million people worldwide would be living with dementia due to AD, and that this will double every 20 years, to 74.7 million people in 2030 and 131 million people in 2050 (World Alzheimer Report 2015). AD pathology often occurs together with vascular pathology (mixed AD) (Schneider 2007; Attems 2014). AD and mixed AD share cognitive and behavioral symptoms (Kapasi 2016). For the purposes of this systematic review, we will therefore use ‘AD’ when we refer to individuals diagnosed with AD or mixed AD.
Neuronal damage and death in the brain of a person with dementia lead to progressive changes in cognition, function and behavior, which negatively impact his or her ability to perform everyday activities of daily living and increase dependence on others. Treating AD is a healthcare and societal priority as the cost of care and the burden on caregivers increases. The governments of countries such as the UK, the USA, Canada, Norway, France and South Korea have developed and implemented national strategic plans to address the changes society will incur as AD prevalence increases (Prince 2013).
Treatment of the symptoms associated with AD is an important aspect of improving the quality of life of people living with AD and their caregivers. One such symptom is apathy which, according to diagnostic criteria outlined by Robert 2009, is defined as a disorder of motivation which includes reduced goal‐directed behavior, goal‐directed cognitive activity and emotions, as well as identifiable functional impairments. Apathy is associated with greater caregiver burden, as individuals diagnosed with apathy require more support to initiate and complete activities even when they may still be capable of doing so themselves. The presence of apathy increases with increasing disease severity, and is associated with increased cognitive and functional deficits (Tagariello 2009; Kales 2015). As such, apathy has been an emerging target of interest for pharmacological interventions.
Apathy and depression share some clinical features, such as diminished interest, psychomotor retardation, fatigue/hypersomnia and lack of insight, although depression is characterized by symptoms of dysphoria, suicidal ideation, self‐criticism, guilty feelings, pessimism and hopelessness which are absent in apathy (Marin 1994). There has been interstudy variability in estimates of the prevalence of apathy in individuals diagnosed with dementia (Landes 2001). This variability may be due in part to the inclusion of apathy symptoms on assessment tools of depression, such as the Hamilton Rating Scale of Depression (HAM‐D). However, in a study that administered the Neuropsychiatric Inventory (NPI), a behavioral scale which contains a depression subscale (with no apathy‐related items), and an apathy subscale (with no depression‐related items), there was no correlation between apathy and depression in dementia (Levy 1998). Several neuroimaging studies support the biological and phenomenological independence of apathy and depression. These studies report that structural relationships with apathy symptoms are functionally and anatomically distinct from structural relationships with depression (Starkstein 2009; Kang 2012).
Description of the intervention
Treatments for apathy include both behavioral and pharmacological interventions, which target psychosocial changes and neurochemical and neuropathological changes in AD, respectively (Landes 2001; Gitlin 2012).
Compared to those without significant apathy, the brains of apathetic individuals with AD show some increased pathological changes, such as increased neuronal loss (Förstl 1993), neurofibrillary tangles (Tekin 2001) and white matter hyperintensities (Starkstein 1997) in the frontal lobes and the associated circuits between frontal and subthalamic structures (Landes 2001). The cholinergic (Kaufer 1998a), dopaminergic (DA) (Roccaforte 1990; Debette 2002; Padala 2007), serotonergic (Hoehn‐Saric 1990; Marin 1995; Barnhart 2004), gamma‐aminobutyric acid (GABA)‐ergic (Lanctôt 2007a) and noradrenergic neurotransmitter systems have been investigated as potential targets for drug intervention, since each system may potentially be associated with the manifestations of apathy in people with AD.
Current pharmacological treatments used in the symptomatic management of apathy in individuals with AD include, but are not limited to, the following:
CNS stimulants.
Antidepressants.
Atypical antipsychotics.
Apomorphine.
Amantadine.
Cholinesterase inhibitors.
DA agonists.
How the intervention might work
Evidence from pharmacological, post mortem and imaging studies suggests that apathy in individuals with AD may be related to abnormalities of cholinergic, DA, serotoninergic, GABA‐ergic and noradrenergic neurotransmitter systems (Lanctôt 2001; Garcia‐Alloza 2005; Lanctôt 2007a; Lanctôt 2007b).
The cholinergic hypothesis of AD suggests that the degeneration of cholinergic neurons in the basal forebrain, and the associated disruption to cholinergic neurotransmission in the cerebral cortex, contribute greatly to the cognitive impairment experienced by individuals with AD (Bartus 1982). Cholinergic deficiency may also limit the neurotransmission between limbic system afferents and neocortical afferents, and hence contributes to the development of apathy in individuals with dementia due to AD. Treatment with acetylcholinesterase inhibitors, such as metrifonate (Kaufer 1998b; Raskind 1999; Cummings 2001) and tacrine (Kaufer 1998a), has shown benefits for apathy, and provides evidence for the involvement of a cholinergic deficiency in the manifestation of apathy. However, multiple studies suggest that deficiencies in other neurotransmitter systems are also involved (Hoehn‐Saric 1990; Herrmann 2004a; Lanari 2006; Lanctôt 2007a; Lanctôt 2007b).
Many studies have identified changes to the DA system in individuals with dementia (Allard 1990; Storga 1996; David 2008). In individuals with AD specifically, there is a reduction in DA neurotransmission between the basal ganglia, anterior cingulate and frontal cortex (Lanctôt 2007b). As these neural circuits comprise the brain reward system, which is highly correlated with apathy in individuals without cognitive impairment, it has been proposed that disruption to DA neurotransmission may contribute to the development and severity of apathy in individuals with dementia (Bressan 2005; Mitchell 2011).
The role of the serotonergic system in neuropsychiatric symptoms (NPS) has also been studied. The serotonin hypothesis of NPS postulates that a serotonin deficiency increases the likelihood of developing NPS, particularly depression and aggression (Vartiainen 1995; De Boer 2005; Albert 2013). In contrast, since selective serotonergic reuptake inhibitors (SSRIs), commonly used in the treatment of depression, have been linked to increased apathy in clinical trials, increased serotonergic neurotransmission has been implicated in the manifestation of apathy in dementia (Hoehn‐Saric 1990; Marin 1995; Barnhart 2004). SSRIs influence the DA system through inhibitory mechanisms that involve 5‐HT2C receptors (Walsh 1997), and stimulatory mechanisms that involve 5‐HT1B and 5‐HT3 receptors (De Deurwaerdère 1998). It has been hypothesized that pharmacological treatments aimed at balancing the serotonin‐dopamine neurotransmitter systems may reduce apathy in those with dementia (Abe 1975; De Boer 2005; Albert 2013).
Post mortem and neuroimaging studies indicate that there is a loss of GABA‐ergic and noradrenergic (NA) neurons in individuals with dementia (Abe 1975; Rossor 1982; Ellison 1986; Lowe 1988). It has been hypothesized that since GABA and NA are co‐transmitters with serotonin, pathological changes to the serotonergic system are accompanied by changes to the GABA‐ergic and noradrenergic systems, and that these may play a role in the manifestation of NPS in dementia (Rossor 1982). Lanctôt 2007a found that higher plasma GABA concentrations were linked to apathy in AD. Noradrenergic changes have not been specifically linked to this symptom (Herrmann 2004b).
Why it is important to do this review
It has been estimated that 97% of individuals with dementia experience one or more NPS over the course of their cognitive impairment, with apathy having a high prevalence of 71% (Steinberg 2008). As apathy has been associated with reduced quality of life and increased functional impairment, caregiver burden, cost of care and risk of institutionalization, it is an important NPS to treat (Boyle 2003; Hurt 2008; Vialta‐Franch 2013)
Behavioral interventions, usually involving caregivers, may be a safe treatment option as they are not accompanied by the adverse effects that can be associated with pharmacological interventions. They have a beneficial impact on the frequency and severity of NPS overall, and on caregivers’ negative reactions towards NPS (Overshott 2004; Brodaty 2012). However, caregiver‐delivered behavioral interventions have not been well investigated in apathetic individuals with AD.
Currently, there are pharmacological recommendations for the management of cognitive and functional impairments and NPS in individuals with AD (Herrmann 2013). However, there are no formal pharmacological recommendations specifically about the treatment of apathy in AD. There have been a few pharmacological studies in which treating apathy has been the primary objective of the study. There have also been many studies, involving several different classes of drugs, in which the primary outcome measures have been cognition or other non‐cognitive symptoms, but which have reported on apathy as a secondary outcome measure. Claims for efficacy against apathy have been made on the basis of this second class of study (Berman 2012). As a result, a systematic review is required, which considers the nature and quality of the evidence, to determine the safety, tolerability and efficacy of current pharmacological options for the treatment of apathy and to identify the most promising drugs to target for future investigation.
Objectives
Objective 1: To assess the safety and efficacy of pharmacotherapies for the treatment of apathy in Alzheimer's disease (AD). Objective 2: To assess the effect on apathy of pharmacotherapies investigated for other primary outcomes in the treatment of AD.
Methods
Criteria for considering studies for this review
Types of studies
We included all placebo‐controlled, parallel and cross‐over randomized controlled trials (RCTs) that investigated medications to treat apathy in AD or mixed AD, or both. Apathy was a primary (Objective 1) or secondary (Objective 2) outcome in the included studies.
We also included parallel and cross‐over RCTs that compared two or more medications for treating apathy in people with AD or mixed AD.
Types of participants
We included participants who met standardized diagnostic criteria for AD or mixed AD (e.g. the Diagnostic and Statistical Manual of Mental Disorders (IV, IV Text Revision, 5) (APA 2013), the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS‐ADRDA) (McKhann 1984), the National Institute on Aging/Alzheimer’s Association (NIA/AA) (McKhann 2011) and the International Classification of Diseases and Related Health Problems 10th Revision (ICD‐10) (WHO 1992)).
We extracted information on baseline scores of apathy in order to determine if study populations had clinically significant apathy. However this was not an inclusion criterion for this meta‐analysis. Although some authors reported on clinically significant apathy using the Apathy Evaluation Scale (AES), Frontal Systems Behaviour Scale (FrSBe) Tscore, or clinical judgement, we classified studies as including participants with clinically significant apathy when the mean NPI‐apathy subscore was more than three at baseline (Mulin 2011).
Types of interventions
We included any pharmacological interventions. We applied no restrictions to duration of treatment or to medication dosage.
Types of outcome measures
For Objective 1 – the assessment of efficacy and safety of drugs being investigated specifically for the treatment of apathy in AD – we included the following outcomes in the review:
Primary outcomes
Apathy measured by a scale which specifically measures apathy, either exclusively or as one of its components. The scales include, but are not limited to, the Apathy Evaluation Scale (AES), the apathy component of the Clinical Global Impressions of Change scale (CGI‐C apathy), the Neuropsychiatric Inventory (NPI) apathy subscale, the Lille Apathy Rating Scale (LARS), the FrSBe–Apathy component, the Nurses’ Observation Scale for Inpatient Evaluation (NOSIE), the Brief Psychiatric Rating Scale (BPRS) and the Sandoz Clinical Assessment‐Geriatric Scale (SCAG).
Adverse effects.
Secondary outcomes
Neuropsychiatric symptoms other than apathy.
Cognition.
Functional performance.
Changes in global disease severity (CGI‐C).
Dropouts due to adverse events (AEs).
For Objective 2, when we considered studies reporting apathy as a secondary outcome measure, we evaluated the effect on apathy only. This was because these studies are likely to form only a small and unrepresentative subset of studies investigating the other outcomes. We considered them a useful source of preliminary information about possible effects on apathy, but an unsuitable dataset to estimate effects on safety or our secondary outcomes.
Search methods for identification of studies
Electronic searches
We searched ALOIS (www.medicine.ox.ac.uk/alois), the Cochrane Dementia and Cognitive Improvement Group Specialized Register, on 15 June 2016 and 05 May 2017. We used the following search terms: apathy, apathetic, BPSD.
The Information Specialist maintains ALOIS, which contains dementia and cognitive improvement studies identified from the following sources:
Monthly searches of a number of major healthcare databases: MEDLINE, Embase, CINAHL, PsycINFO and LILACS.
Monthly searches of a number of trial registers: the metaRegister of Controlled Trials; the Umin Japan Trial Register; the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (which covers ClinicalTrials.gov, ISRCTN, the Chinese Clinical Trials Register, the German Clinical Trials Register, the Iranian Registry of Clinical Trials, the Netherlands National Trials Register and others).
Quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (the Cochrane Library).
Six searches a month of a number of grey literature sources: ISI Web of Knowledge with Conference Proceedings; Index to Theses; and Australasian Digital Theses.
We did not limit the search by language or date of publication. If we found articles in languages other than English, we ensured that these articles were translated and screened for potential inclusion.
We performed separate searches of many of the above‐named sources to ensure that we retrieved the most up‐to‐date results. The search strategy that we used for the retrieval of trial reports from MEDLINE is in Appendix 1.
Searching other resources
We performed electronic searches only.
Data collection and analysis
Selection of studies
Three review authors independently screened the citations identified from the literature search by title and abstract. We identified potentially relevant articles and obtained the full‐text articles for assessment. Three review authors independently assessed these articles according to the previously‐mentioned criteria. We resolved any disagreements by discussion, and involved a third review author if necessary, until we reached consensus. We contacted the study authors for further information when necessary. We identified duplicate citations through author names, institution name or participant data.
For articles which investigated apathy as a primary outcome measure (Objective 1), we contacted the study authors for further information on our primary and secondary outcome measures when necessary. For articles which investigated apathy as a secondary outcome measure (Objective 2), we contacted the study authors for further information about the change in apathy, when necessary.
Data extraction and management
Three review authors independently extracted the data using a data extraction form. We obtained missing data from the study authors when possible. One review author entered the data into Review Manager 5 (RevMan) (Review Manager 2014), and the other review authors checked for accuracy. We resolved any discrepancies by consensus.
Assessment of risk of bias in included studies
Two review authors independently assessed the risks of bias in accordance with the Cochrane 'Risk of bias' assessment tool for assessing quality and risk of bias (Higgins 2011). We compared 'Risk of bias' ratings, and resolved discrepancies through discussion with co‐authors. The tool encourages consideration of how the sequence was generated, how allocation was concealed, the integrity of blinding (participants, raters and personnel), the completeness of outcome data, selective reporting and other potential sources of biases (e.g. carry‐over bias in cross‐over trials, recruitment bias in cluster‐RCTs or bias due to early stopping in specific situations). Where the included study provided inadequate details of randomization and other characteristics of the trials, we contacted the study authors to obtain further information.
We assessed the risk of bias in each domain and categorized it into one of the following.
Low risk of bias: plausible bias that is unlikely to seriously alter the results.
High risk of bias: plausible bias that seriously weakens confidence in the results.
Unclear risk of bias: plausible bias that raises some doubts about the results.
Measures of treatment effect
We analyzed the longer ordinal scales in meta‐analyses as continuous data. We converted shorter ordinal scales into dichotomous data by combining adjacent categories into two groups and defining one of the grouped categories as the event.
For continuous data, the measure of treatment effect was the mean difference (MD) with the 95% confidence interval (CI) if the pooled trials used the same rating scale or test, or the standardized mean difference (SMD) with a 95% CI if the trials used different scales to measure the same outcome.
For dichotomous data, the measure of treatment effect was the relative risk (RR) and its 95% CI.
In order to interpret findings, we used the GRADE approach (Guyatt 2008) to assess the overall quality of evidence for all outcomes with pooled data, rating each one as either high, moderate, low or very low quality. The GRADE ratings take into account risk of bias, imprecision, inconsistency, publication bias and indirectness, and express the degree of confidence one can have that the effect estimate is close to the true effect.
Unit of analysis issues
Carry‐over effects are a concern associated with cross‐over trials. If a study reported significant carry‐over effects, we used only data from the first phase of a cross‐over study. If a study reported no carry‐over effects, then we included data (paired data if possible) from both treatment phases.
Dealing with missing data
In the event of missing information, we requested unreported data from the author(s) of the original study. We preferred intention‐to‐treat (ITT) data when available. In the case of missing data for non‐completers, we recorded any imputation methods used by the study authors and considered the use of sensitivity analyses to assess the impact on the results of different methods of dealing with missing data.
Assessment of heterogeneity
Clinical heterogeneity
We considered separately studies that were related by drug group. Within each group of studies, we noted the obvious sources of heterogeneity and considered these in the analyses.
Statistical heterogeneity
Visual inspection
We inspected graphs to assess the possibility of statistical heterogeneity.
Use of the I2 statistic
We used the I2 statistic to identify heterogeneity across the included studies. If the I2 statistic value was greater than 40%, we took this value to represent significant heterogeneity.
Assessment of reporting biases
We performed a comprehensive search and included trial registries to minimize the risk of reporting bias, which may arise when publication is influenced by the nature and direction of results (Egger 1997).
Data synthesis
We used a fixed‐effect model for analyses with sufficient homogeneity. If there was significant heterogeneity, we used a random‐effects model. If possible, we conducted analyses in accordance with the principles of ITT.
Subgroup analysis and investigation of heterogeneity
Data permitting, we conducted subgroup analyses for each drug group to examine the effect of the following:
Treatment duration.
Disease severity and diagnostic group (i.e. AD or mixed dementia).
If there were high levels of heterogeneity, we explored the plausible causes of heterogeneity. If we identified statistical heterogeneity, we used the strategies recommended by the Cochrane Handbook for Systematic Reviews of Inteventions, Section 9.5.3 and completed a random‐effects meta‐analysis to incorporate heterogeneity among studies (Deeks 2011).
Sensitivity analysis
In order to address the robustness of our results to potential risks of bias, we repeated the previous analyses, excluding studies at high risk of bias. We identified issues suitable for sensitivity analysis during the review process.
Data presentation: 'Summary of findings' tables
We used the GRADE approach to assess the quality of evidence behind each estimate of treatment effect (Schünemann 2011). For each comparison, we presented key findings, including a summary of the amount of data and the magnitude of the effect size. For comparisons in Objective 1, we presented the overall quality of the evidence in a 'Summary of findings' table, created using GRADEpro software (www.gradepro.org). We preselected the following outcomes.
Apathy.
Adverse effects.
Overall behavioral symptoms.
Cognition.
Function.
Clinical global impression.
Dropouts due to AEs.
As described above, apathy was the only outcome measure evaluated for those studies assessing apathy as a secondary outcome measure (Objective 2). We did not produce ‘Summary of findings’ tables for Objective 2 comparisons. However, we described the quality of evidence, using the GRADE approach, along with the results.
Results
Description of studies
Results of the search
Our search of the database and other electronic sources yielded 5295 references and 675 references, respectively. After de‐duplication and first‐assess removal of non‐relevant references by Anna Noel‐Storr (Information Specialist of the CDCIG), MR and EHA, or MR and SC independently assessed the remaining 1566 references for relevance. Of these, we ruled out 1504 references as they either did not investigate apathy as a primary or secondary outcome measure, did not investigate the efficacy of a pharmacological agent, did not conduct a double‐blind, randomized, placebo‐controlled trial, and/or did not include people with AD. This left 62 full‐text articles for assessment by MR, EHA and SC independently. These articles reported on apathy as a primary outcome measure, or used a scale such as the NPI to evaluate apathy as a secondary outcome measure. We contacted authors when data were not sufficiently reported for extraction for this review. We received further information from Ruths 2008 and Tariot 2011. Forty‐one studies which investigated apathy as a secondary outcome measure did not publish or provide upon request sufficient data on apathy; we therefore excluded them from this review. Of the 21 studies included in this meta‐analysis, four investigated apathy as a primary outcome (Objective 1), and 17 studies investigated apathy as a secondary outcome (Objective 2).
See Figure 1 for the study flow diagram.
1.
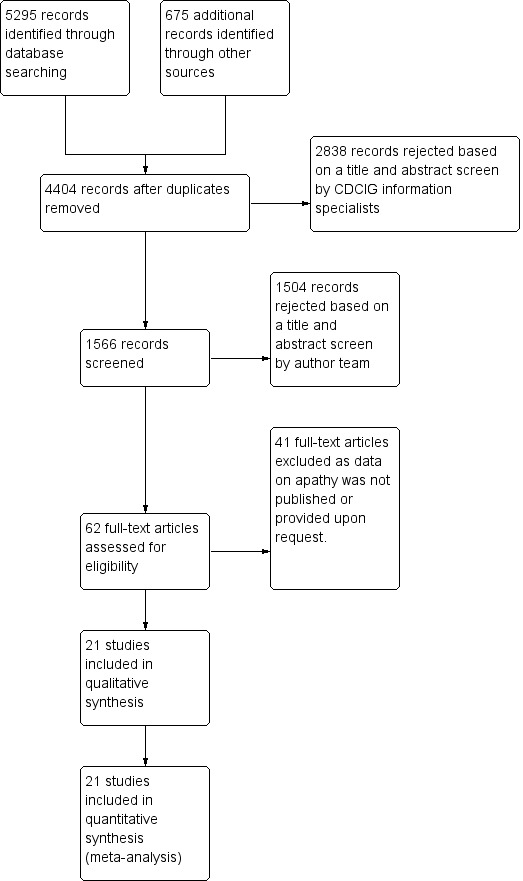
2Study flow diagram.
Included studies
The characteristics of the 21 included studies in this review are summarized in the Characteristics of included studies table.
Twenty‐one studies met our inclusion criteria for this meta‐analysis. We provide further information on study and participant characteristics in Table 3. All of the trials included in this meta‐analysis were randomized, double‐blind and placebo‐controlled. All but three studies (Sival 2002; Frakey 2012; Padala 2017) were multicenter trials. All studies included people with possible or probable AD according to standardized criteria (NINCDS‐ADRDA, DSM‐IV‐TR, or ICD‐10).
1. Study and participant characteristics.
| STUDY DURATION | N OF PARTICIPANTS | Diagnosis | MEAN AGE (YRS) | MEAN MMSE (SD) | MEAN BL NPI‐APATHY (SD) score | COUNTRY | NUMBER OF SITES | TREATMENT GROUPS | |
| METHYLPHENIDATE | |||||||||
| Herrmann 2008 | 2 weeks Cross‐over design: 2 treatment phases of 2 weeks with a 1‐week washout between phases | 13 total | Possible or probable AD (NINCDS‐ADRDA), and apathy (NPI‐apathy subscale ≥ 1) | 77.9 (7.8) | 19.9 (4.7) | 5.9 (3) | Canada | 3 | Group 1: Methylphenidate (10 mg twice a day) Group 2: Placebo |
| Rosenberg 2013 | 6 weeks | 60 participants Group 1: 29 Group 2: 31 | Possible or probable AD (NINCDS‐ADRDA), and clinically significant apathy for at least 4 weeks | 76 (8) | 20 (5) | Group 1: 7 (2) Group 2: 7 (2) | USA, Canada | 3 | Group 1: Methylphenidate (target: 20 mg daily) Group 2: Placebo |
| Padala 2017 | 12 weeks | 60 participants Group 1: 30 Group 2: 30 | Dementia of the AD type (DSM‐IV‐TR), and presence of apathy (AES > 40) | 76.6 (7.9) | 23.8 (2.5) | Not reported (AES only) | USA | 1 | Group 1: Methylphenidate (target: 20 mg daily) Group 2: Placebo |
| MODAFINIL | |||||||||
| Frakey 2012 | 8 weeks | Group 1: 11 Group 2: 11 | Possible or probable AD (NINCDS‐ADRDA criteria) and clinically significant apathy (FrSBe Tscore ≥ 65) | Group 1: 75.3 (8.3) Group 2: 29.4 (7.6) | Not disclosed | Not reported | USA | 1 | Group 1: Modafinil (200 mg daily) Group 2: Placebo |
| CHOLINESTERASE INHIBITORS | |||||||||
| Tariot 2001 | 24 weeks |
Group 1: 103 Group 2: 105 |
Possible or probable AD with cerebrovascular disease (but not vascular dementia) (NINCDS‐ADRDA criteria) | Group 1: 85.4 Group 2: 85.9 | Group 1: 14.4 (5.4) Group 2: 14.4 (5.8) | Not reported | USA | 27 | Group 1: Donepezil ‐ 5 mg/day for 28 days. 10 mg/day after 28 days based on tolerability. Group 2: placebo |
| MSAD trial | 24 weeks | Group 1: 144 Group 2: 146 | AD (DSM‐IV and NINCDS‐ADRDA criteria) moderate‐severe AD | 73.6 | Group 1: 11.7 (0.35) Group 2: 12.0 (0.34) ** | Group 1: 3.48 (0.29) Group 2: 3.48 (0.28) | Canada, Australia, France | 32 | Group 1: Donepezil ‐ 5 mg/day for 28 days. 10 mg/day after 28 days based on tolerability. Group 2: placebo |
| Herrmann 2005 | Range: 3 ‐ 6 months (12 ‐ 24 weeks) | Group 1: 1347 Group 2: 686 | Probable AD (NINCDS‐ADRDA criteria) mild‐moderate AD | 76 | 18 | Group 1: 2.34 (3.2) Group 2: 2.32 (3.3) | USA, Canada, Great Britain, South Africa, Australia, and New Zealand | Multicenter, but number not disclosed | Group 1: Galantamine Group 2: Placebo |
| Kaufer 1998 | 26 weeks | Group 1: 273 Group 2: 135 | Probable AD (NINCDS‐ADRDA criteria) mild‐moderate AD | Not reported | Not reported | Not reported | USA | 25 | Group 1: Metrifonate (2 weeks – 2.0 mg/kg, followed by 0.65 mg/kg) Group 2: Placebo |
| Morris 1998 | 26 weeks | Group 1: 273 Group 2: 135 | Probable AD (NINCDS‐ADRDA criteria) mild‐moderate AD | Group 1: 73.5 (8.1) Group 2: 73.7 (7.3) | Group 1: 18.8 (5) Group 2: 19.4 (4.3) | Not reported | USA | 24 | Group 1: Metrifonate (2 weeks – 2.0 mg/kg, followed by 0.65 mg/kg) Group 2: Placebo |
| Raskind 1999 | 26 weeks | Group 1: 177 Group 2: 87 | Probable AD (NINCDS‐ADRDA criteria) mild‐moderate AD |
Group 1: 74.6 (8.3) Group 2 : 74.5 (7.5) | Group 1: 18.7 (4.76) Group 2: 18.7 (4.97) | Not reported | USA (additional sites are not disclosed) | Multicenter, but number not disclosed | Group 1: 50 mg, OD Group 2: placebo |
| CHOLINESTERASE DISCONTINUATION | |||||||||
| Herrmann 2016 | 8 weeks | Group 1: 21 Group 2: 19 | Probable AD (NINCDS‐ADRDA criteria) moderate‐severe AD | 89.3 | Group 1: 8.1(5.2) Group 2: 10 (5.1) | Group 1: 3.29 (4.0) Group 2: 2.16 (4.0) | Canada | 2 | Group 1: Donepezil, rivastigmine, galantamine (oral only) Group 2: Placebo |
| ATYPICAL ANTIPSYCHOTICS | |||||||||
| De Deyn 2004 | 10 weeks | Group 1: 132 Group 2: 125 Group 3: 134 Group 4: 129 Group 5: 129 | Possible or probable AD (NINCDS‐ADRDA criteria and DSM‐IV‐TR), and clinically significant psychotic symptoms | 76.6 (10.4) | 13.7 (5.1) | Group 1: 3.2 (3.9) Group 2: 3.2 (3.7) Group 3: 3.4 (3.9) Group 4: 3.4 (3.7) Group 5: 3.0 (3.5) | Europe, Australia, Israel, Lebanon, and South Africa | 61 | Group 1: 7.5 mg OLZ Group 2: 5 mg OLZ Group 3: 2.5 mg OLZ Group 4: 1.0 mg OLZ Group 5: Placebo |
| Sultzer 2008 | Up to 36 weeks (12 weeks of treatment) data available | Group 1: 100 Group 2: 94 Group 3: 85 Group 4: 142 | Dementia of the AD type (DSM‐IV) or probable AD (NINCDS‐ADRDA) and daily delusions, hallucinations, agitation, or aggression over 4 weeks prior to study entry | 77.9 (7.5) | Group 1: 15 (5.4) Group 2: 14.9 (6.1) Group 3: 15.7 (6.1) Group 4: 14.7 (5.8) | Not reported | USA | 42 | Group 1: OLZ Group 2: QUE Group 3: RIS Group 4: Placebo |
| ANTIPSYCHOTIC DISCONTUATION | |||||||||
| Ruths 2008 | 4 weeks | Group 1: 28 Group 2: 27 | Dementia diagnosis according to ICD‐10 | 83.4 (6.9) | Not provided | Group 1: 1.4 (1.5)Group 2: 1.9 (1.5) | Norway | 9 | Group 1: Antipsychotics (haloperidol, risperidone, or olanzapine) Group 2: Placebo |
| ANTIDEPRESSANTS | |||||||||
| Lanctôt 2002 | 4 weeks: Cross‐over design: 2 treatment phases of 4 weeks with a 1‐week washout between phases | 22 total | Primary degenerative dementia (DSM‐IV) and probable AD (NINCDS‐ADRDA), and significant behavioral problems (NPI ≥ 8) | 82 (6) | 4.1 (4.7) | Group 1: 1.27 (3.5) Group 2: 1.45 (3.6) | Canada | 3 | Group 1: Sertraline (100 mg daily) Group 2: Placebo |
| CitAD trial | 9 weeks | Group 1: 94 Group 2: 92 | Probable AD (NINCDS‐ADRDA), and significant behavioral problems (NPI ≥ 8), and clinically significant agitation on the NPI > 3 | Group 1: 78 (9) Group 2: 79 (8) | Group 1: 17 (6.2) Group 2: 14.4 (6.9) | Group 1: 6 (0.9) Group 2: 6 (0.9) | USA | 6 | Group 1: Citalopram (30 mg daily) Group 2: Placebo |
| MIBAMPATOR | |||||||||
| Trzepacz 2013 | 12 weeks | Group 1: 63 Group 2: 69 | Probable AD (NINCDS‐ADRDA) (DSM‐IV‐TR), and clinically significant agitation/aggression | Group 1: 77.2 (8.2) Group 2: 77.7 (7.6) | Group 1: 16.0 (6.1) Group 2: 18 (5.3) | Not reported | USA | Multicenter, but number not disclosed | Group 1: Mibampator (target dose: 3 mg daily) Group 2: Placebo |
| VALPROATE | |||||||||
| Herrmann 2007 | 6 weeks: Cross‐over design: 2 treatment phases of 6 weeks with a 2‐week washout between phases | Group 1: 14 Group 2: 13 | Probable AD (NINCDS‐ADRDA), primary degenerative dementia (DSM‐IV) | 85.6 (4.5) | 4.5 (4.6) | Group 1: 2.4 (3.8) Group 2: 3.0(4.3) | Canada | 2 | Group 1: Valproate (mean dose: 1134.6 (400.1) mg daily) Group 2: Placebo |
| Sival 2002 | 3 weeks: Cross‐over design: 2 treatment phases of 3 weeks with a 1‐week washout between phases | Group 1: 42 Group 2: 42 | Senile dementia (NINCDS‐ADRDA)(DSM‐IV) | 80.4 (6.8) | 11.4 (5) | Not reported | Netherlands | 1 | Group 1: Valproate (2 x 240 mg) Group 2: Placebo |
| Tariot 2011 | 24 months (+ 2‐month single‐blind placebo phase) | Group 1: 153 Group 2: 160 | Possible or probable AD (NINCDS‐ADRDA) | Group 1: 74.9 Group 2: 76.6 | Group 1: 16.9 (3.0) Group 2: 16.9 (2.9) | Group 1: 1.1 (2.7) Group 2: 1.2 (2.9) | USA | 46 | Group 1: Valproate (flexible‐dose) (mean modal dose: 250 mg daily) Group 2: Placebo |
| SEMAGACESTAT | |||||||||
| Semgacestat trial | 76 weeks | Group 1: 463 Group 2: 472 Group 3: 473 | Mild‐moderate AD (NINCDS‐ADRDA) | Group 1: 72.7 (7.9) Group 2: 73 (8.5) Group 3: 73.3 (8) | Group 1: 20.9 (3.5) Group 2: 20.8 (3.5) Group 3: 20.9 (3.6) | Not reported | USA | 91 | Group 1: LY100 Group 2: LY140 Group 3: Placebo |
AD: Alzheimer's disease, BL: baseline, DSM: Diagnostic and Statistical Manual of Mental Disoders, FrSBe: Frontal Systems Behavior Scale, ICD: International Classification of Diseases, LY: LY450319 (Eli Lillyand Company study drug), NINCDS‐ADRDA: National Institute of Neurological and Communicative disorders and the Alzheimer's Disease and Related Disorders Association, NPI: Neuropsychiatric Inventory, OLZ: olanzapine, QUE: quetiapine, RIS: risperidone, SD: standard deviation.
Objective 1:
Four studies investigated the effect of a pharmacological treatment on apathy as a primary outcome measure. The drugs studied were methylphenidate and modafinil, both compared to placebo. We treat modafinil and methylphenidate separately in this review and meta‐analysis. Although both are CNS stimulants, their mechanisms of action differ. Modafinil activates glutamatergic circuits while inhibiting GABA neurotransmission (Gerrard 2007). Among other actions, methylphenidate blocks dopamine uptake in central adrenergic neurons by blocking dopaminergic transporter and carrier proteins (Volkow 2002).
Methylphenidate
Three placebo‐controlled studies have investigated the efficacy of methylphenidate for the treatment of apathy in people with AD (Herrmann 2008; Rosenberg 2013; Padala 2017). These studies had similar eligibility criteria, with participants having mild‐to‐moderate AD and clinically significant apathy at baseline. In all three studies, the daily target dose of methylphenidate was 20 mg.
Herrmann 2008 conducted a cross‐over study with two two‐week treatment phases and a one‐week placebo washout between treatment phases. The authors reported no treatment order or carry‐over effects. We extracted paired data from this study. We did not consider the cross‐over design to be a source of bias.
Rosenberg 2013 and Padala 2017 both used a parallel‐group design and investigated the efficacy and safety of methylphenidate in the treatment of apathy over six and 12 weeks respectively.
Modafinil
Frakey 2012 also investigated the effect of modafinil on apathy in people with mild‐to‐moderate AD and clinically significant apathy at baseline (FrSBe apathy Tscore ≥ 65). As Frakey 2012 provided the baseline and final standard deviation (SD) values, we imputed the change SD using methodology provided in the Cochrane Handbook (Section 16.1.3.2).
Objective 2:
Seventeen studies reported the effect of a pharmacotherapy on apathy as a secondary outcome measure. The drugs studied were cholinesterase inhibitors (ChEIs), atypical antipsychotics, antidepressants, mibampator, valproate and semagacestat, versus placebo. Two placebo‐controlled discontinuation studies with ChEIs and antipsychotics also investigated apathy as a secondary outcome measure. Clinically significant apathy was not an inclusion criterion in any of the studies.
Cholinesterase inhibitors
Six studies investigating ChEIs met the inclusion criteria for this review. Two studies (Tariot 2001; MSAD trial) included participants with moderate‐to‐severe AD. Herrmann 2005; Kaufer 1998; Morris 1998; and Raskind 1999 included participants with mild‐to‐moderate AD. Although none of the studies actively recruited participants with clinically significant apathy (considered as an NPI apathy subscore ≥ 3), baseline apathy in the MSAD trial was clinically significant in both treatment groups. In the remaining studies, neither treatment group had clinically significant apathy at baseline.
Tariot 2001, the MSAD trial and Herrmann 2005 included currently approved ChEIs for the treatment of AD (donepezil, galantamine and rivastigmine).
Tariot 2001 and the MSAD trial investigated the efficacy and safety of donepezil (target dose: 5 ‐ 10 mg/daily) over 24 weeks. Both papers reported change scores as least square mean (LSM) change. We considered this to be a potential source of selective reporting bias, as covariates were included in a linear regression which computed adjusted mean change values. We computed the SD values from the provided standard error (SE) values for LSM change using methods provided in the Cochrane Handbook (Section 7.7.3.2).
Herrmann 2005 reported the effect of galantamine on neuropsychiatric symptoms (NPS) in a post hoc analysis of pooled data from three large trials (Tariot 2000; Rockwood 2001; data file from Janssen‐Ortho) which had study durations of three, five and six months, respectively. We included data from this post hoc analysis because each trial met inclusion criteria for this meta‐analysis, and because we were unable to obtain sufficient data from the primary papers. Herrmann 2005 conducted an ITT analysis on the pooled data obtained from the last observation on each participant.
Kaufer 1998, Morris 1998, and Raskind 1999 all investigated the efficacy and safety of metrifonate in AD. Metrifonate is an irreversible organophosphate acetylcholinesterase inhibitor which was not approved for the symptomatic management of AD. All three papers reported LSM change scores which used covariates to create an adjusted mean change score. Again, we considered this to be a potential source of selective reporting bias. We were able to compute SD change values from Raskind 1999 using reported SE change values. However, as neither Kaufer 1998 nor Morris 1998 reported SE or SD change values, we used SD values computed from Raskind 1999 for both these studies, as all studies had participants with similar AD severity, and the same study duration and dosing regimen.
ChEI discontinuation
Herrmann 2016 investigated the efficacy and safety of ChEI discontinuation in people with moderate to severe AD. Continuing treatment with a ChEI was compared to ChEI discontinuation (placebo substitution), and so we included the results of this study in the meta‐analysis. However, this evidence is indirect in terms of our review questions, as all participants were receiving long‐term ChEI treatment (more than a year) prior to study enrollment, and it is unclear how this may influence our findings. Although Herrmann 2016 did not actively recruit participants with clinically significant apathy, those who were randomized to continue ChEI use had clinically significant apathy (NPI‐apathy subscale score ≥ 3) compared to placebo. However, the difference between groups was not statistically significant.
Atypical antipsychotics
We identified 16 RCTs that evaluate the efficacy of atypical antipsychotics for aggression and psychosis in people with AD (Ballard 2006). However, only two of these studies met our inclusion criteria and reported sufficient data on apathy, or provided data upon request, for this meta‐analysis (De Deyn 2004; Sultzer 2008). De Deyn 2004 investigated the efficacy of olanzapine versus placebo in treating NPS over 10 weeks. As participants in this study were randomized into one of five groups (1, 2.5, 5 or 7.5 mg of olanzapine, or placebo), we have combined results from those randomized to olanzapine to prevent a unit‐of‐analysis error due to multiple comparisons (Cochrane Handbook section 16.5.4). The method used for combining groups was provided in the Cochrane Handbook (Section 7.7.3.8). As well as meeting standardized criteria for AD, all participants also had clinically significant psychotic symptoms. Sultzer 2008 investigated the efficacy of atypical antipsychotics (olanzapine, quetiapine and risperidone) versus placebo in treating NPS for up to 36 weeks (phase 1 of the study). In phase 2 of the study, participants could be randomized to a different medication at the clinician’s discretion. Mean change scores were reported over the first 12 weeks of phase 1 of the study, and so we used these results in the meta‐analysis. Participants were randomized to one of four groups (olanzapine, quetiapine, risperidone or placebo). In order to prevent a unit‐of‐analysis error due to multiple comparisons, we combined results from participants receiving all three atypical antipsychotics. In addition to meeting standardized criteria for AD, all participants also had clinically significant psychotic symptoms or agitation/aggression over the four weeks prior to study entry.
Neither study actively recruited people with clinically significant apathy. However, in De Deyn 2004 each treatment group had clinically significant apathy at baseline. As Sultzer 2008 did not provide baseline scores on apathy, we were unable to determine whether participants enrolled in this study had clinically significant apathy.
Antipsychotic discontinuation
We identified nine clinical trials which investigated the efficacy and safety of antipsychotic discontinuation in people with AD, but we were able to include only one study which met our inclusion criteria and provided data on apathy upon request (Ruths 2008).
Ruths 2008 investigated the efficacy of antipsychotic discontinuation in people with AD who had been receiving haloperidol, risperidone or olanzapine (range: 3 to 62 months). Neither treatment group had clinically significant apathy at baseline.
Antidepressants
We identified 12 trials comparing antidepressants with placebo in people with AD. However, only two studies met our inclusion criteria and yielded extractable data (CitAD trial) or provided data upon request (Lanctôt 2002).
Lanctôt 2002 investigated the effect of sertraline on NPS in people with severe AD and clinically significant NPS (NPI ≥ 8). This cross‐over study consisted of two four‐week treatment phases separated by a one‐week placebo washout. Neither treatment group had clinically significant apathy at baseline. As the authors reported that treatment order did not have an effect on treatment response, we did not consider the cross‐over design to be a source of bias. Lanctôt 2002 published results in treatment responders only, but data on all participants were provided upon request.
CitAD trial investigated the effect of citalopram on agitation in people with AD and clinically significant agitation over nine weeks. As CitAD trial provided median and interquartile range (IQR) values for NPI‐apathy subscores and NPI‐total scores, we used methods described in the Cochrane Handbook (Section 7.7.3.5) to validate use of the median to estimate mean values, and to convert IQR to SD values. For mini‐mental state examination (MMSE) and Alzheimer's Disease Cooperative Study Activities of Daily Living (ADCS‐ADL) scores, SD values were provided at baseline, while SE values were provided at study endpoint. We calculated the SD from SE values using methods provided in the Cochrane Handbook (Section 7.7.3.2), and derived change SD values also using methods provided in the Cochrane Handbook (Section 16.1.3.2).
The presence of clinically significant apathy was not an inclusion criterion for either study. However, participants in CitAD trial had clinically significant apathy (NPI‐apathy subscore ≥ 3) at baseline.
Mibampator
Trzepacz 2013 investigated the efficacy of mibampator (LY451396) on agitation/aggression in people with AD over 12 weeks. Trzepacz 2013 reported the LSM change score and SD for overall behavior using the FrSBe total Tscore. Apathy was assessed using the FrSBe apathy T‐subscore. We extrapolated the LSM change score for apathy from a graph. However, as SD change scores for apathy were not provided in the paper, we inferred these values from the FrSBe total change Tscore results. As LSM change scores use covariates to create an adjusted mean change score, we considered this as a potential source of selective reporting bias. Since Trzepacz 2013 did not provide baseline apathy scores, we were unable to determine whether study participants had clinically significant apathy.
Valproate
We identified three studies investigating valproate which met the inclusion criteria for this meta‐analysis (Sival 2002; Herrmann 2007; Tariot 2011). These studies had similar eligibility criteria, with participants having moderate AD and clinically significant agitation/aggression. Tariot 2011 also included participants with clinically significant psychosis. In all three studies, the primary outcome measure was the efficacy of valproate on agitation/aggression (and/or psychosis in Tariot 2011). Herrmann 2007 was a cross‐over study with two six‐week treatment phases separated by a two‐week placebo washout period. Data from this study were provided upon request. Though Herrmann 2007 did not actively recruit people with clinically significant apathy, those randomized to receive placebo had clinically significant apathy (NPI‐apathy subscale score ≥ 3) compared to those receiving valproate. However, the difference between groups was not statistically significant. Sival 2002 was also a cross‐over study with two three‐week treatment phases separated by a one‐week placebo washout period. We were unable to confirm whether participants enrolled in this study had clinically significant apathy at baseline. Although treatment order and carry‐over effects were investigated by Sival 2002 and Herrmann 2007, both papers reported the absence of these effects. As such, we did not consider the cross‐over design to be a source of bias. We extracted paired data from both studies.
Tariot 2011 investigated the efficacy of valproate as a prophylactic treatment for emerging agitation or psychosis in people with moderate AD over 24 months, followed by a two‐month period of single‐blind placebo treatment. Neither treatment group had clinically significant apathy. Data from this study were provided by the Alzheimer's Disease Cooperative Study (ADCS) group upon request.
Semagecestat
Rosenberg 2016 investigated the efficacy of semagecestat for the treatment of AD over 76 weeks. Participants in this study were randomized to one of three groups (100 or 140 mg of semagacestat, or placebo). We combined results from those randomized to both semagacestat groups using the method provided in the Cochrane Handbook (Section 7.7.3.8) in order to prevent a unit‐of‐analysis error due to multiple comparisons (Cochrane Handbook Section 16.5.4). We used methods described in the Cochrane Handbook (Section 7.7.3.2) to calculate SD values for the MMSE and ADCS‐ADL scores from the 95% confidence intervals reported by the authors.
We were unable to confirm whether participants enrolled in this study had clinically significant apathy, as neither Doody 2013 nor Rosenberg 2016, who published the original findings of the study, provided baseline apathy scores.
Interventions
We present relevant details about treatment groups and doses of medication used in each study in Table 3.
Outcomes
All trials included in this meta‐analysis examined apathy as a primary or secondary outcome measure. We summarize the details of outcomes measured and reported in each trial in Table 4. A number of scales were used to measure each outcome.
2. Outcome Measures and Assessments.
| Study | Apathy | AE reported | NPS | Cognition | Function | Global Change | Dropouts due to AEs reported |
| METHYLPHENIDATE | |||||||
| Herrmann 2008 | AES‐Informant NPI‐apathy subscale | Yes | NPI‐total | MMSE | N/A | CGI‐C | Yes |
| Rosenberg 2013 | AES‐Informant NPI‐apathy subscale | Yes | NPI‐total (not reported) | MMSE | N/A | ADCS‐CGIC | Yes |
| Padala 2017 | AES‐Clinician | Yes | N/A | MMSE | ADL IADL | N/A | Yes |
| MODAFINIL | |||||||
| Frakey 2012 | FrSBe‐apathy subscale | Yes | N/A | N/A | ADLQ | N/A | Yes |
| CHOLINESTERASE INHIBITORS | |||||||
| Tariot 2001 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| MSAD trial | NPI‐apathy subscale | ||||||
| Herrmann 2005 | NPI‐apathy subscale | ||||||
| Kaufer 1998 | NPI‐apathy subscale | ||||||
| Morris 1998 | NPI‐apathy subscale | ||||||
| Raskind 1999 | NPI‐apathy subscale | ||||||
| CHOLINESTERASE DISCONTINUATION | |||||||
| Herrmann 2016 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison. | |||||
| ATYPICAL ANTIPSYCHOTICS | |||||||
| De Deyn 2004 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| Sultzer 2008 | BPRS‐withdrawn depression factor score | ||||||
| ANTIPSYCHOTIC DISCONTINUATION | |||||||
| Ruths 2008 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| ANTIDEPRESSANTS | |||||||
| Lanctôt 2002 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| CitAD trial | NPI‐apathy subscale | ||||||
| MIBAMPATOR | |||||||
| Trzepacz 2013 | FrSBe‐apathy T score | These outcomes were not investigated for this drug comparison | |||||
| VALPROATE | |||||||
| Herrmann 2007 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| Sival 2002 | GIP‐apathetic behavior subscore | These outcomes were not investigated for this drug comparison | |||||
| Tariot 2011 | NPI‐apathy subscale | These outcomes were not investigated for this drug comparison | |||||
| SEMAGACESTAT | |||||||
| Semgacestat trial | NPI‐apathy | These outcomes were not investigated for this drug comparison | |||||
ADCS‐CGIC: Alzheimer's Diserase Cooperative Study ‐ Clinical Global Impression of Change, ADL: Activities of Daily Living scale, ADLQ: Lawton and Brody Fucntional Assessment, AES: Apathy Evaluation Scale, CGI‐C: Clinical Global Impression of Change, FrSBe: Frontal Systems Behavior Scale, GIP: The Behavior Observation Scale for Intramural Psychogeriatric Patients, IADL: Instrumental Activities of Daily Living Scale, MMSE: Mini‐Mental State Examination, NPI: Neuropsychiatric Inventory.
Primary efficacy and safety outcomes:
1) Apathy
NPI‐apathy subscale: Apathy is a subscale item on the NPI scale. The apathy score is calculated as the product of frequency and severity of apathy symptoms, with a range of 0 to 12. Higher scores indicate more frequent and/or severe symptoms (Cummings 1994). AES‐Informant (AES‐I) and AES‐Clinician (AES‐C): This is an 18‐item informant (AES‐I) or clinician (AES‐C)‐rated scale which measures apathy severity as defined by simultaneous deficits in the overt behavioral, cognitive and emotional constructs of goal‐directed behavior. The higher the score, the greater the apathy severity (Marin 1991). BPRS Withdrawn depression factor score: The Withdrawn Depression component of the BPRS consists of emotional withdrawal, depressed mood, motor retardation, and blunted affect (Overall 1962). This component of the BPRS has been shown to be fairly associated with scores on the NPI‐apathy subscale (Politis 2004). FrSBE apathy: Apathy is a subscale item on the FrSBE, which measures three frontal systems behavioral syndromes: apathy, disinhibition, and executive dysfunction. The higher the score, the greater the severity of apathy (Grace 2011). Behavior Rating Scale for Psychogeratric Inpatients (GIP): Apathy is one of four components of the 82‐item GIP scale. Higher scores indicate greater severity of apathy (Diesfeldt 2013).
2) Adverse events
As a number of drug classes were included in this meta‐analysis, we chose to use the number of participants who experienced one or more adverse events (AEs) as an indication of safety. This outcome was reported by all studies which reported safety outcomes.
Secondary outcomes:
3) NPS
NPI: The NPI is a widely‐used assessment of 12 behavioral symptoms in dementia, including: delusions, hallucinations, agitation/aggression, apathy, depression, euphoria, aberrant motor behavior, irritability, disinhibition, anxiety, sleeping and eating. The frequency and severity of these symptoms are judged on a four‐point and three‐point scale, respectively (Cummings 1994).
4) Cognition
MMSE: This scale measures global cognition, and assesses orientation to time and place, immediate recall, short‐term verbal memory, calculation, language, and construct ability. The MMSE is scored out of 30, with lower scores indicating greater cognitive impairment (Folstein 1975).
5) Function
IADL scale/ADL‐Q: Although termed differently by Frakey 2012 and Padala 2017, the scale used was the same between both studies. This questionnaire measures functional abilities in elderly people necessary for independent living. Scores range from 0 to 28, with lower scores indicating greater functional impairment (Lawton 1969). ADL scale: This questionnaire assesses independence in performing basic tasks such as bathing, dressing, and feeding. Scores range from 0 to 24, with lower scores indicating greater functional impairment (Katz 1963).
6) Global change
CGIC and ADCS‐CGIC: This scale quantifies disease severity and clinical change (worsening, no change, or improvement), based on information about the person’s medical history, cognition, behavior, and function (Schneider 1997).
7) Dropouts due to AEs
In clinical trials with AD participants, attrition is a common problem attributed to loss to follow‐up, lack of efficacy, violation of study protocol, and the presence of AEs. As we are concerned with tolerability, we report on the number of dropouts due to an AE.
Excluded studies
Double‐blind, placebo‐controlled RCTs which we exclude from this meta‐analysis are presented in the Characteristics of excluded studies. We did not exclude any RCTs which investigated and reported on the efficacy of a pharmacological intervention on apathy as a primary or secondary outcome measure in people with mild, moderate, or severe AD.
Risk of bias in included studies
All studies included in this meta‐analysis were described as double‐blind, randomized, placebo‐controlled trials. We present details concerning the risks of bias of individual studies in the Characteristics of included studies tables; Figure 2; and Figure 3.
2.
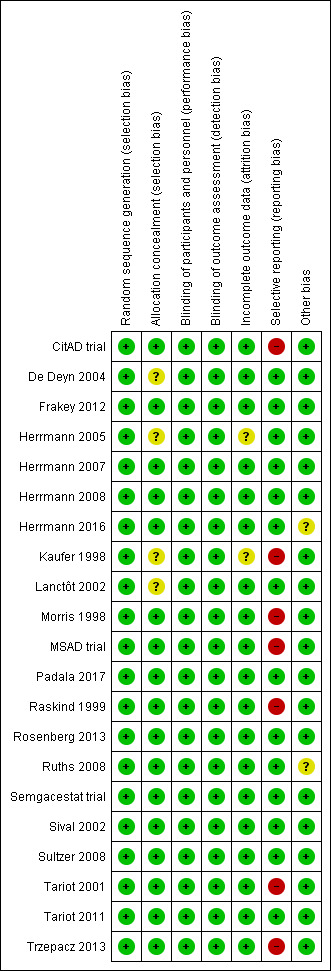
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
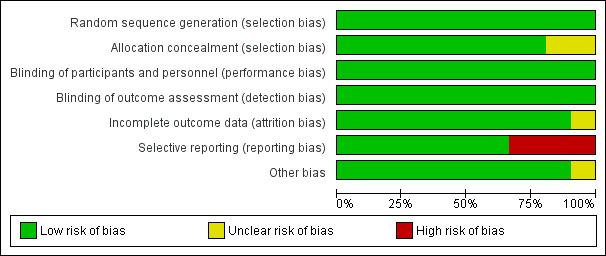
Figure 3 CaptionRisk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Most trials provided an adequate description of treatment allocation, but for some trials we were unable to obtain or locate this information. In these cases, we indicated unclear bias. These trials included the following: Kaufer 1998; Lanctôt 2002; De Deyn 2004; Herrmann 2005; Padala 2017.
Incomplete outcome data
All trials except Kaufer 1998 and Herrmann 2005 adequately described attrition rates in text, or included a figure detailing participant flow, or both. As mentioned previously, Herrmann 2005 investigated the efficacy of galantamine in a pooled post hoc analysis of three placebo‐controlled RCTs. Two of the studies included participant flow diagrams, and described study discontinuations in detail (Tariot 2000; Rockwood 2001). In Rockwood 2001, discontinuations due to AEs were more common in participants who were randomized to galantamine than placebo. However, the authors used ITT analyses, and last observation carried forward (LOCF) analysis as appropriate. Tariot 2000 reported that discontinuations due to AEs were similar between participants randomized to galantamine compared to placebo. We considered that these two studies had a low risk of attrition bias. However, as Herrmann 2005 was not able to confirm whether this also applied to data obtained from Janssen‐Ortho, this was a potential source of bias. In Kaufer 1998, authors mention that reported data included LOCF analysis in an ITT population. However, as no further details on attrition were provided, we included this as a potential source of bias.
Selective reporting
As mentioned previously in the Description of studies, we had concerns with selective reporting, specifically in studies comparing ChEI, citalopram and semagacestat versus placebo. In Kaufer 1998, Morris 1998, Raskind 1999, and the MSAD trial, change scores were reported as LSM values. As LSM values use covariates to generate an adjusted mean change score, this is a possible source of selective reporting bias.
Raskind 1999 and the MSAD trial both reported SE values of LSM change scores. We were able to compute SD values from the SE values provided. However, as Kaufer 1998 and Morris 1998 did not report SE or SD values, we used the SD values we had computed from Raskind 1999, as there were similarities across all three studies in AD severity of participants, study duration, and dosing regimen. Again, we considered this to be a possible source of selective reporting bias.
CitAD trial reported median values for continuous outcome measures. This is a source of selective reporting bias, as CitAD trial also reported that the data for these measures were not normally distributed, and may not be an accurate representation of the raw mean values. Rosenberg 2016 reported on our primary efficacy outcome of apathy, and NPS.
Effects of interventions
Summary of findings for the main comparison. Methylphenidate compared to placebo for apathy in Alzheimer's disease.
| Methylphenidate compared to placebo for apathy in Alzheimer's disease | ||||||
| Patient or population: Apathy in people with mild‐to‐moderate Alzheimer's disease Setting: Multicenter, USA and Canada Intervention: methylphenidate Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Methylphenidate | |||||
| Change in apathy (AES score) assessed with: AES Scale from: 0 to 42 follow‐up: range 2 weeks to 12 weeks | The mean change from baseline in apathy was ‐4.2 to 0.6 | MD 4.99 lower (9.55 lower to 0.43 lower) | ‐ | 145 (3 RCTs) | ⊕⊕⊝⊝ LOW 1, 2 | AES: Limited data on clinically meaningful changes |
| Change in apathy (NPI‐apathy subscale score) assessed with: NPI‐apathy subscale Scale from: 0 to 12 follow‐up: 2 weeks to 6 weeks | The mean change from baseline in apathy ‐2.6 to ‐1.69 | MD 0.08 lower (3.85 lower to 3.69 higher) | ‐ | 85 (2 RCTs) | ⊕⊕⊝⊝ LOW 1, 2 | 1‐ to 2‐point change suggested to be clinically significant in people with a clinically significant apathy (Rosenberg 2013) |
| Adverse events assessed with: Number of participants reporting ≥ 1 adverse event follow‐up: 2 weeks to 12 weeks | Study population | RR 1.28 (0.67 to 2.42) | 145 (3 RCTs) | ⊕⊕⊝⊝ LOW 1, 2 | ‐ | |
| 534 per 1000 | 684 per 1000 (358 to 1,000) | |||||
| Change in NPS assessed with: NPI Scale from: 0 to 144 follow‐up: 2 weeks | The mean change from baseline in NPS was ‐2.08 | MD 0.16 higher (7.89 lower to 8.21 higher) | ‐ | 25 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | 4‐point change suggested to be clinically significant |
| Change in cognition assessed with: MMSE Scale from: 0 to 30 follow‐up: 2 weeks to 12 weeks | The mean change from baseline in cognition was ‐1.08 to ‐0.3 | MD 1.79 higher (0.53 higher to 3.05 higher) | ‐ | 145 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | MMSE: 2‐ to 4‐point change suggested to be clinically significant |
| Change in functional performance assessed with: ADL scale Scale from: 0 to 6 follow‐up: 12 weeks | The mean change from baseline in functional performance was 0.4 | MD 0.50 higher (0.39 lower to 1.39 higher) | ‐ | 60 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | Limited data on clinically meaningful changes |
| Change in functional performance assessed with: IADL scale Scale from: 0 to 8 for women, and 0 to 5 for men, to avoid potential for gender bias follow‐up: 12 weeks | The mean change from baseline in functional performance was ‐0.6 | MD 2.30 higher (0.74 higher to 3.86 higher) | ‐ | 60 (1 RCT) | ⊕⊕⊕⊝ MODERATE 3 | Limited data on clinically meaningful changes |
| Change in global disease severity assessed with: ADCS‐CGIC or CGIC follow‐up: 2 weeks to 6 weeks | Study population | RR 0.56 (0.15 to 2.10) | 85 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | ‐ | |
| 116 per 1000 | 65 per 1000 (17 to 244) | |||||
| Dropouts assessed with: Number of participants who dropped out prior to study completion. follow‐up: 2 weeks to 12 weeks | Study population | RR 2.10 (0.60 to 7.38) | 145 (3 RCTs) | ⊕⊕⊝⊝ LOW 4 | ‐ | |
| 41 per 1000 | 86 per 1000 (25 to 303) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). AD: Alzheimer's disease; AEs: Adverse Events; MMSE: Mini‐Mental Status Examination; MD: Mean Difference; NPS: Neuropsychiatric Symptom, SMD: Standardized Mean Difference, CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Quality downgraded one level due imprecision (wide 95% confidence interval). 2Quality downgraded one level due to inconsistency (substantial heterogeneity was present). 3Quality downgraded one level due to imprecision (only one study, with a relatively small sample size). 4Quality downgraded two levels due to very serious imprecision (very wide 95% confidence interval).
Summary of findings 2. Modafinil compared to placebo for apathy in Alzheimer's disease.
| Modafinil compared to placebo for apathy in Alzheimer's disease | ||||||
| Patient or population: Apathy in people with mild‐to‐moderate Alzheimer's disease Setting: Single site, USA Intervention: modafinil Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with Modafinil | |||||
| Change in apathy assessed with: FrSBe‐apathy subscale (T‐score converted from raw score) Scale from: 14 to 70 (raw score) follow‐up: mean 8 weeks | The mean change from baseline in apathy was ‐6.82 | MD 0.27 higher (3.51 lower to 4.05 higher) | ‐ | 22 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Limited data on clinically meaningful changes on the FrSBe apathy score |
| Adverse Events ‐ reported, but not analyzed in this review | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in NPS ‐ not investigated | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in cognition ‐ not investigated | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change in functional performance assessed with: ADLQ Scale from: 0 to 84 follow‐up: mean 8 weeks | The mean change from baseline in functional performance was 0 | MD 0.54 lower (1.40 lower to 0.32 higher) | ‐ | 22 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Limited data on clinically meaningful changes |
| Change in global disease severity ‐ not investigated | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dropouts ‐ reported, but not analyzed in this review | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Quality downgraded two levels due to small sample size and imprecision (wide 95% confidence interval).
Objective 1:
Comparison of methylphenidate with placebo
See: Table 1.
Primary efficacy and safety outcomes:
1. Apathy
Three included studies investigated and reported on the efficacy of methylphenidate for the treatment of apathy as a primary outcome measure (Herrmann 2008; Rosenberg 2013; Padala 2017). All studies used the AES to assess apathy. Herrmann 2008 and Rosenberg 2013 also used the NPI‐apathy subscale. We conducted separate analyses using results from the AES scale, and results from the NPI‐apathy subscale.
Apathy assessed by the AES:
Based on findings obtained from the AES, we found that methylphenidate may improve apathy compared to placebo (mean difference (MD) ‐4.99, 95% confidence interval (CI) ‐9.55 to ‐0.43, P = 0.03, n = 145, 3 studies, I2 = 83%). However, there was uncertainty associated with this result, which we considered to be of low quality, because of serious concerns with inconsistency due to substantial heterogeneity, and imprecision due to a wide 95% confidence interval.
We conducted an exploratory subgroup analysis in studies with a trial duration of less than 12 weeks, and studies with a trial duration of 12 weeks or more. We had not prespecified this trial duration cut‐off, but chose this duration based on visual inspection of the forest plot which suggested that Padala 2017 had a greater change in apathy scores than Herrmann 2008 and Rosenberg 2013, despite having similar participant characteristics and dosing (Table 3). In studies lasting less than 12 weeks, methylphenidate may improve apathy compared to placebo (MD ‐2.62, 95% CI ‐4.80 to ‐0.44, P = 0.02, n = 85, 2 studies, I2 = 0%). In Padala 2017, the only study with a trial duration longer than 12 weeks, methylphenidate may also improve apathy compared to placebo (MD ‐9.90, 95% CI ‐13.50 to ‐6.30, P < 0.001, n = 60, 1 study). Within each subgroup, there was uncertainty associated with the results, which we considered to be of low quality because of serious concerns with indirectness due to nongeneralizability of results, and to imprecision.
We noted significant differences between subgroups (Chi2(1) = 11.49, P < 0.001, I2 = 91.3%). Trial duration is one possible explanation for the difference identified between subgroups. See Analysis 1.1; Figure 4.
1.1. Analysis.
Comparison 1 Methylphenidate, Outcome 1 Change in apathy from baseline as measured by the AES.
4.
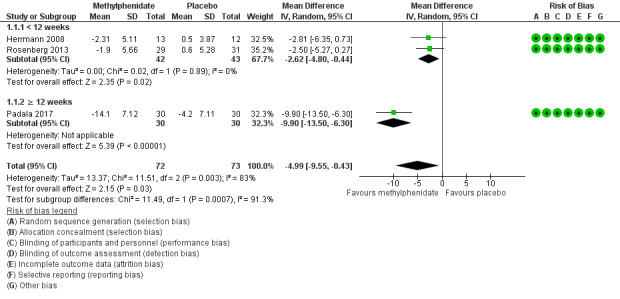
Forest plot of comparison: 7 Methylphenidate, outcome: 7.1 Apathy (AES only).
We could not conduct a subgroup analysis by disease severity, as all three studies enrolled participants with similar AD severity.
Apathy assessed by the NPI‐apathy subscale:
Based on findings obtained from the NPI‐apathy subscale, we found that methylphenidate may have no effect on apathy (MD ‐0.08, 95% CI ‐3.85 to 3.69, P = 0.97, n = 85, 2 studies I2 = 84%). There was uncertainty associated with this result, which we considered to be of low quality because of serious concerns with inconsistency due to substantial heterogeneity, and to imprecision due to a wide 95% confidence interval which may have contributed to an overall null effect. See Analysis 1.2.
1.2. Analysis.
Comparison 1 Methylphenidate, Outcome 2 Change in apathy from baseline as measured by the NPI‐apathy subscore.
As we included only two studies in this meta‐analysis, we did not conduct additional subgroup analyses.
2. Adverse events
Although Rosenberg 2013 reported that there were trends towards increased anxiety and weight loss (> 2%) in those allocated to methylphenidate, there was little or no difference between treatment groups in the risk of developing an AE (RR 1.28, 95% CI 0.67 to 2.42, P = 0.45, n = 145, 3 studies, I2 = 62%). There was uncertainty associated with this result, which we considered to be of low quality due to serious concerns with inconsistency and imprecision.
An exploratory subgroup analysis demonstrated that there was probably little or no difference between treatment groups in the risk of developing an AE in trials with a duration of less than 12 weeks (RR 1.28, 95% CI 0.44 to 3.72, P = 0.65, n = 85, 2 studies, I2 = 38%), or in trials of 12 weeks or longer (RR 1.44, 95% CI 0.73 to 2.86, P = 0.29, n = 60, 1 study). Within each subgroup, there was uncertainty associated with the results, which we considered to be of low quality because of serious concerns with indirectness due to nongeneralizability of results, and to imprecision. There were no significant differences noted between subgroups (Chi2(1) = 0.03, P = 0.85, I2 = 0%). See Analysis 1.3; Figure 5. We did not conduct a subgroup analysis based on disease severity, as all three studies enrolled participants with similar AD severity.
1.3. Analysis.
Comparison 1 Methylphenidate, Outcome 3 Adverse Events.
5.
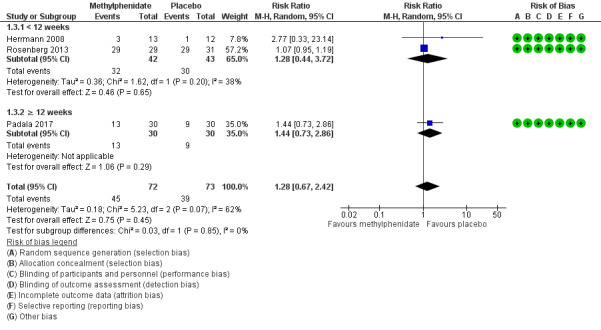
Forest plot of comparison: 7 Methylphenidate, outcome: 7.3 Adverse Events.
Secondary outcomes:
3. Neuropsychiatric symptoms
Only one study investigated and reported on the change in NPS over study duration (Herrmann 2008). There may be little or no difference between treatment groups in the change in NPI total score over two weeks (MD 0.16, 95% CI ‐7.89 to 8.21, P = 0.97, n = 25, 1 study). There was uncertainty associated with this result, which we considered to be of low quality because of serious concerns with imprecision, as there was a wide 95% confidence interval, which may have contributed to the overall null effect, in a single study with a small sample size. See Analysis 1.4.
1.4. Analysis.
Comparison 1 Methylphenidate, Outcome 4 Change in NPS from baseline as measured by the NPI.
4. Cognition
All studies assessed change in cognition using the MMSE. Compared to placebo, methylphenidate probably improves cognition slightly, although this difference may not be large enough to be of clinical importance (MD 1.98, 95% CI 1.06 to 2.91, P < 0.0001, n = 145, 3 studies, I2 = 37%). We considered this evidence to be of moderate quality, because of serious concerns with imprecision due to a wide 95% confidence interval. An exploratory subgroup analysis in studies by trial duration demonstrated that there was probably little or no difference between treatment groups on change in cognition over time in trials with a duration of less than 12 weeks (MD 1.00, 95% CI ‐0.49 to 2.49, P = 0.19, n = 85, 2 studies, I2 = 0%). In Padala 2017, the only study with a trial duration longer than 12 weeks, methylphenidate probably improves cognition compared to placebo (MD 2.60, 95% CI 1.43 to 3.77, P < 0.001, n = 60, 1 study). We rated the evidence for both subgroup analyses as of moderate quality, because of serious concerns with imprecision due to a wide 95% confidence interval.
We found no significant differences between subgroups (Chi2(1) = 2.74, P = 0.10, I2= 63.5%).See Analysis 1.5.
1.5. Analysis.
Comparison 1 Methylphenidate, Outcome 5 Change in cognition from baseline as measured by the MMSE.
We did not conduct a subgroup analysis based on disease severity, as all three studies enrolled participants with similar AD severity.
5. Functional performance
Only one study reported on the change in functional performance using the ADL and IADL (Padala 2017). There was no evidence of a difference between methylphenidate and placebo ADLs over 12 weeks: MD 0.50, 95% CI ‐0.39 to 1.39, P = 0.27, n = 60 patients, 1 study. See Analysis 1.6. However, compared to placebo, methylphenidate probably improves IADLs over 12 weeks: MD 2.30, 95% CI 0.74 to 3.86, P = 0.004, n = 60 patients, 1 study. There was some uncertainty associated with both findings, which we considered to be of moderate quality, as only one study with a small sample size was included in these comparisons. See Analysis 1.7.
1.6. Analysis.
Comparison 1 Methylphenidate, Outcome 6 Change in functional permance from baseline as measured by the ADL.
1.7. Analysis.
Comparison 1 Methylphenidate, Outcome 7 Change in functional performance from baseline as measured by the IADL.
6. Global disease severity
Two studies reported on global disease severity, measured with the CGI. This was expressed in both studies as the number of participants who experienced clinical deterioration over the course of the study (Herrmann 2008; Rosenberg 2013). There was probably little or no difference between treatment groups in the number who experienced clinical deterioration (RR 0.58, 95% CI 0.16 to 2.11, P = 0.40, n = 85, 2 studies I2 = 0%). We considered this evidence to be of moderate quality, because of serious concerns with imprecision due to a wide 95% confidence interval. See Analysis 1.8.
1.8. Analysis.
Comparison 1 Methylphenidate, Outcome 8 Change in global disease severity from baseline as measured by the CGIC and the ADCS‐CGIC.
7. Dropouts due to AEs
There may be little or no difference between treatment groups in the number of dropouts due to an AE (RR 2.18, 95% CI 0.64 to 7.45, P = 0.21, n = 145, 3 studies, I2 = 0%). There was low certainty associated with this result, which we considered to be of low quality due to serious concerns about imprecision. See Analysis 1.9.
1.9. Analysis.
Comparison 1 Methylphenidate, Outcome 9 Dropouts due to adverse events.
Comparison of modafinil with placebo
See: Table 2. Only one very small study (n = 23 randomized, 1 participant excluded from analysis due to AE related dropout) investigated and reported on the efficacy of modafinil for the treatment of apathy as a primary outcome measure (Frakey 2012). In all of the following outcomes, there were no concerns with risk of bias, inconsistency or indirectness. However, we had very serious concerns with imprecision due to the small sample size and wide 95% confidence intervals. As such we rated the quality of evidence for all of the outcomes as low.
Primary efficacy and safety outcomes:
1. Apathy
Apathy was assessed using the FrSBE apathy subscale. Tscores are converted from raw scores which range from 14 to 70, and there is very limited information available on what constitutes a clinically important difference in score. There was no evidence of a difference between treatment groups in the change in apathy over eight weeks (MD 0.27, 95% CI ‐3.51 to 4.05, P = 0.89, n = 22, 1 study). See Analysis 2.1.
2.1. Analysis.
Comparison 2 Modafinil, Outcome 1 Change in apathy from baseline as measured by the FrSBe‐apathy subscale.
2. Adverse events
We did not conduct an analysis for this outcome, as there was only one adverse event reported in the modafinil treatment group and none reported in the placebo group.
Secondary outcomes:
3. Neuropsychiatric symptoms
This outcome was not reported by Frakey 2012.
4. Cognition
This outcome was not investigated by Frakey 2012.
5. Functional performance
There was no evidence of a difference between modafinil and placebo in change in functional status over eight weeks (MD ‐0.54, 95% CI ‐1.40 to 0.32, P = 0.22, n = 22, 1 study). See Analysis 2.2.
2.2. Analysis.
Comparison 2 Modafinil, Outcome 2 Change in functional performance from baseline as measured by the ADL‐Q.
6. Global disease severity
This outcome was not investigated by Frakey 2012.
7. Dropouts due to AEs
There was one dropout from the modafinil treatment group due to an increase in motor tics, and no dropouts from the placebo arm. We did not conduct an analysis for this outcome, as only one individual in the study experienced an AE.
Objective 2:
Included studies of ChEIs, antipsychotics, mibampator and semagacestat were supported by pharmaceutical industry sponsors. Additionally, although a large number of studies with antipsychotics, antipsychotic discontinuation, and antidepressants in AD have been conducted, many of which collected data on NPS including apathy, only a few studies reported this or provided data upon request. We therefore downgraded the overall quality of the evidence due to serious concern about the effect of publication bias. Publication bias was not a concern with studies included in the ChEI comparison, as these trials included a large number of participants, and were less likely to remain unpublished or ignored. Furthermore, these trials may provide a more precise estimate of the treatment effect, whether positive or negative.
We also downgraded the overall quality of the evidence due to serious concerns with indirectness, as none of the studies contributing to Objective 2 comparisons actively recruited participants with clinically significant apathy.
The evidence provided in this section must therefore be considered of low quality at best.
Comparison of cholinesterase inhibitors (ChEIs) with placebo
We included six studies investigating ChEIs for cognition in people with AD, that also included apathy as a secondary outcome measure in this meta‐analysis. All six studies used the NPI‐apathy subscale to assess apathy.
ChEIs may slightly improve apathy compared to placebo (MD ‐0.40, 95% CI ‐0.80 to ‐0.00, P = 0.05, n = 3598, 6 studies, I2= 71%). We considered the available evidence to be of low quality. We downgraded the evidence due to serious concerns with indirectness, inconsistency and selective reporting of adjusted mean values, but upgraded one level due to the large sample size (Kaufer 1998; Morris 1998; Raskind 1999; Tariot 2001; MSAD trial; Herrmann 2005). However, the clinical importance of these findings is uncertain, as apathy was not a primary outcome measure of these studies. We conducted subgroup analyses of studies using currently‐approved ChEIs (donepezil, galantamine or rivastigmine), all of which had a duration 24 weeks or less, and studies using metrifonate which all lasted more than 24 weeks. Currently‐approved ChEIs may have little or no effect on apathy compared to placebo (MD ‐0.21, 95% CI ‐0.85 to 0.43, P = 0.29, n = 2531, 3 studies, I2 = 80%). However, metrifonate may improve apathy compared to placebo (MD ‐0.63, 95% CI ‐0.98 to ‐0.29, P > 0.001, n = 1067, 3 studies, I2 = 0%). Again, we rated the quality of the evidence as low due to serious concerns with indirectness, and selective reporting of adjusted mean value. There were no significant differences between subgroups (Chi2(1) = 1.30, P = 0.25, I2 = 23.2%). See Analysis 3.1; Figure 6. We also conducted subgroup analyses of studies in which participants had moderate or severe AD. In those with moderate AD, ChEIs may slightly improve apathy compared to placebo (MD ‐0.43, 95% CI ‐0.79 to ‐0.07, P = 0.02, n = 3100, 4 studies, I2 = 50%). In participants with severe AD, ChEIs may also slightly improve apathy compared to placebo (MD ‐0.36, 95% CI ‐1.82 to 1.10, P = 0.63, n = 498, 2 studies, I2 = 90%); however this finding was not statistically significant. As mentioned previously, we rated the quality of the evidence as low due to serious concerns with indirectness of the study population, and selective reporting of adjusted mean values. There was no significant difference between subgroups (Chi2(1) = 0.01, P = 0.93, I2 = 0%). See Analysis 3.2.
3.1. Analysis.
Comparison 3 Cholinesterase inhibitors, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore (subgroup analysis with licensed versus unlicensed ChEIs).
6.
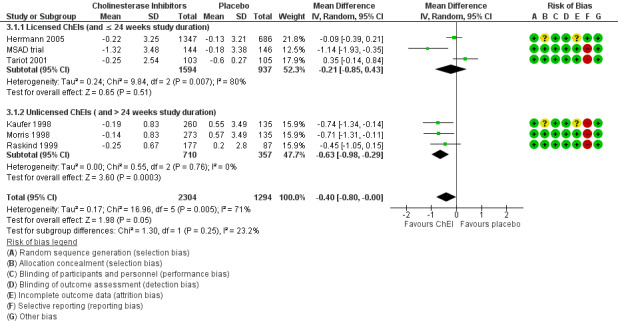
Forest plot of comparison: 3 Cholinesterase Inhibitors, outcome: 3.1 Change in apathy from baseline as measured by the NPI‐apathy subscore (subgroup analysis with licensed versus unlicensed ChEIs).
3.2. Analysis.
Comparison 3 Cholinesterase inhibitors, Outcome 2 Change in apathy from baseline as measured by the NPI‐apathy subscore (subgroup analysis with disease severity).
Comparison of ChEI with ChEI discontinuation (placebo)
Only one study investigating the discontinuation of ChEIs met the inclusion criteria for this meta‐analysis (Herrmann 2016). All participants in this trial were long‐term ChEI users (more than two years), and were considered eligible for this trial if they no longer demonstrated any clinical benefit from their ChEI.
Although not statistically significant, discontinuing ChEIs may slightly improve apathy compared to continued ChEI use, based on the NPI‐apathy subscale (MD 1.11, 95% ‐0.88 to 3.10, P = 0.28, n = 40, 1 study). There was some uncertainty associated with this result, which we considered to be of low quality because of serious concerns with imprecision due to the small sample size and wide 95% confidence interval, and indirectness of results.See Analysis 4.1.
4.1. Analysis.
Comparison 4 Discontinuation of cholinesterase inhibitors, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore.
Comparison of antipsychotics with placebo
All studies in this meta‐analysis that investigated antipsychotics were conducted in people with clinically significant agitation and aggression. We did not perform subgroup analyses by trial duration and disease severity, as only two studies met the inclusion criteria for this particular meta‐analysis. De Deyn 2004 assessed apathy using the NPI‐apathy subscale, while Sultzer 2008 assessed apathy using the BPRS withdrawn depression factor score. Antipsychotic use may slightly worsen apathy compared to placebo (standardized mean difference (SMD 0.14, 95% CI ‐0.00 to 0.28, P = 0.05, n = 1070, 2 studies, I2 = 15%). There is some uncertainty associated with this result, which we considered to be of low quality due to serious concerns with indirectness and publication bias. See Analysis 5.1.
5.1. Analysis.
Comparison 5 Atypical antipsychotics, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore and the BPRS withdrawn depression factor score.
Comparison of antipsychotics with antipsychotic discontinuation (placebo)
Only one study which investigated the discontinuation of antipsychotics met the inclusion criteria for this meta‐analysis (Ruths 2008).
Continued antipsychotic use may slightly improve apathy on the NPI‐apathy subscale compared to antipsychotic discontinuation (placebo): MD ‐0.24, 95% CI ‐0.51 to 0.03, P = 0.08, n = 55, 1 study. There was uncertainty associated with this result, which we considered to be of low quality due to serious concerns with indirectness and publication bias. Additionally, the clinical importance of these findings is uncertain, as apathy was not a primary outcome measure of these studies. See Analysis 6.1.
6.1. Analysis.
Comparison 6 Discontinuation of antipsychotics, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore.
Comparison of antidepressants with placebo
Two studies with antidepressants met the inclusion criteria for this meta‐analysis (Lanctôt 2002; CitAD trial). All participants had clinically significant NPS (Lanctôt 2002) or agitation (CitAD trial) at baseline. Apathy was assessed with the NPI‐apathy subscale.
We are uncertain whether antidepressants improved apathy compared to placebo over the duration of treatment (MD ‐1.24, 95% ‐1.44 to ‐1.04, P < 0.00001, n = 126, 2 studies, I2 = 3%). There was uncertainty associated with this result, which we considered to be of very low quality due to selective reporting (the NPI‐subscores in the CitAD trial were not normally distributed, and the reported medians were used as an estimate of the sample mean), indirectness of results, and publication bias. See Analysis 7.1.
7.1. Analysis.
Comparison 7 Antidepressants, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore.
Comparison of mibampator with placebo
Only one study investigated and reported on the efficacy of mibampator for the treatment of apathy (Trzepacz 2013).
We are uncertain whether mibampator improved apathy, assessed with the FrSBe‐apathy Tscore compared to placebo over the duration of treatment (MD ‐1.20, 95% ‐1.94 to ‐0.46, P = 0.001, n = 132, 1 study). There was uncertainty associated with this result, which we considered to be of very low quality due to selective reporting of adjusted mean values, indirectness of results, and publication bias. See Analysis 8.1.
8.1. Analysis.
Comparison 8 Mibampator, Outcome 1 Change in apathy from baseline as measured by the FrSBe‐apathy T score.
Comparison of valproate with placebo
Three studies investigated and reported on the efficacy of valproate for the treatment of apathy (Sival 2002; Herrmann 2007; Tariot 2011).
There may be little or no difference between treatment groups in the change in apathy over the duration of treatment (SMD 0.02, 95% CI ‐0.23 to 0.26, P = 0.88, n = 257, 3 studies, I2 = 0%). There was uncertainty associated with this result, which we considered to be of low quality due to indirectness and imprecision. See Analysis 9.1.
9.1. Analysis.
Comparison 9 Valproate, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore and GIP‐apathy subscore.
Comparison of semagacestat with placebo
Only one paper (Rosenberg 2016) reported on the efficacy of semagacestat for the treatment of apathy in a secondary analysis of a previously published trial (Semgacestat trial).
Semagacestat may slightly worsen apathy assessed with the NPI subscale compared to placebo over the duration of treatment (MD 0.20, 95% CI 0.15 to 0.25, P < 0.001, n = 939, 1 study), but the effect is probably too small to be of clinical significance. There was some uncertainty associated with this result, which we considered to be of low quality as there were concerns with indirectness of results and publication bias. See Analysis 10.1.
10.1. Analysis.
Comparison 10 Semagacestat, Outcome 1 Change in apathy from baseline as measured by the NPI‐apathy subscore.
Discussion
Summary of main results
The results of this meta‐analysis showed the following main findings:
Methylphenidate may improve apathy compared to placebo, based on AES change scores in people with AD and clinically significant apathy at baseline. However, this finding was not supported by the results on NPI‐apathy subscale change scores. Although this inconsistency may be attributed to differences between the scales, another difference is that the analysis with AE data included three studies and 145 participants (Herrmann 2008; Rosenberg 2013; Padala 2017), whereas the analysis with NPI‐apathy subscale data included only two studies with 85 participants (Herrmann 2008; Padala 2017). Subgroup analysis with AE data indicates that the effect size was larger in a 12‐week study than in six‐ and eight‐week studies, but this finding could be due to reasons other than trial duration. Notably, there was uncertainty with these findings, as they were associated with low quality of evidence. Methylphenidate may also improve cognition and function. Both of these findings were associated with moderate quality of evidence, although the difference in cognition was small and as a result may not be clinically significant. There were no significant differences between treatment groups in the risk of developing an AE.
In people with AD and clinically significant apathy at baseline, low‐quality evidence from one very small study suggested that there was no effect of modafinil on apathy.
All other included studies measured apathy as a secondary outcome. Participants in these studies did not necessarily have clinically significant apathy at baseline.
ChEI use may slightly improve apathy compared to placebo, but there is some uncertainty associated with this result due to low quality of evidence. Furthermore, the clinical importance of this finding is uncertain, as apathy was not a primary outcome measure, and the effect size was small. Significance was maintained and heterogeneity was reduced in studies with metrifonate only. These findings were not replicated in more recent studies with currently‐approved ChEIs. Studies with metrifonate had a longer study duration (more than 24 weeks), and were conducted only in people with mild‐to‐moderate AD. However, as subgroup differences were not significant, we cannot confirm the effect of ChEI type (approved ChEI versus metrifonate) or trial duration on the efficacy of ChEI intervention on apathy. In a subgroup analysis of disease severity, we found similarly‐sized effects in people with moderate and severe AD. However, there is uncertainty associated with this result, due to the low quality of evidence.
Atypical antipsychotics and semagacestat may slightly worsen apathy compared to placebo. Based on the findings of one antipsychotic discontinuation trial, continued antipsychotic use may slightly improve apathy compared to antipsychotic discontinuation. The clinical importance of this finding is uncertain, as apathy was not a primary outcome measure, and the effect size was small. The findings of one ChEI discontinuation trial indicated that ChEI discontinuation may slightly improve apathy compared to ChEI continuation. These findings were limited by a low quality of evidence.
Valproate demonstrated little or no difference in apathy. However, these findings were supported by low quality of evidence.
Our findings on antidepressants and mibampator had very low quality of evidence, due to selective reporting, indirectness of results and publication bias. We are therefore uncertain whether these interventions have any effect on apathy.
Overall completeness and applicability of evidence
By conducting a meta‐analysis we were able to combine findings from multiple clinical trials to evaluate the efficacy of a number of pharmacological interventions for the treatment of apathy in AD. When evaluating the applicability of this review to the clinical setting, outcome measures, participant characteristics, and treatment duration should be taken into consideration.
The studies with modafinil and methylphenidate were directly applicable to our review question. They recruited participants with clinically significant apathy, and included apathy as a primary outcome measure, which was likely chosen prior to designing their respective trials. As such, these trials may have a reduced risk of false‐positive errors resulting from the statistical analysis of many outcomes, and may have a reduced risk of false‐negative errors by providing the foundation for a sample size calculation for an adequately‐powered study (Andrade 2015). For methylphenidate, we found some data on all of our outcome measures. For modafinil, we found no data on cognition, overall NPS, or global disease severity.
All other included studies investigated apathy as a secondary outcome measure. Although this may increase the risk of false‐positive and false‐negative errors, the results of these studies are still of interest given the current absence of approved medications for the treatment of apathy in AD. However, for these interventions, the analyses must be regarded as exploratory. From these studies, we sought data on apathy only. In addition to a standardized diagnosis of AD, studies with antipsychotics, antidepressants, and mibampator also had clinically significant NPS as part of their inclusion criteria; in different studies these were overall NPS burden, agitation/aggression or psychosis. Apathy is one of the most frequently observed NPS in people with mild AD. In the advanced stages of AD, psychosis, agitation and aggression also become more prevalent, but apathy persists. In a systematic review of 59 studies, apathy was reported to be the only NPS with high baseline prevalence, persistence and incidence throughout the course of dementia (Van der Linde 2016). Hence, results from these studies may still be applicable, despite participants having NPS other than apathy. Some but not all of the Objective 2 studies did report apathy scores at baseline in their participants, and in several of these apathy was at levels usually regarded as clinically significant.
Five comparisons contained more than one study and in these study duration varied. The three methylphenidate studies varied from two to 12 weeks. In studies with ChEIs, study duration varied from 12 to 26 weeks. In studies with atypical antipsychotics, study duration varied from four to 36 weeks. In studies with antidepressants, study duration varied from four to nine weeks. Studies with valproate varied in study duration from 3 weeks to 24 months. We had hoped to explore the effect of treatment duration, but in practice the data offered little opportunity to do this due to the small numbers of studies and participants.
For this review, apathy was the only outcome evaluated in Objective 2. However, published systematic reviews of RCTs with ChEIs (Birks 2006), ChEI discontinuation (O'Regan 2015), atypical antipsychotics (Ballard 2006), antipsychotic discontinuation (Declercq 2013), antidepressants (Seitz 2011), and valproate (Lonergan 2009) may provide insight regarding their safety and efficacy in people with AD and apathy.
Quality of the evidence
All the included studies were well conducted and well reported, so that we had few serious concerns about risks of bias. We rated four studies as being at high risk of selective reporting bias because they reported only adjusted and not raw mean scores.
For Objective 1, the overall quality of the evidence was low for our primary efficacy outcome and low or moderate for adverse events and our secondary outcomes. This was largely due to imprecision; there were few studies and they were small. There was also inconsistency between the three methylphenidate studies in the effect on apathy.
For Objective 2, the nature and quality of the evidence meant that our results must be regarded as exploratory only. Many participants in these studies were recruited due to high levels of NPS other than apathy, or had no particular behavioral problems. Hence these studies addressed our review question indirectly. Furthermore, we were able to obtain data on apathy from only a minority of drug trials in AD which have measured it, raising concerns about publication bias. We also considered some of the results to be affected by imprecision, inconsistency or risk of selective reporting bias.
Potential biases in the review process
Ten out of the 21 studies which we included in our meta‐analysis were pharmaceutical industry‐sponsored studies. This may be a source of publication bias, as it has been reported that pharmaceutical‐industry funding is associated with outcomes that are favorable for the funder (Lexchin 2003; Bhandari 2004; Heres 2006). The pharmaceutical industry may not publish negative studies as frequently as positive studies. Publication bias is not necessarily confined to industry‐sponsored trials, as academic researchers may also be more likely to publish positive results, which are more likely to be accepted by editors, reviewed by peers, and more often cited. Clinical trial registries aim to increase transparency and access to information about clinical trials. Although clinical trial registries have been available since 1997, governmental bodies and international organizations have been pushing for the registration and standardization of clinical trial registries since 2005. Since 2008, the revised Declaration of Helsinki has stated that every clinical trial must be registered in a publicly accessible database before recruitment of the first participant (General Assembly of the World Medical Association). However only 10 of the 21 studies included in this meta‐analysis were published in or after 2008. We contacted the authors of the 41 additional studies which met our inclusion criterion of investigating apathy as a primary or secondary outcome measure as they did not provide data on our primary outcome of apathy in an extractable format. However, since we did not receive a response, or since data were no longer available to the authors, we excluded those studies from our meta‐analyses.
The NPI‐apathy subscale and the AES are two of the most widely‐used scales in research related to apathy in AD. In this review, 16 studies used the NPI‐apathy subscale, four studies used the AES, two used the FrSBe apathy Tscore, and one study each used the BPRS‐withdrawn depression factor score, and the GIP apathetic behavior subscore. As different scales were used to assess apathy in the studies included in this review, heterogeneity of results is a concern. However, there is no widely‐accepted gold‐standard scale for assessing apathy, and there is still a lack of consensus about the definition and diagnostic criteria for apathy (Cummings 2015). It is therefore important for future research to focus on validating scales and diagnostic criteria for apathy in AD. This would provide definitive recommendations for future clinical trials on how to appropriately target recipients of pharmacological interventions, and how to assess apathy, which would reduce the heterogeneity between studies.
Agreements and disagreements with other studies or reviews
The results of this meta‐analysis add to the current body of evidence for pharmacological interventions for apathy, as previous reports have been inconclusive in elucidating whether methylphenidate is effective and safe in the treatment of apathy in people with AD. In a meta‐analysis conducted by Sepehry 2017, the authors investigated pharmacological therapy for apathy in AD. Fifteen studies were included, and 11 were quantitatively analyzed. Pharmacological agents included ChEIs, memantine and psychostimulants (modafinil and methylphenidate). That review did not find any significant effect of drug over placebo on apathy. However, the authors disclosed that in the absence of descriptive statistics (mean and SD values), they generated effect sizes using P values and sample sizes. If P values were not reported, the authors assumed an alpha of 0.06 for their analyses. This method of data imputation would have likely biased the findings of their results due to a high risk of selective reporting. A comprehensive review by Harrison 2016 evaluated the evidence of pharmacotherapies for apathy from studies since 2013. However, as they did not require trials to be placebo‐controlled, nor treat apathy as a primary or secondary outcome measure, nor have AD as a standardized diagnosis, their findings were not consistent with ours. With respect to ChEIs, Harrison 2016 reported that previous findings about cognitive enhancers (ChEIs and memantine) were not replicated in more recent studies. This finding was similar to our own, as we found a benefit in the older trials investigating metrifonate, but not with the more recent trials with currently‐approved ChEIs. Additionally, although results with antidepressants were mixed, they found benefits with agomelatine in a non‐placebo‐controlled RCT (Callegari 2016). Their findings with methylphenidate were inconclusive, but they only included one study (Rosenberg 2013). We reported on the efficacy of methylphenidate on apathy in three trials. However, as we did not have high‐quality evidence, we suggest that further studies investigating the efficacy of methylphenidate on apathy in people with AD would increase the quality of evidence and strengthen this finding.
Authors' conclusions
Implications for practice.
Apathy is one of the most prevalent NPS of AD, affecting approximately 20% to 70% of sufferers. It is also associated with a number of negative implications such as increased mortality, increased cognitive and functional deterioration, and increased caregiver burden. However, there are few data available to guide clinicians in treating apathy in AD. Our meta‐analysis is limited by the small number of studies within each drug class, risk of bias, publication bias, imprecision, indirectness of studies included in Objective 2, and inconsistencies between studies. The evidence suggests that methylphenidate may demonstrate a benefit on apathy, although there is limited information available on clinically significant improvement on the AES, and there is some uncertainty regarding the clinical meaningfulness associated with this finding. Methylphenidate may also demonstrate a benefit on cognition in people with AD. However, as methylphenidate has been contraindicated in people with agitation, open‐angle glaucoma, treatment with monoamine oxidase inhibitors, hypertension and other cardiovascular conditions, physicians are encouraged to exercise caution when prescribing methylphenidate in people with AD.
Our findings with ChEIs suggest it may also have a benefit for apathy, but there were no subgroup differences identified with AD severity, or ChEI type (approved versus not approved). Nevertheless, there may be a signal for a benefit of donepezil on apathy (MSAD trial), and when targeting people with moderate AD. There is very low quality of evidence available for antidepressants and mibampator.
Implications for research.
Limitations and challenges encountered in trial design should be addressed to enhance the quality of evidence of future research. For example, while apathy is a well‐defined syndrome with cognitive, affective, and behavioral dimensions, there is a need to refine this definition. Measurements of apathy as diagnostic criteria have been well articulated, but have not yet been fully validated as a treatment target (Cummings 2015). Furthermore, although there are no gold‐standard measurements available for the assessment of apathy, future studies should use scales that have high test/re‐test and interrater reliabilities, such as the AES and the NPI‐apathy subscale (Clarke 2011). This may limit the inconsistency of future findings. Future research should also focus on the subdomains of apathy based on neurobiological, neurochemical and neuroimaging endpoints, as this may assist in identifying new targets for pharmacological intervention. Apathy has also been linked to cognitive and functional deterioration in people with AD. Future studies should therefore include cognitive and functional outcome measures to investigate how targeting apathy may have secondary benefits for cognition and function. Finally, additional studies that target people with AD with clinically‐significant apathy, investigate apathy as a primary outcome measure, and which also have a longer duration and larger sample size, are encouraged. Altogether, this would increase the quality of evidence for methylphenidate, as well as ChEIs, antidepressants and mibamapator, and may justify its future use in clinical practice..
Feedback
Comment on Risk of Bias, 10 May 2018
Summary
Comment written by Martin Vuillème
The authors report assessing the risks of bias in accordance with the Cochrane 'Risk of bias' assessment tool. I am concerned the Handbook guidance wasn't followed appropriately and the risk of bias in included studies was underestimated. As an example, many included RCTs were deemed at low risk of detection bias due to self‐describing themselves as "double‐blinded", yet the Cochrane Handbook guidance points out that [Study reports often describe blinding in broad terms, such as ‘double blind’. This term makes it impossible to know who was blinded (Schulz 2002a). Such terms are also used very inconsistently (Devereaux 2001, Boutron 2005, Haahr 2006), and the frequency of explicit reporting of the blinding status of study participants and personnel remains low even in trials published in top journals (Montori 2002)]. Similarly, many included RCTs were deemed at low risk of selection bias associated with a random sequence generation due to the studies including the sentence "Patients randomly assigned to receive" or "patients randomized" yet the Cochrane Handbook guidances points out that [A simple statement such as ‘we randomly allocated’ or ‘using a randomized design’ is often insufficient to be confident that the allocation sequence was genuinely randomized. It is not uncommon for authors to use the term ‘randomized’ even when it is not justified: many trials with declared systematic allocation are described by the authors as randomized. If there is doubt, then the adequacy of sequence generation should be considered to be unclear.].
Reply
We thank the Publisher and Editor for the opportunity to respond to Martin Vuillème’s letter, which raises concerns about our use of the Cochrane Handbook when evaluating the risk of bias of the studies included in our review. M Vuillème’s concerns are specifically about the blinding and randomization aspects of the included studies.
Though the Cochrane Handbook provides instructions on how to assess risk of bias, it also points out that this assessment involves a degree of subjective judgement. In order to limit subjective bias, we ensured that two authors independently assessed the risk of bias for each of the studies. Any discrepancies were discussed and resolved with the remaining co‐authors. This process is described in the methods section of the review.
M Vuillème is concerned that the included studies may not be truly randomized. The Handbook advises authors to consider the risk of material bias, which is bias of sufficient magnitude to have a notable impact on the results/conclusions of the trial. We judged that the included studies provided sufficient data on randomization and therefore evaluated them as having a low risk of bias on this matter. Additionally, in all of the included trials, the drug and placebo groups were similar across measured covariates, suggesting successful randomization.
The text that M Vuillème quoted directly from the Cochrane Handbook (section 8.12.2) refers to the adequacy of blinding. Fortunately the most recent risk of bias tool on Review Manger allows authors to assess blinding of participants and personnel separately from blinding of outcome assessors. Therefore, we were able to take both into account when evaluating the risk of bias. M Vuillème refers to an old paper which raises concerns about reporting of blinding status in clinical trials (Montori 2002). However, speaking as clinical trialists with a quarter century of experience, we consider that the clinical trial landscape has changed dramatically over the last decade or two in North America and Europe. The expectations of journals, and even more particularly, health regulatory agencies, for the conduct and reporting of results from clinical trials have risen substantially.
M Vuillème’s comments do highlight that there is a subjective element in judgements about risk of bias. However, as the Handbook (section 8.3.1) points out, this is to some extent unavoidable. Overall, our tendency is to believe the statements made by investigators. We would encourage readers to assess the included studies for themselves.
Contributors
Martin Vuillème
For CDCIG:
Authors: Myuri T. Ruthirakuhan, Nathan Herrmann, Krista L. Lanctôt,
Co‐ordinating editor: Jenny McCleery
What's new
| Date | Event | Description |
|---|---|---|
| 10 May 2018 | Amended | Feedback and response incorporated |
| 10 May 2018 | Feedback has been incorporated | Comment on Risk of Bias and response from authors |
Acknowledgements
We would like to thank the Cochrane Dementia and Cognitive Improvement Group for their non‐author contributions to the study appraisal and search strategy development. We would also like to thank Ruths 2008 and the ADCS group (Tariot 2011) for providing data upon request.
Appendices
Appendix 1. Sources searched and search strategies
| Source | Search strategy | Hits retrieved |
| 1. CENTRAL (The Cochrane Library) http://crso.cochrane.org/SearchSimple.php [Date of most recent search: 4 May 2017] |
#1 MeSH descriptor: [Dementia] explode all trees #2 dement* #3 alzheimer* #4 ((lewy* adj2 bod*) or DLB or LBD) #5 LBD #6 "organic brain disease" or "organic brain syndrome" #7 "benign senescent forgetfulness" #8 (cerebr* adj2 deteriorat*) #9 (cerebral* adj2 insufficient*) #10 VCI #11 FTD or FTLD or "fronto‐temporal" or frontotemporal #12 "parkinson* disease dementia" or PDD #13 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 #14 MeSH descriptor: [Mild Cognitive Impairment] explode all trees #15 "cognit* impair*" #16 MCI #17 ACMI #18 ARCD #19 SMC #20 CIND #21 BSF #22 AAMI #23 MD #24 LCD #25 QD #26 AACD #27 MNCD #28 MCD #29 "N‐MCI" or "A‐MCI" or "M‐MCI" #30 "cognit* declin*" or "cognit* los*" or "cognit* deteriorat*" or "cognit* degenerat*" or "cognit* complain*" or "cognit* disturb*" or "cognit* disorder*" or "memory declin*" or "memory los*" or "memory deteriorat*" or "memory degenerat*" or "memory complain*" or "memory disturb*" #31 "memory disorder*" or "cerebr* declin*" or "cerebr* los*" or "cerebr* deteriorat*" or "cerebr* degenerat*" or "cerebr* complain*" or "cerebr* disturb*" or "cerebr* disorder*" or "mental* declin*" or "mental* los*" or "mental* deteriorat*" or "mental* degenerat*" or "mental* complain*" or "mental* disturb*" or "mental* disorder*" #32 "preclinical AD" #33 "pre‐clinical AD" #34 ("preclinical alzheimer*" or "pre‐clinical alzheimer*") #35 (aMCI or MCIa) #36 ("CDR 0.5" or "clinical dementia rating scale 0.5") #37 ("GDS 3" or "stage 3 GDS") #38 ("global deterioration scale" and "stage 3") #39 (AES or "apathy evaluation scale") #40 (NPI or "neuropsychiatric inventory") #41 "mild neurocognit* disorder*" #42 (prodrom* adj2 dement*) #43 (episodic* adj2 memory) #44 ("preclinical dementia" or "pre‐clinical dementia") #45 #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 #46 MeSH descriptor: [Apathy] explode all trees #47 apathy #48 apathetic #49 MeSH descriptor: [Lethargy] explode all trees #50 lethargy #51 lethargic #52 listless* #53 detachment or detached #54 disinterest* #55 dispassion* #56 lack adj3 interest* #57 BPSD or "behav* and psychological symptom*" #58 #57 and #45 in Trials |
17.06.17 ‐133 4.5.17 ‐ 33 |
| 2. MEDLINE Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) 1946 to Present (Ovid SP) [Date of most recent search: 4 May 2017] |
1. exp Dementia/ 2. dement*.mp. 3. alzheimer*.mp. 4. ((lewy* adj2 bod*) or DLB or LBD).ti,ab. 5. (LBD).mp. 6. ("organic brain disease" or "organic brain syndrome").mp. 7. "benign senescent forgetfulness".mp. 8. (cerebr* adj2 deteriorat*).mp. 9. (cerebral* adj2 insufficient*).mp. 10. VCI.ti,ab. 11. (FTD or FTLD or “fronto‐temporal” or frontotemporal).ti,ab. 12. ("parkinson* disease dementia" or PDD).ti,ab. 13. or/1‐12 14. Mild Cognitive Impairment/ 15. "cognit* impair*".mp. 16. MCI.ti,ab. 17. ACMI.ti,ab. 18. ARCD.ti,ab. 19. SMC.ti,ab. 20. CIND.ti,ab. 21. BSF.ti,ab. 22. AAMI.ti,ab. 23. MD.ti,ab. 24. LCD.ti,ab. 25. QD.ti,ab. 26. AACD.ti,ab. 27. MNCD.ti,ab. 28. MCD.ti,ab. 29. ("N‐MCI" or "A‐MCI" or "M‐MCI").ti,ab. 30. ((cognit* or memory or cerebr* or mental*) adj3 (declin* or los* or deteriorat* or degenerat* or complain* or disturb* or disorder*)).ti,ab. 31. "preclinical AD".mp. 32. "pre‐clinical AD".mp. 33. ("preclinical alzheimer*" or "pre‐clinical alzheimer*").mp. 34. (aMCI or MCIa).ti,ab. 35. ("CDR 0.5" or "clinical dementia rating scale 0.5").ti,ab. 36. ("GDS 3" or "stage 3 GDS").ti,ab. 37. ("global deterioration scale" and "stage 3").ti,ab. 38. (AES or “apathy evaluation scale”).ti,ab. 39. (NPI or “neuropsychiatric inventory”).ti,ab. 40. "mild neurocognit* disorder*".ti,ab. 41. (prodrom* adj2 dement*).ti,ab. 42. (episodic* adj2 memory).mp. 43. ("preclinical dementia" or "pre‐clinical dementia").mp. 44. or/14‐43 45. 13 or 44 46. Apathy/ 47. apathy.ti,ab. 48. apathetic.ti,ab. 49. Lethargy/ 50. lethargy.ti,ab. 51. lethargic.ti,ab. 52. listless*.ti,ab. 53. (detachment or detached).ti,ab. 54. disinterest*.ti,ab. 55. dispassion*.ti,ab. 56. (lack adj3 interest*).ti,ab. 57. (BPSD or "behav* and psychological symptom*").ti,ab. 58. or/46‐57 59. 45 and 58 60. randomized controlled trial.pt. 61. controlled clinical trial.pt. 62. randomized.ab. 63. placebo.ab. 64. drug therapy.fs. 65. randomly.ab. 66. trial.ab. 67. groups.ab. 68. or/60‐67 69. (animals not (humans and animals)).sh. 70. 68 not 69 71. 59 and 70 |
17.06.17 ‐1200 4.5.17 ‐ 157 |
| 3. EMBASE 1974 to 03 May 2017 [Date of most recent search: 4 May 2017] |
1 exp Dementia/ 2 Delirium, Dementia, Amnestic, Cognitive Disorders/ 3 ("benign senescent forgetfulness" or ("normal pressure hydrocephalus" and "shunt*") or ("organic brain disease" or "organic brain syndrome") or ((cerebral* or cerebrovascular or cerebro‐vascular) adj2 insufficien*) or (cerebr* adj2 deteriorat*) or (chronic adj2 (cerebrovascular or cerebro‐vascular)) or (creutzfeldt or jcd or cjd) or (lewy* adj2 bod*) or (pick* adj2 disease) or alzheimer* or binswanger* or deliri* or dement* or huntington* or korsako*).tw. 4 dement*.mp. 5 alzheimer*.mp. 6 ((lewy* adj2 bod*) or DLB or LBD).ti,ab. 7 (chronic adj2 cerebrovascular).mp. 8 ("organic brain disease" or "organic brain syndrome").mp. 9 "benign senescent forgetfulness".mp. 10 (cerebr* adj2 deteriorat*).mp. 11 (cerebral* adj2 insufficient*).mp. 12 VCI.ti,ab. 13 (FTD or FTLD or "fronto‐temporal" or frontotemporal).ti,ab. 14 or/1‐13 15 Mild Cognitive Impairment/ 16 "cognit* impair*".mp. 17 MCI.ti,ab. 18 ACMI.ti,ab. 19 ARCD.ti,ab. 20 SMC.ti,ab. 21 CIND.ti,ab. 22 BSF.ti,ab. 23 AAMI.ti,ab. 24 MD.ti,ab. 25 LCD.ti,ab. 26 QD.ti,ab. 27 AACD.ti,ab. 28 MNCD.ti,ab. 29 MCD.ti,ab. 30 ("N‐MCI" or "A‐MCI" or "M‐MCI").ti,ab. 31 ((cognit* or memory or cerebr* or mental*) adj3 (declin* or los* or deteriorat* or degenerat* or complain* or disturb* or disorder*)).ti,ab. 32 "preclinical AD".mp. 33 "pre‐clinical AD".mp. 34 ("preclinical alzheimer*" or "pre‐clinical alzheimer*").mp. 35 (aMCI or MCIa).ti,ab. 36 ("CDR 0.5" or "clinical dementia rating scale 0.5").ti,ab. 37 ("GDS 3" or "stage 3 GDS").ti,ab. 38 ("global deterioration scale" and "stage 3").ti,ab. 39 (AES or "apathy evaluation scale").ti,ab. 40 (NPI or "neuropsychiatric inventory").ti,ab. 41 "mild neurocognit* disorder*".ti,ab. 42 (prodrom* adj2 dement*).ti,ab. 43 (episodic* adj2 memory).mp. 44 ("preclinical dementia" or "pre‐clinical dementia").mp. 45 or/15‐44 46 14 or 45 47 Apathy/ 48 apathy.ti,ab. 49 apathetic.ti,ab. 50 Lethargy/ 51 lethargy.ti,ab. 52 lethargic.ti,ab. 53 listless*.ti,ab. 54 (detachment or detached).ti,ab. 55 disinterest*.ti,ab. 56 dispassion*.ti,ab. 57 (lack adj3 interest*).ti,ab. 58 (BPSD or "behav* and psychological symptom*").ti,ab. 59 or/47‐58 60 46 and 59 61 randomized controlled trial/ 62 controlled clinical trial/ 63 random$.ti,ab. 64 randomization/ 65 intermethod comparison/ 66 placebo.ti,ab. 67 (compare or compared or comparison).ti. 68 ((evaluated or evaluate or evaluating or assessed or assess) and (compare or compared or comparing or comparison)).ab. 69 (open adj label).ti,ab. 70 ((double or single or doubly or singly) adj (blind or blinded or blindly)).ti,ab. 71 double blind procedure/ 72 parallel group$1.ti,ab. 73 (crossover or cross over).ti,ab. 74 ((assign$ or match or matched or allocation) adj5 (alternate or group$1 or intervention$1 or patient$1 or subject$1 or participant$1)).ti,ab. 75 (assigned or allocated).ti,ab. 76 (controlled adj7 (study or design or trial)).ti,ab. 77 (volunteer or volunteers).ti,ab. 78 trial.ti. 79 or/61‐78 80 60 and 79 |
17.06.17 ‐1667 4.5.17 ‐ 284 |
| 4. PSYCINFO 1806 to May Week 1 2017 [Date of most recent search: 4 May 2017] |
1 exp Dementia/ 2 dement*.mp. 3 alzheimer*.mp. 4 ((lewy* adj2 bod*) or DLB or LBD).ti,ab. 5 (chronic adj2 cerebrovascular).mp. 6 ("organic brain disease" or "organic brain syndrome").mp. 7 "benign senescent forgetfulness".mp. 8 (cerebr* adj2 deteriorat*).mp. 9 (cerebral* adj2 insufficient*).mp. 10 VCI.ti,ab. 11 (FTD or FTLD or "fronto‐temporal" or frontotemporal).ti,ab. 12 ("parkinson* disease dementia" or PDD).ti,ab. 13 or/1‐12 14 Mild Cognitive Impairment/ 15 "cognit* impair*".mp. 16 MCI.ti,ab. 17 ACMI.ti,ab. 18 ARCD.ti,ab. 19 SMC.ti,ab. 20 CIND.ti,ab. 21 BSF.ti,ab. 22 AAMI.ti,ab. 23 MD.ti,ab. 24 LCD.ti,ab. 25 QD.ti,ab. 26 AACD.ti,ab. 27 MNCD.ti,ab. 28 MCD.ti,ab. 29 ("N‐MCI" or "A‐MCI" or "M‐MCI").ti,ab. 30 ((cognit* or memory or cerebr* or mental*) adj3 (declin* or los* or deteriorat* or degenerat* or complain* or disturb* or disorder*)).ti,ab. 31 "preclinical AD".mp. 32 "pre‐clinical AD".mp. 33 ("preclinical alzheimer*" or "pre‐clinical alzheimer*").mp. 34 (aMCI or MCIa).ti,ab. 35 ("CDR 0.5" or "clinical dementia rating scale 0.5").ti,ab. 36 ("GDS 3" or "stage 3 GDS").ti,ab. 37 ("global deterioration scale" and "stage 3").ti,ab. 38 (AES or "apathy evaluation scale").ti,ab. 39 (NPI or "neuropsychiatric inventory").ti,ab. 40 "mild neurocognit* disorder*".ti,ab. 41 (prodrom* adj2 dement*).ti,ab. 42 (episodic* adj2 memory).mp. 43 ("preclinical dementia" or "pre‐clinical dementia").mp. 44 or/14‐43 45 13 or 44 46 Apathy/ 47 apathy.ti,ab. 48 apathetic.ti,ab. 49 Lethargy/ 50 lethargy.ti,ab. 51 lethargic.ti,ab. 52 listless*.ti,ab. 53 (detachment or detached).ti,ab. 54 disinterest*.ti,ab. 55 dispassion*.ti,ab. 56 (lack adj3 interest*).ti,ab. 57 (BPSD or "behav* and psychological symptom*").ti,ab. 58 or/46‐57 59 45 and 58 60 exp Clinical Trials/ 61 randomly.ab. 62 randomi?ed.ti,ab. 63 placebo.ti,ab. 64 groups.ab. 65 "double‐blind*".ti,ab. 66 "single‐blind*".ti,ab. 67 RCT.ti,ab. 68 or/60‐67 69 59 and 68 |
17.06.17 ‐ 547 4.5.17 ‐ 53 |
| 5. CINAHL (EBSCOhost) [Date of most recent search: 4 May 2017] |
S1 (MH "Dementia+") S2 TX dement* S3 TX alzheimer* S4 TX "lewy* bod*" S5 TX DLB OR TX LBD S6 TX "organic brain disease" or "organic brain syndrome" S7 TX "benign senescent forgetfulness" S8 TX "cerebr* deteriorat*" S9 TX "cerebral* insufficient*" S10 TX "cerebral* insufficient*" S11 TX VCI S12 TX FTD or FTLD or “fronto‐temporal” or frontotemporal S13 TX "parkinson* disease dementia" or PDD S14 S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 S15 TX "cognit* impair*" S16 TX MCI S17 TX ACMI S18 TX ARCD S19 TX SMC S20 TX CIND S21 TX BSF S22 TX AAMI S23 TX MD S24 TX LCD S25 TX QD S26 TX AACD S27 TX MNCD S28 TX MCD S29 TX "N‐MCI" or "A‐MCI" or "M‐MCI" S30 TX "cognit* declin*" OR "cognit* los*" OR "cognit* deteriorat*" OR "cognit* degenerat*" OR "cognit* complain*" OR "cognit* disturb*" OR "cognit* disorder*" OR "memory declin*" OR "memory los*" OR "memory deteriorat*" OR "memory degenerat*" OR "memory complain*" OR "memory disturb*" OR "memory disorder*" OR "cerebr* declin*" OR "cerebr* los*" OR "cerebr* deteriorat*" OR "cerebr* degenerat*" OR "cerebr* complain*" OR "cerebr* disturb*" OR "cerebr* disorder*" OR "mental* declin*" OR "mental* los*" OR "mental* deteriorat*" OR "mental* degenerat*" OR "mental* complain*" OR "mental* disturb*" OR "mental* disorder*" S31 TX "preclinical AD" S32 TX "pre‐clinical AD" S33 TX ("preclinical alzheimer*" or "pre‐clinical alzheimer*") S34 TX (aMCI or MCIa) S35 TX ("CDR 0.5" or "clinical dementia rating scale 0.5") S36 TX ("GDS 3" or "stage 3 GDS") S37 TX ("global deterioration scale" and "stage 3") S38 TX (AES or “apathy evaluation scale”) S39 TX (NPI or “neuropsychiatric inventory”) S40 TX "mild neurocognit* disorder*" S41 TX "prodrom* dement*" S42 TX "episodic* memory" S43 TX ("preclinical dementia" or "pre‐clinical dementia") S44 (S15 OR S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S32 OR S33 OR S34 OR S35 OR S36 OR S37 OR S38 OR S39 OR S40 OR S41 OR S42 OR S43) S45 S14 OR S44 S46 (MH "Apathy") S47 TX apathy S48 TX apathetic S49 TX Lethargy S50 TX lethargy S51 TX lethargic S52 TX listless S53 TX (detachment or detached) S54 TX disinterest* S55 TX dispassion* S56 TX lack N3 interest* S57 TX (BPSD or "behav* and psychological symptom*") S58 (S46 OR S47 OR S48 OR S49 OR S50 OR S51 OR S52 OR S53 OR S54 OR S55 OR S56 OR S57) S59 (MH "Randomized Controlled Trials") S60 TX randomised S61 TX randomized S62 AB placebo S63 AB randomly S64 AB "double blind* S65 AB "single blind* S66 AB RCT S67 (S59 OR S60 OR S61 OR S62 OR S63 OR S64 OR S65 OR S66) S68 (S45 AND S58 AND S67) |
17.06.17 ‐ 239 4.5.17 ‐ 18 |
| 6. ISI Web of Science – all databases [includes: Web of Science (1945‐present); BIOSIS Previews (1926‐present); MEDLINE (1950‐present); Journal Citation Reports] [Date of most recent search: 4 May 2017] |
((dement* OR alzheimer* OR "vascular cognitive impairment" OR "lew* bod*" OR CADASIL OR "cognit* impair*" OR FTD OF FTLD OR "cerebrovascular insufficienc*" OR AD OR VCI)) AND TOPIC: ((Apathy or apathetic or Lethargy or lethargic or listless* or detachment or detached or disinterest* or dispassion* or "lack of interest" or BPSD)) AND TOPIC:((randomly OR randomised OR randomized OR "random allocat*" OR RCT OR CCT OR "double blind*" OR "single blind*" OR "double blind*" OR "single blind*" OR trial)) | 17.06.17 ‐ 625 4.5.17 ‐ 74 |
| 7. LILACS (BIREME) [Date of most recent search: 4 May 2017] |
(alzheimer OR alzheimers OR alzheimer’s OR dementia OR demenc$ [Words] and Apathy OR apathetic OR Lethargy OR lethargic OR listless$ OR detachment OR detached OR disinterest$ OR dispassion$ [Words] and randomly OR randomised OR randomized OR RCT OR "controlled trial" OR "double blind$" OR placebo [Words] | 17.06.17 ‐ 0 4.5.17 ‐ 0 |
| 8. ClinicalTrials.gov (www.clinicaltrials.gov) [Date of most recent search: 4 May 2017] |
dementia OR alzheimers OR cognition OR cognitive | Apathy OR apathetic OR Lethargy OR lethargic OR listless* OR detachment OR detached OR disinterest* OR dispassion* | 17.06.17 ‐ 51 4.5.17 ‐ 18 |
| 9. ICTRP [Date of most recent search: 4 May 2017] |
Apathy OR apathetic OR Lethargy OR lethargic OR listless* OR detachment OR detached OR disinterest* OR dispassion* AND dementia OR alzheimers | 7 4.5.17 ‐ 0 |
| TOTAL before de‐duplication | 5295 | |
Data and analyses
Comparison 1. Methylphenidate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the AES | 3 | 145 | Mean Difference (IV, Random, 95% CI) | ‐4.99 [‐9.55, ‐0.43] |
| 1.1 < 12 weeks | 2 | 85 | Mean Difference (IV, Random, 95% CI) | ‐2.62 [‐4.80, ‐0.44] |
| 1.2 ≥ 12 weeks | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐9.90 [‐13.50, ‐6.30] |
| 2 Change in apathy from baseline as measured by the NPI‐apathy subscore | 2 | 85 | Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐3.85, 3.69] |
| 3 Adverse Events | 3 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.67, 2.42] |
| 3.1 < 12 weeks | 2 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.44, 3.72] |
| 3.2 ≥ 12 weeks | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [0.73, 2.86] |
| 4 Change in NPS from baseline as measured by the NPI | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.16 [‐7.89, 8.21] |
| 5 Change in cognition from baseline as measured by the MMSE | 3 | 145 | Mean Difference (IV, Fixed, 95% CI) | 1.98 [1.06, 2.91] |
| 5.1 < 12 weeks study duration | 2 | 85 | Mean Difference (IV, Fixed, 95% CI) | 1.00 [‐0.49, 2.49] |
| 5.2 ≥ 12 weeks study duration | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 2.6 [1.43, 3.77] |
| 6 Change in functional permance from baseline as measured by the ADL | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐0.39, 1.39] |
| 7 Change in functional performance from baseline as measured by the IADL | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 2.3 [0.74, 3.86] |
| 8 Change in global disease severity from baseline as measured by the CGIC and the ADCS‐CGIC | 2 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.16, 2.11] |
| 9 Dropouts due to adverse events | 3 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.18 [0.64, 7.45] |
Comparison 2. Modafinil.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the FrSBe‐apathy subscale | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [‐3.51, 4.05] |
| 2 Change in functional performance from baseline as measured by the ADL‐Q | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐0.54 [‐1.40, 0.32] |
Comparison 3. Cholinesterase inhibitors.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore (subgroup analysis with licensed versus unlicensed ChEIs) | 6 | 3598 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.80, ‐0.00] |
| 1.1 Licensed ChEIs (and ≤ 24 weeks study duration) | 3 | 2531 | Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.85, 0.43] |
| 1.2 Unlicensed ChEIs ( and > 24 weeks study duration) | 3 | 1067 | Mean Difference (IV, Random, 95% CI) | ‐0.63 [‐0.98, ‐0.29] |
| 2 Change in apathy from baseline as measured by the NPI‐apathy subscore (subgroup analysis with disease severity) | 6 | 3598 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.80, ‐0.00] |
| 2.1 Moderate AD (MMSE ≥ 18) | 4 | 3100 | Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.79, ‐0.07] |
| 2.2 Severe AD (MMSE < 18) | 2 | 498 | Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐1.82, 1.10] |
Comparison 4. Discontinuation of cholinesterase inhibitors.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | 1.11 [‐0.88, 3.10] |
Comparison 5. Atypical antipsychotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore and the BPRS withdrawn depression factor score | 2 | 1070 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.14 [0.00, 0.28] |
Comparison 6. Discontinuation of antipsychotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | ‐0.24 [‐0.51, 0.03] |
Comparison 7. Antidepressants.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore | 2 | 126 | Mean Difference (IV, Fixed, 95% CI) | ‐1.24 [‐1.44, ‐1.04] |
Comparison 8. Mibampator.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the FrSBe‐apathy T score | 1 | 132 | Mean Difference (IV, Fixed, 95% CI) | ‐1.2 [‐1.94, ‐0.46] |
Comparison 9. Valproate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore and GIP‐apathy subscore | 3 | 257 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.23, 0.26] |
Comparison 10. Semagacestat.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in apathy from baseline as measured by the NPI‐apathy subscore | 1 | 939 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.15, 0.25] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
CitAD trial.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial | |
| Participants | Participant information obtained from Porsteinsson 2014:
|
|
| Interventions | Participants were randomized to receive:
During the first 3 weeks after randomization, clinicians could adjust the medication dosage according to response and tolerability. Caregivers received a standardized practical psychosocial intervention of 3 components: provision of educational materials, 24‐hour availability of crisis management services, and a 20‐ to 30‐minute counseling session at each scheduled study visit Duration: 9‐week treatment phase Enrollment: 186 participants randomized |
|
| Outcomes | Outcomes were obtained from Leonpacher 2016:
Primary:
Secondary:
|
|
| Notes | We have selected Leonpacher 2016 to be the primary paper. However, additional information for this study was obtained from clinicaltrials.gov (NCT00898807) and Porsteinsson 2014. Study dates: July 2009 ‐ September 2013. Eight sites in the USA and Canada were included. Specific site locations not disclosed. Coordinating site: Johns Hopkins University, Baltimore, MD, USA. Funding provided by the National Institute on Aging and NIMH grant R01AG031348, and in part by HIH grant P50‐AG05142 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were randomized to receive…” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “Patients were randomized to receive citalopram at a target dosage of 30 mg/day… or matching placebo.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “…there was no difference in adherence between the citalopram and placebo groups and that side effects were generally modest and consistent with those known to be associated with SSRIs (gastrointestinal complaints, respiratory tract infections and falls). The adverse effects of cognitive worsening (of unknown clinical significance) and QT prolongation, however, raise concerns about 30 mg/day dosage used…” Comment: Study withdrawals and reason for withdrawals have been reported in Porsteinsson 2014 (Safety and Adherence section and Table 3) and appear to be balanced between groups |
| Selective reporting (reporting bias) | High risk | The authors provided medians and interquartile ranges for each NPI item at baseline (Table 1) and at 9 weeks (Table 2), but did not report means ± SD scores. The authors of this meta‐analysis computed these values themselves. However, as CitAD trial disclosed that these values were not normally distributed, there may be a selective reporting bias |
| Other bias | Low risk | No other identified biases |
De Deyn 2004.
| Methods | Multicenter, double‐blind, randomized, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Following placebo lead‐in phase (up to 14 days) participants were randomized to receive either:
Duration: 10‐week treatment phase (+ maximum 14‐day placebo lead in) Enrollment: 652 patients randomized, however 649 included in analysis. |
|
| Outcomes | Primary:
Secondary:
Safety:
|
|
| Notes | Study dates not reported. 61 sites in Europe, Australia, Israel, Lebanon, and South Africa were included. Specific site locations not disclosed. Corresponding author’s institution: Lily Research Laboratories, Indianapolis, IN, USA. Contract/grant sponsor: Eli Lilly and Company. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients randomly assigned to receive…” Comment: Probably done |
| Allocation concealment (selection bias) | Unclear risk | This information has not been made available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “The proportion of patients completing the 10‐week treatment period was not significantly different among the five treatment groups (Table 1)…” Comment: Study withdrawals and reason for withdrawals have been reported in Table 1 and appear to be balanced between groups |
| Selective reporting (reporting bias) | Low risk | The authors reported means ± standard deviations on each of the 12‐item NPI‐NH scores (Table 2), BPRS (total, negative and positive) scores, and CGI scores (Table 3) |
| Other bias | Low risk | No other identified biases |
Frakey 2012.
| Methods | Randomized, double‐blind, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Participants were urn‐randomized into either:
Duration: 8 weeks Enrolment: 23 participants randomized, 1 participant excluded from analysis due to AE‐related drop‐out |
|
| Outcomes | Primary:
Other:
|
|
| Notes | Study dates: July 2005 ‐ September 2007. Study site: Butler Hospital, Providence, RI, USA. Salary support for the corresponding author provided by a National Research Service Award from the National Institute of Mental Health. Cephalon provided study medication, placebo, and $40,000 USD through an unrestricted investigator‐initiated grant | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “A randomized, double‐blind, placebo‐controlled design was employed.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “Participants were urn‐randomized into either the experimental group (modafinil) or the control group (placebo) using apathy severity (mild, moderate, or severe), dementia severity (mild or moderate), presence of antidepressant medication, and presence of memantine as randomization factors.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..."“Both the physician (S.S.) and the clinician (L.L.F.) who performed the assessments were blind to the medication status of the participants.” Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “One experimental group participant was withdrawn from the study after 2 weeks due to an increase in motor tics.” Comment: Though no participant flow diagram was provided in text, participant withdrawal information and reasoning described in text |
| Selective reporting (reporting bias) | Low risk | Quote: “The mean scores and SDs for our 2 groups for each of the outcome measures are presented in Table 2.” Comment: Table 2 provides descriptive statistics for independent and dependent variables assessed in the study, providing means ± standard deviations for each outcome |
| Other bias | Low risk | No other identified biases |
Herrmann 2005.
| Methods | Data pooled from 3 multicenter, double‐blind, placebo‐controlled, parallel‐group studies | |
| Participants | Similar criteria between the 3 studies:
Rockwood 2001:
Tariot 2000:
Data obtained by authors from Janssen‐Ortho Inc.:
|
|
| Interventions |
Rockwood 2001: Participants were randomized to either: ‐ Galantamine (IR) (N = 261) ‐ Placebo (N = 125) Duration: 3‐month treatment phase (+1 month placebo run‐in) Enrollment: 368 participants randomized Tariot 2000: Participants were randomized to either:
Duration: 5‐month treatment phase (+1 month placebo run‐in) Enrollment: 978 participants randomized Data obtained by authors from Janssen‐Ortho Inc. Participants were randomized to either:
Duration: 6‐month treatment phase (+1 month placebo run) Enrollment: 971 participants randomized |
|
| Outcomes | Primary outcome for pooled analysis:
Primary outcome for included studies include changes from baseline in:
Behavioral outcome for included studies include changes from baseline in:
|
|
| Notes | Study dates note reported. Sites of studies included in analyses include the USA, Canada, Great Britain, South Africa, Australia, and New Zealand. Specific site locations not disclosed. Corresponding author’s institution: Sunnybrook Health Sciences Centre, Toronto, Canada.This research was supported by a grant from the American Health Assistance Foundation‐Alzheimer's Disease Research Program (#A2003‐236) and by the Dean's Fund of the University of Toronto. All studies were supported by Jannssen. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Data were pooled from 2033 subjects...who had participated in one of three randomized, double‐blind placebo‐controlled trials". Comment: Probably done |
| Allocation concealment (selection bias) | Unclear risk | This information has not been made available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 of the studies included in this pooled analysis (Tariot 2000; Rockwood 2001) have each included a figure on participant flow. However, as the data from Janssen‐Ortho Inc. has not been made available, we are unsure about the risk of bias |
| Selective reporting (reporting bias) | Low risk | The authors reported on means ± standard deviations on each of the 12‐item NPI scores, at baseline and over treatment duration |
| Other bias | Low risk | No other identified biases |
Herrmann 2007.
| Methods | Randomized, double‐blind, placebo‐controlled cross‐over trial | |
| Participants |
|
|
| Interventions | Participants underwent a placebo washout of all psychotropic drugs based on a minimum of 5 half‐lives of the drug used Participants were then randomized to receive:
Loxapine 2.5 mg maximum 4 doses/week was available as a rescue medication Duration: 14 weeks (6‐week treatment phases of valproate or placebo + 2‐week placebo washout and tapering) Enrollment: 14 patients randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates not reported. Study site: 1) Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada; 2) North York General Hospital, Toronto, Canada. Additional data and information was provided by Herrmann and Lanctôt upon request. Funding provided by the Alzheimer’s Society of Canada (grant No. 01‐07). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were randomized to receive…” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “Patients were randomized to receive valproate liquid suspension or an identical placebo…” Comment: Probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “Twelve patients experienced at least one adverse event during valproate treatment, compared to 8 patients during placebo treatment. Although this difference is not statistically significant, the mean number of adverse events experienced with valproate treatment (4.15 +/‐ 3.67) was significantly greater than with placebo (1.23 +/‐ 1.69) treatment (Z = ‐2.82, p = 0.005).” Comment: Withdrawal numbers and adverse events per group are provided in the Results section and are not balanced between groups |
| Selective reporting (reporting bias) | Low risk | Mean change scores ± SD scores are provided in Table 1 for NPI‐agitation subcategory, total NPI, total CMAI and MMSE scores. NPI‐apathy mean change scores ± SD scores were calculated with data provided by authors upon request |
| Other bias | Low risk | No other identified biases |
Herrmann 2008.
| Methods | Randomized, double‐blind, placebo‐controlled, cross‐over trial | |
| Participants |
|
|
| Interventions | Participants took a dextroamphetamine (D‐amph) challenge test (10 mg D‐amph orally). Following up to a 1‐week washout phase, participants were randomized to receive either: ‐ methylphenidate (initiated at 5 mg orally twice/day for 3 days and increased to 10 mg orally twice/day for 11 days) ‐ placebo Duration: 5 weeks (2‐week treatment phases with a 1‐week placebo washout between phases) + at least 1 week washout after the D‐amph challenge test Enrollment: 13 participants randomized |
|
| Outcomes | Primary:
Other:
|
|
| Notes | Study dates: October 2003 ‐ October 2006. Study site: Sunnybrook Health Sciences Centre, Toronto, Canada. This research was supported by a grant from the American Health Assistance Foundation‐Alzheimer’s Disease Research Program (#A2003‐236) and by the Dean’s Fund of the University of Toronto. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “We conducted a double‐blind, randomized, placebo‐controlled crossover trial...” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “Patients were randomized to receive methylphenidate or an identical placebo...” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind…” Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “Eleven of the 13 patients completed the study, with both dropouts occurring during the methylphenidate treatment phase. One dropout completed all placebo assessments and the baseline methylphenidate assessment, which was carried forward for the efficacy analysis. The second patient dropped out after 8 days of methylphenidate treatment but completed a retrieved dropout assessment. This patient did not participate in the placebo phase. Results are therefore available for 13 patients treated with methylphenidate and 12 patients treated with placebo.” Comment: Though no participant flow diagram was provided in text, withdrawal information and reasoning described in text |
| Selective reporting (reporting bias) | Low risk | Table 1 provides means ± standard deviations for treatment change scores (end of treatment‐ baseline) for AES total, NPI apathy, NPI total and MMSE for participants during the methylphenidate and placebo treatment phases |
| Other bias | Low risk | No other identified biases |
Herrmann 2016.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 8 weeks Enrolment: 40 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates: July 2010 ‐ September 2015. Study sites: 1) Sunnybrook Health Sciences Centre, Toronto, Canada; 2) North York General Hospital, Toronto, Canada. Additional data provided by Herrmann et al. This study was funded by the Alzheimer's Society of Canada (#:12‐74) and the Coleman Fund (internal funding) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “...patients were randomized with a 1:1 balanced by ChEI to continue receiving their ChEI (continuation)...or to receive an identical‐looking placebo substitution.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “Randomization was completed independently by the pharmacy...in permuted blocks using a computer‐generated code.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: 'Patients, family members, nurses, clinicians, outcome assessors, and investigators were unaware of treatment group assignments or block size'. Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The reasons for withdrawal were provided in the participant flow diagram (figure 1). Number of early terminations and time to early termination were balanced between groups |
| Selective reporting (reporting bias) | Low risk | Mean and ± SD of the baseline, endpoint and change scores of all primary and secondary outcome measures were reported in table 3 |
| Other bias | Unclear risk | This was a discontinuation study completed in people who had been receiving long‐term ChEI treatment |
Kaufer 1998.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 26 weeks Enrollment: 408 patients randomized, 393 were included in the analysis as they were a part of the valid intention‐to‐treat population. |
|
| Outcomes | Primary:
|
|
| Notes | Study dates not reported. 25 sites in the USA. Specific site locations not disclosed. Corresponding author’s institution: University of Pittsburgh School of Medicine, Pittsburgh, PA, USA. No funding support reported in paper. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Subjects were randomized to either placebo...or metrifonate...treatment groups'. Comment: Probably done |
| Allocation concealment (selection bias) | Unclear risk | This information has not been made available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: '...double‐blind...' Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: '...double‐blind...' Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | This information has not been made available |
| Selective reporting (reporting bias) | High risk | LSM changes in NPI total and NPI‐subitem scores were provided in table 1. As LSM are adjusted for covariates, there is a risk of bias. Additionally, no standard error or standard deviation was reported in this paper. As a result, the authors of this meta analysis computed a standard deviation based on other studies (Raskind 1999) investigating the use of metrifonate in people with AD |
| Other bias | Low risk | No other identified biases |
Lanctôt 2002.
| Methods | Multicentre, double‐blind, randomized, placebo‐controlled cross‐over study | |
| Participants |
|
|
| Interventions |
Participants were then randomized to:
Duration: 9‐week treatment phase (4 weeks of first treatment phase + 1 week placebo washout, and cross over to 4 weeks of second treatment phase) + approximately 1‐week placebo run‐in Enrollment: 22 participants randomized |
|
| Outcomes | The following are primary outcomes: Behavior:
Function:
Cognition:
|
|
| Notes | Study dates not provided. Study sites: 1) Sunnybrook Health Sciences Centre, Toronto, Canada; 2) Baycrest Centre for Geriatric Care, Toronto, Canada; 3) North York General Hospital, Toronto, Canada. Additional data requested, and provided by Dr. Krista Lanctôt.This study was funded by Physicians' Services Incorporated Foundation (96‐06), Alzheimer Society of Canada Research Program, and Kunin Lunenfeld Applied Research Unite of Baycrest Centre for Geriatric Care | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...administered in a randomized, double‐blind trial." Comment: Probably done |
| Allocation concealment (selection bias) | Unclear risk | This information has not been made available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of those 26, three patients dropped out before randomization and one
dropped out because the caregiver withdrew consent. Of the remaining 22 patients, one was withdrawn due to severe antipsychotic withdrawal dyskinesia and a fall shortly after randomization (not included in efficacy analyses). Therefore, there were 21 patients who completed the entire study." Comment: Although no participant flow diagram was included, attrition was described in detail in the paper |
| Selective reporting (reporting bias) | Low risk | Lanctôt 2002 reported on treatment responders in this paper. Dr. Lanctôt provided relevant data for this meta‐analysis. We were able to extract the mean ± standard deviations, and frequencies for all relevant outcome measures |
| Other bias | Low risk | No other identified biases |
Morris 1998.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 26‐week double‐blind period (+ 2‐week screening period at beginning of the study + follow‐up visit at 8 weeks post‐treatment; 36 weeks total) Enrolment: 408 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates not reported. 24 sites in the USA included. Specific site locations not disclosed. Corresponding author’s institution: Washington University, St. Louis, MO, USA. The data in this report were collected from protocol D95‐018, sponsored by Bayer Corporation. For the purposes of this meta‐analysis, the authors have collected information from the double‐blind phase only. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At the time of enrolment, the 408 patients in this study were randomized to the placebo (N=135) or the metrifonate (N=237) group..." Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: "…according to a randomization code with blocks of six generated by computer at Bayer Corporation…" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “...double‐blind...” “Only the statistician…had access to the randomization code…” Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “...double‐blind...” “…investigators were masked as to random code assignment…” Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The reasons for withdrawal were provided in the participant flow diagram (figure 1) |
| Selective reporting (reporting bias) | High risk | LSM changes in NPI total and NPI‐subitem scores were extracted from figure 4. As LSM are adjusted for covariates, there is a risk of bias. Additionally, no standard error or standard deviation was reported in this paper. As a result, the authors of this meta‐analysis computed a standard deviation based on other studies (Raskind 1999) investigating the use of metrifonate in people with AD |
| Other bias | Low risk | No other identified biases |
MSAD trial.
| Methods | Multi‐center, randomized, double‐blind, placebo controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 24 weeks Enrolment: 290 participants randomized |
|
| Outcomes | Outcomes were obtained from Gauthier 2002:
Primary:
|
|
| Notes | We have selected Gauthier 2002 as the primary paper, but this group (MSAD investigators) have also reported on the study in two other published papers (Feldman 2001; Feldman 2005). Study dates not reported. 32 sites including 22 in Canada, 6 in Australia, and 4 in France. Specific site locations not disclosed. Corresponding author’s: McGill Centre for Studies in Aging, Montreal, Canada. The results of this study are supported by Pfizer, Inc. (New York, NY) and Eisai, Inc. (Teaneck, NJ). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...patients...were randomized to receive...". Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote from Feldman et al "At baseline, eligible patients were randomized in a 50/50 split using a computerized randomization schedule..." Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Quote from Feldman et al "Blinding was established with identical film‐coated tablets within a blister packaged card." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study withdrawals and reason for withdrawals have been reported in Figure 1 in Feldman 2001, and appear to be balanced between groups |
| Selective reporting (reporting bias) | High risk | "The outcome measure of interest was 12‐item Neuropsychiatric Inventory (NPI)." The authors reported on baseline least‐square means (LSM) ± standard errors on each of the 12‐item NPI scores. As LSM are adjusted for covariates, there is a risk of bias. They also included LSM change scores in figure 1. However, they did not include standard errors of these mean change scores. As such, we approximate standard deviation of change scores by calculating the standard deviation from the standard error of baseline scores |
| Other bias | Low risk | No other identified biases |
Padala 2017.
| Methods | Randomized, double‐blind, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Participants were randomized to receive either:
Duration: 12‐week treatment period (+ 2 week discontinuation phase) Enrollment: 60 participants randomized initially |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates: August 2007 ‐ June 2010. Study site: VA Medical Center, Omaha, NE, USA. This study was funded by a VA Merit Review Entry Program grant to Dr. Prasad Padala | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “…randomized double‐blind, placebo‐controlled trial...” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: "...randomized to methylphenidate (N=30) or placebo (N=30) groups using a random block design developed by a statistician using sealed envelopes." Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “…double‐blind…” Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “One subject dropped off from the placebo group.” Comment: Although no participant flow diagram was provided in text, withdrawal information and reasoning described in text |
| Selective reporting (reporting bias) | Low risk | The authors reported mean ± standard deviations for apathy scores in each intervention arm |
| Other bias | Low risk | No other identified biases |
Raskind 1999.
| Methods | Multicenter, randomized, double‐blind, placebo controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 26 weeks (+ 8‐week post‐treatment follow‐up visit) Enrollment: 264 patients randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates not reported. Multicenter study in the USA. Specific number and site locations not disclosed. Corresponding author’s institution: Northwest Mental Illness Research, Education and Clinical Center, Washington, DC, USA.The data in this report were collected from protocol D96‐010, sponsored by Bayer Corporation. For the purposes of this meta‐analysis, we have collected information from the double‐blind phase only. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: '...The 264 patients enrolled in this study were randomly assigned to the placebo...or the metrifonate group...'Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: '...patients...were randomly assigned...according to a computer‐generated randomization code..." Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: '...double‐blind...'. Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: 'The investigators were blinded as to random code assignment' Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The reasons for withdrawal were provided in text (page 322) |
| Selective reporting (reporting bias) | High risk | LSM changes in NPI total and NPI‐subitem scores were provided in figure 2. As LSM are adjusted for covariates, there is a risk of bias. SD was computed from provided SE of change scores |
| Other bias | Low risk | No other identified biases |
Rosenberg 2013.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Participants were randomized to receive either:
Duration: 6 weeks Enrollment: 60 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates: June 2010 ‐ August 2012. Study sites: 1) Johns Hopkins University Baltimore, MD, USA; 2) Medical University of South Carolina, Charleston, SC, USA; 3) Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada. Funding was provided by the National Institute on Aging (R01 AG033032‐01 and 1 K08 AG029157‐01A1). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “…randomized, double‐blind, placebo‐controlled multicenter trial...” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “The randomization scheme, stratified by clinical center with permuted length blocks, assigned participants to methylphenidate or placebo in a 1:1 ration. The coordinating center generated the treatment assignment schedule using a documented, auditable SAS program.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Study drug was supplied as identical‐appearing capsules containing either 5 mg methylphenidate or lactose (placebo).” Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The time to early termination did not differ significantly by group". Comment. Probably done. Additionally, participant flow, CONSORT diagram is provided in figure 1 |
| Selective reporting (reporting bias) | Low risk | Table 2 provides means ± standard errors for measures for the scores, change, and treatment effects of apathy (AES, ADCS‐CGI‐C and NPI) at 6 weeks of methylphenidate and placebo groups |
| Other bias | Low risk | No other identified biases |
Ruths 2008.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Participants were randomly assigned to either:
Duration: 4‐week intervention Enrollment: 55 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates not reported. 13 sites in Bergen and Oslo, Norway. Specific site locations not disclosed. Corresponding author’s institution: University of Kalfarveien, Bergen, Norway. Funding: No funding information provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Participants were consecutively assigned to antipsychotic drug discontinuation (intervention group, IG) or no discontinuation (reference group, RG) by means of computer generated, random, permuted blocks of four.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “All study medications were provided by an independent pharmacy… to maintain blindness.”,“…patients received identically looking capsules…” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was a multicenter double‐blind, controlled four week intervention." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "…double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “Seven patients completed the study prematurely (IG, n = 4; RG, n = 3; X2 = 0.20, p = 0.70), due to un‐blinding for randomization code (IG, n = 1; RG, n = 2), behavioural deterioration (IG, n = 2), restless legs (IG, n = 1) or delirium (RG, n = 1).” Comment: Study withdrawals and reason for withdrawals are described and appear to be balanced between groups |
| Selective reporting (reporting bias) | Low risk | The authors reported the number of IG participants still on antipsychotics at study completion. Mean NPI total score difference were provided from baseline to Week 4. Changes in behavioral symptoms between groups are presented in Table 2. Means ± standard deviations of differences in change in BPSD between groups are presented in Table 3 for NPI total and factor scores. Additional information regarding NPI‐subscores were requested by the authors, and were provided by Dr. Sabine Ruths |
| Other bias | Unclear risk | This is a discontinuation study |
Semgacestat trial.
| Methods | Multinational, randomized, double‐blind, placebo‐controlled trial | |
| Participants | Participant information obtained from Doody 2013:
|
|
| Interventions | Participants were randomized to receive:
Duration: 76‐week treatment phase Enrolment: 1537 patients randomized |
|
| Outcomes | Outcomes were obtained from Rosenberg 2016:
Primary:
Other:
|
|
| Notes | Study dates: March 2008 ‐ May 2011. Multicenter study in the USA. Specific number and site locations not disclosed. Although we have chosen Rosenberg 2016 as the primary paper, additional information regarding participant population was obtained from Doody 2013. Funding provided by Eli Lilly and the University of California at San Diego (the latter as a fiduciary for the Alzheimer’s Disease Cooperative Study), a clinical trials consortium established by the National Institute on Aging. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “participants were randomly assigned to receive…” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | This information has not been made available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “Adverse events were more common in the two semagacestat groups than in the placebo group…The percentage of patients who discontinued the study drug because of adverse events was higher with semagacestat than with placebo (26% with 100 mg and 30% with 140 mg vs. 11% with placebo, P>0.001 for both comparisons).” This quote is from Doody 2013 Comment: Withdrawal percentages by group are provided in Doody 2013 and are not balanced between groups. AEs are presented in Table 3 |
| Selective reporting (reporting bias) | Low risk | Change in apathy subscores are presented by treatment group in Figure C on p. 377 of Rosenberg 2016, but means ± SD scores needed to be computed by the authors of this meta‐analysis |
| Other bias | Low risk | No other identified biases |
Sival 2002.
| Methods | Randomized, double‐blind, placebo‐controlled cross‐over trial | |
| Participants |
|
|
| Interventions | Participants were randomized to receive:
Duration: 8 weeks (1‐week baseline + 3‐week placebo period + 1‐week washout + 3‐week treatment phase with sodium valproate). Extension of the baseline period was allowed once for 1 week in participants who did not show a score ≥ 3 on one of the items of the SDAS‐9 Enrollment: 42 participants randomized |
|
| Outcomes | Primary:
Secondary:
Other:
|
|
| Notes | Study dates not reported. Study site: Parnassia Psycho Medical Center, The Hague, The Netherlands. Funding provided by a grant from the Van Helten Foundation, Royal Netherlands Academy of Arts and Sciences, Amsterdam, The Netherlands (grant number SHV94/AANV/5), a grant from the National Fund for Mental Health, Utrecht, The Netherlands (grant number 4145), and a grant from the Stichting tot Steun VCVGZ, Bennekom, The Netherlands (grant number ST07064BB. VE) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The sequence of the treatment periods was assigned at random. The code was not accessible for the investigators.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: “During the ‘treatment period with placebo’ and during the wash‐out period a placebo suspension was given, identical to the active medication in appearance, quantity, smell and taste.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “…there were three drop‐outs. One patient had high fever of unknown origin during treatment with placebo, one patient was hit by a stroke during treatment with placebo, and one patient broke his hip during the wash‐out period. None of the dropouts could be associated with the intake of sodium valproate.” Comment: Withdrawal numbers and reasons for withdrawal are provided in the Results section |
| Selective reporting (reporting bias) | Low risk | Mean change scores ± SD scores are presented in Table 2, demonstrating the effects of sodium valproate compared to placebo on aggressive behavior and other types of disturbed behavior (including the GIP apathetic behavior subscore) |
| Other bias | Low risk | No other identified biases |
Sultzer 2008.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial | |
| Participants |
|
|
| Interventions | Participants were randomized initially (2:2:2:3 ratio) to either:
Treating physician selected the number of low‐ or high‐dose capsules for initial treatment and could adjust the dosage, as indicated clinically, over 36 weeks of trial. At any time after the first 2 weeks of treatment, the clinician could discontinue the initially‐assigned (Phase 1) medication based on their clinical judgment. Phase 1 would end and the participant could enter Phase 2 and be assigned randomly to masked treatment with an atypical antipsychotic medication not assigned to them in Phase 1 or with citalopram. Participants could also go directly to an open‐choice treatment Duration: Phase 1: 12 weeks; Phase 2: 24 weeks (36 weeks total) Enrolment: 421 participants randomized |
|
| Outcomes | Psychiatric and behavioral symptoms:
Cognitive skills, functional abilities, care needs and quality of life:
|
|
| Notes | For the purposes of this meta‐analysis, data from Phase 1 of the study were used. Phase 2 was not included as there was no placebo control. Study dates: March 2001 ‐ October 2004. 42 sites included. Site locations not disclosed. Principal Investigator institutions: University of Southern Carolina, Columbia, SC, USA; University of Rochester, Rochester, NY, USA. Funding was provided by the National Institute of Mental Health (N01 MH9001) and in part by the Department of Veterans Affairs. Astra‐Zeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutical and Eli Lilly provided medications for the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “This protocol is fundamentally a randomized‐treatment assignment…” Comment: Probably done. This quote is from Schneider 2003, where the CATIE‐AD research design and methods were originally described |
| Allocation concealment (selection bias) | Low risk | Quote: “Medication has been prepared into identically appearing ‘low strength’ capsules containing risperidone 0.5 mg, olanzapine 2.5 mg, quetiapine 25mg, citalopram 10 mg or placebo, or ‘higher strength’ capsules containing 1 mg,5 mg, 50 mg, 20 mg or placebo, respectively, in order to preserve the blind.” Comment: Probably done. This quote is from Schneider 2003 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double blind..." Comment: Probably done. This quote is from Schneider 2003 |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done. This quote is from Schneider 2003 |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote:“There were no significant overall differences among treatment groups with regard to the time to discontinuation of treatment for any reason” “There were no significant differences among the groups with regards to the proportion of patients who had at least one serious adverse event and the proportion who had any adverse event” Comment: Time to treatment discontinuation and adverse events are reported in Tables 2 and 3 and appear to be balanced between groups. These quotes are from Schneider 2006, where the CATIE‐AD time to treatment discontinuation and adverse events data are originally described |
| Selective reporting (reporting bias) | Low risk | Quote: “Two sets of clinical outcomes were measured: 1) psychiatric and behavioural symptoms… 2) cognition, functional skills, care needs and quality of life” Comment: Baselines scores on clinical measures provided, along with mean change ± standard deviation scores for clinical symptoms from baseline to last observation in Phase 1. Mean change ± standard deviations on clinical symptom measures between baseline and treatment week 12 are provided for cognitive measures. |
| Other bias | Low risk | No other identified biases |
Tariot 2001.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study | |
| Participants |
|
|
| Interventions | Participants were randomized to either:
Duration: 24 weeks Enrolment: 208 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates not reported. 27 sites across the USA. Specific site locations not disclosed. Corresponding author’s institution: Departments of Psychiatry, Medicine and Neurology, University of Rochester Medical Center, Monroe Community Hospital, Rochester, NY, USA. The results of this study are supported by Pfizer, Inc. (New York, NY) and Eisai, Inc. (Teaneck, NJ). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...patients...were randomized..." Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | Quote: "...randomized in blocks of four, using a computerized randomization schedule..." Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Blinding was achieved using identical appearing film‐coated tablets of donepezil and placebo..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study withdrawals and reason for withdrawals have been reported in the first paragraph of the Results section of Tariot 2001 |
| Selective reporting (reporting bias) | High risk | LSM changes in NPI‐subitem scores were provided in figure 1. As LSM are adjusted for covariates, there is a risk of bias. SD was computed from provided SE of change scores |
| Other bias | Low risk | No other identified biases |
Tariot 2011.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled, flexible‐dose trial | |
| Participants |
|
|
| Interventions | Participants were assigned to either:
Dose reduction was permitted if clinically warranted, and the target dose could be resumed if appropriate. Adherence of 80% was required Duration: 24‐month double‐blind treatment phase + 2‐month single‐blind placebo treatment period Enrolment: 313 participants randomized |
|
| Outcomes | Primary:
Secondary:
Other:
Safety and tolerability:
Volumetric Magnetic Resonance Image:
|
|
| Notes | Study dates: October 2003 ‐ December 2009. 46 sites included. Site locations not disclosed. Study Director site: University of Rochester Medical Center, Rochester, NY, USA. Data for this study were obtained from the University of California, San Diego Alzheimer's Disease Cooperative Study Legacy Database. Funding provided by National Institute on Aging (U01AG010483). Additional support provided by a research grant and material support from Abbott Laboratories (NCT00071721). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were assigned to 1 of 2 treatment groups in permuted blocks of 4, according to a randomization list created and maintained by the ADCS Data Core.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | “The trial used a 125‐mg enteric‐coated extended‐release divalproex sodium formulation or identical‐appearing placebo…” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “In the valproate group, 61.40% discontinued treatment prematurely, and in the placebo group, 60.6% did so; reasons are shown in Figure 1.” Comment: Study withdrawals and reason for withdrawals have been reported in Figure 1 and all AEs experienced in each group are outlined in Table 3 |
| Selective reporting (reporting bias) | Low risk | The authors reported means ± SD scores of each outcome over time for each study visit in Table 3. Additional data were provided upon request |
| Other bias | Low risk | No other identified biases |
Trzepacz 2013.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial. | |
| Participants |
|
|
| Interventions | Participants were randomly assigned to either:
After 1‐week of treatment twice a day with either 3 mg mibampator or placebo, a one‐time dose reduction to 1 mg twice daily due to intolerability was permitted, which remained their dose for the remainder of the study Duration: 12‐week double‐blind treatment phase (+ 3 ‐ 28 day screening period and 1‐week single‐blind washout) Enrolment: 132 participants randomized |
|
| Outcomes | Primary:
Secondary:
|
|
| Notes | Study dates: February 2009 ‐ June 2011. Multicentre study in USA. Specific number and site location details are not disclosed. Research supported by Eli Lilly and Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The interactive voice response system (IVRS) was used to assign blisterpacks containing double‐blind study drug to each patient.” Comment: Probably done |
| Allocation concealment (selection bias) | Low risk | “The interactive voice response system (IVRS) was used to assign blisterpacks containing double‐blind study drug to each patient.” Comment: Probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "…double‐blind..." Comment: Probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "…double‐blind..." Comment: Probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “There were no significant differences between groups for SAEs, discontinuation due to AEs, TEAEs or TEAEs possibly related to study drug as deemed by the investigator...” Comment: Authors describe all SAEs and reasons for discontinuation reasons due to AE for each treatment group in the ‘Safety Evaluation’ section p. 8 |
| Selective reporting (reporting bias) | High risk | The authors used MMRM analysis to assess the primary outcome, NPI‐4‐A/A least square mean change from baseline after treatment, (Figure 2), as well as secondary efficacy measures including FrSBe total and subscale least square means change from baseline after treatment (Figure 3. |
| Other bias | Low risk | No other identified biases |
AChEI: anti‐cholinesterase inhibitor; AD: Alzheimer’s disease; ADAS‐Cog: Alzheimer’s Disease Assessment Scale‐Cognitive subscale; ADAS‐Cog11: Alzheimer’s Disease Assessment Scale‐ 11‐item cognitive subscale; ADAS‐Cog14: Alzheimer’s Disease Assessment Scale‐ 14‐item cognitive; ADAS‐Noncog: Alzheimer’s Disease Assessment Scale‐Noncognitive subscale; ADCS: Alzheimer’s Disease Cooperative Study; ADCS‐ADL: Alzheimer’s Disease Cooperative Study – Activities of Daily Living; ADCS‐ADL‐sev: Alzheimer’s Disease Cooperative Study‐Activities of Daily Living severity scale; ADCS‐CGI‐C: Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; ADLQ: Activities of Daily Living Questionnaire; ADRQL: Alzheimer’s Disease Related Quality of Life; AE: adverse events; AES: Apathy Evaluation Scale; AIMS: Abnormal Involuntary Movement Scale; ARCI: Addiction Research Centre Inventory; BEHAVE‐AD: Behavioural Pathology in Alzheimer’s Disease Rating Scale; BID: twice daily; BPRS: Brief Psychiatric Rating Scale; BPSD: behavioral and psychological symptoms of dementia; CAS: Caregiver Activity Scale; CATIE‐AD: Clinical Antipsychotic Trials of Intervention Effectiveness‐Alzheimer’s Disease trial; CDR‐SOB: Clinical Dementia Rating sum of boxes; CGI: Clinical Global Impression scale; CGI‐C: Clinical Global Impression‐Change scale; CGI‐S: Clinical Global Impression‐Severity scale; CGI‐S‐AA: Clinical Global Impression‐Severity of Symptoms of Agitation/Aggression; CGI‐S‐GF: Clinical Global Impression‐Severity of Symptoms of Global Functioning; ChEI: Cholinesterase inhibitors; CIBIC‐plus: Clinician’s Interview‐Based Impression of Change plus Caregiver Input; CMAI: Cohen‐Mansfield Agitation Inventory; CPT: Conners’ Continuous Performance Task; CSDD: Cornell Scale for Depression in Dementia; CT: Computerized Tomography; DAD: Disability Assessment in Dementia; DAFS: Direct Assessment of Functional Status; D‐amph: dextroamphetamine challenge; DS: Dependence Scale; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision; FAST: Functional Assessment Staging of Alzheimer’s Disease; FrSBe: Frontal Systems Behaviour Scale; GDS: Global Deterioration Scale; HIS: Hachinski Ischemic Score; IADL: Instrumental Activities of Daily Living; ICD‐10: International Classification of Diseases‐ Tenth Revision; IG: intervention group; IR: immediate release; IVRS: interactive voice response system; kg: kilogram; lbs: pounds; L.L.F.: study neuropsychologist; LSM: least‐square means; LTC: long‐term care; mg: milligram; MMRM: mixed‐effects model repeated measures analysis; MMSE: Mini‐Mental State Exam; MRI: Magnetic Resonance Imaging; MSAD: moderate‐to‐severe AD; N: number; NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association; NJ: New Jersey; NPI: Neuropsychiatric Inventory; NPI‐4 A/A: Neuropsychiatric Inventory‐4 domain subscale, which combines the following domains: agitation/aggression, aberrant motor behavior, irritability/emotional lability and disinhibition; NPI‐10: Neuropsychiatric Inventory‐10 domains; NPI‐NH: Neuropsychiatric Inventory‐Nursing Home Version; NRS: Neurobehavioural Rating Scale; NY: New York; PO: by mouth, in Latin per os; POMA: Performance‐Oriented Mobility Assessment‐II; POMS: Profile of Mood States; QOL‐AD: Quality of Life‐AD; QUALID: Quality of Life in Late‐Stage Dementia; RG: reference group; SAEs: serious adverse events; *SAS: Simpson‐Angus Scale; SAS: SAS version 9.2 (SAS Institute Inc, Cary, North Carolina); SDAS: Social Dysfunction and Aggression Scale; SD: standard deviation; SE: standard error; SIB: Severe Impairment Battery; sMMSE: Standardized Mini‐Mental State Exam; SNRI: serotonin and norepinephrine reuptake inhibitor; S.S.: study neurologist; SSRI: selective serotonin reuptake inhibitor; TEAEs: treatment emergent adverse events; ZBI: Zarit Burden Interview
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ballard 2004 | Apathy not reported |
| Ballard 2005 | Apathy not investigated nor reported |
| Ballard 2008 | Apathy not investigated nor reported |
| Banerjee 2011 | Apathy not reported |
| Breder 2004 | Apathy not investigated nor reported |
| Bridges‐Parlet 1997 | Apathy not investigated nor reported |
| Brodaty 2003 | Apathy not investigated nor reported |
| Burns 1999 | Apathy not investigated nor reported |
| Cohen‐Mansfield 1999 | Apathy not reported |
| De Deyn 1999 | Apathy not investigated nor reported |
| De Deyn 2005 | Apathy not reported |
| De Vasconcelos 2007 | Apathy not investigated nor reported |
| Deberdt 2005 | Apathy not reported |
| Devanand 2011 | Apathy not reported |
| Devanand 2012 | Apathy not reported |
| Findlay 1989 | Apathy not reported |
| Holmes 2004 | Apathy not reported |
| Howard 2012 | Apathy not reported |
| Johannsen 2006 | Apathy not reported |
| Katz 1999 | Apathy not investigated nor reported |
| Lyketsos 2003 | Apathy not reported |
| Magai 2000 | Apathy not investigated nor reported |
| Mintzer 2006 | Apathy not investigated nor reported |
| Nyth 1992 | Apathy not investigated nor reported |
| Petracca 1996 | Apathy not investigated nor reported |
| Petracca 2001 | Apathy not investigated nor reported |
| Raskind 2000 | Apathy not investigated nor reported |
| Reifler 1989 | Apathy not investigated nor reported |
| Rosenberg 2010 | Apathy not reported |
| Rosler 1999 | Apathy not investigated nor reported |
| Roth 1996 | Apathy not investigated nor reported |
| Satterlee 1995 | Apathy not investigated nor reported |
| Schneider 2006 | Apathy not reported |
| Seltzer 2004 | Apathy not reported |
| Street 2000 | Apathy not reported |
| Streim 2004 | Apathy not reported |
| Tariot 2004a | Apathy not investigated nor reported |
| Tariot 2004b | Apathy not investigated or reported |
| Van Reekum 2002 | Apathy not reported |
| Wilcock 2000 | Apathy not investigated nor reported |
| Winblad 2001 | Apathy not reported |
Differences between protocol and review
In the original protocol, primary and secondary outcome measures were going to be analyzed for all drug comparisons. However, many studies investigated apathy as a secondary outcome measure only, and thus did not directly target this symptom in their investigation. Furthermore, as there were a limited number of studies within each drug comparison, investigating apathy as a secondary outcome measure, meaningful results on safety, overall NPS, cognition, function, clinical deterioration, and dropouts due AEs could not be interpreted. As such, we have created two Objectives for this meta‐analysis. Objectives 1 and 2 investigated the efficacy of pharmacotherapies on apathy in studies which investigated this as a primary or a secondary outcome measure respectively.
Contributions of authors
Myuri T Ruthirakuhan (MTR) performed protocol development, correspondence, drafting review versions, selection of randomized controlled trials (RCTs), extraction of data; assessing risk of bias, data entry, data analysis, GRADE, interpretation of data/analyses. Nathan Herrmann (NH) performed supervision and development of the protocol and review versions. Eleanor H Abraham (EHA) assisted with selection of RCTs, extraction of data, assessing risk of bias, and drafting characteristics of included studies. Sarah Chan (SC) assisted with selection of RCTs and extraction of data. Krista L Lanctot (KLL) performed supervision and development of the protocol and review versions.
Sources of support
Internal sources
No sources of support supplied
External sources
-
NIHR, UK.
This review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Dementia and Cognitive Improvement group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health
Declarations of interest
KLL is the executive director of the Medical Outcomes and Research in Economics (MORE®) Research group. She currently receives research funding from AbbVie Laboratories, Novartis Canada, F. Hoffman‐La Roche, and Lundbeck Canada, is funded by grants from the National Institute of Health, the Canadian Institutes of Health Research, Brain Canada, the Alzheimer Society of Canada, the Heart and Stroke Foundation, Weston Brain Institute and the Alzheimer’s Disease Discovery Fund.
NH receives research funding from Lundbeck Canada Inc., Roche, Transition Therapeutics, and holds grants from the National Institute of Health, the Canadian Institutes of Health Research, the Alzheimer Society of Canada, the Alzheimer’s Drug Discovery Foundation, the Heart and Stroke Foundation and the Physicians’ Services Incorporated Foundation. He has received consultation fees from Eli Lilly and AbbVie.
MTR is funded by a CIHR Doctoral Research Award (the Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award). EHA does not have any known conflicts of interest. SC does not have any known conflicts of interest.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
CitAD trial {published data only}
- Leonpacher AK, Peters ME, Drye LT, Makino KM, Newell JA, Devanand DP, et al. Effects of citalopram on neuropsychiatric symptoms in Alzheimer's dementia: evidence from the CitAD Study. American Journal of Psychiatry 2016;173(5):473‐80. [PUBMED: 27032628] [DOI] [PubMed] [Google Scholar]
- Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z, et al. CitAD Research Group. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. The Journal of the American Medical Association 2014;311(7):682‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
De Deyn 2004 {published data only}
- Deyn PP, Carrasco MM, Deberdt W, Jeandel C, Hay DP, Feldman PD, et al. Olanzapine versus placebo in the treatment of psychosis with or without associated behavioral disturbances in patients with Alzheimer's disease. International Journal of Geriatric Psychiatry 2004;19(2):115‐26. [PUBMED: 14758577] [DOI] [PubMed] [Google Scholar]
Frakey 2012 {published data only}
- Frakey LL, Salloway S, Buelow M, Malloy P. A randomized, double‐blind, placebo‐controlled trial of modafinil for the treatment of apathy in individuals with mild‐to‐moderate Alzheimer's disease. Journal of Clinical Psychiatry 2012;73(6):796‐801. [PUBMED: 22687392] [DOI] [PubMed] [Google Scholar]
Herrmann 2005 {published data only}
- Herrmann N, Rabheru K, Wang J, Binder C. Galantamine treatment of problematic behavior in Alzheimer disease: post‐hoc analysis of pooled data from three large trials. American Journal of Geriatric Psychiatry 2005;13(6):527‐34. [PUBMED: 15956273] [DOI] [PubMed] [Google Scholar]
Herrmann 2007 {published data only}
- Herrmann N, Lanctôt KL, Rothenburg LS, Eryavec G. A placebo‐controlled trial of valproate for agitation and aggression in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders 2007;23(2):116‐9. [PUBMED: 17148938] [DOI] [PubMed] [Google Scholar]
Herrmann 2008 {published data only}
- Herrmann N, Rothenburg LS, Black SE, Ryan M, Liu BA, Busto UE, et al. Methylphenidate for the treatment of apathy in Alzheimer disease: prediction of response using dextroamphetamine challenge. Journal of Clinical Psychopharmacology 2008;28(3):296‐301. [PUBMED: 18480686] [DOI] [PubMed] [Google Scholar]
Herrmann 2016 {published data only}
- Herrmann N, O'Regan J, Ruthirakuhan M, Kiss A, Eryavec G, Williams E, et al. A randomized placebo‐controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. Journal of the American Medical Directors Association 2016;17(2):142‐7. [PUBMED: 26482056] [DOI] [PubMed] [Google Scholar]
Kaufer 1998 {published data only}
- Kaufer D. Beyond the cholinergic hypothesis: the effect of metrifonate and other cholinesterase inhibitors on neuropsychiatric symptoms in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders 1998;9 Suppl 2:8‐14. [PUBMED: 9718229] [DOI] [PubMed] [Google Scholar]
Lanctôt 2002 {published data only}
- Lanctôt KL, Herrmann N, Reekum R, Eryavec G, Naranjo CA. Gender, aggression and serotonergic function are associated with response to sertraline for behavioral disturbances in Alzheimer's disease. International Journal of Geriatric Psychiatry 2002;17(6):531‐41. [PUBMED: 12112177] [DOI] [PubMed] [Google Scholar]
Morris 1998 {published data only}
- Morris JC, Cyrus PA, Orazem J, Mas J, Bieber F, Ruzicka BB, et al. Metrifonate benefits cognitive, behavioral, and global function in patients with Alzheimer's disease. Neurology 1998;50(5):1222‐30. [PUBMED: 9595967] [DOI] [PubMed] [Google Scholar]
MSAD trial {published data only}
- Feldman H, Gauthier S, Hecker J, Vellas B, Subbiah P, Whalen E. A 24‐week, randomized, double‐blind study of donepezil in moderate to severe Alzheimer's disease. Neurology 2001;57(4):613‐20. [PUBMED: 11524468] [DOI] [PubMed] [Google Scholar]
- Feldman H, Gauthier S, Hecker J, Vellas B, Xu Y, Ieni JR, et al. Efficacy and safety of donepezil in patients with more severe Alzheimer's disease: a subgroup analysis from a randomized, placebo‐controlled trial. International Journal of Geriatric Psychiatry 2005;20(6):559‐69. [PUBMED: 15920715] [DOI] [PubMed] [Google Scholar]
- Gauthier S, Feldman H, Hecker J, Vellas B, Ames D, Subbiah P, et al. Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer's disease. International Psychogeriatrics 2002;14(4):389‐404. [PUBMED: 12670060] [DOI] [PubMed] [Google Scholar]
Padala 2017 {published data only}
- Padala PR, Padala KP, Lensing SY, Ramirez D, Monga V, Bopp MM, et al. Methylphenidate for apathy in community‐dwelling older veterans with mild Alzheimer's disease: a double‐blind, randomized, placebo‐controlled trial. American Journal of Psychiatry 2017;175:appiajp201717030316. [PUBMED: 28945120] [DOI] [PubMed] [Google Scholar]
Raskind 1999 {published data only}
- Raskind MA, Cyrus PA, Ruzicka BB, Gulanski BI. The effects of metrifonate on the cognitive, behavioral, and functional performance of Alzheimer's disease patients. Metrifonate Study Group. Journal of Clinical Psychiatry 1999;60(5):318‐25. [PUBMED: 10362441] [DOI] [PubMed] [Google Scholar]
Rosenberg 2013 {published data only}
- Rosenberg PB, Lanctôt KL, Drye LT, Herrmann N, Scherer RW, Bachman DL, et al. Safety and efficacy of methylphenidate for apathy in Alzheimer's disease: a randomized, placebo‐controlled trial. Journal of Clinical Psychiatry 2013;74(8):810‐6. [PUBMED: 24021498] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ruths 2008 {published data only}
- Ruths S, Straand J, Nygaard HA, Aarsland D. Stopping antipsychotic drug therapy in demented nursing home patients: a randomized, placebo‐controlled study‐‐the Bergen District Nursing Home study (BEDNURS). International Journal of Geriatric Psychiatry 2008;23(9):889‐95. [PUBMED: 18306150] [DOI] [PubMed] [Google Scholar]
Semgacestat trial {published data only}
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. New England Journal of Medicine 2013;369(4):341‐50. [PUBMED: 23883379] [DOI] [PubMed] [Google Scholar]
- Rosenberg PB, Lanctôt KL, Herrmann N, Mintzer JE, Porsteinsson AP, Sun X, et al. Changes in neuropsychiatric inventory associated with semagacestat treatment of Alzheimer's disease. Journal of Alzheimer's Disease 2016;54(1):373‐81. [PUBMED: 27567808] [DOI] [PubMed] [Google Scholar]
Sival 2002 {published data only}
- Sival RC, Haffmans PM, Jansen PA, Duursma SA, Eikelenboom P. Sodium valproate in the treatment of aggressive behavior in patients with dementia‐‐a randomized placebo controlled clinical trial. International Journal of Geriatric Psychiatry 2002;17(6):579‐85. [PUBMED: 12112183] [DOI] [PubMed] [Google Scholar]
Sultzer 2008 {published data only}
- Sultzer DL, Davis SM, Tariot PN, Dagerman KS, Lebowitz BD, Lyketsos CG, et al. Clinical symptom responses to atypical antipsychotic medications in Alzheimer's disease: phase 1 outcomes from the CATIE‐AD effectiveness trial. American Journal of Psychiatry 2008;165(7):844‐54. [PUBMED: 18519523] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tariot 2001 {published data only}
- Tariot PN, Cummings JL, Katz IR, Mintzer J, Perdomo CA, Schwam EM, et al. A randomized, double‐blind, placebo‐controlled study of the efficacy and safety of donepezil in patients with Alzheimer's disease in the nursing home setting. Journal of the American Geriatrics Society 2001;49(12):1590‐9. [PUBMED: 11843990] [PubMed] [Google Scholar]
Tariot 2011 {published data only}
- Tariot PN, Schneider LS, Cummings J, Thomas RG, Raman R, Jakimovich LJ, et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Archives of General Psychiatry 2011;68(8):853‐61. [PUBMED: 21810649] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trzepacz 2013 {published data only}
- Trzepacz PT, Cummings J, Konechnik T, Forrester TD, Chang C, Dennehy EB, et al. Mibampator (LY451395) randomized clinical trial for agitation/aggression in Alzheimer's disease. International Psychogeriatrics 2013;25(5):707‐19. [PUBMED: 23257314] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Ballard 2004 {published data only}
- Ballard CG, Thomas A, Fossey J, Lee L, Jacoby R, Lana MM, et al. A 3‐month, randomized, placebo‐controlled, neuroleptic discontinuation study in 100 people with dementia: the neuropsychiatric inventory median cutoff is a predictor of clinical outcome. Journal of Clinical Psychiatry 2004;65(1):114‐9. [PUBMED: 14744180] [DOI] [PubMed] [Google Scholar]
Ballard 2005 {published data only}
- Ballard C, Margallo‐Lana M, Juszczak E, Douglas S, Swann A, Thomas A, et al. Quetiapine and rivastigmine and cognitive decline in Alzheimer's disease: randomised double blind placebo controlled trial. The British Medical Journal (Clinical research ed.) 2005;330(7496):874. [PUBMED: 15722369] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ballard 2008 {published data only}
- Ballard C, Lana MM, Theodoulou M, Douglas S, McShane R, Jacoby R, et al. A randomised, blinded, placebo‐controlled trial in dementia patients continuing or stopping neuroleptics (the DART‐AD trial). PLoS Medicine 2008;5(4):e76. [PUBMED: 18384230] [DOI] [PMC free article] [PubMed] [Google Scholar]
Banerjee 2011 {published data only}
- Banerjee S, Hellier J, Dewey M, Romeo R, Ballard C, Baldwin R, et al. Sertraline or mirtazapine for depression in dementia (HTA‐SADD): a randomised, multicentre, double‐blind, placebo‐controlled trial. Lancet 2011;378(9789):403‐11. [PUBMED: 21764118] [DOI] [PubMed] [Google Scholar]
Breder 2004 {published data only}
- Breder C, Swanink R, Marcus R, Kostic D, Iwamoto T, Carson W, et al. Dose‐ranging study of aripiprazole in patients with dementia of Alzheimer's disease. Neurobiology of Aging 2004;25(2):S190. [Google Scholar]
Bridges‐Parlet 1997 {published data only}
- Bridges‐Parlet S, Knopman D, Steffes S. Withdrawal of neuroleptic medications from institutionalized dementia patients: results of a double‐blind, baseline‐treatment‐controlled pilot study. Journal of Geriatric Psychiatry and Neurology 1997;10(3):119‐26. [PUBMED: 9322135] [DOI] [PubMed] [Google Scholar]
Brodaty 2003 {published data only}
- Brodaty H, Ames D, Snowdon J, Woodward M, Kirwan J, Clarnette R, et al. A randomized placebo‐controlled trial of risperidone for the treatment of aggression, agitation, and psychosis of dementia. Journal of Clinical Psychiatry 2003;64(2):134‐43. [PUBMED: 12633121] [DOI] [PubMed] [Google Scholar]
Burns 1999 {published data only}
- Burns A, Rossor M, Hecker J, Gauthier S, Petit H, Moller HJ, et al. The effects of donepezil in Alzheimer's disease ‐ results from a multinational trial. Dementia and Geriatric Cognitive Disorders 1999;10(3):237‐44. [PUBMED: 10325453] [DOI] [PubMed] [Google Scholar]
Cohen‐Mansfield 1999 {published data only}
- Cohen‐Mansfield J, Lipson S, Werner P, Billig N, Taylor L, Woosley R. Withdrawal of haloperidol, thioridazine, and lorazepam in the nursing home: a controlled, double‐blind study. Archives of Internal Medicine 1999;159(15):1733‐40. [PUBMED: 10448776] [DOI] [PubMed] [Google Scholar]
Deberdt 2005 {published data only}
- Deberdt WG, Dysken MW, Rappaport SA, Feldman PD, Young CA, Hay DP, et al. Comparison of olanzapine and risperidone in the treatment of psychosis and associated behavioral disturbances in patients with dementia. American Journal of Geriatric Psychiatry 2005;13(8):722‐30. [PUBMED: 16085789] [DOI] [PubMed] [Google Scholar]
De Deyn 1999 {published data only}
- Deyn PP, Rabheru K, Rasmussen A, Bocksberger JP, Dautzenberg PL, Eriksson S, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology 1999;53(5):946‐55. [PUBMED: 10496251] [DOI] [PubMed] [Google Scholar]
De Deyn 2005 {published data only}
- Deyn P, Jeste DV, Swanink R, Kostic D, Breder C, Carson WH, et al. Aripiprazole for the treatment of psychosis in patients with Alzheimer's disease: a randomized, placebo‐controlled study. Journal of Clinical Psychopharmacology 2005;25(5):463‐7. [PUBMED: 16160622] [DOI] [PubMed] [Google Scholar]
Devanand 2011 {published data only}
- Devanand DP, Pelton GH, Cunqueiro K, Sackeim HA, Marder K. A 6‐month, randomized, double‐blind, placebo‐controlled pilot discontinuation trial following response to haloperidol treatment of psychosis and agitation in Alzheimer's disease. International Journal of Geriatric Psychiatry 2011;26(9):937‐43. [PUBMED: 21845596] [DOI] [PMC free article] [PubMed] [Google Scholar]
Devanand 2012 {published data only}
- Devanand DP, Mintzer J, Schultz SK, Andrews HF, Sultzer DL, Pena D, et al. Relapse risk after discontinuation of risperidone in Alzheimer's disease. New England Journal of Medicine 2012;367(16):1497‐507. [PUBMED: 23075176] [DOI] [PMC free article] [PubMed] [Google Scholar]
De Vasconcelos 2007 {published data only}
- Vasconcelos Cunha UG, Lopes Rocha F, Avila de Melo R, Alves Valle E, Souza Neto JJ, Mendes Brega R, et al. A placebo‐controlled double‐blind randomized study of venlafaxine in the treatment of depression in dementia. Dementia and Geriatric Cognitive Disorders 2007;24(1):36‐41. [PUBMED: 17495474] [DOI] [PubMed] [Google Scholar]
Findlay 1989 {published data only}
- Findlay DJ, Sharma J, McEwen J, Ballinger BR, MaClennan WJ, McHarg AM. Double‐blind controlled withdrawal of thioridazine treatment in elderly female inpatients with senile dementia. International Journal of Geriatric Psychiatry 1989;4(2):115‐20. [Google Scholar]
Holmes 2004 {published data only}
- Holmes C, Wilkinson D, Dean C, Vethanayagam S, Olivieri S, Langley A, et al. The efficacy of donepezil in the treatment of neuropsychiatric symptoms in Alzheimer disease. Neurology 2004;63(2):214‐9. [PUBMED: 15277611] [DOI] [PubMed] [Google Scholar]
Howard 2012 {published data only}
- Howard R, McShane R, Lindesay J, Ritchie C, Baldwin A, Barber R, et al. Donepezil and memantine for moderate‐to‐severe Alzheimer's disease. New England Journal of Medicine 2012;366(10):893‐903. [PUBMED: 22397651] [DOI] [PubMed] [Google Scholar]
Johannsen 2006 {published data only}
- Johannsen P, Salmon E, Hampel H, Xu Y, Richardson S, Qvitzau S, et al. Assessing therapeutic efficacy in a progressive disease: a study of donepezil in Alzheimer's disease. CNS Drugs 2006;20(4):311‐25. [PUBMED: 16599649] [DOI] [PubMed] [Google Scholar]
Katz 1999 {published data only}
- Katz IR, Jeste DV, Mintzer JE, Clyde C, Napolitano J, Brecher M. Comparison of risperidone and placebo for psychosis and behavioral disturbances associated with dementia: a randomized, double‐blind trial. Risperidone Study Group. Journal of Clinical Psychiatry 1999;60(2):107‐15. [PUBMED: 10084637] [DOI] [PubMed] [Google Scholar]
Lyketsos 2003 {published data only}
- Lyketsos CG, DelCampo L, Steinberg M, Miles Q, Steele CD, Munro C, et al. Treating depression in Alzheimer disease: efficacy and safety of sertraline therapy, and the benefits of depression reduction: the DIADS. Archives of General Psychiatry 2003;60(7):737‐46. [PUBMED: 12860778] [DOI] [PubMed] [Google Scholar]
Magai 2000 {published data only}
- Magai C, Kennedy G, Cohen CI, Gomberg D. A controlled clinical trial of sertraline in the treatment of depression in nursing home patients with late‐stage Alzheimer's disease. American Journal of Geriatric Psychiatry 2000;8(1):66‐74. [PUBMED: 10648297] [DOI] [PubMed] [Google Scholar]
Mintzer 2006 {published data only}
- Mintzer J, Greenspan A, Caers I, Hove I, Kushner S, Weiner M, et al. Risperidone in the treatment of psychosis of Alzheimer disease: results from a prospective clinical trial. American Journal of Geriatric Psychiatry 2006;14(3):280‐91. [PUBMED: 16505133] [DOI] [PubMed] [Google Scholar]
Nyth 1992 {published data only}
- Nyth AL, Gottfries CG, Lyby K, Smedegaard‐Andersen L, Gylding‐Sabroe J, Kristensen M, et al. A controlled multicenter clinical study of citalopram and placebo in elderly depressed patients with and without concomitant dementia. Acta Psychiatrica Scandinavica 1992;86(2):138‐45. [PUBMED: 1529737] [DOI] [PubMed] [Google Scholar]
Petracca 1996 {published data only}
- Petracca G, Teson A, Chemerinski E, Leiguarda R, Starkstein SE. A double‐blind placebo‐controlled study of clomipramine in depressed patients with Alzheimer's disease. Journal of Neuropsychiatry and Clinical Neurosciences 1996;8(3):270‐5. [PUBMED: 8854297] [DOI] [PubMed] [Google Scholar]
Petracca 2001 {published data only}
- Petracca GM, Chemerinski E, Starkstein SE. A double‐blind, placebo‐controlled study of fluoxetine in depressed patients with Alzheimer's disease. International Psychogeriatrics 2001;13(2):233‐40. [PUBMED: 11495397] [DOI] [PubMed] [Google Scholar]
Raskind 2000 {published data only}
- Raskind MA, Peskind ER, Wessel T, Yuan W. Galantamine in AD: A 6‐month randomized, placebo‐controlled trial with a 6‐month extension. The Galantamine USA‐1 Study Group. Neurology 2000;54(12):2261‐8. [PUBMED: 10881250] [DOI] [PubMed] [Google Scholar]
Reifler 1989 {published data only}
- Reifler BV, Teri L, Raskind M, Veith R, Barnes R, White E, et al. Double‐blind trial of imipramine in Alzheimer's disease patients with and without depression. American Journal of Psychiatry 1989;146(1):45‐9. [PUBMED: 2643356] [DOI] [PubMed] [Google Scholar]
Rosenberg 2010 {published data only}
- Rosenberg PB, Drye LT, Martin BK, Frangakis C, Mintzer JE, Weintraub D, et al. Sertraline for the treatment of depression in Alzheimer disease. American Journal of Geriatric Psychiatry 2010;18(2):136‐45. [PUBMED: 20087081] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosler 1999 {published data only}
- Rosler M, Anand R, Cicin‐Sain A, Gauthier S, Agid Y, Dal‐Bianco P, et al. Efficacy and safety of rivastigmine in patients with Alzheimer's disease: international randomised controlled trial. The British Medical Journal (Clinical research ed.) 1999;318(7184):633‐8. [PUBMED: 10066203] [DOI] [PMC free article] [PubMed] [Google Scholar]
Roth 1996 {published data only}
- Roth M, Mountjoy CQ, Amrein R. Moclobemide in elderly patients with cognitive decline and depression: an international double‐blind, placebo‐controlled trial. British Journal of Psychiatry 1996;168(2):149‐57. [PUBMED: 8837903] [DOI] [PubMed] [Google Scholar]
Satterlee 1995 {published data only}
- Satterlee WG, Reams SG. Burns PR, Hamilton S, Tran PV, Tollefson GD. A clinical update on olanzapine treatment in schizophrenia and in elderly Alzheimer's disease patients. Psychopharmacology Bulletin 1995;31:534. [Google Scholar]
Schneider 2006 {published data only}
- Schneider LS, Tariot PN, Dagerman KS, Davis SM, Hsiao JK, Ismail MS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. New England Journal of Medicine 2006;355(15):1525‐38. [PUBMED: 17035647] [DOI] [PubMed] [Google Scholar]
Seltzer 2004 {published data only}
- Seltzer B, Zolnouni P, Nunez M, Goldman R, Kumar D, Ieni J, et al. Efficacy of donepezil in early‐stage Alzheimer disease: a randomized placebo‐controlled trial. Archives of Neurology 2004;61(12):1852‐6. [PUBMED: 15596605] [DOI] [PubMed] [Google Scholar]
Street 2000 {published data only}
- Street JS, Clark WS, Gannon KS, Cummings JL, Bymaster FP, Tamura RN, et al. Olanzapine treatment of psychotic and behavioral symptoms in patients with Alzheimer disease in nursing care facilities: a double‐blind, randomized, placebo‐controlled trial. The HGEU Study Group. Archives of General Psychiatry 2000;57(10):968‐76. [PUBMED: 11015815] [DOI] [PubMed] [Google Scholar]
Streim 2004 {published data only}
- Steim J, Breder C, Swanink R, McQuade R, Iwamoto T, Carson W, et al. Flexible‐dose aripiprazole in psychosis of Alzheimer's disease. Neurobiology of Aging 2004;25(2):S191. [Google Scholar]
Tariot 2004a {published data only}
- Tariot PN, Profenno LA, Ismail MS. Efficacy of atypical antipsychotics in elderly patients with dementia. Journal of Clinical Psychiatry 2004;65 Suppl 11:11‐5. [PUBMED: 15264966] [PubMed] [Google Scholar]
Tariot 2004b {published data only}
- Tariot P. Medication helps quell the agitation of dementia. www.urmc.rochester.edu/pr/news/story.cfm?id=593 2004.
Van Reekum 2002 {published data only}
- Reekum R, Clarke D, Conn D, Herrmann N, Eryavec G, Cohen T, et al. A randomized, placebo‐controlled trial of the discontinuation of long‐term antipsychotics in dementia. International Psychogeriatrics 2002;14(2):197‐210. [PUBMED: 12243210] [DOI] [PubMed] [Google Scholar]
Wilcock 2000 {published data only}
- Wilcock GK, Lilienfeld S, Gaens E. Efficacy and safety of galantamine in patients with mild to moderate Alzheimer's disease: multicentre randomised controlled trial. Galantamine International‐1 Study Group. The British Medical Journal (Clinical research ed.) 2000;321(7274):1445‐9. [PUBMED: 11110737] [DOI] [PMC free article] [PubMed] [Google Scholar]
Winblad 2001 {published data only}
- Winblad B, Engedal K, Soininen H, Verhey F, Waldemar G, Wimo A, et al. A 1‐year, randomized, placebo‐controlled study of donepezil in patients with mild to moderate AD. Neurology 2001;57(3):489‐95. [PUBMED: 11502918] [DOI] [PubMed] [Google Scholar]
Additional references
Abe 1975
- Abe T, Morita M, Kawai K, Misawa S, Kanai H, Hirose G, et al. Transmission of a t(13q22q) chromosome observed in three generations with segregation of the translocation D1‐trisomy syndrome. Humangenetik 1975;30(3):207‐15. [DOI] [PubMed] [Google Scholar]
Albert 2013
- Albert PR, Benkelfat C. The neurobiology of depression‐‐revisiting the serotonin hypothesis. II. Genetic, epigenetic and clinical studies. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 2013;368(1615):20120535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Allard 1990
- Allard P, Alafuzoff I, Carlsson A, Eriksson K, Ericson E, Gottfries CG, et al. Loss of dopamine uptake sites labeled with [3H]GBR‐12935 in Alzheimer's disease. European Neurology 1990;30(4):181‐5. [DOI] [PubMed] [Google Scholar]
Andrade 2015
- Andrade 2015. The primary outcome measure and its importance in clinical trials. Journal of Clinical Psychiatry 2015;76(10):e1320‐3. [DOI] [PubMed] [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th Edition. Arlington, VA: American Psychiatric Association, 2013. [Google Scholar]
Attems 2014
- Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer's disease‐‐lessons from pathology. BioMed Central Medicine 2014;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ballard 2006
- Ballard C, Waite J, Birks. Atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer's disease. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD003476.pub2; PUBMED: 16437455] [DOI] [PMC free article] [PubMed] [Google Scholar]
Barnhart 2004
- Barnhart WJ, Makela EH, Latocha MJ. SSRI‐induced apathy syndrome: a clinical review. Journal of Psychiatric Practice 2004;10(3):196‐9. [DOI] [PubMed] [Google Scholar]
Bartus 1982
- Bartus RT, Dean RL 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982;217(4558):408‐14. [DOI] [PubMed] [Google Scholar]
Berman 2012
- Berman K, Brodaty H, Withall A, Seeher K. Pharmacologic treatment of apathy in dementia. American Journal of Geriatric Psychiatry 2012;20(2):104‐22. [PUBMED: 21841459] [DOI] [PubMed] [Google Scholar]
Bhandari 2004
- Bhandari M, Busse JW, Jackowski D, Montori VM, Schunemann H, Sprague S, et al. Association between industry funding and statistically significant pro‐industry findings in medical and surgical randomized trials. Canadian Medical Association Journal 2004;170(4):477‐80. [PUBMED: 14970094] [PMC free article] [PubMed] [Google Scholar]
Birks 2006
- Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD005593; PUBMED: 16437532] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boyle 2003
- Boyle PA, Malloy PF, Salloway S, Cahn‐Weiner DA, Cohen R, Cummings JL. Executive dysfunction and apathy predict functional impairment in Alzheimer disease. American Journal of Geriatric Psychiatry 2003;11(2):214‐21. [PubMed] [Google Scholar]
Bressan 2005
- Bressan RA, Crippa JA. The role of dopamine in reward and pleasure behaviour‐‐review of data from preclinical research. Acta Psychiatrica Scandinavica. Supplement 2005;427:14‐21. [DOI] [PubMed] [Google Scholar]
Brodaty 2012
- Brodaty H, Arasaratnam C. Meta‐analysis of nonpharmacological interventions for neuropsychiatric symptoms of dementia. American Journal of Psychiatry 2012;169(9):946‐53. [DOI] [PubMed] [Google Scholar]
Callegari 2016
- Callegari I, Mattei C, Benassi F, Krueger F, Grafman J, Yaldizli O, et al. Agomelatine Improves apathy in frontotemporal dementia. Neuro‐Degenerative Diseases 2016;16(5‐6):352‐6. [PUBMED: 27229348] [DOI] [PubMed] [Google Scholar]
Clarke 2011
- Clarke DE, Ko JY, Kuhl EA, Reekum R, Salvador R, Marin RS. Are the available apathy measures reliable and valid? A review of the psychometric evidence. Journal of Psychosomatic Research 2011;70(1):73‐97. [PUBMED: 21193104] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cummings 1994
- Cummings JL, Mega M, Gray K, Rosenberg‐Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44(12):2308‐14. [PUBMED: 7991117] [DOI] [PubMed] [Google Scholar]
Cummings 2001
- Cummings JL, Nadel A, Masterman D, Cyrus PA. Efficacy of metrifonate in improving the psychiatric and behavioral disturbances of patients with Alzheimer's disease. Journal of Geriatric Psychiatry and Neurology 2001;14(2):101‐8. [DOI] [PubMed] [Google Scholar]
Cummings 2015
- Cummings J, Friedman JH, Garibaldi G, Jones M, Macfadden W, Marsh L, et al. Apathy in neurodegenerative diseases: recommendations on the design of clinical trials. Journal of Geriatric Psychiatry and Neurology 2015;28(3):159‐73. [PUBMED: 25809634] [DOI] [PubMed] [Google Scholar]
David 2008
- David R, Koulibaly M, Benoit M, Garcia R, Caci H, Darcourt J, et al. Striatal dopamine transporter levels correlate with apathy in neurodegenerative diseases A SPECT study with partial volume effect correction. Clinical Neurology and Neurosurgery 2008;110(1):19‐24. [DOI] [PubMed] [Google Scholar]
De Boer 2005
- Boer SF, Koolhaas JM. 5‐HT1A and 5‐HT1B receptor agonists and aggression: a pharmacological challenge of the serotonin deficiency hypothesis. European Journal of Pharmacology 2005;526(1‐3):125‐39. [DOI] [PubMed] [Google Scholar]
De Deurwaerdère 1998
- Deurwaerdère P, Stinus L, Spampinato U. Opposite change of in vivo dopamine release in the rat nucleus accumbens and striatum that follows electrical stimulation of dorsal raphe nucleus: role of 5‐HT3 receptors. Journal of Neuroscience 1998;18(16):6528‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Debette 2002
- Debette S, Kozlowski O, Steinling M, Rousseaux M. Levodopa and bromocriptine in hypoxic brain injury. Journal of Neurology 2002;249(12):1678‐82. [DOI] [PubMed] [Google Scholar]
Declercq 2013
- Declercq T, Petrovic M, Azermai M, Vander Stichele R, Sutter AIM, Driel ML, et al. Withdrawal versus continuation of chronic antipsychotic drugs for behavioural and psychological symptoms in older people with dementia. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD007726.pub2] [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration. Available from www.cochrane‐handbook.org 2011.
Diesfeldt 2013
- Diesfeldt HF. Interpreting change scores of the Behavioural Rating Scale for Geriatric Inpatients (GIP). Tijdschrift voor Gerontologie en Geriatrie 2013;44(4):166‐74. [PUBMED: 23921986] [DOI] [PubMed] [Google Scholar]
Doody 2013
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. New England Journal of Medicine 2013;369(4):341‐50. [PUBMED: 23883379] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. The British Medical Journal 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ellison 1986
- Ellison DW, Beal MF, Mazurek MF, Bird ED, Martin JB. A postmortem study of amino acid neurotransmitters in Alzheimer's disease. Annals of Neurology 1986;20(5):616‐21. [DOI] [PubMed] [Google Scholar]
Feldman 2001
- Feldman H, Gauthier S, Hecker J, Vellas B, Subbiah P, Whalen E. A 24‐week, randomized, double‐blind study of donepezil in moderate to severe Alzheimer's disease. Neurology 2001;57(4):613‐20. [PUBMED: 11524468] [DOI] [PubMed] [Google Scholar]
Feldman 2005
- Feldman H, Gauthier S, Hecker J, Vellas B, Xu Y, Ieni JR, et al. Efficacy and safety of donepezil in patients with more severe Alzheimer's disease: a subgroup analysis from a randomized, placebo‐controlled trial. International Journal of Geriatric Psychiatry 2005;20(6):559‐69. [PUBMED: 15920715] [DOI] [PubMed] [Google Scholar]
Folstein 1975
- Folstein MF, Folstein SE, McHugh PR. "Mini‐mental state". A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research 1975;12(3):189‐98. [PUBMED: 1202204] [DOI] [PubMed] [Google Scholar]
Förstl 1993
- Förstl H, Burns A, Levy R, Cairns N, Luthert P, Lantos P. Neuropathological correlates of behavioural disturbance in confirmed Alzheimer's disease. British Journal of Psychiatry 1993;163:364‐8. [DOI] [PubMed] [Google Scholar]
Garcia‐Alloza 2005
- Garcia‐Alloza M, Gil‐Bea FJ, Diez‐Ariza M, Chen CP, Francis PT, Lasheras B, et al. Cholinergic‐serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer's disease. Neuropsychologia 2005;43(3):442‐9. [DOI] [PubMed] [Google Scholar]
Gauthier 2002
- Gauthier S, Feldman H, Hecker J, Vellas B, Ames D, Subbiah P, et al. Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer's disease. International Psychogeriatrics 2002;14(4):389‐404. [PUBMED: 12670060] [DOI] [PubMed] [Google Scholar]
General Assembly of the World Medical Association
- General Assembly of the World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. The Journal of the American College of Dentists 2014;81(3):14‐8. [PUBMED: 25951678] [PubMed] [Google Scholar]
Gerrard 2007
- Gerrard P, Malcolm R. Mechanisms of modafinil: A review of current research. Neuropsychiatric Disease and Treatment 2007;3(3):349‐64. [PMC free article] [PubMed] [Google Scholar]
Gitlin 2012
- Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacologic management of behavioral symptoms in dementia. The Journal of the American Medical Association 2012;308(19):2020‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grace 2011
- Grace J. Frontal systems behavior scale. Encyclopedia of Clinical Neuropsychology 2011:1090‐3. [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. The British Medical Journal 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harrison 2016
- Harrison F, Aerts L, Brodaty H. Apathy in dementia: systematic review of recent evidence on pharmacological treatments. Current Psychiatry Reports 2016;18(11):103. [PUBMED: 27726067] [DOI] [PubMed] [Google Scholar]
Heres 2006
- Heres S, Davis J, Maino K, Jetzinger E, Kissling W, Leucht S. Why olanzapine beats risperidone, risperidone beats quetiapine, and quetiapine beats olanzapine: an exploratory analysis of head‐to‐head comparison studies of second‐generation antipsychotics. American Journal of Psychiatry 2006;163(2):185‐94. [PUBMED: 16449469] [DOI] [PubMed] [Google Scholar]
Herrmann 2004a
- Herrmann N, Lanctôt KL, Khan LR. The role of norepinephrine in the behavioral and psychological symptoms of dementia. Journal of Neuropsychiatry and Clinical Neurosciences 2004;16(3):261‐76. [DOI] [PubMed] [Google Scholar]
Herrmann 2004b
- Herrmann N, Lanctôt K, Eryavec G, Reekum R, Khan LR. Growth hormone response to clonidine predicts aggression in Alzheimer's disease. Psychoneuroendocrinology 2004;29(9):1192‐7. [DOI] [PubMed] [Google Scholar]
Herrmann 2013
- Herrmann N, Lanctôt KL, Hogan DB. Pharmacological recommendations for the symptomatic treatment of dementia: the Canadian consensus conference on the diagnosis and treatment of dementia 2012. Alzheimer's Research & Therapy 2013;5(Suppl 1):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne, JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoehn‐Saric 1990
- Hoehn‐Saric R, Lipsey JR, McLeod DR. Apathy and indifference in patients on fluvoxamine and fluoxetine. Journal of Clinical Psychopharmacology 1990;10(5):343‐5. [PubMed] [Google Scholar]
Hurt 2008
- Hurt C, Bhattacharyya S, Burns A, Camus V, Liperoti R, Marriott A, et al. Patient and caregiver perspectives of quality of life in dementia. An investigation of the relationship to behavioural and psychological symptoms in dementia. Dementia and Geriatric Cognitive Disorders 2008;26(2):138‐46. [DOI] [PubMed] [Google Scholar]
Kales 2015
- Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. The British Medical Journal 2015;350:h369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kang 2012
- Kang JY, Lee JS, Kang H, Lee HW, Kim YK, Jeon HJ, et al. Regional cerebral blood flow abnormalities associated with apathy and depression in Alzheimer's disease. Alzheimer Disease and Associated Disorders 2012;26(3):217‐24. [DOI] [PubMed] [Google Scholar]
Kapasi 2016
- Kapasi A, Schneider JA. Vascular contributions to cognitive impairment, clinical Alzheimer's disease, and dementia in older persons. Biochimica et Biophysica Acta 2016;1862(5):878‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Katz 1963
- Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: A standardized measure of biological and psychosocial function. JAMA 1963;185:914‐9. [PUBMED: 14044222] [DOI] [PubMed] [Google Scholar]
Kaufer 1998a
- Kaufer D, Cummings JL, Christine D. Differential neuropsychiatric symptom responses to tacrine in Alzheimer's disease: relationship to dementia severity. Journal of Neuropsychiatry and Clinical Neurosciences 1998;10(1):55‐63. [DOI] [PubMed] [Google Scholar]
Kaufer 1998b
- Kaufer D. Beyond the cholinergic hypothesis: the effect of metrifonate and other cholinesterase inhibitors on neuropsychiatric symptoms in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders 1998;9 Suppl 2:8‐14. [DOI] [PubMed] [Google Scholar]
Lanari 2006
- Lanari A, Amenta F, Silvestrelli G, Tomassoni D, Parnetti L. Neurotransmitter deficits in behavioural and psychological symptoms of Alzheimer's disease. Mechanisms of Ageing and Development 2006;127(2):158‐65. [DOI] [PubMed] [Google Scholar]
Lanctôt 2001
- Lanctôt KL, Herrmann N, Mazzotta P. Role of serotonin in the behavioral and psychological symptoms of dementia. Journal of Neuropsychiatry and Clinical Neurosciences 2001;13(1):5‐21. [DOI] [PubMed] [Google Scholar]
Lanctôt 2007a
- Lanctôt KL, Herrmann N, Rothenburg L, Eryavec G. Behavioral correlates of GABAergic disruption in Alzheimer's disease. International Psychogeriatrics 2007;19(1):151‐8. [DOI] [PubMed] [Google Scholar]
Lanctôt 2007b
- Lanctôt KL, Moosa S, Herrmann N, Leibovitch FS, Rothenburg L, Cotter A, et al. A SPECT study of apathy in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders 2007;24(1):65‐72. [DOI] [PubMed] [Google Scholar]
Landes 2001
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. Journal of the American Geriatrics Society 2001;49(12):1700–7. [DOI] [PubMed] [Google Scholar]
Lawton 1969
- Lawton MP, Brody EM. Assessment of older people: self‐maintaining and instrumental activities of daily living. Gerontologist 1969;9(3):179‐86. [PUBMED: 5349366] [PubMed] [Google Scholar]
Leonpacher 2016
- Leonpacher AK, Peters ME, Drye LT, Makino KM, Newell JA, Devanand DP, et al. Effects of citalopram on neuropsychiatric symptoms in Alzheimer'sdDementia: evidence from the CitAD Study. American Journal of Psychiatry 2016;173(5):473‐80. [PUBMED: 27032628] [DOI] [PubMed] [Google Scholar]
Levy 1998
- Levy ML, Cummings JL, Fairbanks LA, Masterman D, Miller BL, Craig AH, et al. Apathy is not depression. Journal of Neuropsychiatry and Clinical Neurosciences 1998;10(3):314‐9. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. The British Medical Journal (Clinical research ed.) 2003;326(7400):1167‐70. [PUBMED: 12775614] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lonergan 2009
- Lonergan E, Luxenberg J. Valproate preparations for agitation in dementia. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD003945.pub3; PUBMED: 19588348] [DOI] [PubMed] [Google Scholar]
Lowe 1988
- Lowe SL, Francis PT, Procter AW, Palmer AM, Davison AN, Bowen DM. Gamma‐aminobutyric acid concentration in brain tissue at two stages of Alzheimer's disease. Brain 1988;111(Pt 4):785‐99. [DOI] [PubMed] [Google Scholar]
Marin 1991
- Marin RS, Biedrzycki RC, Firinciogullari S. Reliability and validity of the apathy evaluation scale. Psychiatry Research 1991;38(2):143‐62. [PUBMED: 1754629] [DOI] [PubMed] [Google Scholar]
Marin 1994
- Marin RS, Firinciogullari S, Biedrzycki RC. Group differences in the relationship between apathy and depression. Journal of Nervous and Mental Disease 1994;182(4):235‐9. [DOI] [PubMed] [Google Scholar]
Marin 1995
- Marin RS, Fogel BS, Hawkins J, Duffy J, Krupp B. Apathy: a treatable syndrome. Journal of Neuropsychiatry and Clinical Neurosciences 1995;7(1):23‐30. [DOI] [PubMed] [Google Scholar]
McKhann 1984
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34(7):939‐44. [DOI] [PubMed] [Google Scholar]
McKhann 2011
- McKhann GM, Knopman DS, Chertkow H, Hyman, BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer's & Dementia 2011;7(3):263‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mitchell 2011
- Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer's disease. CNS Neuroscience & Therapeutics 2011;17(5):411‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mulin 2011
- Mulin E, Leone E, Dujardin K, Delliaux M, Leentjens A, Nobili F, et al. Diagnostic criteria for apathy in clinical practice. International Journal of Geriatric Psychiatry 2011;26(2):158‐65. [PUBMED: 20690145] [DOI] [PubMed] [Google Scholar]
O'Regan 2015
- O'Regan J, Lanctôt KL, Mazereeuw G, Herrmann N. Cholinesterase inhibitor discontinuation in patients with Alzheimer's disease: a meta‐analysis of randomized controlled trials. Journal of Clinical Psychiatry 2015;76(11):e1424‐31. [PUBMED: 26646039] [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;19:799‐812. [Google Scholar]
Overshott 2004
- Overshott R, Byrne J, Burns A. Nonpharmacological and pharmacological interventions for symptoms in Alzheimer's disease. Expert Review of Neurotherapeutics 2004;4(5):809‐21. [DOI] [PubMed] [Google Scholar]
Padala 2007
- Padala PR, Burke WJ, Bhatia SC. Modafinil therapy for apathy in an elderly patient. Annals of Pharmacotherapy 2007;41(2):346‐9. [DOI] [PubMed] [Google Scholar]
Politis 2004
- Politis AM, Mayer LS, Passa M, Maillis A, Lyketsos CG. Validity and reliability of the newly translated Hellenic Neuropsychiatric Inventory (H‐NPI) applied to Greek outpatients with Alzheimer's disease: a study of disturbing behaviors among referrals to a memory clinic. International Journal of Geriatric Psychiatry 2004;19(3):203‐8. [PUBMED: 15027034] [DOI] [PubMed] [Google Scholar]
Porsteinsson 2014
- Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z, et al. CitAD Research Group. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. The Journal of the American Medical Association 2014;311(7):682‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prince 2013
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer's & Dementia 2013;9(1):63‐75.e2. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Robert 2009
- Robert P, Onyike CU, Leentjens AF, Dujardin K, Aalten P, Starkstein S, et al. Proposed diagnostic criteria for apathy in Alzheimer's disease and other neuropsychiatric disorders. European Psychiatry 2009;24(2):98‐104. [DOI] [PubMed] [Google Scholar]
Roccaforte 1990
- Roccaforte WH, Burke WJ. Use of psychostimulants for the elderly. Hospital & Community Psychiatry 1990;41(12):1330‐3. [DOI] [PubMed] [Google Scholar]
Rockwood 2001
- Rockwood K, Mintzer K, Truyen L, Wessel T, Wilkinson D. Effects of a flexible galantamine dose in Alzheimer's disease: a randomized, controlled trial.. Journal of Neurology, Neurosurgery and Psychiatry 2001;71(5):589‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosenberg 2016
- Rosenberg PB, Lanctôt KL, Herrmann N, Mintzer JE, Porsteinsson AP, Sun X, et al. Changes in neuropsychiatric inventory associated with semagacestat treatment of Alzheimer's disease. Journal of Alzheimer's Disease : JAD 2016;54(1):373‐81. [PUBMED: 27567808] [DOI] [PubMed] [Google Scholar]
Rossor 1982
- Rossor MN, Garrett NJ, Johnson AL, Mountjoy CQ, Roth M, Iversen LL. A post‐mortem study of the cholinergic and GABA systems in senile dementia. Brain 1982;105(Pt 2):313‐30. [DOI] [PubMed] [Google Scholar]
Schneider 1997
- Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, Reisberg B, et al. Validity and reliability of the Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change. The Alzheimer's Disease Cooperative Study. Alzheimer Disease and Associated Disorders 1997;11 Suppl 2:S22‐32. [PUBMED: 9236949] [DOI] [PubMed] [Google Scholar]
Schneider 2007
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology 2007;69(24):2197‐204. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann H, Hill S, Guyatt G, Akl EA, Ahmed F. The GRADE approach and Bradford Hill's criteria for causation. Journal of Epidemiology and Community Health 2011;65(5):392‐5. [DOI] [PubMed] [Google Scholar]
Seitz 2011
- Seitz DP, Adunuri N, Gill SS, Gruneir A, Herrmann N, Rochon P. Antidepressants for agitation and psychosis in dementia. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD008191.pub2; PUBMED: 21328305] [DOI] [PubMed] [Google Scholar]
Sepehry 2017
- Sepehry AA, Sarai M, Hsiung GR. Pharmacological therapy for apathy in Alzheimer's disease: a systematic review and meta‐analysis. Canadian Journal of Neurological Sciences. 2017;44(3):267‐75. [PUBMED: 28148339] [DOI] [PubMed] [Google Scholar]
Starkstein 1997
- Starkstein SE, Sabe L, Vázquez S, Lorenzo G, Martínez A, Petracca G, et al. Neuropsychological, psychiatric, and cerebral perfusion correlates of leukoaraiosis in Alzheimer's disease. Journal of Neurology, Neurosurgery, and Psychiatry 1997;63(1):66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Starkstein 2009
- Starkstein SE, Mizrahi R, Capizzano AA, Acion L, Brockman S, Power BD. Neuroimaging correlates of apathy and depression in Alzheimer's disease. Journal of Neuropsychiatry and Clinical Neurosciences 2009;21(3):259‐65. [DOI] [PubMed] [Google Scholar]
Steinberg 2008
- Steinberg M, Shao H, Zandi P, Lyketsos CG, Welsh‐Bohmer KA, Norton MC, et al. Point and 5‐year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. International Journal of Geriatric Psychiatry 2008;23(2):170‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Storga 1996
- Storga D, Vrecko K, Birkmayer JG, Reibnegger G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neuroscience Letters 1996;203(1):29‐32. [DOI] [PubMed] [Google Scholar]
Tagariello 2009
- Tagariello P, Girardi P, Amore M. Depression and apathy in dementia: same syndrome or different constructs? A critical review. Archives of Gerontology and Geriatrics 2009;49(2):246‐9. [DOI] [PubMed] [Google Scholar]
Tariot 2000
- Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C. A 5‐month, randomized, placebo‐controlled trial of galantamine in AD. The Galantamine USA‐10 Study Group. Neurology 2000;54(12):2269‐76. [DOI] [PubMed] [Google Scholar]
Tekin 2001
- Tekin S, Mega MS, Masterman DM, Chow T, Garakian J, Vinters HV, et al. Orbitofrontal and anterior cingulate cortex neurofibrillary tangle burden is associated with agitation in Alzheimer disease. Annals of Neurology 2001;49(3):355‐61. [PubMed] [Google Scholar]
Van der Linde 2016
- Linde RM, Dening T, Stephan BCM, Prina AM, Evans E, Brayne C. Longitudinal course of behavioural and psychological symptoms of dementia: systematic review. British Journal of Psychiatry 2016;209(5):366‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vartiainen 1995
- Vartiainen H, Tiihonen J, Putkonen A, Koponen H, Virkkunen M, Hakola P, et al. Citalopram, a selective serotonin reuptake inhibitor, in the treatment of aggression in schizophrenia. Acta Psychiatrica Scandinavica 1995;91(5):348‐51. [DOI] [PubMed] [Google Scholar]
Vialta‐Franch 2013
- Vilalta‐Franch J, Calvó‐Perxas L, Garre‐Olmo J, Turró‐Garriga O, López‐Pousa S. Apathy syndrome in Alzheimer's disease epidemiology: prevalence, incidence, persistence, and risk and mortality factors. Journal of Alzheimer's Disease 2013;33(2):535‐43. [DOI] [PubMed] [Google Scholar]
Volkow 2002
- Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. Mechnism of action of methylphenidate: Insights from PET imaging studies. Journal of Attention Disorders 2002;6(1):S31‐S43. [DOI] [PubMed] [Google Scholar]
Walsh 1997
- Walsh SL, Cunningham KA. Serotonergic mechanisms involved in the discriminative stimulus, reinforcing and subjective effects of cocaine. Psychopharmacology (Berlin) 1997;130(1):41‐58. [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. The ICD‐10 Classification of Mental and Behavioural Disorders: clinical descriptions and diagnostic guidelines. 1992. apps.who.int/iris/handle/10665/37958#sthash.Vaxjp53U.dpuf (accessed 05 February 2016).
World Alzheimer Report 2015
- Prince M, Wimo A, Guerchet M, Ali G, Wu Y, Prina M. World Alzheimer Report 2015: The Global Impact of Dementia. www.worldalzreport2015.org/downloads/world‐alzheimer‐report‐2015.pdf (accessed 05 February 2016).
References to other published versions of this review
Ruthirakuhan 2016
- Ruthirakuhan MT, Herrmann N, Abraham EH, Lanctôt KL. Pharmacological interventions for apathy in Alzheimer's disease (Protocol). Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD012197] [DOI] [PMC free article] [PubMed] [Google Scholar]