

Abstract
Background
Many patients with cancer experience moderate to severe pain that requires treatment with strong analgesics. Buprenorphine, fentanyl and morphine are examples of strong opioids used for the relief of cancer pain. Strong opioids are, however, not effective for pain in all patients nor are they well‐tolerated by all patients. The aim of this Cochrane review is to assess whether buprenorphine is associated with superior, inferior or equal pain relief and tolerability compared to other analgesic options for patients with cancer pain.
Objectives
To assess the effectiveness and tolerability of buprenorphine for pain in adults and children with cancer.
Search methods
We searched CENTRAL (the Cochrane Library) issue 12 or 12 2014, MEDLINE (via OVID) 1948 to 20 January 2015, EMBASE (via OVID) 1980 to 20 January 2015, ISI Web of Science (SCI‐EXPANDED & CPCI‐S) to 20 January 2015, ISI BIOSIS 1969 to 20 January 2015. We also searched ClinicalTrials.gov (http://clinicaltrials.gov/; metaRegister of Controlled Trials (mRCT) (http://www.controlled‐trials.com/mrct/), the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal (http://apps.who.int/trialsearch/) and the Proceedings of the Congress of the European Federation of International Association for the Study of Pain (IASP; via European Journal of Pain Supplements) on 16 February 2015. We checked the bibliographic references of identified studies as well as relevant studies and systematic reviews to find additional trials not identified by the electronic searches. We contacted authors of included studies for other relevant studies.
Selection criteria
We included randomised controlled trials, with parallel‐group or crossover design, comparing buprenorphine (any formulation and any route of administration) with placebo or an active drug (including buprenorphine) for cancer background pain in adults and children.
Data collection and analysis
Two review authors independently extracted data pertaining to study design, participant details (including age, cancer characteristics, previous analgesic medication and setting), interventions (including details about titration) and outcomes, and independently assessed the quality of the included studies according to standard Cochrane methodology. As it was not feasible to meta‐analyse the data, we summarised the results narratively. We assessed the overall quality of the evidence for each outcome using the GRADE approach.
Main results
In this Cochrane review we identified 19 relevant studies including a total of 1421 patients that examined 16 different intervention comparisons.
Of the studies that compared buprenorphine to another drug, 11 studies performed comparative analyses between the randomised groups, and five studies found that buprenorphine was superior to the comparison treatment. Three studies found no differences between buprenorphine and the comparison drug, while another three studies found treatment with buprenorphine to be inferior to the alternative treatment in terms of the side effects profile or patients preference/acceptability.
Of the studies that compared different doses or formulations/routes of administration of buprenorphine, pain intensity ratings did not differ significantly between intramuscular buprenorphine and buprenorphine suppository. However, the average severity of dizziness, nausea, vomiting and adverse events as a total were all significantly higher in the intramuscular group relatively to the suppository group (one study).
Sublingual buprenorphine was associated with faster onset of pain relief compared to subdermal buprenorphine, with similar duration analgesia and no significant differences in adverse event rates reported between the treatments (one study).
In terms of transdermal buprenorphine, two studies found it superior to placebo, whereas a third study found no difference between placebo and different doses of transdermal buprenorphine.
The studies that examined different doses of transdermal buprenorphine did not report a clear dose‐response relationship.
The quality of this evidence base was limited by under‐reporting of most bias assessment items (e.g., the patient selection items), by small sample sizes in several included studies, by attrition (with data missing from 8.2% of the enrolled/randomised patients for efficacy and from 14.6% for safety) and by limited or no reporting of the expected outcomes in a number of cases. The evidence for all the outcomes was very low quality.
Authors' conclusions
Based on the available evidence, it is difficult to say where buprenorphine fits in the treatment of cancer pain with strong opioids. However, it might be considered to rank as a fourth‐line option compared to the more standard therapies of morphine, oxycodone and fentanyl, and even there it would only be suitable for some patients. However, palliative care patients are often heterogeneous and complex, so having a number of analgesics available that can be given differently increases patient and prescriber choice. In particular, the sublingual and injectable routes seemed to have a more definable analgesic effect, whereas the transdermal route studies left more questions.
Plain language summary
Buprenorphine for treating people with cancer pain
Buprenorphine produced good pain relief for most people with moderate or severe cancer pain, but its role in the treatment of cancer pain is still unclear.
Many patients with cancer experience moderate‐to‐severe pain that requires treatment with strong pain relief medicines. Buprenorphine and morphine are examples of strong pain relief medicines that are used for the relief of cancer pain. However, strong pain relief medicines are not effective for pain in all patients nor are they well‐tolerated by all patients. The aim of this Cochrane review is to assess whether buprenorphine is associated with better, worse or equal pain relief and tolerability compared to other pain relief medicines for patients with cancer pain.
We searched the literature on 20 Janurary 2015 and found 19 relevant studies with a total of 1421 patients that compared different types of buprenorphine to each other or to other strong pain relief medicines or to placebo. The reported average ages of the patients ranged from 49.1 years to 67.16 years, and the duration of the studies ranged from single dose treatment to six months.
Generally, the studies showed that buprenorphine is an effective strong pain relief medicine that in some cases may be slightly better than other strong pain relief medicines. However, the evidence provided by these studies were of very low quality and on the basis of the available evidence, it is still hard to say where buprenorphine fits in in the treatment of cancer pain with strong opioids. All the strong pain relief medicines examined in the studies are also associated with a number of unwanted effects, such as vomiting, constipation and drowsiness.
Summary of findings
for the main comparison.
| Quality assessment | Summary of findings | |||||||||||||||||
| No of patients | Effect | Quality | ||||||||||||||||
| No of studies | Design | Limitations | Inconsistency | Indirectness | Imprecision | Other considerations | Bup | Comparison | ||||||||||
| SL Bup versus SD Bup injection: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | SL: 10 | SD: 7 | Onset: SL faster Duration: SL = SD |
Very low | ||||||||
| SL Bup versus SD Bup injection: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | SL: 10 | SD: 7 | SL = SD | Very low | ||||||||
| SL Bup versus oral Til+Na: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 20 | 20 | Bup superior | Very low | ||||||||
| SL Bup versus oral Til+Na: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 20 | 20 | Bup = Til+Na | Very low | ||||||||
| SL Bup versus oral Tra: Efficacy | ||||||||||||||||||
| 2 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 1233 | 1283 | Tra superior or similar | Very low | ||||||||
| SL Bup versus oral Tra: Adverse events | ||||||||||||||||||
| 2 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 1233 | 1283 | Tra superior or similar | Very low | ||||||||
| SL Bup versus SL Bup + oral P versus oral P: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | SL: 25 | SL+ P: 25 | P: 25 | SL = SL+P = P | Very low | |||||||
| SL Bup versus SL Bup + oral P versus oral P: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | SL: 25 | SL+P: 25 | P: 25 | SL = SL+P = P | Very low | |||||||
| SL Bup versus oral Pen: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 1204 | 1204 | Bup superior | Very low | ||||||||
| SL Bup versus oral Pen: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 1204 | 1204 | Bup ≠ Pen | Very low | ||||||||
| Bup tablets/fluid versus Pen Tab/F: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | Serious indirectness5 | Imprecision2 | None | Tab | F | Tab | F | Bup superior | Very low | ||||||
| 11 | 11 | 10 | 10 | |||||||||||||||
| Bup tablets/fluid versus Pen Tab/F: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | Serious indirectness5 | Imprecision2 | None | Tab | F | Tab | F | Bup superior or similar | Very low | ||||||
| 11 | 11 | 10 | 10 | |||||||||||||||
| TD Bup versus placebo: Efficacy | ||||||||||||||||||
| 4 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | ‐35 μg/h: 102 ‐52.5 μg/h: 82 ‐70 μg/h: 169 |
189 | Bup better or similar | Very low | ||||||||
| TD Bup versus placebo: Adverse events | ||||||||||||||||||
| 4 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | ‐35 μg/h: 102 ‐52.5 μg/h: 82 ‐70 μg/h: 169 |
189 | Apparently similar | Very low | ||||||||
| TD Bup versus controlled‐release Mor: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 26 | 26 | Bup superior | Very low | ||||||||
| TD Bup versus controlled‐release Mor: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 26 | 26 | Bup superior or similar | Very low | ||||||||
| TD Bup versus TD Fen: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 16 | 16 | Bup = Fen | Very low | ||||||||
| TD Bup versus TD Fen: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 16 | 16 | Fen superior or similar | Very low | ||||||||
| IM Bup injection versus Bup Sup: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | IM: 35 | Sup: 34 | IM = Sup | Very low | ||||||||
| IM Bup injection versus Bup Sup: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | IM: 35 | Sup: 34 | Sup superior or similar | Very low | ||||||||
| IM Bup versus IM Mor: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 376 | 376 | Bup superior or similar | Very low | ||||||||
| IM Bup versus IM Mor: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 274 | 274 | Mor superior or similar | Very low | ||||||||
| IM Bup + SC Bup versus SC Bup versus placebo (Pla) + SC Bup: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | IM+SC: 10 | SC: 10 | Pla+SC: 10 | Not analysed inferentially | Very low | |||||||
| IM Bup + SC Bup versus SC Bup versus placebo + SC Bup: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | IM+ SC: 10 | SC: 10 | Pla+SC: 10 | Pla+SC inferior or similar | Very low | |||||||
| Epidural Bup versus epidural Mor: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 6 | 6 | Bup = Mor | Very low | ||||||||
| Epidural Bup versus epidural Mor: Adverse events | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 6 | 6 | Bup = Mor | Very low | ||||||||
| Intravenous Bup versus intravenous Mor: Efficacy | ||||||||||||||||||
| 1 | RCTs | Very serious1 | No serious inconsistency | No serious indirectness | Imprecision2 | None | 30 + 30 | 30 + 30 | Bup and Mor not compared inferentially | Very low | ||||||||
1The quality of the evidence provided by the included studies was compromised by under‐reporting with or without missing data. 2Low numbers of patients. 3One of the included studies was a crossover trial with a total of 60 patients. These 60 patients are included in the totals for both buprenorphine and tramadol. 4The included study was a crossover trial. The total number of patients are listed in the totals for both interventions. 5Two of the studies included patients with non‐cancer pain. 6Both included studies were crossover trials. The total number of patients are listed in the totals for both intervention.
Abbreviations: Bup = buprenorphine; F = fluid; Fen = fentanyl; IM = intramuscular; Mor = morphine; P = phenytoin; Pen = pentazocine; Pla = placebo; SC = subcutaneous; SD = subdermal; SL = sublingual; Sup = suppository; Tab = tablets; Til+Na = tilidin + naloxone; Tra = tramadol.
Background
Description of the condition
Pain affects approximately 75% of people with advanced cancer (Deandra 2008). According to the World Health Organization (WHO), the incidence of cancer was just under 12.7 million new cases in 2008 and is estimated to reach over 15 million cases in 2020 (Ferlay 2010; Frankish 2003). Unrelieved cancer pain is a cause of major suffering worldwide. Globally, millions of people suffer from unrelieved pain, particularly in low‐ and middle‐income countries (World Bank 2013) where cancer is diagnosed in late stages when pain is often severe (Seya 2011; Ferlay 2010). Estimates of cancer pain prevalence vary widely. This has been due in part to a lack of standardisation in the definition of pain and the measures used to assess it, and because of the heterogeneity of cancer diagnoses. There is also heterogeneity in terms of where and in what setting patients with cancer and cancer pain receive their treatments (e.g., in outpatient clinics, in hospitals or in day units). In general, prevalence of pain at the time of cancer diagnosis and early in the course of disease is thought to be approximately 50%, increasing to 75% in the more advanced stages (Portenoy 1989). According to a systematic review, pain prevalence ranges from 33% in patients after curative cancer treatment, to 59% in patients on anticancer treatment and to 64% in patients with metastatic, advanced or terminal phase disease (van den Beuken‐van Everdingen 2007).
Cancer pain may be acute and chronic and is divided into four physiological types: nociceptive (somatic or visceral), neuropathic and sympathetically maintained pain (Foley 1998). Each of these pain types can result from the tumour itself causing compression or infiltration, or it may be more indirectly related to the cancer and its treatments, e.g., constipation, muscle spasms, post‐surgical scars or lymphoedema. Patients with cancer may have painful concurrent disorders which may be exacerbated by the presence of the cancer, e.g., osteoarthritis.
Description of the intervention
Buprenorphine is prescribed in the management of cancer pain, but is not a typical first‐line opioid. However, it is starting to experience a renaissance in the management of both chronic cancer and non‐cancer pain and it is also used in people with opioid‐dependence (Foster 2013).
The WHO classifies buprenorphine as a step III opioid analgesic (WHO 1996). It has mixed agonistic and antagonistic properties. Its opioid agonistic activity is exerted on µ‐opioid receptors and the ORL‐1 receptor, whilst it is a kappa‐ and delta‐opioid receptor antagonist (Lewis 2004; Rothman 1995; Zaki 2000). It is given either transdermally (via a patch), as an injection or via the oral mucosa (sublingually). Mainly metabolised by the liver, buprenorphine goes through dealkylation and glucuronidation and is excreted predominantly in bile. Buprenorphine pharmacokinetics vary with route of administration. Whilst the sublingual (SL) and intramuscular (IM) routes produce similar outcomes in terms of pain‐relief, when taken orally, buprenorphine undergoes extensive pre‐systemic elimination (Bullingham 1981; Bullingham 1983). Oral bioavailability is therefore low (15%) due to extensive first‐pass metabolism in the gastrointestinal mucosa and liver. However, it is longer‐acting than morphine. Whilst buprenorphine is rapidly absorbed via oral mucosa, absorption into the systemic circulation is slow (tmax is 30 minutes to 3.5 hours after a single dose; one to two hours with repeat dosing; Elkader 2005). However, it subsequently has a long duration of action (six to eight hours), which suggests that SL buprenorphine may be not suited for the management of breakthrough pain. Poulain 2008 has demonstrated the successful use of buprenorphine as a breakthrough analgesic for patients on maintenance transdermal (TD) buprenorphine.
Buprenorphine activity as a partial agonist at the µ receptor means it has agonist and antagonist activity. Its long duration of action is thought to be due to an unusually slow dissociation constant for the drug‐receptor complex. Naloxone appears to be relatively ineffective in reversing opioid effects from buprenorphine, despite naloxone's high affinity for the µ‐opioid receptor (Gal 1989), and this is due to buprenorphine's even stronger receptor affinity (Dahan 2010). In humans, a ceiling effect has been shown with buprenorphine for respiratory depression but not for analgesia (Dahan 2005; Dahan 2006). Whilst buprenorphine has been shown to slow intestinal transit, it possibly does this less than morphine (Bach 1991; Robbie 1979); importantly, constipation as an adverse effect may be less severe (Pace 2007). Buprenorphine also exerts little or no pressure on pancreatic and biliary ducts, distinguishing it from morphine in this respect (Staritz 1986). Compared with other opioids, buprenorphine causes little or no immunosuppression (Budd 2004; Sacerdote 2000; Sacerdote 2008). As a drug, buprenorphine does not accumulate in renal failure and is not removed by haemodialysis. This means that analgesia is unaffected, making it potentially clinically useful in these situations (Filitz 2006; Hand 1990).
Examples of buprenorphine patch preparations are three or seven day TD formulations (5, 10, 20, 35, 52.5, 70 µg/hour). It is a highly lipid‐soluble drug, making it ideal for TD delivery. Within patch formulations it is evenly distributed in a drug‐in‐adhesive matrix and its release is governed by the physical attributes of the matrix and proportional to the surface area of the patch. It is also available as an injection (300 µg). Buprenorphine via either the TD or injectable route is approved for managing moderate to severe chronic pain. SL tablets and a SL film preparation are also available in some countries and are combined with naloxone. Currently these are used for the treatment of opioid addiction, although some SL tablets (200 µg and 400 µg) without naloxone are available for chronic moderate to severe pain. It should not be used for acute pain, e.g., when there is a need for rapid dose titration for severe pain in cancer and palliative care settings.
Buprenorphine is most commonly prescribed as a TD formulation for cancer patients. It is estimated to be 70 to 115 times more potent than oral morphine (Likar 2008; Mercadante 2009; Sittl 2005). In practical terms, the National Institute for Health and Care Excellence (NICE) has suggested using caution when calculating opioid equivalences for TD patches and that a TD buprenorphine patch of 20 µg/hour equates to approximately 30 mg of oral morphine daily (NICE 2012). All opioid conversions have to take into account inter‐individual differences in such factors as pain perception and opioid receptor affinity. Research into genetic, gender and immunological differences in how people respond to opioids will form an ever‐increasing part of pain management in the future.
Why it is important to do this review
Many patients with cancer experience moderate to severe pain that requires treatment with strong analgesics. In 1986, the WHO published the Method for Cancer Pain Relief (WHO analgesic ladder), advocating a stepwise approach to analgesia for cancer pain and revolutionising the use of oral opioids (WHO 1987). It recommended that morphine be used as a first‐line treatment for moderate to severe cancer pain. Observational studies have suggested that this approach results in pain control for 73% of patients (Bennett 2008) with a mean reduction in pain intensity of 65% (Ventafridda 1987).
Buprenorphine, oxycodone (Schmidt‐Hansen 2015), fentanyl (Hadley 2013), hydromorphone (Quigley 2002), methadone (Nicholson 2007) and morphine (Wiffen 2013) are examples of more commonly used opioids used for the relief of cancer pain worldwide. However, Step III opioids are ineffective for treating pain in all patients (Pergolizzi 2008) and are not well‐tolerated by all patients. However, buprenorphine does not accumulate in renal impairment and is not removed by haemodynamics, making it a practical analgesic in some situations where the use of other strong opioids may be more problematic.
The aim of this Cochrane review is to assess whether buprenorphine is associated with superior, inferior or equal pain relief and tolerability compared to other analgesic options for patients with cancer pain.
Objectives
To assess the effectiveness and tolerability of buprenorphine for pain in adults and children with cancer.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), with parallel‐group or cross‐over design, comparing buprenorphine (any formulation and any route of administration) with placebo or an active drug (including buprenorphine) for treating people with cancer background pain. We did not examine studies on breakthrough pain.
Types of participants
Adults and children with cancer pain.
Types of interventions
Buprenorphine (any dose, formulation and route of administration) versus buprenorphine (any dose, formulation and route of administration);
Buprenorphine (any dose, formulation and route of administration) versus other active drug (any dose, formulation and route of administration);
Buprenorphine (any dose, formulation and route of administration) versus placebo.
Types of outcome measures
Primary outcomes
-
Pain intensity and pain relief:
Both outcomes had to be patient‐reported and could be reported in any transparent manner (e.g., by using numerical or verbal rating scales);
We did not consider these outcomes reported by physicians, nurses or carers;
If possible, we aimed to distinguish between nociceptive and neuropathic pain. However, this was not possible on the basis of the included trials.
In line with Wiffen 2013, we looked for outcomes that are equivalent to 'no worse than mild pain' (Moore 2013) operationalised as either one of the following:
No or mild pain;
≤ 3/10 on a numerical rating scale;
≤ 30/100 mm on a visual analogue scale;
Positive ratings on patient measures of satisfaction (usually very satisfied), or treatment success, or global impression of change (very good, excellent).
Secondary outcomes
Side effects or adverse events (e.g., constipation, nausea, vomiting, drowsiness, confusion, respiratory depression), quality of life and patient preference. We considered all of these outcomes as they were reported in the included studies.
Search methods for identification of studies
We did not apply language, date or publication status (published in full, published as abstract or unpublished) restrictions to the search.
Electronic searches
We identified relevant trials by searching the following databases on 20 January 2015:
Cochrane Central Register of Controlled Trials, (CENTRAL; Issue 12 of 12, 2014, the Cochrane Library);
MEDLINE (OVID; 1948 to 20 January 2015);
EMBASE (OVID; 1980 to 20 January 2015);
Web of Science (ISI) (SCI‐EXPANDED & CPCI‐S) to 20 January 2015;
BIOSIS (ISI) (1969 to 20 January 2015).
We have listed the electronic search strategies in Appendix 1.
Searching other resources
We checked the bibliographic references of identified studies, as well as relevant studies and systematic reviews in order to find additional trials not identified by the electronic searches. We also searched ClinicalTrials.gov (http://clinicaltrials.gov/), the metaRegister of Controlled Trials (mRCT) (http://www.controlled‐trials.com/mrct/), the WHO International Clinical Trials Registry Platform (ICTRP) search portal (http://apps.who.int/trialsearch/) and the Proceedings of the Congress of the European Federation of International Association for the Study of Pain (IASP; via European Journal of Pain Supplements) up to 16 February 2015 as complementary sources for related studies. We contacted authors of the included studies to ask if they knew of any other relevant studies.
Data collection and analysis
Selection of studies
Two review authors (MSH, JH) assessed the titles and abstracts of all the studies identified by the search for potential inclusion. We independently considered the full records of all potentially relevant studies for inclusion by applying the selection criteria outlined in the Criteria for considering studies for this review section. We resolved potential disagreements by discussion. We did not restrict the inclusion criteria by date, language or publication status (published in full, published as abstract, unpublished).
Data extraction and management
Using a standardised data extraction form, two review authors extracted data pertaining to study design, participant detail (including age, cancer characteristics, previous analgesic medication and setting), interventions (including details about titration) and outcomes. We resolved potential disagreements by discussion. In studies in which only a subgroup of the participants met the inclusion criteria for this review, we only extracted the data on this subgroup provided randomisation was not broken.
Assessment of risk of bias in included studies
Two review authors independently assessed the methodological quality of each included study by using the Cochrane Collaboration's 'Risk of bias' assessment tool (Higgins 2011). For each study we assessed the risk of bias for the following domains:
Selection bias (study level: random sequence generation, allocation concealment);
Performance bias (outcome level: blinding of patients, blinding of treating personnel);
Detection bias (outcome level: blinding of outcome assessment);
Attrition bias (outcome level: incomplete outcome data);
Reporting bias (study level: selective reporting).
In addition, we included an item that assesses the adequacy of titration. Each of the items from the above domains required a 'low risk', 'high risk' or 'unclear risk' response. We also documented the reasons for each response in accordance with Higgins 2011. We resolved potential disagreements through discussion. In addition to this strategy for 'Risk of bias' assessment in the individual studies, we considered the impact that study size may have on the validity of the results. We assessed the overall quality of the evidence for each outcome using the GRADE approach (Guyatt 2008).
Measures of treatment effect
For continuous outcomes we extracted the means and standard deviations (SDs), where possible, with the intention of using these to estimate the mean difference (MD) between the treatments along with the 95% confidence interval (CI), if the outcome were measured on the same scale in the studies. Where the outcome was measured on different scales, we intended to report the standardised mean difference (SMD) with 95% CIs instead when performing meta‐analyses. However, this was not feasible. For dichotomous outcomes we extracted event rates but did not, as planned, calculate risk ratios (RRs) and number needed to treat for an additional beneficial outcome (NNTB)/number needed to treat for an additional harmful outcome (NNTH), again because no meta‐analyses were performed.
Unit of analysis issues
Our plan to deal with any unit‐of‐analysis issues was to consider the patient the unit of analysis. However, if the data reported in any included cross‐over trials could not be otherwise incorporated into the analyses (see Dealing with missing data), we would include them as if the design had been parallel group. Higgins 2011 points out that this approach, while giving rise to unit‐of analysis error, is nevertheless conservative as it results in an under‐weighting of the data. Moreover, if we included cross‐over trial data in this manner we would perform sensitivity analyses assessing the impact of this strategy. However, as we did not perform any meta‐analyses, this strategy was not used.
Dealing with missing data
In cases where data were missing, we contacted the trial authors to request missing data. However, we received no replies. We planned to limit missing data imputation to the imputation of missing SDs if enough information was available from the studies to calculate the SD according to the methods outlined by Higgins 2011. However no missing data were imputed in this manner as no meta‐analyses were performed. We have recorded the drop‐out/missing data rates in the 'Risk of bias' tables under the items on attrition bias, and we addressed the potential effect of the missing data on the results, not in sensitivity analyses as originally planned, but in the Discussion section. Although we aimed to perform intention‐to‐treat (ITT) analyses, we were unable to do so in all cases as we were unable to perform meta‐analyses.
Assessment of heterogeneity
We planned to assess heterogeneity by using the I2 statistic, with I2 values > 50% representing substantial heterogeneity in line with Higgins 2011. We aimed to assess potential sources of heterogeneity through subgroup analyses as outlined in Subgroup analysis and investigation of heterogeneity. However as we were unable to undertake any meta‐analyses, we did not perform these subgroup analyses.
Assessment of reporting biases
In addition to implementing the comprehensive search strategy outlined in the section Search methods for identification of studies, the risk of outcome reporting bias is included in the 'Risk of bias' summary figures (Figure 1; Figure 2) that we constructed for each study and each type of assessed bias.
1.
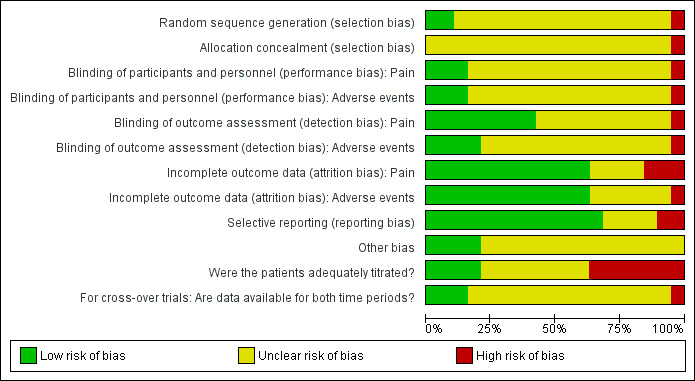
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.
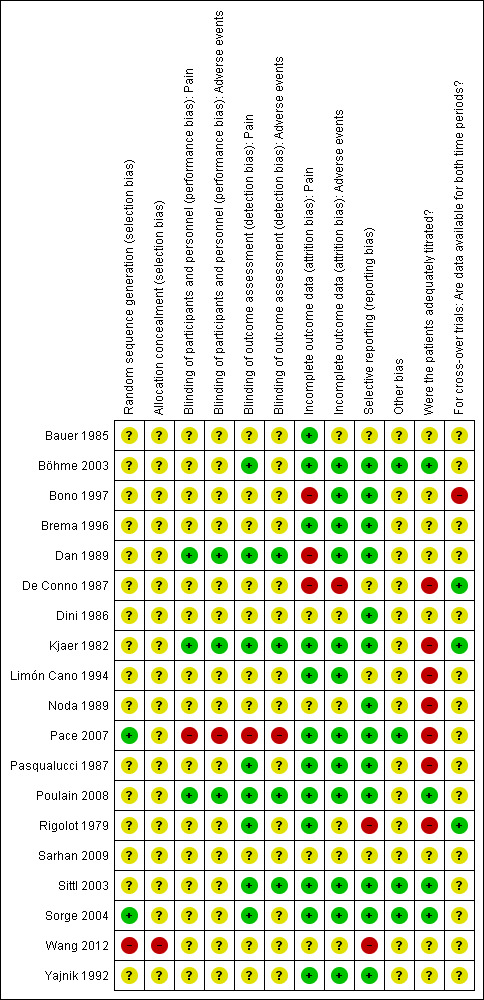
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Data synthesis
We planned to enter the data extracted from the included studies into the Cochrane Collaboration's statistical software, Review Manager 2014, in order to use this for data synthesis. We planned to analyse continuous outcomes using the generic inverse variance method, and dichotomous outcomes using the Mantel‐Haenszel method in accordance with Higgins 2011. If the I2 statistic value was > 50% we planned to use a random‐effects model and consider not reporting a summary estimate of the data (depending on the subgroup analyses; see also the section Subgroup analysis and investigation of heterogeneity). Otherwise we would use a fixed‐effect model for the meta‐analyses. However, as it was not feasible to meta‐analyse the data from the included studies, we summarised the data narratively and in tables. We have also, as planned, summarised the results for all the listed outcomes in a 'Summary of findings' table.
Subgroup analysis and investigation of heterogeneity
Different aspects of the trials are likely to contribute heterogeneity to the proposed main analyses. If there were sufficient data, we planned to perform subgroup analyses based on doses, titration, routes of administration (e.g., SL, TD), length of the trials and populations (e.g., opioid‐naive patients, solid/haematological cancer type, adults/children, co‐morbidities). However, there were insufficient data to perform such analyses.
Sensitivity analysis
If sufficient data were available, we aimed:
To examine the robustness of the meta‐analyses by conducting sensitivity analyses using different components of the 'Risk of bias' assessment, particularly those relating to whether allocation concealment and blinding were adequate;
To conduct further sensitivity analyses to examine the impact of missing data on the results if a large proportion of the studies were at an 'unknown' or 'high risk' of attrition bias; and
To perform sensitivity analyses examining whether publication status and trial size influenced the results.
However, we did not perform any sensitivity analyses because there were insufficient data.
Results
Description of studies
Results of the search
The search identified 473 unique records of which we excluded 419 based on the title/abstract. We retrieved 54 records for full‐text evaluation. Of the 54 records, we included 19 studies published in 23 articles, while we excluded 22 because they were not in PICO (i.e., an RCT conducted in the target population examining the target comparisons as measured by the target outcomes; N = 14), withdrawn (N = 1), or narrative reviews (N = 7) (see Figure 3). In addition to the 19 included studies, we identified four ongoing studies and five potentially relevant studies. We await further information, including study completion and publication, of the latter before we can ascertain their relevance to the current review and classify them accordingly. See also Characteristics of ongoing studies and Characteristics of studies awaiting classification, respectively.
3.
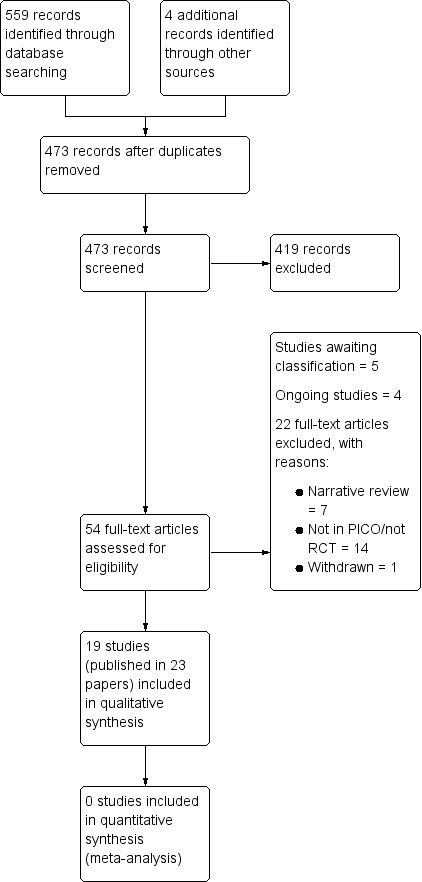
Study flow diagram.
Included studies
The 19 included studies were published between 1979 and 2012 and enrolled/randomised a total of 1421 patients (study range 10 to 189) with 1304 (study range 10 to 188) of these analysed for efficacy and 1216 (study range 12 to 189) for safety (Rigolot 1979; Wang 2012 did not report this outcome). The reported mean ages of the patient populations in the studies ranged from 49.1 years to 67.16 years. Four studies were crossover trials (Bono 1997; De Conno 1987; Kjaer 1982; Rigolot 1979) and the remainder were parallel‐group trials, with six studies conducted in Italy (Bono 1997; Brema 1996; De Conno 1987; Dini 1986; Pace 2007; Pasqualucci 1987), two in Japan (Dan 1989; Noda 1989) and in the following countries: Denmark (Kjaer 1982); Germany (Bauer 1985); Austria, Germany and Hungary (Böhme 2003); Mexico (Limón Cano 1994); Austria, Belgium, Croatia, France, Poland and the Netherlands (Poulain 2008); France (Rigolot 1979); Egypt (Sarhan 2009); Austria, Germany and the Netherlands (Sittl 2003); Germany and Poland (Sorge 2004); China (Wang 2012); and India (Yajnik 1992). The treatment groups in the included studies were either comparable at baseline (Dan 1989; Kjaer 1982; Pace 2007; Poulain 2008; Wang 2012; Yajnik 1992), not comparable at baseline (Böhme 2003 (age); Sittl 2003 (age); Sorge 2004 (disease stage)), or it was unclear (e.g., due to lack of reporting of baseline characteristics whether they differed; Bauer 1985 (age); Bono 1997 (baseline pain); Brema 1996 (gender); De Conno 1987; Dini 1986; Limón Cano 1994 (age, gender); Noda 1989 (cancer type and stage); Pasqualucci 1987; Rigolot 1979; Sarhan 2009). Three of the studies included patients with pain of a both malignant and non‐malignant origin (Böhme 2003; Sittl 2003; Sorge 2004). One of these studies presented some of the results split by pain origin (32.8% of the patients had cancer pain; Sorge 2004). The other two studies did not present the results separately for the patients with cancer pain, but they were still included as the percentage of patients with malignant pain were above 50 in both studies (55% in Böhme 2003; and 77.1% in Sittl 2003). Trial length ranged from single dose treatment to six months, and the studies reported the following comparisons:
SL buprenorphine versus subdermal (SD) buprenorphine injection (Limón Cano 1994);
SL buprenorphine versus oral tilidin + naloxone (Bauer 1985);
SL buprenorphine versus oral tramadol (Bono 1997; Brema 1996);
SL buprenorphine versus SL buprenorphine + oral phenytoin versus oral phenytoin (Yajnik 1992);
SL buprenorphine versus oral pentazocine (De Conno 1987);
Buprenorphine tablets/fluid versus pentazocine tablets/fluid (Dini 1986);
TD buprenorphine versus placebo (Böhme 2003; Poulain 2008; Sittl 2003; Sorge 2004);
TD buprenorphine versus controlled‐release morphine (Pace 2007);
TD buprenorphine versus TD fentanyl (Sarhan 2009);
IM buprenorphine injection versus buprenorphine suppository (Dan 1989);
IM buprenorphine versus IM morphine (Kjaer 1982; Rigolot 1979);
IM buprenorphine + SC buprenorphine versus SC buprenorphine versus placebo + SC buprenorphine (Noda 1989);
Epidural buprenorphine versus epidural morphine (Pasqualucci 1987);
Intravenous buprenorphine versus intravenous morphine (Wang 2012).
See also Characteristics of included studies for further details about the studies.
Excluded studies
We excluded 22 studies because they were not in PICO (i.e., an RCT conducted in the target population examining the target comparisons as measured by the target outcomes; N = 14), withdrawn (N = 1), or narrative reviews (N = 7). One of the studies identified in the search compared buprenorphine in combination with diclofenac against buprenorphine alone (Corli 1988). We excluded this study as it would not answer our primary question which is concerned with the effectiveness of buprenorphine for cancer pain. See also Characteristics of excluded studies.
Risk of bias in included studies
In this section we have described the risk of bias for the included studies. See also Figure 1 and Figure 2 for summaries of the risk of bias judgements.
Allocation
We considered generation of the randomisation sequence to be at low risk of bias in only two included trials (Pace 2007; Sorge 2004). A third study was considered to be at high risk of selection bias because it included only 120 patients that were allocated to one of four treatment groups with stratification for several factors. With each stratification factor it became increasingly conceivable that the group allocation ceased to be truly random or, indeed, concealed given the relatively high number of treatment groups to the relatively low number of patients (Wang 2012). The remaining included studies did not report enough information to enable us to assess the risk of selection bias. Therefore we considered them at unclear risk of selection bias.
Blinding
Lack of reporting was also an issue when assigning risk of bias estimates to the items assessing performance and detection bias, i.e., blinding. Very few trials reported directly who was blinded, so in most cases we inferred on the basis of supplementary information whether we were reasonably certain that blinding had been adequately executed for a given individual (i.e., patient, treating personnel or the outcome assessors, or both, where not the patients themselves). On this basis, we considered the risk of performance bias to be low for the primary outcome of pain and for the secondary outcome of adverse events in three studies (Dan 1989; Kjaer 1982; Poulain 2008), high in one study described as open label (Pace 2007) and unclear in the remaining 13 studies that reported this outcome. We considered eight studies at low risk of detection bias for pain (which according to our criteria had to be patient‐assessed) either because it was clearly stated that the patient was blinded (Dan 1989; Kjaer 1982; Poulain 2008; Rigolot 1979 (although in this study it is not clear whether pain is patient‐assessed)) or because the study was described as double‐blind without stating who was blinded (i.e., patient, treating personnel or outcome assessor) and we considered it sufficiently likely that at least the patient was blinded (Böhme 2003; Pasqualucci 1987; Sittl 2003; Sorge 2004; see also Characteristics of included studies). Apart from Pace 2007, judged at high risk due to being open label, we judged the remaining studies to be at unclear risk of detection bias for the outcome of pain. In the case of adverse events, it was often unclear who reported/assessed this outcome. Therefore we felt unable to assume with sufficient confidence that it had be assessed in a blinded manner (unlike with pain as described above), unless it had been clearly stated. We considered the risk of detection bias for adverse events to below in four studies that all clearly stated that the outcome assessor was blinded (Dan 1989; Kjaer 1982; Poulain 2008; Sittl 2003), high in one open label study (Pace 2007) and unclear in the remaining 12 studies reporting this outcome.
Incomplete outcome data
Overall the data from 91.8% of the total number of enrolled/randomised patients were analysed for pain. We judged the risk of attrition bias as low in most included studies (Bauer 1985; Böhme 2003; Brema 1996; Kjaer 1982; Limón Cano 1994; Pace 2007; Pasqualucci 1987; Poulain 2008; Rigolot 1979; Sittl 2003; Sorge 2004; Yajnik 1992), with three studies considered at high risk (Bono 1997; Dan 1989; De Conno 1987) and four studies considered at unclear risk (Dini 1986; Noda 1989; Sarhan 2009; Wang 2012) of attrition bias, respectively. For adverse events, we analysed the data from 85.6% of the total number of enrolled/randomised patients, and we considered the risk of attrition bias to be low in 12 included studies (Böhme 2003; Bono 1997; Brema 1996; Dan 1989; Kjaer 1982; Limón Cano 1994; Pace 2007; Pasqualucci 1987; Poulain 2008; Sittl 2003; Sorge 2004; Yajnik 1992), high in one study (De Conno 1987) and unclear in the remaining four studies (Bauer 1985; Dini 1986; Noda 1989; Sarhan 2009) that reported this outcome. Rigolot 1979 and Wang 2012 did not report adverse events.
Selective reporting
We considered 13 included studies to be at low risk of reporting bias, with Rigolot 1979 and Wang 2012 considered at high risk of reporting bias as neither reported adverse events. We judged the remaining four studies (Bauer 1985; De Conno 1987; Limón Cano 1994; Sarhan 2009) at unclear risk of reporting bias due to under‐reporting from being available either only in abstract form or in a foreign language.
Other potential sources of bias
Patients appeared to be adequately titrated in only four studies (Böhme 2003; Poulain 2008; Sittl 2003; Sorge 2004), and inadequately or not titrated in a further seven studies (De Conno 1987; Kjaer 1982; Limón Cano 1994; Noda 1989; Pace 2007; Pasqualucci 1987; Rigolot 1979). Titration schedule or adequacy, or both, was unclear in the remaining eight studies.
Apart from five studies which reported to have received commercial funding (Böhme 2003; Kjaer 1982; Poulain 2008; Sittl 2003; Sorge 2004), it was unclear whether the remaining studies received such funding.
Data were available for both cross‐over phases for three of the four crossover trials included (De Conno 1987; Kjaer 1982; Rigolot 1979). We considered these trials to be at low risk of bias, whereas we judged the final trial (Bono 1997) to be at high risk of bias because the pain intensity data did not appear to be inferentially analysed collapsed over phases for any of the seven (per phase) study days, apart from for the first four hours of treatment.
Nine included studies appeared to conduct the analyses according to the ITT principle (Bauer 1985; Brema 1996; Kjaer 1982; Limón Cano 1994; Pace 2007; Pasqualucci 1987; Poulain 2008; Sittl 2003; Sorge 2004), although this was often not clearly stated. In the remaining studies it was either unclear if ITT analyses were performed (Böhme 2003; Dini 1986; Noda 1989; Rigolot 1979; Sarhan 2009; Wang 2012; Yajnik 1992) or they were clearly not performed (Bono 1997; Dan 1989; De Conno 1987).
With the exception of four studies (Böhme 2003; Pace 2007; Sittl 2003; Sorge 2004) which we judged at low risk of 'other bias', for most included studies we were unable to evaluate with sufficient confidence whether they were subject to other kinds of bias due to the very limited reporting that this body of evidence generally suffered from (Bauer 1985; Bono 1997; Brema 1996; Dan 1989; De Conno 1987; Dini 1986; Kjaer 1982; Limón Cano 1994; Noda 1989; Pasqualucci 1987; Poulain 2008; Rigolot 1979; Sarhan 2009; Wang 2012; Yajnik 1992).
Effects of interventions
See: Table 1
SL buprenorphine versus SD buprenorphine
Limón Cano 1994 conducted a placebo‐controlled, parallel‐group study of (it seems) 24‐hour duration comparing SL buprenorphine (N = 10) to SD buprenorphine (N = 7) administered every four to eight hours on a patient‐need basis. Both treatments resulted in a 50% reduction in pain intensity, with faster onset of pain relief observed in the SL group (63 ± 22.1 min) compared to the subdermic group (94.3 ± 22.7 min). The mean duration of analgesia was similar between the SL (7.4 ± 1.2 hours) and the SD (6.8 ± 1.2 hours; P > 0.2) groups, and no significant differences in adverse event rates were reported (see also Table 2).
1. SL buprenorphine comparisons: Adverse events.
| AE | SL Bup versus SD Bup | SL Bup versus oral tilidin‐HCI + naloxone‐HCI | SL Bup versus oral Tra | SL Bup versus SL Bup + oral P versus oral P | SL Bup versus oral Pen | Bup tablets/fluid versus Pen Tab/F | |||||||||||
| Limón Cano 1994 | Bauer 1985 | Bono 1997 | Brema 1996 | Yajnik 1992 | De Conno 1987a | Dini 1986 | |||||||||||
| SL bup | SD bup | SL bup | Til+Na | SL bup | Tra | SL bup | Tra | Bup | Bup + P | P | SL bup | Pen | Bup tablets | Bup fluid | Pen tablets | Pen fluid | |
| Any AEs | 34/60 | 9/60 | 16/63 | 17/68 | 13/25 | 5/25 | 2/25 | 18% | 50% | ||||||||
| Total AEs | 49 | 9 | |||||||||||||||
| Abdominal pain | 0.6 | 1 | |||||||||||||||
| Acidity | 0.5 | 1 | |||||||||||||||
| Agitation | 2 | 2.4 | 0/11 | 0/11 | 0/10 | 2/10 | |||||||||||
| Allergy | 0.1 | 0.2 | |||||||||||||||
| Anorexia/appetite loss | 1/60 | 0/60 | |||||||||||||||
| Blood loss | 0.1 | 0.1 | |||||||||||||||
| Bradycardia | 3/25 | 1/25 | 0/25 | ||||||||||||||
| Confusion | 0/11 | 0/11 | 0/10 | 1/10 | |||||||||||||
| Constipation | Bup = Til+Na | 4/60 | 2/60 | 3/63 | 4/68 | ||||||||||||
| Dizziness/confusion | 6/60 | 1/60 | 3/63 | 4/68 | 1.1 | 2 | |||||||||||
| Drowsiness/somnolence | 14/60 | 5/60 | 5/63 | 7/68 | 4/25 | 1/25 | 0/25 | 2.7 | 2.2 | 0/11 | 2/11 | 3/10 | 1/10 | ||||
| Dry mouth | 1/60 | 0/60 | 2.8 | 2.6 | |||||||||||||
| Fatigue | Bup = Til+Na | ||||||||||||||||
| Giddiness | 0/25 | 0/25 | 1/25 | ||||||||||||||
| Hallucinations | 3/10 | 5/7 | 1/60 | 0/60 | |||||||||||||
| Headache | 0/25 | 0/25 | 1/25 | 0.9 | 1.1 | ||||||||||||
| Heartburn | 0.6 | 0.9 | |||||||||||||||
| Heavy head | 1/60 | 0/60 | |||||||||||||||
| Hiccup | 1/60 | 0/60 | |||||||||||||||
| Hypotension | 1/25 | 1/25 | 0/25 | ||||||||||||||
| Irritability | 1/60 | 0/60 | |||||||||||||||
| Nausea | Bup = Til+Na | 8/60 | 0/60 | 7/63 | 8/68 | 1.4 | 1.6 | ||||||||||
| Nausea and vomiting | SL = SD | SL = SD | 6/60 | 0/60 | 2/25 | 1/25 | 0/25 | ||||||||||
| Pruritus | 1.1 | 0.7 | |||||||||||||||
| Respiratory depression | 1/10 | 1/7 | 3/25 | 1/25 | 0/25 | ||||||||||||
| Sedation | SL = SD | SL = SD | |||||||||||||||
| Sweating | 1/60 | 0/60 | 2.4 | 2.1 | |||||||||||||
| Thirst | |||||||||||||||||
| Tremors | 1.1 | 1.7 | |||||||||||||||
| Vomiting | Bup = Til+Na | 6/60 | 1/60 | 1.1 | 0.8 | 2/11 | 2/11 | 2/10 | 0/10 | ||||||||
| Discontinuation due to AE | 0 | 0 | 19/60 | 2/60 | 7/63 | 6/68 | |||||||||||
Abbreviations: AE = adverse events; SL = sublingual; SD = subdermal; Bup = buprenorphine; P = phenytoin; Pen = pentazocine; SC = subcutaneous; SD = subdermal; SL = sublingual; Tab = tablets; Til+Na = tilidin + naloxone; Tra = tramadol; F = fluid. aAverage.
SL buprenorphine versus oral tilidine‐HCI + naloxone‐HCI
Bauer 1985 conducted a parallel‐group, 28‐day study with 20 women in each group comparing SL buprenorphine to oral tilidine with naloxone. This study found that the pain intensity ratings, which were comparable at baseline (7.16 for buprenorphine versus 7.11 for tilidine + naloxone), were significantly lower for the patients who received buprenorphine on days 1 (4.31 for buprenorphine versus 4.97 for tilidine + naloxone), 7 (3.43 for buprenorphine versus 4.58 for tilidine + naloxone), 14 (3.83 for buprenorphine versus 4.54 for tilidine + naloxone) and 21 (4.06 for buprenorphine versus 4.56 for tilidine + naloxone), but not on day 28, where it was only numerically lower (4.07 for buprenorphine versus 4.42 for tilidine + naloxone). The mean number of drug administrations necessary to achieve satisfactory analgesia was 39 (range = 26 to 52) in the buprenorphine group and 60 (range = 36 to 104) in the tilidine + naloxone group over the 28‐day study period (P < 0.05). The mean interval between drug administrations was 17.6 (range = 12.8 to 25.5) hours in the buprenorphine group and 11.85 (range = 6.4 to 17.7) hours in the tilidine + naloxone group (P < 0.05). The trial authors reported that there did not seem to be any detectable increase in analgesic requirements due to tachyphylaxis for any of the drugs, and all the patients in both treatment groups described the analgesic effectiveness as satisfactory. Treatment‐related side effects such as nausea, vomiting and fatigue, and constipation occurred with the same frequency in both groups, and led to no cases of discontinuation of treatment (see also Table 2).
SL buprenorphine versus oral tramadol
Bono 1997 undertook a cross‐over study with two phases, each lasting 7 days, comparing SL buprenorphine to oral tramadol in 60 patients. The pain intensity data did not appear to be inferentially analysed where it was collapsed over phases for (any of) the study days, apart from for the first four hours of treatment, where no differences were observed between the treatments. Other analyses that appeared to include all patients collapsed across phases showed:
Ratings of pain intensity relative to the pain intensity experienced the previous day did not differ significantly between the treatment groups;
Tramadol treatment was associated with significantly better quality of sleep than buprenorphine on days 6 and 7, but not on days one to five where no differences were observed;
Significantly better patient ratings of tolerability (mean = 80.1, SEM = 2.3) compared to buprenorphine (mean = 41.8, SEM = 4.1);
The number of patients with side effects was also lower during tramadol (9/60 patients) than buprenorphine treatment (34/60 patients; see also Table 2).
In a parallel‐group trial planned to last up to six months, Brema 1996 compared SL buprenorphine (N = 63) to slow‐release tramadol (N = 68). The mean duration of treatment was 50.9 days in the buprenorphine group and 57.7 days in the tramadol group. One patient in the buprenorphine group and four patients in the tramadol group completed the six months of treatment. At baseline, 92% buprenorphine and 98.4% tramadol patients reported 'strong‐to‐unbearable' pain, which reduced to 66.7% and 48.4% respectively at seven days and to 54.5% and 43.1%, respectively, at day 14. These percentages differed statistically significantly at day 7, but this significance had disappeared by day 14, and may have been a result of quicker titration with tramadol than buprenorphine in the early stage of the study. No significant differences in the percentage of patients reporting good deep sleep were observed between the buprenorphine and tramadol patients at baseline (buprenorphine 32.7%, tramadol 37.2%), day 7 (buprenorphine 40%, tramadol 51.1%) or day 14 (buprenorphine 43.9%, tramadol 50%). After two weeks of treatment the overall treatment efficacy was rated as higher in the tramadol (mean 100‐mm VAS = 62.3, SD = 26.7) than in the buprenorphine (mean 100‐mm VAS = 57.2, SD = 25.6) group although not significantly so. This was also the case at the end of treatment and at this stage the difference may have become significant although this cannot be ascertained based on the reported results (tramadol: mean 100‐mm VAS = 60.9, SD = 27.8; buprenorphine: mean 100‐mm VAS = 47.4, SD = 26; P ≤ 0.05). After two weeks of treatment the overall treatment acceptability was rated as significantly higher in the tramadol (mean 100‐mm VAS = 70.7, SD = 19.8) than in the buprenorphine (mean 100‐mm VAS = 58.9, SD = 24.5) group. This was also the case at the end of treatment, although it is apparently only marginally significantly higher at this stage (tramadol: mean 100‐mm VAS = 69.2, SD = 19.1; buprenorphine: mean 100‐mm VAS = 58.3, SD = 22.9; P ≤ 0.05). The trial authors also reported that in the tramadol group 71.4% of the patients reported moderate/no pain in the first month and 80% did so in the second month, with the corresponding percentages for the buprenorphine group at 45.4% after one month and 65.2% after two months, but reported no inferential statistics. We have listed the adverse events in Table 2.
SL buprenorphine versus SL buprenorphine + oral phenytoin versus oral phenytoin
Yajnik 1992 conducted a parallel‐group trial of one month duration comparing treatment with SL buprenorphine, with SL buprenorphine + oral phenytoin and with phenytoin in three groups of 25 patients, and found no significant difference in pain relief rates after one month between the buprenorphine (good: 15/25; moderate: 6/25; poor: 4/25; none: 0/25), the buprenorphine + phenytoin (good: 18/25; moderate: 4/25; poor: 2/25; none: 1/25) and phenytoin (good: 4/25; moderate: 14/25; poor: 5/25; none: 2/25) groups. The groups did not differ significantly in incidence of adverse events (see Table 2).
SL buprenorphine versus oral pentazocine
De Conno 1987 compared SL buprenorphine with oral pentazocine in a cross‐over study lasting 14 days (seven days per phase) in 120 patients, of whom 29 did not complete the study. This study found that buprenorphine was associated with significantly better pain relief compared to pentazocine, reducing the mean daily pain intensity 10 to 25 points more than pentazocine. Patients also slept statistically significantly more (on average one hour) during treatment with buprenorphine compared to treatment with pentazocine. Also, they spent 10 to 30 minutes longer a day standing during the buprenorphine treatment phase than pentazocine treatment, although it is unclear whether this difference is statistically significant. Buprenorphine was associated with significantly more drowsiness, whereas pentazocine was associated with significantly more dizziness and stomach pain. Otherwise the side effects profiles did not differ significantly between the treatments (see also Table 2). Of the 29 patients who did not complete the study, more patients stopped study treatment during the pentazocine phase (N = 16) than during the buprenorphine phase (N = 3; P = 0.03).
Buprenorphine tablets/fluid versus pentazocine tablets/fluid
Dini 1986 conducted a parallel‐group study with four experimental groups (buprenorphine SL tablets and vials, pentazocine tablets and vials) of seven days duration with a total of 42 patients, of whom two (one each treated with buprenorphine and pentazocine tablets) did not complete the course of therapy due to excessive nausea and vomiting. Dini 1986 reported that the final daily average pain intensity was significantly lower after treatment with buprenorphine tablets (mean = 58, SE = 19) compared to pentazocine tablets (mean = 118, SE = 23). This pattern of results was also observed when comparing treatment with buprenorphine vials/fluid (mean = 38, SE = 9) and pentazocine vials/fluid (mean = 115, SE = 15). Treatment with buprenorphine vials/fluid was associated with a longer time spent asleep (mean = 8 hours, SE = 0.6 hours) relative to pentazocine vials/fluid (mean = 6.5 hours, SE = 0.4 hours). However, this effect was not observed after treatment with the tablet forms of buprenorphine (mean = 7.2 hours, SE = 0.6 hours) and pentazocine (mean = 7 hours, SE = 0.6 hours), which did not differ statistically significantly. No differences were observed either in time spent awake in the supine position between treatment with buprenorphine vials/fluid (mean = 15 hours, SE = 1.8 hours) and pentazocine vials/fluid (mean = 17 hours, SE = 2.1 hours) or between treatment with buprenorphine tablets (mean = 13.5 hours, SE = 2.2 hours) and pentazocine tablets (mean = 11.9 hours, SE = 2.1 hours). No differences were observed between the treatment groups in time spent sitting or standing either. The trial authors did not report any formal statistical comparisons for the patient ratings of treatment effectiveness and tolerability, which are therefore only reported descriptively: patients rated effectiveness of treatment after treatment with buprenorphine tablets as excellent (three patients), good (six patients) and fair (one patient); as good (one patient), fair (one patient), poor (six patients) and nothing (two patients) after treatment with pentazocine tablets; as excellent (three patients), good (seven patients) and fair (one patient) after treatment with buprenorphine vials/fluid; and as good (two patients), fair (five patients) and poor (three patients) after treatment with pentazocine vials/fluid. Patients rated tolerability of treatment after treatment with buprenorphine tablets as excellent (9 patients), good (one patient) and poor (one patient); as good (two patients), fair (four patients), and poor (four patients) after treatment with pentazocine tablets; as excellent (five patients), good (five patients) and fair (one patient) after treatment with buprenorphine vials/fluid; and as excellent (one patient), good (six patients), fair (one patient) and poor (two patients) after treatment with pentazocine vials/fluid. We have reported adverse events in Table 2.
TD buprenorphine versus placebo
Böhme 2003 is a six day, four‐arm, parallel‐group trial that included patients with pain from both malignant (55%) and non‐malignant (45%) origin and compared placebo (N = 37) to TD buprenorphine at three different doses, 35 μg/h (N = 35), 52.5 μg/h (N = 41) and 70 μg/h (N = 38). Böhme 2003 found that the number of patients who responded to treatment (i.e., patients who obtained at least satisfactory pain relief at all determination points (excluding the final examination) and who took a mean of 0.2 mg/day or less of SL buprenorphine on days seven to 12) and the mean daily doses of rescue medication (SL buprenorphine) did not differ significantly between the four groups. No significant differences in rates of adverse events were observed between the groups (see Table 3).
2. Transdermal buprenorphine comparisons: Adverse events.
| AE | TD Bup versus placebo | TD Bup versus controlled‐release Mor | TD Bup versus TD Fen | |||||||||||||
| Böhme 2003 | Poulain 2008 | Sittl 2003 | Sorge 2004 | Pace 2007 | Sarhan 2009 | |||||||||||
| Placebo | 35 μg/h Bup | 52.5 μg/h Bup | 70 μg/h Bup | Placebo | 70 μg/h Bup | Placebo | 35 μg/h Bup | 52.5 μg/h Bup | 70 μg/h Bup | Placebo | 35 μg/h Bup | TD Bup | CR Mor | TD Bup | TD Fen | |
| At least one AE | 28/38 | 35/41 | 33/41 | 28/37 | ||||||||||||
| Asthenia | 1/95 | 4/94 | ||||||||||||||
| Central nervous system AE | 20/38 | 23/41 | 19/41 | 20/37 | ||||||||||||
| Confusion | 1/26 | 1/26 | ||||||||||||||
| Constipation | 2/95 | 9/94 | 2/26 | 10/26 | ||||||||||||
| Dizziness/confusion | 0/95 | 0/94 | ||||||||||||||
| Drowsiness/somnolence | 3/26 | 2/26 | Bup > Fen | Bup > Fen | ||||||||||||
| Erythema | 7/38 | 12/41 | 12/41 | 12/37 | 0/19a | 0/26a | ||||||||||
| Exanthema | 0/37a | 0/35a | 1/41a | 0/38a | 1/38 | 5/41 | 5/41 | 1/37 | ||||||||
| Fatigue | 2/95 | 0/94 | ||||||||||||||
| Gastrointestinal AE | 26.30% | 17.10% | 36.60% | 43.20% | ||||||||||||
| Headache | 3/26 | 4/26 | ||||||||||||||
| Nausea | 7/95 | 3/94 | 3/26 | 9/26 | ||||||||||||
| Pruritus | 0/37a | 0/35a | 3/41a | 1/38a | 9/38 | 10/41 | 11/41 | 9/37 | 0/19a | 0/26a | ||||||
| Skin complication, local | Bup > Fen | Bup > Fen | ||||||||||||||
| Swelling, non‐inflammatory | 1/38 | 1/41 | 0/41 | 0/37 | ||||||||||||
| Vertigo | 3/26 | 11/26 | ||||||||||||||
| Vomiting | 6/95 | 5/94 | ||||||||||||||
| Discontinuation due to AE | 6/95 | 1/94 | 6/38 | 3/41 | 5/41 | 3/37 | ||||||||||
Abbreviations: Bup = buprenorphine; F = fluid; Fen = fentanyl; IM = intramuscular; Mor = morphine; P = phenytoin; Pen = pentazocine; Pla = placebo; SC = subcutaneous; SD = subdermal; SL = sublingual; Sup = suppository; Tab = tablets; Til+Na = tilidin + naloxone; Tra = tramadol. aSevere.
In a study similar to Böhme 2003, Sittl 2003 conducted a 15‐day long parallel group trial in 157 patients with both malignant (77.1%) and non‐malignant (22.9%) pain comparing placebo (N = 38) to TD buprenorphine at three different doses, 35 μg/h (N = 41), 52.5 μg/h (N = 41) and 70 μg/h (N = 37). Sittl 2003 found that the two lower doses of buprenorphine (35 μg/h: 36.6%; 52.5 μg/h: 47.5%; 70 μg/h: 33.3%) were found to have a significantly higher percentage of responders (i.e., patients requiring no more than one SL tablet of buprenorphine (rescue medication) per day from day 2 until the end of the study and who recorded at least satisfactory pain relief at each application of a new patch) than placebo (16.2%). The percentage reduction in mean daily dose of rescue medication relative to pre‐study was also significantly larger in all the buprenorphine treatment groups (35 μg/h: ‐56.9%; 52.5 μg/h: ‐61.6%; 70 μg/h: ‐51.6%) compared to placebo (‐8%), but did not differ significantly from each other. The mean overall ratings of pain relief were also higher in the buprenorphine groups (35 μg/h: 2.3/4; 52.5 μg/h: 2.4/4; 70 μg/h: 2.5/4) than in the placebo group (1.9/4), but it is unclear if they are significantly so. Moreover, 8/37 (placebo), 9/41 (35 μg/h), 15/40 (52.5 μg/h) and 8/37 (70 μg/h) patients, respectively, rated their pain relief as satisfactory over the course of the study, with a further 12/37 (placebo), 19/41 (35 μg/h), 16/40 (52.5 μg/h) and 16/36 (70 μg/h) patients, respectively, rating their pain relief as good or complete. The mean ratings of daily pain intensity were 'moderate‐very severe' in 60% (placebo), 52% (35 μg/h), 40% (52.5 μg/h) and 37% (70 μg/h) patients, respectively; 'mild' in 31% (placebo), 29% (35 μg/h), 42% (52.5 μg/h) and 43% (70 μg/h) patients, respectively, and 'none' in 9% (placebo), 19% (35 μg/h), 18% (52.5 μg/h) and 20% (70 μg/h) patients, respectively (no inferential statistical analyses reported). The incidence of none of the reported adverse events differed significantly between the four treatment groups (see Table 3 for the reported adverse events).
Another parallel‐group study of two weeks duration, Poulain 2008, compared placebo (N = 95) to 70 μg/h TD buprenorphine (N = 94) and found that the proportion of responders (i.e., patients with a mean pain intensity < five during the last six days of the maintenance phase and a mean daily SL buprenorphine (rescue medication) intake ≤ 2 tablets over the entire maintenance phase) was significantly higher in the buprenorphine group (70/94) compared to the placebo group (47/94). The baseline‐corrected pain intensity and rescue medication tablet intake at the end of the two‐week maintenance phase were also significantly lower in the buprenorphine group (pain intensity: least square mean = 0.23, SE = 0.15; rescue medication tablet intake: least square mean = ‐0.76, SE = 0.14) than in the placebo group (pain intensity: least square mean = 1.14, SE = 0.17; rescue medication tablet intake: least square mean = ‐0.23, SE = 0.15). Also, 51/94 buprenorphine patients and 39/94 placebo patients rated their global satisfaction with treatment as 'excellent' or 'very good' with a further 32 and 33 patients, respectively, giving 'good' or 'fair' ratings and nine buprenorphine patients and 19 placebo patients giving a rating of 'poor' (see Table 3 for the reported adverse events).
Sorge 2004 is a nine‐day, parallel‐group trial that included patients with pain of both malignant (N = 45) and non‐malignant origin (N = 92), but presented some of the results by pain origin (of which only those relating to malignant pain are included here). Sorge 2004 compared placebo (N = 19) to 35 μg/h TD buprenorphine (N = 26). This study found that the mean (SD) daily requirement for SL buprenorphine (rescue medication) tablets was 1.2 (0.3) in the run‐in phase and 0.4 (0.5) in the double‐blind phase for the buprenorphine group and 1 (0.2) and 0.6 (0.3), respectively, in the placebo group. No inferential statistics and no further efficacy results were reported separately for the patients with cancer pain (see Table 3 for the reported adverse events).
TD buprenorphine versus controlled‐release morphine
Pace 2007 compared TD buprenorphine to controlled‐release morphine in an eight‐week long parallel‐group trial with 26 patients in each arm. Pace 2007 found that buprenorphine was associated with significantly lower pain scores from week 2 of treatment and with less interference with sleep from week 1 of treatment as well as with higher quality of life in terms of 'physical pain', 'mental health' and 'vitality', with significant differences between the groups on the quality of life items of 'physical activity', 'limited activity due to physical problems', 'social activity', 'limited activity due to emotional problems' and 'problems of general health'. The study also showed that buprenorphine treatment was associated with lower anger/aversion, fatigue/inertia and total mood disorder scores and significantly higher strength/activity scores than morphine. Twenty‐five of the 26 buprenorphine patients and 19 of the 26 morphine patients indicated that their global impression of change was 'moderately better' or 'considerable improvement'. Eleven buprenorphine patients and 16 morphine patients needed supplemental analgesia with tramadol, with seven and nine buprenorphine and morphine patients, respectively, needing 100 mg tramadol and the remainder needing 200 mg tramadol. The morphine patients had significantly higher rates of vertigo, constipation and nausea, but no differences were observed between the groups in rates of drowsiness, headache and confusion (see Table 3).
TD buprenorphine versus TD fentanyl
In a parallel‐group study lasting six weeks, Sarhan 2009 compared escalating doses of TD buprenorphine (N = 16) and TD fentanyl (N = 16). Sarhan 2009 reported that the only significant differences that this study revealed were that buprenorphine was associated with significantly higher rates of drowsiness and local skin complications compared to fentanyl. The mean pain scores, mean number of each category patch dose, treatment satisfaction, mean daily dose of diclofenac sodium, mean cost of the treatment and other side effects and complications did not differ significantly between the groups during the six weeks (see Table 3).
IM buprenorphine versus buprenorphine suppository
Dan 1989 is a parallel‐group study consisting of two consecutive treatments six to eight hours apart of the study drugs. Dan 1989 found that after the first administration of buprenorphine, the number of people who rated their pain intensity as "none" or "little" changed from 0 out of 34 patients at baseline to 23, 24 and 19 patients at 2, 4 and 6 hours in the rectal buprenorphine group and to 27, 30, and 28 (out of 35) patients, respectively, in the IM buprenorphine group. Before the second administration of the study drugs, 18 and 19 patients in the rectal and IM buprenorphine groups, respectively, described their pain intensity as "none" or "little". This changed to 26, 26 and 24 (out of 33) patients in the rectal buprenorphine group and to 22, 22, 22 (out of 28) patients in the IM buprenorphine group at 2, 4 and 6 hours after the second drug administration. Pain intensity ratings did not differ significantly between the groups at any point. Only one patient rated their pain intensity as "severe" after either the first (at two and six hours, and just before the second administration) or after the second (six hours) study drug administration (of IM buprenorphine). The overall ratings of pain relief at study end showed that most patients rated the drugs as "effective" (32 out of 34 rectal buprenorphine patients and 31 out of 34 IM buprenorphine patients) with the remaining patients rating the drugs as "minor response" and no patients giving a rating of "ineffective" with no statistically significant differences between the groups observed. While the severity of drowsiness, feeling heavy‐headed, sweating, thirst, urinary retention, euphoria and fatigue did not differ significantly between the groups, Dan 1989 found that the average severity of dizziness, nausea, vomiting, and adverse events as a total were all significantly higher in the IM group relatively to the suppository group (see Table 4).
3. Single study comparisons: Adverse events.
| AE | IM Bup versus Bup Sup | IM Bup versus IM Mor | IM Bup + SC Bup versus SC Bup versus placebo + SC Bup | Epi Bup versus Epi Mor | |||||
| Dan 1989a | Kjaer 1982 | Noda 1989 | Pasqualucci 1987 | ||||||
| IM Bup | Sup Bup | IM Bup | IM Mor | IM + SC Bup | SC Bup | Placebo + SC Bup | Epi Bup | Epi Mor | |
| Local toxicity/abnormal effect at injection/infusion site | 0/10 | 0/10 | 0/10 | ||||||
| Total AEs | 21.80 ± 3.67 | 11.41 ± 1.75 | 80 | 54 | |||||
| Anorexia/appetite loss | 9/31 | 8.5/32.5 | |||||||
| Anxiety | 1/26 | 0/26 | |||||||
| Blurred vision | 3/26 | 0/26 | |||||||
| Chest pain | 0/10 | 0/10 | 0/10 | ||||||
| Decreased memory | 1/26 | 2/26 | |||||||
| Deep respiration | 1/26 | 0/26 | |||||||
| Depression | 0/10 | 0/10 | 0/10 | ||||||
| Dizziness/confusion | 1.63 ± 0.53 | 0.24 ± 0.09 | 18/26 | 7/26 | 2/6 | 0/6 | |||
| Drowsiness/somnolence | 5.29 ± 0.8 | 3.44 ± 0.56 | 0/10 | 2/10 | 1/10 | 1/6 | 0/6 | ||
| Drunken feeling | 1/26 | 0/26 | |||||||
| Eruption | 0/10 | 0/10 | 0/10 | ||||||
| Euphoria | 2.09 ± 0.54 | 2.09 ± 0.56 | 5/26 | 5/26 | |||||
| Fatigue | 0.69 ± 0.43 | 0.26 ± 0.17 | 1/10 | 0/10 | 0/10 | ||||
| Hallucinations | 0/10 | 0/10 | 1/10 | ||||||
| Headache | 2/26 | 1/26 | |||||||
| Heavy head | 1.83 ± 0.53 | 0.91 ± 0.31 | |||||||
| Hypotension | 0/10 | 0/10 | 0/10 | ||||||
| Nausea | 2.89 ± 0.63 | 1.29 ± 0.43 | 11/26 | 4/26 | 0/10 | 0/10 | 2/10 | 3/6 | 1/6 |
| Numbness, hand and feet | 0/26 | 1/26 | |||||||
| Palpitation | 0/10 | 0/10 | 0/10 | ||||||
| Pruritus | 0/10 | 0/10 | 1/10 | 0/6b | 2/6b | ||||
| Remote feeling | 0/26 | 1/26 | |||||||
| Respiratory depression | 0/10 | 0/10 | 0/10 | ||||||
| Sedation | 14/26 | 18/26 | |||||||
| Sweating | 1.31 ± 0.47 | 0.79 ± 0.32 | 10/26 | 3/26 | |||||
| Thirst | 1.94 ± 0.53 | 0.71 ± 0.24 | 2/26 | 7/26 | |||||
| Urinary retention | 1.94 ± 0.72 | 0.91 ± 0.41 | 0/10 | 0/10 | 0/10 | ||||
| Vertigo | 0/10 | 0/10 | 3/10 | ||||||
| Vomiting | 2.20 ± 0.53 | 0.68 ± 0.21 | 11/26 | 5/26 | 0/10 | 0/10 | 1/10 | 2/6 | 1/6 |
| Discontinuation due to AE | 7/35 | 1/34 | |||||||
Abbreviations: Bup = buprenorphine; Fen = fentanyl; IM = intramuscular; Epi = epidural; Mor = morphine; SC = subcutaneous; SD = subdermal; SL = sublingual; Sup = suppository; Tab = tablets; Til+Na = tilidin + naloxone; Tra = tramadol. aAverage. bOf the face.
IM buprenorphine versus IM morphine
In a crossover study lasting a total of eight days Rigolot 1979 compared IM buprenorphine to IM morphine in 10 patients. Rigolot 1979 reported that, when analysed separately, both morphine and buprenorphine are associated with significantly lower pain intensities at hours one to five compared to baseline. When compared directly, no significant differences were observed between the two treatments in analgesic efficacy at any of the measured times. Rigolot 1979 did not report adverse events.
Kjaer 1982 conducted a cross‐over study with 27 patients comparing single IM doses of buprenorphine and morphine, and found no differences in 'maximum pain intensity difference' between the groups. However the 'total pain relief scores' were significantly greater after buprenorphine treatment than morphine treatment, and the time to re‐medication was significantly longer after buprenorphine (mean = 10 hours) than after morphine (mean = eight hours) treatment. There were no differences between the treatments in severity, onset and duration of euphoria, sweating, blurred vision, thirst, sedation, deep respiration, decreased memory, numbness of hands and feet, headache, anxiety, feeling intoxicated and feeling remote. However, dizziness, nausea and vomiting were more severe, had earlier onset and longer duration after treatment with buprenorphine compared to morphine (see Table 4).
IM buprenorphine + SC buprenorphine versus SC buprenorphine versus placebo + SC buprenorphine
In a parallel‐group trial lasting 48 hours, Noda 1989 compared SC buprenorphine (4 μg/kg/day) preceded by an IM injection of buprenorphine (0.004 μg/kg; N = 10) to SC buprenorphine (4 μg/kg/day, N = 10) and to SC buprenorphine (8 μg/kg/day) preceded by placebo infusion (N = 10). However, Noda 1989 unfortunately did not report the results clearly and inferentially analysed between the treatment groups. Descriptively, it appears that pain intensity was lower, and comparable, in the groups receiving the higher dose of SC buprenorphine or buprenorphine preceded by IM buprenorphine compared to the group that received 4 μg/kg/day SC buprenorphine (see Table 4 for the reported adverse events).
Epidural buprenorphine versus epidural morphine
Pasqualucci 1987 compared single epidural doses of buprenorphine and morphine in a parallel‐group trial with six patients in each arm. This trial found no differences in pain scores after treatment with either buprenorphine or morphine (see Table 4 for the reported adverse events).
Intravenous buprenorphine versus intravenous morphine
In a parallel‐group study with treatment lasting 36 hours, Wang 2012 examined pain scores as assessed by a visual assessment scale for patients who were treated with IV buprenorphine and who had P‐gp+ (P‐glycoprotein; group B1; N = 30) and P‐gp‐ (group B2; N = 30) tumours and in patients who had received IV morphine and who had P‐gp+ (group M1; N = 30) and P‐gp‐ (group M2; N = 30) tumours. Wang 2012 reported that the VAS scores were similar between the four groups at baseline (means (SDs): B1 = 7.8 (1.6); B2 = 7.9 (1.2); M1 = 8.0 (1.5); M2 = 8.1 (1.7)), but then only included analyses between the groups that received the same drug. Wang 2012 reported that the VAS scores of groups B1 and B2 all differed significantly from baseline, but did not differ significantly from each other at four hours (means (SDs): B1 = 1.5 (0.9); B2 = 1.6 (0.8)), 12 hours (means (SDs): B1 = 1.6 (0.7); B2 = 1.5 (1)), 24 hours (means (SDs): B1 = 1.4 (0.7); B2 = 1.4 (0.9)) or 36 hours (means (SDs): B1 = 1.5 (1); B2 = 1.4 (1.1)), whereas for morphine the pattern of results was different. The VAS scores for both groups M1 and M2 were significantly lower than baseline at all times, but the pain scores were higher for group M1 compared to group M2 at four hours (means (SDs): M1 = 4.1 (2.4); M2 = 1.7 (1.1)), 12 hours (means (SDs): M1 = 4.4 (1.9); M2 = 1.8 (1.6)), 24 hours (means (SDs): M1 = 4.3 (1.6); M2 = 1.9 (1.4)) and 36 hours (means (SDs): M1 = 4.3 (2.3); M2 = 1.8 (1.4)). Groups M1 and M2 had received an identical dose of morphine (0.75 mg/kg). A second set of analyses comparing group M2 to group M1 after M1 had received a higher dose of morphine (1.1 mg/kg) showed that the increased dose of morphine brought down the pain scores of group M1 to levels that were comparable to those of group M2 at all the times (means (SDs) for group M1 new: 8 (1.7) at baseline; 1.8 (1.4) at four hours; 1.8 (1.9) at 12 hours; 1.7 (1.6) at 24 hours; 1.9 (1.8) at 36 hours), indicating that patients with P‐gp+ tumours are less sensitive to the analgesic effect of morphine than patients with P‐gp‐ tumours. Wang 2012 did not report on adverse events.
Discussion
Summary of main results
In this review we identified 19 relevant studies that included a total of 1421 patients and examined 16 different intervention comparisons. A number of these studies that performed comparative analyses between the randomised groups found that buprenorphine was superior in terms of pain relief/pain intensity reduction to the comparison treatment (Bauer 1985, SL buprenorphine versus oral tilidine with naloxone; De Conno 1987, SL buprenorphine versus oral pentazocine (significantly more drowsiness with buprenorphine, but significantly more dizziness and stomach pain with pentazocine); Dini 1986, buprenorphine tablets (SL) and vials versus pentazocine tablets and vials; Pace 2007; TD buprenorphine versus controlled‐release morphine; Kjaer 1982, IM buprenorphine versus IM morphine (although dizziness, nausea and vomiting were more severe, had earlier onset and longer duration after treatment with buprenorphine compared to morphine)), while three studies found no differences between buprenorphine and the comparison drug (Pasqualucci 1987; epidural buprenorphine versus epidural morphine; Rigolot 1979, IM buprenorphine versus IM morphine; Yajnik 1992, SL buprenorphine versus SL buprenorphine + oral phenytoin versus phenytoin), and yet other studies found treatment with buprenorphine to be inferior to the alternative treatment in terms of the side effects profile (Bono 1997, SL buprenorphine versus oral tramadol; Sarhan 2009, TD buprenorphine versus TD fentanyl) or patient preference/ratings of acceptability (Brema 1996, SL buprenorphine versus slow‐release tramadol).
Of the studies that compared different doses or formulations/routes of administration of buprenorphine, pain intensity ratings did not differ significantly between IM buprenorphine and buprenorphine suppository. However, the average severity of dizziness, nausea, vomiting, and adverse events as a total were all significantly higher in the IM group relatively to the suppository group (Dan 1989). SL buprenorphine was found to be associated with faster onset of pain relief compared to SD buprenorphine, with similar duration analgesia and no significant differences in adverse event rates reported between the treatments (Limón Cano 1994). In terms of TD buprenorphine, two studies found that it was superior to placebo (Sittl 2003, placebo versus TD buprenorphine at 35 μg/h, 52.5 μg/h, 70 μg/h; Poulain 2008, placebo versus 70 μg/h TD buprenorphine), whereas a third study found no difference between placebo and different doses of TD buprenorphine (Böhme 2003 placebo versus TD buprenorphine at 35 μg/h, 52.5 μg/h, 70 μg/h). Finally, the studies that examined different doses of TD buprenorphine did also not report a clear dose‐response relationship (see also Table 1).
Overall completeness and applicability of evidence
The included 19 studies reported on 16 different comparisons, and there were only three comparisons with data from more than one study. More specifically, these comparisons had data from two or three studies, of all them comparing placebo to TD buprenorphine at different doses. However even in this group of four studies, two studies (Böhme 2003; Sittl 2003) included patients with pain of both malignant and non‐malignant origin and did not report the results separately for these two pain populations. The results of Böhme 2003 and Sittl 2003 are therefore only applicable to the current review to the extent that TD buprenorphine at the studied doses and placebo have similar analgesic and safety profiles across these two populations. Across all the studies the total number of included patients ranged from 10 to 189, and all treatment groups included less than 100 patients. The evidence base for the effectiveness of buprenorphine in patients with cancer pain may be considered to span a wide range of potential treatment options, but at the expense of depth provided by large numbers of patients treated within each comparison and replication of any effects observed.
It should also be noted that not all the routes described are in common use in palliative and cancer care today. For instance, a number of trials assessed the IM route, which is now less commonly utilised due to injections causing pain, and this route has been replaced by the SC route more commonly. However, in general trial data including, for example, the TD and SL route is applicable, as these are current acceptable and practical modes of delivery.
Quality of the evidence
It is possible that the quality of the evidence is higher than it appears. This is because the evidence base generally suffers from a large number of unreported details that may or may not indicate a high risk of bias, had these methodological details been reported. It is therefore very difficult to assess the extent that the results are subject to different biases. For example, as inspection of Figure 2 indicates, only two of the included 19 trials clearly used an appropriate randomisation sequence. We could not establish in any of the studies that there had definitely been concealed allocation to the treatment groups. On the other hand, we could only rate one study at high risk of bias on the items dealing with selection bias based on the reported details, which consequently leaves a large amount of uncertainty for the remaining (vast majority of) studies.
With data from 91.8% of the total number of enrolled/randomised patients analysed for pain, and from 85.6% of the patients analysed for safety, the results of this review are at moderate to high risk of attrition bias. The observed attrition did not appear to be selective over and beyond the results we have already reported of any differences in 'discontinuation of treatment rates' due to adverse events. It is therefore conceivable that this bias exerts an equal effect on the studied treatment comparisons.
Both the manner and the extent of outcome reporting observed in the included studies also lead us to conclude that the results are at some risk of reporting bias, with some studies not reporting all expected outcomes and others reporting the outcomes in a format that makes it impossible (or very difficult) to include them in any data syntheses. This bias risk is perhaps not fully realised in this Cochrane review due to the large number of different comparisons that have only been considered by single studies (that are therefore not meta‐analysed). However future updates of this review may include further studies examining the same comparisons and will therefore be subject to a higher risk of reporting bias if all relevant studies cannot be included in any resultant meta‐analyses.
We also cannot exclude the possibility that our results are at some (unknown) risk of publication bias, given the fact that most included studies did not report null results (see also Potential biases in the review process). We did include abstracts and perform a comprehensive search of grey literature in order to minimise the risk of publication bias in our review.
Finally, we note that some included studies had very small sample sizes and are unlikely to be powered to reliably detect any potential real differences between the compared treatments. This may also result in spurious findings that we are wary to treat as true differences.
Potential biases in the review process
This Cochrane review included a number of studies that were published in languages other than English. Although we did not exclude any study for this reason, only one review author/translator extracted data and appraised these studies rather than two review authors, as is the preferred standard for included studies. This practice may have introduced author‐specific bias into the process. However, as already outlined above, in general the included studies were severely compromised by under‐reporting with 'unclear' judgements given in 125 (a total which excludes the 15 'unclear' rating of the item that only applied to the four cross‐over trials) out of 228 bias assessments. It is unlikely that these ratings (of which there are more in the non‐English language papers and in the abstract) would change by double‐reviewing as they reflect a clear absence of information reported, and therefore the least judgment of all the bias assessment ratings. Another potential source of bias comes from the screening of search results process. We identified one included study by chance very late in the review process (Noda 1989). Both review authors who screened the search results missed this study because the title and abstract gave no (or very little) indication that it was a RCT assessing three different buprenorphine treatment strategies. Although we have checked other systematic reviews on buprenorphine for relevant studies, its possible that we have missed more relevant studies for this same reason.
Agreements and disagreements with other studies or reviews
Given the absence of meta‐analyses and the resultant narrative summaries of the results of the included studies, it is perhaps unsurprising that they tend to be in agreement in general with other systematic reviews conducted on different but overlapping questions (e.g., Deandrea 2009 examining the role of TD buprenorphine in managing severe cancer pain; Tassinari 2008 examining the adverse effects of TD opioids compared to long‐acting morphine for moderate‐severe cancer pain; Tassinari 2011 examining the use of TD opioids as front‐line treatment for moderate‐severe cancer pain; Wolff 2012 examining the adverse events of TD buprenorphine and fentanyl for chronic moderate‐severe pain). This is because their conclusions regarding buprenorphine are based on equally limited evidence.
Authors' conclusions
Implications for practice.
In clinical practice, morphine is accepted as the first‐line strong opioid of choice for the relief of cancer pain; other opioid analgesics such as oxycodone and fentanyl are considered as second‐line options (NICE 2012). Side‐effects can mean that one opioid may need to be substituted with another opioid, so having a wider ranging choice is practical. Furthermore, having a choice with regard to route of administration can be of great importance in some patients, who, for example, are unable to swallow. Data on the efficacy of buprenorphine compared to other opioids in this Cochrane review was of varying, mostly low quality. However, the available data demonstrate that it is an effective pain reliever compared to comparison analgesics in the studies that were analysed. It performed less well in terms of side‐effect profile when compared to other analgesics, such as tramadol.
The results provide the treating clinician with limited data on the efficacy of different routes of administration for buprenorphine. Having said that, in those settings where buprenorphine is seen to be an acceptable drug for treating cancer pain, a clinician may decide that the type of pain may suit one delivery mode more than another. For instance, a patient with pain specifically on movement due to bone metastases may find it useful to have access to SL buprenorphine prior to movement. Also, its quicker action would make it a suitable medication to take, compared to having an injection. It would also mean that the patient could use this analgesic strategy at home, whereas the injectable route would be more dependent on being delivered in a healthcare setting such as a hospital ward or hospice.
Two studies found TD buprenorphine to be superior to placebo (placebo versus TD buprenorphine at 35 μg/h, 52.5 μg/h, 70 μg/h; placebo versus 70 μg/h TD buprenorphine). However, a third study found no difference between placebo and different doses of TD buprenorphine (placebo versus TD buprenorphine at 35 μg/h, 52.5 μg/h, 70 μg/h). Studies examining different doses of TD buprenorphine also did not report a clear dose‐response relationship, making recommendations on starting doses very difficult for the TD route. It also makes guidance on the titration of TD buprenorphine difficult, in so far as it would have to be initiated at the lowest dose, very gradually uptitrated and then have varying, unpredictable efficacy, whereas the response from another analgesics may be more easy to predict and control. TD buprenorphine therefore becomes a less attractive choice for a prescriber who wants to resolve cancer pain swiftly and efficiently.
In summary, clinicians who are faced with a choice of analgesics to consider for cancer pain should use morphine as a first‐line on the grounds of price, at least until inferiority of morphine has been established (NICE 2012). This Cochrane review did not find sufficient evidence to make buprenorphine a valid first‐line choice alongside standard therapies like morphine, oxycodone and fentanyl. However it has a place as an analgesic and its different routes of administration may make it a practical option for limited types of cancer pain, for limited numbers of patient in limited types of clinical setting. Where its place is exactly, is still hard to say. It seems reasonable to suggest that it might be considered to rank as a fourth‐line option compared to the more standard therapies like morphine, oxycodone and fentanyl, and even there it would only be suitable for some patients. Having said that, palliative care patients are often heterogeneous and complex, so having a number of analgesics available that can be given differently increases patient and prescriber choice. In particular, the SL and injectable routes seemed to have a more definable analgesic effect, whereas the TD route studies left more questions than they resolved.
Implications for research.
Overall, the evidence was of poor quality. In a large number of studies it was unclear whether an appropriate randomisation sequence had been used. Moreover, we could not establish in any of the included studies whether there had definitely been concealed allocation to the treatment groups. Any future research studies should take this into account. Heterogeneity of methods and outcome reporting was a further problem, which makes it very difficult to apply the current body of evidence to clinical and research settings through further meta‐analytic and summative analyses. There is a need to establish efficacy and safety of buprenorphine in its various formulations and routes, and its dose‐response relationship needs to be analysed further and compared to standard first‐line therapies, such as morphine sulphate, in adequately powered, well‐designed studies of sufficient duration in the setting of cancer pain.
What's new
| Date | Event | Description |
|---|---|---|
| 18 December 2018 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 1, 2012 Review first published: Issue 3, 2015
| Date | Event | Description |
|---|---|---|
| 4 September 2018 | Amended | Corrected reporting of total number of excluded studies. |
| 24 August 2018 | Amended | Typo removed in Abstract. |
| 25 June 2018 | Amended | Updated Other published versions of this review |
| 10 November 2016 | Amended | Contact details updated. |
| 17 October 2016 | Review declared as stable | See Published notes. |
| 16 June 2015 | Amended | Minor changes made to wording in Abstract. |
| 17 May 2013 | New citation required and major changes | New citation: major change. This protocol has been significantly updated by new authors. See Published notes. |
Notes
At December 2018, we found one potentially relevant study (Nosek 2017) but we were unable to assess whether it was likely to change the conclusions due to inadequate reporting of the data (relevant data only reported collapsed across all intervention groups). We have contacted the study authors to request the data, but at the time of publishing we have not received a response. We have now stabilised this review following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is made available to us or published, or if standards change substantially which necessitate major revisions.
Nosek, K., et al (2017). "A comparison of oral controlled‐release morphine and oxycodone with transdermal formulations of buprenorphine and fentanyl in the treatment of severe pain in cancer patients." Drug Design Development and Therapy 11: 2409‐19.
Acknowledgements
This article presents an extension to a systematic review undertaken as part of the 2011 NICE guideline on "Opioids in palliative care" (NICE 2012), which was developed by the National Collaborating Centre for Cancer (NCC‐C). The NCC‐C receives funding from NICE. We thank Yuhong Yuan (Department of Medicine, Division of Gastroenterology, McMaster University, Canada) for help with translation of Chinese papers, Prof Vasiliy V Vlassov (PO Box 13, 109451 Moscow, Russian Federation) for help with the translation of a Russian paper, Dr Erika Ota (Chief, Department of Health Policy, National Center for Child Health and Development, 2‐10‐1 Okura, Setagaya‐ku, 157‐8535 Tokyo, Japan) and Dr Maki Kawasaki (Japanese Branch of the Australasian Cochrane Centre) for help with the translation of Japanese papers, Dr Valeria Martinez (MD PhD, Praticien hospitalier, Anesthésiste‐Algologue, Groupe Hospitalier Raymond Poincaré, 104, Bd Raymond Poincaré, 92380 Garches France) for help with the translation of a French paper, and Joanne Abbot at Cochrane PaPaS Group for her assistance in devising and executing the search strategy. We also thank Phil Wiffen and Scott Strassels for helpful comments on an earlier version of the protocol.
We acknowledge Cho‐Min Naing (Division of Community Medicine, International Medical University, Kuala Lumpur, Malaysia), Kyan Aung (School of Medical Science, International Medical University, Kuala Lumpur, Malaysia) and Peng Nam Yeoh (International Medical University, Kuala Lumpur, Malaysia) for their contributions to a previous version of this review.
Cochrane Review Group funding acknowledgement: the National Institute for Health Research (NIHR) is the largest single funder of the Cochrane PaPaS Group.
Disclaimer: the views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the NIHR, National Health Service (NHS) or the Department of Health.
We acknowledge the support of James Casley Grose in assessing the September 2016 search results.
Appendices
Appendix 1. Search strategies
CENTRAL
#1 MeSH descriptor: [Buprenorphine] this term only
#2 buprenorphine:ti,ab,kw (Word variations have been searched)
#3 (magnogen or temgesic or subutex or transtec or anorfin or bupren or norphin or pentorel or tidigesic or nopan or finibron or brospina or temgesic‐nX or buprex or prefin or suboxone or buprenex or buprine or butrans):ti,ab,kw (Word variations have been searched)
#4 #1 or #2 or #3
#5 MeSH descriptor: [Pain] explode all trees
#6 (pain* or nocicept* or neuropath*):ti,ab,kw (Word variations have been searched)
#7 #5 or #6
#8 MeSH descriptor: [Neoplasms] explode all trees
#9 (cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*):ti,ab,kw (Word variations have been searched)
#10 #8 or #9
#11 #4 and #7 and #10
MEDLINE
1. Buprenorphine/
2. buprenorphine.tw.
3. (magnogen or temgesic or subutex or transtec or anorfin or bupren or norphin or pentorel or tidigesic or nopan or finibron or brospina or temgesic‐nX or buprex or prefin or suboxone or buprenex or buprine or butrans).tw.
4. or/1‐3
5. exp Pain/
6. (pain* or nocicept* or neuropath*).tw.
7. 5 or 6
8. exp Neoplasms/
9. (cancer$ or neoplas$ or tumo$ or carcinoma$ or hodgkin$ or nonhodgkin$ or adenocarcinoma$ or leuk?emia$1 or metasta$ or malignan$ or lymphoma$ or sarcoma$ or melanoma$ or myeloma$ or oncolog$).tw.
10. 8 or 9
11. 4 and 7 and 10
12 randomized controlled trial.pt.
13 controlled clinical trial.pt.
14 randomized.ab.
15 placebo.ab.
16 drug therapy.fs.
17 randomly.ab.
18 trial.ab.
19 or/12‐18
20 exp animals/ not humans.sh.
21 19 not 20
22 11 and 21
EMBASE
1. Buprenorphine/
2. buprenorphine.tw.
3. (magnogen or temgesic or subutex or transtec or anorfin or bupren or norphin or pentorel or tidigesic or nopan or finibron or brospina or temgesic‐nX or buprex or prefin or suboxone or buprenex or buprine or butrans).tw.
4. or/1‐3
5. exp Pain/
6. (pain* or nocicept* or neuropath*).tw.
7. 5 or 6
8. exp Neoplasms/
9. (cancer$ or neoplas$ or tumo$ or carcinoma$ or hodgkin$ or nonhodgkin$ or adenocarcinoma$ or leuk?emia$1 or metasta$ or malignan$ or lymphoma$ or sarcoma$ or melanoma$ or myeloma$ or oncolog$).tw.
10. 8 or 9
11. 4 and 7 and 10
12. random$.tw.
13. factorial$.tw.
14. crossover$.tw.
15. cross over$.tw.
16. cross‐over$.tw.
17. placebo$.tw.
18. (doubl$ adj blind$).tw.
19. (singl$ adj blind$).tw.
20. assign$.tw.
21. allocat$.tw.
22. volunteer$.tw.
23. Crossover Procedure/
24. double‐blind procedure.tw.
25. Randomized Controlled Trial/
26. Single Blind Procedure/
27. or/12‐26
28. (animal/ or nonhuman/) not human/
29. 27 not 28
30. 11 and 29
BIOSIS & Web of Science
# 13 #12 AND #6
# 12 #11 AND #10
# 11 TOPIC: (((human*)))
# 10 #9 OR #8 OR #7
# 9 TOPIC: ((((singl* OR doubl* OR trebl* OR tripl*) SAME (blind* OR mask*))))
# 8 TOPIC: (((controlled clinical trial OR controlled trial OR clinical trial OR placebo)))
# 7 TOPIC: (((randomised OR randomized OR randomly OR random order OR random sequence OR random allocation OR randomly allocated OR at random OR randomized controlled trial)))
# 6 #5 AND #4 AND #3
# 5 TOPIC: ((cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*))
#4 TOPIC: ((pain* or nocicept* or neuropath*))
#3 #2 OR #1
# 2 TOPIC: ((magnogen or temgesic or subutex or transtec or anorfin or bupren or norphin or pentorel or tidigesic or nopan or finibron or brospina or temgesic‐nX or buprex or prefin or suboxone or buprenex or buprine or butrans))
# 1 TOPIC: (buprenorphine)
Trial registers
ClinicalTrials.gov, WHO ICTRP, Current Controlled Trials (inc mRCT), and Proceedings of the Congress of the European Federation of International Association for the Study of Pain (IASP) (via European Journal of Pain Supplements):
(magnogen or temgesic or subutex or transtec or anorfin or bupren or norphin or pentorel or tidigesic or nopan or finibron or brospina or temgesic‐nX or buprex or prefin or suboxone or buprenex or buprine or butrans)
AND
(cancer* or neoplas* or tumo* or carcinoma* or hodgkin* or nonhodgkin* or adenocarcinoma* or leuk?emia* or metasta* or malignan* or lymphoma* or sarcoma* or melanoma* or myeloma* or oncolog*)
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bauer 1985.
| Methods |
Design: Randomised, parallel group trial Year: Not reported Country: Germany |
|
| Participants |
Patients: 40 patients were randomised to one of the following groups:
Inclusion criteria: "40 female patients were treated for severe cancer pain at the women's clinic at Heidelberg University." Exclusion criteria: Patients with severe liver or kidney damage, restriction of breathing or respiratory regulation, increased intracranial pressure, or allergy to tilidin‐HCI with naloxone‐HCl or buprenorphine. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study states it is randomised, but gives no further details. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No details reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No details reported. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No details reported. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No details reported. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from all 40 patients appear to be included. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Data not reported in a manner where this can be ascertained. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes are not well‐reported. |
| Other bias | Unclear risk | It is unclear due to limited reporting whether the study is subject to other bias(es). |
| Were the patients adequately titrated? | Unclear risk | No details reported. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Bono 1997.
| Methods |
Design: Randomised cross‐over trial Year: Not reported Country: Italy |
|
| Participants |
Patients: 60 patients (with the following types of cancer: lung (N = 9), urological (N = 11), gynaecological (N = 3), blood‐based (N = 2), ENT (N = 14), thoracic/oesophageal (N = 8), skin (N = 1), gastrointestinal (N = 12)) were randomised to one of the following 2 groups (order of treatment):
Inclusion criteria: "Sixty adults presenting with advanced tumours no longer responsive to NSAIDs were included". Exclusion criteria: Uncooperative patients, those with known intolerance to the test drugs, with renal, respiratory or hepatic failure, associated chronic pathology, and women in pregnancy or breast‐feeding. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
The study was published in French and lay translated by MSH. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported apart from that it is a randomised study. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | High risk | Nine and 23 of the 60 patients discontinued treatment during treatment with tramadol and buprenorphine, respectively. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data are reported for all included patients. |
| Selective reporting (reporting bias) | Low risk | All major outcomes are reported. |
| Other bias | Unclear risk | It is unclear if the study is subject to high risk of other biases. |
| Were the patients adequately titrated? | Unclear risk | It is unclear whether the patients were adequately titrated based on the available information. |
| For cross‐over trials: Are data available for both time periods? | High risk | The pain intensity data did not appear to be inferentially analysed collapsed over phases for (any of) the study days, apart from for the first 4 hours of treatment, where no differences were observed between the treatments. |
Brema 1996.
| Methods |
Design: Randomised parallel group trial Year: Not reported Country: Italy |
|
| Participants |
Patients: 131 patients were randomised to one of the following 2 groups:
Inclusion criteria: "Adults with [neoplastic] pain no longer responsive to regular treatment with non‐steroidal antiinflammatory drugs (NSAIDs) were admitted." Exclusion criteria: "Uncooperative patients, those with known intolerance to the test drugs, with renal, respiratory or hepatic failure, taking morphine, major analgesics or monoamine oxidase inhibitors, and women in pregnancy or breast‐feeding, were considered ineligible." |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "On the basis of the randomization list, each patient was assigned to treatment". No further information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from all patients appear to be included. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data from all patients appear to be included. |
| Selective reporting (reporting bias) | Low risk | The main outcomes are reported. |
| Other bias | Unclear risk | It is unclear whether the study is at high risk of other bias(es). |
| Were the patients adequately titrated? | Unclear risk | No information is reported. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Böhme 2003.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: Not reported Country: Austria, Germany, and Hungary |
|
| Participants |
Patients: 189 patients entered the run‐in phase, of these 38 were excluded and 151 patients were randomised to one of the following 4 groups:
90 to 93% of the patients in the treatment arms had been prescribed strong opioids (WHO step 3), 0 to 8% in each treatment arm used weak opioids (WHO step 2). Inclusion criteria: "The main inclusion criteria for the double‐blind phase was chronic pain that was at least satisfactorily relieved (according to a verbal rating scale) with 0.8‐1.2 mg/day sublingual buprenorphine after a 5‐day run‐in phase." Exclusion criteria: "Exclusion criteria were alcohol or drug abuse, hypersensitivity towards opioids, compromised respiratory function, a history of convulsions, raised intracranial pressure, and previous extensive damage to the dermis in the patch application area (subclavicular chest or upper back regions). Also excluded were patients receiving local radionucleotide therapy, opioids other than sublingual buprenorphine, or MAO‐inhibitors." |
|
| Interventions |
Buprenorphine arms (3):
Comparison arm:
Apart from study drug everything else was similar to the buprenorphine arms. |
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | Study described as double‐blind. Unclear who is blinded. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above. Pain is patient reported. Probably reasonable to assume that patients were blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | Study described as double‐blind. Unclear who is blinded and whether this outcome is assessed by healthcare professionals also. |
| Incomplete outcome data (attrition bias) Pain | Low risk | The analyses appear to include 149/151 patients. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The analyses appear to include 151/151 patients. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appear to be reported. |
| Other bias | Low risk | No other obvious biases were observed. |
| Were the patients adequately titrated? | Low risk | Probably. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Dan 1989.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: July 1987 to March 1988 Country: Japan |
|
| Participants |
Patients: 73 patients were randomised to one of the following 2 groups:
Inclusion criteria: "Patient who have a cancer pain and pain intensity is 2 (mild) or 3 (moderate)". Exclusion criteria: None reported. |
|
| Interventions |
Buprenorphine suppository:
Buprenorphine injection:
|
|
| Outcomes |
|
|
| Notes |
This study is only published in Japanese, and was kindly translated by Dr. Maki Kawasaki, Japanese Branch of the Australasian Cochrane Centre. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about randomisation sequence to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about allocation to make a judgement. |
| Blinding of participants and personnel (performance bias) Pain | Low risk | Double‐blinded and placebo looks identical to the actual drug. |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | Double‐blinded and placebo looks identical to the actual drug. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Assessed by patients and they were blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | Assessed by healthcare professional and they were blinded. |
| Incomplete outcome data (attrition bias) Pain | High risk | Data reported for 28/35 IM patients and 33/34 rectal buprenorphine patients after the second administration of the study drug. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Most of the adverse events appear to be reported for 34/34 rectal buprenorphine patients and for 35/35 IM buprenorphine patients. |
| Selective reporting (reporting bias) | Low risk | All the main expected outcomes are reported. |
| Other bias | Unclear risk | It is unclear whether the study is at risk of other bias(es) |
| Were the patients adequately titrated? | Unclear risk | It is unclear, but probably not. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
De Conno 1987.
| Methods |
Design: Randomised cross‐over trial Year: Not reported Country: Italy |
|
| Participants |
Patients: 120 patients were randomised and 29 did not complete treatment (10/29 patients suspended both treatments, 16/29 patients suspended pentazocine and 3/29 suspended buprenorphine): No patient characteristics are reported. Inclusion criteria: Patients with cancer pain of moderate to severe intensity aged > 18 years who have given informed consent. Exclusion criteria: Severe renal or hepatic impairment, severe respiratory failure, chronic treatment with high doses of agonist analgesics, increase in intracranial pressure, pregnancy. |
|
| Interventions |
Buprenorphine:
Pentazocine:
|
|
| Outcomes |
|
|
| Notes |
This study is published in two papers, the earlier paper is in English and includes a smaller sample than the later paper, which is published in Italian and lay‐translated by MSH. The data from this newer paper which reports the larger sample have been included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | No information reported. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | No information reported. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | No information reported. |
| Incomplete outcome data (attrition bias) Pain | High risk | Data only available for 91/120 randomised patients. |
| Incomplete outcome data (attrition bias) Adverse events | High risk | See cell above. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes are not well‐reported. |
| Other bias | Unclear risk | It is unclear whether the study is at high risk of other biases. |
| Were the patients adequately titrated? | High risk | The patients were not titrated at the beginning of the actual study. |
| For cross‐over trials: Are data available for both time periods? | Low risk | Yes, for a number of participants. |
Dini 1986.
| Methods |
Design: Randomised parallel‐group trial Year: Not reported Country: Italy |
|
| Participants |
Patients: Patients divided into two group: Group Tablet and group Vial (fluid). Within each of these groups patients were allocated to treatment with either buprenorphine or pentazocine:
Inclusion criteria: 42 patients aged > 18 years with pain of moderate‐severe intensity of neoplastic origin was selected by/from The Center for Pain Management at the National Institute of Cancer Research. Exclusion criteria: Severe renal or hepatic impairment, severe respiratory failure, previous treatment with agonist analgesics, intracranial hypertension, mental confusion, pregnancy/lactation. |
|
| Interventions |
Buprenorphine tablets:
Buprenorphine vials/fluid:
Pentazocine tablets:
Pentazocine vials/fluid:
|
|
| Outcomes |
|
|
| Notes |
This study is published in Italian and lay‐translated by MSH. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information other than that the patients were randomised is reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | It is unclear whether any data are missing. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes appear to be reported. |
| Other bias | Unclear risk | It is unclear whether the study is subject to high risk of other types of bias. |
| Were the patients adequately titrated? | Unclear risk | No information reported. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Kjaer 1982.
| Methods |
Design: Randomised, double‐blind, cross‐over trial Year: Not reported Country: Denmark |
|
| Participants |
Patients: 27 patients were randomised, 26 patients completed the study and were included in the safety analyses (1 patient withdrew to receive treatment for bone metastases), and 25 patients were included in the efficacy analyses (1 patient was excluded due to being remedicated by mistake): 13 males/14 females; mean age (range) = 60 (41 to 71) years; Type of cancer: lung (N = 12), breast (N = 7), female genital system (N = 4), head and neck (N = 3), oesophagus (N = 1). 13/27 received buprenorphine first and 14/27 received morphine first. Inclusion criteria: "None of the patients had previously received regular doses of narcotics. The basis for selection of patients to the study was persistent pain where aspirin, dextro‐propoxyphene or paracetamol were no longer effective in controlling the pain. All patients gave informed consent to participate in the study and agreed to at least 3 full days in hospital." Exclusion criteria: "Patients with severe renal damage (serum creatinine ≥ 120 μmol/L), and severe hepatic damage (serum bilirubin ≥ 17 μmol/L, plasma aspartate aminotransferase ≥ 50 U/L, plasma alkaline phosphatase ≥ 275 U/L, plasma lactate dehydrogenase ≥ 450 U/L) were not included in the study. Neither were patients with marked ventilatory impairment or persistent mental confusion." |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Pain | Low risk | Study described as double‐blind. "Individual treatments were supplied in identical coded ampoules (1 ml) containing either buprenorphine (0.3 mg) or morphine (10 mg). Each treatment pack consisted of two ampoules labelled A and B which were administered in alphabetical order. The order of treatments were randomized both within and between patients." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | The analyses included 25/27 patients for efficacy. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The analyses included 26/27 patients for safety. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes are reported, although not in a manner that allow their inclusion in a meta‐analysis. |
| Other bias | Unclear risk | It is unclear whether the study is subject to other types of bias. |
| Were the patients adequately titrated? | High risk | There was no titration. |
| For cross‐over trials: Are data available for both time periods? | Low risk | The data appear to be available for both periods. |
Limón Cano 1994.
| Methods |
Design: Randomised, double‐blind, parallel‐group trial Year: Not reported Country: Mexico |
|
| Participants |
Patients: 17 patients (11 males/6 females) were randomised to one of the following two groups:
Inclusion criteria: 17 patients with moderate to severe cancer pain that had not responded to treatment with non‐opioid and adjuvant analgesics according to the WHO analgesic ladder. Exclusion criteria: Patients with liver damage, renal or severe cardiorespiratory problems. |
|
| Interventions |
SL buprenorphine arm:
SD buprenorphine arm:
|
|
| Outcomes | "The analgesic response (decrease in pain intensity according to visual analogue scale), latency and duration of analgesia, side effects and difficulties to appreciate the procedure [patient preference?]." | |
| Notes |
Study published in Spanish and lay‐translated by MSH. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The allocation to each treatment group: SL and SD was randomised and the study was double blind. No further information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | Patients and doctors blinded. Blinding "kept closed until the end of the study". Unclear how blinding was achieved. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data appear to be available for all included patients. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | See cell above |
| Selective reporting (reporting bias) | Unclear risk | The outcomes are not well‐reported. |
| Other bias | Unclear risk | It is unclear whether the study is at high risk of other biases. |
| Were the patients adequately titrated? | High risk | The patients were not titrated. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | Not applicable. |
Noda 1989.
| Methods |
Design: Randomised, parallel group trial Year: Not reported Country: Japan |
|
| Participants |
Patients: 30 patients randomised to one of the following groups:
Inclusion criteria: "This study was carried out in patients admitted to Kyoto University Hospital. Informed verbal consent to participate in this study was obtained from all patients, but information regarding what and how much drug was to be administered was not provided to them. Thirty patients included had not undergone any opioid administration for at least 1 week prior to the study." "The study was started when the patients scored their level of pain at VAS 10." Exclusion criteria: No information reported. |
|
| Interventions |
Buprenorphine arm (1):
Buprenorphine arm (2):
Placebo:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomly assigned..." No further information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | "Informed verbal consent to participate in this study was obtained from all patients, but information regarding what and how much drug was to be administered was not provided to them." No further information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | Three patients failed to complete their assigned treatment, and it is unclear if these patients were included in analysis. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Three patients failed to complete their assigned treatment, and it is unclear if these patients were included in analysis. |
| Selective reporting (reporting bias) | Low risk | The expected outcomes are reported. |
| Other bias | Unclear risk | It is unclear whether the study is subject to high risk of other types of bias. |
| Were the patients adequately titrated? | High risk | The patients were not titrated. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Pace 2007.
| Methods |
Design: Randomised, open‐label, parallel group trial Year: Not reported Country: Italy |
|
| Participants |
Patients: 52 patients randomised and completed the study, randomised to one of the following groups:
Inclusion criteria: The outpatients with chronic cancer pain for a period of 1 to 3 years, a diagnosis of abdominal neoplasia and a pain score equal to at least 40 mm on the visual‐analogue scale (VAS) of Short‐Form McGill Pain Questionnaire (SF‐MPQ). Patients with a pain score average equal to at least 4 out of the 11 points on a Likert scale and with at least 4 observations recorded in the daily diary of pain during the previous week, were randomised. All the patients taking part in the study had previously received therapy with NSAIDs or other analgesic agents discontinuously without obtaining successful results. Eligible patients were confirmed during a week‐long screening phase. Exclusion criteria:
|
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The patients eligible for the study were randomized into blocks of 4, according to a computer‐generated randomized code, to receive buprenorphine or morphine. Two groups, matching in age, general baseline conditions and staging degree of abdominal neoplasia, were formed." |
| Allocation concealment (selection bias) | Unclear risk | See cell above. No further information reported. |
| Blinding of participants and personnel (performance bias) Pain | High risk | Open‐label study. |
| Blinding of participants and personnel (performance bias) Adverse events | High risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | High risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | High risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data available for all patients. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data appear to be available for all patients. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes are reported. |
| Other bias | Low risk | The study appears to be free of other obvious biases. |
| Were the patients adequately titrated? | High risk | The patients did not appear to be titrated at all. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Pasqualucci 1987.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: Not reported Country: Italy |
|
| Participants |
Patients: 12 patients randomised and completed the study, randomised to one of the following groups:
Inclusion criteria: "The patients were recruited from different clinical departments of the same hospital, and all were suffering from severe continuous, non‐incident pain (5 cm out of a maximum score of 10 on the visual‐analogue scale), which did not respond to common analgesic drugs (non‐steroidal anti‐inflammatory agents)" "No narcotics had been administered prior to entry to the trial, and none of the patients had mechanical and/or neuromuscular disorders of the chest wall." Exclusion criteria: None reported. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | Study described as double‐blind. Unclear who is blinded. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above. Pain is patient reported. Probably reasonable to assume that patients were blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | Study described as double‐blind. Unclear who is blinded and whether this outcome is assessed by healthcare professionals also. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from all participants appear to be available. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | Data from all participants appear to be available. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes are reported, although not in a manner that allow their inclusion in a meta‐analysis. |
| Other bias | Unclear risk | It is unclear whether the study is subject to other types of bias. |
| Were the patients adequately titrated? | High risk | The patients were not titrated. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Poulain 2008.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: February 2004 to January 2005 Country: Austria, Belgium, Croatia, France, Poland, and the Netherlands |
|
| Participants |
Patients: 289 patients were recruited, and of these 92 patients discontinued BUP TDS treatment and 8 patients died during the run‐in phase. The remaining 189 patients (who responded to BUP TDS treatment) were randomised to one of the following 2 groups:
Inclusion criteria: "Patients with documented malignant disease and insufficient pain relief from their current opioid regimen were eligible. Patients were receiving single opioids or combination therapy, including oral morphine 90‐150 mg/day (n = 105), transdermal fentanyl 25‐50 μg/h (n = 170), tramadol 400‐600 mg (n = 75), hydromorphone 8‐16 mg (n = 6), or oxycodone 40‐60 mg (n = 5). A protocol for chemotherapy or radiotherapy could be applied concomitantly. Adjuvant analgesics (tricyclic antidepressants, benzodiazepines, anticonvulsants, muscle relaxants, and corticosteroids) were allowed providing the dose was stable." Exclusion criteria: None reported. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
Apart from study drug everything else was similar to the buprenorphine arm. |
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomization was performed in blocks with a 1:1 ratio (BUP TDS: placebo)." No more information reported. |
| Allocation concealment (selection bias) | Unclear risk | Not enough information is reported to assess whether there was allocation concealment. |
| Blinding of participants and personnel (performance bias) Pain | Low risk | "Hospital pharmacies received coded study medication from the sponsor and delivered the blinded supply for each patient. BUP TDS and placebo patches were identical in appearance and adhesive properties. The randomization list was stored in a sealed, nontransparent envelope and the code was not broken until the database had been locked." |
| Blinding of participants and personnel (performance bias) Adverse events | Low risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | Patient‐reported. See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | The safety analyses included all 189 patients and the efficacy analyses included 188/189 patients. One patient in the placebo group was excluded from the efficacy analyses due to missing data (no pain assessment). |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | See cell above. |
| Selective reporting (reporting bias) | Low risk | The expected outcomes are reported. |
| Other bias | Unclear risk | It is unclear whether the study results are subject to high risk of other bias(es). |
| Were the patients adequately titrated? | Low risk | Yes, it appears so. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Rigolot 1979.
| Methods |
Design: Randomised, single/double(?)‐blind, cross‐over trial Year: Not reported Country: France |
|
| Participants |
Patients: 10 patients, males and females (no numbers given) aged 18 to 60 years, hospitalised with severe disabling pain from advanced cancer who were lucid and able to communicate with the investigator, and who received insufficient pain relief from non‐opioid analgesics. Inclusion criteria: See above. No further information reported. Exclusion criteria: Pregnant women, patients already receiving high doses of opiates, and, it seems, those with addiction problems, intolerance to morphine or buprenorphine or both (?), asthma, cerebral pathology, and hepatic and renal impairment. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes | Patients were interviewed just before taking the drug (time 0) and every hour for the first 5 hours and then again at hour 9 by a single observer qualified to detect potential side effects and evaluate the intensity of pain, which was measured on a 4‐point verbal rating scale (0 = no pain, 1 = mild pain, 2 = moderate pain, 3 = severe, intense pain). | |
| Notes |
This study was published in French and lay translated by MSH with input from Dr Valeria Martinez MD PhD, Praticien Hospitalier, Anesthésiste‐Algologue, Groupe Hospitalier Raymond Poincaré, 104, Bd Raymond Poincaré, 92380 Garches France, regarding the nature of the "permutation table of Hazard" referred to in the publication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patient is his own control since he/she received the two drugs within eight days: The drugs will each be administered for 4 days, the day of injection being determined in advance using the permutation table of Hazard." No further information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | The study is described as single‐blind (patient blinded), but also reported that although the drugs have not been conditioned anonymously, the person administering the injection is not the same as the interviewer asking about the reported outcomes. The personnel providing care (e.g., injection) is therefore not blinded. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | Outcome not reported. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Patient and interviewer blinded. See also cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | Outcome not reported. |
| Incomplete outcome data (attrition bias) Pain | Low risk | All data appear to be included in the analyses. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Outcome not reported. |
| Selective reporting (reporting bias) | High risk | It appears that adverse event data were collected, but not reported. |
| Other bias | Unclear risk | It is unclear whether the study was subject to high risk of other types of bias. |
| Were the patients adequately titrated? | High risk | No information reported about titration, but unlikely as the dose is set. |
| For cross‐over trials: Are data available for both time periods? | Low risk | Efficacy data are available from both time periods. |
Sarhan 2009.
| Methods |
Design: Randomised, parallel‐group trial Year: Not reported Country: Egypt |
|
| Participants |
Patients: "32 opioid naive patients suffering from chronic cancer pain with visual analogue scale (VAS) ⊇ 7 were randomly allocated to one of two groups 16 patients each". No further information reported. Inclusion criteria: See above. No further information reported. Exclusion criteria: See above. No further information reported. |
|
| Interventions |
Buprenorphine arm:
Comparison arm:
|
|
| Outcomes | "Measurements: were done for 6 weeks by an assessor blinded to the study *severity of pain by VAS every 3 days *Mean number of each category patch dose *treatment satisfaction scale *Mean daily dose of diclofenac sodium *Mean cost of treatment *Side effects and complications." | |
| Notes |
This study was published as an abstract only. We contacted the trial author via email on 19 September 2014 for study details and data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Group assignment described as random. No further information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | No information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | Assessor described as blinded, but no further information reported, and unclear if this outcome was patient reported. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | No actual data presented. Data just described as being statistically significantly different or not. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | See cell above. |
| Selective reporting (reporting bias) | Unclear risk | See cell above. |
| Other bias | Unclear risk | It is unclear if the study is subject to high risk of other biases. |
| Were the patients adequately titrated? | Unclear risk | No information reported. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Sittl 2003.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: Not reported Country: Austria, Germany, and the Netherlands |
|
| Participants |
Patients: 157 patients were enrolled and randomised to one of the following 4 groups:
Inclusion criteria: "Patients aged ≥18 years with chronic, severe pain related to cancer or other diseases were enrolled in the study." Exclusion criteria: "Women using inadequate contraceptive measures or who were pregnant, possibly pregnant, or lactating were excluded from the study, as were patients with elevated intracranial pressure, a history of abuse of centrally acting substances or cerebral convulsions, previous extensive dermal damage in the patch area, or clinically relevant impairment of respiratory function. Patients with known hypersensitivity to opioids, impaired hepatic or renal function, or impaired consciousness, or who were receiving treatment with monoamine oxidase inhibitors also were excluded." |
|
| Interventions |
Buprenorphine arms (3):
Comparison arm:
Apart from study drug everything else was similar to the buprenorphine arms. |
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | Study described as double‐blind, and although it does not detail who was blinded and how blinding was employed, the patient is likely to be among those who were blinded, but unclear whether treating healthcare professionals were blinded. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Patient‐reported. Study described as double‐blind, and although it does not detail who was blinded and how blinding was employed, the patient is likely to be among those who were blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Low risk | Investigator who recording this outcome was blinded to the study/treatment group. |
| Incomplete outcome data (attrition bias) Pain | Low risk | The analyses include 154/157 patients. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | The analyses include 154/157 patients. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appear to be reported. |
| Other bias | Low risk | No other obvious biases were observed. |
| Were the patients adequately titrated? | Low risk | The patients were not titrated, but had access to top‐up fast‐acting strong analgesia to ensure adequate pain control. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Sorge 2004.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: Not reported Country: Germany and Poland |
|
| Participants |
Patients: 174 patients entered the run‐in phase, of these 37 were excluded and 137 patients were randomised to one of the following 2 groups:
Inclusion criteria: "Eligible patients were aged ≥18 years and were receiving outpatient or inpatient hospital care for severe or very severe chronic pain related to cancer or other disorders justifying treatment with strong opioids such as buprenorphine. The probable course of the patient's disease and pain could not be such that they might interfere with adherence to the study protocol, and no surgery could be scheduled during the study period." "The principal criterion for inclusion in the double‐blind phase was that pain had been at least satisfactorily relieved during the run‐in phase." Exclusion criteria: "Exclusion criteria were pregnancy or lactation; abuse of alcohol, hypnotics, analgesics, psychotropic drugs, or other central nervous system‐acting substances; hypersensitivity to opioids; a history of convulsions; compromised respiratory function; elevated intracranial pressure; and previous extensive damage to the site where the patch was to be applied. Patients receiving concomitant opioids apart from buprenorphine SL or monoamine oxidase inhibitors in the 2 weeks before study enrollment were also excluded." |
|
| Interventions |
Buprenorphine arms:
Comparison arm:
Apart from study drug everything else was similar to the buprenorphine arm. |
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was conducted in a 2:1 ratio (buprenorphine TDS:placebo) using permuted block randomisation, with a single block generated according to the urn model. The size of the blocks was provided in the randomisation list and was not imparted to investigators. The assigned patient numbers were documented on all pages of the case‐report form. After the randomisation criteria had been checked within each centre, eligible patients were randomised to receive 3 sequential patches containing buprenorphine 35 μg/h or placebo. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. No further information reported. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | The study is described as double‐blind, but does not detail how blinding was accomplished and who were blinded. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Low risk | Although the study is described as double‐blind, but does not detail how blinding was accomplished and who were blinded, the patient was probably blinded. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | Unclear who was blinded and whether this outcome was patient‐reported. See also cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | "all patients who entered the double‐blind phase were included in the analyses (including premature withdrawals). |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | See cell above. |
| Selective reporting (reporting bias) | Low risk | All obvious outcomes appear to be reported, although not by pain subgroup (cancer/non‐cancer), which would allow their inclusion into the results section. |
| Other bias | Low risk | No other obvious biases were observed. |
| Were the patients adequately titrated? | Low risk | The patients were probably adequately titrated. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Wang 2012.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: 2005 to 2010 Country: China |
|
| Participants |
Patients: 163 patients were screened, of these 43 did not meet the inclusion criteria (declined participation, N = 14; adjuvant chemotherapy, N = 11; elevated levels of serum transaminase N = 9; respiratory tract infection, N = 9) while the remaining 120 patients were randomised to one of the following 2 groups (each consisting of two sub‐groups based on the status of P‐gp expression in their tumour tissues): Buprenorphine:
Morphine:
Inclusion criteria: "Individual patients with histologically confirmed malignant tumours at stage IV, able to communicate effectively with the healthcare providers, regardless of previous chemotherapy and surgical treatment, were included." Exclusion criteria: "individual tumor patients with opioid intolerance, previous usage of strong opioids, severe renal or hepatic dysfunction, predominantly neuropathic pain, or breakthrough pain; or individuals who needed neural block or neuroablative treatment for pain relief, with impaired sensory or cognitive function, with coma or other mental disorders were excluded." |
|
| Interventions |
Buprenorphine arm (B1 and B2):
Morphine arm (M1 and M2):
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No information is reported about the random sequence generation, but 120 patients were randomised into 4 groups with stratification for tumour type, P‐gp expression, and their demographic characteristics, which (due to the relatively large number of stratification factors) increases the risk that the allocation ceases to be random. |
| Allocation concealment (selection bias) | High risk | See above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | The study reports that the analgesics effect was tested in "a double blinded manner", but reports no further details. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | Outcome not reported. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | The study reports that the analgesics effect was tested in "a double blinded manner", but reports no further details. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | Outcome not reported. |
| Incomplete outcome data (attrition bias) Pain | Unclear risk | It is unclear if data are included from all the patients at all the time points. |
| Incomplete outcome data (attrition bias) Adverse events | Unclear risk | Outcome not reported. |
| Selective reporting (reporting bias) | High risk | Adverse events not reported. |
| Other bias | Unclear risk | It is unclear whether the study is subject to high risk of other bias(es). |
| Were the patients adequately titrated? | Unclear risk | It is unclear, based on the reported information, whether the patients were adequately titrated. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Yajnik 1992.
| Methods |
Design: Randomised, double‐blind, parallel group trial Year: 1988 to 1989 Country: India |
|
| Participants |
Patients: 75 patients (aged 20 to 30 years: N = 6; 31 to 40 years: N = 7; 41 to 50 years: N = 29; 51 to 60 years: N = 21; ≥ 61 years: N = 12; only given overall, not per group) randomised to one of the following 3 groups:
Inclusion criteria: "In this study, 75 patients with complaints of malignant pain were assigned at random to 1 of 3 treatment groups, phenytoin (PHT) alone, buprenorphine (Bu) alone, or buprenorphine plus phenytoin, from August 1988 to March 1989. Approximately 9 patients were admitted to the study per month. The patients in this trial had all been treated with surgery and/or radiotherapy, but none had any type of pain therapy. All had moderate‐to‐severe pain levels as determined by use of a visual analogue scale (see below). They were taking no drugs other than those used during the study." Only patients with pain scores of 6 to 10 cm on a 10‐cm VAS (i.e., moderate‐severe pain) and requesting pain medication were included. Exclusion criteria: None reported. |
|
| Interventions |
Buprenorphine arm:
Buprenorphine + phenytoin arm:
Phenytoin arm:
|
|
| Outcomes |
|
|
| Notes | Study free of commercial funding? No information reported. Groups comparable at baseline? The baseline characteristics of the three groups were comparable in terms of gender and cancer type. Otherwise not reported. ITT analyses undertaken? Unclear. Not enough information reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information reported. |
| Allocation concealment (selection bias) | Unclear risk | See cell above. |
| Blinding of participants and personnel (performance bias) Pain | Unclear risk | "Both the patient and observer were blind to their treatment", but no further information reported. |
| Blinding of participants and personnel (performance bias) Adverse events | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Pain | Unclear risk | See cell above. |
| Blinding of outcome assessment (detection bias) Adverse events | Unclear risk | See cell above. |
| Incomplete outcome data (attrition bias) Pain | Low risk | Data from all the patients are included. |
| Incomplete outcome data (attrition bias) Adverse events | Low risk | It appears that data from all the patients are included. |
| Selective reporting (reporting bias) | Low risk | The main expected outcomes are reported. |
| Other bias | Unclear risk | It is unclear whether the study is subject to high risk of other bias(es). |
| Were the patients adequately titrated? | Unclear risk | No information is reported. |
| For cross‐over trials: Are data available for both time periods? | Unclear risk | NA. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Corli 1988 | Comparison not in PICO: buprenorphine versus buprenorphine + diclofenac |
| De Conno 1993 | Not a RCT |
| De Conno 1997 | Not a RCT |
| Deng 2002 | No cancer patients received buprenorphine |
| Enig 1983 | Not a RCT |
| Evans 2003 | Narrative review |
| Gantsev 1994 | Not a RCT |
| Hans 2007 | Narrative review |
| Hans 2009 | Narrative review |
| Hayek 2011 | Narrative review |
| Likar 2006 | Not a RCT |
| Likar 2007 | Mixed population, only 9/49 patients had cancer. Results not presented separately for cancer patients |
| Michel 2011 | Narrative review |
| Radbruch 2003 | Narrative review |
| Ritzman 2008 | Narrative review |
| Robbie 1979 | Not a RCT |
| Taguchi 1982 | Not a RCT |
| Van Dongen 1999 | Comparison not in PICO: Morphine versus morphine + bupivacaine |
| Wallenstein 1986 | Not cancer patients |
| Wirz 2009 | Not randomised allocation to treatment. Patients randomly selected from pools of patients already undergoing treatments under investigation instead |
| Yarlas 2013 | Not cancer patients |
| Zeppetella 2007 | Withdrawn |
Characteristics of studies awaiting assessment [ordered by study ID]
Lari 1985.
| Methods | "Epidural buprenorphine versus morphine in the bone cancer pain". |
| Participants | "With regard to the drugs used, patients were divided (Fig. 1) into two groups, sufficiently homogeneous for number, age, sex, and topography of the neoplastic lesion, treated with morphine and buprenorphine at doses respectively of 0.07 mg/kg and 4 mcg/kg every 12 hours." (Google translate on 24 July 2014). |
| Interventions | See cell above. |
| Outcomes | Pain intensity, side effects. |
| Notes | The study is published in Italian and translation of the article has revealed no mention about allocation to treatment beyond the description of the study outlined in the "Participants" section 3 cells above. It is thus uncertain whether it was a randomised controlled trial. |
NCT01324570.
| Methods | Open‐label, multicentre study of safety, pharmacokinetics and efficacy of buprenorphine TD system (BTDS) in children from seven to 16 years, inclusive, who require continuous opioid analgesia for moderate to severe pain. |
| Participants |
Inclusion criteria:
Exclusion criteria:
Other protocol‐specific inclusion/exclusion criteria may apply. |
| Interventions | Buprenorphine TD system 2.5 mcg/h, 5 mcg/h, 10 mcg/h or 20 mcg/h applied transdermally for 7‐day wear. |
| Outcomes |
|
| Notes | Location: USA. Sponsors: Purdue Pharma LP. Principal investigators/contact: Eduardo Rodenas, MD; 203‐588‐7660; email Eduardo.Rodenas@pharma.com Target enrolment: N = 40. Study starting date: July 2011. Study completion date: June 2015. Other study ID numbers: BUP3031, 2010‐021954‐21. |
Ripamonti 1987.
| Methods | "The objective of this study was to compare the analgesic effect over the quality of analgesic relief and side effects of the two drugs." (Google translate on 24 July 2014). |
| Participants | "Twenty‐five patients suffering from painful advanced cancer were given intramuscular injections of 0,3 mg buprenorphine and 10 mg morphine, the second 24 hours after the first." |
| Interventions | See cell above. |
| Outcomes | "The following parameters were examined: 1) intensity of pain; 2) relief from pain; 3) side effects; 4( vital signs (pulse, respiration, blood pressure)." |
| Notes | The study is published in Italian and translation of the article has revealed no mention about allocation to treatment beyond the description of the study as "double‐blind cross‐over". It is thus uncertain whether drug administration was in a random or fixed order. |
Staquet 1990.
| Methods | Double‐blind, parallel‐group (3 groups), double‐observer comparison of multiple dose regimen of IM ketorolac (10 mg, 30 mg) and buprenorphine (0.3 mg) in patients with cancer pain. |
| Participants | 120 patients with moderate‐severe cancer pain. |
| Interventions | IM ketorolac (10 mg, 30 mg) and buprenorphine (0.3 mg) for 3 days, administered by a nurse (who was not involved in the evaluation of the results) as required by the patients. |
| Outcomes | Patients rated pain severity and pain relief on a standard verbal scale. "Patterns of usage, duration of efficacy, acceptance, side‐effects, global evaluation were also investigated at preselected times." |
| Notes | Location: Belgium. Published as an abstract only. Unclear if treatment group assignment was randomised. |
Wallenstein 1982.
| Methods | Clinical analgesic assays of buprenorphine and morphine: "Limited crossover, randomized, double‐blind comparisons." |
| Participants |
Inclusion criteria: "Hospitalized patients with postoperative or cancer pain". Exclusion criteria: None reported. |
| Interventions | "An assay consisting of five sequentially graded dose comparisons of intramuscular buprenorphine and morphine was carried out in 136 patients. In a second assay, graded sublingual doses of buprenorphine were compared with intramuscular morphine in a single six‐point assay in 150 patients." |
| Outcomes | Hourly subjective reports of pain and pain relief. |
| Notes | Location: USA. Sponsors: "Suported in part by NIDA grant DA‐01707, NCI core grant CA‐08748, and a contribution from Reckitt and Colman, Ltd." Published as abstract only. Population composition unclear. |
Characteristics of ongoing studies [ordered by study ID]
2008‐002273‐12.
| Trial name or title | Long term opioid administration in oncologic chronic pain: open label, prospective study on efficacy, safety and pharmacogenetic factors. |
| Methods | Randomised, parallel‐group, open controlled trial. |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes | Pain reduction at least 40% in VAS scale. |
| Starting date | Not reported. |
| Contact information | Location: Italy. Sponsors: Ospedale Policlinico S. Matteo. Principal investigators: Not reported. |
| Notes | Target enrolment: N = 320. Study completion date: Unknown but of 3‐year duration. Other study ID numbers: None reported, but is it the same as NCT00916890 below? |
2008‐003592‐48.
| Trial name or title | A double‐blind, multi‐centre, reference‐controlled, randomised Phase III study to compare the analgesic efficacy and tolerability of a buprenorphine transdermal system in two different application intervals using three different dosages (35, 52.5 or 70 µg/h) in patients with chronic, severe cancer pain inadequately controlled with other analgesics. |
| Methods | Randomised (parallel group), double‐blind controlled trial. |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Main objective: Primary objective of the study is to demonstrate that the BUP‐TDS applied every 96 hours (Treatment group A) is therapeutically non‐inferior to BUP‐TDS applied every 72 hours (Treatment group B). Primary end point(s): To assess and compare the analgesic efficacy and tolerability of a buprenorphine TD system in two different application intervals using three different dosages (35, 52.5, or 70 µg/h). |
| Outcomes |
|
| Starting date | 25 November 2008. |
| Contact information | Location: Bulgaria. Sponsors: Novosis AG. Principal investigators: Not reported. |
| Notes | Target enrolment: N = 100. Study completion date: Not reported. Other study ID numbers: EUCTR2008‐003592‐48‐BG, BUP/006/C. |
NCT00916890.
| Trial name or title | Chronic Administration of Opioids in Cancer Chronic Pain:an Open Prospective Study on Efficacy, Safety and Pharmacogenetic Factors Influence. |
| Methods | Randomised (parallel group), single‐blind (outcome assessor) controlled trial. |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
Primary outcomes:
"We will define a treatment effective if it will produce a mean reduction of NRS values at least of 40% than basal values. Among all effective treatments, we will identify the best as the one that will have a reduction of NRS to a value of 4 or less in 90% of patients compared to the 70% of the others treatments. To evaluate pharmacological safety the plasma concentrations of the drugs and their metabolites will be measured. We will branch patients population in 3 groups to evaluate the correlation between clinical‐pharmacological response and genetics (responder,partially and not responder)." Secondary Outcome Measures:
|
| Starting date | February 2009. |
| Contact information | Location: Italy. Sponsors/collaborators/investigators : IRCCS Policlinico S. Matteo, University of Pavia, Italy. Principal investigator: Massimo Allegri, IRCCS Foundation Policlinico "San Matteo", Pavia, Italy; email: m.allegri@smatteo.pv.it , Tel: 00390382502627. |
| Notes | Target enrolment: N = 320. Study completion date: December 2015. Other study ID numbers: PT‐SM‐1‐Op‐Cancer. |
NCT01809106.
| Trial name or title | RCT Comparing the Analgesic Efficacy of 4 Therapeutic Strategies Based on 4 Different Major Opioids (Fentanyl, Oxycodone, Buprenorphine vs Morphine) in Cancer Patients With Moderate/Severe Pain, at the Moment of Starting 3rd Step of WHO Analgesic Ladder. |
| Methods | Randomised (parallel? cross‐over?), open‐label controlled trial |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
. |
| Outcomes |
Primary outcomes:
Secondary outcomes:
Other outcomes:
|
| Starting date | April 2011. |
| Contact information | Location: Italy. Sponsors/collaborators: Mario Negri Institute for Pharmacological Research. Principal investigator: Oscar Corli, MD. Mario Negri Institute of Pharmacological Research ‐ IRCCS. Contact: oscar.corli@marionegri.it; anna.roberto@email.it |
| Notes | Target enrolment: N = 600. Study completion date: April 2014. Other study ID numbers: None reported. |
Differences between protocol and review
It was originally planned that we would also search CINAHL and PubMED, however we did not search these databases as it was felt (in consultation with PaPaS) that this level of database searching was not required. In this respect, it was noted that:
CINAHL is a nursing database and is therefore unlikely to yield anything extra that will meet the inclusion criteria; and
There is a great deal of overlap between MEDLINE and PubMED. Therefore, generally one or the other is searched, which was MEDLINE in our case.
In the protocol we stated that two review authors would perform all data extractions from the included studies. However, we only performed double‐reviewing for the studies published in full in English and not for those published as an abstract only or those requiring translation by translators either external or internal to the review author team.
The structure of the 'Summary of findings' table differs from that planned at protocol stage. We felt that the chosen format gave a clearer summary given the nature of the included data and the number of different comparisons.
Contributions of authors
MSH and MT conceived, designed and wrote the review. MSH coordinated the review and devised the analysis strategy. The Cochrane PaPaS Group and SA devised and executed the search strategy. MSH and JH screened the search results, and appraised and extracted data from the included studies. MSH and MT wrote the review, and all review authors approved the final version of the review.
Sources of support
Internal sources
None, Other.
External sources
None, Other.
Declarations of interest
Mia Schmidt‐Hansen has no conflicts of interest to declare. Nathan Bromham has no conflicts of interest to declare. Mark Taubert has no conflicts of interest to declare. Stephanie Arnold has no conflicts of interest to declare. Jennifer S Hilgart has no conflicts of interest to declare.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Bauer 1985 {published data only}
- Bauer M, Schmid H, Schulz‐Wentland R. Gynecologic carcinoma patients with chronic pain. Comparison of sublingual buprenorphine with tilidine plus naloxone. Therapiewoche 1985;35(35):3943‐7. [Google Scholar]
Böhme 2003 {published data only}
- Boehme K. Buprenorphine transdermal system (TDS) (Delivery rates 35 / 52.5 70 mg/h) in comparison to sublingual buprenorphine in chronic pain patients. Journal of Pain and Symptom Management 2000;20:S54. [Google Scholar]
- Böhme K, Likar R. Efficacy and tolerability of a new opioid analgesic formulation, buprenorphine transdermal therapeutic system (TDS), in the treatment of patients with chronic pain. A randomised, double‐blind, placebo‐controlled study. The Pain Clinic 2003;15(2):193‐202. [Google Scholar]
Bono 1997 {published data only}
- Bono AV, Cuffari S. [Effectiveness and tolerance of tramadol in cancer pain. A comparative study with respect to buprenorphine]. Drugs 1997;53(Suppl 2):40‐9. [DOI] [PubMed] [Google Scholar]
Brema 1996 {published data only}
- Brema F, Pastorino G, Martini MC, Gottlieb A, Luzzani M, Libretti A, et al. Oral tramadol and buprenorphine in tumour pain. An Italian multicentre trial. International Journal of Clinical Pharmacology Research 1996;16(4‐5):109‐16. [PubMed] [Google Scholar]
Dan 1989 {published data only}
- Dan K, Yoshitake J, Tashiro H, Kurihara Y, Yoshida T, Fukushima K, et al. Double blind comparative study on 0.2mg suppository and 0.2mg injection preparation of buprenorphine hydrochloride for light to middle degree cancer pain. Igaku no Ayumi 1989;148(6):435‐46. [Google Scholar]
De Conno 1987 {published data only}
- Conno F, Ripamonti C, Tamburini M, Ventafridda V. Buprenorphine in the treatment of cancer pain: Comparison with pentazocine [Buprenorfina nel dolore da cancro: confronto incrociato con la pentazocina]. Minerva Medica 1987;78(15):1177‐81. [PubMed] [Google Scholar]
- Ventafridda V, Conno F, Guarise G, Tamburini M, Savio G. Chronic analgesic study on buprenorphine action in cancer pain. Comparison with pentazocine. Arzneimittel‐Forschung 1983;33(4):587‐90. [PubMed] [Google Scholar]
Dini 1986 {published data only}
- Dini D, Fassio T, Gottlieb A, Gini M. Controlled study of the analgesic effect and tolerability of buprenorphine in cancer patients [Italian]. Minerva Medica 1986;77(3‐4):93‐104. [PubMed] [Google Scholar]
Kjaer 1982 {published data only}
- Kjaer M, Henriksen H, Knudsen EJ. Intramuscular buprenorphine and morphine in the treatment of cancer pain. A controlled study [Danish]. Ugeskrift for Laeger 1982;144(18):1306‐9. [PubMed] [Google Scholar]
- Kjaer M, Henriksen H, Knudsen J. A comparative study of intramuscular buprenorphine and morphine in the treatment of chronic pain of malignant origin. British Journal of Clinical Pharmacology 1982;13(4):487‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Limón Cano 1994 {published data only}
- Limón Cano S, Martínez Gómez JL, Vicencio Serrano RE, García Cuevas M, Carreón García J. Subdermic and sublingual buprenorphine for the control of cancer pain [Buprenorfina sublingual y subdermica para el control del dolor por cancer]. Revista Mexicana de Anestesiología 1994;17(4):170‐2. [Google Scholar]
Noda 1989 {published data only}
- Noda J, Umeda S, Arai T, Harima A, Mori K. Continuous subcutaneous infusion of buprenorphine for cancer pain control. Clinical Journal of Pain 1989;5(2):147‐52. [DOI] [PubMed] [Google Scholar]
Pace 2007 {published data only}
- Pace MC, Passavanti MB, Grella E, Mazzariello L, Maisto M, Barbarisi M, et al. Buprenorphine in long‐term control of chronic pain in cancer patients. Frontiers in Bioscience 2007;12:1291‐9. [DOI] [PubMed] [Google Scholar]
Pasqualucci 1987 {published data only}
- Pasqualucci V, Tantucci C, Paoletti F, Dottorini ML, Bifarini G, Belfiori R, et al. Buprenorphine vs. morphine via the epidural route: a controlled comparative clinical study of respiratory effects and analgesic activity. Pain 1987;29(3):273‐86. [DOI] [PubMed] [Google Scholar]
Poulain 2008 {published data only}
- Poulain P, Denier W, Douma J, Hoerauf K, Samija M, Sopata M, et al. Efficacy and safety of transdermal buprenorphine: a randomized, placebo‐controlled trial in 289 patients with severe cancer pain. Journal of Pain and Symptom Management 2008;36(2):117‐25. [DOI] [PubMed] [Google Scholar]
Rigolot 1979 {published data only}
- Rigolot JC, Vergnes Y, Bouket JL, Gauthier‐Lafaye P. [Comparative study about the analgesic effect of buprenorphine (author's transl)]. Anesthésie, Analgésie, Réanimation 1979;36(7‐8):317‐21. [PubMed] [Google Scholar]
Sarhan 2009 {published data only}
- Sarhan T, Doghem M. A comparison of two trans‐dermal drug delivery systems; Buprenorphine and fentanyl for chronic cancer pain management. European Journal of Pain 2009;13:S199. [Google Scholar]
Sittl 2003 {published data only}
- Sittl R, Griessinger N, Likar R. Analgesic efficacy and tolerability of transdermal buprenorphine in patients with inadequately controlled chronic pain related to cancer and other disorders: a multicenter, randomized, double‐blind, placebo‐controlled trial. Clinical Therapeutics 2003;25(1):150‐68. [DOI] [PubMed] [Google Scholar]
Sorge 2004 {published data only}
- Sorge J. Buprenorphine transdermal system (TDS) (delivery rate 35 mg/h) in comparison to sublingual tablets in chronic pain patients. Journal of Pain and Symptom Management 2000;20:S78. [Google Scholar]
- Sorge J, Sittl R. Transdermal buprenorphine in the treatment of chronic pain: results of a phase III, multicenter, randomized, double‐blind, placebo‐controlled study. Clinical Therapeutics 2004;26(11):1808‐20. [DOI] [PubMed] [Google Scholar]
Wang 2012 {published data only}
- Wang J, Cai B, Huang DX, Yang SD, Guo L. Decreased analgesic effect of morphine, but not buprenorphine, in patients with advanced P‐glycoprotein(+) cancers. Pharmacological Reports: PR 2012;64(4):870‐7. [DOI] [PubMed] [Google Scholar]
Yajnik 1992 {published data only}
- Yajnik S, Singh GP, Singh G, Kumar M. Phenytoin as a coanalgesic in cancer pain. Journal of Pain and Symptom Management 1992;7(4):209‐13. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Corli 1988 {published data only}
- Corli O, Roma G, Battaiotto L, Bernoni M. Buprenorphine and diclofenac in cancer pain relief. Schmerz/Pain/Douleur 1988;9:114‐5. [Google Scholar]
De Conno 1993 {published data only}
- Conno F, Ripamonti C, Sbanotto A, Barletta L. A clinical note on sublingual buprenorphine. Journal of Palliative Care 1993;9:44‐46. [PubMed] [Google Scholar]
De Conno 1997 {published data only}
- Conno F. Analgesic efficacy and side effects of opioides in different routes of administration for terminal cancer patients: A clinical, pharmacokinetic and neurophysiological study. New Trends in Experimental and Clinical Psychiatry 1997;13:49‐51. [Google Scholar]
Deng 2002 {published data only}
- Deng YP, Xu GZ, Wang W, Shen XH, Chen QT, Gao HZ, et al. Clinical evaluation of analgesic effect and safety of scorpion venom injection [Chinese]. Chinese Pharmaceutical Journal 2002;37(6):459‐62. [Google Scholar]
Enig 1983 {published data only}
- Enig B. Sublingual buprenorphine in the treatment of pain caused by cancer. Transition to buprenorphine from other opiates. Ugeskrift for Laeger 1983;145:3236‐3240. [PubMed] [Google Scholar]
Evans 2003 {published data only}
- Evans HC, Easthope SE. Transdermal buprenorphine. Drugs 2003;63:1999‐2010. [DOI] [PubMed] [Google Scholar]
Gantsev 1994 {published data only}
- Gantsev SK, Vaisman MA, Gazizov AA. Prolonged sacral anesthesia with buprenorphine in combination with a local anesthetic in the treatment of chronic pain of oncologic patients with lesions of organs of the small pelvis. Anesteziologiia i Reanimatologiia, Anesteziologiia i Reanimatologiia 1994;4:50‐51. [PubMed] [Google Scholar]
Hans 2007 {published data only}
- Hans G. Buprenorphine ‐ A review of its role in neuropathic pain. Journal of Opioid Management 2007;3:195‐206. [DOI] [PubMed] [Google Scholar]
Hans 2009 {published data only}
- Hans G, Robert D. Transdermal buprenorphine ‐ A critical appraisal of its role in pain management. Journal of Pain Research 2009;2:117‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hayek 2011 {published data only}
- Hayek SM, Deer TR, Pope JE, Panchal SJ, Patel V. Intrathecal therapy for cancer and non‐cancer pain. Pain Physician 2011;14:219‐248. [PubMed] [Google Scholar]
Likar 2006 {published data only}
- Likar R, Kayser H, Sittl R. Long‐term management of chronic pain with transdermal buprenorphine: a multicenter, open‐label, follow‐up study in patients from three short‐term clinical trials. Clinical Therapeutics 2006;28(6):943‐52. [DOI] [PubMed] [Google Scholar]
Likar 2007 {published data only}
- Likar R, Lorenz V, Korak‐Leiter M, Kager I, Sittl R. Transdermal buprenorphine patches applied in a 4‐day regimen versus a 3‐day regimen: a single‐site, Phase III, randomized, open‐label, crossover comparison. Clinical Therapeutics 2007;29(8):1591‐606. [DOI] [PubMed] [Google Scholar]
Michel 2011 {published data only}
- Michel E, Anderson BJ, Zernikow B. Buprenorphine TTS for children ‐ A review of the drug's clinical pharmacology. Paediatric Anaesthesia 2011;21:280‐290. [DOI] [PubMed] [Google Scholar]
Radbruch 2003 {published data only}
- Radbruch L, Vielvoye‐Kerkmeer A. Buprenorphine TDS: The clinical development ‐ Rationale and results. International Journal of Clinical Practice 2003;Supplement:15‐18. [PubMed] [Google Scholar]
Ritzman 2008 {published data only}
- Ritzmann P. Retarded highly effective opioid analgesics [Retardierte hochwirksame opioid‐analgetika]. Pharma‐Kritik 2008;30:61‐62. [Google Scholar]
Robbie 1979 {published data only}
- Robbie DS. A trial of sublingual buprenorphine in cancer pain. British Journal of Clinical Pharmacology 1979;7(Supplement 3):315S‐317S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Taguchi 1982 {published data only}
- Taguchi T. Effect of a long‐acting analgesic, buprenorphine on cancer pain‐‐a single‐blind crossover comparison with pentazocine [Japanese]. Gan To Kagaku Ryoho (Japanese Journal of Cancer & Chemotherapy) 1982;9(2):250‐7. [PubMed] [Google Scholar]
Van Dongen 1999 {published data only}
- Dongen RTM, Crul BJP, Egmond J. Intrathecal coadministration of bupivacaine diminishes morphine dose progression during long‐term intrathecal infusion in cancer patients. Clinical Journal of Pain 1999;15:166‐172. [DOI] [PubMed] [Google Scholar]
Wallenstein 1986 {published data only}
- Wallenstein SL, Kaiko RF, Rogers AG, Houde RW. Crossover trials in clinical analgesic assays: studies of buprenorphine and morphine. Pharmacotherapy:The Journal of Human Pharmacology & Drug Therapy 1986;6:228‐235. [DOI] [PubMed] [Google Scholar]
Wirz 2009 {published data only}
- Wirz S, Wittmann M, Schenk M, Schroeck A, Schaefer N, Mueller M, et al. Gastrointestinal symptoms under opioid therapy: a prospective comparison of oral sustained‐release hydromorphone, trasndermal fentanyl, and transdermal buprenorphine. European Journal of Pain 2009;13(7):737‐43. [DOI] [PubMed] [Google Scholar]
Yarlas 2013 {published data only}
- Yarlas A, Miller K, Wen W, Kowalski M, Lynch S, Dain B, et al. Long‐term maintenance of improvements in patient‐reported functioning, quality‐of‐life, and pain outcomes for moderate‐to‐severe chronic pain patients receiving continuous treatment of Butrans (buprenorphine) Transdermal System (BTDS). Journal of Pain 2013;14:S80. [Google Scholar]
Zeppetella 2007 {published data only}
- Zeppetella G Wiffen PJ. Buprenorphine for cancer pain. Cochrane Database of Systematic Reviews 2007;withdrawn. [Google Scholar]
References to studies awaiting assessment
Lari 1985 {published data only}
- Lari S, Fabbri G, Mattioli R, Elmar K. Epidural buprenorphine versus morphine in bone cancer pain [Italian]. Minerva Anestesiologica 1985;51(11‐12):609‐14. [PubMed] [Google Scholar]
NCT01324570 {published data only}
Ripamonti 1987 {published data only}
- Ripamonti C, Conno F, Tamburini M, Ventafridda V. Acute study of parenteral buprenorphine versus parenteral morphine [Italian]. Minerva Anestesiologica 1987;53(4):135‐9. [PubMed] [Google Scholar]
Staquet 1990 {published data only}
- Staquet M, Wasch G. A double‐blind comparison of multiple dose regiment of ketorolac and buprenorphine in patients with cancer pain. Pain 1990;Suppl 5:s353. [Google Scholar]
Wallenstein 1982 {published data only}
- Wallenstein SL, Kaiko RF, Rogers AG, Houde RW. Clinical analgesic assays of buprenorphine and morphine. Clinical Pharmacology and Therapeutics 1982;31(2):278. [Google Scholar]
References to ongoing studies
2008‐002273‐12 {published data only}
- Long term opioid administration in oncologic chronic pain: open label, prospective study on efficacy, safety and pharmacogenetic factors.. Ongoing study Not reported..
2008‐003592‐48 {published data only}
- A double‐blind, multi‐centre, reference‐controlled, randomised Phase III study to compare the analgesic efficacy and tolerability of a buprenorphine transdermal system in two different application intervals using three different dosages (35, 52.5 or 70 µg/h) in patients with chronic, severe cancer pain inadequately controlled with other analgesics.. Ongoing study 25 November 2008..
NCT00916890 {published data only}
- Chronic Administration of Opioids in Cancer Chronic Pain:an Open Prospective Study on Efficacy, Safety and Pharmacogenetic Factors Influence.. Ongoing study February 2009..
NCT01809106 {published data only}
- RCT Comparing the Analgesic Efficacy of 4 Therapeutic Strategies Based on 4 Different Major Opioids (Fentanyl, Oxycodone, Buprenorphine vs Morphine) in Cancer Patients With Moderate/Severe Pain, at the Moment of Starting 3rd Step of WHO Analgesic Ladder.. Ongoing study April 2011..
Additional references
Bach 1991
- Bach V, Kamp‐Jensen M, Jensen NH, Eriksen J. Buprenorphine and sustained release morphine ‐ Effect and side‐effects in chronic use. Pain Clinic 1991;4:87‐93. [Google Scholar]
Bennett 2008
- Bennett MI. What evidence do we have that the WHO analgesic ladder is effective in cancer pain?. In: McQuay HJ, Moore R, Kalso E editor(s). Systematic Reviews in Pain Research; Methodology Refined. Seattle: IASP Press, 2008:303‐13. [Google Scholar]
Budd 2004
- Budd K, Shipton E. Acute pain and the immune system and opioimmunosuppression. Acute Pain 2004;6:123‐35. [Google Scholar]
Bullingham 1981
- Bullingham RES, McQuay HJ, Dwyer D, Allen MC, Moore RA. Sublingual buprenorphine used post‐operatively: clinical observations and preliminary pharmacokinetic analysis. British Journal of Clinical Pharmacology 1981;12(2):117‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bullingham 1983
- Bullingham RES, McQuay HJ, Moore RA. Clinical pharmacokinetics of narcotic agonist‐antagonist drugs. Clinical Pharmacology 1983;8(4):332‐43. [DOI] [PubMed] [Google Scholar]
Dahan 2005
- Dahan A, Yassen A, Bijl H, Romberg R, Sarton E, Teppema L, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. British Journal of Anaesthesia 2005;94(6):825‐34. [DOI] [PubMed] [Google Scholar]
Dahan 2006
- Dahan A, Yassen A, Romberg R, Sarton E, Teppema L, Olofsen E, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. British Journal of Anaesthetics 2006;96(5):627‐32. [DOI] [PubMed] [Google Scholar]
Dahan 2010
- Dahan A, Aarts L, Smith TW. Incidence, reversal and prevention of opioid‐induced respiratory depression. Anesthesiology 2010;112(1):226‐38. [DOI] [PubMed] [Google Scholar]
Deandra 2008
- Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Annals of Oncology 2008;19(12):1985‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deandrea 2009
- Deandrea S, Corli O, Moschetti I, Apolone G. Managing severe cancer pain: the role of transdermal buprenorphine: a systematic review. Therapeutics and Clinical Risk Management 2009;5(5):707‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elkader 2005
- Elkader A, Sproule B. Buprenorphine: clinical pharmacokinetics in the treatment of opioid dependence. Clinical Pharmacokinetics 2005;44(7):661‐80. [DOI] [PubMed] [Google Scholar]
Ferlay 2010
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International Journal of Cancer 2010;127(12):2893‐917. [DOI] [PubMed] [Google Scholar]
Filitz 2006
- Filitz J, Griessinger N, Sittl R, Likar R, Schüttler J, Koppert W. Effect of intermittent hemodialysis on buprenorphine and norbuprenorphine plasma concentrations in chronic pain patients treated with transdermal buprenorphine. European Journal of Pain 2006;10(8):743‐8. [DOI] [PubMed] [Google Scholar]
Foley 1998
- Foley KM. Pain assessment and cancer pain syndromes. In: Doyle D, Hanks GWC, MacDonald N editor(s). Oxford Textbook of Palliative Medicine. Oxford: Oxford University Press, 1998:310‐31. [Google Scholar]
Foster 2013
- Foster B, Twycross R, Mihalyo M, Wilcock A. Buprenorphine. Journal of Pain and Symptom Management 2013;45(5):939‐49. [DOI] [PubMed] [Google Scholar]
Frankish 2003
- Frankish H. 15 million new cancer cases per year by 2020, says WHO. Lancet 2003;361(9365):1278. [DOI] [PubMed] [Google Scholar]
Gal 1989
- Gal TJ. Naloxone reversal of buprenorphine‐induced respiratory depression. Clinical Pharmacology and Therapeutics 1989;45(1):66‐71. [DOI] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist G, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hadley 2013
- Hadley G, Derry S, Moore RA, Wiffen PJ. Transdermal fentanyl for cancer pain. Cochrane Database of Systematic Reviews 2013, Issue 10. [DOI: 10.1002/14651858.CD010270.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hand 1990
- Hand CW, Sear JW, Uppington J, Ball MJ, McQuay HJ, Moore RA. Buprenorphine disposition in patients with renal impairment: single and continuous dosing, with special reference to metabolites. British Journal of Anaesthesia 1990;64(3):276‐82. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Lewis 2004
- Lewis JW, Husbands SM. The orvinols and related opioids‐high affinity ligands with diverse efficacy profiles. Current Pharmaceutical Design 2004;10(7):717‐32. [DOI] [PubMed] [Google Scholar]
Likar 2008
- Likar R, Krainer B, Sittl R. Challenging the equipotency calculation for transdermal buprenorphine: four case studies. International Journal of Clinical Practice 2008;62(1):152‐6. [DOI] [PubMed] [Google Scholar]
Mercadante 2009
- Mercadante S, Casuccio A, Tirelli W, Giarratano A. Equipotent doses to switch from high doses of opioids to transdermal buprenorphine. Support Care Cancer 2009;17(6):715‐8. [DOI] [PubMed] [Google Scholar]
Moore 2013
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI] [PubMed] [Google Scholar]
NICE 2012
- National Institute for Health and Care Excellence. Opioids in palliative care. Clinical guidelines CG140. Issued: May 2012. www.nice.org.uk/cg140 (accessed 7 November 2013).
Nicholson 2007
- Nicholson AB. Methadone for cancer pain. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD003971.pub3] [DOI] [PubMed] [Google Scholar]
Pergolizzi 2008
- Pergolizzi J, Böger RH, Budd K, Dahan A, Erdine S, Hans G, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Practice 2008;8(4):287‐313. [DOI] [PubMed] [Google Scholar]
Portenoy 1989
- Portenoy RK. Cancer pain. Epidemiology and syndromes. Cancer 1989;63(11 Suppl):2298‐307. [DOI] [PubMed] [Google Scholar]
Quigley 2002
- Quigley C. Hydromorphone for acute and chronic pain. Cochrane Database of Systematic Reviews 2002, Issue 1. [DOI: 10.1002/14651858.CD003447] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Robbie 1979
- Robbie DS. A trial of sublingual buprenorphine in cancer pain. British Journal of Clinical Pharmacology 1979;7(Suppl 3):315S‐7S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rothman 1995
- Rothman R. A review of the binding literature. In: Cowan A, Lewis JW editor(s). Buprenorphine Combating Drug Abuse with a Unique Opioid. New York: Wiley‐Liss, 1995:19‐29. [Google Scholar]
Sacerdote 2000
- Sacerdote P, Bianchi M, Gaspani L, Manfredi B, Maucione A, Terno G, et al. The effects of tramadol and morphine on immune responses and pain after surgery in cancer patients. Anesthesia & Analgesia 2000;90(6):1411‐4. [DOI] [PubMed] [Google Scholar]
Sacerdote 2008
- Sacerdote P, Franchi S, Gerra G, Leccese V, Panerai AE, Somaini L. Buprenorphine and methadone maintenance treatment of heroin addicts preserves immune function. Brain, Behavior, and Immunity 2008;22(4):606‐13. [DOI] [PubMed] [Google Scholar]
Schmidt‐Hansen 2015
- Schmidt‐Hansen M, Bennett MI, Arnold S, Bromham N, Hilgart J. Oxycodone for cancer‐related pain. Cochrane Database of Systematic Reviews 2015, Issue 2. [DOI: 10.1002/14651858.CD003870.pub5] [DOI] [PubMed] [Google Scholar]
Seya 2011
- Seya MJ, Gelders SF, Achara OU, Milani B, Scholten WK. A first comparison between the consumption of and the need for opioid analgesics at country, regional, and global levels. Journal of Pain and Palliative Care Pharmacotherapy 2011;25(1):6‐18. [DOI] [PubMed] [Google Scholar]
Sittl 2005
- Sittl R, Likar R, Nautrup BP. Equipotent doses of transdermal fentanyl and transdermal buprenorphine in patients with cancer and noncancer pain: results of a retrospective cohort study. Clinical Therapeutics 2005;27(2):225‐37. [DOI] [PubMed] [Google Scholar]
Staritz 1986
- Staritz M, Poralla T, Manns M, Meyer Zum Büschenfelde KH. Effect of modern analgesic drugs (tramadol, pentazocine, and buprenorphine) on the bile duct sphincter in man. Gut 1986;27(5):567‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tassinari 2008
- Tassinari D, Sartori S, Tamburini E, Scarpi E, Raffaeli W, Tombesi P, et al. Adverse effects of transdermal opiates treating moderate‐severe cancer pain in comparison to long‐acting morphine: a meta‐analysis and systematic review of the literature. Journal of Palliative Medicine 2008;11(3):492‐501. [DOI] [PubMed] [Google Scholar]
Tassinari 2011
- Tassinari D, Drudi F, Rosati M, Maltoni M. Transdermal opioids as front line treatment of moderate to severe cancer pain: a systemic review. Palliative Medicine 2011;25(5):478‐87. [DOI] [PubMed] [Google Scholar]
van den Beuken‐van Everdingen 2007
- Beuken‐van Everdingen MH, Rijke JM, Kessels AG, Schouten HC, Kleef M, Patijn J. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Annals of Oncology 2007;18(9):1437‐49. [DOI] [PubMed] [Google Scholar]
Ventafridda 1987
- Ventafridda V, Tamburini M, Caraceni A, Conno F, Naldi F. A validation study of the WHO method for cancer pain relief. Cancer 1987;59(4):851‐6. [DOI] [PubMed] [Google Scholar]
WHO 1987
- World Health Organization. Traitement de la douleur cancéreuse. Geneva, Switzerland: World Health Organization, 1987. [Google Scholar]
WHO 1996
- World Health Organization. Cancer pain relief. 1996. http://whqlibdoc.who.int/publications/9241544821.pdf (accessed 07 November 2013).
Wiffen 2013
- Wiffen PJ, Wee B, Moore RA. Oral morphine for cancer pain. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD003868.pub3] [DOI] [PubMed] [Google Scholar]
Wolff 2012
- Wolff RF, Reid K, Nisio M, Aune D, Truyers C, Hernandez AV, et al. Systematic review of adverse events of buprenorphine patch versus fentanyl patch in patients with chronic moderate‐to‐severe pain. Pain Management 2012;2(4):351‐62. [DOI] [PubMed] [Google Scholar]
World Bank 2013
- World Bank. How we classify countries. http://data.worldbank.org/about/country‐classifications (accessed 07 November 2013).
Zaki 2000
- Zaki P, Keith DE Jr, Brine GA, Carroll FI, Evans CJ. Ligand‐induced changes in surface mu‐opioid receptor number: relationship to G protein activation?. Journal of Pharmacology and Experimental Therapeutics 2000;292(3):1127‐34. [PubMed] [Google Scholar]
References to other published versions of this review
Naing 2012
- Naing C‐M, Aung K, Yeoh PN. Buprenorphine for treating cancer pain. Cochrane Database of Systematic Reviews 2012, Issue 1. [DOI: 10.1002/14651858.CD009596.pub3] [DOI] [Google Scholar]
Schmidt‐Hansen 2016
- Schmidt‐Hansen, M, Taubert, M, Bromham, N, Hilgart, J, Arnold, S. The effectiveness of buprenorphine for treating cancer pain: An abridged Cochrane review. BMJ Supportive & Palliative Care 2016;6:292‐306. [DOI: 10.1136/bmjspcare-2015-000939] [DOI] [PubMed] [Google Scholar]