

Summary
Methylation of histone H3 K79 by Dot1L is a hallmark of actively transcribed genes that depends on monoubiquitination of H2B K120 (H2B-Ub), and is an example of histone modification cross-talk that is conserved from yeast to humans. We report here cryo-EM structures of Dot1L bound to ubiquitinated nucleosome that show how H2B-Ub stimulates Dot1L activity and reveal a role for the histone H4 tail in positioning Dot1L. We find that contacts mediated by Dot1L and the H4 tail induce a conformational change in the globular core of histone H3 that reorients K79 from an inaccessible position, thus enabling this side chain to project deep into the active site in a position primed for catalysis. Our study provides a comprehensive mechanism of cross-talk between histone ubiquitination and methylation and reveals a structural plasticity in histones that makes it possible for histone-modifying enzymes to access residues within the nucleosome core.
Keywords: Chromatin, ubiquitin, histones, methylation, Dot1L, nucleosome, cryo-EM, structural biology
Graphical Abstract
eTOC
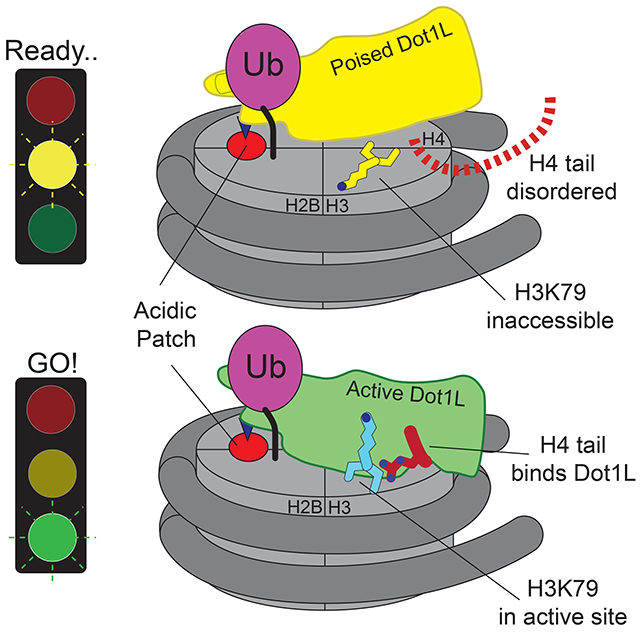
Cryo-EM structures show how H2B monoubiquitination anchors the Dot1L methyltransferase to nucleosomes and stimulates methylation of histone H3K79. H2B-ubiquitin anchors Dot1L at one edge of the nucleosome, allowing the enzyme to adopt a poised or active state. In the active complex, the histone H4 tail binds to a pocket in Dot1L and positions the active site over H3K79. Dot1L and the H4 tail together induce a conformational change in the globular core of histone H3 that inserts K79 into the Dot1L active site.
Introduction
Post-translational modifications of histones play a central role in regulating all cellular processes requiring access to DNA. Cross-talk between histone modifications, in which one histone modification regulates deposition of a second (Suganuma and Workman, 2008), provides an additional layer of regulation and specificity. The dependence of histone H3 Lysine79 (H3K79) methylation on monoubiquitination of histone H2B Lysine120 (humans; lysine 123 in yeast) (Briggs et al., 2002; McGinty et al., 2008; Ng et al., 2002b; Steger et al., 2008; Van Leeuwen et al., 2002) is an example of cross-talk that is conserved across eukaryotes. H3K79 mono-, di- and tri-methylation regulates transcription elongation (Kouskouti and Talianidis, 2005; Krogan et al., 2003; Steger et al., 2008), telomeric silencing (Ng et al., 2002a; Takahashi et al., 2011) and the DNA damage response (Giannattasio et al., 2005; Huyen et al., 2004; Wysocki et al., 2005). H3K79 is methylated by human Dot1L (Feng et al., 2002; McGinty et al., 2008) and yeast Dot1 (Van Leeuwen et al., 2002), which are methyltransferase enzymes that attach up to three methyl groups to the lysine ε amino group using S-adenosylmethionine (SAM) as the methyl donor (Feng et al., 2002; Van Leeuwen et al., 2002). Human Dot1L is a 1,527 amino acid protein that contains an N-terminal methyltransferase domain that is conserved in yeast Dot1p (Sawada et al., 2004), and a C-terminal extension that interacts with proteins that direct Dot1L to specific genomic loci (Kuntimaddi et al., 2014). Mislocalization of Dot1L due to gene fusions between Dot1L-interacting proteins and the histone H4 methyltransferase, Mixed Lineage Leukemia (MLL) can lead to hypermethylation of H3K79 and constitutive gene expression at ectopic loci, which is a causative factor for around 40% of Mixed Lineage Leukemias (MLL) (Bernt et al., 2011; Chen and Armstrong, 2015; Guenther et al., 2008; Krivtsov et al., 2008). Despite multiple studies, the mechanism by which H2B-Ub stimulates Dot1L to methylate H3K79 remains unknown.
The dependence of H3K79 methylation on prior monoubiquitination of histone H2B (H2B-Ub) has been established both in vivo and in vitro (Briggs et al., 2002; McGinty et al., 2008; Ng et al., 2002c). Methylation of H3K79 in yeast is abolished in strains lacking either the E2 or E3 that ubiquitinate H2B-K123, Rad6 and Bre1, or in strains in which histone H2B bears an arginine in place of K123 (Briggs et al., 2002; Ng et al., 2002c; Wood et al., 2003). Knockdown of human RNF20 or RNF40, which form the RNF20/40 heterodimeric E3 ligase that ubiquitinates H2BK120, leads to a decrease in H3K79 methylation in human cells (Wang et al., 2013; Zhu et al., 2005). In vitro, Dot1L is directly stimulated by the presence of H2B-Ub in mononucleosomes (McGinty et al., 2008), which increases the apparent catalytic rate of H3K79 methylation (McGinty et al., 2009). Both Dot1L and yeast Dot1p require a nucleosomal substrate and are not active on isolated histone H3, suggesting that these methyltransferases may recognize other portions of the nucleosome (Feng et al., 2002; Van Leeuwen et al., 2002). In another layer of regulation, the flexible histone H4 tail has been implicated in contacting Dot1p/Dot1L and is required for H3K79 methylation by both human Dot1L (McGinty et al., 2009) and yeast Dot1p (Altaf et al., 2007; Fingerman et al., 2007). Deletions and point substitutions in H4 residues R17-H18-R19 dramatically reduce methylation of H3K79 in yeast (Altaf et al., 2007; Fingerman et al., 2007) and abolish in vitro activity of both yeast Dot1p (Fingerman et al., 2007) and human Dot1L (McGinty et al., 2009). Although structures of the Dot1p (Sawada et al., 2004) and Dot1L (Min et al., 2003; Richon et al., 2011) catalytic domains have been determined, it is still not known how the enzyme recognizes ubiquitin or contacts other portions of its nucleosomal substrate. Due to the limited accessibility of the H3K79 side chain, which is oriented along the nucleosome surface (Lu et al., 2008), it has not been possible to construct a plausible model of Dot1L bound to a nucleosome in which the K79 side chain can enter the active site and attack the bound SAM cofactor (Min et al., 2003).
We report here three cryo-EM structures of Dot1L bound to ubiquitinated nucleosomes that reveal the molecular basis for cross-talk between histone H2B ubiquitination and H3K79 methylation, and uncover unexpected allostery in both enzyme and substrate that stimulates catalysis. We have captured two complexes of Dot1L bound to the H2B-Ub nucleosome and SAM in a catalytically competent state, which were determined at resolutions of 3.54 Å and 2.96 Å. The structure of Dot1L bound to H2B-Ub nucleosomes in a poised state, in which the active site is too far from H3K79 for catalysis, was determined at a resolution of 3.95 Å. The structures reveal that Dot1L contains structural elements that contact the H2B-conjugated ubiquitin and the nucleosome H2A/H2B acidic patch, which establish a pivot point about which Dot1L can rotate. In the active complex, the histone H4 tail binds to a previously uncharacterized groove in Dot1L, tethering Dot1L to the opposing edge of the nucleosome and precisely positioning the catalytic center above H3K79. Remarkably, a conformational change is induced within the globular core of histone H3, which repositions K79 from an inaccessible conformation to one that inserts the side chain into Dot1L’s active site in a position to initiate the methyl transfer reaction. The altered conformation of histone H3 is stabilized by contacts with both the H4 tail and with Dot1L, which itself undergoes conformational changes that accommodate binding of the H4 tail and enclose the substrate lysine in a hydrophobic channel. The structure accounts for all prior observations regarding the role of histone H2B ubiquitination, as well as point mutations in Dot1L, Dotlp and histone H4, in activating Dot1L/Dot1p and suggests new avenues for designing inhibitors that are specific for the Dot1L catalytic domain.
Results
Structural overview of Dot1L bound to nucleosomes containing H2B-Ub
We determined cryo-EM structures of Dot1L bound to nucleosomes in which ubiquitin was conjugated to histone H2B-K120 via a non-hydrolyzable dichloroacetone (DCA) linkage (Morgan et al., 2016). For the poised state structure, we formed complexes between Dot1L (2-416) and H2B-Ub nucleosomes in the presence of an analogue of the SAM methyl donor (see Methods), although we see no bound cofactor in our maps. To trap Dot1L in the active state, we formed complexes between Dot1L, SAM, and H2B-Ub nucleosomes containing histone H3 in which K79 was replaced with norleucine (Nle) (see Methods), a non-native amino acid that has been shown to increase the affinity of methyltransferases for peptides in a SAM-dependent manner (Brown et al., 2014; Jayaram et al., 2016; Van Hest et al., 2000). Norleucine has the same number of aliphatic carbons as lysine but lacks the charged ε amino group. The structure of the complex in the poised state was determined to a resolution of 3.90 Å and the structures of the complex in the active state, in which H3K79 extends into the Dot1L active site, were determined to a resolution of 3.54 Å and 2.96 Å (Figures 1A-C, S1–2 and Table 1).
Figure 1: Structures Dot1L bound to H2B-ubiquitin nucleosome.
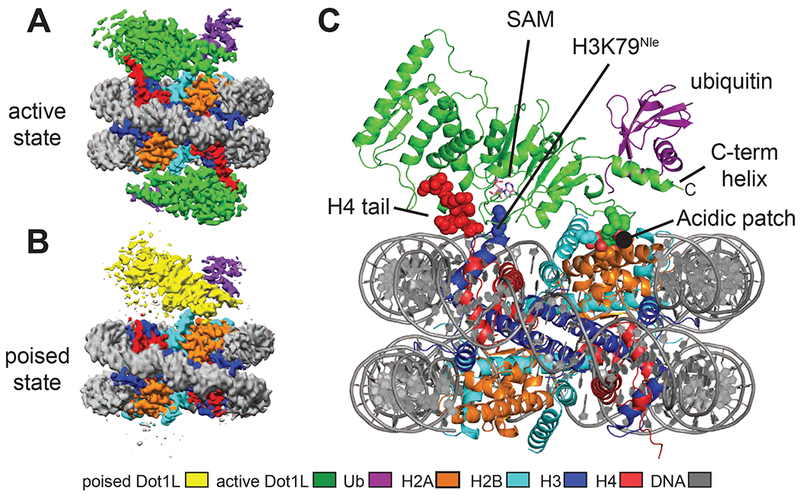
A, Cryo-EM reconstruction of the 2-to-1 active state complex. B, Cryo-EM reconstruction of the poised state complex. C, Atomic model of the active state complex between Dot1L and the H2B-Ub nucleosome. The H4 tail, H3K79Nle and the acidic patch are depicted as spheres and the SAM cofactor is depicted in stick representation. See also Figures S1–S4.
Table 1:
Cryo-EM data collection, refinement and validation statistics
| Active State: 2:1 complex (EMD-9384) (PDB 6NJ9) | Active State: 1:1 complex (EMD-0480) (PDB 6NQA) | Poised State (EMD-0468) (PDB 6NOG) | |
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 130,000 | 130,000 | 130,000 |
| Voltage (kV) | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 50 | 50 | 50 |
| Defocus range (μm) | −1 to −2.5 | −1 to −2.5 | −1 to −2.5 |
| Pixel size (Å) | 0.532 | 0.532 | 0.532 |
| Symmetry imposed | C2 | ||
| Initial particle images (no.) | 800727 | 800727 | 868027 |
| Final particle images (no.) | 237780 | 220681 | 108658 |
| Map resolution (Å) | 2.96 | 3.54 | 3.9 |
| FSC threshold | (0.143) | (0.143) | (0.143) |
| Refinement | |||
| Initial model used (PDB code) | 1KX3, 1NW3, 1UBQ | 1KX3, 1NW3, 1UBQ | 1KX3, 3QOW 1UBQ |
| Model resolution (Å) | 3.2 | 4.0 | 4.4 |
| FSC threshold | (0.5) | (0.5) | (0.5) |
| Model resolution range (Å) | 272.4-3.3 | 272.4-4.0 | 272.4-4.4 |
| Map sharpening B factor (Å2) | −73 | −96.5 | −180 |
| Model composition | |||
| Non-hydrogen atoms | 15510 | 15322 | 14957 |
| Protein residues | 1186 | 1164 | 1126 |
| Nucleotides | 292 | 292 | 292 |
| Ligands | 1 | 1 | 0 |
| B factors (Å2) | |||
| Protein | 71.6 | 72.8 | 113.1 |
| Nucleotide | 19.47 | 24.5 | 134.4 |
| Ligand | 77.42 | 68.1 | - |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.013 | 0.007 | 0.009 |
| Bond angles (°) | 1.04 | 0.853 | 1.35 |
| Validation | |||
| MolProbity score | 1.46 | 1.59 | 1.41 |
| Clashscore | 2.72 | 4.11 | 4.29 |
| Poor rotamers (%) | 0.99 | 0.51 | 0.94 |
| Ramachandran plot | |||
| Favored (%) | 94.08 | 94.23 | 96.73 |
| Allowed (%) | 5.92 | 5.77 | 3.27 |
| Disallowed (%) | 0 | 0 | 0 |
Two classes of active state complexes were identified: one (36% of particles), a 2:1 complex in which Dot1L is bound to both faces of the nucleosome (Figures 1A and S1B) and one (34% of particles), a 1:1 complex in which one Dot1L is bound to the nucleosome (Figure S1B). Aside from the presence of an extra copy of Dot1L, the 2:1 complex is nearly identical to the 1:1 complex. All complexes contain a fragment of human Dot1L (residues 2-416) that comprises the catalytic domain and a C-terminal DNA binding region (Figure 2A), and that can be fully stimulated by nucleosomes containing H2B-Ub (McGinty et al., 2008; Min et al., 2003). In all structures, only Dot1L residues ~4-332 are visible in the maps, indicating that the C-terminal DNA binding region of Dot1L is either bound to the DNA in multiple orientations, disordered or highly mobile.
Figure 2 : Analysis of Dot1L binding and activity.
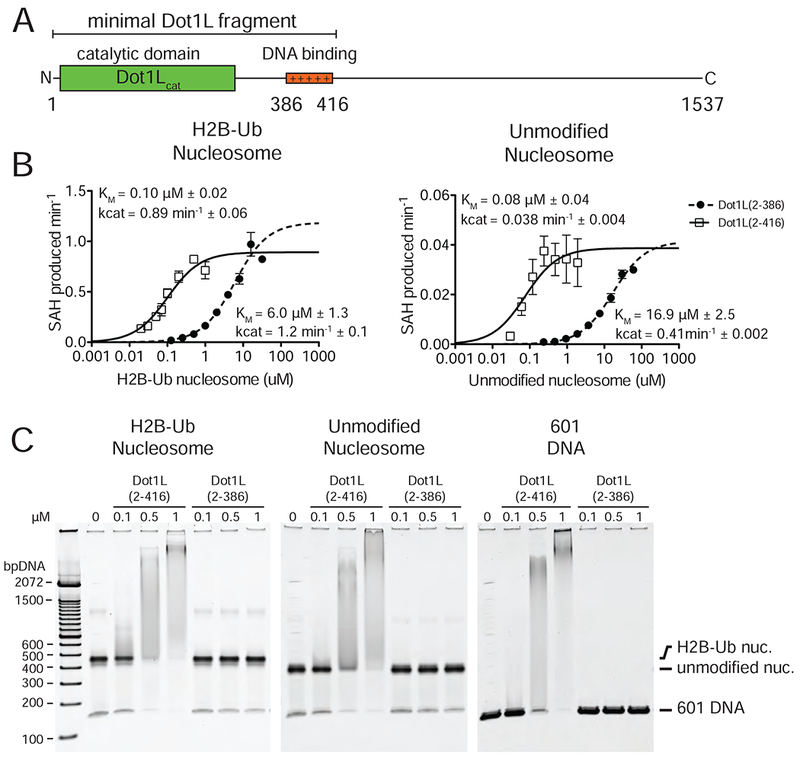
A, Schematic of the Dot1L domain architecture. B Michaelis Menten titrations of two variants of Dot1L. Left, titration with H2B-Ub nucleosome. Right, titration with unmodified Nucleosome. Error bars correspond to the standard error of three replicate measurements. The kcat and Km of the fitted data are reported in the graph field. The reported errors of the fitted Km and kcat correspond to the standard error. C Electrophoretic Mobility Shift Assay of Dot1L variants binding to different nucleosome substrates and to free DNA.
To test whether the previously identified DNA-binding region of Dot1L (McGinty et al., 2008; Min et al., 2003) contributes to the ability of H2B-Ub to stimulate Dot1L activity, we assayed the kinetics of Dot1L fragments with (2-416) and without (2-286) the DNA-binding region on both unmodified and H2B-Ub nucleosomes (Figure 2B). While Dot1L(2-416) has a much lower KM for methyl transfer than Dot1L(2-386) on both nucleosome substrates (Figure 2B), both Dot1L(2-416) and Dot1L(2-386) had nearly identical kcat values for each substrate and were stimulated by H2B-Ub to a similar extent (Figure 2B). We also compared the ability of both Dot1L fragments to bind to H2B-Ub nucleosomes in an electrophoretic mobility shift assay and could only detect complex formation for the Dot1L fragment containing the DNA interaction region (Figure 2C). These data are consistent with previous studies (McGinty et al., 2008; McGinty et al., 2009; Min et al., 2003) and confirm that the DNA-binding region that is not visible in our maps does not play a role in mediating H2B-Ub stimulation of Dot1L.
In both the active and poised state complexes, the Dot1L C-terminus contacts ubiquitin and the nucleosome acidic patch formed by histones H2A and H2B, anchoring Dot1L to one edge of the nucleosome (Figures 1C, 3A and S3). As compared to the poised state, Dot1L in the active state is rotated clockwise by 22° about the ubiquitin and pivots down towards the nucleosome face by 25 Å at its N-terminus (Figure 3A,B). The active state is further stabilized by binding of histone H4 tail residues 15-19 in a previously unknown groove in the N-terminal portion of Dot1L (Figure 1C). The greater number of contacts between Dot1L and the nucleosome in the active complex appear to stabilize the enzyme, as evidenced by higher map resolution and well-resolved density throughout the catalytic domain (Figure S4). Importantly, the H3K79Nle side chain projects into the active site immediately adjacent to the SAM methyl group (Figure 1C). This close approach is facilitated by an unprecedented conformational change in the histone fold of H3 that shifts the K79 side chain from an inaccessible orientation, enabling it to enter the Dot1L active site in a position poised for catalysis. With the exception of conformational changes in two Dot1L loops described below, the structure of Dot1L is essentially identical to that previously reported (Figure S5A,C) (Min et al., 2003; Richon et al., 2011).
Figure 3: Dot1L transitions between the Poised and Active States.
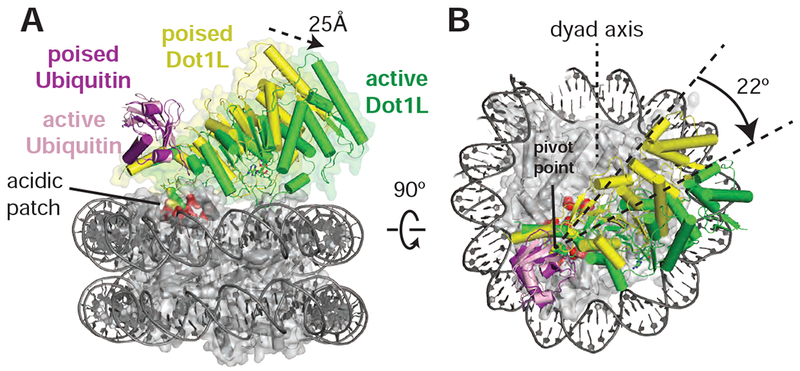
Dot1L movements from the poised state to the active state shown as from the side , A, and top, B. Poised state (yellow) and active state (green) Dot1L are depicted as cylinders and the histone octamer is shown as a transparent gray surface. H2A/H2B acidic patch residues are shown as red spheres. Arrows indicate movements made by Dot1L in the switch to the active state. See also Figure S3 and S5.
The poised structure represents a catalytically incompetent state in which Dot1L cannot methylate H3K79 (Figures 1B, and S3) and likely exists directly before or after methyl-transfer. Dot1L contacts ubiquitin and the acidic patch in essentially the same manner as seen in the active state structure (Figures S5D and S3A-D). In the poised state, the N-terminal portion of the Dot1L catalytic domain does not contact the nucleosome and the Dot1L active site is ~21 Å away from H3K79 (Figure S3E). The relatively weak density for the N-terminal portion of Dot1L, as compared to the C-terminal portion that contacts ubiquitin and the nucleosome (Figures 1B and S3A), suggests that Dot1L can occupy a continuum of conformations when the N-terminal part of the enzyme does not contact the nucleosome. Due to the higher resolution and better density of the 2:1 active state structure, all images and discussion below focuses on the 2:1 active state structure unless otherwise noted.
Contacts with ubiquitin and the H2A/H2B acidic patch orient Dot1L on the nucleosome
The structure reveals that Dot1L contains a ubiquitin-binding hydrophobic cradle comprising the C-terminal helix (residues 320-330) of the catalytic domain and a loop between β-strands 8-9, which forms a convex surface that binds ubiquitin (Figures 4A,B and S5D). The interface between Dot1L and ubiquitin buries 637Å2 of surface area. While nearly all ubiquitin-binding motifs bind to a canonical patch centered on L8 and I44 of ubiquitin (Komander and Rape, 2012), Dot1L contacts a hydrophobic patch comprising ubiquitin residues I36, L71, and L73 (Figure 4B). Residues L322 and F326 in the ubiquitin-binding helix abut ubiquitin residues I36 and L71, while Dot1L residues L284, I290 and L322 cradle ubiquitin residue L73, which is located at the start of the ubiquitin C-terminal tail (Fig. 4B).
Figure 4: Dot1L interactions with Ubiquitin and the acidic patch.
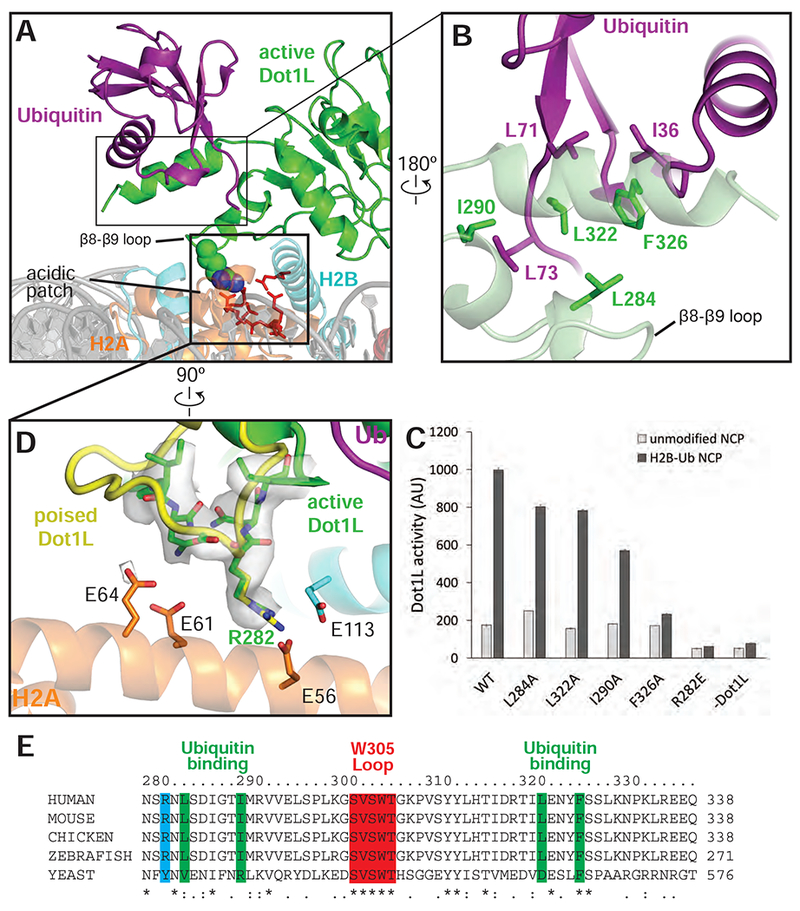
A, Overview of the interaction between Dot1L, Ubiquitin and the nucleosome in the active state structure. Residues important for Dot1L interaction with the nucleosome acidic patch residues are depicted as red sticks. B, Detailed view of the contacts made between Dot1L and ubiquitin. Dot1L is depicted as a semi-transparent green cartoon. Important residues at the Dot1L-Ubiquitin interface are shown as sticks. C, Endpoint H3K79 methylation activity assays using Dot1L mutants with either unmodified or H2B-Ub nucleosomes. Error bars correspond to the standard deviation of 3 replicate experiments. D, Detailed view of interactions between Dot1L and the H2B/H2A acidic patch. Residues at the interface are depicted as sticks and the EM density for Dot1L in the active state is shown as a semi-transparent gray surface. A superimposed poised state Dot1L is depicted in yellow. E, Multiple Sequence alignment of Dot1L from different species. The alignment was performed with Clustal Omega (Sievers et al., 2011). See also Figures S3, S5 and Table S1.
The observed interactions with ubiquitin explain previous findings that the C-terminal helix in yeast Dot1p is required for H3K79 methylation in vivo and in vitro (Fingerman et al., 2007) and that a patch containing ubiquitin residues L71 and L73 is critical for ubiquitin-dependent activation of Dot1L (Holt et al., 2015; Zhou et al., 2016), rather than the canonical L8/I44 patch contacted by the majority of ubiquitin-binding proteins (McGinty et al., 2009) (prior findings summarized in Table S1). To probe the contributions of individual Dot1L residues to ubiquitin-dependent stimulation, we generated alanine substitutions in Dot1L residues that contact ubiquitin and assessed the ability of the mutant proteins to be stimulated by H2B-Ub in vitro (Figure 4C). Whereas alanine substitutions L284A, I290A, L322A and F326A did not impair methylation of unmodified nucleosomes, all of these Dot1L substitutions impaired simulation by H2B-Ub-containing nucleosomes (Figure 4C). The most severe defects resulted from an alanine substitution at F326, which was not stimulated at all by nucleosomes containing H2B-Ub. F326 is a highly conserved Dot1L residue (Figure 4E) that contacts I36 of ubiquitin, and which is clearly resolved in our maps (Figures 4B and S4C). An alanine substitution at Dot1L residue I290, which is in van der Waals contact with ubiquitin residue L73, reduced stimulation by H2B-Ub to an intermediate level, while the L284A and L322A substitutions had modest effects. Taken together, the effects of both Dot1L (Figure 4B,C) and ubiquitin (Holt et al., 2015) mutations on the ability of H2B-ubiquitinated nucleosomes to stimulate Dot1L confirm the presence of a ubiquitin binding element that is specific for the hydrophobic base and tail region of ubiquitin.
The pivot point for the switch between the poised and active state is centered on Dot1L residue R282, which contacts the nucleosome acidic patch in both states (Figures 4D and S3D). R282 projects into the H2A/H2B acidic patch and interacts with residues H2A-E56 and H2B-E113 (Figures 4D, and S3C,D). Density for the Dot1L R282 side chain is well-resolved in both active (Figure 4D) and poised states (Figure. S3D), reflecting the stability of this contact. The interaction mediated by R282 is reminiscent of other nucleosome binding proteins that employ an “arginine anchor” to interact with the nucleosome acidic patch (Figure S5E-J) (Armache et al., 2011; Barbera et al., 2006; Makde et al., 2010; McGinty et al., 2014). However, whereas all reported arginine anchor-containing proteins bind to the acidic patch near H2A D90-E92, the Dot1L R282 interaction occurs at a distinct location in the acidic patch (Figure S5F-J). Consistent with its buried position in the poised and active states, a Dot1L R282E substitution completely abrogated the ability of Dot1L to methylate both unmodified and ubiquitinated nucleosomes (Figure 4C).
The H4 tail binds Dot1L and orients the active site over H3K79
In the active complex, we found that a stretch of basic residues in the tail of H4, 15-19, binds within a groove in the N-terminal portion of Dot1L, tethering the enzyme to the opposite edge of the nucleosome (Figure 5A,B). These H4 residues are not ordered in the poised structure (Figure S6A) and are either unstructured or closely associated with the DNA in most nucleosome structures (Figure S6B). The Dot1L loop 22-32 moves by ~5 Å as compared to the Dot1L crystal structure (Min et al., 2003), opening a cleft in Dot1L that enables the H4 tail to bind (Figures 5B and S6C,D), burying 770Å2 of surface area. The sidechain of histone H4 R19 is well ordered in the structure (Figure 5B) and is in a position to interact with the backbone atoms of H3 residues T80, Q76, and K79Nle (Figure 5C). The sidechain of H18 is positioned close to Dot1L residues N126 and S304 and may form hydrogen bonds with these residues (Figure 5C). Histone H4 residue R17 is oriented in a position to project into a deep acidic pocket containing highly conserved residues E138 and Y115 (Figures 5B-D, F). The position of R17 is model built based on clear density for the β carbon and spatial constraints in the acidic pocket. Concurrent binding of the H4 tail and ubiquitin to opposing ends of Dot1L position the enzyme with its catalytic site directly over H3K79, thus ensuring specificity for this residue.
Figure 5: H4 tail interactions with Dot1L.
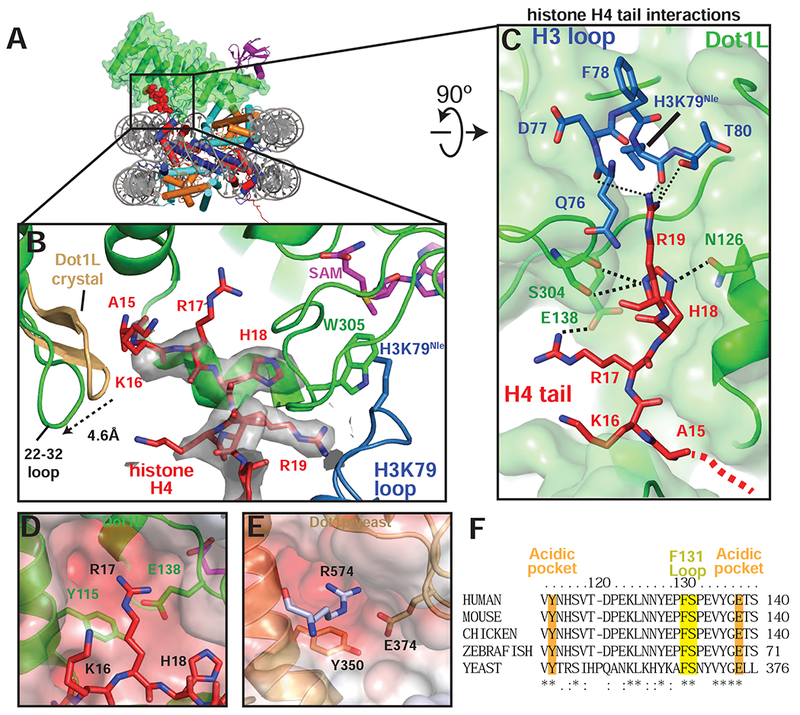
A, Overview of the active Dot1L structure. Dot1L is shown as a transparent green surface, ubiquitin as purple ribbon, H4 tail in red spheres. B, H4 tail (red) interaction with the Dot1L binding groove. EM density for the H4 tail is shown as a semi-transparent gray surface. The 22-32 loop from the crystal structure of Dot1L alone (PDB ID 1NW3) is colored tan. C, H4 tail (red) interactions between Dot1L (green) and the H3K79 loop (blue). The surface of Dot1L is shown in semi-transparent green. Potential hydrogen bonding or van der Waals interactions are shown as black dashed lines. Red dashed lines illustrate the direction of the H4 mainchain in the Dot1L binding groove. D, Modeled position of H4 R17 binding in the Dot1L acidic pocket. Conserved residues in the binding pocket are depicted as sticks and the surface of Dot1L is shown and colored according the electrostatic potential. E, Close-up view yeast Dot1p (PDB ID 1U2Z) showing arginine from a neighboring Dot1p molecule in the crystal bound in the conserved acidic pocket. Electrostatic surface potential shown as in D. Electrostatic potential in D, E, was calculated using the APBS tool (Baker et al., 2001). F, Multiple Sequence alignment of Dot1L from different species. The alignment was performed with Clustal Omega (Sievers et al., 2011). See also Figure S6 and Table S1.
The observed interactions between Dot1L and the H4 tail explain previous studies showing that H4 residues R17-H18-R19 are required for H3K79 methylation by both Dot1L (McGinty et al., 2009) and yeast Dot1p (Altaf et al., 2007; Fingerman et al., 2007). In vitro experiments have shown that a Dot1L R17A/R19A mutant is defective in methylating both ubiquitinated and unmodified nucleosomes (McGinty et al., 2009). Furthermore, deletion of the RHR motif abolishes binding of the H4 tail to Dot1p (Fingerman et al., 2007). Deletion of the RHR motif or alanine substitutions at H4 residues R17 or R19 completely eliminate H3K79 di- and tri-methylation by yeast Dot1p and severely reduce mono-methylation of H3K79 both in vivo and in vitro (Altaf et al., 2007; Fingerman et al., 2007). Due to conservation in the Dot1L catalytic domain and histone residues between yeast and human, these H4 residues are expected to form similar interactions in yeast Dot1p that we observe in the structure of Dot1L. Remarkably, the structure of yeast Dot1p (Sawada et al., 2004) contains a fortuitous interaction in the crystal lattice that appears to mimic binding of H4 R17 to Dot1L (Figure 5E). In the yeast Dot1p crystal structure, an arginine (R574) from the flexible C-terminal tail of a neighboring Dot1p protein in the crystal lattice docks in the conserved acidic pocket of yeast Dot1p (Figure 5E). This interaction had been proposed to reflect a potential interaction with histone H3 (Sawada et al., 2004), but instead likely reflects binding of the H4 tail as observed in our structure of Dot1L. Consistent with the importance of the acidic pocket in which this arginine binds, substitutions in yeast Dot1p residues Y350 and E374, which correspond to human Dot1L Y115 and E138 (Figure 5D-F), dramatically decrease Dot1p methyltransferase activity (Sawada et al., 2004). The structure of the active Dot1L complex, combined with the effects of histone H4 mutations on both Dot1L and yeast Dot1p, explains the importance of H4 tail interactions in Dot1L activity.
An induced conformational change in histone H3 orients H3K79 for catalysis
Previous attempts to model a catalytically competent complex by docking the Dot1L crystal structure on the nucleosome were unsuccessful because no orientation of Dot1L can accommodate the H3K79 substrate lysine without significant steric clashes with the core histones (Min et al., 2003). These clashes occur because, in the poised state structure and in other nucleosome structures, the H3K79 side chain is in an inaccessible conformation that lies parallel to the nucleosome surface (Figures 6A,B and S7A) and is unable to adopt a rotamer capable of entering the Dot1L active site. Our structure of the active complex shows that binding of Dot1L in the active state induces a dramatic conformational change in the H3K79Nle loop that reorients both the backbone and side chain of H3K79Nle by ~90°, reorienting the side chain to point away from the nucleosome surface and into the Dot1L active site (Figures 6A-E and 7A). The result of this transition is that the H3K79 lysine sidechain rotates by about 90° and moves up and away from the nucleosome surface, which moves the ε amino group by ~10 Å (Figure 6B). A modeled H3K79 sidechain based on the position of the aliphatic side chain in the H3K79Nle substitution (Figure 6E) shows that, in the active state, the ε-amino group of lysine comes within 3.0 Å of the SAM methyl donor and is positioned for nucleophilic attack on the SAM methyl group (Figure 7A). Importantly, the conformational change in the loop containing H3K79Nle is not due to substitution of lysine with norleucine, since the 1:1 active state complex with just a single Dot1L bound (Figure S1B) shows that H3K79Nle on the unoccupied face of the nucleosome is in the normal, inaccessible conformation (Figure S7C-E). The unprecedented change observed here in the core histone fold has, to our knowledge, never been observed before in intact nucleosomes and has important implications for how other histone-modifying enzymes may access inaccessible or buried histone residues.
Figure 6: Conformational change in histone H3 reorients K79.
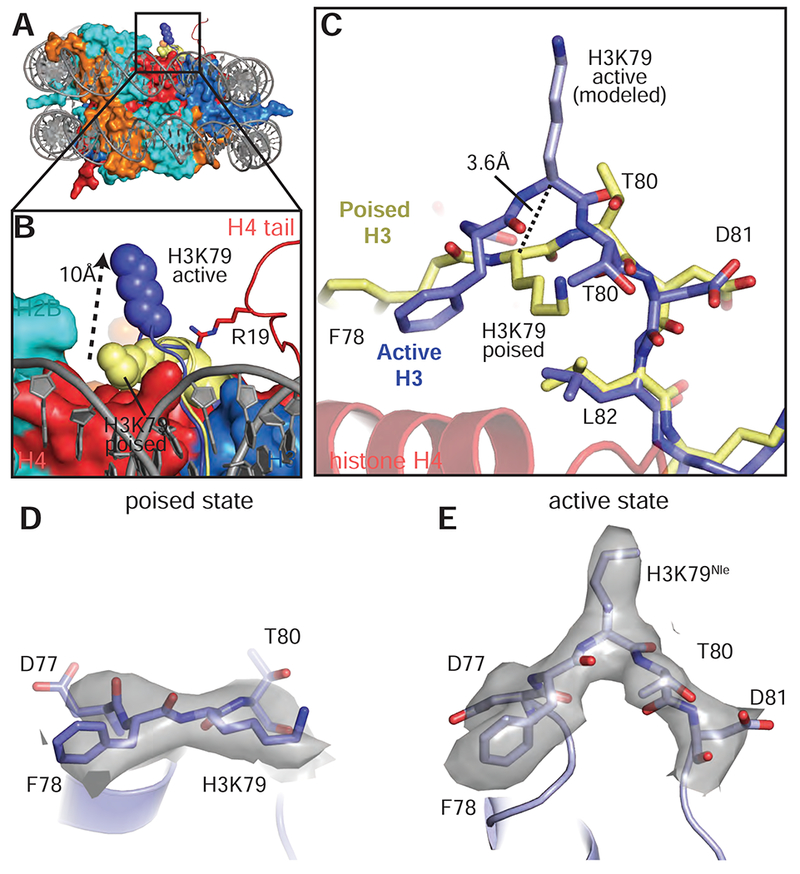
A, Superimposition of the active state and poised state nucleosomes. The active state histone octamer is depicted in surface representation and colored as in Figure 1. The H3K79 loops from the active state (blue) and poised state (yellow) structures are shown as cartoons and H3K79 is shown as spheres. B, Close up view of H3K79 in the active state (blue spheres) and poised state (yellow spheres). The H3K79 sidechain moves ~10 Å (measured from ε-amino groups) from the poised state to the active state. C, Superimposition of the histone H3K79 loop from the active (blue) and poised state (yellow) structures showing the conformational change that occurs in the transition from the poised state to the active state. In the transition from the poised state to the active state the H3K79 backbone moves up by 3.6 Å (measured from the Cα carbons of H3K79) and rotates by an angle of ~90°. D, EM density (gray surface) for the H3K79 loop in the poised state. E, EM density (gray surface) for the H3K79 loop in the active state. See also Figure S7 and Table S1.
Figure 7: Formation of the Dot1L active site enclosure.
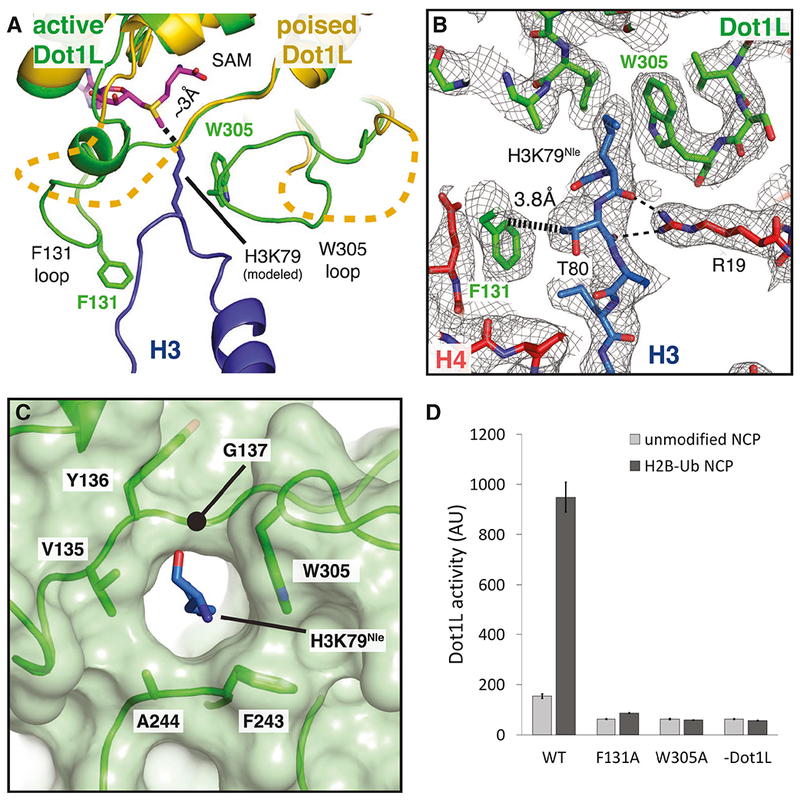
A, Superimposition of active state (green) and poised state (yellow) Dot1L. The H3K79 loop from the active state structure is shown as a blue cartoon and the modeled H3K79 sidechain is shown in stick representation. The disordered F131 and W305 loops from the poised state structure are depicted as yellow dashed lines. The ε- amino group of lysine comes within 3 Å of the SAM methyl donor. B, Close up view of the Dot1L active site enclosure with H3 (blue), Dot1L (green) and H4 (red) shown in stick representation. Sharpened experimental EM density is shown as a gray mesh. A van der Waals contact between Dot1L F131 and H3 T80 is shown as thick a dashed black line and the hydrogen bonds between R19 and the H3K79 loop are shown as thin black dashed lines. C, Formation of the Dot1L H3K79 lysine binding channel. Dot1L is depicted as a green cartoon surrounded by a semi-transparent green surface. H3K79Nle is shown as blue sticks. D, Endpoint H3K79 methylation activity assays using Dot1L mutants with either unmodified or H2B-Ub nucleosomes. Error bars correspond to the standard deviation of 3 replicates. See also Figure S7 and Table S1.
The tail of histone H4 and the Dot1L loops containing F131 and W305 play a critical role in stabilizing the active conformation of histone H3 (Figure 7A,B). The loop containing the H3K79 residue is sandwiched between Dot1L residues on one side and H4 tail residues on the other, thus helping to stabilize the altered conformation of histone H3 (Figure 7A,B). Histone H4 R19 hydrogen bonds with backbone atoms of H3 residues K79, T80 and Q76 on one side, while Dot1L residue F131 is in van der Waals contact with the side chain of H3 residue, T80, on the other side of the altered H3 segment (Figures 5C and 7B). Upon transition from the poised state to the active state, the highly conserved F131 residue inserts into the same location on the nucleosome that is occupied by the H3K79 side chain in the poised state, further favoring the H3 conformation seen in the active state (Figures 7B and 5F). Additional van der Waals contacts between W305 and the aliphatic side chain of H3K79Nle further stabilize the lysine and orient it towards the SAM methyl group. Both F131 and W305 are invariant and reside in loops that are disordered in the poised state but become structured in the transition to the active complex (Figures 7A and 4E, 5F), although we note that the F131 loop adopts the active state conformation in a crystal structure of Dot1L(Min et al., 2003). Previous studies have shown that the conformation of these loops can be modulated by binding to SAM and SAM-like inhibitors (Yu et al., 2012). To test the importance of these residues for activity, we tested the effects of F131A or W305A substitutions on Dot1L methyltransferase activity. As shown in Figure 7D, these substitutions completely abrogated Dot1L activity on both H2B-ubiquitinated and unmodified nucleosomes, consistent with an important role for these contacts in histone methylation.
The repositioning of W305 from a disordered loop completes formation of a narrow, hydrophobic lysine-binding tunnel into which H3K79 inserts (Figure 7C). The other hydrophobic residues that surround the aliphatic portion of the side chain are V135 and F243, thus completely surrounding H3K79 in a hydrophobic environment. All three of these hydrophobic residues are invariant (Figure 4E, 5F). Interestingly, the residue corresponding to W305 in yeast Dot1p (W543) is in the active conformation even in the absence of a bound substrate lysine (Figure S7B), thus preforming the tunnel in the yeast enzyme. Consistently, previous studies have shown that W543A and W543F mutations in yeast Dot1p completely abolish activity in vitro (Sawada et al., 2004). The methyl group of the bound SAM cofactor caps the end of the tunnel, further encapsulating the attacking H3K79 side chain in a completely hydrophobic environment.
The very hydrophobic lysine-binding tunnel, capped by the hydrophobic methyl group of the bound SAM cofactor, suggests a resolution to the long-standing problem of how Dot1L activates H3K79 for attack on the SAM methyl group, which is the first step in the methylation reaction. The lysine must first be deprotonated; however, previous studies have failed to identify an active site residue that deprotonates the lysine (Min et al., 2003). We see no candidate ionizable residues in the active Dot1L complex. Instead, we speculate that the hydrophobic nature of the tunnel and narrow dimensions that exclude water may be sufficient to lower the pKa of the lysine sufficiently to remove the proton from the ε amino group. Burial of lysine in a hydrophobic environment has been shown to lower its pKa and favor the uncharged amino group (Isom et al., 2011). The very hydrophobic nature of this tunnel also explains why substitution of H3K79 with norleucine, which lacks the charged ε amino group but has the same 4-carbon aliphatic side chain, stabilizes the active complex.
Discussion
Our study reveals the basis for cross-talk between histone H2B ubiquitination and methylation of H3K79 by Dot1L and uncovers an unexpected remodeling of a core histone that is induced with the assistance of the tail of histone H4. Our structures suggest that H2B-Ub increases the catalytic rate of Dot1L methylation in part by restricting the position of Dot1L on the face of the nucleosome. Ubiquitin conjugated to H2B-K120 greatly reduces the orientations Dot1L can sample, while positioning the active site of Dot1L in an orientation that faces the nucleosome (Figure 1). The rigid nature of the Dot1L ubiquitin-binding element and its contacts with a region of ubiquitin that includes tail residue I73 (Figures 4B and S5D) help to limit Dot1L’s range of motion by partly immobilizing the flexible C-terminal ubiquitin tail. The range of motion is further limited by the contact between R282 and the nucleosome acidic patch (Figure 4D) which can occur in both the poised and active states. Conformational restriction of Dot1L thus increases its catalytic rate (McGinty et al., 2009) by greatly limiting the enzyme’s search for its substrate lysine. Binding of the H4 tail to the opposing end of Dot1L further reduces movement of the N-terminal portion of Dot1L and positions the active site over H3K79.
Residues in both Dot1L (F131 and W305) and histone H4 (R19) appear to play a role in stabilizing the active conformation of H3K79. The importance of these contacts is underscored by the fact that mutations in Dot1L F131, W305 and H4 R19 all abolish Dot1L activity (Figures 7D) (Fingerman et al., 2007; McGinty et al., 2009), presumably by disfavoring the active state conformation of H3K79. The critical role of the H4 tail in particular is, to our knowledge, unprecedented, but it explains why previous studies found this region to be so critical for Dot1L and Dot1p methyltransferase activity (Altaf et al., 2007; Fingerman et al., 2007; McGinty et al., 2009). It is not possible to discern from our structural study whether Dot1L and H4 actively induce the conformational change observed around H3K79 or if the multiple contacts made between Dot1L, ubiquitin, the acidic patch and the H4 tail serve to position the enzyme so that it can trap rare fluctuations of the structure around H3K79. Given the consistent positioning of H3K79 observed in many structures of the nucleosome (Figure S7A), we speculate that the additional binding energy conferred by ubiquitin may, in part, help pay the energetic cost of distorting histone H3 and lowering the pKa of the substrate lysine. This energetic trade-off may also explain why H2B ubiquitination does not strongly decrease the apparent KM of the reaction, but instead significantly increases kcat (Figure 2) (McGinty et al., 2009).
Conformational restriction could also account for the ability of H2B-Ub to stimulate methylation of H3K4, which lies in the long flexible N-terminal tail of histone H3 (Davey et al., 2002; Sun and Allis, 2002). The H3K4 methyltransferase belongs to the Set domain family, which is unrelated to the Dot1 family, and is part of the multiprotein yeast COMPASS and human SET1/MLL complexes (Miller et al., 2001). The ubiquitin L70-L73 patch required to stimulate Dot1L is also required to stimulate COMPASS (Holt et al., 2015), although mutations in the basic H4 tail residues that are required for Dot1 activity have no effect on H3K4 methylation (Fingerman et al., 2007). Structures of a 5- and 6-protein COMPASS subcomplex were recently reported (Hsu et al., 2018; Qu et al., 2018), but it is not known how this complex recognizes ubiquitin or the nucleosome. We speculate that contacts with ubiquitin may anchor COMPASS to the nucleosome surface and reduce the conformational search for H3K4 which, unlike H3K79, lies in a flexible and highly accessible histone tail.
The ability of Dot1L to reconfigure residues in the globular histone core raises the possibility that other histone-modifying enzymes may induce local conformational changes to access core residues that are fully or partly inaccessible. While histones can partially unfold when they are outside the histone octamer and bound to histone chaperones (English et al., 2006) or histone-modifying enzymes (Zhang et al., 2018), histones incorporated into nucleosomes adopt stable, virtually invariant conformations. However, recent methyl-TROSY NMR and cross-linking studies (Sinha et al., 2017) have suggested that the histone octamer can be distorted due to the action of ATP-dependent nucleosome remodeling enzymes. Our structural study of Dot1L has, to our knowledge, captured the first structural snapshot of a distortion in the globular histone core of an intact nucleosome. The view of the nucleosome core as a rigid substrate and binding partner should thus be modified to take into account structural variations, particularly in residues that lie at the ends of histone fold helices and in loops.
Because of its causative role in MLL Leukemia, Dot1L is actively pursued as a drug target (Chen and Armstrong, 2015). Most Dot1L inhibitors described to date are derived from S-adenosyl-L-homocysteine (SAH), a product of the methyltransferase reaction that binds to many other cellular enzymes, including other methyltransferases (Chen and Armstrong, 2015). Structures of inhibitors bound to Dot1L reveal that many inhibitors characterized to date induce dramatic restructuring of the F131 loop, thereby occluding the SAM-binding site and the lysine-binding tunnel (Figure S7F) (Basavapathruni et al., 2012; Chen et al., 2016; Daigle et al., 2013; Mobitz et al., 2017; Scheufler et al., 2016; Yi et al., 2015; Yu et al., 2012). In light of our structure and the importance of F131 in Dot1L activity, it would appear that at least some of the inhibitory potential of these inhibitors can be attributed to defects in stabilizing the active conformation of H3K79. The binding groove for the H4 tail revealed by our study suggests another avenue to designing more specific inhibitors that bind to both the H4 tail binding region and the active site tunnel, thereby yielding Dot1L inhibitors that could serve as effective chemotherapeutic agents.
STAR Methodsn
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cynthia Wolberger (cwolberg@jhmi.edu).
All plasmids used in this study have been deposited in and can be requested from Addgene.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Histone genes were isolated from Xenopus Laevis. Ubiqutiin and Dot1L genes were isolated from Homo Sapiens. All genes were cloned into expression plasmids and expressed in Escherichia coli.
METHOD DETAILS
Protein and DNA preparations
Purification of Dot1L variants
A plasmid bearing the gene for human Dot1L, MSCB-hDot1Lwt, was a gift from Yi Zhang (Addgene plasmid # 74173 ; http://n2t.net/addgene:74173 ; RRID: Addgene_74173). A truncated Dot1L, Dot1L(2-416), was amplified by PCR from MSCB-hDot1Lwt using primers that encoded KpnI and EcoRI restriction sites and a TEV protease site. The resulting PCR product was cloned into pET32-a (Novagen) using the KpnI and EcoRI restriction sites to produce the expression constructs, which contained an N-terminal fusion of thioredoxin (Trx), a hexahistidine tag and a cleavage site for Tobacco Etch Virus (TEV) protease (Trx-His-TEV-Dot1L(2-416)). Dot1L point mutants were cloned in the Trx-His-TEV-Dot1L(2-416) plasmid using around-the-horn mutagenesis. Rosetta2(DE3) cells were transformed with the Trx-His-TEV-Dot1L(2-416) plasmid and grown in 2XYT media at 37°C while shaki ng. When the cell density reached an OD600 of 0.8-1.0, the growth temperature was reduced to 18°3, 1 mM IPTG was added to induce protein expression, and the cells were grown overnight. Cells were harvested by centrifugation and the cell pellet was resuspended in 20 ml of Dot1L lysis buffer (50 mM Tris pH 8.0, 750 mM NaCl, 5% glycerol, 0.5 mM dithiothreitol (DTT), 15 mM imidazole, 100 μM Phenylmethylsulfonyl fluoride (PMSF) per liter of growth medium, flash frozen in liquid nitrogen, and stored at −80°C until needed.
To purify Dot1L, the frozen pellet was thawed at 25°C and cooled on ice. The cells were lysed using an LM10 Microfluidizer (Microfluidics) and the clarified lysate was added to 10 ml of Ni-NTA beads (Qiagen) that had been equilibrated in NiA buffer (50 mM Tris pH 8.0, 750 mM NaCl, 5% glycerol, 0.5 mM DTT, 15 mM imidazole). The beads were incubated with the lysate at 4°C for 45 minutes, poured into a gravity flow column and washed with 200 ml of NiA buffer. Protein was eluted using ~ 70ml of NiB buffer (50 mM Tris pH 8.0, 750 mM NaCl, 5% glycerol, 0.5 mM DTT, 250 mM imidazole). After elution the absorbance at 280nm was measured and 6mg of TEV protease was added directly to the eluate and dialyzed overnight at 4°3 against Dot1L Dialysis buffer (50 mM HEPES, pH 7.0, 100 mM NaCl, 5% glycerol, 1 mM DTT). Precipitate was removed by centrifugation and filtration through a 0.22 μm Millex syringe filter (Millipore). To separate Dot1L from TEV and the cleaved Trx-His tag, the sample was loaded onto a 5 ml SP HP column (GE Healthcare) cation exchange column equilibrated in ion exchange buffer (50 mM HEPES, pH 7.0, 50 mM NaCl, 5% Glycerol, 1 mM DTT) and eluted with a 50 mM - 1000 mM linear NaCl gradient of 15 column volumes. Peak fractions corresponding to Dot1L were pooled and the buffer exchanged into Dot1L storage buffer (30 mM Tris pH 8.0, 200 mM NaCl, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 20% glycerol) by repeated concentration and dilution using an Amicon Ultra spin concentrator (Millipore). The pure concentrated protein (~500 μM) was flash frozen and stored at −80°C unti l use.
Dot1L(2-386) was purified in the same manner as Dot1L(2-416) except that, after TEV cleavage, the protein was concentrated using an Amicon Ultra spin concentrator (Millipore) and separated from TEV and the Trx-His tag using a Superdex 200 16/600 size exclusion chromatography column (GE Healthcare) that was pre-equilibrated with SEC buffer (30 mM Tris pH 8.0, 200 mM NaCl, 2 mM DTT, 5% glycerol). Peak fractions were pooled, the buffer exchanged into Dot1L storage buffer and frozen as described for Dot1L(2-416).
Purification of His-TEV-ubiquitin(G76C)
His-TEV-Ubiquitin(G76C) was purified as described previously (Morgan et al., 2016). Briefly, His-TEV-ubiquitin was expressed in Rosetta2 cells and purified by Ni-affinity chromatography, followed by dialysis into ammonium acetate, pH 4.5, overnight. Dialyzed protein was loaded onto tandem SP-HP cation exchange columns (GE) that were pre-equilibrated with ammonium acetate pH 4.5. Ubiquitin was eluted using a step gradient of 750mM NaCl, dialyzed into Ubiquitin storage buffer (10 mM Tris pH 7.8, 1 mM TCEP), flash frozen and stored at −80°C.
Purification of histone proteins
Unmodified Xenopus laevis histone expression plasmids containing H2A, H2B, H3 and H4 were a generous gift from Greg Bowman. Histone expression and purification were conducted following standard protocols described in Luger et. al.(Dyer et al., 2004).
To produce histone H3 with the unnatural amino acid, norleucine, in place of K79, histone H3 was cloned into pQE-81L and K79 was mutated to methionine around-the-horn PCR. For protein expression, the methionine auxotroph, E. coli B834(DE3)pLysS (Novagen) was transformed with the pQE-81L(H3K79M) plasmid and grown at 37°C in M9 minimal media supplemented with 60 mg/L of all amino acids except for 100 mg/L of lysine and 500 mg/L of methionine. Cells were grown to an OD600 of 0.6, pelleted by centrifugation and then washed three times with M9 medium. The cells were resuspended in fresh M9 media supplemented with all amino acids except methionine and supplemented with 500 mg/L of norleucine. 1mM IPTG was added to induce expression and the cells were grown at 37°C for 2-3 hours. Cells were harvested by centrifugation and the H3K79Nle histone was purified as described in Luger et. al.(Dyer et al., 2004)
Purification of 601 DNA
The Widom 601 strong positioning DNA sequence (Lowary and Widom, 1998) was produced in the E.coli strain XL-1 Blue using the pST55-16x601 plasmid (Makde et al., 2010). The pST55-16x601 plasmid was purified and 601 DNA sequence was isolated from the plasmid as described previously (Dyer et al., 2004).
Preparation of H2B-Ubiquitin
H2B-Ub containing a dichloroacetone linkage between the ubiquitin C-terminus and H2BK120 was prepared as described (Morgan et al., 2016). Briefly, 100 μM His-TEV-Ubiquitin(G76C) and 100 μM H2B(K120C) were mixed in crosslinking buffer (50 mM Borate pH 8.1, 1 TCEP) and incubated at 50°C for one hour to reduce cysteines. The mixture was cooled on ice and then crosslinking was initiated by the addition of 100uM dichloroacetone and allowed to proceed for 1 hour. The reaction was then quenched with 50 mM β-mercaptoethanol (BME), dialyzed into water and lyophilized. The product was then resuspended in NiNTA-6MU buffer (6M urea, 50mM Tris pH7.8, 500 mM NaCl, 10 mM BME, 20mM Imidazole, 0.1 mM PMSF) and incubated with 20 ml of Ni-NTA resin. Unreacted H2B and H2B-H2B crosslinks were removed in the flow through. The desired H2B-Ub product along with unreacted Ubiquitin and Ubiquitin-Ubiquitin crosslinks were eluted in NiNTA-6MU buffer supplemented with 250mM Imidazole and dialyzed into TEV cleavage buffer (50 mM Tris-HCl pH 8.0, 0.5 mM EDTA, and 1 mM DTT) and the his-tag on ubiquitin was removed by the addition of TEV protease. The material was dialyzed into water, lyophilized, re-dissolved in NiNTA-6MU buffer and uncleaved protein and TEV were removed by passage over Ni-NTA resin. The flow through was dialyzed into water and lyophilized again. Finally, the dry protein was dissolved in 7M Guanidine-HCl and separated on a Proto 300 C4 column (Higgins Analytical) in 0.1% TFA and a gradient of 0.1%TFA + 90% acetonitrile. Fractions corresponding to pure H2B-Ub were pooled and lyophilized.
Reconstitution of nucleosome core particles
Nucleosome core particles containing either unmodified, wild type H2B, H2B-Ub, or norleucine-substituted H2B-Ub were reconstituted as described previously(Dyer et al., 2004; Luger et al., 1999). Briefly, histone octamers were prepared from individual histone proteins by refolding and salt dialysis. Histone octamers were isolated from H2A/H2B dimers and H3/H4 tetramers by size exclusion chromatography, concentrated using an Amicon Ultra spin concentrator (Millipore) and stored at 4°C until use. Nucleosomes were prepared by mixing histone octamers and purified 601 DNA under high salt followed by salt gradient deposition over 18 hours. Fully formed nucleosomes were separated from misfolded nucleosomes by separation on TSK DEAE-5PW HPLC column(Luger et al., 1999) (TOSOH biosciences).
Dot1L assays
Kinetic assays
For Michaelis-Menten kinetic assays of Dot1L methyltransferase activity on nucleosome, a 20 nM stock of Dot1L(2-416) containing 40 μM S-adenosyl methionine (SAM) was prepared in Dot1L reaction buffer (20 mM Tris pH 8.0, 50 mM NaCl, 1 mM DTT). A 2-fold dilution series of H2B-Ub from 2000 nM – 40 nM was prepared in Dot1L reaction buffer. To initiate the reaction, 22.5 μl of Dot1L and 22.5 μl of nucleosome were mixed at 25°C, giving a final concentration of 10 n M Dot1L, 1000 nM - 20 nM H2B-Ub nucleosome and 20 μM SAM. 10 μl time points were taken at 5, 10, 15 and 20 min and quenched by adding 2.5 μl 0.5% trifluoroacetic acid (TFA). 10 μl of each quenched time point was transferred to a white 384-well microplate and methyltransferase activity was evaluated using the MTase-Glo assay (Promega), which detects the production of S-adenosyl homocysteine (SAH) as a luminescent signal. The raw luminescence values were correlated to SAH concentration using an SAH standard curve and initial rates of the reaction were determined by a linear regression fit of the data. Kinetic parameters of the methyltransferase reaction were determined by fitting the initial rates directly to the Michaelis-Menten equation using GraphPad Prism. All reactions were done in triplicate with the same preparation of protein and the standard error of the fitted Michalis Menten coefficients are reported. Titration of Dot1L(2-416) with unmodified nucleosomes was done identically to what is described here, except that a higher concentration of Dot1L(2-416) (35 nM) was used due to the very slow kinetics of Dot1L on unmodified nucleosomes.
Kinetic assays of Dot1L(2-386) were done similarly to those described for Dot1L(2-416) except that higher concentrations of nucleosome and Dot1L were used due to the much higher KM values with this truncation. For the titration of H2B-Ub nucleosome, a stock of 75 nM Dot1L(2-386) plus 40 μM SAM was prepared and a 2-fold dilution series of H2B-Ub ranging from 64 μM – 500 nM NCP-Ub nucleosome was prepared in Dot1L reaction buffer. For the titration of unmodified wild type nucleosome, a 1 μM stock of Dot1L(2-386) was used and the nucleosome dilution series ranged from 120 μM – 0.94 μM. Since quenching reactions with TFA caused significant aggregation at high nucleosome concentrations, these samples were first centrifuged to pellet insoluble protein before measuring SAH with the MTase Glo assay.
Endpoint methylation assays
Stocks of 100 nM of each mutant Dot1L(2-416) plus 40 μM S-adenosyl methionine SAM and 2 μM H2B-Ub nucleosome or unmodified wild type nucleosome were prepared in assay buffer (20 mM Tris pH 8.0, 50 mM NaCl, 1 mM DTT, 0.1mg/ml bovine serum albumin). The reactions were initiated by mixing 6 μl of Dot1L and 6μl of nucleosome for a final concentration of 50 nM Dot1L and 1 μM nucleosome. The reaction proceeded for 30 min at 25°C and was quenched by addition of 3μl 0.5% TFA. 10 μl of the quenched reaction was transferred to a white 384 well plate and S-adenosyl homocysteine (SAH) production was detected using the MTase Glo assay (Promega). Each reaction was done in triplicate with the same preparation of protein and the standard deviation of the raw data is reported.
Electrophoretic mobility shift assay
For the electrophoretic mobility shift assays 100nM of nucleosome or free 601 DNA was mixed with Dot1L(2-416) or Dot1L(2-386) at the indicated concentrations and incubated at room temperature for 30minute in EMSA buffer (20 mM Tris pH 7.5, 50mM NaCl, 1mM DTT). After the incubation, the samples were diluted into an equal volume of 2× EMSA sample buffer (40 mM HEPES pH 7.6, 100 mM NaCl, 10% Sucrose, 2 mM DTT) and 10ul was loaded onto a 6% native TBE gel at 4C. After the experiment the sample was stained with SybrGold DNA stain (Thermo) and visualized.
Cryo-EM sample preparation and structure determination
Poised state Dot1L
A 4:1 complex between Dot1L(2-416) (60 μM) and H2B-Ub nucleosome (15 μM) was prepared in crosslinking buffer (50 mM HEPES, pH8.0, 50 mM NaCl, 1mM EDTA, 0.2 mM DTT) in the presence of 1 mM 5’-(diaminobutyric acid)-N-iodoethyl-5’-deoxyadenosine ammonium hydrochloride (AAI), an N-mustard SAM analog inhibitor(Weller and Rajski, 2006) that was a generous gift from Dr. Lindsay Comstock-Ferguson (Wake Forest). The mixture was incubated for 1 hour at 30°C and then diluted to 100 nM nucleosome in crosslinking buffer. Glutaraldehyde crosslinking was initiated by adding an equal volume of 0.06% glutaraldehyde to the mixture to yield a reaction containing 50 nM H2B-Ub nucleosome, 200 nM Dot1L and 0.03% glutaraldehyde. The reaction was incubated for 1 hour on ice and then quenched by addition of 100 mM Tris, then dialyzed overnight against quenching buffer (50mM Tris pH 8.0, 50mM NaCl, 1mM DTT) which removed the AAI inhibitor. The complex was then concentrated using an Amicon Ultra spin concentrator (Millipore) and purified on a Superdex 200 16-60 size exclusion column (GE Healthcare) that was pre-equilibrated with EM buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM DTT). Peak fractions were concentrated to a final nucleosome concentration of 3.4 uM nucleosome. A sample at 1 mg/ml (2.43 uM) was frozen on 400 mesh C-Flat grids (Electron Microscopy Sciences) with 2 um holes using a Vitrobot (Thermo Fisher Scientific) with settings at 4°C, 100% humidity and 3 sec blot time.
Active state Dot1L
A 4:1 complex containing 60 uM Dot1L(2-416) 15 uM H2B-Ub nucleosomes containing the H3K79Nle mutation was prepared in crosslinking buffer supplemented with 200 mM SAM. The resulting complex was diluted to 100 nM using reaction buffer and 2 mM SAM was added to a final concentration of 160 μM. An equal volume of 0.064% glutaraldehyde in reaction buffer was added to the complex to yield a crosslinking reaction containing 50 nM H2B-Ub nucleosome, 200 nM Dot1L and 0.032% glutaraldehyde. After 1 hour on ice, the reaction was quenched with 100 mM Tris and dialyzed against quenching buffer. The crosslinked complex was purified and frozen as described for the poised complex except that the concentration of the active complex on the grid was 0.75 mg/ml.
Refinement and model building
All data were collected at the National Cryo-Electron Microscopy Facility (NCEF) at the National Cancer Institute on a Titan Krios at 300 kV utilizing a K2 direct electron detector in Super resolution electron counting-mode at a nominal magnification of 130,000 and a pixel size of 0.532 Å. For both the poised and active state Dot1L datasets, data were collected at a nominal dose of 50 e−/Å2 with 40 frames per movie and 1.25 e−/frame. A total of 2,143 super resolution movies were collected for the poised state dataset and 2,284 movies were collected for the active state dataset.
For the poised Dot1L dataset, all movies were motion-corrected, dose-weighted and binned by a factor of 2 using MotionCorr2 (Zheng et al., 2017) which resulted in a pixel size of 1.064 Å. Contrast transfer function (CTF) correction was performed with Ctffind4 (Rohou and Grigorieff, 2015). Particle picking utilizing the ab-initio particle picking mode was performed in cisTEM (Grant et al., 2018), which resulted in 868,027 particles that were subjected to 2D classification to remove junk particles. A cleaned dataset of 578,660 particles was then transferred to Relion 2.0 (Kimanius et al., 2016) for 3D classification, yielding a single class with 187,014 particles that showed density for a well-resolved Dot1L in the poised state after refinement (Figure S1). Focused classification without alignment using a mask that encompassed the Dot1L-ubiquitin complex further reduced the dataset to 108,658 particles, which were refined to 3.9 Å resolution as given by the Fourier shell correlation (FSC) 0.143 criterion (Rosenthal and Henderson, 2003; van Heel and Schatz, 2005) (4.4 Å at FSC 0.5). The final map was sharpened with an automatically calculated B-factor of −180 Å2 using the Relion post-processing tool.
The active Dot1L dataset was processed in Relion 3 (Zivanov et al., 2018). All movies were motion-corrected, dose-weighted and binned by a factor of 2 using the Relion 3 implementation of MotionCorr2 (Zivanov et al., 2018), which resulted in a pixel size of 1.064 Å. A initial batch of 143 micrographs were selected based on CTF fit statistics to pick 84,534 particles using the Laplacian auto-picking feature. Initial class averages were calculated and used to perform template-based particle picking on the entire dataset, yielding 800,727 particles. Next, 2D-classification was performed to remove junk particles, and 652,532 particles were retained and 3D-classified (Figure S1). In total two well resolved classes emerged from the 3D classification, a 2:1 complex with 2 Dot1L molecules per nucleosome and a 1:1 complex with 1 Dot1L molecule per nucleosome. The best resolving, highest-populated class corresponded to the 2:1 Dot1L-nucleosome complex and contained 237,780 particles. A masked refinement of the 2:1 structure including the nucleosome and the upper, better resolved Dot1L and imposed C2 symmetry yielded a 3.5 Å reconstruction. The 2:1 Dot1L complex particle stack was then subjected to 2 iterations of CTF refinement, beam tilt correction and Bayesian polishing as implemented in Relion 3.0. Refinement of the shiny particles using C2 symmetry and a mask that encompassed the nucleosome and the upper Dot1L and yielded a higher resolving reconstruction which was filtered using the Relion post-processing tool and sharpened with an automatically calculated B-factor of −73 Å2. The final resolution of the 2:1 active state structure according to the FSC 0.143 criterion (Rosenthal and Henderson, 2003) is 2.96 Å (3.3 Å at FSC 0.5). A consensus refinement of the 1:1 active state complex with no symmetry applied, yielded a 3.9 Å resolution structure according to the FSC 0.143 criterion. The 1:1 Dot1L complex particle stack was then subjected to 1 iteration of CTF refinement, beam tilt correction and Bayesian polishing as implemented in Relion 3.0. Gold standard refinement of the shiny particles with no imposed symmetry resulted in the final 3.5 Å structure (0.143 criterion). The final reconstruction of the 1:1 complex was sharpened using a soft mask encompassing the entire density and a an automatically calculated B-factor of −96.5 Å2
The local resolution of all three reconstructions was assessed using ResMap (Kucukelbir et al., 2014) by supplying the unfiltered half-maps as inputs (Figure S2).
Model Building and Refinement
PDB crystal structures for the core histones (1kx3), isolated Dot1L residues 5-332 (1nw3), 4-330 (3qow), 601 DNA (3MVD) and ubiquitin (1ubq) were rigid-body fitted in the maps for the poised structure (3qow) and active state 2:1 structure (1nw3) using UCSF Chimera (Goddard et al., 2007). Next, both Dot1L-nucleosome structures were manually rebuilt using COOT (Emsley et al., 2010) and extended in several regions where the density allowed. Both models were subjected to alternating local and global real-space refinement using PHENIX (Adams et al., 2010) and COOT (Emsley et al., 2010). Due to imposing C2 symmetry on the 2:1 active state complex, only one of the 2 Dot1L and ubiquitin molecules was modeled in the final structure. After initial refinement of the active state atomic model into the high resolution 2:1 structure, the complete active state model was rigid body fit into the density for the 1:1 structure, rebuilt using COOT and refined using PHENIX. To assess for overfitting and avoid overinterpretation, we refined the models of poised state structure, the 2:1 active state structure and the 1:1 active state structure against one of the sharpened, masked half-maps of the respective dataset (mapwork) and validated the structure against the second half-map (maptest), similarly sharpened and masked. Specifically, the poised Dot1L reconstruction was refined using reference restraints from ubiquitin (PDB ID 4XOF), Dot1L (PDB ID 3QOW) and histone H2A (PDB ID 1KX3) as well as secondary structure restraints to better account for the lower resolved Dot1L density. We also calculated individual atomic displacement parameters (ADP’s) by optimizing the real-space cross correlation between the model and the experimental map as implemented in phenix.real_space_refine (Afonine et al., 2018). The active Dot1l reconstructions were iteratively refined against the sharpened half-maps applying alternating rounds of local and global refinements. Additional secondary structure restraints and reference restraints for the ubiquitin (1UBQ) were applied and ADP’s were calculated. The final structures were validated against the second half-map of the respective reconstruction (maptest) (Figure S2) and the FSC between the model and each half map (FSCwork and FSCtest) and the full map (FSCFull) was determined. The computed FSC between the model and the second half-map (FSCtest) agrees well with the FSC of the refined model against the target map used for refinement (FSCwork). The resolution estimate of the model/map FSCFull agrees well with the FSC 0.5 cutoff applied between the two half maps (Table 1 and Figure S2) and in all model/map FSC calculations, no significant correlation is measured that exceeds the calculated map resolutions. Both observations indicate that the built structures were not overfit nor overinterpreted versus their refinement targets. The final structures show excellent stereochemistry (see Table 1), as assessed by Molprobity (Chen et al., 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Michaelis Menten Data were fit using the Graphpad Prism curve fitting software. Fits were calculated from 3 replicate titrations and the reported standard errors of the fitted Michaelis Menten coefficients were determined using the Graphpad Prism software. For endpoint Methylation assays the raw results were averaged and the standard deviation of the data is reported.
DATA AND SOFTWARE AVAILABILITY
The cryo-EM maps described in this study have been deposited in the EMDB with accession codes EMD-9384 (2.96 A active 2:1 Dot1L-nucleosome), EMD-0480 (3.5 A active 1:1 Dot1L-nucleosome) and EMD-0468 (3.9 Å poised Dot1L-nucleosome). Coordinates of the corresponding atomic models were deposited in the Protein Data Bank under accession code 6NJ9 (active 2:1 Dot1L-nucleosome), 6NQA (active 1:1 Dot1L-nucleosome) and 6NOG (poised Dot1L-nucleosome).
The raw 2D micrographs of the active state and poised state datasets will be uploaded to the EMPIAR database.
Supplementary Material
Figure S1: Cryo-EM processing pipelines, Related to Figure 1
A, EM processing pipeline for the Poised state structure. The displayed micrograph is low-pass filtered to 20Å for improved particle visibility. B, EM processing pipline for the Active state structures. The displayed micrograph is low-pass filtered to 20Å for improved particle visibility. The Relion 3 particle-based CTF-refinement, beamtilt correction and Bayesian polishing tools were applied in an iterative manner until there was no further change in the resolution or maps.
Figure S2: Cryo-EM map and model validation, Related to Figure 1
A, Left, Fourier Shell Correlation (FSC) and, right, local resolution assessment by Resmap for the poised Dot1L-nucleosome reconstruction. The final resolution as determined by FSC 0.143 criterion is 3.9 Å. The local resolution in Å is displayed on the sharpened full map (middle = surface, right = cross section). B, FSC and local resolution assessment for the active 2-to-1 Dot1L-nucleosome structure depicted as in A. The final resolution is 2.96Å as determined by the FSC 0.143 criterion. C, FSC and local resolution assessment for the active 1-to-1 Dot1L-nucleosome structure depicted as in a and B. The final resolution is 3.5Å as determined by the FSC 0.143 criterion. D-E, Model-Map FSC curves calculated between the refined atomic models and the masked, sharpened half maps used for refinement (Red, FSCwork), the second masked, sharpened half-map not used in refinement (Blue, FSCtest) and the full, sharpened map (Black, FSCFull). Model-Map FSCs are shown for the Poised structure, D, the 2-to-1 active structure, E, and the 1-to-1 active structure, F. The close agreement between the FSCwork and FSCtest curves and the absence of significant correlation beyond the calculated map resolution for all three curves indicates absence of overfitting and model bias.
Figure S3: Details of the poised state structure, Related to Figure 1, 3 and 4
A, EM density for the poised-state structure. The unsharpened density is shown as a transparent surface and the sharpened density is shown as an opaque surface colored as in the main text. The N- and C-terminal parts of Dot1L are denoted with C and N. B, The poised state structure atomic model colored as in the main text. C, Close up view of Dot1L in the poised state showing that the only direct contact between Dot1L and the nucleosome is through the acidic patch. The F131 and W305 loops do not have defined density in the poised state and are depicted as yellow dashed lines. D, Detailed view of the poised state interaction with the acidic patch superimposed with the active state structure. The poised state structure is colored yellow and the active state structure is colored green. Sharpened EM density for the poised state structure is shown as a transparent gray surface and acidic patch residues of H2A and H2B are shown as sticks. E, Close up view of the poised state Dot1L active site showing that a modeled SAM cofactor is too far for the methyl transfer reaction to occur.
Figure S4: Example density of the 2:1 active-state EM structure, Related to Figure 1
A, A vertical slice through the 2:1 active state structure centered on H3K79Nle. Atomic models of all chains are shown as sticks and the EM density is shown as a gray mesh. Areas highlighted in the boxes are denoted with letters b, c, d and e. B, Example density of the SAM cofactor. C, Example density of the interaction between ubiquitin and the Dot1L C-terminal helix. D,E, Example EM density of the DNA.
Figure S5: Structural Comparisons of Dot1L in different states, Related to Figure 3 and 4
A Superimposition between the active state Dot1L and the Dot1L crystal structure (PDB ID: 1NW3). B, Superimposition between the active state and poised sate Dot1L. The disordered Dot1L F131 and W305 loops in the poised state are depicted as dashed yellow lines. C, Close up view of loop restructuring that occurs in Dot1L during the transition to the active state. The active state loops are colored red and the loops from the crystal structure of Dot1L (PDB ID: 1NW3) are colored blue. D, Close up view of the superimposed Dot1L-ubiquitin interaction in the poised and active states showing that the contact surface does not change. The convex surface made up by the Dot1L C-terminal helix and the β8- β9 loop is depicted as a dashed line. E, Surface representation of the nucleosome colored by electrostatic potential. The highly charged acidic patch is indicated by the black square. F-J, Close up views of the arginine anchor interaction with the acidic patch for different protein-nucleosome complexes. F, Dot1L, G, LANA peptide (PDB: 1ZLA), H, RCC1 (PDB: 3MVD), I, Sir3 (3TU4), J, PRC1 Ubiquitylation Module (4R8P).
Figure S6: Details of the H4 tail interaction with Dot1L, Related to Figure 5
A, Example density of the H4 tail in the poised state structure is depicted as a gray mesh and shows that residues after K20 are not structured. B The active state Dot1L structure (red) is superimposed with several high resolution nucleosome crystal structures (PDB ID: 1KX3,1KX5, 5YOD, 1S32, 3UTA, 5Y0C, 3C1B, 3UT9 and 3UTB) colored tan showing that the H4 tail is usually unstructured or associated with DNA. C, The Dot1L crystal structure (PDB ID: 1NW3) is depicted in cartoon representation and colored brown with a semi-transparent gray surface. The groove for H4 tail is closed in this state. D, The active Dot1L structure is depicted in cartoon representation and colored green with a semi-transparent gray surface. The groove for the H4 tail is open in this state.
Figure S7: Details of the H3K79 conformational change, Related to Figure 6 and 7
A, The active state Dot1L structure (blue) is superimposed with the poised Dot1L structure (yellow) and several high resolution nucleosome crystal structures (PDB ID: 1KX3,1KX5, 5YOD, 1S32, 3UTA, 5Y0C, 3C1B, 3UT9 and 3UTB) colored tan showing that the sidechain of H3K79 is always held in an inaccessible state. B, Superimposition of the active state Dot1L (green) the yeast Dot1p crystal structure (PDB ID: 1U2Z, gray) showing the relative conformations of the W305 and F131 loops. The yeast Dot1p residue W543 is in a similar position as W305 in Dot1L. C, Overview of the 1-to-1 active state structure. D, Close up view of the H3K79Nle loop on the side of the nucleosome that is bound by Dot1L showing that H3K79Nle is oriented upward. E, Close up view of the H3K79Nle loop on the side of the nucleosome that is not bound by Dot1L, showing that H3K79Nle is oriented in the inaccessible conformation. F, superimposition between active state Dot1L (green) and several structures of inhibitor bound Dot1L (gray, PDB IDs: 4EKG, 4EKI, 3UWP, 4EQZ, 4ER0, 4ER3, 4ER5, 4ER6, 4ER7,4HRA, 5MW3, 4WV1, 5DTM, 5DTQ, 5DTR, 5DRT, 5DRY, 5DSX, 5DT2, 5MVS, 5MW4) showing that the F131 loop (blue) restructures upon inhibitor binding. F131 in the various inhibitorbound structures is shown in red stick representation.
Highlights.
Cryo-EM structures of Dot1L methyltransferase bound to H2B-ubiquitinated nucleosomes
Dot1L bound in poised and active states contacts the ubiquitin I36 hydrophobic patch
The tail of histone H4 binds in a cleft in Dot1L.
A conformational change is induced in histone H3 that enables Dot1L to access H3K79
Editor’s suggested In Brief:
Unanticipated conformational plasticity in the globular core of histone H3 underlies crosstalk between histone modifications.
Acknowledgements
We thank Xiangbin Zhang for purifying mutants and Duncan Sousa for advice. This research was supported, in part, by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. E.J.W. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG 2308-17), N.A.H. is supported by an EMBO Long-term Fellowship (EMBO-ALTF 309-2017), and C.W.H. is supported by training grant T32-GM007445. Supported by grant GM095822 from the National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
C.W. is a member of the scientific advisory board of ThermoFisher Scientific.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaf M, Utley RT, Lacoste N, Tan S, Briggs SD, and Côté J (2007). Interplay of Chromatin Modifiers on a Short Basic Patch of Histone H4 Tail Defines the Boundary of Telomeric Heterochromatin. Molecular Cell 28, 1002–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armache KJ, Garlick JD, Canzio D, Narlikar GJ, and Kingston RE (2011). Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 A resolution. Science 334, 977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K , and Kaye KM (2006). The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861. [DOI] [PubMed] [Google Scholar]
- Basavapathruni A, Jin L, Daigle SR, Majer CRA, Therkelsen CA, Wigle TJ, Kuntz KW, Chesworth R, Pollock RM, Scott MP, et al. (2012). Conformational Adaptation Drives Potent, Selective and Durable Inhibition of the Human Protein Methyltransferase DOT1L. Chemical Biology and Drug Design 80, 971–980. [DOI] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. (2011). MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Xiao T, Sun Z-W, Caldwell JA, Shabanowitz J, Hunt DF, Allis CD, and Strahl BD (2002). Trans-histone regulatory pathway in chromatin. Nature 418, 498. [DOI] [PubMed] [Google Scholar]
- Brown ZZ, Müller MM, Jain SU, Allis CD, Lewis PW, and Muir TW (2014). Strategy for “Detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. Journal of the American Chemical Society 136, 13498–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhu H, Stauffer F.d.r., Caravatti G., Vollmer S, Machauer R., Holzer P., M??bitz H., Scheufler., Klumpp M., et al. (2016). Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Medicinal Chemistry Letters 7, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CW, and Armstrong SA (2015). Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Experimental Hematology 43, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. (2013). Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, and Richmond TJ (2002). Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, and Luger K (2004). Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods in enzymology 375, 23–44. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English CM, Adkins MW, Carson JJ, Churchill ME, and Tyler JK (2006). Structural basis for the histone chaperone activity of Asf1. Cell 127, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, and Zhang Y (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Fingerman IM, Li HC, and Briggs SD (2007). A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: Identification of a new trans-histone pathway. Genes and Development 21, 2018–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Plevani P, and Muzi-Falconi M (2005). The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem 280, 9879–9886. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, and Ferrin TE (2007). Visualizing density maps with UCSF Chimera. Journal of Structural Biology 157, 281–287. [DOI] [PubMed] [Google Scholar]
- Grant T., Rohou A., and Grigorieff N. (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, Croce CM, Nakamura T, Canaani E, and Young RA (2008). Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes and Development 22, 3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt MT, David Y, Pollock S, Tang Z, Jeon J, Kim J, Roeder RG, and Muir TW (2015). Identification of a functional hotspot on ubiquitin required for stimulation of methyltransferase activity on chromatin. Proceedings of the National Academy of Sciences of the United States of America 112, 10365–10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PL, Li H, Lau H-T, Leonen C, Dhall A, Ong S-E, Chatterjee C, and Zheng N (2018). Crystal Structure of the COMPASS H3K4 Methyltransferase Catalytic Module. Cell 0, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio R.a., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston E.a., Mellert HS, Stavridi ES, and Halazonetis TD (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411. [DOI] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, and Garcia-Moreno B (2011). Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci U S A 108, 5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, Allis CD, Cummings R, Bellon S, Lewis PW, et al. (2016). S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Pnas 113, 6182–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Kim R, Park G, Park JW, and Kim JE (2012). Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem 287, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, and Rape M (2012). The Ubiquitin Code. Annual Review of Biochemistry 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Kouskouti A, and Talianidis I (2005). Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J 24, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, et al. (2008). H3K79 Methylation Profiles Define Murine and Human MLL-AF4 Leukemias. Cancer Cell 14, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, et al. (2003). The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11, 721–729. [DOI] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, Adli M, Zeleznik-Le NJ, and Bushweller JH (2014). Degree of Recruitment of DOT1L to MLL-AF9 Defines Level of H3K79 Di- and Tri-methylation on Target Genes and Transformation Potential. Cell Reports 11, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of Molecular Biology 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, and Luger K (2008). The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nature structural & molecular biology 15, 1122–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, and Richmond TJ (1999). Preparation of nucleosome core particle from recombinant histones. Methods in enzymology 304, 3–19. [DOI] [PubMed] [Google Scholar]
- Makde RD, England JR, Yennawar HP, and Tan S (2010). Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Henrici RC, and Tan S (2014). Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Kim J, Chatterjee C, Roeder RG, and Muir TW (2008). Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Köhn M, Chatterjee C, Chiang KP, Pratt MR, and Muir TW (2009). Structure-activity analysis of semisynthetic nucleosomes: Mechanistic insights into the stimulation of Dot1L by ubiquitylated histone H2B. ACS Chemical Biology 4, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, and Shilatifard A (2001). COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proceedings of the National Academy of Sciences of the United States of America 98, 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, and Xu RM (2003). Structure of the catalytic domain of human Dot1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723. [DOI] [PubMed] [Google Scholar]
- Mobitz H, Machauer R, Holzer P, Vaupel A, Stauffer F, Ragot C, Caravatti G, Scheufler C, Fernandez C, Hommel U, et al. (2017). Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach. ACS Med Chem Lett 8, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MT, Haj-Yahya M, Ringel AE, Bandi P, Brik A, and Wolberger C (2016). Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science 351, 725–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, and Struhl K (2002a). Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev 16, 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Xu RM, Zhang Y, and Struhl K (2002b). Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. Journal of Biological Chemistry 277, 34655–34657. [DOI] [PubMed] [Google Scholar]
- Ng HH, Xu RM, Zhang Y, and Struhl K (2002c). Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem 277, 34655–34657. [DOI] [PubMed] [Google Scholar]
- Qu Q, Takahashi Y. h., Yang Y, Hu H, Zhang Y, Brunzelle JS, Couture J-F, Shilatifard A, and Skiniotis G (2018). Structure and Conformational Dynamics of a COMPASS Histone H3K4 Methyltransferase Complex. Cell 0, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva F, Scott MP, and Copeland RA (2011). Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des 78, 199–210. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, and Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Sawada K, Yang Z, Horton JR, Collins RE, Zhang X, and Cheng X (2004). Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. The Journal of biological chemistry 279, 43296–43306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Mobitz H, Gaul C, Ragot C, Be C, Fernandez C, Beyer KS, Tiedt R, and Stauffer F (2016). Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med Chem Lett 7, 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha KK, Gross JD, and Narlikar GJ (2017). Distortion of histone octamer core promotes nucleosome mobilization by a chromatin remodeler. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim J.-e., Chen J, Lazar MA, et al. (2008). DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Molecular and cellular biology 28, 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, and Workman JL (2008). Crosstalk among Histone Modifications. Cell 135, 604–607. [DOI] [PubMed] [Google Scholar]
- Sun Z-W, and Allis CD (2002). Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108. [DOI] [PubMed] [Google Scholar]
- Takahashi YH, Schulze JM, Jackson J, Hentrich T, Seidel C, Jaspersen SL, Kobor MS, and Shilatifard A (2011). Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell 42, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M, and Schatz M (2005). Fourier shell correlation threshold criteria. J Struct Biol 151, 250–262. [DOI] [PubMed] [Google Scholar]
- Van Hest JCM, Kiick KL, and Tirrell DA (2000). Efficient incorporation of unsaturated methionine analogues into proteins in vivo. Journal of the American Chemical Society 122, 1282–1288. [Google Scholar]
- Van Leeuwen F, Gafken PR, and Gottschling DE (2002). Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756. [DOI] [PubMed] [Google Scholar]
- Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, Zhu J, Roeder RG, and Vakoc CR (2013). Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proceedings of the National Academy of Sciences of the United States of America 110, 3901–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RL, and Rajski SR (2006). Design, synthesis, and preliminary biological evaluation of a DNA methyltransferase-directed alkylating agent. ChemBioChem 7, 243–245. [DOI] [PubMed] [Google Scholar]
- Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, Dean K, Golshani A, Zhang Y, Greenblatt JF, et al. (2003). Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell 11, 267–274. [DOI] [PubMed] [Google Scholar]
- Wysocki R, Javaheri A, Allard S, Sha F, Cote J, and Kron SJ (2005). Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol 25, 8430–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Federation AJ, Qi J, Dhe-Paganon S, Hadler M, Xu X, St Pierre R, Varca AC, Wu L, Marineau JJ, et al. (2015). Structure-guided dOt1L probe optimization by label-free ligand displacement. ACS Chem Biol 10, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, et al. (2012). Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 3, 1288. [DOI] [PubMed] [Google Scholar]
- Zhang L, Serra-Cardona A, Zhou H, Wang M, Yang N, Zhang Z, and Xu RM (2018). Multisite Substrate Recognition in Asf1-Dependent Acetylation of Histone H3 K56 by Rtt109. Cell 174, 818–830 e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Holt MT, Ohashi N, Zhao A, Möller MM, Wang B, and Muir TW (2016). Evidence that ubiquitylated H2B corrals hDot1L on the nucleosomal surface to induce H3K79 methylation. Nature communications 7, 10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Zheng Y, Pham A.-d., Mandal SS, Erdjument-bromage H, Tempst P, and Reinberg D (2005). Monoubiquitination of Human Histone H2B : The Factors Involved and Their Roles in HOX Gene Regulation. 20, 601–611. [DOI] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Cryo-EM processing pipelines, Related to Figure 1
A, EM processing pipeline for the Poised state structure. The displayed micrograph is low-pass filtered to 20Å for improved particle visibility. B, EM processing pipline for the Active state structures. The displayed micrograph is low-pass filtered to 20Å for improved particle visibility. The Relion 3 particle-based CTF-refinement, beamtilt correction and Bayesian polishing tools were applied in an iterative manner until there was no further change in the resolution or maps.
Figure S2: Cryo-EM map and model validation, Related to Figure 1
A, Left, Fourier Shell Correlation (FSC) and, right, local resolution assessment by Resmap for the poised Dot1L-nucleosome reconstruction. The final resolution as determined by FSC 0.143 criterion is 3.9 Å. The local resolution in Å is displayed on the sharpened full map (middle = surface, right = cross section). B, FSC and local resolution assessment for the active 2-to-1 Dot1L-nucleosome structure depicted as in A. The final resolution is 2.96Å as determined by the FSC 0.143 criterion. C, FSC and local resolution assessment for the active 1-to-1 Dot1L-nucleosome structure depicted as in a and B. The final resolution is 3.5Å as determined by the FSC 0.143 criterion. D-E, Model-Map FSC curves calculated between the refined atomic models and the masked, sharpened half maps used for refinement (Red, FSCwork), the second masked, sharpened half-map not used in refinement (Blue, FSCtest) and the full, sharpened map (Black, FSCFull). Model-Map FSCs are shown for the Poised structure, D, the 2-to-1 active structure, E, and the 1-to-1 active structure, F. The close agreement between the FSCwork and FSCtest curves and the absence of significant correlation beyond the calculated map resolution for all three curves indicates absence of overfitting and model bias.
Figure S3: Details of the poised state structure, Related to Figure 1, 3 and 4
A, EM density for the poised-state structure. The unsharpened density is shown as a transparent surface and the sharpened density is shown as an opaque surface colored as in the main text. The N- and C-terminal parts of Dot1L are denoted with C and N. B, The poised state structure atomic model colored as in the main text. C, Close up view of Dot1L in the poised state showing that the only direct contact between Dot1L and the nucleosome is through the acidic patch. The F131 and W305 loops do not have defined density in the poised state and are depicted as yellow dashed lines. D, Detailed view of the poised state interaction with the acidic patch superimposed with the active state structure. The poised state structure is colored yellow and the active state structure is colored green. Sharpened EM density for the poised state structure is shown as a transparent gray surface and acidic patch residues of H2A and H2B are shown as sticks. E, Close up view of the poised state Dot1L active site showing that a modeled SAM cofactor is too far for the methyl transfer reaction to occur.
Figure S4: Example density of the 2:1 active-state EM structure, Related to Figure 1
A, A vertical slice through the 2:1 active state structure centered on H3K79Nle. Atomic models of all chains are shown as sticks and the EM density is shown as a gray mesh. Areas highlighted in the boxes are denoted with letters b, c, d and e. B, Example density of the SAM cofactor. C, Example density of the interaction between ubiquitin and the Dot1L C-terminal helix. D,E, Example EM density of the DNA.
Figure S5: Structural Comparisons of Dot1L in different states, Related to Figure 3 and 4
A Superimposition between the active state Dot1L and the Dot1L crystal structure (PDB ID: 1NW3). B, Superimposition between the active state and poised sate Dot1L. The disordered Dot1L F131 and W305 loops in the poised state are depicted as dashed yellow lines. C, Close up view of loop restructuring that occurs in Dot1L during the transition to the active state. The active state loops are colored red and the loops from the crystal structure of Dot1L (PDB ID: 1NW3) are colored blue. D, Close up view of the superimposed Dot1L-ubiquitin interaction in the poised and active states showing that the contact surface does not change. The convex surface made up by the Dot1L C-terminal helix and the β8- β9 loop is depicted as a dashed line. E, Surface representation of the nucleosome colored by electrostatic potential. The highly charged acidic patch is indicated by the black square. F-J, Close up views of the arginine anchor interaction with the acidic patch for different protein-nucleosome complexes. F, Dot1L, G, LANA peptide (PDB: 1ZLA), H, RCC1 (PDB: 3MVD), I, Sir3 (3TU4), J, PRC1 Ubiquitylation Module (4R8P).
Figure S6: Details of the H4 tail interaction with Dot1L, Related to Figure 5
A, Example density of the H4 tail in the poised state structure is depicted as a gray mesh and shows that residues after K20 are not structured. B The active state Dot1L structure (red) is superimposed with several high resolution nucleosome crystal structures (PDB ID: 1KX3,1KX5, 5YOD, 1S32, 3UTA, 5Y0C, 3C1B, 3UT9 and 3UTB) colored tan showing that the H4 tail is usually unstructured or associated with DNA. C, The Dot1L crystal structure (PDB ID: 1NW3) is depicted in cartoon representation and colored brown with a semi-transparent gray surface. The groove for H4 tail is closed in this state. D, The active Dot1L structure is depicted in cartoon representation and colored green with a semi-transparent gray surface. The groove for the H4 tail is open in this state.
Figure S7: Details of the H3K79 conformational change, Related to Figure 6 and 7
A, The active state Dot1L structure (blue) is superimposed with the poised Dot1L structure (yellow) and several high resolution nucleosome crystal structures (PDB ID: 1KX3,1KX5, 5YOD, 1S32, 3UTA, 5Y0C, 3C1B, 3UT9 and 3UTB) colored tan showing that the sidechain of H3K79 is always held in an inaccessible state. B, Superimposition of the active state Dot1L (green) the yeast Dot1p crystal structure (PDB ID: 1U2Z, gray) showing the relative conformations of the W305 and F131 loops. The yeast Dot1p residue W543 is in a similar position as W305 in Dot1L. C, Overview of the 1-to-1 active state structure. D, Close up view of the H3K79Nle loop on the side of the nucleosome that is bound by Dot1L showing that H3K79Nle is oriented upward. E, Close up view of the H3K79Nle loop on the side of the nucleosome that is not bound by Dot1L, showing that H3K79Nle is oriented in the inaccessible conformation. F, superimposition between active state Dot1L (green) and several structures of inhibitor bound Dot1L (gray, PDB IDs: 4EKG, 4EKI, 3UWP, 4EQZ, 4ER0, 4ER3, 4ER5, 4ER6, 4ER7,4HRA, 5MW3, 4WV1, 5DTM, 5DTQ, 5DTR, 5DRT, 5DRY, 5DSX, 5DT2, 5MVS, 5MW4) showing that the F131 loop (blue) restructures upon inhibitor binding. F131 in the various inhibitorbound structures is shown in red stick representation.
Data Availability Statement
The cryo-EM maps described in this study have been deposited in the EMDB with accession codes EMD-9384 (2.96 A active 2:1 Dot1L-nucleosome), EMD-0480 (3.5 A active 1:1 Dot1L-nucleosome) and EMD-0468 (3.9 Å poised Dot1L-nucleosome). Coordinates of the corresponding atomic models were deposited in the Protein Data Bank under accession code 6NJ9 (active 2:1 Dot1L-nucleosome), 6NQA (active 1:1 Dot1L-nucleosome) and 6NOG (poised Dot1L-nucleosome).
The raw 2D micrographs of the active state and poised state datasets will be uploaded to the EMPIAR database.