

Structural modularity and positive selection govern functional evolution of doublesex, a master regulator of insect polymorphisms.
Abstract
doublesex regulates early embryonic sex differentiation in holometabolous insects, along with the development of species-, sex-, and morph-specific adaptations during pupal stages. How does a highly conserved gene with a critical developmental role also remain functionally dynamic enough to gain ecologically important adaptations that are divergent in sister species? We analyzed patterns of exon-level molecular evolution and protein structural homology of doublesex from 145 species of four insect orders representing 350 million years of divergence. This analysis revealed that evolution of doublesex was governed by a modular architecture: Functional domains and female-specific regions were highly conserved, whereas male-specific sequences and protein structures evolved up to thousand-fold faster, with sites under pervasive and/or episodic positive selection. This pattern of sex bias was reversed in Hymenoptera. Thus, highly conserved yet dynamic master regulators such as doublesex may partition specific conserved and novel functions in different genic modules at deep evolutionary time scales.
INTRODUCTION
Critical developmental genes such as transcription factors regulate evolutionary adaptations in myriad organisms (1, 2). Transcription factors are highly connected and usually conserved genes that act as hubs where genetic networks are integrated (3). How do these highly conserved master regulators also facilitate rapidly evolving ecological adaptations that are often divergent in sister species? Some of these adaptations evolve as a consequence of co-option of gene regulatory networks and spatial or temporal modulation of expression (4, 5). However, evolution of sequences and structures of transcription factors themselves are increasingly shown to regulate divergent phenotypic variation and adaptations (1, 6, 7). Do they accommodate these conflicting dual evolutionary roles by partitioning different conserved and rapidly evolving functions across different genic modules?
doublesex (dsx) is a transcription factor that regulates the last step of sex differentiation in all holometabolous insects during early development (8–10). It splices into male- and female-specific isoforms in response to sex-determination cues (Fig. 1A) and triggers a sex-specific developmental cascade by regulating several genes involved in male and female reproductive functions such as development of genitalia, yolk proteins, and deposition of fat bodies in females (11). Apart from this well-characterized conserved role, recent studies have shown that dsx may also be involved in regulating sex-specific, sex-limited, or morph-specific adaptive phenotypes during late developmental stages (Fig. 1B) (12–16). These co-opted functions of dsx are now known from a wide range of insect species, which may be broadly classified into primary and secondary sexual traits such as genitalia, pheromone production, and courtship behavior (Fig. 1C).
Fig. 1. Functional roles of dsx during development of holometabolous insects.
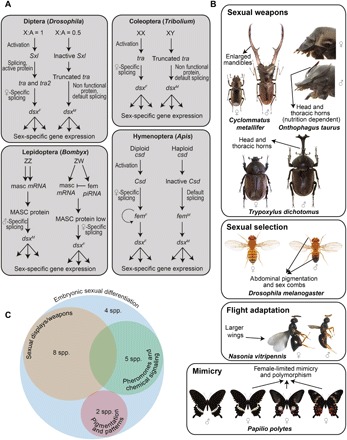
(A) The activity of dsx in sex-determination pathways illustrated in a representative genus of each insect order. (B) Examples of sexual weapons (large mandibles and horns in male beetles), sexual ornaments (sex combs in male Drosophila), large wings associated with dispersal in female Nasonia, and female-limited mimetic polymorphism in swallowtail butterflies that are developmentally regulated by dsx (12, 13, 15). (C) Developmental outcomes regulated by dsx fall into three broad functional categories apart from early embryonic sexual differentiation. masc, masculinizer; fem, feminizer. [Photo credit: N. Gompel (Drosophila melanogaster), A. P. Moczek (Onthophagus taurus), R. R. Choudhury (Nasonia vitripennis), and M. Yago (Cyclommatus metallifer and Trypoxylus dichotomous), used with permission, and K. Kunte (P. polytes)].
The versatile dsx functions can be attributed to multiple sex-, tissue-, and form-specific isoforms and differential expression in various tissues at critical developmental stages (11). Although the function of dsx as a transcription factor is well established, its downstream targets and interacting partners have been characterized only in a few model organisms where dsx regulates novel adaptive phenotypes. For instance, in Drosophila, Anopheles, Tribolium, and Bombyx, dsx targets AbdB, ylp1-ylp2, and dsat to regulate reproductive structures and pheromones (11, 15–18). In several cases, DsxF(female isoform) and DsxM (male isoform) seem to interact with the same binding sites with the help of different binding partners, resulting in sex-specific molecular cascades (11). Therefore, evolutionary changes not only in the dsx gene but also in the sequences or structures of its interacting partners could result in lineage-specific, sexually dimorphic novelties.
dsx is able to perform these diverse functions perhaps because it is a complex gene that is made up of up to six alternatively spliced exons. Two of these exons contain conserved oligomerization domains—OD1 and OD2—that are responsible for DNA binding and protein-protein interactions, respectively (8, 19). OD1 is the DM domain that is characteristic of all doublesex and mab-3 related transcription factor (DMRT) genes, which, along with OD2, controls sex differentiation (8, 19). These two domains are non–sex specific and common across insect orders and isoforms. Moreover, the OD2 and sex-specific sequences together enable Dsx to interact with other proteins. The remaining exons are spliced in a variable and clade-specific manner according to sex, form, developmental stage, and tissue (Fig. 2 and table S1) (8). The structural implications of various isoforms have not yet been studied. Hence, their effect on the activity of dsx remains to be elucidated. However, the diverse functional roles and structural complexity of this gene make it an ideal candidate to study conflicting evolutionary dynamics where both purifying and diversifying selection are acting on the same gene to produce divergent adaptive phenotypes while maintaining its core conserved function.
Fig. 2. Phylogenetic relationships and exon usage among insect orders in relation to dsx evolution.
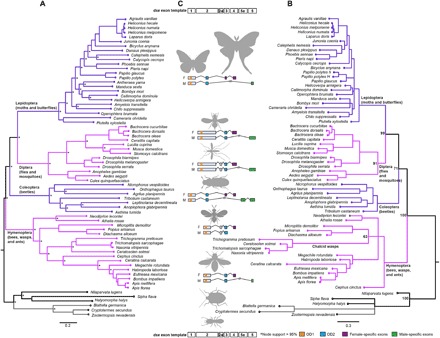
(A) A mito-nuclear phylogeny of the four orders sampled. (B) dsx gene tree based on its coding sequence. In (A) and (B), adjacent clades are colored pink and purple, and the outgroups are colored gray and black, for contrast. (C) Exon usage of dsx across insect orders. Exons are numbered arbitrarily on the basis of mRNA initiation as per scientific convention and indicate a generalized exonic organization of dsx in holometabolous insects. Order-wise exon organization of the translated product of dsx is depicted in the center with domains and sex-specific regions colored based on sequence homology. Only those exons that are translated are shown, but 5′ and 3′ untranslated exons (not shown) may have poorly understood regulatory functions. OD1 is a DM DNA-binding domain, and OD2 is a DSX dimerization domain. Exon 5a, unique to Coleoptera, is homologous to OD2.
To study how genetic modularity might be facilitated by differential selection on dsx, we traced its molecular evolution and structural diversity in 145 insect species from orders Lepidoptera, Diptera, Coleoptera, and Hymenoptera. These orders span the 350-million-year evolutionary history and diversity of holometabolous insects in which dsx has a well-characterized, conserved function in early sex differentiation (9, 10, 20, 21). We predicted that genic regions that control the early embryonic, conserved function will be under evolutionary constraints. On the other hand, genic regions that show elevated rates of molecular substitutions and protein structural evolution may represent regions that control the late-developmental, sex- and morph-specific novel functions.
RESULTS
Molecular evolution of dsx is highly structured acrossexonic partitions
We divided the dsx dataset from 145 species into six groups based on alignment, phylogeny, and exon usage: Lepidoptera (moths and butterflies), Diptera (flies), Diptera (mosquitoes), Coleoptera (beetles), Hymenoptera (only chalcid wasps), and Hymenoptera (remaining wasps and bees) (Figs. 2 and 3). The numbering of exons of dsx in each order depends on the position at which mRNA begins; however, one or more exons may be untranslated (e.g., Diptera and Hymenoptera exon 1). Thus, the exact numbering of exons is arbitrary but used here as per scientific convention (Fig. 2 and table S1). However, the relative positions of exons in the gene sequence and the genetic architecture of domains remain conserved across orders and were aligned here by homology (Fig. 3). The dsx gene tree mirrored the species tree to a large extent and was clustered by orders rather than exon usage. This is despite the fact that exon usage is similar in Lepidoptera and Coleoptera and between Diptera and Hymenoptera (Fig. 2). On the basis of the pattern of conservation and variation, we divided coding sequences of dsx into different genic regions (Fig. 4) and independently estimated the rate of synonymous (dS) and nonsynonymous (dN) substitution for each. The group-wise exon-level analysis of average substitution rates and dN/dS ratios revealed that dsx on the whole was under strong purifying selection (Fig. 4B). This was especially evident in the domain regions that were depauperate in nonsynonymous substitutions, as expected of highly conserved genic regions (Figs. 3 and 4). dS alone was a poor predictor of dN (pseudo R2 = 0.05, 0.018, 0.004, and 0.01 for Lepidoptera, Diptera, Coleoptera, and Hymenoptera, respectively). However, species and genic region were good predictors of dN across all orders (regression model: dN ∼ Species + Region; pseudo R2 = 0.61, 0.64, 0.68, and 0.80 for Lepidoptera, Diptera, Coleoptera, and Hymenoptera, respectively). Among the genic regions, post-OD1 and male-specific regions of Lepidoptera, Diptera, and Coleoptera (P < 0.001) and pre-OD1 and female-specific regions of Hymenoptera (P < 0.001) contributed significantly to dN (table S2).
Fig. 3. Exon-level molecular evolution and functional partitioning of dsx across insect orders.
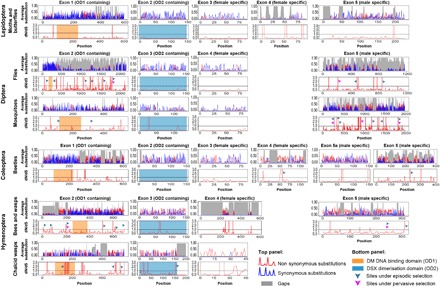
Rates of molecular evolution (average substitutions; top panels) and signatures of selection with reference to nonsynonymous and synonymous substitutions (dN/dS ratios; bottom panels) per codon for each dsx exon are shown separately for each insect group. The exons are arranged according to sequence homology, and the two domain regions are highlighted. Arrows point toward sites under episodic (green arrows) and/or pervasive (magenta arrows) positive selection.
Fig. 4. Molecular evolution across genic regions.
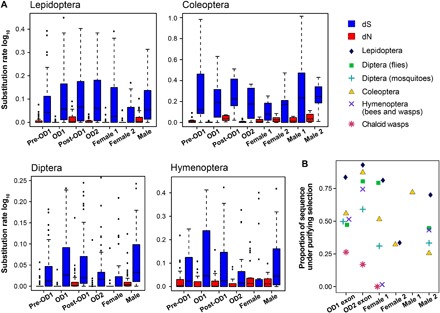
Rate of molecular evolution across different genic regions (A) and proportion of sites under purifying selection (B), in each insect group.
The regression analysis also revealed that female- and male-specific dsx regions evolved at significantly different rates, with the dN of male-specific exons being much greater than that of female-specific exons (table S2). Similar to the domain regions, female-specific dsx regions (largely exons 3 and 4, depending on insect order; Fig. 2) were under intense purifying selection (Figs. 3 and 4). On the other hand, male-specific regions (exons 5a and 5; Fig. 2) showed significantly high rates of nonsynonymous substitutions and indels, which were comparable to non-OD regions that generally evolve faster (Figs. 3 and 4 and table S2). This pattern of conservation in female-specific regions and high variation in male-specific regions was reversed in the Hymenoptera (Figs. 3 and 4 and table S2). Thus, sequence conservation and rates of nonsynonymous evolution were prominently partitioned across structural components (sex-specific exons and OD regions) of dsx. Such partitioning presumably resolves the intrinsic evolutionary conflict, facilitating simultaneous purifying and diversifying selection acting on different functional components of the same master regulator.
dsx has evolved under episodic and pervasivepositive selection
dsx regulates novel adaptive phenotypes such as exaggerated sexual weapons in male beetles (14–16), sex combs and abdominal melanization in male Drosophila, and naturally selected female-limited mimetic polymorphism in Papilio polytes and Papilio memnon (12, 13, 22). These adaptations are divergent even among sister species. To understand the detailed evolutionary history of dsx, we used fixed effects likelihood (FEL) (23) and mixed effects model of evolution (MEME) (24) to detect sites that are under pervasive and episodic positive selection, respectively. A considerable proportion of dsx codons were under strong purifying selection, again as expected of a highly conserved gene (Fig. 4B). Lepidoptera and Coleoptera showed no sites under pervasive positive selection, whereas Diptera and Hymenoptera had a few sites under pervasive positive selection (Fig. 3 and table S3). Lepidoptera also did not show any sites under episodic positive selection, whereas several sites in Diptera, Coleoptera, and Hymenoptera showed signature of episodic positive selection (Fig. 3 and table S3). A significant fraction of these sites was present in non-OD region and male-specific exons, further strengthening evidence for functional partitioning within dsx. Among the lineages where dsx is involved in regulating novel adaptive phenotypes (12–16, 18, 22, 25), branch-site unrestricted statistical test for episodic diversification (BUSTED) (26) detected sites under positive selection in only a single case: the mimetic allele of dsx in P. polytes (codon 102, P < 0.05). This is consistent with a recent study that showed a strong association between the interdomain region of dsx and mimicry (27). In lineages such as P. memnon (22), Cyclommatus (14), Onthophagus (25), and Nasonia (18) where dsx regulates novel traits such as mimicry, exaggerated mandibles, exaggerated horns, and elongated wings, respectively, there were no sites under positive selection. Apart from studying selection at individual sites, we used a hypothesis-testing framework, RELAX (28), to test whether the strength of natural selection was constant during the evolution of dsx across insect groups. There was an increase in the strength of positive selection in two cases: Heliconius exon 1 (K = 1.34, P < 0.01) and Anopheles exons 2 (K = 1.60, P < 0.05) and 3 (K = 4.69, P < 0.05). Increase in the strength of positive selection in these lineages warrants a closer developmental genetic investigation.
Dsx protein shows accelerated, male-biasedstructural evolution
Although molecular evolution of gene sequences that are under strong selection has been studied in a number of genes and organisms, there is limited understanding of how sequence evolution influences protein evolution at large phylogenetic distances and deep time scales. We performed homology modeling and structural alignment of Dsx protein structures across holometabolous insects, which showed patterns parallel to sex- and exon-specific sequence evolution. Functional units of Dsx protein showed a graded level of structural conservation: DM DNA-binding and DSX dimerization domains (OD1 and OD2, respectively) showed highly conserved structures, in which tertiary structures closely superimposed over 350 million years of divergence across all the orders of holometabolous insects (Fig. 5A and fig. S1). Female-specific regions also showed highly conserved structures, which largely superimposed across species, except in the Hymenoptera (Fig. 5A and fig. S1). In contrast, male-specific regions of Dsx showed little structural conservation. This was true not only across orders but also across closely related genera within each insect order (Fig. 5A and fig. S1). The root mean square deviation (RMSD), which measures structural deviation between protein backbones, was significantly greater in male-specific regions compared to OD and female-specific regions (Fig. 5B and fig. S1). Thus, male-specific regions of dsx have undergone rapid molecular sequence as well as protein structural evolution, which might link to the great diversity of secondary sexual traits seen in male insects.
Fig. 5. Protein structural conservation in various domain and sex-specific regions of dsx in insects.
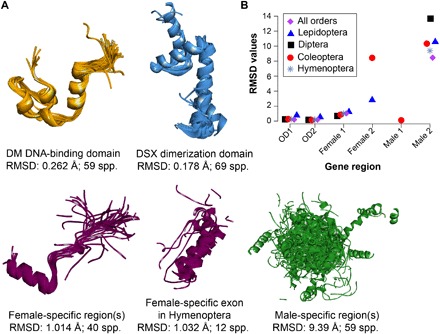
Drosophila melanogaster crystal structures for OD1 and OD2 were used as a reference for protein modeling from the dsx sequences. (A) Superimposition of protein structures in each exonic region. RMSD as a measure of structural deviation between protein backbones and the number of species used for protein modeling are also shown. (B) Comparison of protein structural differences across exonic regions and insect orders, which highlights male-biased evolution of the Dsx protein. Since protein structures are relatively similar in sister species, only representatives of each genus were chosen for this comparison (see table S1 for the list of species used for modeling and fig. S1 for structures of domains and sex-specific regions of each insect order).
DISCUSSION
It is increasingly recognized that the genetics of adaptation can take myriad forms, where complex genetic architecture, sequence evolution, and developmental regulation may strongly influence the tempo and mode of adaptation. On the basis of recent discoveries, it appears that several pleiotropic genes accommodate diverse functions that should be under contrasting selection pressures (5). We addressed this problem with a key developmental gene, dsx, that performs both conserved early developmental and highly divergent late developmental functions (11–18, 22, 29). Our analysis of nucleotide sequence and protein structure of dsx revealed modular molecular evolution in its gene organization. Although evolution of new functions in many conserved genes is associated with evolution of noncoding regulatory regions (4, 5), we found strong signatures of purifying as well as positive evolution within coding regions of dsx. We propose that structural and functional partitioning, as evident in dsx coding sequence, may explain contrasting functions of dsx in producing critical adaptations that are, in parts, sex-limited, polymorphic, developmentally conserved, and rapidly evolving even across closely related species. This partitioning is achieved by different protein-coding regions themselves becoming modular with up to a 1000-fold difference in the rate of molecular evolution (Fig. 4).
As a consequence of modular architecture, genic regions within dsx have a highly skewed rate of molecular evolution (Figs. 3 and 4) with several sites and regions (Fig. 3 and table S2) that show strong signatures of episodic and/or pervasive positive selection or show prominent nonsynonymous substitutions, indicating rapid evolutionary change. This modular architecture holds true at multiple levels of taxonomic organization ranging from a few species in Chalcid wasps to more than 60 species in the Lepidoptera (Figs. 3 and 4B). These findings suggest a widespread role of dsx beyond currently known functions in producing sexual dimorphisms and polymorphisms of ecological significance. This is irrespective of diverse mechanisms of sex determination, heterogamety, and exon usage that are evident across insect orders. Most of the insect groups we analyzed have not been studied at a developmental genetic level, and the role of dsx in sexual dimorphism in these groups is largely unknown. The sites that we identified as positively selected should thus be good targets for developmental genetic manipulations in these species groups. It was not possible in this study to model the entire protein structure of Dsx because its three-dimensional crystal structure is unknown. However, it will be important in the future to study the structural implications of sex-specific dsx isoforms and sites under pervasive, episodic, and/or lineage-specific positive selection.
Among lineages with known novel adaptations regulated by dsx, a single case—the mimetic allele of P. polytes—had sites under positive selection. This species has two known alleles of dsx between which recombination is suppressed due to an inversion (13). The presence of two nonrecombining alleles in the gene pool of this species may have facilitated independent accumulation of mutations, enabling differential evolution of this gene in the two female forms. The absence of signatures of selection in the coding sequence of dsx in other insect lineages hints at modifications in regulatory elements and/or binding partners aiding the evolution of novel adaptive phenotypes.
dsx had a prominent sex bias in rates of substitutions (Fig. 4)—as was previously known in some Diptera (30)—and protein structural evolution (Fig. 5 and fig. S1). However, this sex bias revealed an important ecological correlate: Male-specific regions of dsx evolved much faster than female-specific regions in Lepidoptera, Diptera, and Coleoptera, but this trend was reversed in Hymenoptera (Figs. 4 and 5 and fig. S1). This pattern was consistent with ecological and social selection for sexual dimorphism and polymorphisms across insect groups. In Lepidoptera, Diptera, and Coleoptera, males display great diversity in secondary sexual traits, which is potentially related to sexual selection on male traits that are governed by developmental regulation by dsx. On the other hand, the evolution of female-dominated eusociality and female-specific caste differentiation may be linked to greater rates of evolution in female-specific regions in the Hymenoptera. This clade-specific sex bias suggests that there is an underappreciated role for intense ecological and social selection on sex-limited traits that may also produce bouts of rapid adaptive evolution in specific parts of the genome (31, 32).
Exon-level modularity and alternative splicing, as shown by dsx, are widespread in eukaryotic genomes. This is especially true of developmental genes and transcription factors that perform a wide array of functions. Our work offers a paradigm to understand sequence and protein evolution in these structurally complex and functionally diverse genes. It also provides a rare benchmark against which broad evolutionary comparisons in such genes may be performed.
MATERIALS AND METHODS
dsx sequences and multiple sequence alignment
We downloaded insect-specific dsx gene sequences from GenBank (http://www.ncbi.nlm.nih.gov/, last accessed December 2017). From the available genomes and individual sequences, we shortlisted sequences from 145 insect species across four holometabolous insect orders (Lepidoptera, Diptera, Hymenoptera, and Coleoptera) based on the availability of and the ability to annotate intron-exon boundaries of this gene (table S1). We used dsx homolog sequences from three species of Isoptera and three species of Hemiptera as outgroups (Fig. 2A). We used NCBI (National Center for Biotechnology Information) BLAST+ to find the location of dsx within genomes of species where the gene was not annotated. We used SAMtools V0.1.19 (33) for extracting dsx exon sequences from genomes. We obtained sex-specific variants of dsx in most of the shortlisted species and used the corresponding exon sequences for further analysis. We performed multiple sequence alignment of dsx exons from insect species within each order and across all orders using the codon aligner PRANK v150803 (34). Note that the numbering of exons in each sequence of dsx depends on the position at which mRNA begins; however, one or more exons in each sequence may be untranslated. Thus, the exact numbering of exons is arbitrary but used here as per scientific convention (table S1). However, their relative positions and the genetic architecture of domains remain conserved across orders (Fig. 2). Thus, the alignment used for further analysis (Figs. 3 and 4) is as per sequence homology.
Phylogenetic analyses
We constructed a dsx gene tree and a species-level molecular phylogeny of 61 representative species from the four ingroup orders and six species from two outgroup orders (Fig. 2). We used four nuclear markers (elongation factor I-alpha, wingless, DNA-directed RNA polymerase II subunit RPB2, and DNA polymerase delta catalytic subunit) and one mitochondrial marker (cytochrome c oxidase I) that are commonly used in insect molecular phylogenies. For the dsx gene tree, we used the common exons that are conserved and the male-specific exon that shows a high degree of variation within each order, for estimation of molecular evolution (Fig. 3). We used Partition Finder to choose the best partition schemes for mitochondrial markers, nuclear markers, and the dsx gene, along with the corresponding models of sequence evolution. We used the greedy algorithm and models = MrBayes and a Bayesian information criterion to compare the best-fit models (35). We performed a partitioned Bayesian analysis using MrBayes 3.2.7 (36). We used a split frequency below 0.01 to assess stationarity and to set the burn-in in MrBayes and then built a consensus tree using the remaining trees.
Site-specific estimation of molecular evolution
From our alignments, we calculated average synonymous substitutions, nonsynonymous substitutions, and gaps for each site within the dsx gene (Fig. 3). We used the FEL (23) method to estimate the dN/dS ratio per site (Fig. 3) as well as to estimate sites that have experienced pervasive diversifying or purifying selection (Fig. 3). We also used MEME (24) to estimate the sites subjected to episodic positive or diversifying selection (Fig. 3).
dsx is involved in regulating novel adaptive phenotypes in several insect species, which may be associated with sites within the dsx sequence that have undergone positive selection. We used BUSTED (26) to test for positive selection by asking whether dsx has experienced positive selection in at least one site in species of our interest. We used hypothesis testing using phylogenies (HyPhy) 2.3.14 (37) for this estimation and parsed the results using in-house scripts.
Estimation of synonymous (dS)and nonsynonymous (dN) substitutions
Our analysis of regions of conservation and variation within dsx (Fig. 3) revealed that different regions of this gene experience different evolutionary pressures. We therefore divided the gene into multiple partitions (pre-OD1, OD1, post-OD1, OD2, female-specific, and male-specific region) and estimated dN and dS separately using the AnalyzeCodonData function of HYPHY 2.3.14 (Fig. 4). We performed beta regression using betareg (v3.1-1) (38) package in R (v3.3.1) on dN using dS, gene partitions, and taxonomy as predictor variables.
Estimating the strength of natural selection
We used RELAX (28), a hypothesis-testing framework, to assess whether the dsx sequences in certain monophyletic lineages of biological interest had experienced relaxed or intensified selection. RELAX is useful for identifying trends and/or shifts in the stringency of natural selection on a given gene. RELAX estimates a relaxation/intensification parameter (K). K > 1 in test lineages indicates intensified positive selection whereas K < 1 indicates relaxed selection. We used HYPHY 2.3.14 (37) for this estimation.
Homology modeling
The modeling of dsx protein structure posed a challenge due to the overwhelming presence of loops, owing to which most protein databases only contained the two functional domains of this protein. We modeled these functional domains and sex-specific exons using Phyre2 (39) (protein homology/analogy recognition engine). We performed structural alignment and visualization in PyMOL (2.0.6) (40) and Discovery Studio Visualizer 4.5 (Fig. 4) (41). For this analysis, we used a subset of sequences from 29 species of Lepidoptera, 14 species of Diptera, 10 species of Coleoptera, and 18 species of Hymenoptera (table S1). The reason is that (i) sex-specific sequences were not known from every species for which some dsx sequences were available and (ii) protein structure within genera was often similar; therefore, representative sampling of Dsx protein across insect genera was adequate to study protein structure evolution. We also did not model the protein structure of the Female 2 exon region of Coleoptera and Lepidoptera because these were present only in a few transcripts of a few species, where comparison of homologous regions was not feasible.
Supplementary Material
Acknowledgments
We thank R. Sowdhamini, S. Mahajan, and F. Kondrashov for advice on bioinformatic methods; D. Agashe, A. S. N. Seshasayee, and R. Sowdhamini for comments on the manuscript; and N. Gompel, A. P. Moczek, R. R. Choudhury, and M. Yago for images of organisms used in Fig. 1. Funding: This work was partially funded by an NCBS research grant to K.K., an NCBS student fellowship to S.B., an NCBS Bridging Fellowship to G.A., and a CSIR SPM Fellowship to R.D. Author contributions: S.B., R.D., and K.K. conceived the study. S.B. and G.A. assembled the sequence dataset and performed bioinformatic analyses. S.B., R.D., and K.K. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Data are available on GenBank (see table S1 for accession numbers). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau3753/DC1
Fig. S1. Protein structural conservation in various domain and sex-specific regions of Dsx across insect orders.
Table S1. Species, GenBank accession numbers, and exons of dsx sequences used in this study.
Table S2. Regression log file.
Table S3. Sites under episodic and pervasive positive selection in different insect groups and specific lineages.
REFERENCES AND NOTES
- 1.Lynch V. J., Wagner G. P., Resurrecting the role of transcription factor change in developmental evolution. Evolution 62, 2131–2154 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Voordeckers K., Pougach K., Verstrepen K. J., How do regulatory networks evolve and expand throughout evolution? Curr. Opin. Biotechnol. 34, 180–188 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Johnson A. D., The rewiring of transcription circuits in evolution. Curr. Opin. Genet. Dev. 47, 121–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wray G. A., The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Deshmukh R., Baral S., Gandhimathi A., Kuwalekar M., Kunte K., Mimicry in butterflies: Co-option and a bag of magnificent developmental genetic tricks. Wiley Interdiscip. Rev. Dev. Biol. 7, e291 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Powder K. E., Cousin H., McLinden G. P., Craig Albertson R., A nonsynonymous mutation in the transcriptional regulator lbh is associated with cichlid craniofacial adaptation and neural crest cell development. Mol. Biol. Evol. 31, 3113–3124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nadimpalli S., Persikov A. V., Singh M., Pervasive variation of transcription factor orthologs contributes to regulatory network evolution. PLOS Genet. 11, e1005011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price D. C., Egizi A., Fonseca D. M., The ubiquity and ancestry of insect doublesex. Sci. Rep. 5, 13068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker B. S., Sex in flies: The splice of life. Nature 340, 521–524 (1989). [DOI] [PubMed] [Google Scholar]
- 10.Prakash A., Monteiro A., Molecular mechanisms of secondary sexual trait development in insects. Curr. Opin. Insect Sci. 17, 40–48 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Verhulst E. C., van de Zande L., Double nexus—Doublesex is the connecting element in sex determination. Brief. Funct. Genomics 14, 396–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunte K., Zhang W., Tenger-Trolander A., Palmer D. H., Martin A., Reed R. D., Mullen S. P., Kronforst M. R., doublesex is a mimicry supergene. Nature 507, 229–232 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa H., Iijima T., Kajitani R., Yamaguchi J., Ando T., Suzuki Y., Sugano S., Fujiyama A., Kosugi S., Hirakawa H., Tabata S., Ozaki K., Morimoto H., Ihara K., Obara M., Hori H., Itoh T., Fujiwara H., A genetic mechanism for female-limited Batesian mimicry in Papilio butterfly. Nat. Genet. 47, 405–409 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Gotoh H., Zinna R. A., Warren I., DeNieu M., Niimi T., Dworkin I., Emlen D. J., Miura T., Lavine L. C., Identification and functional analyses of sex determination genes in the sexually dimorphic stag beetle Cyclommatus metallifer. BMC Genomics 17, 250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotoh H., Ishiguro M., Nishikawa H., Morita S., Okada K., Miyatake T., Yaginuma T., Niimi T., Molecular cloning and functional characterization of the sex-determination gene doublesex in the sexually dimorphic broad-horned beetle Gnatocerus cornutus (Coleoptera, Tenebrionidae). Sci. Rep. 6, 29337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito Y., Harigai A., Nakata M., Hosoya T., Araya K., Oba Y., Ito A., Ohde T., Yaginuma T., Niimi T., The role of doublesex in the evolution of exaggerated horns in the Japanese rhinoceros beetle. EMBO Rep. 14, 561–567 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotoh H., Miyakawa H., Ishikawa A., Ishikawa Y., Sugime Y., Emlen D. J., Lavine L. C., Miura T., Developmental link between sex and nutrition; doublesex regulates sex-specific mandible growth via juvenile hormone signaling in stag beetles. PLOS Genet. 10, e1004098 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loehlin D. W., Oliveira D. C. S. G., Edwards R., Giebel J. D., Clark M. E., Cattani M. V., van de Zande L., Verhulst E. C., Beukeboom L. W., Muñoz-Torres M., Werren J. H., Non-coding changes cause sex-specific wing size differences between closely related species of Nasonia. PLOS Genet. 6, e1000821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matson C. K., Zarkower D., Sex and the singular DM domain: Insights into sexual regulation, evolution and plasticity. Nat. Rev. Genet. 13, 163–174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shukla J. N., Palli S. R., Sex determination in beetles: Production of all male progeny by parental RNAi knockdown of transformer. Sci. Rep. 2, 602 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gempe T., Hasselmann M., Schiøtt M., Hause G., Otte M., Beye M., Sex determination in honeybees: Two separate mechanisms induce and maintain the female pathway. PLOS Biol. 7, e1000222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iijima T., Kajitani R., Komata S., Lin C.-P., Sota T., Itoh T., Fujiwara H., Parallel evolution of Batesian mimicry supergene in two Papilio butterflies, P. polytes and P. memnon. Sci. Adv. 4, eaao5416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosakovsky Pond S. L., Frost S. D. W., Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Murrell B., Wertheim J. O., Moola S., Weighill T., Scheffler K., Kosakovsky Pond S. L., Detecting individual sites subject to episodic diversifying selection. PLOS Genet. 8, e1002764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kijimoto T., Moczek A. P., Andrews J., Diversification of doublesex function underlies morph-, sex-, and species-specific development of beetle horns. Proc. Natl. Acad. Sci. U.S.A. 109, 20526–20531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murrell B., Weaver S., Smith M. D., Wertheim J. O., Murrell S., Aylward A., Eren K., Pollner T., Martin D. P., Smith D. M., Scheffler K., Kosakovsky Pond S. L., Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W., Westerman E., Nitzany E., Palmer S., Kronforst M. R., Tracing the origin and evolution of supergene mimicry in butterflies. Nat. Commun. 8, 1269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wertheim J. O., Murrell B., Smith M. D., Kosakovsky Pond S. L., Scheffler K., RELAX: Detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams T. M., Selegue J. E., Werner T., Gompel N., Kopp A., Carroll S. B., The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134, 610–623 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes A. L., Runaway evolution of the male-specific exon of the doublesex gene in Diptera. Gene 472, 1–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng C., Kirkpatrick M., Sex-specific selection and sex-biased gene expression in humans and flies. PLoS Genet. 12, e1006170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campos J. L., Johnston K. J. A., Charlesworth B., The effects of sex-biased gene expression and X-linkage on rates of sequence evolution in Drosophila. Mol. Biol. Evol. 35, 655–665 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Löytynoja A., Goldman N., An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. U.S.A. 102, 10557–10562 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanfear R., Calcott B., Ho S. Y. W., Guindon S., PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Huelsenbeck J. P., Ronquist F., MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Pond S. L. K., Frost S. D. W., Muse S. V., HyPhy: Hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Cribari-Neto F., Zeileis A., Beta Regression in R. J. Stat. Softw. 34, 1–24 (2010). [Google Scholar]
- 39.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. E., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.L. Schrödinger, The PyMOL molecular graphics system, version 2.0.6.
- 41.Dassault Systèmes BIOVIA, Discovery studio modeling environment, release 4.5 (Dassault Systemes, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau3753/DC1
Fig. S1. Protein structural conservation in various domain and sex-specific regions of Dsx across insect orders.
Table S1. Species, GenBank accession numbers, and exons of dsx sequences used in this study.
Table S2. Regression log file.
Table S3. Sites under episodic and pervasive positive selection in different insect groups and specific lineages.