

ABSTRACT
Macroautophagy/autophagy is a lysosome-dependent catabolic process for the turnover of proteins and organelles in eukaryotes. Autophagy plays an important role in immunity and inflammation, as well as metabolism and cell survival. Diverse immune and inflammatory signals induce autophagy in macrophages through pattern recognition receptors, such as toll-like receptors (TLRs). However, the physiological role of autophagy and its signaling mechanisms in microglia remain poorly understood. Microglia are phagocytic immune cells that are resident in the central nervous system and share many characteristics with macrophages. Here, we show that autophagic flux and expression of autophagy-related (Atg) genes in microglia are significantly suppressed upon TLR4 activation by lipopolysaccharide (LPS), in contrast to their stimulation by LPS in macrophages. Metabolomics analysis of the levels of phosphatidylinositol (PtdIns) and its 3-phosphorylated form, PtdIns3P, in combination with bioinformatics prediction, revealed an LPS-induced reduction in the synthesis of PtdIns and PtdIns3P in microglia but not macrophages. Interestingly, inhibition of PI3K, but not MTOR or MAPK1/3, restored autophagic flux with concomitant dephosphorylation and nuclear translocation of FOXO3. A constitutively active form of FOXO3 also induced autophagy, suggesting FOXO3 as a downstream target of the PI3K pathway for autophagy inhibition. LPS treatment impaired phagocytic capacity of microglia, including MAP1LC3B/LC3-associated phagocytosis (LAP) and amyloid β (Aβ) clearance. PI3K inhibition restored LAP and degradation capacity of microglia against Aβ. These findings suggest a unique mechanism for the regulation of microglial autophagy and point to the PI3K-FOXO3 pathway as a potential therapeutic target to regulate microglial function in brain disorders.
Abbreviations: Atg: autophagy-related gene; Aβ: amyloid-β; BafA1: bafilomycin A1; BECN1: beclin 1, autophagy related; BMDM: bone marrow-derived macrophage; CA: constitutively active; CNS: central nervous system; ZFYVE1/DFCP1: zinc finger, FYVE domain containing 1; FOXO: forkhead box O; ELISA:enzyme-linked immunosorbent assay; HBSS: Hanks balanced salt solution; LAP: LC3-associated phagocytosis; MAP1LC3B: microtubule-associated protein 1 light chain 3; LPS: lipopolysaccharide; LY: LY294002; MTOR: mechanistic target of rapamycin kinase; Pam3CSK4: N-palmitoyl-S-dipalmitoylglyceryl Cys-Ser-(Lys)4; PtdIns: phosphatidylinositol; PtdIns3P: phosphatidylinositol-3-phosphate; PLA: proximity ligation assay; Poly(I:C): polyinosinic-polycytidylic acid; qRT-PCR: quantitative real-time polymerase chain reaction; RPS6KB1: ribosomal protein S6 kinase, polypeptide 1; TLR: Toll-like receptor; TNF: tumor necrosis factor; TFEB: transcription factor EB; TSPO: translocator protein.
KEYWORDS: Amyloid, FOXO3, LC3-associated phagocytosis, microglia, PI3K, PtdIns3K
Introduction
Autophagy is an evolutionarily conserved intracellular degradation process of cytosolic constituents through formation of autophagosomes followed by their fusion with lysosomes, which results in autolysosome formation [1,2]. In addition to the conventional homeostatic and adaptive function of autophagy, recent studies have shown that autophagy also plays critical roles in immunity by regulating cytokine production and release, inflammasome activation, antigen presentation, and clearance of invading pathogens [3–6].
Accumulating lines of evidence indicate that autophagy is necessary for optimal immune function. For example, autophagy removes invading pathogens such as mycobacteria or toxic materials [7]. Mutations in genes involved in autophagy, such as those encoding ATG16L1 (autophagy-related protein 16 like 1), NOD2, and IRGM, are reproducibly associated with Crohn disease, a chronic inflammatory bowel disease, because these mutations impair defense against bacteria and lead to overproduction of proinflammatory cytokines [8–10]. Autophagy also controls the processing and transit of antigen-presenting major histocompatibility complex classes I and II to the cell surface to promote T cell responses to infection [11]. In addition, autophagy influences the transcription and release of several cytokines [12–14].
Autophagy is activated by inflammatory stimuli in various types of immune cells; among the latter, the best-studied in this respect are macrophages, the principal phagocytic cells at the front line of host defense. The activity of autophagy in macrophages contributes the phagocytic ability and degradation through MAP1LC3B/LC3-associated phagocytosis (LAP). The LAP process uses a subset of autophagy molecules to conjugate MAP1LC3B directly to the phagosome membrane and is required for the efficient clearance of pathogens, dead cells and misfolded protein [15–18]. Activation of toll-like receptors (TLRs), especially TLR4 and TLR7, induces autophagy in macrophages [19,20]. TLRs belong to the pattern recognition receptor family, and activation of TLR signaling upon exposure to microorganisms or their derivatives is crucial for immune cell activation and host defense [21]. Several proteins, such as RIP1, MAPK11/12/13/14 (formerly referred to as the p38 family), and HMOX1 (heme oxygenase 1), regulate lipopolysaccharide (LPS)-induced autophagy in macrophages [19,22,23]. LPS-induced autophagy is also observed in other cell types including hepatocytes, cardiomyocytes, and peritoneal mesothelial cells [24–26].
Microglia are principal immune cells in the central nervous system (CNS) that are important for normal brain development and homeostasis [27]. Microglia release inflammatory mediators and neurotrophic factors, engulf and degrade microbes, and phagocytose cellular debris and apoptotic cells [28]. As the brain’s professional phagocytes, microglia are also able to dispose of other brain-derived materials including myelin fragments, axonal and synaptic structures, as well as protein deposits, such as amyloid β (Aβ) plaques [29]. Through these capabilities, microglia respond to CNS damage and promote repair of injury and restoration of homeostasis in the brain [27]. However, in the wake of pathological insults or in the presence of developmental defects, activated microglia secrete multiple proinflammatory cytokines and other inflammatory mediators that are detrimental to nearby neurons. Thereby, microglia activation is one of the key pathogenic pathways causing tissue injury in various neurodegenerative diseases, such as Alzheimer disease (AD), Parkinson disease, and amyotrophic lateral sclerosis [30].
Ontogenically, microglia are myeloid cells of embryonic hematopoietic, not neural, origin; they migrate to the developing nervous system from the embryonic yolk sac at the very early days of development, as recently established in several species including humans and rodents [31]. Therefore, microglia are ontogenic relatives of peripheral tissue macrophages, and the 2 cell types share striking functional similarities and activation phenotypes. However, despite these physiological resemblances, whether immune signaling and autophagy interact similarly in microglia and macrophages has not yet been explored.
In this study, we examined the effects of TLR signaling activation on microglial autophagy. Interestingly, the autophagic responses elicited by LPS in microglia and macrophages were opposite. We found that LPS markedly repressed autophagy through inhibition of FOXO3 and impaired the phagocytic capability of microglia. Our findings reveal differential regulation of autophagy between activated microglia and macrophages and we propose the class I phosphoinositide 3-kinase (PI3K)-FOXO3 pathway as a potential therapeutic target for treatment of brain disorders through regulation of microglial function.
Results
Autophagy is suppressed by LPS in microglia
Autophagy induction by TLR ligands in macrophages is well established [20]. To examine the effect of TLR ligands on autophagy in microglia, we treated primary microglia with N-palmitoyl-S-dipalmitoylglyceryl Cys-Ser-(Lys)4 (Pam3CSK4), polyinosinic-polycytidylic acid (poly I:C), LPS, or viral single-stranded RNA, which are ligands specific to TLR1/2, TLR3, TLR4, and TLR7, respectively (Figure S1(a)). As a readout of the inflammatory response, release of TNF (tumor necrosis factor), a proinflammatory cytokine, was measured in the medium (Figure S1(b)). Autophagy was assessed by western blotting with an antibody detecting microtubule-associated protein 1 light chain 3 beta (MAP1LC3B), which is the most widely used marker for autophagosome formation [1]. Following autophagy induction, the soluble form of MAP1LC3B (MAP1LC3B-I) is covalently conjugated to phosphatidylethanolamine and becomes the lipidated form (MAP1LC3B-II) on elongating phagophore membranes. Therefore, an increase in the conversion of MAP1LC3B-I to MAP1LC3B-II can be used as an indicator of autophagosome formation [1,32]. Interestingly, all 4 tested ligands greatly decreased the MAP1LC3B-II level (Figure S1(a)). A decrease in MAP1LC3B-II level sometimes indicates its rapid consumption due to accelerated autophagic flux [1]. This possibility can be tested based on the amount of accumulated MAP1LC3B-II in the absence and presence of an autophagic flux blocker, such as bafilomycin A1 (BafA1), which is a selective inhibitor of the vacuolar-type H+-ATPase and blocks autophagy [33]. In the presence of BafA1, all TLR ligands still decreased MAP1LC3B-II accumulation compared to BafA1 only, indicating that the decrease was caused by a reduced autophagic flux rather than by accelerated autophagy (Figure S1(a)).
To study the underlying molecular mechanism, we selected LPS, a TLR4 ligand and prototypical potent immune stimulator. LPS is an endotoxin component derived from Gram-negative bacteria and is widely used to activate immune cells. We observed a remarkable decrease in MAP1LC3B-II level following LPS treatment in a wide range of doses, and BafA1 treatment confirmed that the decrease was due to a reduced autophagic flux (Figure 1(a)). For quantification of MAP1LC3B-II levels, we used 2 control samples, lane 1 (LPS− and BafA1−) and lane 5 (LPS− and BafA1+) as the references, and first compared control sample (lane 1) with BafA1-added samples (lane 5–8). Then, due to very low MAP1LC3B-II abundance in LPS-treated samples without BafA1 (lane 2–4), we increased exposure time and compared their levels with control (lane 1). Next, based on the ratio between 2 controls (lane 1 and 5), the relative MAP1LC3B-II band density of LPS-treated samples without and with BafA1 conditions was calculated. Autophagy suppression following LPS treatment was also detected in the murine microglial cell line BV-2 (Figure 1(c)). To compare these findings to those in macrophages, we examined the effect of LPS on autophagic flux in bone marrow-derived macrophages (BMDMs). In line with previous studies that reported autophagy induction by LPS in macrophages, LPS increased the MAP1LC3B-II level and autophagic flux in BMDMs (Figure 1(b)). Likewise, LPS treatment induced autophagy in the murine macrophage cell line Raw264.7 (Figure 1(d)). MAP1LC3B-II levels in BMDMs were quantified following the same way as in primary microglia.
Figure 1.
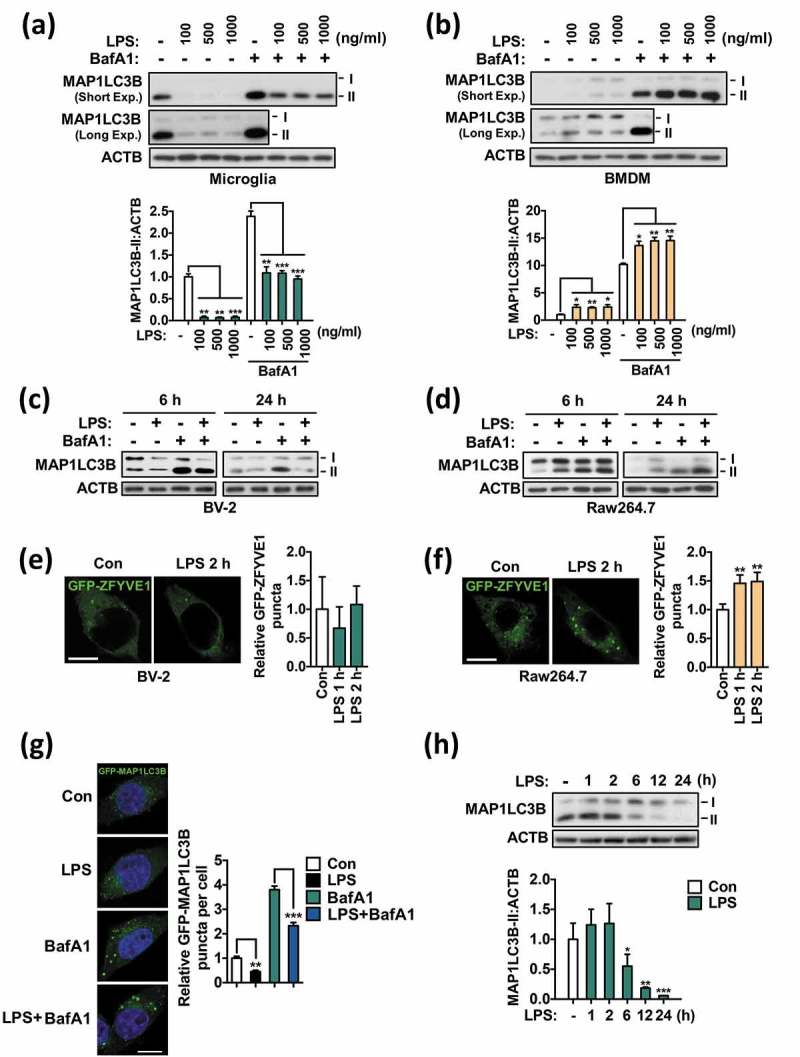
Autophagy is suppressed by LPS in microglia. (a) Analysis of autophagic flux in primary microglia after treatment with the indicated dosages of LPS for 24 h. Graph, quantification of MAP1LC3B-II after normalization to ACTB (n = 3). (b) Analysis of autophagic flux in BMDMs after treatment with the indicated dosages of LPS for 24 h. Graph, quantification of MAP1LC3-II after normalization to ACTB (n = 3). (c) Analysis of autophagic flux in BV-2 microglial cells after treatment with LPS (1 μg/mL) for 6 or 24 h. (d) Analysis of autophagic flux in Raw264.7 macrophages after treatment with LPS (1 μg/mL) for 6 and 24 h. (e) Quantification of ZFYVE1 puncta following LPS treatment in BV-2 cells (n > 30 cells per condition). (f) Quantification of ZFYVE1 puncta following LPS treatment in Raw264.7 cells (n > 40 cells per condition). (g) Quantification of GFP-MAP1LC3B puncta in BV-2 cells 24 h after LPS (1 μg/mL) treatment (n > 30 cells per condition). (h) Time course analysis of MAP1LC3B-II level in primary microglia. Cells were treated with LPS (1 μg/mL) for 1, 2, 6, 12, or 24 h. Graph, quantification of MAP1LC3B-II after normalization to ACTB (n = 4). In all experiments where BafA1 was used, BafA1 (20 nM) was added 2 h before sampling. Scale bars: 10 μm. All data are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the control (Con) unless indicated otherwise.
ZFYVE1/DFCP1 (zinc finger, FYVE domain containing 1) is a phosphatidylinositol-3-phosphate (PtdIns3P)-binding protein that is recruited to the PtdIns3P-enriched omegasome structure, promoting the nucleation step of autophagosome formation [34]. Thus, ZFYVE1 puncta are an early autophagic marker downstream of the activation of the class III phosphatidylinositol 3-kinase (PtdIns3K)-ATG14 complex [35]. To investigate whether LPS treatment differently affects the initial autophagosome formation in microglia and macrophages, we transfected BV-2 and Raw264.7 cells with ZFYVE1-GFP and measured the formation of GFP puncta. Intriguingly, LPS treatment noticeably increased ZFYVE1 puncta formation in Raw264.7 cells, consistent with autophagy induction, but not in microglia (Figure 1(e,f)). Next, we monitored autophagosome formation in BV-2 cells after transfection with GFP-MAP1LC3B and observed a great reduction in the number of MAP1LC3B puncta upon LPS treatment (Figure 1(g)). LPS also decreased BafA1-induced accumulation of MAP1LC3B puncta (Figure 1(g)). Consistent with these imaging analyses of autophagosome formation, a time-course study of MAP1LC3B protein levels in microglia by western blot analysis showed that the suppression of autophagy by LPS was significant from 6 h and lasted until at least 24 h (Figure 1(h)). These data suggest that LPS suppresses autophagy in microglia, in contrast to its effect in macrophages.
As a positive control for autophagy induction in microglia, we induced autophagy by incubating the cells in Hanks balanced salt solution (HBSS), which causes amino acid and serum starvation. Incubation in HBSS greatly increased autophagic flux, as indicated by higher accumulation of MAP1LC3B-II in starved than in control cells following BafA1 treatment (Figure S2). These data suggest that autophagy is induced normally in response to nutrient starvation in microglia and is specifically suppressed by TLR-triggered immune signaling.
LPS downregulates the expression of Atg genes in microglia
We further examined whether LPS regulates the expression of Atg genes. Analyses by quantitative real-time polymerase chain reaction (qRT-PCR) revealed that LPS significantly suppressed the expression of several Atg genes including Map1lc3b, Atg4c, Becn1, Atg7, Atg13, Atg14, and Atg16l1 24 h after LPS treatment (Figure 2(a)). Therefore, together with great reduction in MAP1LC3B turnover and in the number of MAP1LC3B-positive autophagosomes observed in LPS-treated microglia (Figure 1), these data suggest that autophagy suppression is sustained and is not merely a result of a kinetic delay in autophagosome formation.
Figure 2.
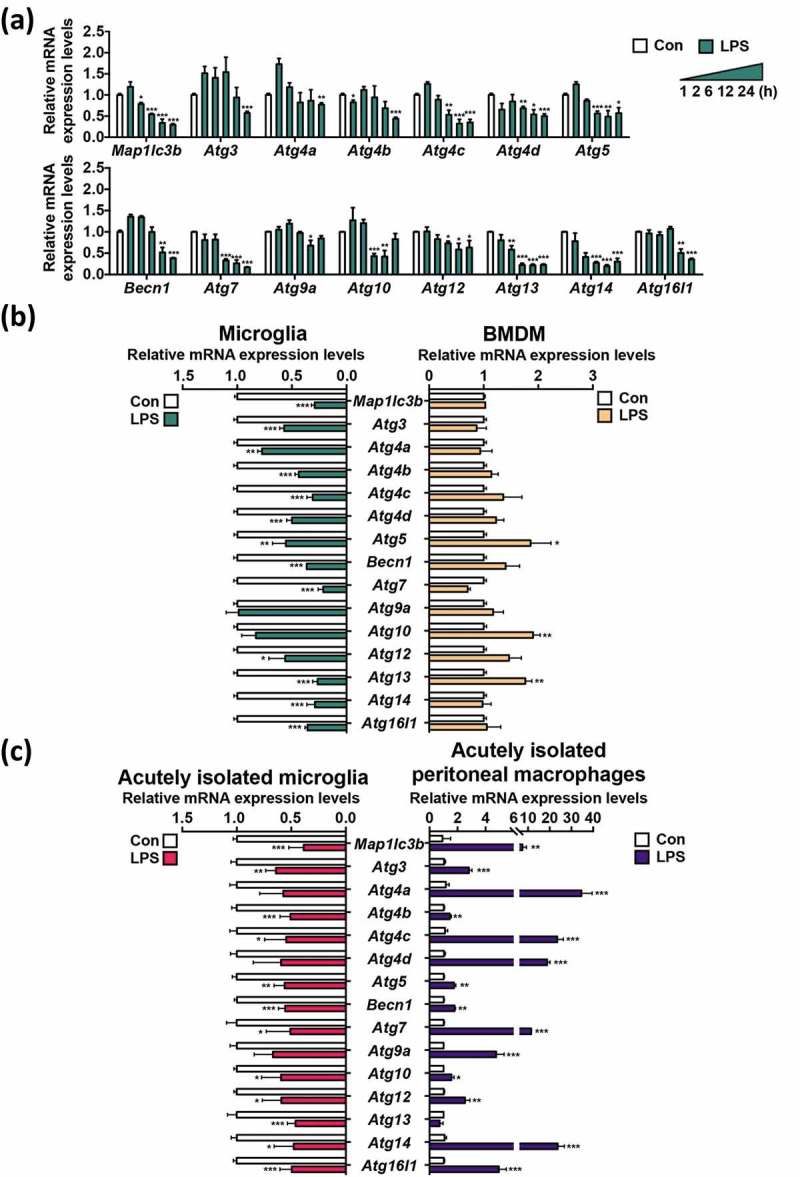
LPS decreases the expression of Atg genes in primary microglia. (a) Time course analysis of the mRNA levels of Atg genes by qRT-PCR following LPS (1 μg/mL) treatment (n = 3). (b) Comparison of relative mRNA levels of Atg genes between primary microglia (n = 5) and BMDMs (n = 4) 24 h after LPS (1 μg/mL) treatment. (c) Comparison of relative mRNA levels of Atg genes, between microglia and peritoneal macrophages acutely isolated from the same LPS-injected mice (5 mg/kg, intraperitoneally), 24 h post-injection (n = 3). In all experiments, mRNA levels were normalized to Actb. All data are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the control (Con).
Next, we compared changes in the expression of Atg genes between microglia and macrophages following LPS treatment. In contrast to microglia, no Atg genes were downregulated in BMDMs; rather, Atg5, Atg10, and Atg13 were significantly upregulated (Figure 2(b)). To validate our results observed in primary cell cultures and monitor the differential modulation of autophagy genes between the 2 immune cell types under more physiological conditions, we performed acute isolation of microglia and peritoneal macrophages from the same mice 24 h after LPS injection. Both microglia and macrophages expressed high levels of the proinflammatory cytokines Tnf and Il1b (interleukin 1 beta) (Figure S3). One of the hallmarks of glial cell activation is the upregulation of TSPO (translocator protein) [36,37]. As such, the transcriptional levels of Tspo were highly elevated in both cell types (Figure S3). These data indicated efficient activation of both microglia and macrophages following LPS injection in vivo. Microglia from LPS-injected mice showed a decrease in the expression of all examined Atg genes, with remarkable suppression of Map1lc3b, Atg4b, Atg5, Becn1, Atg13, and Atg16l1 (Figure 2(c)). Conversely, acutely isolated peritoneal macrophages displayed strong upregulation of Atg genes (Figure 2(c)). This comparative profiling of Atg gene expression in microglia and macrophages derived from the same animals further confirmed the opposite outcomes of LPS treatment in terms of autophagy in these 2 types of immune cells. Taken together, the results of experiments with primary cell cultures and acutely isolated cells indicate that LPS suppresses autophagy in microglia in contrast to autophagy induction in their macrophage counterparts.
Suppression of autophagy by LPS is TLR4-dependent
To check whether the suppression of autophagy by LPS in microglia was TLR4-dependent, we added LPS to microglia isolated from tlr4−/− mice. The tlr4−/− genotype was confirmed by PCR (Figure 3(a)) following our published protocol [38]. The level of autophagic flux in tlr4-knockout microglia was not significantly affected by LPS (Figure 3(b)). Furthermore, there were no noticeable changes in the expression of any of the tested Atg genes following LPS treatment in tlr4−/− microglia (Figure 3(c)). These data demonstrate that LPS-induced autophagy suppression was through the key upstream regulator, TLR4, but not due to other side effects of LPS.
Figure 3.
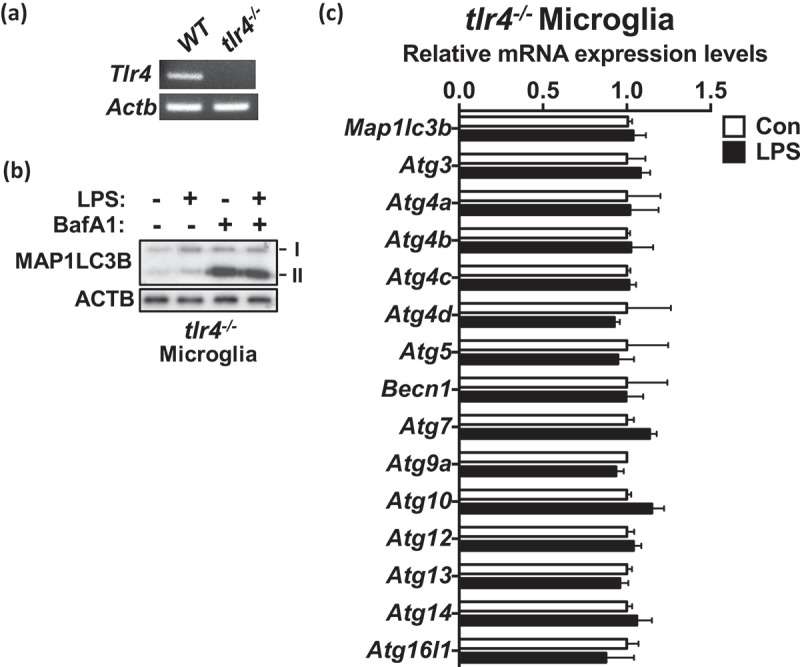
Suppression of autophagy by LPS is TLR4-dependent. (a) The genotyping of tlr4−/− mice by RT-PCR. (b) Autophagic flux analysis in tlr4−/− microglia after treatment with LPS (1 μg/mL) for 24 h. BafA1 (20 nM) was added 2 h before sampling. (c) Analysis of the mRNA levels of Atg genes in tlr4−/− microglia by qRT-PCR 24 h after LPS (1 μg/mL) treatment (n = 4).
Phosphatidylinositol measurements
Phosphoinositides, which include phosphatidylinositol (PtdIns), are inositol lipids that represent a distinct family of phospholipids [39]. PtdIns is a substrate of class III phosphatidylinositol 3-kinase (PtdIns3K); its phosphorylation product, PtdIns3P, is essential for autophagy induction [35]. PtdIns3P binds to its effector proteins such as ZFYVE1 or WIPI (WD-repeat protein interacting with phosphoinositides) protein family, and mediates formation of omegasomes, which function as a platform for the formation of the phagophore [34]. For this reason, we speculated that changes in the PtdIns level might underlie the suppression of autophagy in LPS-stimulated microglia. First, we performed pathway-based analysis of publicly available data from gene expression profiling of microglia acutely isolated from LPS-injected mice [40]. This analysis revealed that genes involved in the generation of the PtdIns precursors myo-inositol and CDP-diacylglycerol such as Impa1, Impa2, Cds1, and Cds2, were downregulated (Figure 4). Moreover, genes involved in the synthesis of Ins3P, Ins4P, phosphatidic acid, diacylglycerol, and Ins1,4,5P3 such as Isyna1, Inpp1, Dgk, and Plc (Plcb1, Plcb2, Plcb3, Plcb4, Plcd1, Plcd3, Plcd4, Plce1, Plcg1, Plcg2, Plcz1) were also downregulated (Figure 4) [40–46]. Therefore, these bioinformatics data suggested that autophagy suppression by LPS might result from decreased levels of PtdIns and PtdIns-derived phospholipids leading to decreased autophagosome biogenesis.
Figure 4.
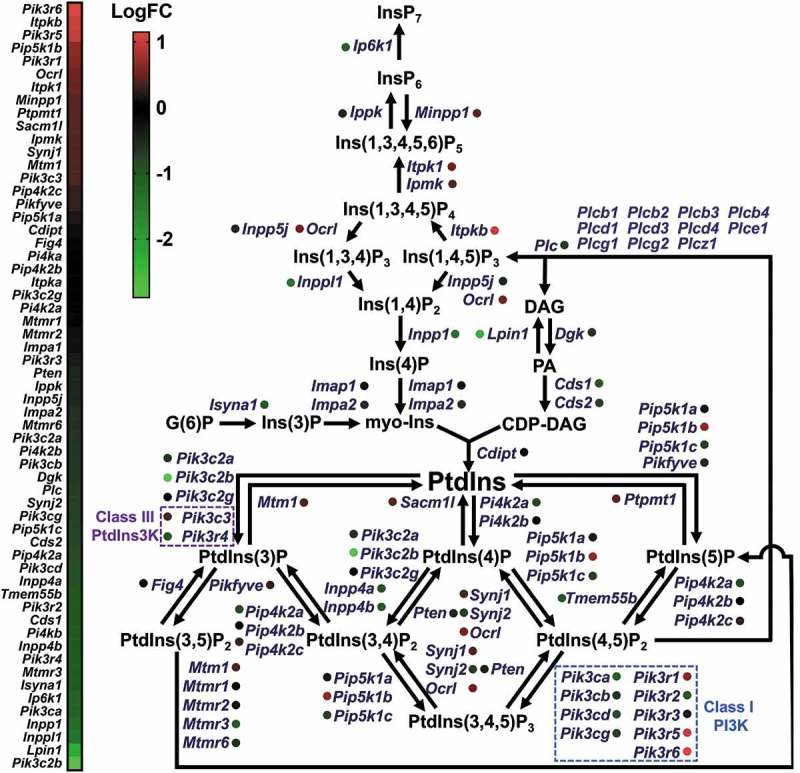
Bioinformatics analysis of RNA sequencing data for microglia activated by LPS. LogFC (fold change) values from 0 to 1 (red) indicate upregulation of gene expression and values below 0 (green) indicate downregulation.
Predicted downregulation of PtdIns synthesis encouraged us to examine the metabolomic correlates of PtdIns amounts and autophagy levels in microglia and macrophages. To validate our bioinformatics prediction by a metabolomics approach, we measured the intracellular amounts of PtdIns in microglia and BMDMs using Q-TOF LC/MS 16 h after LPS treatment. Among various molecular structures of detected PtdIns, PtdIns 38:4 was most abundant in both microglia and BMDMs (Figure 5(a,b)). After LPS treatment, the amount of PtdIns 38:4 in microglia significantly decreased, while in BMDMs, this significantly increased (Figure 5(a,b)). Reserpine, an indole alkaloid widely used as a standard metabolite for LC/MS analysis, was used as the internal standard to ensure the accuracy of LC/MS measurements (Figure S4).
Figure 5.
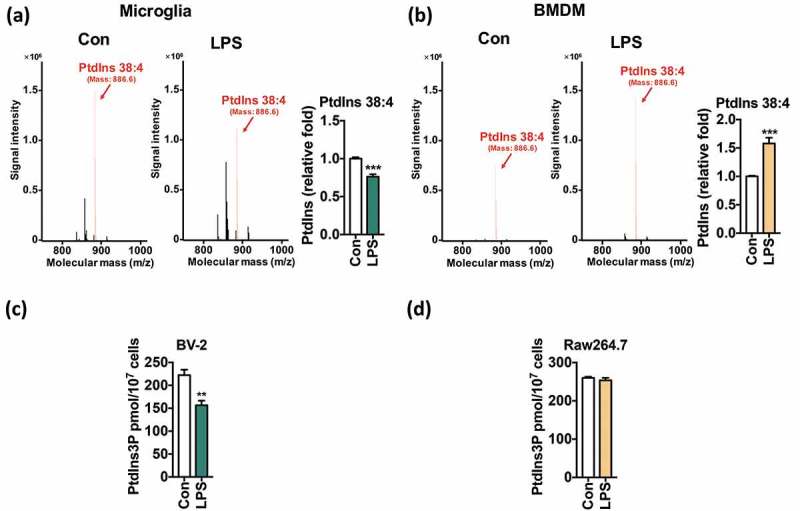
LPS reduces the synthesis of PtdIns and PtdIns3P in microglia. (a, b) LC/MS metabolomics analysis of PtdIns 38:4 amounts in primary microglia (a) and BMDMs (b) 16 h after LPS (1 μg/mL) treatment. (c, d) ELISA analyses of PtdIns3P amounts in BV-2 (c) and Raw264.7 cells (d) 16 h after LPS (1 μg/mL) treatment. All data are mean ± SEM. **P < 0.01 and ***P < 0.001 compared to the control (Con).
Because PtdIns is the substrate for generation of PtdIns3P, a decrease in the PtdIns content in microglia may lead to a reduction in the PtdIns3P level. To test this idea, we extracted lipids and determined the amount of PtdIns3P using enzyme-linked immunosorbent assay (ELISA). At 12 h after LPS treatment, we found a 25% decrease in BV-2 cells (Figure 5(c)), but no change in Raw264.7 cells (Figure 5(d)). Therefore, LPS reduces synthesis of PtdIns and PtdIns3P in microglia but not in macrophages.
Taken together, one possible mechanism to explain the difference in autophagic response to LPS between microglia and macrophages can be the difference in the regulation of PtdIns and PtdIns3P biosynthesis.
LPS suppresses autophagy via the PI3K-AKT1 signaling pathway
To identify the signaling pathway responsible for the downregulation of autophagy genes following LPS treatment, we focused on MTOR, MAPK1/3, and PI3K-AKT1. All these kinases can be readily activated by LPS and inhibit autophagy. LPS markedly increased the phosphorylation of MAPK1/3 at T203/Y205, MTOR at S2448, ribosomal protein S6 kinase, polypeptide 1 (RPS6KB1; an MTOR substrate) at T389, and AKT1 at T308 and S473 (Figure 6(a)). Treatment with PD98059, a MAPK1/3 inhibitor, did not affect LPS-induced autophagy suppression (Figure 6(b) and Figure S5(a)). Rapamycin and Torin-1, both well-known MTOR inhibitors, also failed to reverse autophagy suppression in LPS-treated microglia (Figure 6(b)), while they completely dephosphorylated RPS6KB1, indicating efficient inhibition of MTOR (Figure S5(b)). On the other hand, the PI3K inhibitors LY294002 (LY) and wortmannin (Wort) which also have inhibitory activity against the PtdIns3K counteracted the decrease in MAP1LC3B turnover in LPS-treated cells (Figure 6(b)). We further characterized the effects of LY on autophagic flux using LY. LY remarkably rescued the MAP1LC3B-II level in LPS-treated microglia as high as in the control; notably, LY also elevated basal autophagic flux (Figure 6(c,d)). Phosphorylation of AKT1 was almost completely blocked by LY, confirming inhibition of PI3K (Figure 6(c) and Figure S5(c)). Collectively, these data indicate that the PI3K-AKT1 signaling plays a pivotal role in LPS-induced autophagy suppression in microglia.
Figure 6.
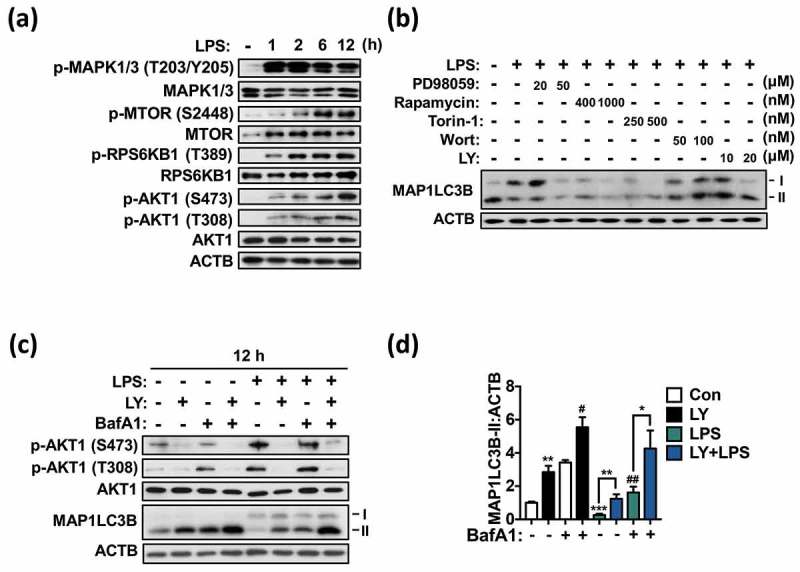
LPS suppresses autophagy via the PI3K-AKT1 signaling pathway in primary microglia. (a) Western blotting analyses of phosphorylation of AKT1 (T308, S473), MTOR (S2448), MAPK1/3 (T203/Y205), and RPS6KB1 (T389) following LPS treatment (1 μg/mL) in primary microglia. (b) Western blotting analysis of MAP1LC3B-II level after inhibition of MAPK1/3 (with PD98059), MTOR (with rapamycin or Torin-1) or PI3K (with LY294002; LY or wortmannin; Wort) in primary microglia treated with LPS (1 μg/mL) for 12 h. (c) Analysis of autophagic flux after inhibition of PI3K with LY (20 μM) in primary microglia treated with LPS (1 μg/mL) for 12 h. (d) Quantitative analysis of MAP1LC3B-II levels in primary microglia. In all experiments, the blots shown are representative of at least 3 experiments with similar results. All inhibitors were added 1 h prior to LPS treatment. All data are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the control (Con) unless indicated otherwise. #P < 0.05 and ##P < 0.01 compared to the control treated with BafA1 only.
LY294002 rescues microglial autophagy by preventing LPS-induced FOXO3 phosphorylation
Given the major role of the PI3K-AKT1 pathway in microglial autophagy, we questioned which downstream targets mediate the inhibitory effects of TLR4-PI3K-AKT1. We reasoned that, among AKT1 downstream targets, FOXO3 may be a good candidate for autophagy suppression because this transcription factor regulates the expression of core autophagy genes such as Map1lc3b, Gabarapl1, Atg12, and Bnip3 in many tissues and cell types under various conditions [47–49], and is negatively regulated by AKT1 [50,51]. Phosphorylation of FOXO3 by AKT1 results in its export from the nucleus and subsequent degradation in the cytosol, and therefore abrogation of its transcriptional activity [50]. On the other hand, unphosphorylated FOXO3 remains in the nucleus to activate target gene expression. Therefore, we hypothesized that LPS-induced activation of AKT1 suppresses autophagy by inhibiting FOXO3 activity through phosphorylation and thus suppresses transcriptional activation of Atg genes. As expected, LPS increased FOXO3 phosphorylation at S253 in a time-dependent manner (Figure 7(a)), whereas treatment with LY reduced FOXO3 phosphorylation (Figure 7(b)). Next, we examined the subcellular localization of endogenous FOXO3 in control vs. LPS-treated microglia in the absence or presence of LY using an antibody specific to FOXO3. To avoid false detection of nuclear localization, 4 images of FOXO3 from different layers of a single cell were taken and of these collapsed stacks were presented (Figure 6(c)). Intensities of FOXO3 from each layer were measured and summed up using Zen software (Carl Zeiss) (Figure S6(a,b)). In control microglia, FOXO3 was predominantly localized in the cytosol; LPS treatment induced a robust export of FOXO3 from the nucleus, further increasing cytosolic localization (Figure 7(c)). PI3K inhibition restored FOXO3 nuclear retention in LPS-treated microglia with a concurrent increase in the expression of the Atg genes in both control and LPS-treated cells (Figure 7(c,e)).
Figure 7.
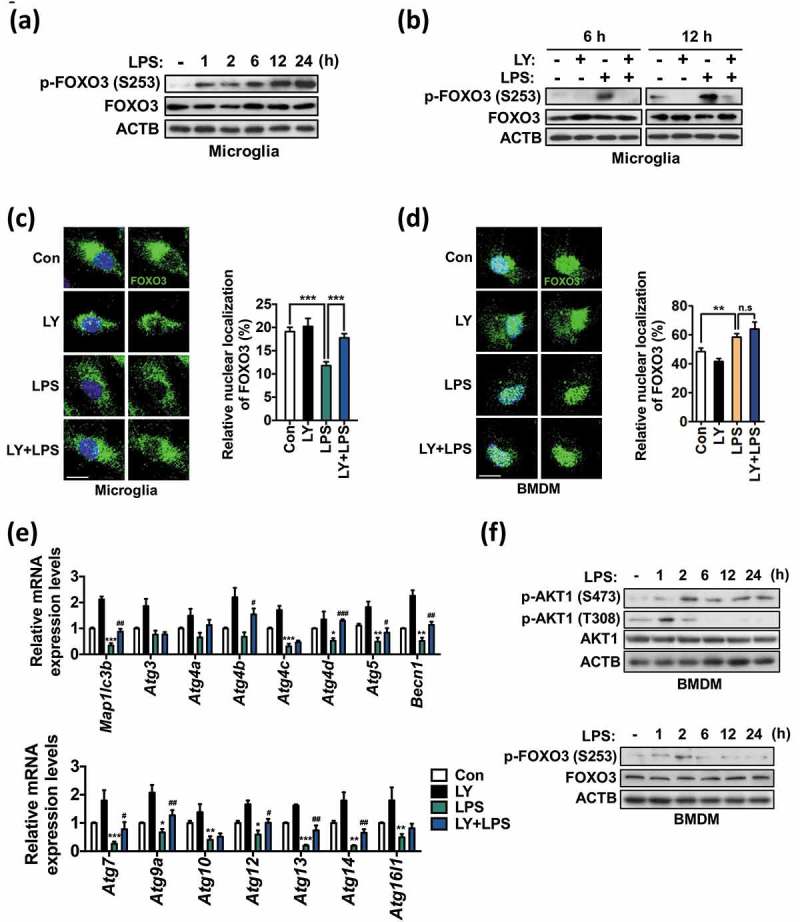
Phosphorylation of FOXO3 by PI3K-AKT1 suppresses autophagy in primary microglia. (a) Time course analysis of phosphorylation of FOXO3 (S253) in primary microglia following LPS treatment (1 μg/mL). (b) Phosphorylation of FOXO3 (S253) in primary microglia after LY294002 (LY, 20 μM) treatment at 6 and 12 h following LPS (1 μg/mL) treatment. The blots shown are representative of 3 experiments with similar results. (c, d) Analyses of nuclear localization of FOXO3 in primary microglia (n > 30 cells per condition) (c) and BMDMs (n = 30 cells per condition) (d) 12 h after LPS (1 μg/mL) treatment with 1 h pretreatment of LY (20 μM). Endogenous FOXO3 (green) was visualized by immunocytochemistry. Scale bars: 10 μm. Graphs, quantitative analysis of FOXO3 nuclear localization. (e) Analysis of the mRNA levels of Atg genes in primary microglia treated with LPS (1 μg/mL) for 12 h with 1 h pretreatment of LY (20 μM). Relative mRNA levels were quantified by qRT-PCR and normalized to ACTB (n = 3). (f) Time-course analyses of phosphorylation of FOXO3 (S253) and AKT1 (T308, S473) in BMDMs following LPS treatment (1 μg/mL). All data are mean ± SEM. n.s., not significant. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the control (Con) unless indicated otherwise. #P < 0.05, ##P < 0.01, and ###P < 0.001 compared to the LPS-treated cells.
Although PI3K activation and subsequent FOXO3 inhibition seemed to be important for autophagy suppression in microglia, it did not readily explain why autophagy is suppressed specifically in microglia but not in macrophages, since PI3K is also activated after stimulation with TLR ligands in macrophages [52]. Therefore, we examined whether FOXO3 localization in macrophages differs from that in microglia. Interestingly, in comparison with microglia, a larger proportion of endogenous FOXO3 in BMDMs was localized in the nucleus at basal state (20% in microglia vs. 45% in BMDMs in control cells), and LPS treatment significantly increased nuclear FOXO3 localization rather than decreased it (Figure 7(d)).
Next, we performed parallel analyses of AKT1 and FOXO3 phosphorylation in BMDMs for comparison with those in microglia. Interestingly, LPS-induced phosphorylation of FOXO3 peaked at the 2-h time point, but returned to the basal level soon (Figure 7(f)). Similarly, phosphorylation of AKT1 T308 was induced as early as 1 h following LPS treatment, however, it decreased quickly. On the other hand, AKT1 S473 remained phosphorylated until 24 h (Figure 7(f)). Overall, time-course analyses of phosphorylation in LPS-treated BMDMs revealed a transient increase of phosphorylation of FOXO3 and AKT1 T308, which was quite different from persistent and robust phosphorylation of these residues in LPS-treated microglia. These data support our claim that distinct regulation of FOXO3 may underlie the different regulation of autophagy in microglia and BMDMs in response to LPS. These differences between microglia and macrophages in the distribution and phosphorylation time-course of FOXO3 at basal and activated states suggest a deep integration of FOXO3 at the interface of autophagy and immunity in microglia.
To directly ascertain the role of FOXO3 in microglia autophagy, we expressed a constitutively active (CA) form of FOXO3, which has 3 mutations (T32A, S253A, and S315A) and does not undergo inhibitory phosphorylation by AKT1 [50]. Expression of GFP-tagged FOXO3-CA (FOXO3-CA-GFP) in BV-2 cells resulted in its robust nuclear location and increased the number of MAP1LC3B dots (Figure 8(a,b)). The number of MAP1LC3B dots was further increased by BafA1 treatment, demonstrating the recovery of autophagic flux in FOXO3-CA-expressing cells (Figure 8(b)). These results support our conclusion that FOXO3 is the main target in suppression of autophagy in LPS-activated microglia. All together, these data point to the TLR4-triggered PI3K-AKT1 activation and subsequent FOXO3 inhibition as the key effector steps required for the transduction of inflammatory cues into suppression of autophagy in microglia.
Figure 8.
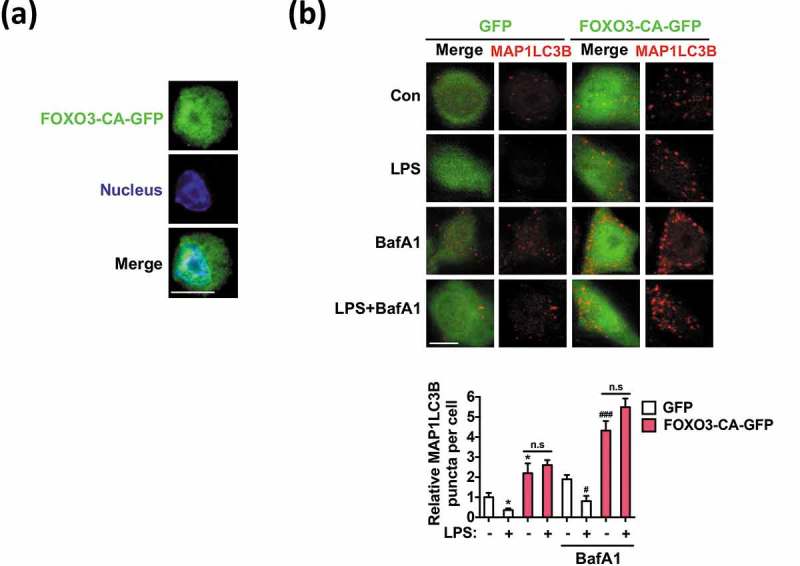
Overexpression of constitutively active FOXO3 (FOXO3-CA) increases the number of MAP1LC3B puncta under basal conditions and following LPS treatment. (a) Nuclear localization of FOXO3-CA-GFP in BV-2 cells. (b) Increased number of MAP1LC3B dots in FOXO3-CA-GFP-transfected BV-2 cells compared to control cells transfected with the GFP-only control vector. LPS (1 μg/mL) was added for 12 h and MAP1LC3B puncta (red) were visualized by immunocytochemistry with an antibody against endogenous MAP1LC3B. Scale bar: 10 μm. Graph, quantification of the puncta from at least 30 cells per condition, using an LSM700 confocal microscope (Carl Zeiss). BafA1 (20 nM) was added 2 h before sampling. All data are mean ± SEM. n.s., not significant. *P < 0.05 compared to the control in the absence of BafA1. #P < 0.05 and ###P < 0.001 compared to the control in the presence of BafA1.
LPS reduces MAP1LC3B-associated phagocytosis and Aβ degradation via the PI3K-AKT1 signaling pathway in microglia
Microglia serve as professional phagocytes in the brain [27,53]. To understand the physiological implications of suppressed autophagy in microglial function, we estimated the phagocytic capacity of LPS-treated microglia. First, we analyzed MAP1LC3B/LC3B-associated phagocytosis (LAP) in activated microglia. Recent reports have illustrated the intersection of phagocytosis and autophagy in the context of microbial invasion or activation of TLR signaling in macrophages. Following TLR stimulation in macrophages, an increase in MAP1LC3B translocation to the phagosome and enhanced digestion of an ingested organism are observed in an ATG5-, ATG7-, or BECN1-dependent manner [15,16]. However, the interplay of TLR signaling and phagocytosis in microglia has not been studied yet. Therefore, we examined the effect of LPS on LAP in microglia, based on our finding that LPS significantly suppressed autophagy. Interestingly, only 15% of zymosan particles entered a MAP1LC3B-positive autophagosome in LPS-treated primary microglia, compared with 73% at basal state, indicating that markedly fewer MAP1LC3B-associated phagosomes were formed following LPS treatment (Figure 9(a)). LY treatment highly upregulated the formation of MAP1LC3B-positive phagosomes surrounding zymosan (to 55%; Figure 9(a)). In BV-2 cells, LAP occurred at a much lower rate than in primary microglia. Nonetheless, the knockdown of Atg7 in BV-2 cells significantly lowered MAP1LC3B-associated phagosome formation at basal state in comparison with the control cells and also prevented the LY-induced recovery of LAP (Figure 9(b)), suggesting that canonical autophagy is required for LAP in microglia. To further investigate the pathological significance of autophagy suppression by LPS, we assessed the association of autophagosomes with fibrillary Aβ by proximity ligation assay (PLA). Deposition of Aβ is a characteristic of AD, and dysfunctional microglial activity is thought to lead to impaired Aβ clearance in AD, although the detailed mechanisms linking activated microglia and reduced clearance of Aβ in a neuroinflammatory environment remain unclear [54,55]. PLA allows quantitative visualization of the direct association between MAP1LC3B-containing autophagosomes and engulfed FITC-conjugated fibrillary Aβ, since the combination of antibodies against MAP1LC3B and Aβ produces fluorescent signals only when they are in close contact. A subpopulation of internalized Aβ was targeted by autophagy, as shown by the PLA signals (red in Figure 9(c)). LPS treatment significantly decreased PLA signals, which were restored by LY (Figure 9(c)). Aβ degradation rates in LPS-treated and (LY+LPS)-treated cells corresponded well to the Aβ and MAP1LC3B proximity anticipated by PLA assay. More Aβ remained in LPS-treated microglia, whereas (LY+LPS)-treated cells were as efficient as control cells in Aβ degradation (Figure 9(d)). These data indicate that LPS-induced PI3K-AKT1 signaling downregulates autophagy-associated phagocytic capability of microglia and this downregulation can be reversed by PI3K inhibition.
Figure 9.
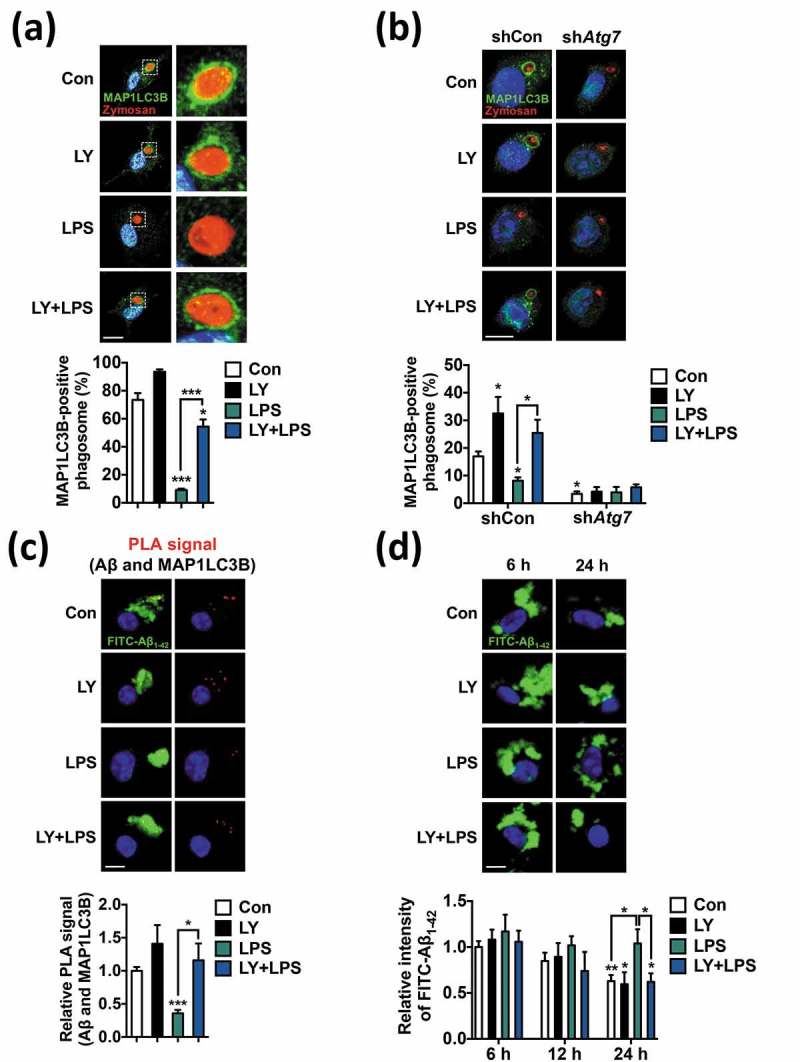
LPS suppresses MAP1LC3B-associated phagocytosis (LAP) and Aβ degradation via the PI3K-AKT1 signaling pathway in microglia. (a) Analysis of LAP in primary microgliawhere stimulated with LPS (1 μg/mL) for 12 h and then incubated with zymosan for another 2 h. LY (20 μM) was added 1 h prior to LPS. Immunocytochemistry images show MAP1LC3B (green), zymosan (red), and nucleus (blue). Graph, quantitative analysis of MAP1LC3B-positive phagosomes from 3 independent experiments (n = 45 to 113 cells). (b) Analysis of LAP in Atg7 knockdown BV-2 cells (n = 35 to 63 cells). BV-2 cells were infected with lentivirus expressing Atg7-targeting (shAtg7) or control (shCon) shRNA for stable knockdown. Experimental conditions were the same as (a). (c) Analysis of PLA signal between MAP1LC3B and Aβ in primary microglia after treatment with LPS (1 µg/mL) for 12 h. LY (20 µM) was added 1 h prior to LPS treatment. Graph, quantification of PLA signal (n = 82 to 135 cells). (d) Measurement of Aβ degradation in LPS-stimulated primary microglia. FITC-conjugated Aβ1−42 fibrils were added to the cells and the fluorescence intensity of the remaining FITC-Aβ1−42 was measured using a fluorescence microscope 6, 12, or 24 h later. Graph, quantification of Aβ fibril intensities from 3 independent experiments (n = 82 to 106 cells). *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the control (Con) unless indicated otherwise.
Discussion
An interesting finding of this study is that, unlike in macrophages, autophagic flux is significantly suppressed by TLR activation in microglia. A decrease in the expression was observed for nearly all tested Atg genes, demonstrating sustained suppression of autophagy in LPS-treated microglia. The results of metabolomic correlation in PtdIns amounts and autophagy levels conjectured from bioinformatics prediction also strongly support our observations. Since this was in sharp contrast to autophagy induction in LPS-treated macrophages and other immune cells, we sought to elucidate the signaling mechanisms underlying autophagy suppression in TLR-triggered microglia. To this end, we examined the roles of several signaling pathways known to be activated by TLR4 activation, and identified the PI3K-AKT1-FOXO3 pathway as a key suppressor of autophagy in activated microglia. FOXO3 has not been previously associated with autophagy in microglia and, to the best of our knowledge, our report is the first to show the critical role of FOXO3 in the regulation of autophagy in microglia. Other notable findings of our study are the first demonstration of LAP and diminished phagocytic capability in activated microglia.
Most of the previous studies on the role of autophagy in immunity and inflammation have focused on peripheral tissue macrophages or other types of cells, and little is known about the roles and regulatory mechanisms of autophagy in microglia. The few available studies on autophagy in microglia show inconsistent results. Song et al. [56] have studied microRNA-mediated autophagy gene regulation in microglia and observe a large reduction in BECN1 and ATG3 protein levels in LPS-treated BV-2 cells. In line with this report, an in vivo study reveals autophagy impairment in the brains of LPS-injected mice, as evidenced by substantial reductions in BECN1 and MAP1LC3B-II levels and an increase in SQSTM1/p62 in the cortex and hippocampus, but these authors do not perform cell type-specific analyses [57]. However, neither of the 2 studies examine autophagic flux. In contrast, Han et al. [58] observe activation of a nonclassical autophagy pathway and an increase in the formation of MAP1LC3B puncta following LPS treatment in BV-2 cells, which is different from our results (Figure 1(c,g)). We did not further examine the different effects of LPS on MAP1LC3B puncta formation because we did not observe autophagy activation following LPS treatment in BV-2 cells. However, it is noteworthy that autophagy suppression by LPS was stronger in primary microglia than in BV-2 cells.
The mechanisms of autophagy inhibition may vary depending on cell type and even on the state of the cells of the same type. Signaling cascades involved in TLR-triggered autophagy are known to follow cascades similar to those involved in nutritional stress-induced autophagy signaling [59]. However, while starvation inhibits MTOR, which is the principal negative regulator of autophagy, MTOR is robustly activated in both microglia (Figure 6) and macrophages following TLR4 activation, as shown by others [60] and us (data not shown). This seemingly paradoxical activation of MTOR in activated macrophages with induced autophagy exemplifies the complicated signaling pathways distinct from starvation-induced autophagy signaling. There are also reports of MTOR-independent autophagy inhibition. In skeletal muscle, rapamycin does not induce autophagy, and FOXO3 activates autophagy independently of MTOR complex 1 activity [61]. Similarly, we observed no noticeable changes in autophagy level upon MTOR inhibition in LPS-treated microglia. Therefore, though the activation of MTOR might have partially affected autophagy level, the major player in LPS-induced autophagy suppression in microglia is PI3K-AKT1 signaling.
Transcriptional regulation plays a critical role in modulating autophagy. TFEB (transcription factor EB) is a master regulator of Atg gene expression. MTOR and MAPK1/3 are the key kinases that negatively regulate TFEB [62,63] by phosphorylating and holding it on lysosomes, which results in autophagy inhibition [64]. When dephosphorylated, TFEB translocates to the nucleus and activates various autophagy genes to induce autophagy [62]. In our study, we were not able to detect any differences in the nuclear location of TFEB in primary microglia between basal state and LPS treatment (data not shown). We did not further examine whether rapamycin or PD98059 induced TFEB translocation. However, since they were not effective in inducing autophagy in activated microglia, it is plausible that autophagy relies more on PI3K-AKT1-FOXO3 signaling than on MTOR-MAPK1/3-TFEB in microglia. It should be mentioned that, although PI3K inhibition restored the expression levels of Atg genes in LPS-treated microglia, some Atg genes examined do not have known FOXO3˗binding sequences in their promoter regions. Therefore, it remains to be determined whether these genes have yet unknown FOXO3-binding sequences, or other transcription factors are also affected by LPS in microglia.
Despite the lack of knowledge on the autophagy regulatory mechanisms in microglia, the importance of autophagy in microglial function is gaining recognition [18,29,65]. For example, mice lacking microglial Atg7 show various neural deficits, including hindered synaptic pruning, defective social behavior, impaired Aβ clearance, and enhanced inflammasome activation [18,65]. These lines of evidence suggest that proper regulation of microglial autophagy is essential for normal brain function and demonstrate the physiological importance of microglial autophagy in brain function and pathology [29,66].
Suppression of autophagy by TLR signaling observed in this study can provide a clue to understanding how impaired microglial function can cause the loss of brain homeostasis and contribute to the pathogenesis of neurodegenerative diseases. Notably, in the brains of patients with neurodegenerative diseases such as AD, the ability of microglia to phagocytose and degrade Aβ is reduced [54,67,68]. However, the elaborate mechanisms of how microglial phagocytic activity is impaired in AD remain to be elucidated. Previously, it has been assumed that excessive production of Aβ in AD patients first overloads the autophagy-lysosome pathway and subsequently causes the malfunction of the autophagy system and ‘burning-out’ of microglia [69,70]. However, our data suggest that suppression of autophagy and downregulation of phagocytic degradation capability is already eminent in microglia activated at the onset of neuroinflammation. In line with our observations, proinflammatory cytokines are reported to reduce Aβ-elicited phagocytosis, but Aβ treatment does not induce overt microglia activation or reduce phagocytic capacity [71]. Another interesting study reports a reduced BECN1 level and inefficient phagocytic degradation of Aβ in microglia derived from human AD patients [55]. Although the level of autophagy is not examined in this study, autophagy suppression is expected in AD patient-derived microglia. The authors discuss that, although it is unknown why BECN1 level is reduced in AD microglia, BECN1 deficiency is not simply the result of amyloid accumulation but might have other causes [55]. Therefore, our results explain well these previous observations made in AD patients and mouse models.
Our findings are also in line with a recent conceptual shift regarding the role of inflammation in AD. Recent studies have not only demonstrated an intimate link between alterations in the immune system and AD pathogenesis, but also suggest a much earlier timing of immune system involvement in AD progression than previously anticipated [72–76]. Therefore, immune functions in the brain, which are mainly mediated by CNS-resident microglia and other myeloid cells, should be studied as one of the primary driving factors in the pathogenesis of AD more than the accompanying event following Aβ overproduction and deposition [77]. In that regard, changes in microglial physiology in the early stage of AD warrant more in-depth analysis.
Given the opposite consequences emanating from the same stimuli in microglia and macrophages, we are far from understanding the mechanisms that regulate microglial autophagy in the neuroinflammatory milieu of the degenerating brain. Nevertheless, our observation of LPS-induced autophagy impairment and decreased degradative capacity in microglia can shed new light on the contribution of neuroinflammation to neurodegeneration and provide novel mechanistic insight into neurodegenerative pathogenesis including AD, pointing to the PI3K-FOXO3 pathway as a potential therapeutic target to regulate microglial function in brain disorders.
Materials and methods
Reagents and antibodies
Antibodies against the following proteins were used: ACTB (sc-47778, HRP-conjugated) from Santa Cruz Biotechnology; MAP1LC3B (NB100-2220) from Novus Biologicals; phosphorylated FOXO3 (S253, ab47285) from Abcam; phosphorylated AKT1 (S473, 9271; T308, 4056), phosphorylated MTOR (S2448, 2971), phosphorylated MAPK1/3 (T203/Y205, 4370), phosphorylated RPS6KB1 (T389, 9206), and FOXO3 (99199) from Cell Signaling Technology; FOXO3, Clone 648716 (MAB6165) from R&D Systems. The followings reagents were used: LPS from Escherichia coli 0111:B4 (L4391) was purchased from Sigma-Aldrich and diluted in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4 and 1.4 mM KH2PO4, pH 7.4); BafA1 (B1793), wortmannin (W1628), LY294002 (L9908), hexadimethrine bromide (107689), and trichloroacetic acid (T6399) were purchased from Sigma-Aldrich. Rapamycin (BML-A275-0005) was purchased from Life Sciences Advanced Technologies. FITC-labeled Aβ (M-2585) was purchased from Bachem. PD98059 (9900) was purchased from Cell Signaling Technology. A TLR ligand kit (Tlrl-kit 1mw) was purchased from Invivogen.
Cell culture
All procedures for the care and use of laboratory animals were approved by the Institutional Animal Care and Use Committees of DGIST and Seoul National University. For primary microglia culture, whole brains were isolated from C57BL/6 wild-type or tlr4−/− mice at postnatal days 1 or 2. The brains were dissected in serum-free high-glucose Dulbecco Modified Eagle Medium (DMEM, Corning 10-013-CVR) with sterile scissors and incubated with 0.25% trypsin-EDTA solution for 7 min at 37°C. An equal volume of culture medium composed of DMEM with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, SH30084.03) and 1% (100 U/ml) penicillin–streptomycin (Hyclone, SV30010) was added to stop trypsinization, and the brains were dissociated by gentle pipetting. The dissociated cells were passed through a 70-μm pore mesh and pelleted at 800 × g for 10 min. Each cell pallet was suspended in culture medium and the cells were seeded in a 100-mm dish per brain. Five days after plating, the medium was changed to fresh medium. Thereafter, half of the medium in each plate was changed every other day to fresh medium. Primary microglia were confluent after 10–14 dand were separated from mixed astrocytes by tapping the plate. High enrichment with primary microglia (>95%, data not shown) was verified by immunostaining with anti-AIF1/Iba-1 (Wako, 019-19741) and anti-GFAP (Novus Biologicals, NBP1-05198) antibodies (specific microglia and astrocyte markers, respectively), as we previously reported [37].
BMDMs were obtained from femurs and tibias of 6- to 7-week-old C57BL/6 mice as previously described [78]. BMDMs were grown in RPMI 1640 (GIBCO, 11875-093) supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin.
BV-2 cells (kindly provided by Dr. Kyoungho Suk (Kyungpook National University, Republic of Korea) are murine immortalized microglia cell line [79]. The murine macrophage cell line, Raw 264.7 cells were purchased from Korea Cell Line Bank (Republic of Korea). BV-2 and Raw264.7 cells were grown in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin.
Generation of Atg7 knockdown BV-2 cells
shCon and shAtg7 lentiviruses (TRCN0000092164, Sigma-Aldrich) were produced by following a published protocol [80]. To obtain the control and stable Atg7 knockdown cell lines, BV-2 cells were infected with the lentiviruses in the presence of hexadimethrine bromide (8 µg/ml) for 24 h and then the medium was replaced with fresh culture medium. After 72 h, cells were selected using puromycin (8 µg/ml) for 2–3 d, and puromycin-resistant cells were re-seeded and grown in fresh culture medium.
Acute isolation of primary microglia and peritoneal macrophages
Brains were removed from 8-week-old C57BL/6 mice after isolating peritoneal macrophages. Subsequently, brains were washed with 1× HBSS, homogenized by razor-blade chopping and transferred to microcentrifuge tubes. Tissue was dissociated with a neural dissociation kit containing papain (Miltenyi Biotec, 130-092-628) according to the manufacturer’s instructions. Myelin was removed using myelin removal beads (Miltenyi Biotec, 130-096-733). Suspended cells, with the beads, were passed through MACS separation columns (Miltenyi Biotec, 130-042-401) and centrifuged for 3 min at 1200 × g. Each pellet was resuspended in MACS buffer, transferred to a new microcentrifuge tube, and microglia were isolated using ITGAM+/CD11b+ microbeads (Miltenyi Biotec, 130-049-601).
Peritoneal macrophages were obtained from 8-week-old C57BL/6 mice as previously described [81].
Plasmids and transfection
pEGFP-MAP1LC3B (21073) and a plasmid encoding a constitutively active (CA) form of human HA-FOXO3 with 3 mutations (T32A, S253A, and S315A) were purchased from Addgene (1788). FOXO3-CA was cloned into the pGFP-N1 vector to generate FOXO3-CA-GFP. For magnetofection, DNA was mixed with Lipofectamine 2000 (Invitrogen, 11668-019) for 5 min. Magnetic nanoparticles CombiMAG (Chemicell, 9005) were then added to the mixture, which was incubated for an additional 30 min at 37°C for stabilization and efficient combination of magnetic particles with the DNA–Lipofectamine 2000 complex. The mixture was then delivered into BV-2 cells in DMEM without FBS and antibiotics for 3 h. The medium was then replaced with normal culture medium for 24 h and cells were re-seeded onto glass coverslips for immunocytochemistry experiments.
Immunocytochemistry
Primary microglia were fixed and permeabilized with absolute methanol. Cells were blocked with antibody diluent reagent solution (Thermo Fisher Scientific, 003218) for 30 min and incubated with appropriate primary antibodies for 2 h at room temperature, rinsed 3 times with PBS containing 0.1% Tween-20 (Duchefa Biochemie, 9005-64-5) and incubated with appropriate fluorescent dye-conjugated secondary antibodies for 1 h at room temperature. Nuclei were stained with Hoechst 33342. Images were obtained using an LSM700 confocal microscope (Carl Zeiss, Oberkochen, Germany).
LAP assay with zymosan
Primary mouse microglia and BV-2 cells were pretreated with LY294002 for 1 h and LPS was treated for 12 h. Cells were then incubated with zymosan (Invitrogen, Z2843; 1 µg/ml for 2 h). After incubation, cells were fixed with absolute methanol at −20°C for 20 min. Representative images of zymosan, MAP1LC3B, and nuclei were obtained using a LSM700 confocal microscope, and cells with MAP1LC3B-positive phagosomes were counted using a fluorescence microscope (Axiovert 40 CFL, Carl Zeiss, Oberkochen, Germany).
PLA and measurement of Aβ degradation
FITC-conjugated Aβ1−42 fibrils were prepared as previously described [18]. The fibrils (0.3 µM) were incubated with primary microglia for 2 h. After fixation and permeabilization with methanol, cells were incubated with mouse anti-Aβ (Cell signaling technology, 15126) and rabbit anti-MAP1LC3B antibodies and then with a pair of PLA probes (Sigma, DUO92002 and DUO92004); probe ligation, signal amplification (Sigma, DUO92007), and mounting (Sigma, DUO82040) were performed according to the manufacturer’s instructions. Representative images of FITC-Aβ and PLA signals were obtained using an LSM700 confocal microscope, and the number of PLA puncta was estimated using a fluorescence microscope.
Degradation of FITC-Aβ1-42 fibrils (0.3 µM) was assessed at 6, 12, or 24 h under a LSM700 confocal microscope and the intensity of FITC fluorescence was analyzed using Zen software (Carl Zeiss, Oberkochen, Germany).
PtdIns analysis
Cells were washed in Dulbecco PBS and then chilled solvent mixture (chloroform:methanol, 1:2 (v:v), 900 µl per well of 6-well plate) containing 5 µM internal standard was added on dry ice for quenching. The sample was transferred into a 2 ml microcentrifuge tube, vortexed, and incubated with agitation for 1 h at 4°C in the dark. After incubation, 300 µl of chilled chloroform and 350 µl of chilled water were added to the sample. The mixture was then vortexed for 1 min, centrifuged at 8000 × g for 10 min at 4°C, and the lower organic phase was transferred to another tube. The remaining aqueous phase was re-extracted with 500 µl of chilled chloroform. After vortexing for 1 min, the mixture was centrifuged at 8000 × g for 10 min at 4°C. The lower organic phase was combined with the first organic extract. The pooled organic phase was evaporated under nitrogen gas, and the residue was reconstituted with 100 µl of methanol.
All samples were analyzed using an Agilent 1290 Ultra-Performance Liquid Chromatography system coupled with an Agilent 6530 Q-TOF LC/MS (Agilent Technologies, Santa Clara, CA, USA) in negative ESI mode. The instrument parameters were as follows: gas temperature, 325°C; drying gas flow rate, 11 L/min; nebulizer pressure, 30 psig; fragment voltage, 170 V; skimmer voltage, 60 V; nozzle voltage, 500 V; and capillary voltage, −3500 V. Internal mass calibration was performed using reference standards during the runs.
Samples (10 µl) were injected onto a Phenomenex Kinetex C18 column (100 × 2.1 mm, 2.6 µm) which was kept at 40°C in a column oven. The mobile phase consisted of 1:1 (v:v) water/methanol containing 10 mM ammonium acetate and 0.2% acetic acid (A) and 1:1 (v:v) isopropanol/acetone (B). Flow rate was 300 µl/min. Gradient program, time (min):solvent A (%):solvent B (%): 0:70:30, 3:50:50, 24:10:90, 24.1:70:30, 30:70:30. The autosampler temperature was 4°C.
Metabolic features were extracted and characterized using the MassHunter Qualitative Analysis Software B.07.00 (Agilent Technologies, Santa Clara, CA). Quantitative analysis of PtdIns was performed using the MassHunter Quantitative Analysis Software B.07.00 (Agilent Technologies).
Measurement of PtdIns3P
Cells were collected and incubated in cold 5% trichloroacetic acid. Precipitate was subjected to sequential extraction with methanol:chloroform (2:1 [v:v]) and methanol:chloroform:12N HCl (80:40:1 [v:v:v]) for neutral and acidic lipids, respectively. Neutral lipids were discarded and the amount of PtdIns3P in the acidic lipid fraction was measured with a Mass ELISA kit (K-3300, Echelon Biotechnology) following the manufacturer’s instructions.
Western blotting
Cells were harvested and lysed in lysis buffer containing 250 mM sucrose (Duchefa Biochemie, 57-50-1), 50 mM NaCl, 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% Triton X-100, 1× protease and phosphate inhibitor cocktail (Thermo Fisher Scientific, 78440), 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride for 15 min on ice. After incubation, lysates were centrifuged (16,100 × g, 10 min) and each supernatant was collected. BCA protein assay reagents (Thermo Fisher Scientific, 23225) were used to measure protein concentration. Samples were separated by SDS gel electrophoresis and electro-transferred onto polyvinylidene difluoride membrane in a semi-dry electrophoretic transfer cell (Bio-Rad). Membranes were blocked with 5% nonfat dry milk powder dissolved in Tris-buffered saline (20 mM Tris base, 0.5 M NaCl, pH 7.5) with 0.1% Tween-20 (TBST) for 1 h at RT. The membranes were then incubated with primary antibodies overnight at 4°C in a shaking incubator, washed with TBST 3 times (10 min each) and incubated for 1 h at room temperature with peroxidase-conjugated secondary antibodies diluted in blocking solution. After washing, proteins of interest were detected using either Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32106) or WesternBright ECL (Advansta, K-12045-D50).
qRT-PCR
Cells were lysed using the QIAzol Lysis Reagent (Qiagen, 79306), and RNA was isolated following the manufacturer’s instructions. cDNA was synthesized using the ImProm-II Reverse Transcriptase kit (Promega, A3802) and oligo-dT primers. TOPreal qPCR 2× PreMIX (SYBR Green with low ROX; Enzynomics, RT500M) containing nTaq-HOT DNA polymerase was used for qRT-PCR, which was performed in a CFX96 Real-Time System (Bio-Rad, Hercules, CA) with primers listed in Table S1. Actb was used as the reference gene for normalization.
ELISA of TNF
Cells were seeded onto a 24-well plate; the culture supernatant from each well was collected at the end of scheduled experiments and used to measure TNF concentration by ELISA according to the manufacturer’s instructions (R&D Systems, DY410-05).
Statistical analysis
At least 3 independent experiments were performed for each condition, and the data were presented as mean ± standard error of the mean (SEM) values. Statistical analysis was performed by either unpaired Student t test and statistical significance was evaluated using GraphPad Prism (GraphPad Software).
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grants (2013M3C7A1056099, 2016M3C7A1905074, 2017R1A2B4004289 and 2017R1A2B2004483) and the DGIST Convergence Science Center Program (18-BD-04) of the Ministry of Science and ICT of Korea.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental material for this article can be accessed here.
References
- [1].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMCPMC4835977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yorimitsu T, Klionsky DJ.. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005. November;12(Suppl 2):1542–1552. PubMed PMID: 16247502; PubMed Central PMCID: PMCPMC1828868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016. November;16(11):661–675. PubMed PMID: 27694913; PubMed Central PMCID: PMCPMC5343289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Harris J. Autophagy and cytokines. Cytokine. 2011. November;56(2):140–144. PubMed PMID: 21889357. [DOI] [PubMed] [Google Scholar]
- [5].Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011. March;12(3):222–230. PubMed PMID: 21151103; PubMed Central PMCID: PMCPMC3079381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Munz C. autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016. November;37(11):755–763. PubMed PMID: 27667710. [DOI] [PubMed] [Google Scholar]
- [7].Singh SB, Davis AS, Taylor GA, et al. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006. September 8;313(5792):1438–1441. PubMed PMID: 16888103. [DOI] [PubMed] [Google Scholar]
- [8].Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for crohn disease in ATG16L1. Nat Genet. 2007. February;39(2):207–211. PubMed PMID: 17200669. [DOI] [PubMed] [Google Scholar]
- [9].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008. November 13;456(7219):264–268. PubMed PMID: 18849965. [DOI] [PubMed] [Google Scholar]
- [10].Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007. May;39(5):596–604. PubMed PMID: 17435756; PubMed Central PMCID: PMCPMC2757939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010. January;16(1):90–97. PubMed PMID: 19966812. [DOI] [PubMed] [Google Scholar]
- [12].Crisan TO, Plantinga TS, van de Veerdonk FL, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One. 2011. April 07;6(4):e18666 PubMed PMID: 21490934; PubMed Central PMCID: PMCPMC3072416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012. January 29;13(3):255–263. PubMed PMID: 22286270; PubMed Central PMCID: PMCPMC4116819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 2011. November 8;30(23):4701–4711. PubMed PMID: 22068051; PubMed Central PMCID: PMCPMC3243609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011. October 18;108(42):17396–17401. PubMed PMID: 21969579; PubMed Central PMCID: PMCPMC3198353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007. December 20;450(7173):1253–1257. PubMed PMID: 18097414. [DOI] [PubMed] [Google Scholar]
- [17].Boyle KB, Randow F. Rubicon swaps autophagy for LAP. Nat Cell Biol. 2015;17(7):843. [DOI] [PubMed] [Google Scholar]
- [18].Cho MH, Cho K, Hj K, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014. October 1;10(10):1761–1775. PubMed PMID: 25126727; PubMed Central PMCID: PMCPMC4198361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xu Y, Jagannath C, Liu XD, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007. July;27(1):135–144. PubMed PMID: 17658277; PubMed Central PMCID: PMCPMC2680670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. Embo J. 2008. April 9;27(7):1110–1121. PubMed PMID: 18337753; PubMed Central PMCID: PMCPMC2323261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461 PubMed PMID: 25309543; PubMed Central PMCID: PMCPMC4174766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu Y, Liu XD, Gong X, et al. Signaling pathway of autophagy associated with innate immunity. Autophagy. 2008. January;4(1):110–112. PubMed PMID: 18059159. [DOI] [PubMed] [Google Scholar]
- [23].Waltz P, Carchman EH, Young AC, et al. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011. March 7;(3):315–320. DOI: 10.4161/auto.7.3.14044 PubMed PMID: 21307647. [DOI] [PubMed] [Google Scholar]
- [24].Chen C, Deng M, Sun Q, et al. Lipopolysaccharide stimulates p62-dependent autophagy-like aggregate clearance in hepatocytes. Biomed Res Int. 2014;2014:267350 PubMed PMID: 24683544; PubMed Central PMCID: PMCPMC3934718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yuan H, Perry CN, Huang C, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009. February;296(2):H470–9. PubMed PMID: 19098111; PubMed Central PMCID: PMCPMC2643899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Feng X, Zeng Y, et al. Lipopolysaccharide (LPS)-induced autophagy is involved in the restriction of Escherichia coli in peritoneal mesothelial cells. BMC Microbiol. 2013;13:255 PubMed PMID: 24219662; PubMed Central PMCID: PMCPMC3833177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007. November;10(11):1387–1394. PubMed PMID: 17965659. [DOI] [PubMed] [Google Scholar]
- [28].Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11(11):775. [DOI] [PubMed] [Google Scholar]
- [29].Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017. March 09;18(3). DOI: 10.3390/ijms18030598 PubMed PMID: 28282924; PubMed Central PMCID: PMCPMC5372614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010. March 19;140(6):918–934. PubMed PMID: WOS:000275746600014; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014. May;15(5):300–312. PubMed PMID: 24713688. [DOI] [PubMed] [Google Scholar]
- [32].Dancourt J, Melia TJ. Lipidation of the autophagy proteins LC3 and GABARAP is a membrane-curvature dependent process. Autophagy. 2014. August;10(8):1470–1471. PubMed PMID: 24991828; PubMed Central PMCID: PMCPMC4203524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Klionsky DJ, Elazar Z, Seglen PO, et al. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008. October;4(7):849–850. PubMed PMID: 18758232. [DOI] [PubMed] [Google Scholar]
- [34].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008. August 25;182(4):685–701. PubMed PMID: 18725538; PubMed Central PMCID: PMCPMC2518708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matsunaga K, Morita E, Saitoh T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010. August 23;190(4):511–521. PubMed PMID: 20713597; PubMed Central PMCID: PMCPMC2928018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bae KR, Shim HJ, Balu D, et al. Translocator protein 18 kDa negatively regulates inflammation in microglia. J Neuroimmune Pharmacol. 2014. June;9(3):424–437. PubMed PMID: 24687172. [DOI] [PubMed] [Google Scholar]
- [37].Lee JW, Nam H, Yu SW. Systematic analysis of translocator protein 18 kDa (TSPO) ligands on toll-like receptors-mediated pro-inflammatory responses in microglia and astrocytes. Exp Neurobiol. 2016. October;25(5):262–268. PubMed PMID: 27790060; PubMed Central PMCID: PMCPMC5081472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Min H, Lee H, Lim H, et al. TLR4 enhances histamine-mediated pruritus by potentiating TRPV1 activity. Mol Brain. 2014. August 21;7:59 PubMed PMID: 25139109; PubMed Central PMCID: PMCPMC4237911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006. October 12;443(7112):651–657. PubMed PMID: 17035995. [DOI] [PubMed] [Google Scholar]
- [40].Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016. March 22;113(12):E1738–46. PubMed PMID: 26884166; PubMed Central PMCID: PMCPMC4812770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell. 2015. January 22;57(2):219–234. PubMed PMID: 25578879; PubMed Central PMCID: PMCPMC4306530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, et al. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010. May;11(5):329–341. PubMed PMID: 20379207. [DOI] [PubMed] [Google Scholar]
- [43].Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012. February 23;13(3):195–203. PubMed PMID: 22358332. [DOI] [PubMed] [Google Scholar]
- [44].Rapoport SI, Primiani CT, Chen CT, et al. Coordinated expression of phosphoinositide metabolic genes during development and aging of human dorsolateral prefrontal cortex. PLoS One. 2015;10(7):e0132675 PubMed PMID: 26168237; PubMed Central PMCID: PMCPMC4500567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007. July;39(7):830–832. PubMed PMID: 17554261; PubMed Central PMCID: PMCPMC2628541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zolov SN, Bridges D, Zhang Y, et al. In vivo, pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci USA. 2012. October 23;109(43):17472–17477. PubMed PMID: 23047693; PubMed Central PMCID: PMCPMC3491506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Warr MR, Binnewies M, Flach J, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013. February 21;494(7437):323–327. PubMed PMID: 23389440; PubMed Central PMCID: PMCPMC3579002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang S, Xia P, Huang G, et al. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun. 2016. March 24;7:11023 PubMed PMID: 27010363; PubMed Central PMCID: PMCPMC4820827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009. October 09;284(41):28319–28331. PubMed PMID: 19696026; PubMed Central PMCID: PMCPMC2788882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999. March 19;96(6):857–868. PubMed PMID: 10102273. [DOI] [PubMed] [Google Scholar]
- [51].Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004. May 07;14(3):395–403. PubMed PMID: 15125842. [DOI] [PubMed] [Google Scholar]
- [52].Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007. September;30(9):1617–1623. PubMed PMID: 17827709. [DOI] [PubMed] [Google Scholar]
- [53].Zhu C, Herrmann US, Falsig J, et al. A neuroprotective role for microglia in prion diseases. J Exp Med. 2016. May 30;213(6):1047–1059. PubMed PMID: 27185853; PubMed Central PMCID: PMCPMC4886355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010. December 24;330(6012):1774 PubMed PMID: 21148344; PubMed Central PMCID: PMCPMC3073454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lucin KM, O’Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013. September 04;79(5):873–886. PubMed PMID: 24012002; PubMed Central PMCID: PMCPMC3779465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Song J, Oh Y, Lee JE. miR-Let7A modulates autophagy induction in LPS-activated microglia. Exp Neurobiol. 2015. June;24(2):117–125. PubMed PMID: 26113790; PubMed Central PMCID: PMCPMC4479807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Francois A, Terro F, Quellard N, et al. Impairment of autophagy in the central nervous system during lipopolysaccharide-induced inflammatory stress in mice. Mol Brain. 2014;7:56 PubMed PMID: 25169902; PubMed Central PMCID: PMCPMC4237961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Han HE, Kim TK, Son HJ, et al. Activation of autophagy pathway suppresses the expression of iNOS, IL6 and cell death of LPS-stimulated microglia cells. Biomol Ther (Seoul). 2013. January;21(1):21–28. PubMed PMID: 24009854; PubMed Central PMCID: PMCPMC3762303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013. October;13(10):722–737. PubMed PMID: 24064518; PubMed Central PMCID: PMCPMC5340150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fang W, Bi D, Zheng R, et al. Identification and activation of TLR4-mediated signalling pathways by alginate-derived guluronate oligosaccharide in RAW264.7 macrophages. Sci Rep. 2017. May 10;7(1):1663 PubMed PMID: 28490734; PubMed Central PMCID: PMCPMC5431981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007. December;6(6):458–471. PubMed PMID: 18054315. [DOI] [PubMed] [Google Scholar]
- [62].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012. June;8(6):903–914. PubMed PMID: 22576015; PubMed Central PMCID: PMCPMC3427256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011. June 17;332(6036):1429–1433. PubMed PMID: 21617040; PubMed Central PMCID: PMCPMC3638014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012. March 07;31(5):1095–1108. PubMed PMID: 22343943; PubMed Central PMCID: PMCPMC3298007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kim HJ, Cho MH, Shim WH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. 2016. July 12 DOI: 10.1038/mp.2016.103 PubMed PMID: 27400854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Su P, Zhang J, Wang D, et al. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016. April 05;319:155–167. PubMed PMID: 26827945. [DOI] [PubMed] [Google Scholar]
- [67].Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008. August 13;28(33):8354–8360. PubMed PMID: 18701698; PubMed Central PMCID: PMCPMC2597474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8(4):e60921 PubMed PMID: 23577177; PubMed Central PMCID: PMCPMC3620049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wolfe DM, Lee JH, Kumar A, et al. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013. June;37(12):1949–1961. PubMed PMID: 23773064; PubMed Central PMCID: PMCPMC3694736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang DS, Stavrides P, Mohan PS, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011. July;7(7):788–789. PubMed PMID: 21464620; PubMed Central PMCID: PMCPMC3359468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005. September 07;25(36):8240–8249. PubMed PMID: 16148231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013. January 10;368(2):107–116. PubMed PMID: 23150908; PubMed Central PMCID: PMCPMC3677583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013. January 10;368(2):117–127. PubMed PMID: 23150934; PubMed Central PMCID: PMCPMC3631573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bradshaw EM, Chibnik LB, Keenan BT, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013. July;16(7):848–850. PubMed PMID: 23708142; PubMed Central PMCID: PMCPMC3703870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Krstic D, Madhusudan A, Doehner J, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012. July 2;9:151 PubMed PMID: 22747753; PubMed Central PMCID: PMCPMC3483167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tarkowski E, Andreasen N, Tarkowski A, et al. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003. September;74(9):1200–1205. PubMed PMID: 12933918; PubMed Central PMCID: PMCPMC1738668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015. June;16(6):358–372. PubMed PMID: 25991443. [DOI] [PubMed] [Google Scholar]
- [78].Marim FM, Silveira TN, Lima DS Jr., et al. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One. 2010. December 17;5(12):e15263 PubMed PMID: 21179419; PubMed Central PMCID: PMCPMC3003694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Blasi E, Barluzzi R, Bocchini V, et al. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27(2–3):229–237. [DOI] [PubMed] [Google Scholar]
- [80].Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1(1):241–245. PubMed PMID: 17406239. [DOI] [PubMed] [Google Scholar]
- [81].Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008. November;Chapter 14:Unit14 1 DOI: 10.1002/0471142735.im1401s83 PubMed PMID: 19016445; PubMed Central PMCID: PMCPMC2834554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.