

Abstract
Benzosuberene analogues (1 and 2) and dihydronaphthalene analogues (3 and 4) function as potent inhibitors of tubulin polymerization, demonstrate pronounced cytotoxicity (low nM to pM range) against human cancer cell lines, and are promising vascular disrupting agents (VDAs). As such, these compounds represent lead anticancer agents with potential translatability towards the clinic. Methodology previously established by us (and others) facilitated synthetic access to a variety of structural and functional group modifications necessary to explore structure activity relationship considerations directed towards the development of these (and related) molecules as potential therapeutic agents. During the course of these studies it became apparent that the availability of synthetic methodology to facilitate direct conversion of the phenolic-based compounds to their corresponding aniline congeners would be beneficial. Accordingly, modified synthetic routes toward these target phenols (benzosuberene 1 and dihydronaphthalene 3) were developed in order to improve scalability and overall yield [45–57% (1) and 32% (3)]. Moreover, benzosuberene-based phenolic analogue 1 and separately dihydronaphthalene-based phenolic analogue 3 were successfully converted into their corresponding aniline analogues 2 and 4 in good yield (>60% over three steps) using a palladium catalyzed amination reaction.
Keywords: Benzosuberene analogues, Dihydronaphthalene analogues, Direct conversion of phenolic moieties to aniline moieties, Small-molecule inhibitors of tubulin polymerization
Graphical Abstract
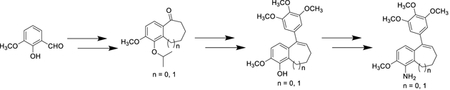
The discovery and development of small-molecule inhibitors of tubulin polymerization as anticancer therapeutics represents an important field of research inquiry that often draws structural inspiration from natural products. We have previously reported benzosuberene (1 and 2) and dihydronaphthalene (3 and 4) analogues (phenolic and aniline-based, respectively) that function as potent inhibitors of tubulin polymerization and demonstrate enhanced cytotoxicity against a variety of human cancer cell lines [low nM to pM range (Fig. 1)].1–7 The structures of these molecules are reminiscent of the natural products colchicine and combretastatin A-4 (Fig. 1),8–10 which are potent inhibitors of tubulin polymerization.11 These molecules interact with the tubulin-microtubule protein system at the colchicine binding site (situated on the tubulin heterodimer) and inhibit microtubule formation. Associated with their tubulin-based mechanism of action, these compounds also disrupt tumor-associated vasculature and thus function as vascular disrupting agents (VDAs).1,3,4,12,13 Solid tumors increasingly require nutrients and oxygen provided by a network of vasculature, which has distinct structural and architectural differences compared with vasculature associated with normal healthy tissue.14 Tumor-associated vasculature is highly disorganized with abnormal bulges, blind ends and shunts.15 It is also characterized as leaky and discontinuous. Collectively, these physiological dissimilarities offer a therapeutic advantage for the selective targeting and disruption of tumor-associated vasculature with VDAs.13
Figure 1.

Natural products (colchicine and combretastatin A-4), and synthetic benzosuberene and dihydronaphthalene analogues as small-molecule inhibitors of tubulin polymerization.
Synthetic routes to each of the four key molecules (1 – 4) were previously established by our laboratory.1,3–6 The synthesis of compounds 1 and 3 was later reported by other groups utilizing different synthetic approaches to these molecular scaffolds.16–18 Our reported1,3,7 synthetic routes towards compounds 1 and 2 were quite similar and involved a Wittig reaction followed by Eaton’s reagent mediated intramolecular Friedel-Crafts acylation to provide the six-seven fused ring system (Scheme 1). One of the critical steps in the synthesis of compound 1 involved microwave assisted regioselective demethylation of an aromatic methoxy group using an ionic liquid [TMAH][Al2Cl7] to generate the corresponding free phenol. In the case of amino functionalized benzosuberene 2, a nitro group was carried through the synthesis and ultimately reduced to reveal the aniline functionality. While these synthetic routes proved to be robust and reproducible, they proceeded in relatively low overall yield [compound 1 (12% over seven steps) and compound 2 (18% over six steps)]. Our original synthetic routes (Scheme 1) to the functionalized dihydronaphthalene analogues were somewhat laborious and involved regioselective oxidation of 6-methoxytetralin to sequentially introduce a hydroxy group and a benzylic ketone in the case of phenolic-based dihydronaphthalene 3. In the case of amino-dihydronaphthalene 4, a regioselective nitration of 6-methoxytetralone was followed by reduction. The overall reported yields for the dihydronaphthalene series were also fairly low [compound 3 (8% over ten steps) and compound 4 (17% over three steps)]. Therefore, we sought to modify our methodology to improve both overall yield and atom economy. We recently described one example of an improved synthesis of dihydronaphthalene 3 that proceeds through an aryl bromide intermediate.19
Scheme 1.

Previously reported synthetic route by Pinney and co-workers toward benzosuberene and dihydronaphthalene analogues 1, 2, 3, and 4.1,3,6,7
Our modified methodology directed towards the synthesis of benzosuberene 1 along with methodology to facilitate the direct conversion of phenolic benzosuberene 1 to its corresponding aniline congener 2 (in three steps using a palladium catalyzed amination reaction) is detailed in Scheme 2. In this revised methodology an isopropyl protecting group replaced the methoxy group utilized previously. The isopropyl group undergoes facile deprotection using fairly mild reaction conditions whereas demethylation requires relatively harsh reaction conditions.
Scheme 2.

Modified synthetic routes to compounds 1 and 2. Reagents and reaction conditions: (a) compound 5 (1.0 eq.), K2CO3 (1.5 eq.), 2-iodopropane (2.0 eq.), DMF, 50–60 °C, 20 h, 98%. (b) (i) 3-(carboxypropyl)triphenylphosphonium bromide (1.5 eq.), potassium tert-butoxide (3.5 eq.), compound 6 (1.0 eq.), DMF, 0 °C to RT, 24 h; (ii) Pd-C (0.1 eq., 10 wt%), H2 balloon, CH3OH, 24 h, 85–94% (two steps). (c) (i) compound 7 (1.0 eq.), oxalyl chloride (2.0 eq.), DMF (0.2 eq.), CH2Cl2, RT, 2 h; (ii) SnCl4 (1.2 eq.), CH2Cl2, −10 °C, 40 min, 75–80% (two steps). (d) (i) 5-bromo-1,3,4-trimethoxybenzene (2.0 eq.), n-BuLi (2.0 eq.), THF, −78 °C, 30 min. (ii) compound 8 (1.0 eq.), −78 °C, 4 h, RT, 16 h, 84%. (e) compound 9 (1.0 eq.), boron trichloride (1.1 eq.), CH2Cl2, 0 °C, 2 h, 85–92%. (f) compound 1 (1.0 eq.), triethylamine (2.0 eq.), triflic anhydride (1.5 eq.), CH2Cl2, 0 °C to RT, 5 h, 80–92%. (g) (i) compound 10 (1.0 eq.), benzophenone imine (1.5 eq.), Cs2CO3 (1.5 eq.), palladium(II) acetate (0.1 eq.), racemic-BINAP (0.15 eq.), toluene, 110–115 °C, 36 h; (ii) 2 M HCl, RT, 1 h, THF, 79–93% (two steps).
Benzaldehyde 6 was prepared by protection of the free phenolic moiety of commercially available (and inexpensive) benzaldehyde 5 by a nucleophilic substitution reaction with 2-iodopropane in quantitative yield. Installation of the side chain using a Wittig reaction proceeded in an analogous fashion to our original synthetic route with minor improvements in yield realized by switching the solvent from THF to DMF and carrying the Wittig reaction product forward to the next step prior to purification. Benzaldehyde 6, upon treatment with (3-carboxypropyl)-triphenylphosphonium bromide generated a mixture of alkenes (E and Z) that were subjected to hydrogenation to afford carboxylic acid 7 (up to 94% yield over two steps without the need for purification of the intermediate alkene mixture). The core benzosuberone 8 was obtained over two steps in a one-pot reaction. Initially, carboxylic acid 7 was converted to its corresponding acyl chloride using oxalyl chloride. Subsequently, an intramolecular Friedel-Crafts acylation reaction utilizing the crude acyl chloride under mild Lewis acid conditions (SnCl4) generated the requisite benzosuberone 8 (80% yield over two steps) with preservation of the isopropyl protecting group.20 This one-pot, two-step reaction sequence was preferred over the one step cyclization using Eaton’s reagent (in our previously reported route), which resulted in concomitant (undesirable) cleavage of the isopropyl group (up to 30% yield of deprotected phenolic ketone, from a reaction mixture that included deprotected phenolic carboxylic acid). There were also practicality and safety considerations (due to highly exothermic work up procedure) associated with the use of Eaton’s reagent on large scale. Compound 9 was obtained (84% yield) through an addition-elimination reaction between in situ generated 3, 4, 5-trimethoxyphenyllithium and ketone 8, which was beneficial since it obviated the need for a separate elimination reaction (through acidic work-up or a separate AcOH mediated elimination) of the intermediate tertiary alcohol that was necessary in our original procedure. Finally, target compound 1 was obtained (92% yield) by selective deprotection of the isopropyl group with boron trichloride followed by acidic (HCl) work-up.. This modified procedure facilitated the preparation of phenolic-based benzosuberene 1 in an overall yield of 45–57% which is approximately four times higher than our previously reported procedure.1,7
Compound 1 was successfully converted (via triflate 10) directly to compound 2 through an optimized palladium catalyzed amination reaction. Triflate 10 was initially synthesized by reaction of compound 1 with triflic anhydride in 92% yield. Subsequently, triflate 10 was converted to the target aniline-based benzosuberene analogue 2 through application of the well-known Buchwald-Hartwig cross coupling reaction, which we optimized for our specific substrate.21 Triflate 10 was heated (toluene at 110 °C) with benzophenone imine in the presence of catalytic palladium acetate and racemic-BINAP under basic conditions for 36 h in a sealed tube. The corresponding benzophenone-based imine was formed initially, which under acidic condition hydrolyzed to generate compound 2 in 79–93% yield (over two steps). The overall yield of compound 2 from compound 1 varied from 63–86% (over three steps) which is three to four times higher than the original procedure (Scheme 1).
While our original synthetic methodology towards dihydronaphthalene analogues involved reactions that initiated from existing fused aromatic-cycloalkane or -alkanone six-membered ring systems,5,6 we subsequently envisioned the synthesis of dihydronaphthalene analogues 3 and 4 through ring-forming methodology similar to that depicted in Scheme 1 for the related benzosuberene analogues.3,4 Unfortunately, efforts to coax the requisite Wittig reaction (Scheme 3) to proceed using (2-carboxyethyl)triphenylphosphonium bromide with NaH or KOtBu in various solvents (THF, CH3OH, and DMF) mimicking our previous methodology1 proved unsuccessful. The reaction was typically complicated by the formation of a mixture of various by-products which appeared to result primarily from decomposition of the Wittig salt under basic conditions. The reported yield (by Aubé and co-workers) for a similar type of Wittig reaction using (2-carboxyethyl)triphenylphosphonium bromide was also low (23%), which further confirmed our observations.22
Scheme 3.

Unsuccessful Wittig reaction using (2-carboxyethyl)triphenylphosphonium bromide.
Although we did not fully characterize each of these various decomposition by-products, one by-product was identified as acrylic acid, which was obtained by elimination of triphenylphosphine. We postulated that a possible driving force for the elimination was the formation of stable α, β-unsaturated acrylic acid. Therefore, to solve the problem, instead of using (2-carboxyethyl)triphenylphosphonium bromide, we employed (3-hydroxypropyl)triphenylphosphonium bromide as the Wittig salt 12 (Scheme 4), which was readily synthesized in high yield (96%) by treatment of 3-bromo-1-propanol with triphenylphosphine at reflux in toluene.23 Wittig salt 12 (as its corresponding in situ TMS protected ylide) was reacted with benzaldehyde 6 following a similar protocol as reported.24 The TMS group was deprotected under acidic workup conditions to generate the intermediate alkene (mixture of E/Z isomers) that contained a primary alcohol moiety. The crude alkene was subjected to hydrogenation (hydrogen gas, Pd-C catalyst) to afford compound 13 (92% over two steps). Alcohol 13 was oxidized (Oxone® and IBX) to its corresponding carboxylic acid 14 (71% yield). Cyclic ketone 15 was prepared (92% yield) under Friedel-Crafts acylation conditions by conversion of carboxylic acid 14 to its corresponding acyl chloride (using oxalyl chloride) followed by exposure to SnCl4. The remaining steps (Scheme 4) to prepare dihydronaphthalene analogues 3 and 4 are akin to that described for the synthesis of related benzosuberene analogues 1 and 2 (Scheme 2). Phenolic dihydronaphthalene analogue 3 was obtained in an overall yield of 32% over seven steps. Amino dihydronaphthalene analogue 4 was obtained directly from phenolic compound 3 in three steps with an overall yield of 58–72%. These modified procedures proved highly scalable, which facilitated the preparation of the majority of these target molecules and intermediates in 2–5 gram amounts without any significant change in overall yields.
Scheme 4.

New synthetic routes to compounds 3 and 4. Reagents and reaction conditions: (a) compound 11 (2.0 eq.), triphenylphosphine (1.0 eq.), toluene, reflux, 24 h, 96%. (b) (i) compound 12 (1.5 eq.), n-BuLi (3.0 eq.), THF, 0 °C, 15 min; TMSCl (1.5 eq.), 0 °C, 30 min; compound 6 (1.0 eq.), 0 °C, 1 h, RT, 2 h (ii) Pd-C (0.1 eq., 10 wt%), H2 balloon, CH3OH, 24 h, 92% (two steps, compound 13). (c) compound 13 (1.0 eq.), Oxone® (1.5 eq.), IBX (0.3 eq.) ACN/H2O, 70 °C, 20 h, 71%. (d) (i) compound 14 (1.0 eq.), oxalyl chloride (2.0 eq.), DMF (0.2 eq.), CH2Cl2, RT, 2 h; (ii) SnCl4 (1.2 eq.), CH2Cl2, −10 °C, 40 min, 92% (two steps). (e) (i) 5-bromo-1,3,4-trimethoxybenzene (2.0 eq.), n-BuLi (2.0 eq.), THF, −78 °C, 30 min. (ii) compound 15 (1.0 eq.), −78 °C, 4 h, RT, 16 h, 65%. (f) compound 16 (1.0 eq.), boron trichloride (1.1 eq.), CH2Cl2, 0 °C, 3 h, 85%. (g) compound 3 (1.0 eq.), Triethylamine (2.0 eq.), triflic anhydride (1.5 eq.), 0 °C to RT, 5 h, 83–100%. (h) (i) compound 17 (1.0 eq.), benzophenone imine (1.5 eq.), Cs2CO3 (1.5 eq.), palladium(II) acetate (0.1 eq.), racemic-BINAP (0.15 eq.), toluene, 110–115 °C, 36 h; (ii) 2 M HCl, RT, 1 h, THF, 72% (two steps).
In conclusion, a general and highly efficient synthetic route has been developed for functionalized benzosuberene (1 and 2) and dihydronaphthalene (3 and 4) analogues. Importantly, an efficient three steps procedure was optimized and employed to synthesize aniline-based congeners 2 and 4 directly from their corresponding phenolic-based counterparts using a well-established palladium catalyzed amination reaction. These synthetic methodologies should be amendable to the synthesis of a wide variety of benzosuberene and dihydronaphthalene analogues with potential extension to other fused ring systems.
Supplementary Material
Highlights:
Efficient synthetic methodology for benzosuberene and dihydronaphthalene analogues
Methodology accommodated larger scale reactions
Palladium catalyzed amination converted phenolic analogues to aniline congeners
Acknowledgements:
The authors are grateful to the Cancer Prevention and Research Institute of Texas (CPRIT, Grant #RP140399 and Grant #RP170696 to K.G.P., the National Cancer Institute of the National Institutes of Health (Grant # 5R01CA140674 to K.G.P.), and Mateon Therapeutics, Inc. (grant to K.G.P.) for their financial support of this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- (1).Sriram M; Hall JJ; Grohmann NC; Strecker TE; Wootton T; Franken A; Trawick ML; Pinney KG Design, Synthesis and Biological Evaluation of Dihydronaphthalene and Benzosuberene Analogs of the Combretastatins as Inhibitors of Tubulin Polymerization in Cancer Chemotherapy. Bioorg. Med. Chem 2008, 16 (17), 8161–8171. [DOI] [PubMed] [Google Scholar]
- (2).Pinney KG; Pettit GR; Trawick ML; Jelinek C; Chaplin DJ The Discovery and Development of the Combretastatins. Anticancer Agents Nat. Prod 2012, 27–63. [Google Scholar]
- (3).Tanpure RP; George CS; Sriram M; Strecker TE; Tidmore JK; Hamel E; Charlton-Sevcik AK; Chaplin DJ; Trawick ML; Pinney KG An Amino-Benzosuberene Analogue That Inhibits Tubulin Assembly and Demonstrates Remarkable Cytotoxicity. MedChemComm 2012, 3 (6), 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tanpure RP; George CS; Strecker TE; Devkota L; Tidmore JK; Lin C-M; Herdman CA; MacDonough MT; Sriram M; Chaplin DJ; et al. Synthesis of Structurally Diverse Benzosuberene Analogues and Their Biological Evaluation as Anti-Cancer Agents. Bioorg. Med. Chem 2013, 21 (24), 8019–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Devkota L; Lin C-M; Strecker TE; Wang Y; Tidmore JK; Chen Z; Guddneppanavar R; Jelinek CJ; Lopez R; Liu L; et al. Design, Synthesis, and Biological Evaluation of Water-Soluble Amino Acid Prodrug Conjugates Derived from Combretastatin, Dihydronaphthalene, and Benzosuberene-Based Parent Vascular Disrupting Agents. Bioorg. Med. Chem 2016, 24 (5), 938–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pinney K; Sriram M; George C; Tanpure R Efficient Method for Preparing Functionalized Benzosuberenes. WO/2012/068284, May 25, 2012. [Google Scholar]
- (7).Herdman CA; Devkota L; Lin C-M; Niu H; Strecker TE; Lopez R; Liu L; George CS; Tanpure RP; Hamel E; et al. Structural Interrogation of Benzosuberene-Based Inhibitors of Tubulin Polymerization. Bioorg. Med. Chem 2015, 23 (24), 7497–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Boyland E; Boyland ME Studies in Tissue Metabolism: The Action of Colchicine and B. Typhosus Extract. Biochem. J 1937, 31 (3), 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).McGown AT; Fox BW Structural and Biochemical Comparison of the Anti-Mitotic Agents Colchicine, Combretastatin A4 and Amphethinile. Anticancer. Drug Des 1989, 3 (4), 249–254. [PubMed] [Google Scholar]
- (10).Pettit GR; Singh SB; Hamel E; Lin CM; Alberts DS; Garcia-Kendal D Isolation and Structure of the Strong Cell Growth and Tubulin Inhibitor Combretastatin A-4. Experientia 1989, 45 (2), 209–211. [DOI] [PubMed] [Google Scholar]
- (11).Lin CM; Ho HH; Pettit GR; Hamel E Antimitotic Natural Products Combretastatin A-4 and Combretastatin A-2: Studies on the Mechanism of Their Inhibition of the Binding of Colchicine to Tubulin. Biochemistry (Mosc.) 1989, 28 (17), 6984–6991. [DOI] [PubMed] [Google Scholar]
- (12).Pinney K; Mocharla V; Chen Z; Garner C; Ghatak A; Hadimani M; Kessler J; Dorsey J; Edvardsen K; Chaplin D; et al. Tubulin Binding Agents and Corresponding Prodrug Constructs. US20040043969A1, March 4, 2004. [Google Scholar]
- (13).Mason RP; Zhao D; Liu L; Trawick ML; Pinney KG A Perspective on Vascular Disrupting Agents That Interact with Tubulin: Preclinical Tumor Imaging and Biological Assessment. Integr. Biol 2011, 3 (4), 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Baluk P; Hashizume H; McDonald DM Cellular Abnormalities of Blood Vessels as Targets in Cancer. Curr. Opin. Genet. Dev 2005, 15 (1), 102–111. [DOI] [PubMed] [Google Scholar]
- (15).Siemann DW The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev 2011, 37 (1), 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen Z; O’Donnell CJ; Maderna A Synthesis of 3-Methoxy-9-(3,4,5-Trimethoxyphenyl)-6,7-Dihydro-5H-Benzo[7]Annulen-4-Ol, a Potent Antineoplastic Benzosuberene Derivative for Anti-Cancer Chemotherapy. Tetrahedron Lett. 2012, 53 (1), 64–66. [Google Scholar]
- (17a).Rasolofonjatovo E; Provot O; Hamze A; Rodrigo J; Bignon J; Wdzieczak-Bakala J; Lenoir C; Desravines D; Dubois J; Brion J-D; et al. Design, Synthesis and Anticancer Properties of 5-Arylbenzoxepins as Conformationally Restricted Isocombretastatin A-4 Analogs. Eur. J. Med. Chem 2013, 62, 28–39. [DOI] [PubMed] [Google Scholar]
- (17b).Rasolofonjatovo E; Provot O; Hamze A; Rodrigo J; Bignon J; Wdzieczak-Bakala J; Desravines D; Dubois J; Brion J-D; Alami M Conformationnally restricted naphthalene derivatives type isocombretastatin A-4 and isoerianin analogues: Synthesis, cytotoxicity and antitubulin activity. Eur. J. Med. Chem 2012, 52, 22–32. [DOI] [PubMed] [Google Scholar]
- (18).Chen Z; Maderna A; Sukuru SCK; Wagenaar M; O’Donnell CJ; Lam M-H; Musto S; Loganzo F New Cytotoxic Benzosuberene Analogs. Synthesis, Molecular Modeling and Biological Evaluation. Bioorg. Med. Chem. Lett 2013, 23 (24), 6688–6694. [DOI] [PubMed] [Google Scholar]
- (19).Maguire CJ; Chen Z; Mocharla VP; Sriram M; Strecker TE; Hamel E; Zhou H; Lopez R; Wang Y; Mason RP; et al. Synthesis of Dihydronaphthalene Analogues Inspired by Combretastatin A-4 and Their Biological Evaluation as Anticancer Agents. MedChemComm 2018, 9 , 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Walsh JJ; Sha R; McCormack EM; Hudson GJ; White M; Stack GD; Moran BW; Coogan A; Breen EC Tubulin Binding Agents. US20150018566A1, August 27, 2012. [Google Scholar]
- (21).Wolfe JP; Åhman J; Sadighi JP; Singer RA; Buchwald SL An Ammonia Equivalent for the Palladium-Catalyzed Amination of Aryl Halides and Triflates. Tetrahedron Lett. 1997, 38 (36), 6367–6370. [Google Scholar]
- (22).Motiwala HF; Vekariya RH; Aubé J Intramolecular Friedel–Crafts Acylation Reaction Promoted by 1,1,1,3,3,3-Hexafluoro-2-Propanol. Org. Lett 2015, 17 (21), 5484–5487. [DOI] [PubMed] [Google Scholar]
- (23).Couturier M; Dory YL; Rouillard F; Deslongchamps P Studies Directed towards the Total Synthesis of Aldosterone and Naturally Occurring Analogues. A Unified Approach Using the Transannular Diels-Alder Reaction. Tetrahedron 1998, 54 (8), 1529–1562. [Google Scholar]
- (24).Hanessian S; Tremblay M; Petersen JFW The N-Acyloxyiminium Ion Aza-Prins Route to Octahydroindoles: Total Synthesis and Structural Confirmation of the Antithrombotic Marine Natural Product Oscillarin. J. Am. Chem. Soc 2004, 126 (19), 6064–6071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.