

SUMMARY
Common fragile sites (CFSs) are genomic regions that display gaps and breaks in human metaphase chromosomes under replication stress and are often deleted in cancer cells. We studied an ~300-bp subregion (Flex1) of human CFS FRA16D in yeast and found that it recapitulates characteristics of CFS fragility in human cells. Flex1 fragility is dependent on the ability of a variable-length AT repeat to form a cruciform structure that stalls replication. Fragility at Flex1 is initiated by structure-specific endonuclease Mus81-Mms4 acting together with the Slx1–4/Rad1–10 complex, whereas Yen1 protects Flex1 against breakage. Sae2 is required for healing of Flex1 after breakage. Our study shows that breakage within a CFS can be initiated by nuclease cleavage at forks stalled at DNA structures. Furthermore, our results suggest that CFSs are not just prone to breakage but also are impaired in their ability to heal, and this deleterious combination accounts for their fragility.
Graphical Abstract
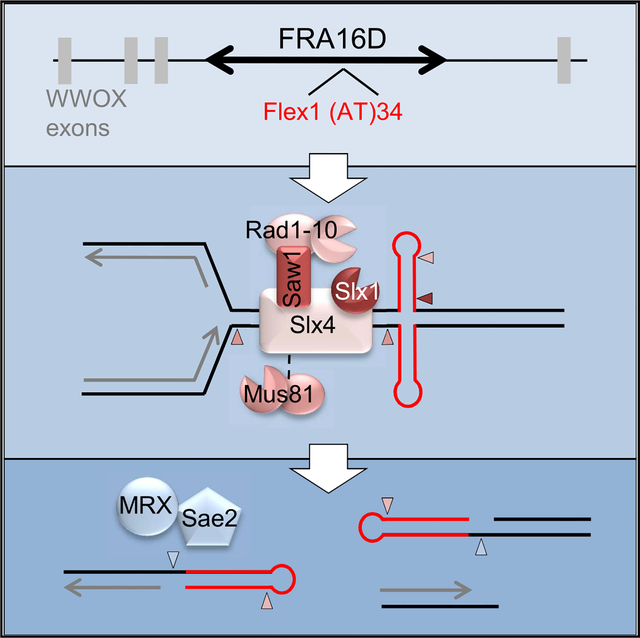
In Brief
Kaushal et al. found that a subregion of fragile site FRA16D forms an abnormal DNA structure that stalls DNA polymerase, resulting in DNA breakage and difficulty healing. They identify nucleases that cleave this structure to cause the DNA breaks and postulate that difficulty healing makes these sites prone to rearrangements.
INTRODUCTION
Common fragile sites (CFSs) are highly unstable human chromosomal regions that are prone to breakage and the formation of cancer-associated deletions. CFS breakage, or expression, can be induced in most individuals, and therefore they are considered a normal part of chromosome structure. The molecular basis for their fragility is still not well understood, although an inability to complete replication during S phase is an important component. CFS expression can be induced by drugs that inhibit polymerase progression, such as aphidicolin and hydroxyurea (HU) (Glover et al., 2017). The two most commonly expressed CFSs in human cells, FRA3B and FRA16D, replicate late in S and into G2 phase (Debatisse and Rosselli, 2019). CFSs can undergo mitotic DNA synthesis (MiDAS) in order to finish replicating these regions before nuclear division occurs (Minocherhomji et al., 2015).
CFS expression varies by cell type (Le Tallec et al., 2013), and there is evidence that gene expression levels may correlate with fragility levels (Helmrich et al., 2011; Le Tallec et al., 2013). FRA16D, located within a large intron of the WWOX tumor suppressor gene, is one of the most breakageprone CFSs because it was expressed in all four cell types tested (Le Tallec et al., 2013), suggesting that FRA16D is inherently fragile even under varied levels of transcription. Recently, it was shown that the protein FANCD2 facilitates replication through FRA16D by suppressing DNA:RNA hybrid formation and inducing dormant origin firing (Madireddy et al., 2016).
CFSs are frequently the locations of homozygous and hemizygous deletions in many cancer cell lines (Finnis et al., 2005; Bignell et al., 2010). CFSs are also hotspots of de novo copy number variations (CNVs) in many tumor types, likely occurring because of replication stress followed by aberrant repair (Glover et al., 2017). Breakage at CFSs is an early event in tumor progression (Halazonetis et al., 2008; Tsantoulis et al., 2008). In addition, oncogene overexpression leads to replication stress (oncogene-induced replication stress) that can then result in CFS breakage, deletions, and rearrangements (Macheret and Halazonetis, 2015; Miron et al., 2015; Glover et al., 2017). CFSs tend to be AT-rich, making their DNA easier to unwind to form unusual or non-B DNA secondary structures, which could play a role in their fragility. Computational analysis of CFSs has identified a higher density of sequences with potential to form stable secondary structures compared with controls, and secondary structures at both rare and CFSs have connections to human disease and cancer (Thys et al., 2015; Kaushal and Freudenreich, 2019).
Flex1 is an ~300-bp AT-rich subregion of human CFS FRA16D. Flex1 contains a polymorphic perfect AT repeat that ranges from 11 to 88 copies in humans tested and is frequently deleted in tumor cell lines (Finnis et al., 2005). AT repeats can nucleate an unwound region of double-stranded supercoiled (SC) DNA to form cruciforms in vivo once they exceed a length of around 22 repeat units (McClellan et al., 1990; Dayn et al., 1991; Bowater et al., 1991). Cruciform cleavage and resolution have been shown to cause multiple common chromosomal translocations (Kato et al., 2012). AT repeats and short inverted repeats are prevalent in the human genome and have been implicated in driving genomic rearrangements in evolution, and they are enriched near cancer translocation and deletion breakpoints (Lu et al., 2015; Bacolla et al., 2016).
Our lab has previously shown that the Flex1 sequence caused chromosome fragility when inserted into an artificial chromosome in S. cerevisiae, and that a Flex1 sequence containing (AT)34 caused replication fork stalling (Zhang and Freudenreich, 2007). It was hypothesized that a secondary structure at Flex1 is causing replication fork stalling and contributing to FRA16D breakage.
In this study, we show that Flex1 is a significant contributor to overall FRA16D breakage, and that Flex1 fragility increases with AT repeat length in a nonlinear fashion. AT repeat lengths that exhibit fragility also show sensitivity to a single-stranded DNA (ssDNA) nuclease at the predicted cruciform loops and cause pausing of human polymerase δ in vitro. Our data support the hypothesis that fork stalling at cruciform structures formed by longer AT lengths causes chromosome fragility. Structure-specific endonuclease (SSE) Mus81-Mms4 is required for Flex1 fragility, in agreement with the known requirement of human MUS81 for FRA16D expression in human cells (Naim et al., 2013; Ying et al., 2013). Importantly, we find that Mus81 induces Flex1 fragility only at AT lengths long enough to form a cruciform, implying that MUS81 is specifically acting at secondary structures in FRA16D, cleaving either the cruciform or a resulting structure such as a stalled replication fork. Mus81-Mms4 basal activity is sufficient to induce cleavage at Flex1 because Mms4 activation by phosphorylation is not required. Slx1-Slx4 and Rad1-Rad10 nucleases also play a role in causing Flex1 fragility, and our data are consistent with the existence of a Slx1-Slx4, Mus81-Mms4, Rad1-Rad10 (SMR) super complex in S. cerevisiae. In contrast, Yen1, which acts in late mitosis, has a role in preventing (rather than causing) fragility at Flex1. Our data suggest that coordinated cleavage by SSEs of forks stalled by DNA structures at CFSs may account for their characteristic expression of gaps and breaks in mitosis. Finally, we identify that it is not only the AT repeat length but also the flanking Flex1 sequences that play a role in the expression of breaks at Flex1, because they inhibit efficient healing of the broken DNA. Therefore, we propose a theory that the DNA sequences at CFSs have both an increased tendency to break and a reduced ability to heal following breakage, contributing to their persistence into M phase and their propensity to instigate large deletions and translocations.
RESULTS
Flex1 Is a Crucial Element Causing FRA16D Fragility In Vivo
Because Flex1 is often deleted in tumor cells, induces fork stalling in yeast, and is predicted to form an alternative secondary structure, we hypothesized that it plays an important role in FRA16D expression. To determine whether Flex1 was responsible for a substantial amount of FRA16D fragility, we deleted the Flex1 sequence from YAC 801B6, which contains 1.4 Mb of human chromosome 16 sequence including FRA16D (Figures 1A, S1A, and S1C). Despite deleting only ~300 bp of the 1.4-Mb human sequence (0.02%), we observed a significant decrease in frequency of YAC end loss (measured by 5-fluoroorotic acid [FOA]R) (Figure 1A). These results indicate that Flex1 accounts for a significant and measurable fraction of breakage events within 801B6, highlighting the importance of the Flex1 sequence in contributing to overall FRA16D fragility. Nonetheless, based on the level of fragility of the adjacent sequence (972D3) (Figures 1A and S1C), we speculate that other fragile elements may combine with Flex1 to account for the full fragility of the entire region.
Figure 1. Flex1 Is Important for FRA16D Fragility and Is Fragile in an AT Length and Structure-Dependent Manner.

(A) Two YACs from the CEPH YAC library containing indicated amounts of human chromosome 16 including FRA16D (801B6) with or without Flex1 (Flex1D) or sequence adjacent to FRA16D (972D3) were assayed for frequency of URA3 marker loss (percent of FOA resistance [% FOAR]). Starting YAC integrity was verified by pulsed-field gel electrophoresis and PCR of subregions (Figure S1). ***p < 0.001 compared with 972D3;^p < 0.05 compared with 801B6 with Flex1, by unpaired t test; see also Table S1.
(B) Schematic of the DDRA fragility assay. See Figure S2B for details. Recombination rate was measured for constructs containing the control (ctrl; see Figure S1A) or indicated Flex1 AT repeats; strains were tested for significant deviation from the control using an unpaired t test; ****p < 0.0001. Orientation 1 data are shown. Rates are reported above the appropriate bar with fold over the control in parentheses; see also Table S2.
(C) Schematic depicting S1 nuclease cleavage assay on plasmids containing cruciform-forming sequences. Hairpin heads are substrates for S1 cleavage, converting supercoiled (SC) plasmids to open circular (OC) and linear (L). Further digestion of plasmids with Eco53kI (blue line, cleavage site) results in a fragment of ~170 or 200 bp for (AT)23 and (AT)34, respectively, if S1 cleavage occurred at the center of the hairpin. Middle: a representative 1% agarose gel showing S1 nuclease cleavage titration (0U, 1U, 1.75U, 2.5U, and 5U) of plasmids containing indicated Flex1 or control sequences; see also Figure S1D.
(D) Flex1 cleavage by S1 nuclease and Eco53kI compared with S1 cleavage alone; left gel 0.8% agarose, and right gel 3% Metaphor showing the fragment released in duplicates.
Flex1 Is Fragile in an (AT)n Repeat Length-Dependent Manner
Because the Flex1 sequence contains a polymorphic (AT)n repeat in humans, it was important to address the role that AT repeat length plays in Flex1 fragility. Our group previously demonstrated an increase in fork stalling with increasing AT length, and there was a trend of increasing fragility as Flex1 AT length increased from 5 to 14 to 23 (Zhang and Freudenreich, 2007). Due to the severity of the replication fork stalling at Flex1(AT)34, it was hypothesized that the AT repeats of this length formed a cruciform, although the sequence could also form hairpins on either strand. To evaluate the role of AT tract length in Flex1 fragility, we inserted three different sequences of varying AT lengths but standardized short 5′ and short 3′(S5′ and S3′, respectively) flanking sequences from the human Flex1 region into a genetic system to measure fragility on yeast chromosome II, the direct duplication recombination assay (DDRA) (Figures 1B and S2B). Breakage can stimulate recombination between flanking homologous ADE2 sequences via single-strand annealing (SSA), which results in loss of the intervening URA3 gene and 5-FOA-resistant, Ade+ cells (Freudenreich et al., 1998; Paeschke et al., 2011; Polleys and Freudenreich, 2018). Recombination rates were also measured for a control sequence, which is a roughly 380-bp sequence from FRA16D that is not predicted to form a stable secondary structure (Figure S1A) (Zhang and Freudenreich, 2007). This assay mimics the types of deletion events that have been shown to occur naturally in cancer cells and other cells under replication stress (Finnis et al., 2005; Durkin et al., 2008), with the benefit of the deletions being selectable. In these constructs, the recombination rate increased significantly with increasing Flex1 AT length, consistent with a repeat length-dependent increase in fragility (Figure 1B). The significant increase in recombination rate of (AT)23 and (AT)34 coincides with the known propensity of AT repeats to form a cruciform structure with much higher frequency when their length exceeds roughly 22 repeats (McClellan et al., 1990; Dayn et al., 1991; Bowater et al., 1991). The dramatic increase in fragility upon adding only 11 additional repeats together with the severity of the Flex1 (AT)34 replication fork stalling in vivo (Zhang and Freudenreich, 2007) strongly supports that a fork-blocking DNA structure is frequently forming at this length.
To test whether a cruciform or slipped-strand hairpins are forming, we performed an in vitro S1 nuclease cleavage assay using plasmids containing the same Flex1 and control sequences used in the fragility assay. Single-stranded hairpin loops will be preferentially cleaved by S1 nuclease, and higher concentrations will introduce random nicks because of helix breathing (von Hippel et al., 2013; Higgins and Vologodskii, 2015). A single nick will convert the SC form to open circular (OC), and two concerted nicks in close proximity on opposite strands will linearize the plasmid (Figure 1C). Increasing concentrations of S1 nicked the control and Flex1(AT)14 plasmids, with linear forms appearing only at the highest S1 concentration; the conversion to OC was somewhat faster for (AT)14 compared with the control, suggesting the formation of some hairpin structures. For the (AT)23- and (AT)34-containing plasmids, OC and linear (L) species formed even at the lowest S1 concentration, with equal parts OC and L by the second concentration for (AT)23 and mostly linear species for (AT)34 (lanes 13, 18), indicative of increased secondary structure formation with increasing AT repeat length. The quick transition to the linear product for (AT)34 and about half of the (AT)23 plasmids is consistent with cruciform formation. We also noted double-banding and aberrant migration for these forms consistent with structured DNA species; for (AT)34 lane 17, some of the OC species were even retained in the well (Figure S1D). Cleavage with restriction enzyme Eco53kl released distinct linear fragments of 170 or 200 bp for (AT)23 and (AT)34 plasmids, respectively, mapping the sites of S1 sensitivity to the center of the AT repeat on both strands, as predicted for a cruciform structure containing two opposite hairpin loops (Figure 1D). These results are most consistent with in vivo formation of cruciform structures in double-stranded DNA (dsDNA) for Flex1 (AT)23 and (AT)34 sequences, with (AT)34 forming a structure on both strands a higher percentage of the time.
An (AT)34 Repeat Causes Human Polymerase Delta Stalling and Replication Termination
Previously, Zhang and Freudenreich (2007) found that the Flex1 (AT)34 sequence causes replication fork stalling during plasmid replication in S. cerevisiae. To test the site of stalling and extend this result to the human enzyme, human four-subunit polymerase δ holoenzyme (Pol δ4) DNA synthesis through either the control or Flex1 ssDNA with various AT repeat lengths in the presence of replication factor C (RFC)-loaded proliferating cell nuclear antigen (PCNA) was measured using an in vitro primer extension assay as described in Shah et al. (2010); Walsh et al. (2013), and Barnes et al. (2017). Pausing was identified as sites of accumulated primer extension reaction products. Note that in this assay, only template hairpins would be able to form, not cruciforms that require dsDNA. As highlighted, human polymerase δ struggles to replicate the AT repeats, and significant stalling is detected throughout the AT tract at all lengths tested (Figure 2A, red lines). The probability of termination within the AT tract increases with AT length (Figure 2B). Stalling was also observed during polymerase δ synthesis using the opposite Flex1 (TA)34 strand as a template (Figure S4). We conclude that the Flex1 (AT)34-dependent replication fork stalling previously observed in vivo is due to pausing specifically at the AT repeat. These data support the hypothesis that the observed AT length-dependent fragility is due, in part, to DNA polymerase stalling at the repeat.
Figure 2. Flex1 AT Repeats Cause Pausing by Human Polymerase δ and Fragility Increases with Replication Stress.

(A) Representative gel of in vitro DNA synthesis of control or Flex1 with various AT lengths by 200 fmol of the four-subunit human polymerase δ holoenzyme (Pol δ4), showing pause sites at the AT repeat (red line). Sequence outside of the marked area is composed of the plasmid backbone. Triangles, increase in time from 5 to 15 min.
H, percent hybridization control for determining the amount of utilizable primer-template; P, no polymerase control; TA, dideoxy sequencing ladder of the DNA template.
(B) Quantification of AT run termination probability of Flex1 with various tract lengths. Data are the mean ± SEM of three replicates with similar amounts of primertemplate utilization (90–114%; see STAR Methods) for all templates. ***p < 0.001 using one-way ANOVA with Tukey’s multiple comparison; see also Table S6.
(C) DDRA assay rates for cells grown in the presence or absence of 100 mM HU. Recombination rates were tested for significant deviation from the same strain grown in non-HU conditions using an unpaired t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The fold increase upon HU treatment is reported above each pair of rates; see also Table S2.
To determine the effect of replication stress on fragility of the FRA16D Flex1 sequence, we measured the rate of FOAR in the DDRA assay after treatment with HU (Figure 2C). HU causes replication fork stalling by depleting dNTP pools, and thus is expected to further exacerbate fragility at all DNA sequences. Interestingly, the effect of HU was much stronger for sequences predicted to form no DNA structure or a weak DNA structure, such as the control (7.2-fold over no HU) and Flex1 (AT)14 (5.8-fold over no HU), compared with sequences predicted to form a stable hairpin or cruciform structure [~2-fold over no HU for Flex1 (AT)23 and Flex1 (AT)34]. These data show that additional replication stress is not required for fragility of sequences that can form a stable-enough structure to stall replication in their normal cellular context and are consistent with stalling by a pre-formed cruciform structure. Because replication stress further increases the likelihood of chromosome breakage at Flex1 (AT)34, it may additionally allow fork remodeling or hairpin formation on separated template strands, consistent with the pronounced pausing of polymerase δ within (AT)34 observed on single-stranded template DNA.
SSE Mus81-Mms4 Causes Fragility at Flex1
In human cells, the SSEs MUS81-EME1 and XPF-ERCC1 were shown to promote FRA16D expression in mitotic cells that had experienced replication stress, presumably by causing cleavage of a persistent replication intermediate (Ying et al., 2013; Naim et al., 2013). However, these results were obtained using whole chromosomes; thus, it was unclear where the cleavage was occurring. SSEs can act on substrates at stalled or reversed forks that form in S phase (and possibly persist into G2/M), and in G2 and M phases SSEs can act on homologous recombination (HR) intermediates or unreplicated DNA to allow chromosome separation (Symington et al., 2014; Dehé and Gaillard, 2017). Because our data indicate that longer AT repeats within the Flex1 sequence form cruciform structures that could resemble SSE substrates and also stall replication forks, it was of interest to determine whether SSEs act at Flex1. SSEs could either cause the observed fragility by directed cleavage or, alternatively, protect against fragility by allowing proper resolution of stalled forks or recombination intermediates.
Upon deletion of the MUS81 gene, Flex1 (AT)34 fragility significantly decreases in both the DDRA fragility assay (Figure 3A) and the previously used YAC end loss assay (Figure 4B) (Zhang and Freudenreich, 2007). Using the DDRA fragility assay, we evaluated the effect of mus81Δ on Flex1 containing various AT tract lengths and the control. The recombination rate is significantly decreased in mus81Δ only when Flex1’s AT stem length exceeds 22 bp, the threshold for forming cruciforms; mus81Δ had no effect on the recombination rate of the control or Flex1 (AT)14 strains (Figure 3A). We conclude that Mus81 is specifically acting to cleave either directly at a DNA structure formed by the AT repeat or at a substrate caused by the structure. These data suggest that the requirement for MUS81 for FRA16D expression in human cells is due to structure-mediated events at Flex1, or perhaps at Flex1 in combination with other fork stalling regions.
Figure 3. Mus81 Causes Fragility at Flex1, and Breaks Are Not Dependent on Recombination or Mus81-Mms4 Hyperactivation.

(A) The DDRA fragility assay was used to measure recombination rates of Flex1 orientation 1 with various AT lengths in mus81Δ strains.
(B) DDRA fragility assay rates for indicated mutants. Statistical difference compared with WT values using an unpaired t test are indicated: ^p < 0.05, ^^p < 0.01, ^^^p < 0.001, ^^^^p < 0.0001.
See also Table S2.
Figure 4. Fragility of AT Repeat-Dependent Structures Is Dependent on Mus81-Mms4, Slx1-Slx4, and Rad1-Rad10 Nucleases, but Not Yen1.

(A) Effect of deleting SSEs in Flex1 (AT)34 or control DDRA fragility assay strains; see also Table S2.
(B) Effect of deleting nucleases in indicated YAC end loss assay strains; see also Table S3. Statistical analysis as in Figure 3.
Mus81-Mms4 nucleolytic activity is hyperactivated by Dbf4-dependent kinase (DDK)/Cdc5/Cdc28 phosphorylation of Mms4 at the G2/M boundary, which is facilitated by Rtt107 binding (Wild and Matos, 2016; Princz et al., 2017). We sought to test whether the basal activity of Mus81-Mms4 present in S phase is sufficient for Flex1 cleavage, or if the hyperactivated form of Mus81-Mms4 is needed in order to induce fragility. Flex1 (AT) 34 with a phosphorylation-defective mutant, mms4–9A (Gallo-Fernández et al., 2012), had the same rate of recombination as a wild-type strain (Figure 3B). The mms4–9A mutant still retains some potential phosphorylation sites and a low level of activity similar to the basal S phase activity (Gallo-Fernández et al., 2012). Therefore, we combined it with an rtt107Δ because Rtt107 mediates the association of DDK and Cdc5 kinases with Mus81-Mms4 and stimulates Mms4 hyperphosphorylation (Gallo-Fernández et al., 2012). Similarly, rtt107Δ single and rtt107Δ mms4–9A double mutants exhibited either minimal or no change in recombination rates (Figure 3B). Therefore, the G2/M hyperactivation of Mus81-Mms4 is not necessary for causing Flex1 fragility. A full deletion of MMS4 (mms4Δ) gave a significant decrease in Flex1 (AT) 34 recombination rate, confirming the importance of the Mms4 component of the Mus81-Mms4 nuclease (Figure 3B). These results imply that the S phase level of Mus81-Mms4 activity is enough to cause fragility at Flex1, although they do not indicate the timing of the activity.
We next wondered what substrate Mus81-Mms4 is acting upon: directly at a secondary structure, at a stalled fork caused by a secondary structure, or at a recombination intermediate formed during the resolution of the stalled fork. In a mus81Δ rad51Δ double mutant, in which recombination intermediates cannot be formed, the FOAR Ade+ rate is no different from a mus81Δ mutant, indicating that Mus81 is not acting upon recombination intermediates to cause Flex1 fragility. These data suggest that Mus81 is acting either directly at a secondary structure or at a stalled, reversed, or converged fork caused by Flex1 (Figure 3B).
The Slx4 Complex Coordinates Cleavage at Flex1
In human cells, SLX4 is a scaffolding protein that recruits multiple enzymes, including MUS81-EME1, SLX1, and XPF-ERCC1, to enhance their activity and coordinate SSE action timing and pathway choice (Dehé and Gaillard, 2017). There is evidence for a super complex of SLX1-SLX4, MUS81-EME1, and XPF-ERCC1 in mammals, called the SMX DNA repair tri-nuclease (Wyatt et al., 2017). We reasoned that stalled or converged forks induced by Flex1 may be substrates for other SSEs acting with Mus81, and tested this hypothesis by creating mutants in the yeast homologs of these proteins. In S. cerevisiae, Slx4 interacts directly with the Slx1 endonuclease and also binds the Saw1 scaffold protein, which in turn binds the Rad1-Rad10 nuclease (XPF-ERCC1) (Sarangi et al., 2014; Cussiol et al., 2017). A direct interaction with Slx4 and Mus81-Mms4 has not been demonstrated, but there is evidence for an indirect interaction through Rtt107 and Dpb11 (Gritenaite et al., 2014).
Strains lacking either Slx4 or Rad1 showed a significant decrease in fragility for both the control and Flex1 (AT)34 sequences in the DDRA fragility assay (Figure S2A). The decrease in recombination in the control strain in the slx4Δ or rad1Δ backgrounds indicates that both Slx4 and Rad1 are required for SSA, as shown previously (Freudenreich et al., 1998; Mimitou and Symington, 2009; Dehé and Gaillard, 2017). However, the deletion of each of these proteins had a more dramatic effect on the Flex1 (AT)34 sequence compared with the control sequence (decreases compared to wild type [WT] were 12-fold versus 3.2-fold for slx4Δ; 6.1-fold versus 1.8-fold for rad1Δ) (Figure S2A). These data indicate that the Slx4 complex and the Rad1-Rad10 nuclease may have an additional role in induction of fragility at Flex1, aside from their role in SSA. We confirmed this conclusion by deleting SLX4 and RAD1 in the Flex1 (AT)34 YAC end loss assay strain, because healing in this assay should not require Slx4 or Rad1. Healing in the YAC assay occurs by resection to the G4T4 telomere seed sequence and subsequent telomere addition, which results in loss of selectable markers distal to the break site (Polleys and Freudenreich, 2018). Indeed, fragility was significantly decreased in both backgrounds, verifying the importance of these proteins in preventing Flex1 fragility (Figure 4B). A mus81Δ rad1Δ strain had about the same level of fragility as each single mutant (Figure 4B), suggesting that they are working in the same pathway to cause Flex1 (AT)34 fragility.
In both yeast and human cells, the Slx1 nuclease associates with Slx4 and targets branched DNA structures (Fricke and Brill, 2003; Svendsen et al., 2009). Therefore, Slx1 may also be required to process structures formed by or because of Flex1. Indeed, slx1Δ mutants had a decrease in fragility to a level similar to mus81Δ in the YAC end loss assay (Figure 4B), although the decrease was less dramatic at the internal chromosome location in the DDRA assay; the control rate was unchanged in the slx1Δ mutant (Figure 4A). The mus81Δ slx1Δ fragility rate is similar to that of a mus81Δ single mutant in the DDRA fragility assay (Figure 4A). Also, the slx1Δ rad1Δ double mutant and the slx1Δ mus81Δ rad1Δ triple mutant showed similar Flex1 (AT)34 fragility levels as each single mutant in the YAC end loss assay (Figure 4B). Overall, these results are consistent with Mus81-Mms4, Slx4-Slx1, and Rad1-Rad10 working in the same pathway to cause fragility at Flex1 (AT)34, suggesting that they are functioning together to cause cleavage of a structure induced by this sequence.
Yen1 Protects Flex1 against Fragility
S. cerevisiae Yen1 (human GEN1) is an SSE that gains access to the DNA only in mitosis and prefers perfect four-way junctions such as Holliday junctions (Minocherhomji and Hickson, 2014). Because Yen1 and Mus81 have overlapping substrates, Yen1 could act as a backup for Mus81 to cleave the Flex1 (AT) 34 sequence. Surprisingly, removal of YEN1 results in a significant increase in Flex1 (AT)34 fragility in the DDRA assay system (Figure 4A). This result prompted us to investigate the order of action of Mus81 and Yen1 by creating a double mutant. In the mus81Δ yen1Δ double mutant, the recombination rate was reduced to mus81Δ levels, indicating that Mus81 acts upstream of Yen1 (Figure 4A). These results suggest that Mus81 acts before anaphase to cleave the Flex1 (AT)34 sequence, whereas Yen1 has an entirely different role, for example, to resolve problems persisting into anaphase. Interestingly, a yen1Δ had no effect on Flex1(AT)34 fragility in the YAC assay, where the repeat is near the end of a chromosome with no converging replication fork (Figure 4B). This result suggests that Yen1 resolves a structure created from two ends, such as two replication forks that have not merged or a two-ended recombination structure.
Flex1 Is Transcribed in the Locations Used to Test Fragility
Because AT cruciform structures are known to form more readily in conditions of negative supercoiling (McClellan et al., 1990; Dayn et al., 1991; Bowater et al., 1991), we investigated transcript levels at Flex1 using qRT-PCR. Indeed, Flex1 is transcribed at both locations studied (Figure S3). Interestingly, transcript levels at the repeat were 2.3-fold higher at the chromosome II compared with the YAC locus, which parallels the greater effect of mus81Δ on Flex1 (AT)34 fragility at that location (2.8-fold decrease from WT compared with 1.9-fold decrease). Thus, transcription could be a source of generating increased structure formation in cells containing Flex1 at the chromosome II locus, resulting in a larger need for SSE cleavage.
Sae2 Is Required for Healing of Flex1
The AT-rich nature of CFSs makes them more likely to form secondary structures, which could inhibit healing after breakage has occurred. If true, proteins involved in end resection should be important for healing breaks at CFSs. Sae2 is required to stimulate the MRX nuclease to cleave hairpin-capped DNA ends to facilitate resection and prevent palindromic gene amplification (Mimitou and Symington, 2009; Cejka, 2015). The human homolog, CtIP, is also needed at hairpin-capped ends (Makharashvili et al., 2014). In the DDRA fragility assay, a sae2Δ mutant had decreased healing specifically for Flex1-containing constructs, but the recombination rate of the control was unchanged (Figure 5A). These data indicate that Sae2 is not required for repair of non-structured DNA but is crucial for repair of Flex1 (AT)34-induced breaks. MRX-Sae2 activity could be required to respond to a number of hairpin-capped structures that could form at Flex1 (see Figure 6 and Discussion).
Figure 5. Flex1 Healing Is Dependent on Sae2 and Flanking Sequences Present.

(A) DDRA fragility assay data for Flex1 (AT)34 orientation 1 and control strains with sae2Δ; strains were tested for significant deviation from the WT with the same Flex1 AT tract length using an unpaired t test.
(B) DDRA fragility assay data for Flex1 (AT)34 in orientation 2. L3′ strains were tested for significant deviation from the same orientation S3′strain using an unpaired t test.
(C) Schematic and YAC end loss assay data showing Flex1 with its 5′ and 3′ flanking sequences in orientations 1 and 2.
(D) Schematic of three I-SceI strains created. Either an I-SceI recognition sequence only or an I-SceI recognition sequence with Flex1 flanking sequences was inserted into the DDRA fragility assay locus in orientation 1. DDRA fragility assay data for all three I-SceI strains under ~50% galactose in-duction of I-SceI breaks; p compared with I-SceI with S3′ flanking sequence using an unpaired t test.
Figure 6. A Model for Mus81 and Slx4 Complex Cleavage of Stalled Fork Substrates Formed by Secondary Structures at Flex1.

Secondary structure forming sequences at Flex1 cause replication fork stalling, which can potentially result in a reversed fork and/or convergence of the approaching fork. A DNA structure and/or the stalled fork is cleaved by SSEs, acting together or sequentially. Cleavages indicated by arrows are color-coded according to the nuclease as depicted. Mus81 cleavage at a stalled fork approaching from the left (arrow 1) will produce a one-ended break (left pathway). The broken end, which may be processed by MRX-Sae2, can invade the intact sister (repaired by gap filling) to initiate repair by homologous recombination, which could proceed by break-induced replication (BIR) or broken fork repair (BFR), synthesis-dependent strand annealing (SDSA), or second-end capture and double Holliday junction resolution by Yen1. Alternatively, if cleavage at stalled forks on either side of the cruciform occurs (arrows 1 and 3) or at the cruciform four-way junction (by coordinated Slx1-Mus81 cleavage, arrows 1 and 2), four ends will be produced. Cleavage of both strands of a single stalled fork will produce three ends, two of which will be hairpin capped if cleavage occurs at the cruciform base. The hairpin-capped ends will be processed by MRX-Sae2. Rad1-Rad10 could also process hairpin loops. Recombinants are recovered by SSA at homologous sequences (DE region of homology denoted by gray box), resulting in deletion of intervening sequences. 3′ non-homologous flaps created during SSA require Rad1-Rad10 and Slx4 nucleases for processing.
Flex1 Flanking Sequences Affect Healing in Two Different Fragility Assay Systems
In the Zhang and Freudenreich (2007) study, Flex1 with (AT)34 gave a significantly lower level of FOAR than the control, which was unexpected. The data suggested that a lower efficiency of healing could be the cause of the decreased recovery of YAC end loss events, and it was hypothesized that the longer 3′ (L3′) flanking sequence in the Flex1 (AT)34 construct compared with the other constructs could play a role (Zhang and Freudenreich, 2007). The L3′ flanking sequence is 102 bp longer than the short 3′ flanking sequence (S3′), and the Mfold program (Zuker, 2003) predicts that this extra 102 bp can form a stable hairpin with a ΔG of −6.7 (Figure S5). Human polymerase δ paused at the L3′sequence, providing evidence that a secondary structure is forming at that sequence (Figure S4, arrow).
To better understand the role of flanking sequences, we evaluated the rate of FOAR of Flex1 (AT)34 strains with L3′ and S3′ flanking sequences in the DDRA fragility assay. The strain with the L3′ flank has a significantly decreased level of FOAR compared with the S3′ strain, supporting the hypothesis that the L3′ flanking sequence inhibits healing (Figure 5B). In the YAC assay, if the additional sequence in the L3′ flanking sequence forms a hairpin that inhibits this leftward resection to the telomere seed sequence, the rate of FOAR should decrease upon the presence of the L3′ only in orientation 1, because left-ward resection proceeds through the 3′ flanking sequence after breakage at or near the AT repeat only in this orientation (Figures 5C and S2C). Indeed, the presence of the L3′ sequence inhibits healing in orientation 1, because FOAR His− rates are significantly decreased compared with the orientation 1 S3′ strain (Figure 5C). However, the identity of the 3′ sequence did not affect recovery in the YAC assay when it was to the right of the AT repeat in orientation 2, consistent with breakage occurring at the AT repeat, followed by leftward resection. Altogether, this evidence supported that the L3′ flanking sequence forms a hairpin that inhibits healing after breakage in our assays. However, it was also possible that the presence of the L3′ flanking sequence actually reduces fragility. To distinguish these possibilities, the Flex1 AT repeat was replaced by an I-SceI recognition sequence in the DDRA assay system so that double-strand breaks (DSBs) could be induced adjacent to the Flex1 flanking sequences. Three strains were created: (1) one with only the I-SceI recognition sequence (breakage without any expected healing impairments), (2) one with the I-SceI recognition sequence flanked by the S5′ and the L3′ Flex1 flanking sequences (breakage with healing impairment by L3′ hairpin/s), and (3) one with the I-SceI recognition sequence flanked by the Flex1 S5 ′and S3′ sequences (breakage without much healing impairment by flanks) (Figure 5D). The S5′-I-SceI-L3′ strain had a reduced recombination rate compared with the I-SceI or S5′-I-SceI-S3′strains (Figure 5D). These results further support the conclusion that the hairpin structure(s) present in the long 3′ flanking sequence reduces healing by inhibiting resection.
DISCUSSION
Flex1 Is an Important Component of FRA16D Fragility
We have demonstrated that Flex1, a roughly 300-bp subregion, is an important determinant of FRA16D breakage in vivo, because large FRA16D-containing YACs with Flex1 replaced go from 18.0% to 12.6% chromosome end loss. This indicates that Flex1 could account for roughly 30% of the breaks happening at FRA16D, even though it accounts for only 0.02% of the sequence on the large FRA16D-containing YAC. Thus, Flex1 is a major determinant of fragility at FRA16D, consistent with the finding that it is contained within the most frequently deleted region of FRA16D in cancer cell lines (Finnis et al., 2005). Nonetheless, there are likely other elements that also contribute to FRA16D breakage that could include other sequence elements or other mechanisms such as transcription-replication collisions. We recently proposed that fork stalling at Flex1 could increase the probability of downstream transcription-replication collisions in human cells because it would allow approach of the converging replication fork, which would be oriented head-on with WWOX transcription direction (Kaushal and Freudenreich, 2019).
Long Uninterrupted AT Repeats Cause Chromosome Fragility and Polymerase Stalling
We find that Flex1 is fragile in an AT repeat length-dependent manner when the flanking sequences are standardized in our DDRA fragility assay. Because cruciforms form in dsDNA, they need to overcome the energy of base-pairing to form, and they exhibit non-linear properties. The dramatic AT length dependence of fragility correlates well with our findings (Figures 1C and 1D) and previous studies showing that AT repeats form cruciforms on plasmids in vivo when the AT stem exceeds 22 bp (McClellan et al., 1990; Dayn et al., 1991; Bowater et al., 1991; Coté and Lewis, 2008). In contrast, hairpin formation typically occurs in ssDNA and is therefore governed more by the pairing strength (ΔG) of the base pairs in the stem. Human polymerase δ holoenzyme exhibited significant stalling at the Flex1 (AT)34 repeat tract in vitro. Because these assays were performed on ssDNA, they indicate that AT hairpins can also be a significant replication barrier. The model of a fork encountering a preformed cruciform is strengthened by the result that HU treatment is not required for fragility or fork stalling at Flex1 (AT)34 sequences (Figure 2C) (Zhang and Freudenreich, 2007). A cruciform could arise during transcription because of increased negative supercoiling caused by passage of RNA polymerase, and then block replication without the need for an additional stressor. Indeed, higher fragility rates and SSE dependence were found in the DDRA system, where there are also higher levels of transcription. The strong AT length dependence observed suggests that individuals with longer AT repeats at Flex1 will be at a significantly greater risk for chromosome fragility and associated deletions or rearrangements at FRA16D. Recently, an unbiased screen of breaks induced by aphidicolin and ataxia telangiectasia and Rad3-related protein (ATR) inhibition showed that most of the breaks form at structure-forming repetitive sequences, with AT repeats most highly represented in human genome (Shastri et al., 2018). Thus, our results at Flex1 could be applicable to many fragile sites in the human genome.
Mus81-Mms4, Slx1-Slx4, and Rad1-Rad10 SSEs Cause AT-Repeat Length-Dependent Cleavage
Flex1 fragility is dependent on AT length and the Mus81-Mms4, Slx1-Slx4, and Rad1-Rad10 nucleases. Notably, Flex1 is acting very similarly to FRA16D in the context of a whole human chromosome, where human MUS81-EME1, SLX4, and XPF-ERCC1 are required for full CFS expression (Naim et al., 2013; Ying et al., 2013; Minocherhomji et al., 2015). Our data suggest that Flex1 could be one of the major regions targeted by MUS81-EME1 nuclease activity at FRA16D in humans.
A fork stalled by a cruciform structure presents several potential SSE substrates (Figure 6). First, the cruciform base or loops could be targeted. A cruciform formed by a perfect AT repeat on a plasmid is cleaved by Mus81 in S. cerevisiae (Coté and Lewis, 2008). Slx4, via its interaction with Slx1 and (indirectly) with Mus81, could facilitate an Slx1-mediated nick followed by a Mus81-mediated counter nick to cleave at the base of the cruciform (Figure 6), as found for their human counterparts (Wyatt et al., 2013, 2017). However, this nick-counter-nick mechanism is likely not a major pathway of Flex1 fragility, because slx1Δ mutants do not have a decrease in fragility as dramatic as mus81Δ mutants, especially in the situation of a converged fork (Figure 4). Alternatively, the three-way junction of a stalled fork or resected reversed fork could be targeted for cleavage. Because Rad51 is not required for Mus81-induced fragility, it is likely acting on a stalled or converged fork rather than a recombination intermediate. This result is consistent with the recruitment of SLX4 and MUS81-EME1 to CFSs in early mitosis before POLD3 (Naim et al., 2013; Minocherhomji et al., 2015) and with a demonstrated role for the yeast Slx4 complex in repair of stalled forks induced by methyl methanesulfonate (MMS) (Gritenaite et al., 2014; Balint et al., 2015). Interestingly, the basal level of Mus81-Mms4 nuclease activity present in S phase is sufficient for induction of Flex1 fragility, suggesting that the cleavage could be occurring in either S/G2 or M phase in the yeast system and does not require activation by cell-cycle kinases at the G2/M boundary. Consistently, our previous analysis of breakage by physical analysis of the 801B6 YAC showed that the major cleavage product mapping to FRA16D appeared during S phase (Zhang and Freudenreich, 2007).
Potential Action of an SMR Tri-nuclease Complex at DNA Structures
The reduction in fragility of slx4Δ mutants in both genetic assays and the similar effect of the double- and triple-nuclease mutants in the YAC end loss fragility assay suggest that the nuclease action at Flex1 could be coordinated by Slx4. These data support that the nucleases are working in cooperation in the same pathway, and suggest the existence of an “SMR” super complex in yeast similar to the SMX DNA repair tri-nuclease complex that has been characterized in human cells (Wyatt et al., 2017). Slx4 has been shown to interact with Rad1-Rad10 (Flott et al., 2007) and responds to stalled forks in S. cerevisiae, S. pombe, and mouse embryonic stem cells (Kaliraman and Brill, 2002; Coulon et al., 2004; Balint et al., 2015; Willis et al., 2017). Sumoylation of Saw1 coordinates Slx1-Slx4 and Rad1-Rad10 cleavage in response to UV in S. cerevisiae (Sarangi et al., 2014), suggesting that they can work together. Also, there is evidence that Mus81-Mms4 interacts with Slx4 indirectly through the Dpb11 protein, and Slx4-Dpb11 and Mus81-Mms4 contribute to the resolution of joint molecules created by replication stress in the same pathway (Gritenaite et al., 2014; Princz et al., 2017). Mus81-dependent cleavage of a resected fork is expected to have a lesser dependence on Slx1 compared with cleavage of an intact four-way junction, which lends support to a stalled fork as the relevant substrate and could provide an Slx1-independent pathway for cleavage when two converged forks are present at the chromosome II location. Consistently, slx1Δ also had no effect on joint molecules at replication forks stalled by MMS that required Slx4 and Mus81-Mms4 for resolution (Gritenaite et al., 2014).
Yen1 Protects Flex1 against Fragility
Yen1 (human GEN1) is sometimes considered a backup nuclease to Mus81. Like mus81Δ, the yen1Δ effects are specific to Flex1 (Figure 4). However, unlike the other SSEs tested, yen1Δ mutants had an increase in fragility, indicating that Yen1 protects against Flex1-induced fragility. Interestingly, the effect of deleting Yen1 was much more evident when the Flex1 (AT)34 sequence was in the middle of chromosome II (DDRA assay) compared with the end of a chromosome (YAC assay). Yen1 could function to resolve a double Holliday junction (dHJ), a situation more likely to arise when there is a second end capture, which is not available when there is no incoming fork as on the end of the YAC. On chromosome II, Flex1 (AT)34 yen1Δ mus81Δ mutant fragility is equivalent to mus81Δ fragility levels, indicating that Mus81 acts upstream of Yen1 in the same pathway, consistent with the known timing of Mus81/MUS81 action earlier in the cell cycle than Yen1/GEN1 (Dehé and Gaillard, 2017). If Mus81-Mms4/SMR cleaves a fork stalled by a secondary structure at Flex1 in such a way as to create a two-ended break, it could be healed by either SSA (Figure 6, right pathway) or recombination (Figure 6, left pathway). If recombination occurs, a dHJ intermediate may result, requiring cleavage by Yen1. Alternatively, incomplete HR might leave connected sister chromatids that would require Yen1 resolution, and in its absence mechanical chromosome breakage could occur. SSA is a pathway that could rescue these breaks, resulting in deletions and recovery in our assay, consistent with the observed increase in SSA in the yen1Δ mutant.
Pathways to Heal Nuclease-Induced Breaks
Once a nuclease cleavage occurs to produce a one-ended or two-ended break, the cell will employ a break repair pathway to heal the broken end (Symington et al., 2014). Either coordinated cleavage at the cruciform base or cleavage of both forks would result in a two-ended break (Figure 6, right pathway). This pathway would favor SSA and recovery in our assay. Rad1-Rad10 could target the hairpin loop, as was recently shown for human XPF-ERCC1 at an inverted repeat structure (Lu et al., 2015), and would also be required for cleavage of non-homologous flaps at hairpin-capped ends or during SSA (Figure 6).
Alternatively, if Mus81 cleaves only one side of a stalled or reversed fork, this would result in a one-ended break (Figure 6, left pathway). After processing, the broken end could invade the sister chromatid to be repaired by HR. This pathway requires DNA repair synthesis, which could occur in G2 phase or M phase (MiDAS). If a second end becomes available (e.g., from the converging fork), the free 3′ end could also participate in synthesis-dependent strand annealing (SDSA) or second-end capture and dHJ resolution. The dHJ could be dissolved by the Sgs1-Top3-Rmi1 complex (human BTR) or branch migrated and resolved by Yen1 (human GEN1) in late mitosis, consistent with our finding of a requirement for Yen1 to protect against fragility in a pathway dependent on Mus81. Note that the DDRA assay measures only the SSA pathway, so mutants that reduce the HR pathway may not have an effect unless the reduction leads to increased SSA, although they could have a phenotype in the YAC assay where a one-ended break is more likely because of the lack of a converging fork.
Structures that Flank a Fragile Site Impair Resection and Alter Repair Outcomes
Our data show that a hairpin predicted to form in the flanking sequence of Flex1 inhibits healing in both of our genetic assays or when placed adjacent to an induced DSB, and causes polymerase δ pausing in a primer extension assay. These data bring about the following hypothesis for CFS fragility: CFS expression could be a combination of cleavage and processing of stalled forks, and inefficient healing due to the presence of multiple contiguous sequences that form secondary structures.
Sae2 is required to process breaks that occur at Flex1, and the absence of Sae2 severely reduced recovery of broken chromosomes. These results are consistent with the known activity of Sae2 in stimulating Mre11 nuclease processing of hairpin-capped ends (Mimitou and Symington, 2009; Cejka, 2015). These ends could result from SSE cleavage near the base of the cruciform to produce AT hairpin-capped ends, or from fold-back of flanking hairpins (for example, on a reversed fork end) (Figure 6). In a similar assay in mammalian cells, CtIP was found to be essential for recovering breaks at Flex1 by SSA but was not required at clean I-SceI DSBs (Wang et al., 2014), and CtIP also functions as a co-factor of MRN nuclease in mammalian cells (Anand et al., 2016). Therefore, this appears to be a conserved pathway and is likely operating at naturally occurring breaks at FRA16D in human cells. Indeed, deletion of the Rad50 component of yeast MRX caused increased death of cells containing FRA16D on the large 801B6 YAC that was exacerbated by replication stress (Zhang and Freudenreich, 2007). Because Sae2 prevents translocations in yeast (Deng et al., 2015), MRN-CtIP could prevent genomic rearrangements at Flex1 in FRA16D.
Our data suggest that the propensity for the Flex1 region of FRA16D to be deleted in cancer cell lines (Finnis et al., 2005) is due to its ability to form a secondary structure. Because resection is an important feature of almost all cellular DSB repair mechanisms, our results predict that breaks that occur within structure-forming DNA in human cells will have a reduced efficiency of healing, which may favor alternative and less conservative repair pathways that generate translocations or large deletions.
Implications for Genome Stability and Cancer Initiation
Our data show that nuclease cleavage is relevant only for Flex1 sequences with 23 or more AT repeats, which corresponds to a size that can form a cruciform and stall replication forks in vivo. This predicts that individuals with longer AT alleles at Flex1 will have a greater risk for genome instability at the FRA16D locus and will be more reliant on the SSEs to process stalled forks and prevent deleterious translocations and deletions at this locus. Overall, the ability to respond to replication stress caused by DNA structures by the regulated action of nucleases may be an important cancer-protective mechanism (Fragkos and Naim, 2017). Cleavage of other naturally occurring palindromes in human cells has been shown to occur in vivo, leading to translocations (Kato et al., 2012), which have also been found at FRA16D in multiple myeloma patients (Ried et al., 2000). Thus, the mechanisms described here could be generally applicable to many cruciform-forming structures in the human genome.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to corresponding author, Catherine H. Freudenreich (Catherine.freudenreich@tufts.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains, oligonucleotides, and plasmids used in this study are listed in the Key Resources Table, Tables S4 and S5, respectively. All yeast strains were grown at 30°C and all bacterial strains were grown at 37°C. Large FRA16D YAC strains were as used as previously described (Zhang and Freudenreich, 2007). Overall YAC length was confirmed using pulsed field gel electrophoresis followed by a Southern blot using a TRP1 probe (Figure S1B). Flex1 was replaced in FRA16D with the KANMX marker, which was confirmed by PCR. Intact YAC structure was also verified using PCR of subregions across FRA16D (Figure S1A).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| R408 Helper Phage | Promega | Cat# P2291 |
| SURE 2 Supercompetent Cells | Agilent Technologies | Cat# 200152 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EcoRI-HF | NEB | Cat# R3101S |
| BamHI-HF | NEB | Cat# R3136S |
| AhdI | NEB | Cat# R0584S |
| XbaI | NEB | Cat# R0145S |
| S1 nuclease | Thermo Fisher | Cat# EN0321 |
| Eco53kI | NEB | Cat# R0116S |
| Polyethylene Glycol (Avg Mol Wt of 8000) | Sigma | Cat# P5413; CAS 25322-68-3 |
| Equilibrated Phenol, pH 8.0, Ultrapure | Affymetrix/Thermo-Fisher | Cat# AAJ75829AN; CAS 108-95-2 |
| γ32P ATP (6000Ci/mmol) | Perkin-Elmer | Cat# BLU002Z001MC |
| T4 Polynucleotide Kinase | Thermo-Fisher | Cat# 18004010 |
| Recombinant human PCNA | Laboratory of Marietta Lee | Biochemistry 2001 40: 4512–4520 |
| Recombinant yeast RFC | Laboratory of Linda Bloom | The Journal of Biological Chemistry 2012 287: 2203–9 |
| Sequenase 2.0 | Affymetrix/Thermo-Fisher | Cat# 70775Y |
| Exo- Klenow Polymerase | Affymetrix/Thermo-Fisher | Cat# 70057Z |
| Deposited Data | ||
| % FOAR colonies in large FRA16D YACs | This paper; Mendeley Data | Table S1;https://doi.org/10.17632/hh5rhpswsf.1" |
| DDRA fragility assay data | This paper; Mendeley Data | Table S2;https://doi.org/10.17632/hh5rhpswsf.1 |
| YAC fragility assay data | This paper; Mendeley Data | Table S3; https://doi.org/10.17632/hh5rhpswsf.1 |
| AT series termination probability | This paper; Mendeley Data | Table S6; https://doi.org/10.17632/hh5rhpswsf.1 |
| RT-qPCR data | This paper; Mendeley Data | Table S7; https://doi.org/10.17632/hh5rhpswsf.1 |
| Experimental Models: Organisms/Strains | ||
| 972D3 YAC (Background: AB1380) | MATa, ura3–52, his5, trp1–289, lys2–1, can1–100, ade2–1 YAC: LEU2 C4A4 URA3 TRP1 | [CFY #1087] (Albertsen et al., 1990; Zhang and Freudenreich, 2007) |
| 801B6 YAC (Background: AB1380) | YAC: LEU2 C4A4 URA3 TRP1 | [CFY #1086] (Albertsen et al., 1990; Zhang and Freudenreich, 2007) |
| 801B6 YAC Flex1D (Background: CFY# 1086) | YAC: LEU2 C4A4 URA3 TRP1 Flex1 ::KANMX6 | [CFY #3076, 3077] this study |
| lys2::ADE2 (Background: YPH499) | MATa, leu2-D1, ura3–52, his3-D200, trp1-D63, ade2D::hisG (salmonella), lys2::ADE2 | [CFY #2268] this study |
| ctrl (Background: CFY# 2268) | lys2::ADE2::URA3-no repeat control | [CFY #2863, 2864] this study |
| Flex1 (AT)14 (Background: CFY# 2268) | lys2::ADE2:: URA3-Flex1(AT)14 | [CFY #3917, 3921] this study |
| Flex1 (AT)23 (Background: CFY# 2268) | lys2::ADE2:: URA3-Flex1(AT)23 | [CFY #3445, 3473] this study |
| Flex1 (AT)34 (Background: CFY# 2268) | lys2::ADE2:: URA3-Flex1(AT)34 | [CFY #2525, 2712] this study |
| ctrl mus81Δ (Background: CFY# 2863) | mus81::KANMX4 | [CFY #3375] this study |
| Flex1 (AT)14 mus81Δ (Background: CFY# 3917) | mus81::KANMX4 | [CFY #4326, 4327] this study |
| Flex1 (AT)23 mus81Δ (Background: CFY# 3445) | mus81::KANMX4 | [CFY #3799, 3800] this study |
| Flex1 (AT)34 mus81Δ (Background: CFY# 2525) | mus81::KANMX4 | [CFY #3377, 3378] this study |
| Flex1 (AT)34 mms4Δ (Background: CFY# 2525) | mms4::KANMX | [CFY #4743, 4744] this study |
| Flex1 (AT)34 mms4–9A (Background: CFY# 2525) | pADH1–3HA-mms4D::URA3::mms4-np (S55A; S56A; S184A; S201A; S221A; S222A; S301A; T302A; S403A)-HIS3 | [CFY #4586, 4587] this study; modified from (Gallo-Fernández et al., 2012) |
| Flex1 (AT)34 rtt107Δ (Background: CFY# 2525) | rtt107::KANMX | [CFY #4666, 4667] this study |
| Flex1 (AT)34 rtt107Δ mms4–9A (Background: CFY# 4666) | pADH1–3HA-mms4Δ::URA3::mms4-np (S55A; S56A; S184A; S201A; S221A; S222A; S301A; T302A; S403A)-HIS3 | [CFY #4668, 4669] this study; modified from (Gallo-Fernández et al., 2012) |
| Flex1 (AT)34 rad51Δ (Background: CFY# 2525) | rad51::NATMX | [CFY #4705, 4708] this study |
| Flex1 (AT)34 rad51Δ mus81Δ(Background: CFY#3377) | mus81::KANMX | [CFY #4706, 4707] this study |
| Flex1 (AT)34yen1Δ (Background: CFY# 2525) | yen1::TRP1 | [CFY #3987, 3988, 4063] this study |
| Flex1 (AT)34 slx1Δ (Background: CFY# 2525) | slx1::KANMX4 | [CFY #4138, 4139] this study |
| Flex1 (AT)34 rad1Δ (Background: CFY# 2525) | rad1::TRP1 | [CFY #4584/4585] this study |
| Flex1 (AT)34 slx4Δ (Background: CFY# 2525) | slx4::KANMX6 | [CFY #4022, 4023] this study |
| Flex1 (AT)34 mus81Δ yen1Δ (Background: CFY# 4063) | mus81::KANMX4, yen1::TRP1 | [CFY #4203, 4204] this study |
| Flex1 (AT)34 mus81Δ slx1Δ (Background: CFY# 4139) | slx1::KANMX4, mus81::TRP1 | [CFY #4238, 4239] this study |
| ctrl yen1Δ (Background: CFY# 2863) | yen1::TRP1 | [CFY #4125, 4126] this study |
| ctrl slx1Δ (Background: CFY# 2864) | slx1::KANMX4 | [CFY #4340, 4341] this study |
| ctrl rad1Δ (Background: CFY# 2863) | rad1::TRP1 | [CFY #4582, 4583] this study |
| ctrl slx4Δ (Background: CFY# 2863) | slx4::KANMX6 | [CFY #4328, 4329] this study |
| ctrl sae2Δ (Background: CFY# 2863) | sae2::KANMX6 | [CFY #3607, 3608] this study |
| Flex1 (AT)34 sae2Δ (Background: CFY# 3106) | sae2::KANMX6 | [CFY #3520, 3521] this study |
| Flex1 (AT)34 S3′ o2 (Background: CFY# 2268) | lys2::ADE2:: URA3-Flex1 (AT)34 S3′ o2 | [CFY #3106, 3202–3204] this study |
| Flex1 (AT)34 L3′ o2 (Background: CFY# 2268) | lys2::ADE2:: URA3-Flex1 (AT)34 L3′ o2 | [CFY #2372– 2375] this study |
| no-I-SceI cut site (Background: CFY# 2268) | ILV1::pGAL-I-SceI nuclease | [CFY #3518] this study |
| I-SceI only (Background: CFY# 3518) | lys2::ADE2::URA3-I-SceI only | [CFY #4439, 4440, 4342] this study |
| I-SceI S3′ (Background: CFY# 3518) | lys2::ADE2 ::URA3-I-SceI-S3′ | [CFY #3989, 4323] this study |
| I-SceI L3′ (Background: CFY# 3519) | lys2::ADE2 ::URA3-I-SceI-L3′ | [CFY #3991, 3992] this study |
| WT strain with YAC CF1 (no Flex1) (Background: BY4705) | MAT α, leu2Δ0, ura3Δ0, his3Δ200, trp1Δ63, ade2Δ::hisG, lys2Δ0, met15Δ0,YAC CF1: ade3–2p ARS1 CEN4 LEU2 (G4T4)13 URA3 | [CFY #765] (Callahan et al., 2003) |
| Flex1 (AT)34 S3′ o1 on YAC (Background: CFY #765) | YAC: LEU2 Flex1(AT)34 HIS3 URA3 (this and all YACs in this study are modified from YAC CF1; only relevant markers and added sequence are listed) | [CFY #3457, 3458] this study |
| Flex1 (AT)34 on YAC yen1Δ (Background: CFY# 3457) | yen1::TRP1 | [CFY #4315, 4316] this study |
| Flex1 (AT)34 on YAC mus81Δ (Background: CFY# 3458) | mus81::KANMX4 | [CFY #4284, 4285] this study |
| Flex1 (AT)34 on YAC slx1Δ (Background: CFY# 3458) | slx1::KANMX4 | [CFY #4313, 4314] this study |
| Flex1 (AT)34 on YAC rad1Δ (Background: CFY# 3458) | rad1::KANXMX6 | [CFY #4351, 4352] this study |
| Flex1 (AT)34 on YAC slx4Δ (Background: CFY#3457) | slx4::KANMX4 | [CFY #4550, 4551] this study |
| Flex1 (AT)34 on YAC mus81Δ rad1Δ (Background: CFY# 4284) | rad1::TRP1 mus81::KANMX4 | [CFY #4408, 4409] this study |
| Flex1 (AT)34 on YAC slx1Δ rad1Δ (Background: CFY# 4313) | rad1::TRP1 slx1:: KANMX4 | [CFY #4425, 4426] this study |
| Flex1 (AT)34 on YAC slx1Δ mus81Δ rad1Δ (Background: CFY#4425) | rad1::TRP1 slx1:: KANMX4 mus81::NATMX | [CFY #4759, 4760] this study |
| Flex1 (AT)23 S3′ o1 on YAC (Background: CFY #765) | YAC: LEU2 Flex1(AT)34 HIS3 URA3 | [CFY #1239, 1240] (Zhang and Freudenreich, 2007) |
| Flex1 (AT)34 L3′ on YAC (Background: CFY #765) | YAC: LEU2 Flex1(AT)34 HIS3 URA3 | [CFY #1241, 1242] (Zhang and Freudenreich, 2007) |
| Oligonucleotides, see Table S4. | This study | N/A |
| Recombinant DNA | ||
| Plasmids, see Table S5. | This study | N/A |
| pGEM-3Zf(−) Vector | Promega | Cat# P2261 |
| Software and Algorithms | ||
| FALCOR | Hall et al., 2009 | N/A |
| ImageQuant version 5.2 | GE Healthcare | N/A |
| Other | ||
| Illustra Microspin G-50 column | GE Healthcare | Cat# 27-5330-1 |
Chromosome II Flex1 strains were created by modifying the pBL007 plasmid, which has a URA3 marker and nucleotides 512–1480 of ADE2 (designated DE in diagrams). The FRA16D subregions of interest were inserted into the EcoRI only or BamHI and EcoRI sites in the MCS of pBL007. Orientation was confirmed by PCR and sequencing. Plasmids were digested with XbaI to linearize them for transformation into lys2::ADE2 yeast strains, replacing ADE2 with the ADE-URA3-Flex1-DE2 cassette. All chromosome II yeast strains were checked by PCR of the pBL007 cassette junctions and sequencing to confirm correct sequence and orientation.
The Flex1 subregion YACs (AT)23-S3′ and (AT)34-L3′ in orientation 1 were created previously (Zhang and Freudenreich, 2007). Flex1 (AT)34-S3′ in o1 and o2 and Flex1 (AT)34-L3′o2 YAC strains were made by modifying the pHZ-HIS3MX6 plasmid. The Flex1 subregion of interest was inserted by EcoRI-based subcloning into the MCS of pHZ-HIS3MX6. Correct Flex1 sequence insertion in the right orientation was confirmed by PCR and sequencing. Plasmids were digested with AhdI to linearize them for transformation into CFY #765 BY4705 yeast strains containing URA3 marked YAC CF1 (Callahan et al., 2003) and selecting for His+ transformants. Correct structure of the Flex1 YACs was confirmed by PCR of the pHZ-HIS3MX6 cassette junctions (primers 375 and 832 in Table S4) and sequencing to confirm Flex1 sequence and orientation.
Chromosome II I-SceI strains were created by modifying the pBL007 plasmid. The I-SceI only insert was created by PCR with primers 1511 and 1512, whose 3′ ends anneal to one another at the I-SceI recognition sequence; that PCR product was then used as a template for PCR with primers 1513 and 1514 to complete generation of the insert. S5′-I-SceI-S3′ and S5′-I-SceI-L3′ inserts were synthesized as gBlocks (Table S4) (Integrated DNA Technologies, Coralville, Iowa) flanked by EcoRI restriction sites and contained S5′ and S3′ from Flex1 flanking sequences and an I-SceI restriction site, or S5′ and L3′ from Flex1 flanking sequences and an I-SceI restriction site. The inserts were cloned into the EcoRI site of pBL007. Correct I-SceI recognition sequence insertion into the plasmid was confirmed by PCR and sequencing. A yeast strain with a galactose-inducible I-SceI nuclease was created by transformation of a PCR product from the pGSHU plasmid (Storici et al., 2003) into the ILV1 locus in a lys2::ADE2 strain (CF stock #2268). Insertion of the galactose-inducible I-SceI nuclease was confirmed by hygromycin resistance and PCR of the 5′ junction of the cassette. XbaI-linearized pBL007+I-SceI DNA was transformed into the lys2::ADE2 strain with the galactose-inducible I-SceI nuclease. Yeast strains were confirmed by PCR and sequencing as stated above.
All gene deletion mutants were created using one-step gene replacement. Primers with homology to regions directly upstream and downstream of ORF for gene replacement were used to amplify gene replacement fragments from either the pFA plasmid series or yeast genomic DNA of a previously made gene replacement strain. Proper gene replacement was confirmed by PCR using primer sets: (1) that hybridize to the marker gene and a genomic region outside of the gene to be replaced and (2) are located within the open reading frame (ORF) to be replaced to confirm ORF absence. Sequences of primers used are available upon request.
METHOD DETAILS
Large FRA16D YAC Breakage Assay
Large FRA16D YAC strains with confirmed YAC structure were patched onto YC-Ura-Leu-Trp plates and then plated for single colonies on YC-Ura-Leu-Trp and grown for 2 days at 30°C. A portion of 10 single colonies was used to inoculate ten 1 mL YC-Leu cultures at 0.02–0.04 OD which were grown at 30 C for 6–7 divisions (~16 hours). 100 uL of a 10−4 dilution of each culture was plated on FOA-Leu to query for cells that had lost URA3 gene function, potentially by breakage within FRA16D and YAC end loss. 100 uL from each culture were combined, diluted to 10−4, and plated on YC-Leu media to obtain a total cell count. Plates were grown for 3 days at 30°C. Breakage frequency was calculated. PCR of representative 801B6 strains using primers 3 and 1223 (Table S4) confirmed URA3 loss in 60/60 independently derived FOAR colonies tested. For all yeast strains and primers used in this study, see Key Resources Table and Table S4, respectively.
DDRA Fragility Assay
DDRA fragility assay strains were patched onto YC-Ura to maintain selection for the ADE2 recombination assay cassette in the starting strains. Cells from a YC-Ura patch were plated for single colonies on YEPD non-selective media for 3 days at 30 C to allow breakage to occur. Individual colonies were resuspended in 400 uL diH2O, diluted as appropriate (varies by strain and mutant), and plated on FOA-Ade media to select for cells that have undergone breakage and recombination of the chromosome II cassette. 100 uL from each colony suspension were combined, diluted to either 10−4 or 10−5, and plated on YEPD media to obtain a total cell count. A rate of FOAR Ade+ was calculated using the method of the median using the FALCOR online calculator (Hall et al., 2009). PCR using primers 4 and 5 (Table S4) confirmed URA3 loss consistent with SSA in 49/50 independently derived FOAR Ade+ colonies from representative Flex1(AT)34 strains.
I-SceI DDRA fragility assays were performed in the same manner, except all media was supplemented with 10x isoleucine, 10x leucine, and 10x valine to compensate for disruption of the ILV locus. YEP plates were made with 1.5% galactose and 0.5% glucose to induce ~50% cutting of I-SceI. For hydroxyurea DDRA fragility assays, the YEPD plates were supplemented with 100 mM HU.
Flex1 Subregion YAC End Loss Fragility Assay
Fragility assays were performed on the YACs as previously described (Zhang and Freudenreich, 2007). Cells were plated onto YC-Leu-Ura plates in order to select for both arms of the YAC. Ten 1 mL YC-Leu liquid cultures of 0.02–0.04 starting OD600 were inoculated from YC-Leu-Ura patches and grown overnight at 30°C for 6–7 divisions (~16 hours for wild-type strains; longer for some mutants). A portion of each culture (100 uL for WT strains; less for strains with high fragility rates) was plated on FOA-Leu to query for cells that had lost URA3 gene function, potentially by breakage within Flex1 and YAC end loss. Plates were grown for 5 days at 30°C. Total cell counts were obtained by combining 100 uL from each YC-Leu overnight culture and plating 10−4 and 10−5 dilutions on YC-Leu. FOA-Leu plates were replica plated onto YC-His; any colonies growing on YC-His did not lose the right arm of the YAC and were removed from colony counts. A rate of FOAR His− was calculated using the method of the median using the Fluctuation Analysis Calculator (FALCOR). End loss PCR was performed using primers 4 and 5 (Table S4) to confirm URA3 absence in 32/36 independently derived FOAR colonies from representative Flex1(AT)34 strains.
S1 Nuclease Cleavage and Mapping
pBL007 constructs containing control and FRA16D sub-regions in orientation1 (S5′(AT)14 L3′, S5′(AT)23 L3′ and S5′(AT)34 L3′) were used for the S1 nuclease assay. The plasmids were incubated in 5X S1 nuclease cleavage buffer with increasing concentration of S1 nuclease (1U, 1.75 U, 2.5 U and 5 U) for 10 minutes at 37°C. The reactions were stopped by addition of 2ml of 0.5 M EDTA and icing. The cleavage products were resolved on 1% Agarose gel and post-stained with ethidium bromide and visualized using UV transilluminator. For mapping S1 nuclease cleavage site, two pBL007 constructs were used (S5′AT23L3′ and S5′AT34L3′). S1 nuclease assay was carried out as described above with 10 U of enzyme per reaction to ensure complete cleavage of supercoiled plasmids (SC) into linear products (L), which were then resolved on 1% Agarose gel, excised and purified. The purified products were digested with Eco53kI at 37°C for 16 hours and run on 1% agarose gel and 3% MetaPhor gel. For chemical details, see Key Resources Table. (Barnes et al., 2017)
In Vitro Polymerase δ Pausing Assay
pBL007 constructs containing control and FRA16D sub-regions (S5′(AT)14 S3′, S5′(AT)23 S3′ and S5′(AT)34 S3′) were used as templates for primer extension analyses in Figure 2. Templates for polymerase reactions in Figure S4 were created by cloning the 315 bp Flex1 sequence (S5′AT34 L3′) into the MCS/BamH1 site of the pGEM3Zf(−) vector (Promega, P2261) (Table S5). Inserts in two orientations were isolated in order to purify ssDNA templates of both strands. For each construct, single-stranded DNA was isolated after R408 helper phage (Promega, P2291) infection of plasmid-bearing SURE cells (e14-(McrA-), Δ(mcrCB-hsdSMR-mrr)171, endA1, gyrA96, thi-1, supE44, relA1, lac, recB, recJ, sbcC, umuC::Tn5 (Kanr) uvrC [F’ proAB lacIqZΔDM15 Tn10 (Tetr) Amy Camr]; Agilent Technologies, 200152). Log phase plasmid-bearing SURE cells in 2XYT media were infected with 1/50th volume of R408 (titer of phage stock was > 1 × 1011 plaque forming units (pfu)/mL) and incubated in a 37°C shaker for 4–8 hours. After pelleting the bacterial cells, virus particles in the supernatant were precipitated on ice for 30 min with a polyethylene glycol (Sigma, P5413)/ammonium acetate solution at final concentrations of 4% and 0.75 M, respectively. Virus was pelleted and resuspended in an appropriate volume of Phenol Extraction Buffer (PEB; 100 mM Tris, pH 8.0, 300 mM NaCl, 1 mM EDTA, pH 8.0). DNA was extracted one time with two volumes of phenol (Affymetrix/Thermo-Fisher, AAJ75829AN) saturated with PEB, one time with one volume of phenol, and one time with half volume 24:1 chloroform: isoamyl alcohol. After extraction, DNA was precipitated with ammonium acetate at 2.0 M final concentration and 2 volumes of ethanol and resuspended in 10 mM Tris and 1 mM EDTA, pH 8.0.
DNA synthesis templates were created by 32P end-labeling (γ32P ATP (6000Ci/mmol); Perkin-Elmer, BLU002Z001MC) either the M13 Forward (−20) 16-mer (Thermo-Fisher, N52002) or a PAGE-purified 16-mer oligonucleotide (G40, Integrated DNA Technologies) using T4 Polynucleotide Kinase (Thermo-Fisher, 18004010) according to the manufacturer’s instructions and hybridizing to ssDNA at a 1:1 molar ratio in 1X SSC buffer (150 mM NaCl and 15 mM sodium citrate). The M13 Forward oligonucleotide initiates synthesis 69 nucleotides downstream of the Flex1 inserts in pBL007. The G40 oligonucleotide initiates synthesis 14 nucleotides downstream of the S5′ AT34 L3′ Flex1 insert in pGEM3Zf(−). To remove unincorporated radionucleotide, the hybridized primer-templates were purified over illustra Microspin G-50 columns (GE Healthcare, 27-5330-01). Primer extension reactions contained 100 fmol of primed ssDNA substrate, 400 fmol human recombinant PCNA (Xu et al., 2001), 1700 fmol yeast RFC (Thompson et al., 2012), 20 mM Tris HCl, pH 7.5, 8 mM MgCl2, 5 mM DTT, 40 mg/ml BSA, 150 mM KCl, 5% glycerol, 0.5 mM ATP, and 250 uM dNTPS, and were preincubated at 37°C for 3 min. Synthesis was initiated upon addition of the indicated fmol purified 4-subunit recombinant human Pol δ4 (Zhou et al., 2012). Aliquots were removed at 5 and 15 minutes, quenched in 1 volume STOP dye (Formamide, 5 mM EDTA pH 8.0, 0.1% xylene cylanol, 0.1% bromophenol blue) and reaction products were separated on an 8% denaturing polyacrylamide gel, scanned using a GE Healthcare Typhoon FLA 9500 and quantified using ImageQuant v5.2 software. A control for the percent of primers productively hybridized to each template substrate (% Hyb) was performed using excess Exo-Klenow polymer-ase (Affymetrix/Thermo-Fisher, 70057Z), and a background control for primer impurities (no Pol) was performed by incubating unextended primer-template substrate in reaction buffer without addition of polymerase. Dideoxy sequencing reactions were carried out simultaneously with the Pol δ4 reactions, using the same primer-template substrates and Sequenase 2.0 (Affymetrix/Thermo-Fisher, 70775Y). For chemical details, see Key Resources Table. For AT series termination probability data, see Table S6.
Pulsed Field Gel Electrophoresis
Large FRA16D YAC length was verified using CHEF gels (Bio-Rad) and Southern blot hybridization. Cells were grown to early log phase in YC-Leu-Ura-Trp media and whole chromosomal DNA was isolated in 0.8% agarose plugs (Bio-Rad Clean Cut agarose). Plugs were run on a 1.2% gel, 5V/cm, 60–120 switch, for 48 hours. The Southern blot was performed using a TRP1 probe to the YAC (see Figure 1C for relative TRP1 location on the YAC).
RT-qPCR
RNA was extracted using the GE illustra RNAspin Mini Isolation Kit using the manufacturer’s instructions from log-phase cultures of strains grown in yeast complete media for chromosome II strains. YAC strains were grown in YC-Leu-Ura media, except for one RNA prep from YC-Leu for the YAC strains. cDNA was generated using the ThermoFisher Superscript First Strand Synthesis System for RT-PCR and random hexamers as primers. qPCR was performed on cDNA using primers 1254 and 1255 and the POWER SYBR Green Master Mix from Thermo Scientific. See Table S7 for raw data.
QUANTIFICATION AND STATISTICAL ANALYSIS
DDRA fragility and YAC end loss assays were all a minimum of 3 assays, usually from 2 independently created strains. Strains were tested for significant deviation from the appropriate control using a t test. Average rates are graphed with error bars indicating the standard error of the mean (see Tables S1, S2, and S3). (Albertsen et al., 1990; Wach et al., 1994)
DATA AND SOFTWARE AVAILABILITY
Data are published on Mendeley (https://doi.org/10.17632/hh5rhpswsf.1).
Supplementary Material
Highlights.
FRA16D subregion Flex1 causes fragility in an AT length-dependent manner
Longer AT repeats form a cruciform and stall replication by human Pol δ
Flex1 is targeted by the Mus81 nuclease, whereas Yen1 protects against breakage
Fragility at Flex1 is due to a combination of increased breaks and impaired healing
ACKNOWLEDGMENTS
We thank Brian Lenzmeier for sharing reagents to construct the ADE2 version of the DDRA assay; Francesca Storici for the inducible I-SceI system; Ryan McGinty and Michael Sigouros for help with yeast strain construction, verification, and some assays; and Allen Su, Keerthana Gnanapradeepan, and Adam Snider for help with cloning. Funding was provided by NSF grants MCB1330743 and MCB1817499, NIH grants GM105473 and GM122880, and a Tufts Dean’s Fund award (to C.H.F.), an Arnold and Mabel Beckman Foundation award (to C.H.F. and C.E.W.), and a Tufts Graduate Student Research Award (to S.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.103.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albertsen HM, Abderrahim H, Cann HM, Dausset J, Le Paslier D, and Cohen D (1990). Construction and characterization of a yeast artificial chromosome library containing seven haploid human genome equivalents. Proc. Natl. Acad. Sci. USA 87, 4256–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Ranjha L, Cannavo E, and Cejka P (2016). Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 64, 940–950. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Tainer JA, Vasquez KM, and Cooper DN (2016). Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 44, 5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, Yimit A, Ou J, Nakato R, Gurevich A, Shirahige K, et al. (2015). Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J. 34, 2182–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RP, Hile SE, Lee MY, and Eckert KA (2017). DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst.) 57, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al. (2010). Signatures of mutation and selection in the cancer genome. Nature 463, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowater R, Aboul-ela F, and Lilley DM (1991). Large-scale stable opening of supercoiled DNA in response to temperature and supercoiling in (A + T)-rich regions that promote low-salt cruciform extrusion. Biochemistry 30, 11495–11506. [DOI] [PubMed] [Google Scholar]
- Callahan JL, Andrews KJ, Zakian VA, and Freudenreich CH (2003). Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Mol. Cell. Biol. 23, 7849–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P (2015). DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J. Biol. Chem 290, 22931–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coté AG, and Lewis SM (2008). Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol. Cell 31, 800–812. [DOI] [PubMed] [Google Scholar]
- Coulon S, Gaillard PH, Chahwan C, McDonald WH, Yates JR 3rd, and Russell P (2004). Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol. Biol. Cell 15, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JR, Dibitetto D, Pellicioli A, and Smolka MB (2017). Slx4 scaffolding in homologous recombination and checkpoint control: lessons from yeast. Chromosoma 126, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayn A, Malkhosyan S, Duzhy D, Lyamichev V, Panchenko Y, and Mirkin S (1991). Formation of (dA-dT)n cruciforms in Escherichia coli cells under different environmental conditions. J. Bacteriol 173, 2658–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatisse M, and Rosselli F (2019). A journey with common fragile sites: From S phase to telophase. Genes Chromosomes Cancer 58, 305–316. [DOI] [PubMed] [Google Scholar]
- Dehé PM, and Gaillard PH (2017). Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 18, 315–330. [DOI] [PubMed] [Google Scholar]
- Deng SK, Yin Y, Petes TD, and Symington LS (2015). Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol. Cell 60, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, and Glover TW (2008). Replication stress induces tumor-like microdeletions in FHIT/ FRA3B. Proc. Natl. Acad. Sci. USA 105, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnis M, Dayan S, Hobson L, Chenevix-Trench G, Friend K, Ried K, Venter D, Woollatt E, Baker E, and Richards RI (2005). Common chromosomal fragile site FRA16D mutation in cancer cells. Hum. Mol. Genet 14, 1341–1349. [DOI] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, Toth R, Sugawara N, Campbell DG, Haber JE, Pasero P, and Rouse J (2007). Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol 27, 6433–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, and Naim V (2017). Rescue from replication stress during mitosis. Cell Cycle 16, 613–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenreich CH, Kantrow SM, and Zakian VA (1998). Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856. [DOI] [PubMed] [Google Scholar]
- Fricke WM, and Brill SJ (2003). Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17, 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo-Fernández M, Saugar I, Ortiz-Bazán MA, Vázquez MV, and Tercero JA (2012). Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 40, 8325–8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Wilson TE, and Arlt MF (2017). Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, and Pfander B (2014). A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 28, 1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, and Bartek J (2008). An oncogeneinduced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hall BM, Ma CX, Liang P, and Singh KK (2009). Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25, 1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, and Tora L (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. [DOI] [PubMed] [Google Scholar]
- Higgins NP, and Vologodskii AV (2015). Topological Behavior of Plasmid DNA. Microbiol. Spectr 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, and Brill SJ (2002). Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr. Genet 41, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kurahashi H, and Emanuel BS (2012). Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev 22, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, and Freudenreich CH (2019). The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 58, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, and Debatisse M (2013). Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428. [DOI] [PubMed] [Google Scholar]
- Lu S, Wang G, Bacolla A, Zhao J, Spitser S, and Vasquez KM (2015). Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 10, 1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, García-Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S, et al. (2016). FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 64, 388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. (2014). Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell 54, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan JA, Boublíková P, Palecek E, and Lilley DM (1990). Superhelical torsion in cellular DNA responds directly to environmental and genetic factors. Proc. Natl. Acad. Sci. USA 87, 8373–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, and Symington LS (2009). DNA end resection: many nucleases make light work. DNA Repair (Amst.) 8, 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, and Hickson ID (2014). Structure-specific endonucleases: guardians of fragile site stability. Trends Cell Biol. 24, 321–327. [DOI] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. [DOI] [PubMed] [Google Scholar]
- Miron K, Golan-Lev T, Dvir R, Ben-David E, and Kerem B (2015). Oncogenes create a unique landscape of fragile sites. Nat. Commun 6, 7094. [DOI] [PubMed] [Google Scholar]
- Naim V, Wilhelm T, Debatisse M, and Rosselli F (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. [DOI] [PubMed] [Google Scholar]
- Paeschke K, Capra JA, and Zakian VA (2011). DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleys EJ, and Freudenreich CH (2018). Methods to Study Repeat Fragility and Instability in Saccharomyces cerevisiae. Methods Mol. Biol 1672, 403–419. [DOI] [PubMed] [Google Scholar]
- Princz LN, Wild P, Bittmann J, Aguado FJ, Blanco MG, Matos J, and Pfander B (2017). Dbf4-dependent kinase and the Rtt107 scaffold promote Mus81-Mms4 resolvase activation during mitosis. EMBO J. 36, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, et al. (2000). Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet 9, 1651–1663. [DOI] [PubMed] [Google Scholar]
- Sarangi P, Altmannova V, Holland C, Bartosova Z, Hao F, Anrather D, Ammerer G, Lee SE, Krejci L, and Zhao X (2014). A versatile scaffold contributes to damage survival via sumoylation and nuclease interactions. Cell Rep. 9, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SN, Opresko PL, Meng X, Lee MY, and Eckert KA (2010). DNA structure and the Werner protein modulate human DNA polymerase deltadependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 38, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri N, Tsai YC, Hile S, Jordan D, Powell B, Chen J, Maloney D, Dose M, Lo Y, Anastassiadis T, et al. (2018). Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol. Cell 72, 222–238.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Durham CL, Gordenin DA, and Resnick MA (2003). Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. Proc. Natl. Acad. Sci. USA 100, 14994–14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, and Harper JW (2009). Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, and Lisby M (2014). Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, Marzahn MR, O’Donnell M, and Bloom LB (2012). Replication factor C is a more effective proliferating cell nuclear antigen (PCNA) opener than the checkpoint clamp loader, Rad24-RFC. J. Biol. Chem 287, 2203–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thys RG, Lehman CE, Pierce LC, and Wang YH (2015). DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics 16, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, and Gorgoulis VG (2008). Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 27, 3256–3264. [DOI] [PubMed] [Google Scholar]
- von Hippel PH, Johnson NP, and Marcus AH (2013). Fifty years of DNA “breathing”: Reflections on old and new approaches. Biopolymers 99, 923–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, and Philippsen P (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Walsh E, Wang X, Lee MY, and Eckert KA (2013). Mechanism of replicative DNA polymerase delta pausing and a potential role for DNA polymerase kappa in common fragile site replication. J. Mol. Biol 425, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, et al. (2014). CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 54, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, and Matos J (2016). Cell cycle control of DNA joint molecule resolution. Curr. Opin. Cell Biol. 40, 74–80. [DOI] [PubMed] [Google Scholar]
- Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, Hasty EP, Liu ET, Alt FW, and Scully R (2017). Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, Sarbajna S, Matos J, and West SC (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247. [DOI] [PubMed] [Google Scholar]
- Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, and West SC (2017). The SMX DNA Repair Tri-nuclease. Mol. Cell 65, 848–860.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhang P, Liu L, and Lee MY (2001). A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 40, 4512–4520. [DOI] [PubMed] [Google Scholar]
- Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, and Hickson ID (2013). MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Zhang H, and Freudenreich CH (2007). An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meng X, Zhang S, Lee EY, and Lee MY (2012). Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS ONE 7, e39156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are published on Mendeley (https://doi.org/10.17632/hh5rhpswsf.1).