

Summary
Acute respiratory distress syndrome (ARDS) induces a strong local infiltration of regulatory T‐cells (Tregs) in the lungs. However, at present, there remains a lack of adequate evidence showing the direct effect of Tregs on pulmonary repair and the related mechanisms of ARDS. Therefore, in this project, we studied the impact of Tregs on lipopolysaccharide (LPS)‐induced ARDS and pulmonary inflammation. Surprisingly, we found that depletion of Tregs by injection of PC61 anti‐CD25 antibody not only interfered with the inflammation resolution, such as inhibited total cell infiltration into the alveolar space, downregulated neutrophils, upregulated macrophages, but also impaired pulmonary epithelium and endothelial cell proliferation. Consistent with the attenuation of pulmonary repair, we found that the Th1 and Th17 immune responses were also impaired in Treg‐depleted mice, suggesting that the presence of Tregs is vital for tissue repair, as Tregs modulate and promote the Th immune response in LPS‐induced pulmonary inflammation.
Keywords: acute respiratory distress syndrome, regulatory T‐cells, T helper cell immune responses
Abbreviations
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BALF
bronchoalveolar lavage fluid
- LPS
lipopolysaccharide
- Th
T helper
- Tregs
regulatory T‐cells
Introduction
Acute respiratory distress syndrome (ARDS) is a syndrome of respiratory failure. Although ARDS can be triggered by a variety of predisposing conditions, including pneumonia, aspiration, sepsis, trauma, blood transfusion and pancreatitis, the most common cause of ARDS is bacterial infection. This syndrome is associated with high mortality and poor prognosis. With in‐hospital mortality ranging from 30% to 70%, on average, 86·2 out of 100 000 people acquire ARDS, and 75 000 deaths each year are attributed to ARDS in the USA.1 Although extensive investigations have been conducted, the underlying mechanisms remain elusive. Thus, it is necessary to further explore the mechanism of ARDS and find a new therapeutic approach to enhance the survival of patients with ARDS.
Inappropriate accumulation of leucocytes and platelets, dysregulated inflammation, and altered permeability of the alveolar endothelial–epithelial barrier contribute to the development of ARDS. While neutrophils, monocytes and macrophages have been extensively studied, the involvement of lymphocytes (other immune cells) recruited to the lungs has largely been ignored. Lymphocyte migration into the lungs was observed in ARDS during the resolution of inflammation.2 Among the lymphocytes, regulatory T‐cells (Tregs) appear to promote the resolution of ARDS and accelerate tissue repair.3, 4
Regulatory T‐cells, a distinct population of lymphocytes, play a critical role in maintaining immune homeostasis. Classically, Tregs have been associated with various types of immune responses but, recently, they were verified to affect diverse non‐immunological contexts as well. For example, Tregs promote the regeneration of acute and chronic muscle injury, improve healing after myocardial infarction, and play a major role in asthma and chronic obstructive pulmonary disease.5, 6 At the sites of damage, Tregs exert a direct effect on their progenitors, exhibit an indirect influence by regulating macrophage activity7, 8 and the balance of other T‐cell subpopulations, or prevent tissue damage through the production of Amphiregulin by alarmin IL‐18/IL‐33, which is distinct from immune responses suppression.4
Other specialized subpopulations of CD4+ T lymphocytes, including T helper (Th)1, Th2 and Th17 cells, have been reported to play a significant role in the initiation of immune responses by interacting with other cells9 and to be involved in several inflammatory immune‐mediated disorders, mostly chronic inflammatory disorders. However, regarding bacteria‐induced infectious acute inflammation, the activities of these Th cells are ill defined. Th2 cells have been found to promote acute and chronic inflammatory responses against various allergens and helminths.10 In addition, a previous study confirmed that IFN‐ɣ+ Th1 cells are involved in the early stage of acute lung injury (ALI)‐induced inflammation.11 Th1 cells produce high levels of IFN‐ɣ, are responsible for phagocyte activation and play an important role in protecting against intracellular pathogens. T helper‐17 (Th17) cells produce abundant inflammatory cytokines, including IL‐17a, IL‐17f and IL‐22, and are key mediators in host defence, inflammatory disorders and autoimmune conditions.12 Th1 and Th17 lymphocytes also appear to play a significant role in neutrophilic airway inflammation. Activation of these immune cells is closely modulated by counterregulatory circuits governed by Tregs, which attenuate the overactive immune responses.13
The resolution of pulmonary inflammation and the process of tissue repair rely on a variety of immune and non‐immune cells and signalling molecules. Until now, there has been little evidence of the direct effect of Tregs on inflammation resolution and pulmonary repair in ARDS. In addition, the role of Tregs in the pathophysiology of ARDS has not been elucidated, and the reciprocal relationship between Tregs and other subpopulations of Th cells has remained elusive. Reviewing various factors of ALI/ARDS, bacterial infection is the leading cause, and lipopolysaccharide (LPS) as a cell wall component of gram‐negative bacteria is considerably important in the pathogenesis of ALI/ARDS. Intratracheal instillation of LPS is a well‐accepted pathway to establish experimental lung injury. Therefore, in this study, we used a mouse model of LPS‐induced ARDS to investigate the role of Tregs in modulating the host response and tissue repair in ARDS, and to comprehensively explore the mechanism. We explicitly demonstrated that Tregs promote tissue repair and the resolution of LPS‐induced pulmonary inflammation by promoting Th1 and Th17 cell responses. In addition, we showed that in vivo depletion of Tregs by injection of PC61 anti‐CD25 antibody resulted in diminished Th1 and Th17 immune responses after LPS administration.
Animals and methods
Animals
Adult male BALB/c mice at 6–8 weeks old were purchased from the Animal Center of Peking Union Medical College Hospital (Beijing, China). All animals were kept in a specific‐pathogen‐free environment and maintained on standard mouse chow at an environmental temperature of 22–24°, with 12‐hr light and 12‐hr dark cycles.
Male mice were randomly allocated into six groups as follows: sham group; LPS‐12‐hr group (L12 h); LPS‐1‐day group (L1); LPS‐2‐day group (L2); LPS‐4‐day group (L4); and LPS‐7‐day group (L7). All mice were anaesthetized with intraperitoneal injection of 2% pentobarbital sodium (45 mg/kg body weight), then, the mice received an intratracheal instillation of LPS (from Escherichia coli serotype O55:B5; Sigma‐Aldrich, St Louis, MO, USA) at a dose of 3 mg/kg. Mice in the sham group received only sterile saline (1·5 ml/kg).
This study was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Animal Management Rules of the Chinese Ministry of Health. All experiments were approved by the Animal Care Committee of Peking Union Medical College.
Treg depletion
To deplete Tregs, mice were intraperitoneally injected with 100 μg of anti‐CD25 antibody (PC61; Biolegend, San Diego, CA, USA) 10 days before LPS exposure, and repeatedly treated every 7 days for continuous Treg depletion. IgG was used as a control. Male mice were randomly divided into 2‐day‐old and 4‐day‐old groups. Every group was then further divided into four subgroups: Saline + IgG; Saline + anti‐CD25; LPS + IgG; and LPS + anti‐CD25.
Bronchoalveolar lavage
The experimental procedure is shown in Fig. 2a. The mice were killed, and bronchoalveolar lavage fluid (BALF) was collected by lavage of the left lung. BALF was centrifuged for 10 min at 300 g. The supernatant was removed and stored at −80° for further detection. Total BALF cells were counted using a haematocytometer.
Histopathological analysis
The middle lobe of the right lung was fixed in 4% paraformaldehyde. The tissue was completely embedded in paraffin, cut into small sections, stained with haematoxylin and eosin (HE), and measured by optical microscopy. Lung injury histopathological scores were measured according to a previous study.14
Isolation of lung and spleen cells
The right lung was removed, dissected into small sections and incubated at 37° in RPMI 1640 medium containing 1 mg/ml collagenase I (Gibco/Invitrogen, Carlsbad, CA, USA) and 50 μg/ml DNase (Sigma‐Aldrich) for 1 hr. Then, the tissues were passed through a 70‐μm nylon cell strainer (BD Falcon). The spleen was collected, ground and mechanically dissociated in cold phosphate‐buffered saline (PBS). Samples were centrifuged at 300 g for 6 min at 4°, washed and resuspended in PBS after lysis of red blood cells.
Flow cytometry
The lung and spleen cells were stimulated with Leucocyte Activation Cocktail (BD Pharmingen, San Jose, CA, USA) for 6 hr when intracellular cytokines were detected. Cell staining was performed with CD16/CD32 Fc, CD3, CD4, CD25, CD31, CD326, CD45, F4/80 (Biolegend), Ly6C, Ly6G, CD11b (eBioscience, San Diego, CA, USA). Cells were fixed and permeabilized using a fixation/permeabilization kit (eBioscience) or the BD Cytofix/Cytoperm TM Fixation/Permeabilization Solution Kit (BD Pharmingen) according to the manufacturer's instructions. Then, cells were stained for 30 min at 4° with IFN‐ɣ, IL‐17A, IL‐4 or Foxp3 (Biolegend), Ki‐67 (eBioscience). Stained cells were washed twice and resuspended in 4% paraformaldehyde. Analysis of cell marker expression was performed using Accuri C6 (BD, Franklin Lakes, NJ, USA). Data were analysed with Flowjo software.
Bead‐based immunoassays
Secreted soluble protein in BALF was detected by bead‐based immunoassays using a Th panel kit (Biolegend) according to the instruction. Samples were collected by Accuri C6 (BD). Data were analysed with Biolegend LEGENDplex™ software.
RNA extraction and real‐time polymerase chain reaction
According to the manufacturer's instructions, total RNA was collected from lung homogenates using the Eastep® Super Total RNA Extraction Kit (Promega, Madison, WI, USA). The RNA concentration and the A260/A280 ratio were determined using a UV spectrophotometer. Total RNA (1 μg) was reverse‐transcribed to cDNA using GoScript Reverse Transcriptase (Promega). Real‐time quantitative polymerase chain reaction (qPCR) was performed using GoTaq qPCR mix (Promega) on the Applied Biosystems 7500 Fast system (Applied Biosystems, Foster City, CA, USA). The relative expression levels of target genes were quantified using the ΔΔCt method and normalized to GAPDH genes (β‐actin or GAPDH) as described previously. The primer sequences are shown in Table 1.
Table 1.
Primer sequences applied in real‐time PCR
| IL‐6 | Forward 5′‐3′ | CAACGATGATGCACTTGCAGA |
| Reverse 5′‐3′ | CTCCAGGTAGCTATGGTACTCCAGA | |
| TNF‐α | Forward 5′‐3′ | ACTCCAGGCGGTGCCTATGT |
| Reverse 5′‐3′ | GTGAGGGTCTGGGCCATAGAA | |
| IFN‐γ | Forward 5′‐3′ | AAGCGTCATTGAATCACACCTG |
| Reverse 5′‐3′ | TGACCTCAAACTTGGCAATACTC | |
| IL‐10 | Forward 5′‐3′ | GGGGCCAGTACAGCCGGGAA |
| Reverse 5′‐3′ | CTGGCTGAAGGCAGTCCGCA | |
| T‐bet | Forward 5′‐3′ | TCAACCAGCACCAGACAGAGA |
| Reverse 5′‐3′ | TCCACCAAGACCACATCCAC | |
| GATA3 | Forward 5′‐3′ | GGATGTAAGTCGAGGCCCAAG |
| Reverse 5′‐3′ | ATTGCAAAGGTAGTGCCCGGTA | |
| Il‐4 | Forward 5′‐3′ | ACGGAGATGGATGTGCCAAAC |
| Reverse 5′‐3′ | AGCACCTTGGAAGCCCTACAGA | |
| ROR‐γT | Forward 5′‐3′ | ACGGCCCTGGTTCTCATCA |
| Reverse 5′‐3′ | CCAAATTGTATTGCAGATGTTCCAC | |
| IL‐17a | Forward 5′‐3′ | GCAAAAGTGAGCTCCAGAAGG |
| Reverse 5′‐3′ | TCTTCATTGCGGTGGAGAGTC | |
| TGF‐β | Forward 5′‐3′ | TGTGGAACTCTACCAGAAATATAGC |
| Reverse 5′‐3′ | GAAAGCCCTGTATTCCGTCTC | |
| Foxp3 | Forward 5′‐3′ | CACCTATGCCACCCTTATCCG |
| Reverse 5′‐3′ | CATGCGAGTAAACCAATGGTAGA | |
| GAPDH | Forward 5′‐3′ | AGGTCGGTGTGAACGGATTTG |
| Reverse 5′‐3′ | GGGGTCGTTGATGGCAACA |
Statistical analysis
Data are presented as the mean ± SEM. All data were analysed using one‐way analysis of variance with a Bonferroni post hoc test for multiple t‐tests. A value of P < 0·05 was considered statistically significant.
Results
Tregs accumulation is increased in LPS‐induced ARDS
Regulatory T‐cells represent approximately 5% of the CD4+ T‐cell compartment in uninjured lungs of BALB/c mice. ARDS generated by intratracheal administration of LPS in mice leads to the dynamic accumulation of a distinct population of Tregs within 1 week. The number of pulmonary Tregs significantly increased on day 1 after LPS administration and remained high on day 2, followed by a decrease on day 4. After 7 days of recovery, pulmonary Tregs were essentially normalized (Fig. 1a–c). In addition, irrespective of pulmonary injury, the increase in splenic Treg number showed similar dynamic variation (Fig. 1a,d,e). Tregs in the spleen reflected a parallel change in the lung, and the disturbance of Tregs recovered within 1 week in the lungs and spleen.
Figure 1.

Regulatory T‐cell (Treg) accumulation is increased in lipopolysaccharide (LPS)‐induced acute respiratory distress syndrome (ARDS). BALB/c mice were intratracheally injected with LPS. The experiment was repeated twice. (a) Cytofluorometric dot plots of Tregs within days after injury. Numbers depict the fraction of CD4 + T‐cells within the designated gate. (b) Summary data for the percentage and (c) number of Tregs in the lungs depicted in (a). (d) Summary data for the percentage and (e) number of Tregs in the spleen depicted in (a). n = 8, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; and ****P ≤ 0·0001.
Impairment of the resolution of pulmonary injury in Tregs‐depleted mice
Next, we chose 2‐day and 4‐day time points to further identify the role of Tregs in LPS‐induced ARDS. A Treg‐depleted mouse model was established by injection of anti‐CD25 antibody every 7 days (Fig. 2a). First, flow cytometry was performed to confirm the Treg depletion effects after anti‐CD25 treatment (Fig. S1a–d), and the numbers of CD4+ effector T‐cells in the lungs and spleen were not interfered (Fig. S1e–i). Then, the histopathological changes of mouse lungs were observed after HE staining. No obvious abnormalities were observed in the saline group and the saline + anti‐CD25 group. When compared with those in the LPS group, mice in the LPS + anti‐CD25 group did not demonstrate exacerbated inflammatory histopathological changes and histopathological score on days 2 or 4 (Fig. 2b,c).
Figure 2.

Impairment of the resolution of pulmonary injury in regulatory T‐cell (Treg)‐depleted mice. (a) Schematic diagrams of the experimental design. Mice were intraperitoneally injected with 100 μg of anti‐CD25 antibody or IgG 10 days before lipopolysaccharide (LPS) exposure and repeatedly treated every 7 days for continuous Treg depletion. Mice were killed on 2 or 4 days after LPS administration. (b) Pulmonary histopathological changes in mice as determined with haematoxylin and eosin (HE) staining. Representative images of the lungs (magnification 100× and 200×). (c) Pulmonary histopathological score of (b). (d) Total cell counts in bronchoalveolar lavage fluid (BALF) were evaluated by a haemocytometer. (e) Schematic sorting of CD11b+Ly6C+ Ly6G+F4/80‐ neutrophil and CD11b+Ly6C+Ly6G‐F4/80 + macrophages in lung suspension. (f) Percentage of neutrophils, (g) macrophages and (h) CD3 + T lymphocytes in lung suspension were determined by flow cytometry. n = 6–8, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; and ****P ≤ 0·0001.
Total cell infiltration in BALF was detected after LPS exposure by using haematocyte counter. Total cell counts peaked on day 2 and decreased on day 4 after LPS administration, Treg depletion attenuated the total cell infiltration both on day 2 and day 4 after LPS administration, especially on day 2 (Fig. 2d). The cell types were further differentiated via flowcytometry sorting of neutrophils, macrophages in lung suspension followed by previous study15 (Fig. 2e), and T lymphocytes were also detected. Treg depletion downregulated neutrophils and upregulated macrophages on day 2 and day 4 after LPS administration, without influencing T lymphocytes (Fig. 2f–h).
Pulmonary epithelium is impaired and endothelial proliferation is abrogated in Treg‐depleted mice
Multicolour flow cytometry was used to identify specific pulmonary epithelium and vascular endothelium in single lung cell suspensions as follows: epithelium (CD45− CD326+ CD31−); endothelium (CD45− CD326− CD31+). In addition, the proliferation of epithelium or endothelium was detected by marker of ki67. In our study, we detected that the number of epithelial cells was decreased on day 4 but not on day 2 in the LPS + anti‐CD25 group (Fig. 3a–c), and no significant difference in the number of endothelial cells was observed in Treg‐depleted mice after LPS administration (Fig. 3d,e). After injection of anti‐CD25 antibody, although epithelial proliferation was not affected when mice received an intratracheal instillation of LPS, endothelial proliferation was impaired on day 4 (Fig. 3f,g). The schematic gating of ki67 + epithelium or endothelium was presented by flow cytometry (Fig. 3h).
Figure 3.
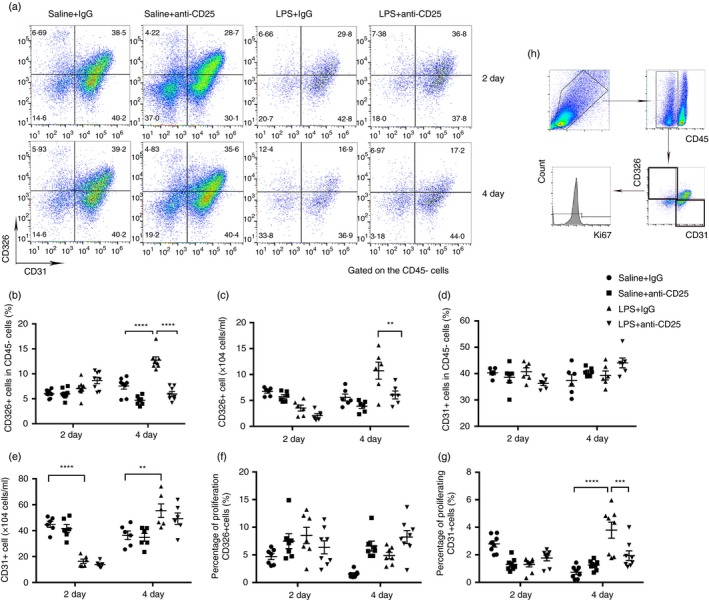
Pulmonary epithelium is impaired and endothelial proliferation is abrogated in regulatory T‐cell (Treg)‐depleted mice. (a) Cytofluorometric dot plots of epithelial and endothelial cells within days after injury. Numbers depict the fraction of CD45‐ cells within the designated gate. (b) Summary data for the percentage and (c) number of CD326 + epithelial cells in the lungs depicted in (a). (d) Summary data for the percentage and (e) number of CD31 + endothelial cells in the lungs depicted in (a). (f) Percentage of proliferating CD326 + cells and (g) percentage of proliferating CD31 + cells in the lungs. (h) Schematic gating of ki67 + epithelium or endothelium. n = 6, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; and ****P ≤ 0·0001.
Tregs promote Th1 and Th17 immune responses during LPS‐induced pulmonary inflammation
To explore whether Tregs regulated LPS‐induced pulmonary inflammation by modulating immune responses, the phenotype of CD4+ T lymphocytes in the lungs or spleen and the levels of cytokines were evaluated. As shown in Fig. 4, the percentage of IFN‐γ‐producing Th1 cells was decreased in the lungs or spleens of anti‐CD25‐treated mice on day 4 after LPS administration (Fig. 4a–d). Anti‐CD25 treatment reduced the concentration of IFN‐γ in BALF on day 4 after LPS exposure but not on day 2 (Fig. 4e). The variation of expression of IFN‐γ and T‐bet (Th1 transcription factor) determined by real‐time PCR was in accordance with the dynamic changes of IFN‐γ in BALF (Fig. 4f,g).
Figure 4.
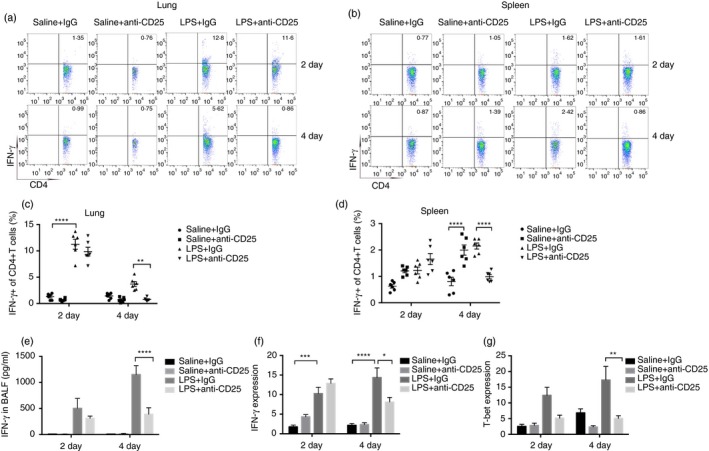
Regulatory T‐cells (Tregs) promote Th1 immune responses during lipopolysaccharide (LPS)‐induced pulmonary inflammation. (a) Cytofluorometric dot plots of IFN‐ɣ+Th1 cells in the lungs. Numbers depict the fraction of Th1 cells within the designated gate. (b) Cytofluorometric dot plots of IFN‐ɣ+ Th1 cells in the spleen. Numbers depict the fraction of Th1 cells within the designated gate. (c) Summary data for the percentage of Th1 cells in the lungs depicted in (a). (d) Summary data for the percentage of Th1 cells in the spleen depicted in (b). (e) Concentration of IFN‐ɣ in bronchoalveolar lavage fluid (BALF) was detected by bead‐based Th immunoassays kit. (f) IFN‐ɣ mRNA expression and (g) T‐bet mRNA expression in the lungs were assayed by real‐time polymerase chain reaction (PCR). n = 6, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; and ****P ≤ 0·0001.
The Th17 response was also studied in mice treated with anti‐CD25 (Fig. 5). Although the flow cytometry results showed a marked increase in Th17 cells in the spleen of the LPS + anti‐CD25 group compared with that of the LPS group on day 2, the percentage of IL‐17‐producing Th17 cells was markedly lower in the lungs of the LPS + anti‐CD25 group than in those of the LPS group, especially on day 4 (Fig. 5a–c). The same results were also observed in the spleen on day 4 (Fig. 5b,d). The concentrations of both IL‐17a and IL‐22 in the BALF were clearly lower in the LPS + anti‐CD25 group than in the LPS group, and the decrease was especially profound on day 4 (Fig. 5e,f). The level of IL17a was clearly lower in anti‐CD25 treatment after LPS administration on day 4 (Fig. 5g). A similar observation was noted regarding the expression of ROR‐γt, a Th17 transcription factor (Fig. 5h).
Figure 5.
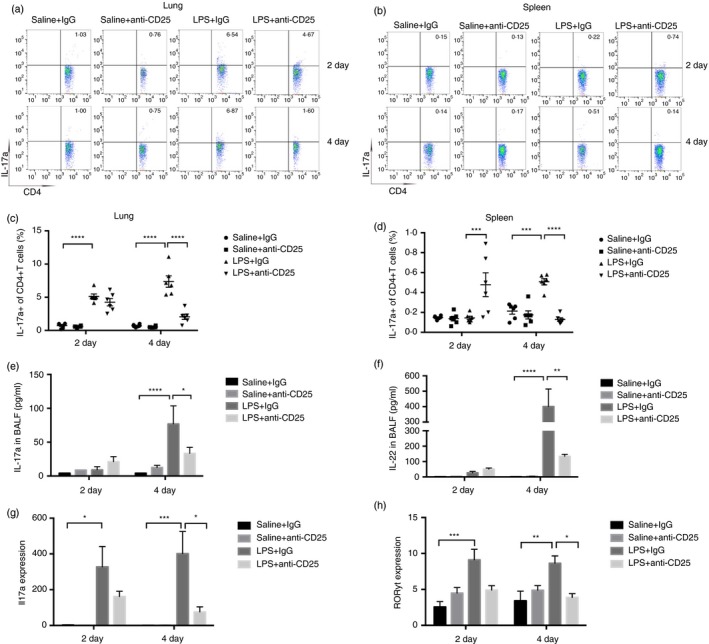
Regulatory T‐cells (Tregs) promote Th17 immune responses during lipopolysaccharide (LPS)‐induced pulmonary inflammation. (a) Cytofluorometric dot plots of IL17a+ Th17 cells in the lungs. Numbers depict the fraction of Th17 cells within the designated gate. (b) Cytofluorometric dot plots of IL17a+ Th17 cells in spleen. Numbers depict the fraction of Th17 cells within the designated gate. (c) Summary data for the percentage of Th17 cells in the lungs depicted in (a). (d) Summary data for the percentage of Th17 cells in the spleen depicted in (b). (e) Concentration of IL‐17a, (f) IL‐22 in bronchoalveolar lavage fluid (BALF) was detected by bead‐based Th immunoassays kit. (g) Il17a mRNA expression and (h) ROR‐ɣT mRNA expression were assayed by real‐time polymerase chain reaction (PCR) in the lungs. n = 6, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; and ****P ≤ 0·0001.
Finally, we examined the Th2 response and found Tregs did not regulate Th2 immune responses during LPS‐induced pulmonary inflammation (Fig. S2).
Tregs might modulate Th responses via TGF‐ß1 signalling
To further explore the mechanism of Tregs in the modulation of Th responses, the representative proinflammatory cytokines IL‐6, TNF‐α, TGF‐β1 and IL‐10 were examined by real‐time PCR and bead‐based immunoassays. LPS administration led to increased expression and secretion of both IL‐6 and TNF‐α in mouse lungs and BALF on day 2. However, the expression and release of IL‐10 in the lungs were not statistically significant (data not shown). The expression of IL‐6/ TNF‐α and release of IL‐6/ TNF‐α into the BALF in the LPS + anti‐CD25 group was markedly increased compared with that in the LPS group on day 2 (Fig. 6a–d). However, the expression and release of TGF‐β1 were reduced in the LPS + anti‐CD25 group compared with the LPS group on day 4 (Fig. 6e,f).
Figure 6.

Regulatory T‐cells (Tregs) might modulate Th responses via TGF‐ß1 signalling. (a) Secretion of cytokine IL‐6 in bronchoalveolar lavage fluid (BALF). (b) IL‐6 mRNA expression in the lungs. (c) Secretion of cytokine TNF‐α in BALF. (d) TNF‐α mRNA expression in the lungs. (e) Secretion of cytokine TGF‐ß1 in BALF. (f) TGF‐ß1 mRNA expression in the lungs. n = 6, Mean ± SEM. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; and ****P ≤ 0·0001.
Discussion
In our study, Tregs promoted tissue repair and the resolution of LPS‐induced pulmonary inflammation by modulating the Th immune response. In vivo depletion of Tregs by injection of PC61 anti‐CD25 antibody resulted in parallelly diminished Th1 and Th17 immune responses after LPS administration.
Tregs were only recently identified, and participated in the regulation of several T‐cell‐associated diseases, such as inflammatory bowel disease and asthma.16, 17 Foxp3 is a key transcription factor and CD25 is a critical surface marker regulating the development and maturation of Tregs. Previous studies on Treg depletion used Foxp3DTR mice and intraperitoneal administration of diphtheria toxin1, 18 or anti‐CD25 neutralization. As CD25 mAb (PC61 mAb) has been confirmed to deplete Foxp3+ Tregs19, 20, 21 and PC61 has been shown to cause a significant reduction in Foxp3+ Tregs by phagocytes via Ab‐dependent cellular cytotoxicity.22 In our study, we employed the widely used and easily approachable strategy of PC61 anti‐CD25 mAb treatment to deplete CD4+ FOXP3+ Tregs prior to LPS administration in mice. Although PC61 does not lead to complete elimination of Foxp3+ Tregs, the partial depletion of Tregs induced by this method has been shown to adequately alter the immunological balance for further exploration of the role of Tregs.23 When considering CD25 is also expressed on most activated T‐cell subsets, we also explored the CD4+ effector T‐cells in both lung and spleen. Our study confirmed that 100 μg of anti‐CD25 treatment contributed to limit Foxp3+ Tregs generation without influencing CD4+ effector T‐cells.
Acute bacterial inflammation usually recruits neutrophils with a subsequent activation of macrophages, which are critically involved in the first line of defence against microbes. Neutrophils release inflammatory cytokines, or proteinases, and cause the formation of extracellular traps (NETs). Mitigated neutrophil infiltration induced delayed resolution of cutaneous wound infection with advanced age in rat,24, 25 and blocking NETs formation reduced the capture of circulating bacteria and aggravated the pathology that followed by a polymicrobial sepsis in vivo, resulting in increased dissemination to distant organs, even impaired vascular remodelling.26 Treg depletion inhibited total cell infiltration, ameliorated neutrophil infiltration and promoted macrophages recruitment, which might manifest impairment of inflammation resolution and pulmonary repair.
The histopathological score of pulmonary section was mainly aimed at exploring the inflammation response and inflammation cells in the lungs. When considering pulmonary repair, epithelial or endothelial cells in the lungs were further detected by flow cytometry. In our study, Treg depletion attenuated pulmonary repair by impairing pulmonary CD326+ epithelial cells, which was consistent with a previous study.1 The peak epithelial proliferation was reported to occur on day 7 after I.T. LPS exposure, this might be why we did not observe evident epithelial proliferation when our experiment time was up to 4 days. Although Treg depletion did not interfere with the CD31+ endothelial cells, it damaged the proliferation of endothelium in the lungs, which was in accordance with reported elevation of pulmonary permeability when Tregs were eliminated. However, we found there is a large percentage of CD45− cells that co‐stain for both CD31 and CD326, as from a previous study.27 This is maybe related with activation of epithelium‐to‐endothelium transition by enzyme digestion. Subgrouping of type 1 or 2 alveolar epithelium is possible by adding additional marker to further explore the co‐stain cells.
Tregs influence the balance of Th1/Th2/Th17 during various inflammatory diseases. Th1 immune responses are considered to be crucial in orchestrating the host defence against intracellular pathogens and in the pathogenesis of many immune‐mediated diseases.28, 29 Th1 cytokines, such as IFN‐ɣ, TNF‐a and IL‐2, are released in response to many innate stimuli, and can regulate both innate and adaptive immune activities.30 T‐bet is a critical regulator of Th1 differentiation, and controls Th1 cell cytokine production or migration by regulating the expression of chemokines and their receptors.31 Tregs are generally viewed as a suppressor of Th1 immune responses in various inflammatory diseases.32, 33 However, our finding contradicts the result acquired by Liu et al.20 Differences in the genetic background or the microbiome in the animal may lead to these divergent results; moreover, the discrepant results may also be related to Th1 lymphocyte plasticity.34, 35 Although we observe a decrease in Th1 immune responses following LPS‐induced pulmonary damage in Treg‐depleted mice, we could not exclude the possibility that the kinetics of their elevation presence in the lungs when the observed time points were prolonged.
Recent evidence has shown that Th17 immune responses are crucial in protecting mucosal surfaces against microbial pathogens, including bacteria, fungi and viruses.36, 37, 38 Moreover, Tregs were shown to be tightly correlated with the promotion of the Th17 immune response in various models of inflammation. The levels of Th17‐related cytokines, such as IL‐17a, IL‐17f and IL‐22, were markedly increased in the LPS‐induced mouse model of ALI, inducing the pulmonary epithelium to produce chemokines that favour neutrophil infiltration, which is regarded as a proinflammatory mediator and contributor to airway remodelling.16,17 Blockade of IL‐17 may facilitate pulmonary fibrosis in asthma.39 However, the relevance of Tregs in the induction of Th17 responses against bacterial infection remains elusive. In a murine model of Chlamydia muridarum‐induced genital tract infection, Tregs were determined to serve as a prominent inducer of and direct contributor to IL‐17/Th17 responses.40 Tregs were also shown to stimulate the Th17‐mediated inflammatory response in a CTLA4‐dependent manner in colon inflammation.41 During oral Candida albicans infection in mice, Tregs were shown to promote acute Th17 cell responses to suppress mucosal fungus infections and maintain immune homeostasis.42 In accordance with those findings, we confirmed that Tregs could promote the Th17 immune response in LPS‐induced ARDS.
Th2 cells are recognized for their role in host defence against multicellular parasites, and are involved in allergies and atopic illnesses. Th2 cells release various pathophysiological cytokines (IL‐4, IL‐5, IL‐13) and stimulate type 2 immunity, such as in human asthma, as well as in an animal model of allergic airway inflammation.43, 44 However, type 2 immunity appeared to be lacking in the bacterial infection mouse model. Consistent with our observations, Treg depletion in bacterial infection has been shown to have a limited impact on Th2 responses.
As CD4+ Th subsets possess the characteristics of plasticity, Foxp3+ Tregs have been shown to convert into Th1 cells or Th17 cells under certain conditions.45 Moreover, Th17 cells highly expressing aryl hydrocarbon receptors can convert into Tregs during the resolution of inflammation in the presence of TGF‐β.46 Th1 cells can also convert into Th17 cells under inflammatory conditions in the intestine mediated by TGF‐ß‐induced Runx1 expression.47 Therefore, Treg depletion might impair the reciprocal conversion among Treg, Th1 and Th17 cells in LPS‐induced pulmonary inflammation and lead to reduced Th1 and Th17 subsets.
Importantly, Treg depletion after LPS administration had an effect on the Th1 and Th17 immune responses on day 4 in LPS‐induced ARDS. Therefore, we concluded that the time frame in which Tregs promote Th1 and Th17 cell development and their function is probably not short.
To investigate the mechanism by which Tregs regulate the Th subpopulation, we detected the proinflammatory cytokines IL‐6, TNF‐α, IL‐10 and TGF‐ß. IL‐6 and TNF‐α are two significant proinflammatory factors used to evaluate inflammation. The elevated level of IL‐6 and TNF‐α confirmed that Treg depletion aggravated pulmonary inflammation after LPS administration. The elevation was detected on day 2, which is inconsistent with the time frame of changes in Th1 and Th17. IL‐10 and TGF‐ß1 are two crucial factors secreted by Tregs and play vital roles in promoting the differentiation of Th1 and Th17 cells in the proinflammatory cytokine‐enriched microenvironment after infection. TGF‐ß1 was highly expressed by activated T‐cells, more importantly implicated in Treg‐suppressive activity and Foxp3 expression,3 facilitated the differentiation of Th17 cells and controlling inflammatory diseases,48, 49 and enhanced effector Th1 cell activation via indirect pathway.50 Although there was no significant difference in the level of IL‐10 in BALF and the lungs, we found that the expression and release of TGF‐β1 were decreased in Treg‐depleted mice 4 days after LPS administration, paralleling the time at which Th1 and Th17 variations are observed. Therefore, we predicted that Tregs might likely modulate Th1 and Th17 differentiation in a TGF‐β1‐dependent manner in a mouse model of LPS‐induced ARDS. As we lack the further verified evidence, the TGF‐ß1‐related mechanism involved in both cell and animal experiments would be performed in our further study.
Both innate and adaptive immunity are required for ALI, and their respective role in pulmonary repair is well established. In our study, we found Treg depletion can also affect innate immune cells, including neutrophils and macrophages. Mounting evidence has been implicated that macrophages especially the M2 subset could induce Tregs51, 52 and, inversely, Treg cells can also promote macrophage efferocytosis during inflammation resolution in zymosan‐induced peritonitis and LPS‐induced lung injury.53 Although few studies have demonstrated the direct effect between Treg and neutrophil, it is still reported that Treg can induce il‐10‐producing neutrophils.54 At present, there is a lack of adequate evidence to explicitly clarify the intricate interaction between various immune cells. Consequently, the crosstalk between the innate and adaptive immune response will become an area of emerging exploration.
Tregs have been ascribed beneficial or detrimental roles during infection depending on the nature of the infectious agent or whether the infection is acute or chronic. Taken together, the data show that Tregs promote tissue repair and the resolution of LPS‐induced pulmonary inflammation by promoting Th1 and Th17 responses. Moreover, our study provided vital supplemental evidence for immune cell involvement and revealed the Th response profile in LPS‐induced pulmonary inflammation. In addition, our study showed the modulation of the Th response by Tregs.
Disclosures
The authors declare that there are no financial or commercial conflicts of interest.
Supporting information
Figure S1. Anti‐CD25 antibody contributed to the depletion of Tregs without influencing CD4+ effector T‐cells.
Figure S2. Tregs do not regulate Th2 immune responses during LPS‐induced pulmonary inflammation.
Acknowledgements
Wen Tan performed the experiments and wrote the paper, Chaoji Zhang and Jianzhou Liu analysed the data, and Qi Miao designed the study. This work was supported by a grant from the CAMS initiative for innovation medicine (NO. CAMS‐I2M‐1‐003).
References
- 1. Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J, Limjunyawong N, Singer BD et al Foxp3 + regulatory T cells promote lung epithelial proliferation. Mucosal Immunol 2014; 7:1440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang L, Yuan R, Yao C, Wu Q, Christelle M, Xie W et al Effects of resolvin D1 on inflammatory responses and oxidative stress of lipopolysaccharide‐induced acute lung injury in mice. Chin Med J (Engl) 2014; 127:803–9. [PubMed] [Google Scholar]
- 3. D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF et al CD4 + CD25 + Foxp3 + Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest 2009; 119:2898–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S et al A distinct function of regulatory T cells in tissue protection. Cell 2015; 162:1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohm L, Maxeiner J, Meyer‐Martin H, Reuter S, Finotto S, Klein M et al IL‐10 and regulatory T cells cooperate in allergen‐specific immunotherapy to ameliorate allergic asthma. J Immunol 2015; 194:887–97. [DOI] [PubMed] [Google Scholar]
- 6. Hou J, Sun Y, Hao Y, Zhuo J, Liu X, Bai P et al Imbalance between subpopulations of regulatory T cells in COPD. Thorax 2013; 68:1131–9. [DOI] [PubMed] [Google Scholar]
- 7. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A et al Foxp3 + CD4 + T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res 2014; 115:55–67. [DOI] [PubMed] [Google Scholar]
- 8. Tonkin DR, Haskins K. Regulatory T cells enter the pancreas during suppression of type 1 diabetes and inhibit effector T cells and macrophages in a TGF‐beta‐dependent manner. Eur J Immunol 2009; 39:1313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu J, Zhang PS, Yu Q, Liu L, Yang Y, Guo FM et al Losartan inhibits conventional dendritic cell maturation and Th1 and Th17 polarization responses: Novel mechanisms of preventive effects on lipopolysaccharide‐induced acute lung injury. Int J Mol Med 2012; 29:269–76. [DOI] [PubMed] [Google Scholar]
- 10. Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T helper cells plasticity in inflammation. Cytometry A 2014; 85:36–42. [DOI] [PubMed] [Google Scholar]
- 11. Xu S, Xu M, Li GG, Wang C, Song H, Bai J. Early recruitment of IL‐10‐producing B cells into alveoli improved the resolution of acute lung injury. Cell Physiol Biochem 2016; 38:1752–60. [DOI] [PubMed] [Google Scholar]
- 12. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 Cells. Annu Rev Immunol 2009; 27:485–517. [DOI] [PubMed] [Google Scholar]
- 13. Kushwah R, Hu J. Dendritic cell apoptosis: regulation of tolerance versus immunity. J Immunol 2010; 185:795–802. [DOI] [PubMed] [Google Scholar]
- 14. Yaxin W, Shanglong Y, Huaqing S, Hong L, Shiying Y, Xiangdong C et al Resolvin D1 attenuates lipopolysaccharide induced acute lung injury through CXCL‐12/CXCR4 pathway. J Surg Res 2014; 188:213–21. [DOI] [PubMed] [Google Scholar]
- 15. Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W et al IL‐22‐producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA 2013; 110:12768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang H, Wu X, Zhu H, Xie Y, Tang S, Jiang Y. FOXP3(+)Treg/Th17 cell imbalance in lung tissues of mice with asthma. Int J Clin Exp Med 2015; 8:4158–63. [PMC free article] [PubMed] [Google Scholar]
- 17. Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A et al The autophagy gene Atg16 l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife 2016; 5:e12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Christiaansen AF, Boggiatto PM, Varga SM. Limitations of Foxp3(+) Treg depletion following viral infection in DEREG mice. J Immunol Methods 2014; 406:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak‐Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature 2011; 480:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu F, Lu X, Dai W, Lu Y, Li C, Du S et al IL‐10‐producing B cells regulate T helper cell immune responses during 1,3‐beta‐glucan‐induced lung inflammation. Front Immunol 2017; 8:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okeke EB, Okwor I, Mou Z, Jia P, Uzonna JE. CD4 + CD25 + regulatory T cells attenuate lipopolysaccharide‐induced systemic inflammatory responses and promotes survival in murine Escherichia coli infection. Shock 2013; 40:65–73. [DOI] [PubMed] [Google Scholar]
- 22. Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4 + FOXP3 + Treg cells by the PC61 anti‐CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol 2010; 40:780–6. [DOI] [PubMed] [Google Scholar]
- 23. Tenorio EP, Fernandez J, Olguin JE, Saavedra R. Depletion with PC61 mAb before Toxoplasma gondii infection eliminates mainly Tregs in BALB/c mice, but activated cells in C57BL/6J mice. FEMS Immunol Med Microbiol 2011; 62:362–7. [DOI] [PubMed] [Google Scholar]
- 24. Ebaid H. Neutrophil depletion in the early inflammatory phase delayed cutaneous wound healing in older rats: improvements due to the use of un‐denatured camel whey protein. Diagn Pathol 2014; 9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meng W, Paunel‐Gorgulu A, Flohe S, Hoffmann A, Witte I, MacKenzie C et al Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care 2012; 16:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baluk P, Phillips K, Yao L‐C, Adams A, Nitschké M, McDonald DM. Neutrophil dependence of vascular remodeling after mycoplasma infection of mouse airways. Am J Pathol 2014; 184:1877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singer BD, Mock JR, D'Alessio FR, Aggarwal NR, Mandke P, Johnston L et al Flow‐cytometric method for simultaneous analysis of mouse lung epithelial, endothelial, and hematopoietic lineage cells. Am J Physiology Lung Cell Mol Physiol 2016; 310:L796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M et al The microRNA miR‐29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon‐γ . Nat Immunol 2011; 12:861–9. [DOI] [PubMed] [Google Scholar]
- 29. Tsolis RM, Barquero‐Calvo E, Martirosyan A, Ordoñez‐Rueda D, Arce‐Gorvel V, Alfaro‐Alarcón A et al Neutrophils exert a suppressive effect on Th1 responses to intracellular pathogen Brucella abortus . PLoS Pathog 2013; 9:e1003167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doherty TM, Watanabe S, Inoue J. Intracellular delivery of lipopolysaccharide induces effective Th1‐immune responses independent of IL‐12. PLoS ONE 2013; 8:e68671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szabo SJ, Kim ST, Costa GL, Zhang XK, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell 2000; 100:655–69. [DOI] [PubMed] [Google Scholar]
- 32. Joller N, Lozano E, Burkett Patrick R, Patel B, Xiao S, Zhu C et al Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014; 40:569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paust HJ, Riedel JH, Krebs CF, Turner JE, Brix SR, Krohn S et al CXCR3 + regulatory T cells control TH1 responses in crescentic GN. J Am Soc Nephrol 2016; 27:1933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N et al Atherosclerosis‐driven treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs Novelty and significance. Circ Res 2016; 119:1190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA et al Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 2011; 186:3918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holley MM, Kielian T. Th1 and Th17 cells regulate innate immune responses and bacterial clearance during central nervous system infection. J Immunol 2012; 188:1360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bystrom J, Al‐Adhoubi N, Al‐Bogami M, Jawad AS, Mageed RA. Th17 lymphocytes in respiratory syncytial virus infection. Viruses 2013; 5:777–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y, Liu W, Russell MW. Suppression of host adaptive immune responses by Neisseria gonorrhoeae: role of interleukin 10 and type 1 regulatory T cells. Mucosal Immunol 2013; 7:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Camargo LDN, Righetti RF, Aristoteles L, Dos Santos TM, de Souza FCR, Fukuzaki S et al Effects of anti‐IL‐17 on inflammation, remodeling, and oxidative stress in an experimental model of asthma exacerbated by LPS. Front Immunol 2017; 8:1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moore‐Connors JM, Fraser R, Halperin SA, Wang J. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 responses and genital tract inflammation upon intracellular Chlamydia muridarum infection. J Immunol 2013; 191:3430–9. [DOI] [PubMed] [Google Scholar]
- 41. Watanabe N, Kaminuma O, Kitamura N, Hiroi T. Induced treg cells augment the Th17‐mediated intestinal inflammatory response in a CTLA4‐dependent manner. PLoS ONE 2016; 11:e0150244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez‐Santos N et al CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 2011; 34:422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fahy JV. Type 2 inflammation in asthma — present in most, absent in many. Nat Rev Immunol 2015; 15:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansbro PM, Scott GV, Essilfie AT, Kim RY, Starkey MR, Nguyen DH et al Th2 cytokine antagonists: potential treatments for severe asthma. Expert Opin Investig Drugs 2013; 22:49–69. [DOI] [PubMed] [Google Scholar]
- 45. Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev 2014; 259:159–72. [DOI] [PubMed] [Google Scholar]
- 46. Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW et al Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015; 523:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu H‐P, Cao AT, Feng T, Li Q, Zhang W, Yao S et al TGF‐β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol 2015; 45:1010–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor‐beta1 promotes in vivo Th17 cell differentiation. Immunity 2011; 34:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL et al Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med 2009; 206:2407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huss DJ, Winger RC, Peng H, Yang Y, Racke MK, Lovett‐Racke AE. TGF‐beta enhances effector Th1 cell activation but promotes self‐regulation via IL‐10. J Immunol 2010; 184:5628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Savage NDL, de Boer T, Walburg KV, Joosten SA, van Meijgaarden K, Geluk A et al Human anti‐inflammatory macrophages induce Foxp3 + GITR+CD25 + regulatory T cells, which suppress via membrane‐bound TGF ‐1. J Immunol 2008; 181:2220–6. [DOI] [PubMed] [Google Scholar]
- 52. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB et al IL‐33 contributes to sepsis‐induced long‐term immunosuppression by expanding the regulatory T cell population. Nat Commun 2017; 8:14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr, Sozen E, Subramanian M et al Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity 2018; 49:666–77 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lewkowicz N, Mycko MP, Przygodzka P, Ćwiklińska H, Cichalewska M, Matysiak M et al Induction of human IL‐10‐producing neutrophils by LPS‐stimulated Treg cells and IL‐10. Mucosal Immunol 2015; 9:364–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Anti‐CD25 antibody contributed to the depletion of Tregs without influencing CD4+ effector T‐cells.
Figure S2. Tregs do not regulate Th2 immune responses during LPS‐induced pulmonary inflammation.