

Until now, knowledge of the utilization of ethanolamine in Streptomyces was limited. Our work represents the first attempt to reveal a novel ethanolamine utilization pathway in the actinobacterial model organism S. coelicolor through the characterization of the key enzyme gamma-glutamylethanolamide synthetase GlnA4, which is absolutely required for growth in the presence of ethanolamine. The novel ethanolamine utilization pathway is dissimilar to the currently known ethanolamine utilization pathway, which occurs in metabolome. The novel ethanolamine utilization pathway does not result in the production of toxic by-products (such as acetaldehyde); thus, it is not encapsulated. We believe that this contribution is a milestone in understanding the ecology of Streptomyces and the utilization of alternative nitrogen sources. Our report provides new insight into bacterial primary metabolism, which remains complex and partially unexplored.
KEYWORDS: Streptomyces, ethanolamine utilization, gamma-glutamylation, nitrogen metabolism
ABSTRACT
Streptomyces coelicolor is a Gram-positive soil bacterium with a high metabolic and adaptive potential that is able to utilize a variety of nitrogen sources. However, little is known about the utilization of the alternative nitrogen source ethanolamine. Our study revealed that S. coelicolor can utilize ethanolamine as a sole nitrogen or carbon (N/C) source, although it grows poorly on this nitrogen source due to the absence of a specific ethanolamine permease. Heterologous expression of a putative ethanolamine permease (SPRI_5940) from Streptomycespristinaespiralis positively influenced the biomass accumulation of the overexpression strain grown in defined medium with ethanolamine. In this study, we demonstrated that a glutamine synthetase-like protein, GlnA4 (SCO1613), is involved in the initial metabolic step of a novel ethanolamine utilization pathway in S. coelicolor M145. GlnA4 acts as a gamma-glutamylethanolamide synthetase. Transcriptional analysis revealed that expression of glnA4 was induced by ethanolamine and repressed in the presence of ammonium. Regulation of glnA4 is governed by the transcriptional repressor EpuRI (SCO1614). The ΔglnA4 mutant strain was unable to grow on defined liquid Evans medium supplemented with ethanolamine. High-performance liquid chromatography (HPLC) analysis demonstrated that strain ΔglnA4 is unable to utilize ethanolamine. GlnA4-catalyzed glutamylation of ethanolamine was confirmed in an enzymatic in vitro assay, and the GlnA4 reaction product, gamma-glutamylethanolamide, was detected by HPLC/electrospray ionization-mass spectrometry (HPLC/ESI-MS). In this work, the first step of ethanolamine utilization in S. coelicolor M145 was elucidated, and a putative ethanolamine utilization pathway was deduced based on the sequence similarity and genomic localization of homologous genes.
INTRODUCTION
Ethanolamine belongs to a class of aliphatic amino alcohols, and it represents both a primary amine and a primary alcohol. Ethanolamine is widely distributed in mammalian and bacterial cell membranes (1, 2), where it is a constituent of phospholipids such as phosphatidylethanolamine. Phosphatidylethanolamine composes 25% to 45% of all phospholipids in a cell and is an important substrate and precursor in several biological pathways (2). As a principal phospholipid in bacteria, phosphatidylethanolamine plays a major role in scattering the negative charge caused by the presence of anionic membrane phospholipids. It enables the proper functioning of transporters and acts as a chaperone to help membrane proteins fold correctly (3, 4). In most bacteria, phosphatidylethanolamine is synthetized via phosphatidylserine decarboxylation (5). Ethanolamine can serve as a precursor of phosphatidylethanolamine in a pathway that includes phosphatidylserine (6) and ethanolamine phosphate (7, 8).
Ethanolamine could have originally formed from NH3/NH4+ in hydrothermal environments on early Earth (9). Since ethanolamine is a common constituent of phospholipids, its omnipresence in all life kingdoms is unsurprising. Ethanolamine can be released into the soil during the decomposition of the organic matter derived from dead plants, animals, fungi, and microorganisms, or it can arrive into the soil as a component of fertilizers or as an unintentionally introduced pollutant (9, 10). Ethanolamine has an antimicrobial effect due to its surface-active properties, and in high concentrations it can effectively inhibit growth of some microorganisms in the soil (9). However, the antimicrobial effect is more significant to autotrophic nitrifying bacteria than to heterotrophic bacteria (11), which seem benefit from this alternative C-/N-source, ensuring their survival under fluctuating nutritional conditions in the soil.
A variety of bacteria, including Gram-negative bacteria such as Salmonella, Escherichia (12), Klebsiella (12), Erwinia, Flavobacterium, Achromobacter, Pseudomonas (13), and Vibrio (14) and Gram-positive bacteria such as Enterococcus (15), Arthrobacter, Corynebacterium (12), Clostridium, Listeria (16, 17), Streptococcus (15), and Mycobacterium (6–8), can utilize ethanolamine as a sole source of carbon and/or nitrogen (18). Bacteria are not able to synthesize ethanolamine de novo; however, extracellular ethanolamine can enter bacterial cells through diffusion or carrier-mediated transport (19). Ethanolamine catabolic genes are located together in an ethanolamine utilization operon (eut). Phylogenetic analysis of almost 100 fully sequenced bacterial genomes revealed the presence of differently organized eut operons (18). Some Actinobacteria and Proteobacteria species have short eut operons containing only the following three genes: eutB and eutC (eutBC; encoding a central enzyme of the ethanolamine utilization process—ethanolamine ammonia lyase) and eat (encoding an ethanolamine transporter). Some Proteobacteria may also contain eutR (encoding a transcriptional regulator EutR of the eut operon). Members of Enterobacteriaceae, which includes Salmonella enterica serovar Typhimurium and Escherichiacoli, as well as members of Firmicutes, possess considerably different and long eut operons (18, 20, 21).
Ethanolamine utilization has been extensively studied in the model organism S. Typhimurium for over 40 years (22). S. Typhimurium possesses a eut operon containing 17 genes encoding proteins involved in ethanolamine transport, metabolism, and regulation. All essential enzymes involved in ethanolamine utilization in this bacterium are located in a metabolosome—a multiprotein complex (carboxysome-like complex). Utilization of ethanolamine involves splitting this compound with an ethanolamine ammonia lyase (EutBC) into ammonia and acetaldehyde, which is further converted into acetyl-coenzyme A (acetyl-CoA) (23). The resulting end products, ammonium and acetyl-CoA, serve as a cellular supply of the easily assimilable N and C sources, respectively (24). Encapsulation of this metabolic pathway prevents loss of the volatile intermediate acetaldehyde and protects the cell from its potential toxicity (25, 26). Although ethanolamine utilization in a metabolosome has been broadly studied in evolutionarily diverse bacteria, the ethanolamine utilization pathway is not universal for all microorganisms.
Hardly anything is known about ethanolamine utilization and its regulation in the genus Streptomyces. The model organism Streptomyces coelicolor is a filamentous Gram-positive, nonmotile, obligate aerobic soil-dwelling bacterium with high G-C content and belongs to the genus Streptomyces, phylum Actinobacteria. Soil, a natural habitat of S. coelicolor, exhibits highly diverse levels of nutrient availability ranging from nutrient-poor to nutrient-rich conditions, depending on the soil type and seasonal changes. This bacterium can assimilate nitrogen from a variety of nitrogen sources, such as ammonium, nitrate/nitrite, amino acids, peptides, urea, and amino sugars. The utilization of alternative nitrogen sources reflects the elaborative survival strategy of Streptomycetes spp. in their natural environment. The capacity of S. coelicolor to utilize alternative nitrogen sources such as polyamines (putrescine, cadaverine, and spermidine) has been recently demonstrated (27).
The high metabolic potential of Streptomycetes ensures the adaptation of these bacteria to a wide variety of ecological niches and successful competition with other microorganisms for space and resources in their habitat. Distinct ecological niches occupied by Streptomyces exerted specific evolutionary pressure on glutamine synthetase (GS) genes (glnA), resulting in the evolution of diverse but thus far uncharacterized glnA-like genes (27, 28). Indeed, Streptomyces coelicolor A3(2) harbors two genes, glnA (SCO2198) and glnII (SCO2210), encoding glutamine synthetases GSI and GSII, whose function and regulation have been extensively studied (29–31), as well as three other genes, glnA2 (SCO2241), glnA3 (SCO6962), and glnA4 (SCO1613), annotated as encoding putative GS-like enzymes (32). As reported by Krysenko et al. (27), the glnA-like genes may encode proteins annotated as glutamine synthetase-like (GS-like) enzymes that may represent as-yet-unrecognized enzymes that catalyze the gamma-glutamylation of different substrates. In this work, we demonstrated the involvement of the GlnA-like enzyme GlnA4, a gamma-glutamylethanolamide synthetase, in a novel pathway for ethanolamine degradation in S. coelicolor M145.
RESULTS
Ethanolamine is not an optimal nitrogen source for S. coelicolor M145.
In this study, the ability of Streptomyces coelicolor M145 to grow on ethanolamine and utilize it as a C or N source was assessed. For this purpose, S. coelicolor M145 was grown in defined medium (Evans), supplemented either with ethanolamine (as the sole N source) and glucose (C source) or with ethanolamine (as the only C source) and ammonium (N source). As a control, S. coelicolor M145 was incubated in defined medium (Evans) supplemented with ammonium (as the sole N source) and glucose (C source). The concentration of ethanolamine and ammonium was 25 mM. The biomass accumulation was determined after 7 days of incubation at 30°C and 180 rpm. S. coelicolor M145 demonstrated very low biomass accumulation in the cultures containing ethanolamine either as the C source or N source. The biomass accumulation in the control culture with ammonium as the sole N source was 5 times higher than that in the culture supplemented with ethanolamine as the sole N source (Fig. 1). This result indicates that ethanolamine is definitely not the preferred nutrient source for S. coelicolor M145. The reason for the low biomass accumulation during growth on ethanolamine might have been either the incapability of efficient ethanolamine uptake or the impaired ethanolamine utilization under the tested conditions.
FIG 1.

Effect of ethanolamine (25 mM) as a sole nitrogen or carbon source on the biomass accumulation of the M145 parental strain, glnA4 mutant, epuRI mutant, and putative ethanolamine permease overexpression strain M145pRM4spri_5940 in defined Evans medium after 7 days of incubation at 30°C. Error bars indicate standard errors of results from n = 3 biological replicates.
The conserved ethanolamine permease gene is absent from the S. coelicolor genome.
To search for the presence of an ethanolamine uptake gene(s) in the S. coelicolor M145 genome-derived proteome, amino acid sequences of the previously extensively characterized ethanolamine uptake proteins EutH and Eat from E. coli and S. Typhimurium were used as targets for the BLASTp analysis. No strong homologs for ethanolamine permease EutH and Eat were identified in S. coelicolor M145 or other Streptomyces spp. However, independent screening of several annotated Streptomyces genomes revealed genes encoding proteins annotated as ethanolamine/amino acid permeases with no similarity to EutH and Eat. Interestingly, some Streptomyces spp. contain a putative ethanolamine/amino acid permease gene in their genomes (SVEN1207 in Streptomyces venezuelae, SPRI_5940 in S. pristinaespiralis, SGR_6475 in S. griseus, XNR_5217 in S. albus, and SCAB_73831 in S. scabies). The putative ethanolamine/amino acid permease gene is located in a thus-far-uncharacterized gene cluster that is conserved to a considerable degree across Streptomyces genomes. Although the gene cluster is present in the S. coelicolor genome (SCO1610 to SCO1615), the gene annotated as encoding a putative ethanolamine/amino acid permease is absent from the gene cluster (Fig. 2). Comparative analysis of the putative ethanolamine permease homologs from 18 Streptomyces spp. by BLASTp revealed a high level of amino acid similarity (83% to 98%). Consequently, if the genome of S. coelicolor M145 contained this homolog, it would have revealed high similarity. However, screening for the putative ethanolamine permease gene in S. coelicolor M145 allowed in silico identification of two putative ethanolamine/amino acid permease genes, SCO6014 and SCO5977, with rather low similarity to the putative ethanolamine permease target from other Streptomycetes spp. (43% to 45%). To verify whether the expression of SCO6014 and SCO5977 might be influenced by ethanolamine, their expression patterns were analyzed in the presence of ethanolamine and ammonium (control) as the sole N source. Transcriptional analysis of SCO6014 and SCO5977 revealed strong expression of these genes in the presence of ammonium, whereas no expression was observed in the presence of ethanolamine under the tested conditions (see Fig. S1 in the supplemental material). These analyses led us to assume that the S. coelicolor M145 genome does not contain any eutH or eat homologs or the putative ethanolamine permease gene (found in other Streptomyces). S. coelicolor M145 seems to be ethanolamine permease deficient; however, small amounts of ethanolamine could possibly enter the cell by diffusion, consistent with our observation of slow growth on ethanolamine.
FIG 2.

Identification and organization of the novel ethanolamine utilization gene cluster in representative Streptomyces sp. genomes (S. coelicolor, S. venezuelae, S. scabies, S. albus, and S. pristinaespiralis). The proposed gene product functions are as follows: orf1, membrane protein; orf2, short-chain dehydrogenase; orf3, aldehyde dehydrogenase; glnA4, gamma-glutamylethanolamide synthetase; etuRI, ethanolamine utilization pathway regulator; orf4, putative ethanolamine transporter; orf5, glutamine amidotransferase.
Transcriptional analysis of SCO6014 and SCO5977. Reverse transcriptase PCR was performed with SCO6014, SCO5977, and hrdB (control) from S. coelicolor M145 cultivated in defined Evans medium with ammonium chloride (25 mM) or ethanolamine (25 mM). Expression of SCO6014 and SCO5977 was not induced by ethanolamine or by ammonium. Download FIG S1, TIF file, 0.1 MB (91KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heterologous expression of the putative ethanolamine permease gene spri_5940 from S. pristinaespiralis promoted biomass accumulation during growth on ethanolamine.
Our analysis showed that ethanolamine is not a preferred N/C source and that its presence might have been a consequence of the absence of an ethanolamine permease in S. coelicolor M145. To verify whether the putative ethanolamine permease gene from S. pristinaespiralis spri_5940 is functional and whether its activity might restore the transport of ethanolamine and improve the growth of S. coelicolor M145 on ethanolamine, heterologous expression analysis was performed. For this purpose, the putative ethanolamine permease gene spri_5940, located on the pRM4 plasmid under the control of the constitutively expressed promoter PemrE, was transferred into S. coelicolor M145. As a negative control, S. coelicolor M145 with pRM4 without an insertion was used. Transcriptional analysis of spri_5940 confirmed its constitutive expression in S. coelicolor M145 pRM4spri_5940 independently of the nitrogen source used (data not shown).
The growth of the S. coelicolor M145 pRM4spri_5940 strain was monitored in defined Evans medium supplemented with ethanolamine (25 mM) as the sole nitrogen source after 7 days of incubation at 30°C. The heterologous expression of spri_5940 in S. coelicolor M145 almost doubled the biomass accumulation during growth on ethanolamine as a N/C source (Fig. 1). Our results show that this gene might encode an ethanolamine permease that is needed for effective ethanolamine uptake in S. coelicolor.
The deletion of glnA4 caused a growth defect in the presence of ethanolamine.
A close inspection of the conserved genomic region comprising SCO1610 to SCO1615 in the S. coelicolor M145 genome revealed a glutamine synthetase-like gene, glnA4 (SCO1613) (Fig. 2). Recently, we showed that this gene encodes neither a glutamine synthetase nor a gamma-glutamylpolyamine synthetase (27); nevertheless, the function of GlnA4 remained unclear. Since the glnA4 gene is most closely located to a putative ethanolamine permease gene in Streptomycetes genomes, we concluded that it might possibly encode a protein involved in the ethanolamine utilization pathway. As previously reported by Krysenko et al. (27), the glnA4 mutant was able to grow on defined Evans agar supplemented with ammonium chloride, sodium nitrate, l-glutamine, monosodium l-glutamate, and polyamines (putrescine, cadaverine, and spermidine) as the sole nitrogen source. However, growth of the glnA4 mutant had never been tested in the presence of ethanolamine. In the current study, the growth of the glnA4 mutant was monitored after 3 to 12 days of incubation at 30°C on defined Evans agar supplemented with 25 mM ethanolamine hydrochloride. As a control, the parental strain was grown under the same conditions. Growth of S. coelicolor M145 on an ethanolamine plate resulted in delayed mycelium development, the absence of spore formation, and slight actinorhodin production, visible as a blue pigment in the agar (Fig. 3). The phenotypic analysis of the glnA4 mutant revealed strongly delayed growth, confirming our hypothesis that GlnA4 might be involved in ethanolamine utilization. The complementation of the glnA4 mutant with the glnA4 gene under the control of its native promoter restored growth on an ethanolamine plate (Fig. 3).
FIG 3.

Physiological role of the glnA4 gene product in S. coelicolor M145 cells grown in the presence of ethanolamine. Phenotypic analysis of S. coelicolor M145 (A), the ΔglnA4 mutant (B), and the complemented mutant ΔglnA4pRM4glnA4 (C) was performed on defined Evans medium with ethanolamine hydrochloride (25 mM) (A) as the sole nitrogen source. Deletion of the glnA4 gene resulted in compromised growth on ethanolamine, whereas complementation with pRMglnA4 under the control of its native promoter restored growth on ethanolamine.
To quantify biomass accumulation and observe cell morphology, the glnA4 mutant and the parental strain were also grown in liquid defined Evans medium supplemented either with 25 mM ethanolamine hydrochloride or with 25 mM ammonium as a control. The biomass yield was determined after 7 days of incubation. The glnA4 mutant was not able to grow in the culture supplemented with ethanolamine. However, in defined Evans medium supplemented with ammonium, both the glnA4 mutant and the parental strain grew well (Fig. 1). The parental strain formed pellets composed of a long and branched mycelium independently of the nitrogen source (ethanolamine or ammonium) added to the medium, whereas the glnA4 mutant revealed no mycelial growth in the culture supplemented with ethanolamine (Fig. 4). Interestingly, only swollen and sporadic germinating spores were present in the glnA4 mutant culture (Fig. 4C).
FIG 4.

Effect of glnA4 deletion on cell morphology in the presence of ethanolamine. The S. coelicolor M145 parental strain (A and B) and ΔglnA4 mutant (C and D) were cultivated in defined Evans medium with ethanolamine hydrochloride (EA; 25 mM) (left panels) or ammonium chloride (AM; 25 mM) (right panels). Phase-contrast microscopic pictures of the M145 parental strain and ΔglnA4 mutant mycelium were taken (under ×400 magnification) after 96 h of growth. Bar = 5 μm.
To verify whether the glnA4 mutant was able to utilize ethanolamine in the presence of other nitrogen sources, both the mutant and parental strains were cultivated in complex medium (yeast extract-malt extract [YEME]–Trypticase soy broth [TSB] [YEME-TSB] [33]) supplemented with 25 mM ethanolamine. Although the mutant was able to grow well in YEME-TSB supplemented with ethanolamine, its growth was inhibited compared to that of the parental strain. To confirm that the inhibition might have been a consequence of impaired ethanolamine utilization, the level of ethanolamine remaining in the culture medium after 24 h and 96 h of cultivation was determined using high-performance liquid chromatography (HPLC). Extracellular ethanolamine levels measured in cultures cultivated for 4 days with ethanolamine (25 mM) demonstrated significant differences. The level of ethanolamine determined in the supernatant from the glnA4 mutant culture remained high during the entire incubation time, which was not the case for the level of ethanolamine in the supernatant from the parental strain culture (Fig. 5). These data indicated that the glnA4 mutant was not able to utilize ethanolamine from the complex medium whereas the parental strain utilized more than half of the initial amount of ethanolamine. These results show that the absence of GlnA4 disallowed growth on ethanolamine, confirming a major role of GlnA4 in ethanolamine utilization in S. coelicolor.
FIG 5.

HPLC-based detection of ethanolamine in the supernatant from the 24-h and 96-h cultures of the M145 parental strain and ΔglnA4 mutant. Both strains were grown in defined Evans medium supplemented with ethanolamine (25 mM) as the sole N source. The HPLC chromatogram data are presented as follows: A, ethanolamine standard; B and C, detection of ethanolamine remaining in the supernatant of the M145 culture after 24 h (B) and 96 h (C) of growth; D and E, detection of ethanolamine remaining in the supernatant of the ΔglnA4 mutant culture after 24 h (D) and 96 h (E). The results indicate that only the S. coelicolor M145 parental strain was able to utilize more than one-third of ethanolamine from the medium after 96 h of incubation. mAU, milli-absorbance units.
Expression of glnA4 is induced under ethanolamine and starvation conditions.
The glnA4 gene, annotated as “glnA-like,” encodes a protein that shows significant similarity to the glutamine synthetase (GlnA = GSI) and the gamma-glutamylpolyamine synthetase, GlnA3. The glnA gene is expressed constitutively (34, 35), and, as reported by Amin et al. (36), the expression of glutamine synthetase glnA is enhanced by low ammonium concentrations. In contrast, the expression of glnA3 is induced by polyamines as well as under conditions of low N and C source concentrations (27). To test whether the expression pattern of glnA4 might reflect some similarities to the glnA or glnA3 expression patterns, a transcriptional analysis of glnA4 was performed under various N conditions. For this purpose, S. coelicolor parental strain M145 was grown in complex S-medium for 4 days and was subsequently transferred into Evans medium supplemented with ethanolamine hydrochloride, ammonium chloride, or polyamines (putrescine, cadaverine, and spermidine) as the sole nitrogen source (25 mM each) and incubated further for 24 h at 30°C. Total RNA was isolated from S. coelicolor M145 and used to generate cDNA for reverse transcriptase PCR (RT-PCR) analysis with primers for glnA4 (and hrdB as an internal control). Transcriptional analysis of glnA4 revealed elevated expression levels of the glnA4 gene in the presence of ethanolamine, a putative substrate of the encoded enzyme. glnA4 gene expression was also enhanced under conditions of N and C starvation, namely, under conditions of small amounts of glucose (2.5 g/liter) and ammonium (5 mM) (Fig. 6). The level of glnA4 gene expression was low in the presence of low ammonium concentrations and high glucose concentrations as well as under conditions of high polyamine concentrations and glucose proficiency, showing that the glnA4 expression pattern is dissimilar to the previously reported expression patterns of glnA (36) and glnA3 (27) under the same tested conditions.
FIG 6.

Transcriptional analysis of glnA4 in the presence of ammonium, polyamines, and ethanolamine as sole nitrogen sources. Data represent results of reverse transcriptase PCR analysis of glnA4 and hrdB (control) from S. coelicolor M145 cultivated in defined Evans medium with 5 mM (Am-) or 50 mM (Am+) concentrations of ammonium chloride, polyamines (Put, putrescine; Cad, cadaverine; Spd, spermidine, 25 mM each) or ethanolamine (EtAm; 25 mM) and glucose as the sole carbon source at a high concentration (Glc+; 25 g/liter) or a low concentration (Glc-; 2.5 g/liter). Total RNA was isolated from mycelium harvested after 24 h of cultivation in defined Evans medium.
The expression of glnA4 is regulated by the negative transcriptional regulator EpuRI.
Analysis of glnA4 genomic localization revealed a gene encoding a putative regulatory protein (SCO1614 [epuRI]). This regulatory gene is positioned upstream of glnA4 in an orientation opposite that of glnA4. To verify whether this gene encodes a potential regulator of glnA4, a mutant with a knockout of the epuRI gene was generated. The growth of the epuRI mutant was monitored in liquid Evans medium supplemented with 25 mM ethanolamine hydrochloride or 25 mM ammonium chloride (control) as the sole nitrogen source. The biomass was determined after 3 to 7 days of incubation at 30°C and 180 rpm. The epuRI mutant demonstrated higher biomass accumulation in the culture with ethanolamine than parental S. coelicolor M145 strain. In the epuRI mutant culture grown on ethanolamine as the sole N source, the biomass doubled compared to the biomass of the parental strain grown under the same conditions. The epuRI mutant culture grown on ethanolamine as the sole C source revealed biomass three times as high as that of the parental strain grown under the same conditions. These results indicate that the deletion of epuRI positively influenced growth on ethanolamine (Fig. 1).
To study the effect of the epuRI deletion on the expression of glnA4 in the presence of ammonium or ethanolamine, the cells of the M145 parental strain and the epuRI mutant were incubated in complex S-medium at 30°C for 4 days. Then, the pellets were washed twice with Evans medium to remove complex nitrogen sources and transferred into Evans medium with ammonium chloride (25 mM), ethanolamine hydrochloride (25 mM), or polyamines as the only nitrogen source and incubated for 24 h at 30°C. Total RNA was isolated and used to generate cDNA, which was then subjected to RT-PCR analysis using internal primers for epuRI and hrdB. Transcriptional analysis revealed strong expression of glnA4 in the M145 parental strain in the presence of ethanolamine and in the epuRI deletion mutant in the presence of ethanolamine and ammonium (Fig. S8). This result indicates that epuRI encodes a repressor of glnA4 because deletion of this regulator allows glnA4 expression in the presence of ammonium.
Transcriptional analysis of glnA4 in the presence of ethanolamine and ammonium as the sole nitrogen source. Reverse transcriptase PCR analysis of glnA4 and hrdB (control) from S. coelicolor M145 and the epuRI mutant cultivated in defined Evans medium with ammonium chloride (25 mM) or ethanolamine (25 mM) was performed. Total RNA was isolated from mycelium harvested after 24 h of cultivation in defined Evans medium. Download FIG S8, TIF file, 1.0 MB (1.1MB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GlnA4, a predicted gamma-glutamylethanolamide synthetase in S. coelicolor M145.
The GlnA4 protein, deduced from the glnA4 DNA sequence, has been classified as a ligase that might form carbon-nitrogen bonds in an ATP-dependent manner, corresponding to class 6.3.1. This class includes 20 different subclasses of enzymes, e.g., glutamine synthetases (GS) (class 6.3.1.2), glutamate-ethylamine ligases (class 6.3.1.6), and glutamate-putrescine ligases (6.3.1.11). Analysis of the GlnA4 protein sequence by InterProScan predicted two enzymatic domains that resemble the N-terminal GS, beta-Grasp domain (IPR008147) and the C-terminal catalytic domain (IPR008146) of GS. Searching for functionally and structurally characterized GlnA4 homologs in the Protein Data Bank (PDB) revealed gamma-glutamylmonoamine/polyamine synthetase PauA7 (PA5508) from Pseudomonasaeruginosa and GSISt from S. Typhimurium as possible templates for a GlnA4 structural model. PauA7 is involved in monoamine/polyamine gamma-glutamylation in P. aeruginosa (37). The crystal structures of GS GSISt from S. Typhimurium (Protein Data Bank entry: 1FPY) and Pau7 (Protein Data Bank entry: 4HPP) were used as templates to generate a GlnA4 structural model. Superposition of the GS and PauA7 template structures and the GlnA4 model structure revealed conserved residues coordinating Mn++/Mg++ (N1 and N2), glutamate, and ATP. GSISt (as well as other eukaryotic and bacterial GS) and PauA7 require two divalent metal ions per subunit for activity. These ions have structural and catalytic roles. Comparison of the conserved catalytic residues with respect to the binding of divalent metal ions revealed six conserved residues, E156, E158, E219, E226, H274, and E359, in the GlnA4 model structure (corresponding to E131, E133, E180, E187, H236, and E322 in PauA7 and to E129, E131, E212, E220, H269, and E357 in GSISt).
Moreover, five conserved residues involved in the coordination of glutamate binding in GSISt and PauA7 were found in the GlnA4 model structure. These GlnA4 residues (E158, E269, G270, R325, and R361) correspond to E133, G234, N233, R290, and R334 in PauA7 as well as to E131, N264, G265, R321, and R359 in GSISt. Finally, two conserved residues (H276 and R348), both corresponding to H271 and R344 in GSISt as well as to H238 and R313 in PauA7, that are important for coordination of the beta-phosphate and alpha-phosphate groups of ADP were found in the GlnA4 model. This comparative in silico analysis demonstrated that GlnA4 possesses conserved residues for the binding of two metal ions, l-glutamate and ATP (Fig. S2 to S6).
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the Mg++/Mn++ binding pockets. Download FIG S2, TIF file, 0.4 MB (417.2KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
An essential element of the GSISt catalytic pocket is a loop termed “the E327 flap” (GSISt). The GSI active site is located between two subunits, and the E327 flap closes the catalytic site by interaction of E335 with D50 from an adjacent subunit within the same ring, shielding the gamma-glutamyl phosphate intermediate from hydrolysis. The second conserved residue, D50, is involved in the deprotonation of ammonium for attack on γ-glutamyl phosphate. Interestingly, these key acidic residues (E327 and D50) that are essential for the catalytic synthesis of glutamine in GSISt are not conserved in PauA7 or in GlnA4 (Fig. S2 to S6). In the PauA7 structure and the GlnA4 model, these loops are much larger, and instead of E327, a nonpolar F331 residue occupies the analogous position in the GlnA4 model structure (corresponding to W296 in PauA7). The conserved D50 residue that increases the affinity for ammonium binding in GSISt is replaced by G40 and D81/D69 (depending on the model) in PauA7 and GlnA4, respectively. Moreover, the conserved Y179 in GSISt that coordinates the ammonium binding pocket is substituted by A147 in PauA7 and by V189/D186 (depending on the model) in GlnA4, providing much more space for a substrate larger than ammonium, such as ethanolamine (Fig. S2 to S6). The significant overall similarity of GlnA4 to the gamma-glutamylmonoamine/polyamine synthetase PauA7, the lack of conserved residues for ammonium binding, and our observations strongly suggest that GlnA4 may function as a gamma-glutamylethanolamine synthetase catalyzing the first step of ethanolamine utilization.
GlnA4 catalyzes the gamma-glutamylation of ethanolamine in vitro.
To elucidate the function of GlnA4 and to show that GlnA4 is able to catalyze the predicted glutamylation reaction, we developed an HPLC/mass spectrometry (HPLC/MS)-based assay designed to detect a gamma-glutamylated product formed by GlnA4. GlnA4 was heterologously produced as a His-tagged protein in E. coli BL21(DE3) and purified by nickel ion affinity chromatography. Purified His-GlnA4 was used in an in vitro assay as described in Materials and Methods. The reaction conditions were as follows. A 10-μg volume of His-GlnA4 was incubated at 30°C in a reaction mixture composed of glutamate, ethanolamine, MgCl2, ATP, and HEPES buffer (pH 7.2). The product of the GlnA4-catalyzed reaction was detected using HPLC/MS analysis in positive MS mode. The results indicated that GlnA4 was able to use glutamate (mass-to-charge ratio of 146 m/z) (Fig. 7A and B) and ethanolamine as substrates in an ATP-dependent reaction, generating a product with a mass-to-charge ratio of 189 m/z, corresponding to the calculated mass of the predicted gamma-glutamylethanolamide (Fig. 7D) reaction product. The results of the HPLC/MS analysis confirm that GlnA4 is a functional gamma-glutamylethanolamide synthetase that uses glutamate and ethanolamine as substrates to generate gamma-glutamylethanolamide.
FIG 7.
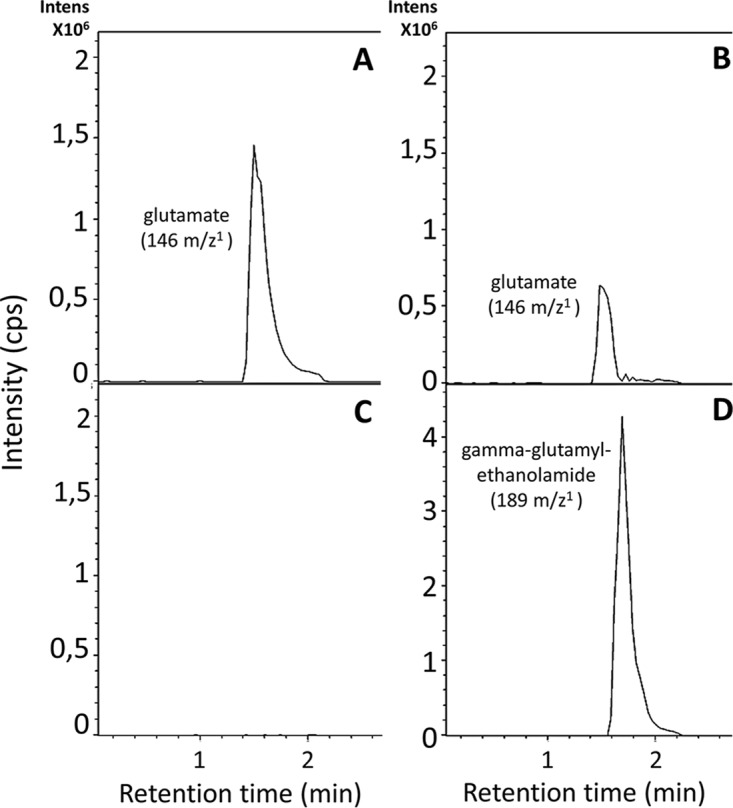
HPLC/MS analysis of the GlnA4-catalyzed reaction. Samples were taken after 0 (A and C) and 10 min (B and D) and analyzed in −MS mode. Extracted ion chromatograms for glutamate with a mass-to-charge ratio of 146 m/z (A and B) and for gamma-glutamylethanolamide with 189 m/z (D) are shown. Intens, intensity. (C) In the absence of the GlnA4 no product (gamma-glutamylethanolamide) could be detected.
Biochemical characterization of GlnA4.
To determine the kinetic parameters of GlnA4 and its substrate specificity, an adapted GS activity assay based on the inorganic phosphate (Pi) released from ATP was used (38). Pi released from ATP was measured using a colorimetric ammonium molybdate-based detection method. Phosphate that formed a complex with molybdate could be reduced in a strong acidic solution to form molybdenum blue. This change in color was measured by reading the absorption at 655 nm.
A total of 10 micrograms of purified His-GlnA4 was used, which was sufficient to produce between 50 and 100 nmol Pi in an incubation performed for 5 min at 30°C. For the quantification of the released inorganic phosphate, a calibration curve was prepared with potassium phosphate dibasic K2PO4 at concentrations of 20, 10, 5, 2.5, 1.25, 0.63, 0.31, 0.16, 0.08, 0.04, 0.02, and 0 mM. After the kinetic parameters of ATP were described, solution E was used to create an additional calibration curve with the same K2PO4 concentrations.
To test the substrate specificity of GlnA4, this enzyme was incubated with different substrates at a concentration of 50 mM. To calculate the activity of GlnA4, the amount of released inorganic phosphate was quantified and expressed in nanomoles of Pi per minute per microgram of enzyme (Fig. 8; see also Fig. S7).
FIG 8.
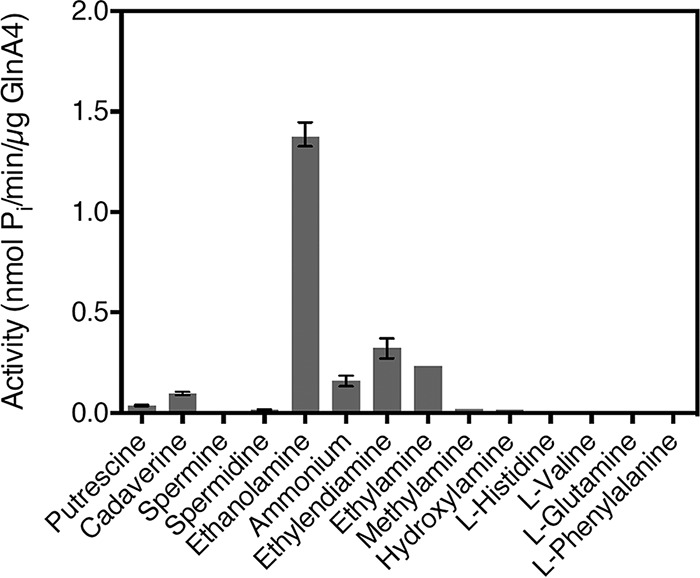
Effect of different amino group-containing compounds on the activity of GlnA4. All potential substrates of GlnA4 were present at a concentration of 50 mM. Mean values of n = 3 measurements are shown with standard deviations. GlnA4 is specific for ethanolamine.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the glutamate binding pockets. Download FIG S3, TIF file, 0.4 MB (440.7KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ADP binding pockets. Download FIG S4, TIF file, 0.4 MB (424KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ammonium binding pockets. Download FIG S5, TIF file, 0.4 MB (377.5KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ammonium binding pockets. Download FIG S6, TIF file, 0.4 MB (441KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Activity of GlnA4 with various concentrations of ethanolamine (A), glutamate (B), and ATP (C) and in the presence of MSO (D). A nonlinear regression (solid black line) was made using a least-squares fit of n = 3 data sets (white bullets) and assuming the Michaelis-Menten model. Download FIG S7, TIF file, 0.1 MB (131.3KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The biochemical analysis revealed that GlnA4 accepts ethanolamine, glutamate, and ATP as substrates. It catalyzes the ATP-dependent synthesis of gamma-glutamylethanolamide from ethanolamine and glutamate. The formation of gamma-glutamylethanolamide and simultaneous depletion of glutamate were detected by HPLC/MS. Assuming the Michaelis-Menten model, the Km values were determined to be 0.47 mM for ethanolamine, 85.41 mM for glutamate, and 2.5 mM for ATP. The highest Vmax value of 4.18 nmol Pi per min per 1 μg of enzyme was determined for glutamate, while Vmax values of 1.48 and 0.76 nmol Pi per min per 1 μg of GlnA4 were determined for ethanolamine and ATP, respectively (Fig. S7).
Incubation of GlnA4 with the reaction mixture containing methionine sulfoximine (MSO; a potent inhibitor of glutamine synthetases) only slightly lowered the activity of GlnA4 (Fig. S7). The estimated Ki value (11 mM) was approximately 10 to 5,000 times higher than the Ki values reported for glutamine synthetases (39–42), indicating that MSO is not a potent inhibitor of GlnA4.
DISCUSSION
Streptomycetes spp. have a vast metabolic potential and are able to assimilate nitrogen from a variety of minerals (e.g., ammonium, nitrate, and nitrite) and organic sources (e.g., urea, amino acids, peptides, amino sugars, and polyamines). However, a nitrogen source such as ethanolamine, which is also omnipresent in nature, has never been reported to be utilized by S. coelicolor. In this study, we demonstrated that Streptomyces coelicolor M145 can utilize ethanolamine as a sole carbon and nitrogen source. However, the slow growth of Streptomyces coelicolor M145 on medium with ethanolamine as the sole N/C source showed that ethanolamine was not a preferred N/C source for growth under the tested conditions.
Depending on conditions, extracellular ethanolamine can enter the cell through diffusion, carrier-mediated transport (19), or, alternatively, with the help of transport proteins, such as permease-like proteins EutH in E. coli and S. Typhimurium (19, 43) and Eat in Actinobacteria and most Proteobacteria (18). Screening of Streptomyces genomes revealed no strong homologs of EutH or Eat. However, genes annotated as ethanolamine/amino acid permeases with no similarity to EutH or Eat were found. Screening of the S. venezuelae, S. pristinaespiralis, S. griseus, S. albus, and S. scabies genomes revealed a gene encoding a putative ethanolamine/amino acid permease located in the gene cluster containing the glnA4 gene. Introduction of one of the predicted ethanolamine permease genes (spri_5940 from S. pristinaespiralis) into S. coelicolor positively influenced the growth of S. coelicolor-pRMspri_5940 on defined medium supplemented with ethanolamine as the sole N source. It is likely that an ethanolamine permease-encoding gene was lost in the S. coelicolor genome during the evolution process; however, the rest of the ethanolamine utilization cluster was maintained, presumably to ensure intracellular ethanolamine recycling. S. coelicolor has a plethora of sugar, organic anion, and amino acid uptake systems of very specific types (total, 658 transporter proteins) (44). The possibility of ethanolamine uptake occurring via an alternative transporter with low affinity cannot be excluded. This kind of transporter might belong to the amino acid-polyamine-organocation (APC) superfamily consisting of 17 predicted members (44).
Intracellular ethanolamine content can increase as a result of glycerophospholipid polar head recycling, as shown in Mycobacterium tuberculosis (45). Phosphodiesterase (Rv3842) breaks down lipid polar heads of glycerophosphoethanolamine, glycerophosphoglycerol, or glycerophospho-1-d-myo-inositol, releasing glycerol phosphate (45). The released by-product of glycerophosphoethanolamine cleavage is ethanolamine, which can be further metabolized as a C and N source. The ethanolamine level in the cell must be strictly controlled due to its potential toxic effect when present in excess. Intracellular ethanolamine accumulation might lead to alkalization of the cellular milieu, unspecific binding to negatively charged molecules such as DNA and RNA, compromised cellular homeostasis, and, ultimately, cell death. Since the expression of glnA4 is not only induced by ethanolamine but also under the control of nutrient limitation, it can be concluded that GlnA4 may play a supporting role during starvation. Under such conditions, cellular membranes likely undergo degradation, increasing extra and intracellular ethanolamine concentrations.
The parental strain S. coelicolor M145 is able to grow on ethanolamine as a sole N/C source. Relating to our studies, this ability is due to the catalytic activity of GlnA4. The deletion of glnA4 resulted in a growth defect for S. coelicolor M145 in liquid medium, strongly impaired growth on solid medium, and affected cell morphology in the presence of ethanolamine. In vitro analysis of the enzymatic activity of GlnA4 revealed that GlnA4 functions as a gamma-glutamylethanolamide synthetase. Our study showed that GlnA4 is specific to ethanolamine and catalyzes its glutamylation using glutamate and ATP. Thus, GlnA4 can be classified in a large class of ligases that can form a carbon-nitrogen bond in an ATP-dependent manner (class 6.3). Although GlnA4 resembles features of glutamine synthetases (GSs), it is not able to catalyze glutamine biosynthesis. Currently, there are few examples described in the scientific literature in reports of glutamine synthetase-like enzymes that catalyze the glutamylation of various N compounds but lack glutamine synthetase activity. For instance, Kurihara et al. (46) described gamma-glutamylputrescine synthetase PuuA, which catalyzes ATP-dependent gamma-glutamylation of putrescine in E. coli. Krysenko et al. (27) demonstrated that gamma-glutamylpolyamine synthetase GlnA3 catalyzes the glutamylation of putrescine, cadaverine, spermidine, and spermine in S. coelicolor. Furthermore, seven gamma-glutamylpolyamine/monoamine/aromatic amine synthetases (PauA1 to PauA7) have been described in Pseudomonas aeruginosa PAO1 (37, 47). PauA7 is the only structurally characterized glutamine-like enzyme with the ability to glutamylate aromatic monoamines. PauA7 exhibits a structure different from that seen with the common glutamine synthetase archetype, forming only a single hexameric ring instead of two (37). Whether GlnA4 forms one or two rings and how its catalytic pocket differs from that of glutamine synthetases are under investigation.
The glutamylation reaction is common in nature. For example, gamma-glutamyltransferases can catalyze the transfer of gamma-glutamyl groups from glutathione to various amino acids or peptides (48); polyglutamylases catalyze the glutamylation of proteins (49); and gamma-glutamylpolyamine/monoamine or aromatic amine synthetases catalyze the glutamylation of various amines (27, 37, 46, 47). Furthermore, gamma-glutamyl compounds, e.g., peptidoglycan, glutathione (gamma-glutamylcysteinylglycine), poly-gamma-glutamic acid, glutamylated proteins, gamma glutamylpolyamines/monoamines, and gamma-glutamyltaurine, are not rare in nature (50). The glutamylation of compounds was shown to positively influence their chemical properties, for instance, to increase solubility, change the taste or aroma, increase stability in solution or half-life in blood serum, and reduce toxicity (50). Therefore, the biotechnological application of gamma-glutamylating enzymes is of high interest. For example, theanine (l-gamma-glutamylethylamide), which is a major aromatic “umami” component of tea and which has thus far been extracted from tea plants, might be able to be biotechnologically synthesized using only enzymatic properties of GlnA4. Altering the binding affinity of GlnA4 through protein engineering such that ethylamine would be preferred as a substrate instead of ethanolamine would lead to the effective and sustainable synthesis of theanine.
The glnA4 gene is annotated as a glutamine synthetase-like gene and is one of the three glnA-like genes in S. coelicolor. The glutamine synthetase-like genes are also found multiple times in the phylum Actinobacteria, including the human pathogen Mycobacterium tuberculosis (28). Sequence analysis showed that glnA1- and glnA2-encoded glutamine synthetase sequences were inherited from an Actinobacteria ancestor whereas glnA4 was sequentially acquired during Actinobacteria speciation. In particular, in Mycobacteria, a reductive evolution of glnA4 can be observed, while glnA1 and glnA2 seem to be more extensively conserved. However, to date, there has been no description of a glutamine-synthetase-like enzyme in Actinobacteria, making the characterization of GlnA4 as a gamma-glutamylethanolamide synthetase the first such characterization.
In this work, novel insights into the regulatory network of ethanolamine utilization genes, including glnA4, were developed. The issue of the regulation of glnA4 has remained unsolved thus far, since it was shown that GlnR and GlnRII, the global transcriptional regulators of nitrogen assimilation genes, do not bind the promoter regions of glnA-like genes (34, 35, 51). Additionally, a genome-wide analysis of GlnR-regulated genes in M. smegmatis demonstrated that a homolog of GlnA4 was not regulated by the global nitrogen-response regulator GlnR (52). The epuRI (sco1614) gene is annotated as a putative transcriptional regulator in the genome of S. coelicolor (53) and is localized upstream of glnA4. Our studies suggest that EpuRI can act as a negative transcriptional regulator of glnA4.
The ability of S. coelicolor to utilize ethanolamine as an N and C source with the help of GlnA4 allowed the prediction of the ethanolamine utilization pathway. S. coelicolor does not possess a canonical ethanolamine utilization pathway that involves eut genes. Additionally, no homologs of eut genes of E. coli or S. Typhimurium were found in the S. coelicolor genome. However, the ethanolamine utilization pathway of S. coelicolor may look similar to the previously unknown ethanolamine utilization pathway in Chromohalobacter salexigens, which was predicted in an in silico approach, using the EFI-GNT Web tool, and is based on glutamylation (54). In S. coelicolor, the gene encoding GlnA4 is localized near four genes that may be involved in the utilization of ethanolamine. After gamma-glutamylethanolamide is generated by GlnA4, it may be oxidized to a gamma-glutamylacetaldehyde by a predicted dehydrogenase (SCO1611). Subsequently, gamma-glutamylacetaldehyde might be converted to gamma-glutamylglycine by a predicted gamma-glutamylaldehyde dehydrogenase (SCO1612). The gamma-glutamylglycine may then be hydrolyzed to glycine and l-glutamate by a member of the formylglutamate amidohydrolase family (e.g., by a predicted gamma-glutamylglycine amidohydrolase [SCO1615]) (see Fig. S9 in the supplemental material). Alternative pathways which do not require a metabolosome and do not result in the production of toxic by-products (such as acetaldehyde) were also reported in Mycobacterium spp. and C.salexigens (7, 8, 54–56). For instance, in Mycobacterium, ethanolamine can be converted via glycoaldehyde and glyoxalate intermediates to glycine, which can be subsequently converted to serine and alanine (7, 55).
Combined model of bacterial ethanolamine metabolism (7, 20, 54–56). ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, phosphate; CoASH, coenzyme A; Glu, glutamate; NAD+/NADH, nicotinamide adenine dinucleotide. Dashed arrows represent transport by diffusion. Download FIG S9, TIF file, 1.7 MB (1.8MB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In this work, we demonstrated the function of GlnA4 as a gamma-glutamylethanolamide synthetase and its importance for ethanolamine catabolism in S. coelicolor. A better understanding of ethanolamine utilization in Streptomyces is of fundamental importance since this pathway allows the survival of Actinobacteria spp. living in diverse habitats. Our work represents the first attempt to reveal the ethanolamine utilization pathway in the actinobacterial model organism S. coelicolor through the characterization of GlnA4, a key enzyme that is absolutely required for survival under conditions of high ethanolamine concentrations. The ethanolamine utilization pathway is of great benefit for Streptomyces spp. that live under constantly unstable nutritional conditions and are forced to compete with other soil microorganisms and plants for the nitrogen available in the soil.
MATERIALS AND METHODS
Strains and growth conditions.
The S. coelicolor M145 parental strain and all mutants (Table 1) were incubated for 4 to 5 days at 30°C on defined Evans agar base (modified from the version described previously by Evans et al. [57]) supplemented with the following different nitrogen sources: 50 mM ammonium chloride, l-glutamine, sodium nitrate, monosodium glutamate, urea, and ethanolamine hydrochloride (in appropriate concentrations). For growth experiments in liquid culture, complex S-medium (58), YEME-TSB (33), or chemically defined Evans medium (57) was used. If appropriate, media were supplemented with ethanolamine hydrochloride or ammonium chloride as a sole nitrogen source. Strains were cultivated for 4 to 5 days at 30°C on a rotary shaker (180 rpm). Genetic manipulation of S. coelicolor M145 was performed as described by Kieser et al. (59) and Gust et al. (60). For genomic DNA preparation, S. coelicolor M145 was grown for 4 days in S-medium, and DNA was isolated by the use of a NucleoSpin tissue kit (Macherey-Nagel, Düren, Germany).
TABLE 1.
Strains and plasmids used in this work
| Strain or plasmid | Genotype or descriptiona | Source or reference |
|---|---|---|
| E. coli XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F0 proAB lacIqZ1M15 Tn10 TetR] | 63 |
| E. coli S17-1 | recA pro mod+ res− tra genes from plasmid RP4 integrated in the chromosome | 64 |
| S. coelicolor M145 | Streptomyces coelicolor A3(2) without native plasmids: spc1− and spc2− | 59 |
| S. pristinaespiralis ATCC 25486 | Streptomyces pristinaespiralis strain ATCC 25486 | Fischbach et al.b |
| S. coelicolor M145 ΔglnA4 | glnA4 mutant strain of S. coelicolor M145; glnA4 replaced by an aac(3)IV cassette; Aprr | This work |
| S. coelicolor M145 ΔglnA4pRM4glnA4 | Complemented glnA4 mutant strain of S. coelicolor M145; Aprr and Kmr | This work |
| S. coelicolor M145 ΔepuRI | epuRI mutant strain of S. coelicolor M145; epuRI replaced by an aac(3)IV cassette; Aprr | This work |
| S. coelicolor M145 pRM4spri_5940 | Heterologous overexpression strain of S. coelicolor M145 with pRM4spri_5940; Aprr | This work |
| pRM4 | pSET152ermEp* with artificial RBS; Aprr | 65 |
| pIJ10700 | pBluescript II KS(C) containing hyg-oriT cassette; Hygr | 60 |
Aprr, apramycin resistance; Hygr, hygromycin resistance; Kmr, kanamycin resistance; RBS, ribosome binding site.
Sequence submitted to the EMBL/GenBank/DDBJ databases by M. Fischbach, P. Godfrey, D. Ward, S. Young, Q. Zeng, M. Koehrsen, 21 October 2009.
Construction of the ΔglnA4 mutant.
The glnA4 gene was replaced by the apramycin resistance gene using plasmid pK18 carrying homologous regions of glnA4. The construct was confirmed by sequencing and subsequently introduced into S. coelicolor M145 by conjugation using the E. coli S17 strain. Conjugants with an apramycin-resistant phenotype were then selected. The correct integration of pRM4sco1613 was confirmed by PCR and sequencing.
Construction of the M145pRM4spri_5940 overexpression mutant.
To enable the heterologous expression of ethanolamine permease from S. pristinaespiralis in S. coelicolor M145, the spri_5940 gene without its native promoter was amplified by PCR using SPRI_5940_F and SPRI_5940_R primers (Table 2) and cloned into the multiple-cloning site of the pRM4 plasmid between the NdeI and HindIII restriction sites. The construct was confirmed by sequencing and subsequently introduced into S. coelicolor M145 by conjugation using the E. coli S17 strain. Conjugants with an apramycin-resistant phenotype were then selected. The correct integration of pRM4-spri_5940 was confirmed by PCR and sequencing.
TABLE 2.
Oligonucleotides used in this work
| Oligonucleotide | Sequence (5′–3′) | Source or reference |
|---|---|---|
| SPRI_5940_F | CGCCCATATGGCTGAAGGCACCACATC | This work |
| SPRI_5940_R | TCCAAGCTTTCACTTCCGTTCCAGTTC | This work |
| pRM_GenF | CTGCAAGGCGATTAAGTTGG | This work |
| pRM_GenR | TTATGCTTCCGGCTCGTATG | This work |
| spri_5940F_RT | GCTGTAGAAGGCGAAGTAGG | This work |
| spri_5940R_RT | CGATCAGCTACGCGCTGATG | This work |
| hrdB-qrt1 | TGACCAGATTCCGGCCACTC | This work |
| hrdB-qrt2 | CTTCGCTGCGACGCTCTTTC | This work |
| rt-glnA4-fw | CGCCTGGGACGCGAACTAC | 66 |
| rt-glnA4-rev | CTGGGCGGCGATCTCCTTG | 66 |
| RT1614N4F | GGACACGATGCTGCACCTGA | This work |
| RT1614N4R | TGCAGTCCAGCAGGTCGTTC | This work |
| rt_sco1612up | TCACGCTCCTCGAACCACTC | This work |
| rt_sco1612dw | CGTCGGCGAAGACGATGTTG | This work |
| RT5977F | TTCCAGGACGGCAACCTCAC | This work |
| RT5977R | AAGACCACGCCGACGATCAG | This work |
| RT6014F | GTCAAGAGCGCCAACTAC | This work |
| RT6014R | TACAGGAGGGTGCAGATG | This work |
| SCO1614_f | AAAAAGCTTCAGCACCTGGAACAGGTCCT | This work |
| SCO1614_r | AAAATCTAGAACCGGCTCATCGTCGTCACT | This work |
| 1614_BamHI | AAAGGATCC TCAGTGGTGGTGGTGGTGG | This work |
| 1614-NdeI | AAAAAACATATGACGGACCGGCTGGCGC | This work |
| SCO1614-XhoI | AAAACTCGAGCGTCAGAAACCCGCGCAGCA | This work |
| SCO1614-NcoI | AAAACCATGGGCACGGACCGGCTGGCGCCG | This work |
Construction of the ΔglnA4 complementation mutant.
For complementation of the mutant, the glnA4 gene with its native promoter was amplified by PCR using glnA4_XbaI and glnA4_EcoRI primers (Table 2) and cloned into the multiple-cloning site of the pRM4 plasmid between the XbaI and EcoRI restriction sites in the opposite orientation to the promoter PemrE, which is located on the plasmid. Subsequently, the kanamycin resistance cassette was amplified by PCR using the pK18 plasmid as a template and primers aphIIupperEcoRI and aphIIlowerHindIII and introduced into the multiple-cloning site of recombinant plasmid pRM4-glnA4C between the EcoRI and HindIII restriction sites. The correct construct was confirmed by sequencing and subsequently introduced into the ΔglnA4 mutant by conjugation using the E. coli S17 strain. Clones with a kanamycin-resistant and apramycin-resistant phenotype were then selected. The correct integration of pRM4-glnA4 was confirmed by PCR and sequencing.
Construction of the ΔepuRI mutant.
To generate the in-frame deletion of the epuRI (sco1614) gene, the REDIRECT gene replacement procedure (60) was employed. The epuRI gene was replaced by the apramycin resistance gene. The construct was confirmed by sequencing and subsequently introduced into S. coelicolor M145 by conjugation using the E. coli S17 strain. Conjugants were then selected on a resistant phenotype against apramycin. The correct integration of pRM4-epuRI was confirmed by PCR and sequencing.
Analysis of gene expression by reverse transcriptase PCR.
For transcriptional analysis experiments, S. coelicolor M145, the glnA4 mutant, and the epuRI mutant were cultured in S-medium. After 4 days of incubation, cells were washed twice with defined Evans medium and cultivated for 24 h in defined Evans medium supplemented with 25 mM ammonium chloride, 25 mM polyamine (cadaverine, spermidine, or putrescine), or 25 mM ethanolamine. RNA was isolated with an RNeasy kit (Qiagen, Venlo, The Netherlands) and treated twice with DNase (Thermo Fisher Scientific, Waltham, MA, USA). First, an on-column digestion was carried out for 30 min at 24°C, and then RNA samples were treated with DNase for 1.5 h at 37°C. RNA concentrations and quality were checked using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). For the generation of cDNA from 3 μg RNA, random nonamer primers (Sigma-Aldrich Chemie GmbH, Munich, Germany) and reverse transcriptase and cofactors (Thermo Fisher Scientific, Waltham, USA) were used. PCRs were performed with the primers listed in Table 2. The PCR conditions were 95°C for 5 min; 35 cycles of 95°C for 15 s, 55 to 60°C for 30 s, and 72°C for 30 s; and 72°C for 10 min. As a positive control, cDNA was amplified from the major constitutively produced vegetative sigma factor (hrdB) transcript. Negative controls were carried out by using total RNA as a template for each RT-PCR to exclude DNA contamination. RT-PCRs were performed three times using RNA isolated from three different cultures.
Cloning, expression, and production and purification of His-GlnA4.
The gene encoding GlnA4 (SCO1613) was amplified by PCR from S. coelicolor M145 genomic DNA and ligated into expression vector pJOE2775 between the NdeI and HindIII restriction sites under the control of rhamnose-inducible promoter Prha. His-GlnA4 was produced in E. coli strain BL21(DE3) grown in LB medium with ampicillin. Cells were grown initially at 37°C overnight, shaken at 180 rpm on a rotary shaker, transferred into a main culture, and incubated until the culture density reached an optical density of ∼0.5. Subsequently, induction with 20% rhamnose and incubation for an additional 5 h were performed. The cells were harvested and stored at −20°C until needed. His-GlnA4 was purified by nickel ion affinity chromatography essentially as directed by the resin manufacturer (GE Healthcare, Munich, Germany). Purified His-GlnA4 was dialyzed against 20 mM Tris and 100 mM NaCl (pH 8) and immediately used for further analysis.
GlnA4 in vitro assay and HPLC/MS detection of the glutamylated product.
The HPLC/MS method was used to evaluate ethanolamine and glutamate as GlnA4 substrates as well as the generated gamma-glutamylated ethanolamine as a product. Reaction mixtures typically contained 20 mM HEPES (pH 7.2), 150 mM glutamate sodium monohydrate, 150 mM ethanolamine hydrochloride, 20 mM MgCl2 × 6H2O, and 10 mM ATP mixed with 10 μg of purified His-GlnA4 (or, as a control, in the absence of GlnA4) and were incubated at 30°C for 10, 20, and 30 min and 1 h. The reaction was stopped by incubation of the reaction mixture at 100°C for 5 min. HPLC/ESI-MS analysis of the glutamylated product generated by GlnA4 was performed on an Agilent 1200 HPLC series system using a Reprosil 120 C18 AQ column (5-μm pore size, 200 by 2 mm) fitted with a precolumn (Dr. Maisch GmbH, Ammerbuch, Germany) (10 by 2 mm) coupled to an Agilent LC/MSD XCT 6330 Ultra Trap system (Agilent, Waldbronn, Germany). Analysis was carried out using 0.1% formic acid as solvent A and acetonitrile with 0.06% formic acid as solvent B at a flow rate of 0.4 ml min−1. The gradient was as follows: t0 to t5 = 0% B and t20 = 40% B (where “t” represents time in minutes). The injection volume was 2.5 μl, and the column temperature was 40°C. ESI ionization was performed in positive mode with a capillary voltage of 3.5 kV and a drying gas temperature of 350°C.
Enzymatic activity assay.
To elucidate the enzymatic reaction catalyzed by GlnA4, a modified GS activity assay previously described by Gawronski and Benson (38) was adopted. Solutions A, B, C, and F and a reaction mixture containing enough protein that 35 to 50 mM Pi would be produced in 5 min were prepared. The pH was adjusted, and a 95-μl volume was loaded into PCR strips for each reaction. Solution D (or solution E, when kinetic parameters of ATP were to be determined) was prepared, and the reaction was initiated by adding 5 μl substrate to the reaction mixture. Additionally, deionized H2O (blank) and a phosphate standard (ranging from 0 to 20 mM) were included. The reaction mixture was incubated at 30°C for 5 min in a thermocycler. Meanwhile, 150 μl of solution D (or solution C) for each reaction was loaded into wells of a 96-well plate, and 50 μl of the reaction mixture was transferred to previously prepared solution D (or solution C) in the 96-well plate. The solutions were mixed well and incubated for 5 min at room temperature. The low pH of the solution terminated the enzymatic reaction, while 150 μl of solution F was added to stop color development. The final reaction mixture was incubated for 15 min at room temperature, and the absorbance was measured at 655 nm using a microplate reader. Raw absorbance readings were put into Excel (Microsoft), and the least-squares fit to the Michaelis-Menten equation was calculated with Prism 6 (GraphPad Software, Inc.).
To test the inhibitory effect of methionine sulfoximine on the enzymatic activity of GlnA4, a reaction mixture containing 20 mM HEPES (pH 7.2), 25 mM glutamate sodium monohydrate, 50 mM ethanolamine hydrochloride, 20 mM MgCl2 × 6H2O, 2,5 mM ATP, 2 μM purified His-GlnA4, and MSO at 0.5 to 5.000 μM was used. Raw absorbance readings were normalized to GlnA4 activity in the absence of MSO, and the 50% inhibitory concentration (IC50) value was determined using 4PL-fit implemented in Prism 6. From this value, the Ki value was calculated using an equation previously described by Cheng and Prusoff (61).
Estimation of the extracellular ethanolamine level using HPLC.
Extracellular ethanolamine levels were measured by the reverse-phase high-performance liquid chromatography (RT-HPLC) method previously described by Potter and Paton (62), which was optimized for S. coelicolor. The cells were centrifuged (6,000 × g, 10 min, 10°C), and the supernatant was transferred into new tubes. For protein precipitation, trichloroacetic acid (TCA) was added to reach a final concentration of 10%, and the mixture was incubated for 5 min on ice. Then, the mixture was cleared by centrifugation (13,000 × g, 10 min at 4°C), and the pH was optimized using HCl or NaOH. The samples were stored at −20°C until analysis. Ethanolamine was derivatized using precolumn derivatization with ortho-phthalaldehyde (OPA)/mercaptoethanol (MCE) (Dr. Maisch GmbH, Ammerbuch, Germany). OPA-derivatized ethanolamine was separated on a Reprosil OPA column (150 mm by 4.0 mm, 3 mm) fitted with a precolumn (Dr. Maisch GmbH, Ammerbuch, Germany) (10 mm by 4 mm, same stationary phase) using an HP1090 liquid chromatograph equipped with a diode-array detector, a thermostated autosampler, and an HP Kayak XM 600 ChemStation (Agilent, Waldbronn, Germany). UV detection was performed at 340 nm. The following gradient was used at a flow rate of 1.1 ml/min with solvent A (25 mM sodium phosphate buffer [pH 7.2] containing 0.75% tetrahydrofuran) and solvent B (25 mM sodium phosphate buffer [pH 7.2] [50%], methanol [35%], acetonitrile [15%] by volume): t0 = 35% B, t16 = 100% B, t22 = 100% B, and t23 = 35% B (where “t” represents time in minutes). Ethanolamine hydrochloride was purchased from Sigma and used as a standard. Solutions of standards were dissolved in distilled water.
Survival assay.
The survival ability of S. coelicolor M145 and the ΔglnA4 mutant in the presence of ethanolamine was examined by estimation of the dry weight biomass after 72 and 144 h of cultivation in the YEME-TSB (1:1)-rich complex medium. The M145 parental strain and ΔglnA4 mutants were inoculated into YEME-TSB supplemented with ethanolamine (25 mM) and incubated for 3 days at 30°C. As a control, strain M145 and the ΔglnA4 mutant were also cultivated in YEME-TSB without ethanolamine supplementation but with ammonium. Samples (1 ml) were harvested by centrifugation (16,200 × g, 15 min, 4°C). The supernatant was removed, and pellets were dried for 24 h at 100°C. Means of values from three technical replicates were used.
ACKNOWLEDGMENTS
We acknowledge support from German Research Foundation (Deutsche Forschungsgemeinschaft [DFG])-funded Research Training Group GRK1708 (I and II). We acknowledge support from Deutsche Forschungsgemeinschaft (DFG) and the Open Access Publishing Fund of University of Tübingen. S.K. and A.B. are members of DFG Research Training Group GRK1708. This project as well as the PhD work of S.K. was funded by DFG Research Training Group GRK1708. O.T. was funded by DAAD. We appreciate support from the Bundesministerium für Bildung und Forschung (BMBF; GPS-TBT) and EraSysApp (SYSTERACT). C.S.M. was funded by the grant from the GPS-TBT project (BMBF).
We declare that we have no conflict of interest.
S.K. performed growth, survival, and viability assays; microscopic analysis of cells; extracellular ethanolamine analysis; and bioinformatics analysis of DNA and protein sequences. S.K. constructed and analyzed the M145pRM4spri_5940 overexpression strain. S.K. and N.O. performed phenotypic analysis of all mutants and the M145 parental strain on Evans medium with different nitrogen sources. S.K. and A.M. performed the reverse transcriptase PCR analysis. A.K. performed the HPLC and HPLC/MS analyses. A.M. cloned, overexpressed, and purified the GlnA4 protein and performed the in vitro GlnA4 enzymatic activity assay. C.S.M. confirmed the enzymatic activity of GlnA4 in vitro and analyzed the MSO effect on GlnA4 activity. O.T. constructed the ΔepuRI mutant. S.K. and M.G. constructed the ΔglnA4pRM4glnA4 mutant. N.O. constructed the ΔglnA4 mutant and was involved in technical assistance. A.B. formulated the original question and provided direction and guidance, designed the study, and developed the methodology. A.B. generated and analyzed the model of the GlnA4 structure. W.W. and F.H. provided helpful feedback on an early draft of the paper and assisted with data analysis. S.K., A.B., and C.S.M. prepared figures for the manuscript. S.K. and A.B. contributed to writing the manuscript and resolved the final approval of the version to be published.
Footnotes
Citation Krysenko S, Matthews A, Okoniewski N, Kulik A, Girbas MG, Tsypik O, Meyners CS, Hausch F, Wohlleben W, Bera A. 2019. Initial metabolic step of a novel ethanolamine utilization pathway and its regulation in Streptomyces coelicolor M145. mBio 10:e00326-19. https://doi.org/10.1128/mBio.00326-19.
REFERENCES
- 1.White DA. 1973. In Ansel GB, Hawthorne JN, Dawson RMC (ed), Form and function of phospholipids, 2nd ed, p 441–483. Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- 2.Vance JE, Guergana T. 2013. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 3.Bogdanov M, Dowhan W. 1995. Phosphatidylethanolamine is required for in vivo function of the membrane-associated lactose permease of Escherichia coli. J Biol Chem 270:732–739. doi: 10.1074/jbc.270.2.732. [DOI] [PubMed] [Google Scholar]
- 4.Gbaguidi B, Hakizimana P, Vandenbussche G, Ruysschaert JM. 2007. Conformational changes in a bacterial multidrug transporter are phosphatidylethanolamine-dependent. Cell Mol Life Sci 64:1571–1582. doi: 10.1007/s00018-007-7031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowhan W. 1997. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem 66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- 6.Shukla SD, Turner JM. 1980. Biosynthetic utilization of ethanolamine for lipid synthesis by bacteria. Biochem J 186:13–19. doi: 10.1042/bj1860013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nandedkar AK. 1974. Report on the utilization of ethanolamine-1-14C by Mycobacterium 607. Biochem Med 11:67–70. doi: 10.1016/0006-2944(74)90096-9. [DOI] [PubMed] [Google Scholar]
- 8.Nandedkar A. 1975. Biosynthesis of phosphatidyl ethanolamine in Mycobacterium 607. Biochem Med 12:116–122. doi: 10.1016/0006-2944(75)90102-7. [DOI] [PubMed] [Google Scholar]
- 9.Kim D-J, Lim Y, Cho D, Rhee IH. 2010. Biodegradation of monoethanolamine in aerobic and anoxic conditions. Korean J Chem Eng 27:1521–1526. doi: 10.1007/s11814-010-0285-5. [DOI] [Google Scholar]
- 10.Bakalova S, Mincheva V, Doycheva A, Groudeva V, Dimkov R. 2008. Microbial toxicity of ethanolaminens. Biotechnol Biotechnol Equip 22:716–720. doi: 10.1080/13102818.2008.10817540. [DOI] [Google Scholar]
- 11.Gernaey K, Verschuere L, Luyten L, Verstraete W. 1997. Fast and sensitive acute toxicity detection with an enrichment nitrifying culture. Water Environ Res 69:1163–1169. doi: 10.2175/106143097X125911. [DOI] [Google Scholar]
- 12.Blackwell CM, Scarlett FA, Turner JM. 1976. Ethanolamine catabolism by bacteria, including Escherichia coli. Biochem Soc Trans 4:495–497. doi: 10.1042/bst0040495. [DOI] [PubMed] [Google Scholar]
- 13.Lundgren BR, Sarwar Z, Pinto A, Ganley JG, Nomura CT. 2016. Ethanolamine catabolism in Pseudomonas aeruginosa PAO1 is regulated by the enhancer-binding protein EatR (PA4021) and the alternative sigma factor RpoN. J Bacteriol 198:2318–2329. doi: 10.1128/JB.00357-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khatri N, Khatri I, Subramanian S, Raychaudhuri S. 2012. Ethanolamine utilization in Vibrio alginolyticus. Biol Direct 7:45. doi: 10.1186/1745-6150-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Papa MF, Perego M. 2008. Ethanolamine activates a sensor histidine kinase regulating its utilization in Enterococcus faecalis. J Bacteriol 190:7147–7156. doi: 10.1128/JB.00952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fox KA, Ramesh A, Stearns JE, Bourgogne A, Reyes-Jara A, Winkler WC, Garsin DA. 2009. Multiple posttranscriptional regulatory mechanisms partner to control ethanolamine utilization in Enterococcus faecalis. Proc Natl Acad Sci U S A 106:4435–4440. doi: 10.1073/pnas.0812194106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joseph B, Przybilla K, Stühler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W. 2006. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol 188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsoy O, Ravcheev D, Mushegian A. 2009. Comparative genomics of ethanolamine utilization. J Bacteriol 191:7157–7164. doi: 10.1128/JB.00838-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penrod JT, Mace CC, Roth JR. 2004. A pH-sensitive function and phenotype: evidence that EutH facilitates diffusion of uncharged ethanolamine in Salmonella enterica. J Bacteriol 186:6885–6890. doi: 10.1128/JB.186.20.6885-6890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garsin DA. 2010. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol 8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garsin DA. 2012. Ethanolamine: a signal to commence a host-associated lifestyle? mBio 3:e00172. doi: 10.1128/mBio.00172-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang GW, Chang JT. 1975. Evidence for the B12-dependent enzyme ethanolamine deaminase in Salmonella. Nature 254:150–151. doi: 10.1038/254150a0. [DOI] [PubMed] [Google Scholar]
- 23.Bradbeer C. 1965. The clostridial fermentations of choline and ethanolamine. II. Requirement for a cobamide coenzyme by an ethanolamine deaminase. J Biol Chem 240:4675–4681. [PubMed] [Google Scholar]
- 24.Jones PW, Turner JM. 1984. Interrelationships between the enzymes of ethanolamine metabolism in Escherichia coli. J Gen Microbiol 130:299–308. doi: 10.1099/00221287-130-2-299. [DOI] [PubMed] [Google Scholar]
- 25.Brinsmade SR, Paldon T, Escalante-Semerena JC. 2005. Minimal functions and physiological conditions required for growth of Salmonella enterica on ethanolamine in the absence of the metabolosome. J Bacteriol 187:8039–8046. doi: 10.1128/JB.187.23.8039-8046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penrod JT, Roth JR. 2006. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krysenko S, Okoniewski N, Kulik A, Matthews A, Grimpo J, Wohlleben W, Bera A. 2017. Gamma-glutamylpolyamine synthetase GlnA3 is involved in the first step of polyamine degradation pathway in Streptomyces coelicolor M145. Front Microbiol 8:726. doi: 10.3389/fmicb.2017.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayward D, van Helden PD, Wiid IJ. 2009. Glutamine synthetase sequence evolution in the mycobacteria and their use as molecular markers for Actinobacteria speciation. BMC Evol Biol 9:48. doi: 10.1186/1471-2148-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hillemann D, Dammann T, Hillemann A, Wohlleben W. 1993. Genetic and biochemical characterization of the two glutamine synthetases GSI and GSII of the phosphinothricyl-alanyl-alanine producer, Streptomyces viridochromeogenes Tu494. J Gen Microbiol 139:1773–1783. doi: 10.1099/00221287-139-8-1773. [DOI] [PubMed] [Google Scholar]
- 30.Fink D, Falke D, Wohlleben W, Engels A. 1999. Nitrogen metabolism in Streptomyces coelicolor A3(2): modification of glutamine synthetase I by an adenylyltransferase. Microbiology 145:2313–2322. doi: 10.1099/00221287-145-9-2313. [DOI] [PubMed] [Google Scholar]
- 31.Weisschuh N, Fink D, Vierling S, Bibb MJ, Wohlleben W, Engels A. 2000. Transcriptional analysis of the gene for glutamine synthetase II and two upstream genes in Streptomyces coelicolor A3(2). Mol Gen Genet 264:461–469. doi: 10.1007/s004380000315. [DOI] [PubMed] [Google Scholar]
- 32.Rexer HU, Schaberle T, Wohlleben W, Engels A. 2006. Investigation of the functional properties and regulation of three glutamine synthetase like genes in Streptomyces coelicolor A3(2). Arch Microbiol 186:447–458. doi: 10.1007/s00203-006-0159-8. [DOI] [PubMed] [Google Scholar]
- 33.Hopwood DA, Bibb MJ, Chater K, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Ward JM, Schrempf H. 1985. Genetic manipulation of Streptomyces: a laboratory manual. John Innes Foundation, Norwich, England. [Google Scholar]
- 34.Reuther J, Wohlleben W. 2007. Nitrogen metabolism in Streptomyces coelicolor: transcriptional and post-translational regulation. J Mol Microbiol Biotechnol 12:139–146. doi: 10.1159/000096469. [DOI] [PubMed] [Google Scholar]
- 35.Fink D, Weissschuh N, Reuther J, Wohlleben W, Engels A. 2002. Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol Microbiol 46:331–347. doi: 10.1046/j.1365-2958.2002.03150.x. [DOI] [PubMed] [Google Scholar]
- 36.Amin R, Franz-Wachtel M, Tiffert Y, Heberer M, Meky M, Ahmed Y. 2016. Post-translational serine/threonine phosphorylation and lysine acetylation: a novel regulatory aspect of the global nitrogen response regulator GlnR in S. coelicolor M145. Front Mol Biosci 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ladner JE, Atanasova V, Dolezelova Z, Parsons JF. 2012. Structure and activity of PA5508, a hexameric glutamine synthetase homologue. Biochemistry 51:10121–10123. doi: 10.1021/bi3014856. [DOI] [PubMed] [Google Scholar]
- 38.Gawronski JD, Benson DR. 2004. Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal Biochem 327:114–118. doi: 10.1016/j.ab.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 39.Leason M, Cunliffe D, Parkin D, Lea PJ, Miflin BJ. 1982. Inhibition of pea leaf glutamine synthetase by methionine sulphoximine, phosphinothricin and other glutamate analogues. Phytochemistry 21:855–857. doi: 10.1016/0031-9422(82)80079-4. [DOI] [Google Scholar]
- 40.Logusch EW, Walker DM, McDonald JF, Franz JE. 1989. Substrate variability as a factor in enzyme inhibitor design: inhibition of ovine brain glutamine synthetase by α- and γ-substituted phosphinothricins. Biochemistry 28:3043–3051. doi: 10.1021/bi00433a046. [DOI] [PubMed] [Google Scholar]
- 41.Logusch EW, Walker DM, McDonald JF, Franz JE, Villafranca JJ, DiIanni CL, Colanduoni JA, Li B, Schineller JB. 1990. Inhibition of Escherichia coli glutamine synthetase by α- and γ-substituted phosphinothricins. Biochemistry 29:366–372. doi: 10.1021/bi00454a009. [DOI] [PubMed] [Google Scholar]
- 42.Jeitner TM, Cooper AJ. 2014. Inhibition of human glutamine synthetase by L-methionine-S, R sulfoximine-relevance to the treatment of neurological diseases. Metab Brain Dis 29:983–989. doi: 10.1007/s11011-013-9439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stojiljkovic I, Baumler AJ, Heffron F. 1995. Ethanolamine utilization in Salmonella Typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA, cchB, eutE, eutJ, eutG, eutH gene cluster. J Bacteriol 177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Getsin I, Nalbandian GH, Yee DC, Vastermark A, Paparoditis PCG, Reddy VS, Saier MH. 2013. Comparative genomics of transport proteins in developmental bacteria: Myxococcus xanthus and Streptomyces coelicolor. BMC Microbiol 13:279. doi: 10.1186/1471-2180-13-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larrouy-Maumus G, Biswas T, Hunt DM, Kelly G, Tsodikov OV, de Carvalho LP. 2013. Discovery of a glycerol 3-phosphate phosphatase reveals glycerophospholipid polar head recycling in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 110:11320–11325. doi: 10.1073/pnas.1221597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurihara S, Oda S, Tsuboi Y, Kim HG, Oshida M, Kumagai H, Suzuki H. 2008. Gamma-glutamylputrescine synthetase in the putrescine utilization pathway of Escherichia coli K-12. J Biol Chem 283:19981–19990. doi: 10.1074/jbc.M800133200. [DOI] [PubMed] [Google Scholar]
- 47.Yao X, He W, Lu C-D. 2011. Functional characterization of seven glutamylpolyamine synthetase genes and the bauRABCD locus for polyamine and alanine utilization in Pseudomonas aeruginosa PAO1. J Bacteriol 193:3923–3930. doi: 10.1128/JB.05105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitfield JB. 2001. Gamma glutamyl transferase. Crit Rev Clin Lab Sci 38:263–355. doi: 10.1080/20014091084227. [DOI] [PubMed] [Google Scholar]
- 49.Janke C, Rogowski K, van Dijk J. 2008. Polyglutamylation: a fine-regulator of protein function? “Protein modifications: beyond the usual suspects” review series. EMBO Rep 9:636–641. doi: 10.1038/embor.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki H, Yamada C, Kato K. 2007. Gamma-glutamyl compounds and their enzymatic production using bacterial gamma-glutamyltranspeptidase. Amino Acids 32:333–340. doi: 10.1007/s00726-006-0416-9. [DOI] [PubMed] [Google Scholar]
- 51.Tiffert Y, Supra P, Wurm R, Wohlleben W, Wagner R, Reuther J. 2008. The Streptomyces coelicolor GlnR regulon: identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol Microbiol 67:861–880. doi: 10.1111/j.1365-2958.2007.06092.x. [DOI] [PubMed] [Google Scholar]
- 52.Jenkins VA, Barton GR, Robertson BD, Williams KJ. 2013. Genome wide analysis of the complete GlnR nitrogen-response regulon in Mycobacterium smegmatis. BMC Genomics 14:301. doi: 10.1186/1471-2164-14-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bentley SD, Chater KF, Cerdeño-Tárraga A-M, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C-H, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream M-A, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete Genome Sequence of the Model Actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 54.Gerlt JA. 2016. Tools and strategies for discovering novel enzymes and metabolic pathways. Perspect Sci (Neth) 9:24–32. doi: 10.1016/j.pisc.2016.07.001. [DOI] [Google Scholar]
- 55.Goldman DS, Wagner MJ. 1962. Enzyme systems in the mycobacteria. XIII. Glycine dehydrogenase and the glyoxylic acid cycle. Biochim Biophys Acta 65:297–306. doi: 10.1016/0006-3002(62)91048-X. [DOI] [PubMed] [Google Scholar]
- 56.San Francisco B, Zhang X, Whalen K, Gerlt K. 2015. A novel pathway for bacterial ethanolamine metabolism, abstr no. 573.45. FASEB J https://www.fasebj.org/doi/abs/10.1096/fasebj.29.1_supplement.573.45. [Google Scholar]
- 57.Evans CGT, Herbert D, Tempest DW. 1970. Chapter XIII the continuous cultivation of micro-organisms: 2. construction of a chemostat. Methods Microbiol 2:277–327. doi: 10.1016/S0580-9517(08)70227-7. [DOI] [Google Scholar]
- 58.Okanishi M, Suzuki K, Umezawa H. 1974. Formation and reversion of Streptomycete protoplasts: cultural condition and morphological study. J Gen Microbiol 80:389–400. doi: 10.1099/00221287-80-2-389. [DOI] [PubMed] [Google Scholar]
- 59.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces Genetics. Norwich: John Innes Foundation [Google Scholar]
- 60.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A 100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng YC, Prusoff WH. 1973. Mouse ascites sarcoma 180 thymidylate kinase. General properties, kinetic analysis, and inhibition studies. Biochemistry 12:2612–2619. doi: 10.1021/bi00738a010. [DOI] [PubMed] [Google Scholar]
- 62.Potter AJ, Paton JC. 2014. Spermidine biosynthesis and transport modulate pneumococcal autolysis. J Bacteriol 196:3556–3561. doi: 10.1128/JB.01981-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bullock WO, Fernandez JM, Short JM. 1987. Xl1-blue—a high-efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. Biotechniques 5:376–378. [Google Scholar]
- 64.Simon R, Priefer V, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 65.Menges R, Muth G, Wohlleben W, Stegmann E. 2007. The ABC transporter Tba of Amycolatopsis balhimycina is required for efficient export of the glycopeptide antibiotic balhimycin. Appl Microbiol Biotechnol 77:125–134. doi: 10.1007/s00253-007-1139-x. [DOI] [PubMed] [Google Scholar]
- 66.Nentwich M. 2010. Die Funktion von GlnA2 in der Transkriptionellen und Posttranslationalen Kontrolle des Stickstoffmetabolismus in Streptomyces coelicolor M145. PhD dissertation. Eberhard Karls Universität Tübingen, Tübingen, Germany. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcriptional analysis of SCO6014 and SCO5977. Reverse transcriptase PCR was performed with SCO6014, SCO5977, and hrdB (control) from S. coelicolor M145 cultivated in defined Evans medium with ammonium chloride (25 mM) or ethanolamine (25 mM). Expression of SCO6014 and SCO5977 was not induced by ethanolamine or by ammonium. Download FIG S1, TIF file, 0.1 MB (91KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transcriptional analysis of glnA4 in the presence of ethanolamine and ammonium as the sole nitrogen source. Reverse transcriptase PCR analysis of glnA4 and hrdB (control) from S. coelicolor M145 and the epuRI mutant cultivated in defined Evans medium with ammonium chloride (25 mM) or ethanolamine (25 mM) was performed. Total RNA was isolated from mycelium harvested after 24 h of cultivation in defined Evans medium. Download FIG S8, TIF file, 1.0 MB (1.1MB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the Mg++/Mn++ binding pockets. Download FIG S2, TIF file, 0.4 MB (417.2KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the glutamate binding pockets. Download FIG S3, TIF file, 0.4 MB (440.7KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ADP binding pockets. Download FIG S4, TIF file, 0.4 MB (424KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ammonium binding pockets. Download FIG S5, TIF file, 0.4 MB (377.5KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Superposition of the GSSt (1FPY) and PauA7 (4HPP) templates with the GlnA4 structural model and comparison of the ammonium binding pockets. Download FIG S6, TIF file, 0.4 MB (441KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Activity of GlnA4 with various concentrations of ethanolamine (A), glutamate (B), and ATP (C) and in the presence of MSO (D). A nonlinear regression (solid black line) was made using a least-squares fit of n = 3 data sets (white bullets) and assuming the Michaelis-Menten model. Download FIG S7, TIF file, 0.1 MB (131.3KB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Combined model of bacterial ethanolamine metabolism (7, 20, 54–56). ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, phosphate; CoASH, coenzyme A; Glu, glutamate; NAD+/NADH, nicotinamide adenine dinucleotide. Dashed arrows represent transport by diffusion. Download FIG S9, TIF file, 1.7 MB (1.8MB, tif) .
Copyright © 2019 Krysenko et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.