

Abstract
The nuclear pore is the gatekeeper of nucleocytoplasmic transport and signaling through which a vast flux of information is continuously exchanged between the nuclear and cytoplasmic compartments to maintain cellular homeostasis. A unifying and organizing principle has recently emerged that cements the notion that several forms of amyotrophic lateral sclerosis (ALS), and growing number of other neurodegenerative diseases, co-opt the dysregulation of nucleocytoplasmic transport and that this impairment is a pathogenic driver of neurodegeneration. The understanding of shared pathomechanisms that underpin neurodegenerative diseases with impairments in nucleocytoplasmic transport and how these interface with current concepts of nucleocytoplasmic transport is bound to illuminate this fundamental biological process in a yet more physiological context. Here, I summarize unresolved questions and evidence and extend basic and critical concepts and challenges of nucleocytoplasmic transport and its role in the pathogenesis of neurodegenerative diseases, such as ALS. These principles will help to appreciate the roles of nucleocytoplasmic transport in the pathogenesis of ALS and other neurodegenerative diseases, and generate a framework for new ideas of the susceptibility of motoneurons, and possibly other neurons, to degeneration by dysregulation of nucleocytoplasmic transport.
Keywords: Neurodegeneration, Nucleocytoplasmic transport, Amyotrophic lateral sclerosis (ALS), Motor neurons, Ran GTPase, Ran-binding protein 2 (Ranbp2), Exportin-1/CRM1, Importin, Karyopherin, Nucleoporin
Introduction
Amyotrophic lateral sclerosis (a.k.a. Lou Gehrig’s disease) as first coined and described by Jean-Martin Charcot in the mid-nineteenth century for its neuropathology [1, 2] is a neurodegenerative disease of motoneurons that has typically bulbar or spinal onsets owing to the respective dysfunction and loss of upper corticospinal or lower spinal cord somatic motoneurons [3–5]. ALS is fatal and with a prognosis of life expectancy between 1.5 and 5 years after diagnosis [3, 4]. Notably, some motoneurons, such as ocular-motoneurons, are spared in ALS for reasons that are elusive [6–8]. ALS is a genetically heterogeneous disease with sporadic (sALS) and familial origins (fALS) [9, 10]. sALS in aggregate comprises ~ 90% of ALS, and the genetic and environmental causes of sALS remain largely unknown. fALS is caused by ~ 20 genes [9, 10], but genotype–phenotype correlations also support allelic heterogeneity between ALS, ALS syndromes and other forms of motoneuron diseases [4, 11, 12]. For example, mutations in the motor domain of the microtubule-based motor protein isoform of kinesin-1, KIF5A, cause hereditary spastic paraplegia and Charcot–Marie–Tooth disease type 2 [13–15]. In contrast, mutations affecting the cargo-binding tail domain of KIF5A cause neonatal intractable myoclonus or ALS [16–19]. Mutations in Cu/Zn superoxide dismutase (SOD1), chromosome 9 open reading frame 72 (C9ORF72), TAR DNA-binding protein (TARDBP), which encodes the TDP-43 protein, and fused in sarcoma (FUS) cause the most common forms of fALS [5, 9]. The dominant inheritance of fALS has supported the notion that ALS mutations promote gain-of-function and neurotoxicity of ALS gene products [10]. To date, there are no effective treatments to ALS [20].
Current challenges in ALS
In spite of significant advances in the identification of ALS genes and overt pathological hallmarks shared by several forms of ALS that can be heralded during the past decade, several fundamental questions about ALS and its pathogenesis remain unresolved. These outstanding issues have hampered therapeutic developments against ALS.
First, ALS genes have ubiquitous expression and diverse functions [9]. This genetic heterogeneity and pleiotropy complicate the understanding of the molecular pathogenesis of ALS. For example, several ALS genes are expressed in motoneurons and glial cells, such as astrocytes and microglia, where ALS genes play an important role in disease onset and progression [20–25]. Second, it remains obscure why motoneurons are preordained to dysfunction and degeneration by mutations in ALS genes that are ubiquitously expressed as well as by potential environmental insults. Likewise, it is obscure why fast and large motoneurons are most vulnerable to dysfunction and degeneration, while small and slow motoneurons are the most resistant to degeneration at the end-stage of disease [26, 27]. Third, pathological hallmarks shared by several forms of ALS have been uncovered [5], but the pathophysiological roles of many pathological markers are incomplete or obscure. For example, TDP-43 inclusions are present in 97% of ALS; however, TDP-43 pathology is not pathognomonic for ALS, because TDP-43 inclusions are also present in several other neurological diseases and even in some healthy brains of the elderly [5]. The concept that intracellular inclusions, which are hallmark to ALS and many other neurodegenerative diseases, are neurotoxic has been challenged by the notion that protein nucleation processes leading to protein aggregation subsumes a series of detoxifying responses by neurons against the detrimental effects of soluble misfolded protein species [28–32]. Further, several ALS mouse models of C9ORF72 recapitulate ALS pathologies without untoward pathophysiological effects, such as ALS motor behaviors [33–35], while other C9ORF72 models develop pathologies and motor deficits linked to ALS [36, 37]. Hence, some ALS pathologies appear to blur the lines of phenotypic demarcation of ALS. Finally, the genetic heterogeneity of ALS and its rapid progression complicates the development of surrogate and predictive pathophysiological measures of the onset and progression of ALS [38–40]. Isolating motor and non-motor endophenotypes that offer pathognomonic signs of sALS and fALS during the preclinical stage of the disease is critical to neurodegenerative diseases with rampant progression, such as ALS. Answers to these complex and challenging but fundamental questions will likely lead to unifying biological and mechanistic principles underpinning ALS and motoneuron biology, and possibly other neurodegenerative diseases.
An emerging and unifying principle in ALS
In spite of the aforementioned and unresolved questions surrounding ALS, a unifying pathobiological and organizing principle has recently emerged that cements the notion that several forms of ALS, and other neurodegenerative diseases, co-opt the dysregulation of nucleocytoplasmic transport [41–55]. Embedded in this principle is the notion that mutations in several ALS genes lead to aberrant nucleocytoplasmic partition of ALS-causing gene products and that this impairment subsumes the subcellular mislocalization and genesis of cytoplasmic inclusions of ALS gene products and accessory factors. Before expanding on some recent findings that lend support to this principle, these developments are encumbered with their own intricate constrains and complexities that forces consideration of basic and current principles underpinning nucleocytoplasmic transport. Some of these principles have been overlooked in past reviews and models attempting to explain impairments in nucleocytoplasmic transport caused by mutations in ALS genes. These principles will help to appreciate the roles and intricacies of nucleocytoplasmic transport in the pathogenesis of ALS and other neurodegenerative diseases, and in the susceptibility of motoneurons to degeneration by impairments in nucleocytoplasmic transport.
Principles of nucleocytoplasmic transport
Nuclear-cytoplasmic gradient of nucleotide-bound Ran GTPase
Ran GTPase is a small Ras-related nuclear protein, which is highly abundant and conserved between yeast and humans [56]. Although Ran GTPase alone harbors very low intrinsic rates of GTPase activity, Ran GTPase switches between GTP and GDP-bound conformational states in the nuclear and cytosolic compartments, respectively [57–60]. The asymmetric nucleocytoplasmic distribution of GTP and GDP-bound states of Ran GTPase is attained by the distinct subcellular localization of two critical regulators of Ran GTPase (Fig. 1). The nuclear localization of the chromatin-associated guanine nucleotide exchange factor (GEF), called the regulator of chromosome condensation 1 (RCC1), stimulates the GDP to GTP exchange of Ran GTPase by 100,000-fold in the nucleus [61–64]. By contrast, the cytoplasmic localization of the Ran GTPase-activating protein-1 (RanGAP1) stimulates the hydrolysis of Ran-GTP by 100,000-fold [64–67]. The resulting asymmetric gradient of Ran-GTP and Ran-GDP between the nucleus and cytoplasm is critical to impart unidirectional transport to Ran nucleotide-bound ensembles between the nuclear and cytoplasmic compartments [57–60, 68–70]. Ran-GDP is imported from the cytoplasm to the nucleus by the accessory factor, nuclear transport factor 2 (NTF2) [71, 72]. Nuclear trafficking occurs through nuclear pore, the gatekeeper of nucleocytoplasmic transport [73], in an energy-independent manner [74–77]. The nucleocytoplasmic shuttling of molecules < 40 kDa or less than 5–10 nm of diameter occurs by passive diffusion, whereas those of greater masses proceed by facilitated translocation in a manner which depends on nuclear transport receptors and interactions with nucleoporins, the components of the nuclear pore complex [78–85]. Importantly, passive and facilitated transports across the nuclear pore are not dynamically coupled [86–88]. It is estimated that a single nuclear pore has a mass flow of 100 MDa/s, translocation rates of 103 s−1 and a translocation speed through the central channel of the pore of ~ 0.5 µm s−1 [82].
Fig. 1.

The Ran GTPase cycle. The nucleocytoplasmic gradient of the guanine-nucleotide-bound state of Ran GTPase is determined by the asymmetric localizations of the chromatin-associated guanine nucleotide exchange factor (GEF), regulator of chromosome condensation 1 (RCC1), in the nucleus, and of Ran GTPase-activating protein-1 (RanGAP1), in the cytoplasm. RCC1 stimulates the GDP to GTP exchange of Ran GTPase, whereas RanGAP promotes the hydrolysis of Ran-GTP. In vertebrates, RanGAP is also post-translationally modified by SUMOylation (SUMO-RanGAP) and recruited to Ranbp2, which localizes at cytoplasmic filaments of the nuclear pore and captures Ran-GTP-bound cargoes exiting the nuclear pore. The nuclear transport factor 2 (NTF2) binds to and transports Ran-GDP from the cytoplasm to the nucleus
Nuclear transport receptors (NTRs)
NTRs in the human comprise 20 structurally related members of the importin-β/karyopherin-β-related nuclear transport receptors. They consist of 8 nuclear export receptors (exportins) and 10 nuclear import receptors (importins) depending on the directionality of cargoes that they transport, but few exceptions to this rule exist for at least three receptors with bidirectional properties (e.g., exportin-4, exportin-7/Xpo7 and importin-13) [89–91]. Exportins have broad (e.g., exportin-1/Xpo1/CRM1) [92] or high substrate specificity toward cargoes (e.g., exportin-2/CAS/Xpo2) [93] exported from the nucleus, whereas importins mediate the nuclear import of cargoes from the cytoplasm (Fig. 2). These NTRs are characterized by a conserved Ran-GTP-binding motif at the N-terminal region, whereas their C-terminal regions are diverse and are implicated in substrate recognition [94, 95]. Several mammalian NTRs lack orthologues in lower organisms, such as yeast, and apparently, they evolved late in evolution for the recognition of selective and species-specific substrates. Importantly, importin-β-like receptors protect Ran-GTP from hydrolysis by RanGAP1 alone [96, 97]. This is an important feature of these receptors and it critically distinguishes them from two other structural and functional unrelated and high-affinity Ran-GTP-binding proteins, Ran-binding protein 1 (Ranbp1) and Ranbp2 (a.k.a Nup358; discussed next section). As a side note, it is important to note that some importin-β/karyopherin-β-related nuclear transport receptors were confusingly termed also Ranbpn (e.g., Ranbp16/exportin-7) [98, 99], but these are structurally and functionally unrelated to Ranbp1 and Ranbp2. Importins display high affinity (low nM) towards Ran-GTP [100–102], whereas exportins in the absence of a nuclear export substrate have intermediate affinities for Ran-GTP in the micromolar range [93, 103–108]. Exportin and importin family members present different specificities toward the nuclear export and import of cargoes. For example, exportin-1 (a.k.a. CRM1/Xpo1) recognizes cargoes with the nuclear export sequence (NES) that comprise a leucine-rich export signal [92, 103, 104, 109–112]. Importin-β1 imports cargoes by binding these directly [113, 114] or indirectly via one of the seven importin-α isoforms of adaptors that recognizes cargoes with the classical nuclear localization sequence (NLS) [115–118], or by heterodimerization with importin 7 (e.g., histone H1) [119]. Importin-β2 (a.k.a., transportin-1) and importin 3 (a.k.a. transportin-2) recognize cargoes with proline–tyrosine (PY)-NLS [120, 121]. The structural context of recognition of signal sequences in cargoes is regulated at multiple levels, such as primary and secondary structural and functional contexts, formation of high-order ensembles and post-translational modifications [51, 90, 122–127]. These and other features may play important roles in the regulation of transport of cargoes. Finally, the nuclear import and export rates of model NES and NLS-substrates have similar flux rates (~ 56 molecules/s/nuclear pore, assuming ~ 2000 nuclear pores/nucleus), and translocation efficiencies (18 and 11 ms/event for nuclear export and import, respectively) [82, 88, 128]. These rates were close to the transport rates observed independently for other substrates, such as importin-β and NTF2-Ran-GDP complex (~ 5 ms) [129, 130]. Together, these results indicate that the gating properties of the nuclear pore (and likely the interactions between cargoes and NTRs) determine the dynamics of transport across the nuclear pore [131, 132].
Fig. 2.

Nuclear export and import of protein substrates. Nuclear export (left) of proteins is mediated by nuclear export receptors, exportins, such as exportin-1 (Exp1), which recognizes cargoes containing nuclear export sequences (NES). Cooperative association of exportin-1, Ran-GTP and NES-containing substrates promotes the nuclear export of nuclear cargo ensembles (1). Disassembly of nuclear export ensembles occurs when they encounter Ran-binding protein 2 (Ranbp2; a.k.a. Nup358) (2), which forms the cytoplasmic filaments of the nuclear pore, or Ranbp1 in the cytosol (3). Ranbp2 has a domain with zinc-finger motifs (ZnF), which binds exportin-1, and four Ran-GTP-binding domains (RBDs; only a single domain is represented). Ranbp1 has a single Ran-GTP-binding domain. The RBDs of Ranp2 and Ranbp1 destabilize Ran-GTP from its cargo ensemble and promotes the hydrolysis of Ran-GTP by RanGAP in the cytosol or by SUMOylated RanGAP (SUMO-RanGAP) at Ranbp2. Finally, free exportin-1 (Exp1) diffuses back to the nuclear compartment (4). Nuclear import (right) of proteins is mediated by the nuclear import receptors, importins, such as importin-β (Impβ), or its adaptors, such as importin-α (Impα), which recognizes cargoes containing nuclear localization sequences (NLS) (1). Nuclear import ensembles are transported to the nucleus (2), where they are disassembled upon encountering a high concentration of Ran-GTP, which binds to importin-β (Impβ) (3). Importin-β (Impβ)-bound to Ran-GTP is recycled back to the cytoplasm, where it is released from Ran-GTP by the RBDs of Ranbp2 (4) or Ranbp1 (5). RanGAP in the cytosol and SUMOylated RanGAP (SUMO-RanGAP) at Ranbp2 promote the hydrolysis of Ran-GTP
Ran GTPase effectors
Importin-β-like receptors exit the nuclear pore bound to Ran-GTP. The release of Ran-GTP from importin-β enables its binding to substrates destined for nuclear import, whereas the release of Ran-GTP from exportin-1 bound to NES-containing substrates unloads these cargoes from exportin-1. Two cytosolic proteins that were introduced earlier, Ranbp1 and Ranbp2, play central roles in displacing Ran-GTP from importin-β-like receptors after they exit the nuclear pore [97, 133, 134]. Ranbp1 is a single domain 26 kDa protein, which is highly conserved from yeast to man (but it is absent in D. melanogaster and C. elegans) [135–137]. In contrast, Ranbp2 is a large 358 kDa and multi-domain protein [138–141], which comprises the cytoplasmic filaments emanating from the nuclear pore [139, 140, 142]. Ranbp2 is absent in yeast and it is well conserved in mammals and some vertebrates, but not in other metazoans [143, 144]. Ranbp2 contains four highly conserved Ran-GTP-binding domains (RBDs) that are highly homologous to Ranbp1 and they are interspersed between other structurally and functionally unrelated domains. Among these domains is a domain with several zinc-finger motifs (ZnF) that binds specifically exportin-1 in a Zn2+-dependent manner [145]. The displacement of Ran-GTP from importin-β-like receptors by Ranbp1 and the RBDs of Ranbp2 in the cytosol enables Ran-GTP hydrolysis by RanGAP, which renders the disassembly reaction irreversible (Fig. 2) [97, 133, 134, 146, 147]. In vertebrates, RanGAP is SUMOylated and this modification causes the recruitment of SUMOylated Ran GAP to Ranbp2 at the cytoplasmic filaments of the nuclear pore (Fig. 2) [148–151]. Hence, Ranbp1 in the cytosol and the RBDs of Ranbp2 at the cytoplasmic face of the nuclear pore act as effectors of Ran GTPase by promoting the disassembly importin-β-like receptors from Ran-GTP. Finally, another protein, Ranbp3, has been described as a Ran-binding protein. The terminology used for Ranbp3 is misleading however, because Ranbp3 has poor homology (< 25%) to Ranbp1 and RBDs of Ranbp2, Ranbp3 has very weak binding activity for Ran-GTP (Kd ~ 10 µM) and Ranbp3 is best known to act as a non-essential cofactor of exportin-1 in the assembly of cargoes for nuclear export in the nucleus, where Ran-GTP concentration is very high (> 10 µM) [152–155].
Nuclear export of mRNA
In contrast to most protein cargo ensembles, tRNA and pre-microRNAs, whose nuclear export is dependent on Ran-GTP and importin-β/karyopherin-β-related nuclear transport receptors (e.g., exportin-t and exportin-5) [156, 157], the nuclear export of the bulk mRNA appears to be independent of the nucleocytoplasmic gradient of nucleotide-bound Ran GTPase (Fig. 3) [158]. Instead, the nuclear export of bulk mRNA in metazoans is orchestrated by the mRNA export receptor heterodimers, TAP (a.k.a., nuclear export factor 1—Nxf1) and p15 (a.k.a., NTF2-related export protein 1—Nxt1) [159–161]. These receptors are recruited to the messenger ribonucleoprotein (mRNP) particle, which is loaded with components of the transcription–export (TREX) complex, such as ALY (a.k.a. REF), and various heterogeneous nuclear ribonucleoproteins (hnRNPs) with various roles in RNA processing and export [162–168]. There are four major steps in the co-transcriptional maturation of mRNA in preparation for its export. These comprise: (i) the 5′ capping with 7′-methylguanosine and the CBP20 and CBP80 of the cap-binding complex (CBC) of the nascent pre-mRNA; these prevent the degradation of the pre-mRNA and promote its multi-step maturation and nuclear export [169–172]; (ii) the splicing of the pre-mRNA by proteins of the exon junction complex (EJC) that are recruited to the sites of exon fusions [163, 173, 174]; (iii) the 3′-end cleavage of the pre-mRNA downstream of its polyadenylation site [175]; (iv) the polyadenylation of the pre-mRNA [175, 176]. Notably, the capping and splicing steps of pre-mRNA processing promote the formation of nuclear export-competent mRNAs by recruiting the TREX complex. Among other factors, this complex includes the ATP-dependent DEAD-box RNA helicase and UAP56 (a.k.a. HEL), which recruits ALY to the mRNP [177]. Finally, pre-mRNAs and mRNAs appear to form “closed-loops” owing to the crosstalk and physical association between factors of the CBC/cap complex and those at the poly(A)+ site [178].
Fig. 3.

Nuclear export of messenger ribonucleoproteins (mRNPs). Two distinct nuclear export pathways of mRNA are represented on the left and right diagrams. The left diagram (1) shows that the nuclear export of mRNAs containing alternative mRNA nuclear export (ALREX)-promoting elements in the signal sequence-coding region (SSCR) at the 5′-end is dependent on the nuclear export receptors of mRNAs, TAP/p15, and poly(A)-binding proteins (PABP), but independent of transcription export (TREX) complex, and splicing and 5′cap structure of pre-mRNAs. The nuclear export factor(s) (Exp?) that mediate export of SSCR mRNAs are not known. The SSCR sequence interacts directly with a domain of Ranbp2 that contains zinc-finger motifs (ZnF) and this interaction potentiates the synthesis (or targeting) of secreted proteins from mRNAs co-translationally targeted to the endoplasmic reticulum (ER) by mechanisms not understood. It is possible that the nucleoporin, Gle1, at the cytoplasmic face of the nuclear pore and its partner, DEAD-box and RNA-dependent ATPase helicase, DDX19, promote also the nuclear export of SSCR mRNAs by catalyzing the remodeling (uncoating) of mRNPs. The right diagram (2) shows that the nuclear export of mRNAs is dependent on the cap-binding complex (CBC), which contains importin-α, TAP/p15, TREX and exon junction complexes (EJC) and PABP. Gle1 and DDX19 promote the nuclear export and remodeling of mRNPs as soon as the 5′-capped mRNP exits the nuclear pore. Release of the CBC and exchange with eIF4E is stimulated by the association of importin-α in the CBC to free importin-β after its regeneration by the Ran-GTP-binding domains (RBDs) of Ranbp2 (and SUMOylated RanGAP). The pioneer round of translation by ribosomes promotes the uncoating of mRNPs in preparation for steady-state translation. The top diagram (3) is like the mRNA export of diagram 2 with the exception that the remodeling the 5′-cap (CBC) takes place in the cytoplasm away from the filaments of the nuclear pore and it is catalyzed by the regeneration of importin-β by Ranbp1 (and RanGAP)
Elegant studies by Daneholt and coworkers and that were extended also by other groups have detailed the highly ordered escort process of Balbiani ring mRNP particles from the chromatin to the nuclear basket of the nuclear pore, its translocation through the central pore and extrusion at the cytoplasmic face of the nuclear pore [179–183]. This process involves the docking of the 5′ cap of the mRNP to the nuclear basket followed by its exit at the cytosolic face of the nuclear pore. The binding of nuclear export receptors of mRNA, TAP/p15 (a.k.a. Nxf1/Nxt1), to FG-repeats of some nucleoporins (Nups) of the nuclear pore appears to mediate the docking of mRNP to the nuclear pore and its translocation across the pore [166, 184–186]. Although two distinct hypotheses have been proposed to explain the transport of mRNPs from the transcription sites to the nuclear pores [187–191], the movement of mRNPs in the nuclear matrix and to the nuclear pore is governed by thermal/Brownian motion (passive diffusion) and thus this process depends on the size of the mRNP particles and viscosity of the nucleoplasm [192]. In contrast, the translocation through the nuclear pore is facilitated by the interaction of the nuclear export receptors with intrinsically disordered and flexible stretches of phenylalanine–glycine (FG)-repeats of ~ 10 nucleoporins that face the central channel of the pore and form a permeability barrier [131, 193, 194].
After exiting the nuclear pore, mRNPs encounter the nucleoporins, Nup214 (a.k.a., CAN), Nupl2/hcG1 (Nup42), Gle1 (Gle1B isoform), and the DEAD-box and RNA-dependent ATPase helicase, DDX19 (a.k.a. Dbp5 in yeast) at the cytosolic face of the nuclear pore [195–202]. Gle1 binds to hCG1 (a.k.a. Nup42) and DDX19 helicase and it stimulates DDX19 ATPase activity [201–203]. Nup214 enhances the Gle1-mediated ATPase activity of DDX19 and promotes its tethering to the cytoplasmic face of the nuclear pore [202]. The Gle1-mediated helicase activity of DDX19 stimulates the remodeling of mRNPs by removing proteins, such as TAP/p15 and the poly(A)+-binding protein Nab2, from the mRNP complex [197, 199, 202, 204–206]. This process also ensures that mRNAs cannot reenter the nuclear pore. Perturbations in the loading or removal of these receptors to or from mRNA decrease the efficiency of nuclear export and may even result in the retrograde transport of the mRNP from the pore [206]. Hence, the asymmetric localization of Gle1 and DDX19 at the cytosolic face of nuclear pore is critical to the directionality of the release of mRNPs in the cytosolic compartment (Fig. 3). Finally, the total translocation time of mRNAs, such as actin, across the nuclear pore (docking, transport, and release) is estimated to be ~ 180 ms [207], and only one-third of mRNPs are estimated to be successfully exported from the nucleus [208].
Another critical feature of the remodeling of mRNP as its 5′ cap exits the nuclear pore involves the replacement of the cap-binding complex (CBC) in mRNAs by eIF4E (and eIF4G and eIF4A) [172, 209, 210]. eIF4E is critical for the recruitment of the small ribosomal subunit and initiation of translation even before the export of the mRNP from the pore is completed [182]. In this regard, Gle1 and DDX19 may also play a role in translation and gene expression (this topic is discussed further below) [211, 212]. In addition to the classical roles of importin-β/α in nucleocytoplasmic transport, importin-β and importin-α exert important roles in cap-binding activity. Importin-α partakes in CBC in the nucleus and importin-β triggers the replacement of CBC-importin-α complex by eIF-4E at mRNA caps when they exit the nuclear pore [213–215]. Ranbp1 and/or Ranbp2, and possibly with the cooperation of RanGAP, triggers the liberation of importin-β from Ran-GTP after exiting the nuclear pore and thus allows importin-β to engage with the capped mRNP-CBC-importin-α complex in the cytosolic compartment. Hence, a non-canonical role of some importins is to regulate the capping and remodeling of mRNPs (Fig. 3). The remodeling of the 5′cap serves also two critical functions. First, CBC-capped mRNPs undergo a pioneer round of translation, which serves as a quality control mechanism to survey mRNAs destined for premature termination of translation and/or nonsense-mediated mRNA decay [216–219]. This step also promotes the remodeling of mRNPs by uncoating spliced mRNAs from other components, such as exon junction complex proteins [214, 220]. Second, the replacement of the CBC-capped mRNA by eIF-4E promotes the steady-state initiation of translation and protein synthesis. Finally, CBC is strongly regulated by growth factors and environmental stressors that by this mechanism regulate gene expression (e.g., suppression of translation) [215, 221–223].
Thematic variations in nucleocytoplasmic transport
In spite of the canonical roles of NTRs and TAP/p15 in nucleocytoplasmic transport that were described in the preceding sections, there are important and mounting variations to this theme. Due to space constraints, few examples are highlighted next that may provide rich mechanistic hints and insights to fundamental and interconnected biological and pathophysiological processes. I envisage that these non-canonical modalities of nucleocytoplasmic transport will play important roles in chartering the refinement, integration, and extension of organizing principles of nucleocytoplasmic transport in normal and disease states.
First, CRM1/exportin-1 also mediates the nuclear export of mRNAs and other RNAs that are released in the cytoplasm by hydrolysis of Ran-GTP. However, the nuclear export of RNA by exportin-1 depends on adaptors with NES signals, because exportin-1 does not bind RNA [196, 224–228]. Initially cited in this context is the nuclear export of the signal recognition particle (SRP) by exportin-1 in S. cerevisiae [229, 230]. The SRP is a cytosolic RNP comprised of a RNA molecule and core SRP proteins. According to the classical view, which has been recently refined [231], the SRP recognizes signal sequences at the amino-terminal end of proteins destined to the plasma membrane and secretion and thereby enabling their co-translational delivery and translocation to the endoplasmic reticulum. Hence, these early studies hint that exportin-1-mediated nuclear export regulates the biogenesis of proteins destined for secretion or to the plasma membrane. In vertebrates, however, it appears that exportin-5 mediates the nuclear export of SRP RNA [232, 233] and that mRNA export by exportin-1 is dependent at least on three different adaptors, such as RNA-binding protein human antigen (HuR), leucine-rich pentatricopeptide repeat protein (LRPPRC) and nuclear export factor 3 (Nxf3) [224–228].
Second, accumulating studies indicate that the large, multi-domain and peripheral nucleoporin, Ran-binding protein 2 (a.k.a. Nup358) serves as a scaffold and multidimensional platform to orchestrate the coupling of nuclear export and translation of mRNAs of secretory and plasma membrane proteins [234]. This effect enhances the translational potential of these mRNAs possibly by remodeling messenger ribonucleoproteins (mRNPs) when they exit the nuclear pore. Ranbp2 comprises the cytoplasmic filaments emanating from the nuclear pore [140, 142]. Exportin-1 docks to a domain of Ranbp2 containing several zinc-finger motifs (ZnF) [145] and this and the N-terminal leucine-rich domain of Ranbp2 are capable to interact directly with single-stranded RNA [234, 235]. The signal sequence-coding region (SSCR) of mRNAs coding secreted proteins contains RNA elements that stimulate an alternative mRNA nuclear export (ALREX) pathway and in particular of a subset of mRNAs characterized by adenine-less tracts in SSCR of ALREX-elements [234, 236]. This pathway appears dependent on TAP/p15, but independent of TREX, and splicing and 5′cap structure of pre-mRNAs [236]. The ALREX-promoting elements in SSCR potentiate the synthesis of reporter and native secreted proteins by mechanisms not understood yet [234, 236]. Interestingly, mutations of ALREX elements in SSCR lead to inhibition of nuclear export and formation of stress granules in the cytoplasm that include the mutated mRNAs and the translational initiation factor, eIF3B [234]. The ALREX-promoting elements interact selectively with the ZnF-containing domain of Ranbp2, while knockdown of Ranbp2 suppresses the synthesis of secreted and membrane-bound proteins, and possibly also of mitochondrial proteins [234]. Parenthetically, a recent study, which revisited the role of SRP in protein translation and targeting, found that loss of SRP leads to the mistargeting of mRNAs from the ER to mitochondria and thereby causing mitochondrial dysfunction [231].
Third, Ranbp2 was independently discovered as a multi-modular and cyclophilin-related protein with strong expression in cone photoreceptor neurons and as a candidate with biological functions similar to the cyclophilin encoded by ninaA of Drosophila that mediates the biogenesis of a subset of opsins [138]. Subsequent studies on the chaperone activity of the cyclophilin domain of Ranbp2 in cell culture and mice have shown that Ranbp2 controls the production of a subset of transmembrane G protein-coupled receptors, such as red/green opsin, in cone photoreceptor neurons, and without impairing the levels of the coding mRNAs [237–240]. Recent genetic studies in mice have also found that loss of Ranbp2 leads to pronounced accumulation of the levels of selective mRNAs that encode the secretory protein, chemokine ligand 14 (Cxcl14) in spinal motoneurons, and acetyl-CoA carboxylase 1 (Acc1) in the optic nerve (P. Ferreira, unpublished observations) [241]. The pronounced increase of Cxcl14 mRNA was accompanied by a sharp decrease of the levels of Cxcl14 protein, thus suggesting the uncoupling between export/targeting and translation of Cxcl14 and possibly of other mRNA species [241].
Fourth, exportin-1 mediates the nuclear export of eukaryotic translational initiation factor (elF4E)-dependent mRNAs with an elF4E-sensitive element (4E-SE) in their 3′UTR [242]. In this instance, the level of elF4E controls the remodeling of nuclear pore components at the cytosolic face, such as of Ranbp2, Nup214, Ranbp1, Gle1 and DDX19 [242]. As described earlier, these factors mediate the nuclear export and translational potential of mRNAs. The elF4E-mediated effects arise by changing indirectly the localizations of Ranbp2 and Nup214 (and the levels of Ranbp2) and by directly affecting the nuclear export of Ranbp1, Gle1 and DDX19 mRNAs containing 3′UTR 4E-SE and that are targets of elF4E [242]. Hence, a regulatory feedback loop appears to exist between factors regulating the translation initiation of selective mRNAs and their nuclear export.
Fifth, apparently conflicting experiments support and exclude the involvement of Ranbp2 in the nuclear export of bulk mRNA. In insect cells of fruit flies (D. melanogaster), which lacks Ranbp1 and express a Ranbp2 isoform with a much simplified molecular architecture than mammalian Ranbp2, knockdown of Ranbp2 promotes the nuclear accumulation of bulk poly(A)+ mRNA and loss of Nxf1 localization at the nuclear pore without changes in nuclear export mediated by exportin-1 [143]. Mouse embryonic fibroblasts lacking Ranbp2 also accumulate bulk poly(A)+ mRNA in the nucleus, but without nucleocytoplasmic mislocalization of Nxf1 [243]. By contrast, knockdown of Ranbp2 in human HeLa and U2OS cells does not affect the nucleocytoplasmic partitioning of bulk poly(A)+ mRNA [234, 244]. The reasons for these apparent discrepancies remain elusive, but they may be rooted on cell-type, compensatory and species-specific differences that underlie the orchestration of nucleocytoplasmic transport by Ranbp2 and its accessory partners. This notion is supported by genetic complementation studies in mice that found loss of Ran-GTP-binding activity of the Ran-binding domains, RBD2 and RBD3, of Ranbp2 is essential to the survival of selective cell types only, and thus, they are not biologically equivalent to other RBDs of Ranbp2 (e.g., RBD1 and RBD4) [241, 245]. Further, some accessory factors or substrates undergo species and cell type-specific recruitment to Ranbp2 (and Ranbp1) because Ranbp1, several domains of Ranbp2, and accessory factors thereof (e.g., SUMOylated RanGAP) are poorly conserved between species or some do not even exist in lower organisms (e.g., Ranbp2) [143]. These non-conservation attributes extend also to several nucleoporins that are absent or have poorly conserved primary structures in several species (e.g., Nup153) [194]. Finally, loss of Ranbp2 in spinal motoneurons (and retinal ganglion neurons; Ferreira, P. unpublished observations) cause perturbations in a restricted and largely non-overlapping set of transcripts [241]; thus supporting that Ranbp2 co-opts accessory factors for the cell type-dependent orchestration of nucleocytoplasmic transport of mRNAs perhaps by combinatorial mechanisms. Together, these distinctions and limitations will likely extend also to other nucleoporins [194]. Notably, these shared and unique attributes of nucleocytoplasmic transport have been overlooked in models of neurodegenerative diseases in which impairment of nucleocytoplasmic transport is emerging as a pathogenic driver of neurodegeneration.
Finally, alternate components of the cap-binding complex (CBC) exist and they play critical roles in mRNA export under stress stimuli. Another conserved and alternate, but less understood complex, TREX-2, which is mostly localized to nuclear pores, appears also to bridge the transcription and transfer of mRNAs to Nxf1 [246, 247]. A component of the TREX-2 complex is the germinal-center associated nuclear protein (GANP; a.k.a. Sac3 in yeast), which binds mRNA and interacts with RNA polymerase II and facilitates its recruitment to selective genes [248].
Collectively, these findings only scratch the surface of the intricacies and complexities of the molecular mechanisms underpinning nucleocytoplasmic transport and its regulation. As the functions, mechanisms, and regulations of many NTRs, nucleoporins and accessory factors are distilled in greater detail and in pathophysiological settings that are undergoing rapid flux, they will serve to validate, refine and extend the physiological significance of existing concepts across cell types and diseases in the human and model organisms. These advances are anticipated to eclipse in the near future the current knowledge of nucleocytoplasmic transport in cell functions and disease and harness their therapeutic potential.
Bridging nuclear export with anterograde transport in neurons
Neurons epitomize the polarization and compartmentalization of cellular architecture. The linear length of an axon of a lower motoneuron of the spinal cord can reach a meter in size in the human [249], whereas the total length of axons of striatal dopaminergic neurons is estimated to be ~ 8000 m in the rat [250, 251]. The exquisite sizes and polarized morphologies of many neural cell types are thought to preordain neurons to neurodegeneration by dysregulation of intracellular transport. In line with this point, it remains largely unresolved how cargoes exported from the nucleus are captured by the intracellular transport machinery, such as microtubule-based motors, for the “fast” and polarized delivery of cargoes to distal compartments, such as dendrites, axons or synapses. The coordination of the coupling of nuclear export and intracellular transport of cargoes poses another dimension of complexity to nucleocytoplasmic trafficking events, whose molecular underpinnings remain incomplete but are of unique significance to neural function and survival. In other words, foremost here are the spatiotemporal challenges intrinsic to neurons and that these need to overcome to prevent the entrapment of cargoes and accessory factors in somata after they exit the nuclear pore. Another point of critical importance that needs consideration is how the roles by moonlighting proteins partaking in nucleocytoplasmic and intracellular transport machineries are discriminated and segregated spatially and temporally. As expanded later in this review, mounting evidence indicates that dysregulation of these processes underpins the manifestation of pathological traits and most importantly, the pathomechanisms of several forms of neurodegenerative diseases, such as ALS.
The cytoplasmic nucleoporin, Ranbp2/Nup358, and the microtubule-based motor protein of the kinesin superfamily proteins (KIF), such as kinesin-1 (KIF5), have emerged as prime candidates to couple nuclear export with fast anterograde transport of cargoes exiting the nuclear pore. Ranbp2 associates directly via a non-conserved kinesin-binding domain (KBD) with the conventional kinesin-1 isoforms, KIF5B and KIF5C [252]. This domain of Ranbp2 jump-starts the motor activity of kinesin-1 [253, 254]. Further, the Ran-GTP-binding domains, RBD2 and RBD3, of Ranbp2 that flank its KBD, enhance the Ran-GTP-independent association of Ranbp2 with kinesin-1 and boost its motor activity [253, 254]. To date, Ranbp2 is still the only native and known cargo that can directly activate and boost motor activity of a kinesin in a minimal reconstitution system of purified components. In retinal ganglion neurons, whose nuclear envelopes are crowded with nuclear pores, Ranbp2 is found at the nuclear rims, where it colocalizes with Ran GTPase [255] (Fig. 4). Ranbp2 and Ran GTPase also colocalize to a subpopulation of discrete granules present along “highways” that radiate from the nuclear rim toward the axon hillock where they coalesce (Fig. 4) [255]. This implies that Ranbp2 can be released from the cytoplasmic filaments of nuclear pore. In this regard, genetic ablation of Ranbp2 has shown that the half-life of Ranbp2 at the nuclear pore is much shorter than the lifespans of many other nucleoporins (e.g., < 2.5 days vs months) [241, 256]. The compositions of the Ranbp2 and Ran-GTPase-containing granules in the cytoplasm are unknown, but independent studies indicate that they may consist of mRNPs transported by kinesin-1. For example, Kanai and coworkers found that in mouse brain and dendrites of hippocampal neurons kinesin-1 binds directly and transports mRNP granules of 1000S [257]. These granules are composed mRNAs, such as Arc and CamKIIα, and 42 proteins, such as RNA helicases, ALY, hnRNPs (e.g., hnRNPA/B, hnRNP-U), eIF factors, Fragile X mental retardation protein and related members (e.g., FMRP1, FXR1, and FXR2) and Staufen [257]. Other studies have also found that kinesin-1 mediates the transport of Staufen-2 (Stau-2), FRMP-containing RNPs and the ribosome receptor, p180 [258–260].
Fig. 4.
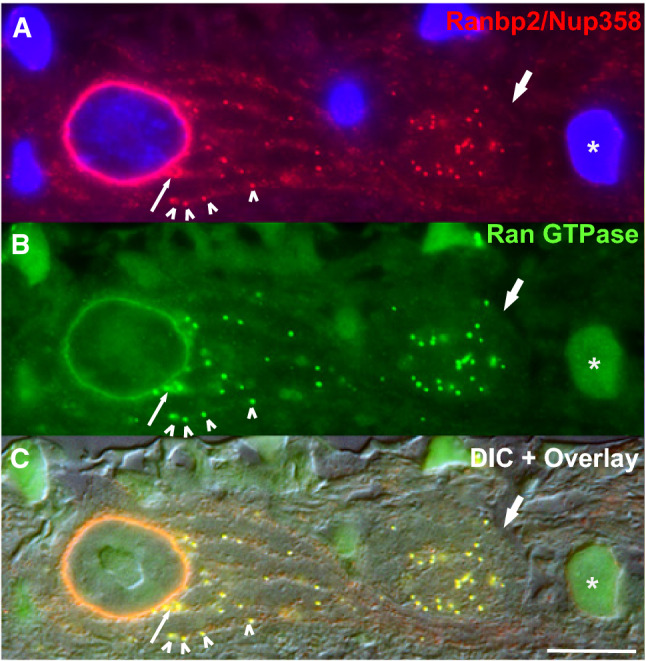
Complete co-localization of Ran-binding protein 2 (Ranbp2) and Ran GTPase at the nuclear rim and to a subset of granules (arrowheads) in threads (“highways”) radiating from the nuclear rim and toward the axon hillock of large retinal ganglion neuron (magnocellular neurons). Thin arrow on the left shows the co-extrusion or co-displacement of Ranbp2 and Ran GTPase from the nuclear rim. Thick arrow on the right points to an axon of an unmyelinated retinal ganglion neuron in which there is complete co-localization of Ranbp2 and Ran GTPase. Top (a) and middle panels (b) show the immunostaining of the retina ganglion cell layer of a cryosection of bovine retina immunostained for Ranbp2 (top) and Ran GTPase. The lower panel (c) is an overlay image of a Nomarski (differential interference contrast—DIC) image and the image a and b. Numerous granules organized along longitudinal bundles (“highways”) are visible and a subset of these co-immunolabel with Ranbp2 and Ran GTPase. These granules likely constitute RNPs. Cell labeled with a “asterisk” shows the nucleus of a small ganglion neuron with intense nuclear immunolabeling of Ran GTPase, but lacking Ranbp2 at the nuclear envelope/pores.
Image adapted from reference [255]
mRNPs illustrate a class of nuclear export cargoes whose long-distance transport and compartmentalized localization are critical to enable the spatial and temporal orchestration of localized protein synthesis. This function is critical for the maintenance of distal subcellular structures and functions of neurons and responses to multiple stimuli. In contrast to other cell types, the translational repression, transport, remodeling, and disassembly of mRNPs are competing imperatives that need to be precisely balanced in a spatial and temporal manner by neurons to ensure neural homeostasis. For example, elF4AIII, is a component of the exon junction complex (EJC), which is localized primarily to the nuclear compartment of HeLa and glial feeder cells [261–263], where mRNPs are remodeled and mRNAs are translated soon after exiting the nuclear pore. By contrast, elF4AIII is localized to somata and dendrites of hippocampal and cortical pyramidal neurons, where it associates presumably with 3′-UTR introns of translationally silent mRNAs in granules containing other RNPs, such as FMRP and Stau-1 [263]. Down-regulation of elF4AIII stimulates quantal synaptic transmission by increasing the levels of Glur1 receptor and Arc, which is required for long-term potentiation at synapses [263]. elF4AIII regulates Arc protein synthesis by degradation of Arc mRNA upon translation by a nonsense-mediated decay (NMD) mechanism and termed, translation-dependent mRNA decay [263]. Another critical implication of these findings is that EJC proteins in mRNPs provide a chronological window of the translational status of mRNAs in neurons [263]. In non-neural cells, EJC proteins are displaced during the pioneer round of translation when NMD also occurs and soon after the mRNP exits the nuclear pore. Hence, a localized translational control mechanism in neural compartments is emerging as a common feature in neural function and with consequential pathophysiological implications.
Finally, two discrete roles were cited previously for importin-β. A third emerging role is that importin-β acts as a sensor of axonal injury [264, 265]. In this regard, importin-β mRNA is transported to axons via a cis-acting regulatory sequence in its 3′-UTR [265]. This mechanism allows the segregation of nucleocytoplasmic transport functions of importin-β with its local translation in axons. Here, importin-β acts a sensor for retrograde signaling in dorsal root ganglion neurons of mice with sciatic nerve injury by mechanisms that are not fully eluciated yet. In light of the preceding discussion, it is tempting to hypothesize that importin-β’s role in axons is inextricably linked to the remodeling of mRNP in a manner reminiscent to that of pioneer round of translation, but that involves the regulation of local translation of mRNPs in axons [264]. Parenthetically, Ran GTPase, Ranbp1 and the nuclear shuttling factor, Stat3, may also partake in the local regulation of protein synthesis elicited by the importin-β-dependent retrograde injury signaling [266, 267]. Although axonal protein synthesis has been a contentious topic [268], these and other studies, such as local translation in axons of commissural neurons and retinal ganglion neurons, support the importance of protein synthesis in axons [269–272]. Importin-β is also localized prominently at the nuclear rim, lower fibers and synaptic pedicles of cone photoreceptor neurons, where it is abundantly expressed [255]. Ranbp2 also associates with importin-β and Stat3 [97, 142, 239]. Stat3 and Ranbp2 mediate the neuroprotection of photoreceptor neurons against physical and/or genetic injury in mice [273–275], and Ranbp2 regulates the trans-activation potential of Stat3 in gene expression [240]. Hence, it is likely that the importin-β-dependent injury-signaling pathway is shared by different neurons in stress signaling.
Dysregulation of nucleocytoplasmic transport: a pathogenic driver in ALS and neurodegeneration
While the guiding principles described in the preceding sections articulate general and alternate concepts of nucleocytoplasmic transport, we are now beginning to appreciate its intricacies and complexities to rationalize another dimension of complexity linked to neurodegenerative diseases, such as sALS and fALS. While this field is still replete with unresolved issues as cited at the beginning of this review, a theme is beginning to emerge that cements the notion that mutations in several ALS genes impair nucleocytoplasmic transport of their products and accessory partners. This impairment is emerging as a major pathogenic driver of neurodegeneration in ALS as well as in other neurodegenerative diseases. Next, I highlight some ALS targets with shared and unique attributes in defects of nucleocytoplasmic transport.
Cu/Zn superoxide dismutase 1 (SOD1)
The first study of nucleocytoplasmic dislocation of NTRs and accessory substrates linked to ALS was reported by Zhang et al. in motoneurons of the anterior horn in a mouse model of fALS with the G93A mutation in SOD1 [41]. In this study, Tg-SODG93A mice presented subcellular redistribution of importin-β and importin-α from the nuclear to the cytosolic compartments and immunoreactivity of Lewy body-like hyaline inclusions to these NTRs and their substrates, such as histone H1 [41]. The same laboratory subsequently reported that sALS patients lacked importin-β in nuclei of motoneurons, that there were irregular nuclear contours of the nuclear envelope immunolabeled with Nup62, even at presymptomatic stages of the disease, and that worsened at later stages of the disease [42]. Although SOD1 inclusions are found in ALS patients with SOD1 mutations, there is controversy surrounding the presence of misfolded SOD1 in the central and peripheral nervous system (e.g., spinal cord and cortex tissues) of sALS and non-SOD1 fALS patients using conformationally sensitive antibodies [276]. Regardless, a provocative study indicates that nuclear clearance of mutant SOD1 exerts neuroprotective effects against cytotoxicity caused by gain-of-function mutations in SOD1 [51]. This study showed that wild-type SOD1 was distributed between the nuclear and cytoplasmic compartments, whereas mutant forms of SOD1 were found primarily in the cytoplasm. Misfolded SOD1 and ALS-causing mutations in SOD1 expose a normally buried NES-like sequence that leads to the exportin-1-mediated nuclear export of misfolded SOD1. Hence, this study suggests that an exportin-1-mediated nuclear export mechanism promotes nuclear proteostasis against the accumulation of misfolded SOD1 in the nucleus. Alternatively, it is possible that saturation or equilibrium shifts of a nuclear export and saturable pathway caused by changes of exportin-1 association to misfolded SOD1, which is an abundant protein, promotes pathogenicity owing to the dysregulation of other cargoes of exportin-1 and/or to rearrangements or displacement of selective nucleoporins (e.g., Nup62).
Chromosome 9 open reading frame 72 (C9orf72)
The subcellular redistribution of importin-β were also confirmed in sALS and fALS patients with noncoding hexanucleotide (G4C2) repeat expansions of C9orf72 [46]. In particular, spinal motoneurons of these patients had loss of localizations of importin-β, Ran GTPase and a short protein isoform encoded by a splice variant of C9orf72, C9-S, at the nuclear rim [46]. Notably, isoform-specific antibodies showed that C9-S relocated from the nuclear to the plasma membrane of spinal neurons of ALS patients with and without mutations in C9orf72 [46]. It is possible that dipeptide repeat proteins (DPRs) expressed from the repeat-associated non-AUG (RAN) translation of bidirectionally transcribed expansion repeats in C9orf72 (C9Ran) cause also the sequestration of importin-β, Ran GTPase and other proteins, such as RanGAP. Expression of DPRs reduces the nuclear-cytoplasmic distribution of Ran GTPase in C9-ALS iPSC neurons [48]. Notably, TDP-43 was also redistributed from the nuclear to the cytosolic compartment in sALS and fALS with expansion of C9orf72 [5, 36, 46]. In mice, DPRs form intranuclear and cytoplasmic inclusions that immunostain for ubiquitin primarily in neurons [36]. In transgenic mice and C9-ALS iPSC-derived motoneurons, the C9Ran transcripts formed RNA foci and colocalized with hnRNPA1 and Pur-α [37, 277]. Finally, genetic screens in yeast and photoreceptor neurons of the compound eye of Drosophila melanogaster against DPR toxicity identified Aly/REF of the TREX complex, Gle1, and selective nucleoporins and substrates of the Ran GTPase cycle, such as importins and exportins, as strong genetic modifiers of the proteotoxicity of DPRs [47, 48, 278, 279].
Transactive response (Tar) DNA-binding protein 43 (TDP-43)
Mutations in TDP-43 cause ALS and frontotemporal dementia (FTD) [280, 281]. Although initial studies found that TDP-43 acts a splicing and transcriptional regulator [282–285], TDP-43 is a nucleocytoplasmic shuttling hnRNP, whose steady-state localization appears to be primarily nuclear [286, 287]. ALS mutations in TDP-43 shift its localization to the cytosol where it aggregates and becomes ubiquitinated [288–290]. Studies have shown that TDP-43 associates with introns of pre-mRNAs, and its own 3′-UTR and that of Ran GTPase, and thereby regulating the levels and splicing of thousands of pre-mRNAs [45, 291–294]. As a splicing regulator, TDP-43 was also found to modulate the splicing of multiple pre-mRNAs, such as hnRNPA1, that leads to the production of an hnRNPA1 variant, hnRNPA1B, with a longer prion-like domain and an increased propensity to aggregation [295]. The cytotoxicity of hnRNPA1B may be enhanced by TDP-43 mislocalization to the cytosol. TDP-43 is also transported as mRNPs via microtubules in axons of primary cortical neurons [296, 297]. ALS mutations in TDP-43 suppress the axonal trafficking of mRNP granules without apparently affecting mitochondrial transport and thus, supporting the notion that this inhibition promotes distal axonopathy as observed in ALS [297, 298]. Although the molecular basis of TDP-43 dysfunction in axonal transport of mRNPs is obscure, this may result from the dysregulation of microtubule-based motors associated to hnRNPs and/or accessory factors that bridge interactions between motors and one or more components of the mRNP granule.
The nuclear shuttling of TDP-43 is carried out by importin-β/karyopherin-β-related NTRs [299]. TDP-43 harbors a classical NLS at its N-terminal domain, but surprisingly none of the ALS mutations affect the NLS [43]. Knockdowns of importin-β and cellular apoptosis susceptibility (CAS) receptor (a.k.a. exportin-2), which acts as a nuclear export receptor for importin-α, result in the cytoplasmic localization and aggregation of TDP-43 [299]. FTD-ALS patients have also reduced levels of CAS in the brain, but not spinal cord [299]. In brains of sporadic FTD and C9ALS/FTD, importin-α2 (KPNA2) was depleted from the nucleus and accumulated in the cytoplasm and there was an overall decrease of its expression levels [54]. Importantly, this pathological manifestation was frequently observed without pTDP-43 inclusions in sporadic FTD and C9ALS/FTD or DPR inclusions in C9ALS/FTD patients, thus suggesting that soluble rather than aggregate forms of pTDP-43 and DPR exert neurotoxicity [54].
As it relates to the nuclear export of TDP-43, recent studies with primary cortical neurons indicate that redundant nuclear export pathways by multiple export receptors, such as exportin-1, exportin-7 and Nxf1, mediate the nuclear egress of TDP-43 and that selective inhibitors of nuclear export targeting exportin-1 partially rescue motor deficits in a rat model of TDP-43-induced paralysis [300]. This study appears to support the observation that NES mutations in TDP-43 ameliorate TDP-43 toxicity caused by its over-expression in fruit flies and primary rat cortical neurons [301, 302]. However, other studies have produced conflicting results. For example, a study found that the nuclear egress of TDP-43 and FUS (discussed see next section) in HeLa cells are independent of CRM1/exportin-1, exportin-5, the mRNA export factor ALY/REF, and of its RNA-binding function(s) [303]. In contrast, artificial enlargement of TDP-43 (and FUS) causes the impairment of their nuclear export, whereas overall inhibition of transcription stimulated TDP-43 nuclear egress [303]. Hence, passive diffusion appears to mediate the nuclear export of TDP-43. The exportin-1-independent nuclear export of TDP-43 and its nuclear egress by passive diffusion was also concordant with another independent study with HeLa and primary hippocampal neurons [304]. The reasons behind these conflicting reports are unclear, but they may partially arise from the cell-type dependent expression of nuclear export accessory factors.
Finally, recent studies found that ectopic expression of mutants of TDP-43 and a 25 kDa C-terminal fragment of TDP-43 in cultured cells sequestered or compromised the localization of nucleoporins and NTRs and promoted the nuclear retention of poly(A)+ mRNA presumably via prion-like domains shared by these proteins [305]. However, it is important to inject a cautionary note that TDP-43 pathology is not pathognomonic for ALS, because TDP-43 inclusions are also present in several other diseases, such as Alzheimer’s, Lewy body disease, Parkinsonism, chronic traumatic encephalopathy and even in healthy brains of the elderly [5].
Fused in sarcoma (FUS)
Mutations in FUS cause ALS and FTD [306, 307]. Unlike TDP-43, FUS harbors a non-classical NLS with a PY motif (PY-NLS) and about half of the known mutations in ALS and FTD affects this sequence [43, 306, 307]. Like TDP-43, FUS localization is primarily nuclear and ALS mutations promote FUS relocation to the cytoplasm, where FUS inclusions are also formed [43, 306–308]. Notably, there is a correlation between the degree of impairment of nuclear import by FUS mutations and the age of disease onset [43]. Nuclear import of FUS is mediated by importin-β2 (a.k.a. transportin) and impairment of this nuclear import pathway promotes the relocation of FUS to the cytoplasm, where it is sequestered into stress granules of cultured cells and of spinal neurons of patients with fALS and sFTD-FUS [43]. Like TDP-43, FUS binds thousands of mRNAs [309] and it appears to mediate mRNA transport to neural dendrites [310, 311].
Heterogeneous nuclear ribonucleoproteins (hnRNPs)
Mutations in hnRNPA1 and hnRNPA2B1 (this gene produces two isoforms, hnRNPA2 and hnRNPB1) cause a rare and dominant form of ALS and multisystem proteinopathy (MSP), which is a degenerative disorder affecting motoneurons, brain, muscle and bone [312]. The disease mutations fall within a prion-like-domain (PrLD) that is common to other hnRNPs, and they appear to increase the propensity to fibrillization of hnRNPA1 and hnRNPA2 [312]. Although hnRNPA1 and hnRNPA2B1 have primarily nuclear localization, the pathogenic D290V mutation in hnRNPA2 promotes its relocation and accumulation in cytoplasmic inclusions of the mouse muscle [312].
hnRNPA1 harbors a 38 residue non-canonical NES and NLS domain, termed M9, that is implicated in nuclear import and export and thus, M9 alone mediates the nuclear shuttling of hnRNPA1 [313–315]. hnRNPA2/B1 appear also to share the M9 signal sequence [314]. Importin-β2 mediates the nuclear import of hnRNPA1 [316]. hnRNPA1 and hnRNPA2B1 are implicated in multiple facets of RNA metabolism, such as splicing, trafficking and ribostasis of mRNAs [317–319]. For example, cis-acting hnRNPA2 response elements (A2RE) that bind hnRNPA2B1 are found in mRNAs (e.g., myelin basic protein and Arc) and they are involved in mRNA trafficking to neural dendrites, where hnRNPA2B1 is also found [320]. Further, hnRNPA2B1 expression and transport to synaptic dendrites are stimulated by BDNF [321].
Recently, transcriptome-wide screening of targets of hnRNPA2B1 in human iPSC-derived motoneurons and mouse spinal cord found a restricted number of transcripts with a shared UAG(G/A) signature in their 3′-UTR even though there was little overlap between the human and mouse mRNAs [322]. The alternative polyadenylation and splicing of some of the mRNAs was affected by hnRNPA2B1 depletion [322]. Further, the pathogenic D290V mutation appears to exert a toxic and perhaps gain-of-function by promoting exon exclusion; however, it produced divergent alternative splicing events between fibroblast and iPSC-derived motor neurons of affected and unaffected individuals as well as from those caused by hnRNPA2B1 depletion [322]. Regardless, these findings contrast to the widespread transcriptomal effects caused by TDP-43 or FUS depletion reported by other studies [291, 323]. However, the pathophysiological relevance of the heterogeneous transcriptomal changes controlled by hnRNPA2B1, TDP-43 or FUS are obscure. Another transcriptomal screening found that hnRNPA2B1 acts as nuclear “reader” of a consensus motif comprising N6-methyladenosine modification of mRNA and that impacts alternative splicing as well as primary miRNA processing [324]. Sumoylation of hnRNPA2B1 also controls its binding to Exomotifs in miRNAs and their loading to exosomes [325].
Gle1 and other nucleporins
As cited earlier, Gle1 is a ubiquitous nucleoporin at the cytosolic face of the nuclear pore that mediates mRNA export [200–202]. Autosomal recessive mutations in GLE1 caused by a 3-residue PFQ-insertion or compound heterozygous mutations, V617M and I684T, lead to the fetal motoneuron diseases, such as lethal congenital contracture syndrome 1 (LCCS1) or lethal arthrogryposis with anterior horn cell disease (LAAHD) [326]. This insertion in Gle1 appears to affect the self-association of Gle1 and its dysfunction at the nuclear pore [200]. Subsequent studies found that heterozygous mutations in GLE1 caused ALS (e.g., R697C) [327]. Notably, while the autosomal recessive mutations in Gle1 do not affect Gle1 localization at the nuclear pore, ALS-associated Gle1 mutations caused a reduction of wild-type Gle1 and loss of the mutant Gle1 at the nuclear pore that results from the inability of mutant Gle1 to bind the scaffold nucleoporin, Nupl2/hcG1, likely due to loss of Gle1stability [202, 327]. Hence, differential losses of Gle1 activities promote distinct pathological outcomes likely by mechanisms that differentially affect the remodeling of mRNPs as they exit the nuclear pore.
The Ran-binding protein 2 (a.k.a., Nup358) is a peripheral nucleoporin that forms the cytoplasmic filaments of the nuclear pore [142]. Although no human mutations linked to ALS have been found in RANBP2 yet, recent studies have shown that mice with loss of Ranbp2 in motoneurons develop rapidly progressive ALS-like motor traits, such as paralysis, dysphagia, and respiratory distress that culminate in the death of mice [241]. These traits are accompanied by axonopathy without TDP-43 pathology and by the disruption of nucleocytoplasmic transport, such as nucleocytoplasmic mislocalization of exportin-1, importin-β and Ran GTPase, and accessory substrates, such as HDAC4 [241]. hnRNPH3 was a new substrate affected by loss of Ranbp2 and its immunogenicity was lost in situ in motoneurons, but not in immunoblots of homogenates, thus indicating the misfolding or aggregation of hnRNPH3 by loss of Ranbp2 [241]. Further, spinal motoneurons presented dysregulation of chemokine signaling, which was characterized by the formation of intracellular deposits of components of this signaling pathway, such as its Cxcl14 ligand, Cxcr4 receptor and latent and activated Stat3 effector [241]. As described in the preceding sections conflicting results have implicated Ranbp2 in the nucleocytoplasmic export of bulk mRNA. However, differential transcriptomal analysis of sciatic nerve of mice with and without Ranbp2 in motoneurons showed that a limited number of mRNAs were affected by Ranbp2 loss [241]. Among these, there was pronounced accumulation of Cxcl14 mRNA both in the spinal cord and sciatic nerve but paradoxically this was accompanied by a decrease of Cxcl14 protein levels [241]; thus, it is possible that loss of Ranbp2 promotes the uncoupling of translation from Cxcl14 mRNA due to improper mRNP assembly or remodeling after exiting the pore. Finally, a novel intranuclear and long-lived Ranbp2 isoform, which is unique to spinal motoneurons, relocates to the cytosolic compartment where it localizes to the mitochondria after losses of Ranbp2 and of the short-lived Ranbp2 isoform at the nuclear pores [241]. In light of the findings that Ranbp2 controls the activation of kinesin-1, which is the primary motor for mitochondrial transport [254, 328–330], it will be important to define the role of this nuclear sequestered Ranbp2 isoform in mitochondrial transport and motor behavior. In this regard, a novel and small isoform of Ranbp2 that co-purifies and colocalizes with the mitochondria has been recently identified [331]. Finally, hnRNPA2B1 is a substrate for the cyclophilin domain (CY) of Ranbp2 and its cis–trans peptidyl–prolyl isomerase (PPIase)/chaperone activity [239]. Loss of the PPIase activity of Ranbp2 in mice promotes a post-transcriptional decline of hnRNPA2B1 levels [239], whereas novel small molecules against the PPIase pocket of CY of Ranbp2 promote a decline of hnRNPA2B1 in HeLa cells [240]. Hence, Ranbp2 emerges as a therapeutic target to control the pathogenicity of mutations in hnRNPA2B1 and the neuroprotection potential of other substrates, such as Stat3, in ALS [240, 332].
Recent studies also indicate that mutations in the nucleoporin, NUP88, cause lethal fetal akinesia deformation sequence (FADS), which involves congenital malformations related to impaired fetal movement [333]. Like Gle1, Nup88 is also involved in mRNA export. Hence, Nup88 appears to be another promising nucleoporin candidate gene for ALS.
ALS genes with unrelated nucleocytoplasmic transport functions
The roles of several other ALS genes, whose functions were once thought not to be involved in nucleocytoplasmic transport and/or RNA metabolism, may also present functions related to nucleocytoplasmic transport events. These include the vesicle-associated membrane protein-associated protein B and C (VAPB) [334, 335], optineurin [336, 337] and profilin 1 [338, 339]. ALS mutations in VAPB cause nuclear envelope defects possibly caused by the disruption of transport of nuclear envelope proteins from the ER-Golgi intermediate compartment [340]. Optineurin, which is implicated in ALS and glaucoma, translocates to the nucleus in response to an apoptotic stimulus and intranuclear inclusions of optineurin are found in patients with neuronal intranuclear inclusion disease (NIID) [336, 337, 341, 342]. Finally, ALS mutations in profilin 1, a small actin-binding protein which controls actin dynamics, influences stress granule dynamics [343]. Profilin 1 is exported from the nucleus by exportin-6, which appears to act as a constitutive suppressor of actin polymerization in the nucleus [344], where its accumulation may be pathogenic. This may represent a back sorting mechanism of extrusion of cytoplasmic proteins from the nucleus by exportins owing to leakage of otherwise cytosolic proteins into the nucleus (e.g., Ranbp1, RanGAP) [84, 345–347].
Concluding remarks
A unifying theme is emerging from provocative and accumulating evidence and hints in the literature that nucleocytoplasmic transport plays a central role in the pathogenesis of several forms of ALS. This affords a novel opportunity and fertile ground to illuminate and interface the underpinnings of a fundamental biological process with disease manifestations that compromise proteostasis, ribostasis, and neural functions in a yet more physiological context. Accumulating evidence also support that alterations in nucleocytoplasmic flux or partition of disease-causing substrates may constitute pathogenic drivers for other diseases other than ALS. For example, formation of cytoplasmic aggregates of mutant huntingtin, TDP-43 and artificial-β-sheets impairs the localization and sequesters several components of the nuclear import and export machinery and affects the integrity of the nuclear pore [49, 52–54]. Likewise, similar findings have been recently extended to tau, whose intracellular aggregation in neurofibrillary tangles is a hallmark of Alzheimer’s disease and tauopathies [55]. The findings indicate that nuclear pore and nucleocytoplasmic transport dysfunctions may promote neurotoxicity induced by tau. Finally, the nucleocytoplasmic export pathway has become a major therapeutic target for cancer and viral diseases and more recently, neurological diseases causing axonal damage, such as ALS [48, 348, 349]. Specifically, a growing armamentarium of reversible and irreversible and structure-based designed inhibitors directed toward exportin-1/CRM1 have been developed. These exportin-1/CRM1 inhibitors [a.k.a. selective inhibitors of nuclear export (SINE)], appear to hold therapeutic promise in preclinical models (e.g., KPT-276 and KPT-350) [48, 348], even though off-target effects, and thus toxicity, are a potential concern due to the broad-spectrum substrate specificity of exportin-1/CRM1 [92]. In spite of these advancements, the challenges ahead are considerable, given the spectrum of biological processes that intersect the biogenesis and pathogenic potential linked to the misprocessing of mRNAs and formation of protein aggregates and inclusions. Looking ahead, I can envision that mechanistic-guided concepts and approaches will illuminate in greater detail the mysteries on how moonlighting proteins of the nucleocytoplasmic transport machinery intersect with multitasking substrates of pathogenic potential to produce neural type-restricted and deleterious manifestations. The emerging new concepts will enable to harness regulatory mechanisms of nucleocytoplasmic transport into therapeutic approaches of ALS and other neurodegenerative diseases that are currently lacking.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- Ranbp2
Ran-binding protein 2
- Ranbp1
Ran-binding protein 1
- RBDs
Ran-GTP-binding domains
- ZnF
Zinc-finger motif
- Nup
Nucleoporin
- CY
Cyclophilin
- RCC1
Regulator of chromosome condensation 1
- RanGAP1
Ran GTPase-activating protein-1
- NTRs
Nuclear transport receptors
- NES
Nuclear export sequence
- NLS
Nuclear localization sequence
- mRNP
Messenger ribonucleoprotein
- TREX
Transcription–export complex,
- hnRNPs
Heterogeneous nuclear ribonucleoproteins
- CBC
Cap-binding complex
- EJC
Exon-junction complex
- eIF
Eukaryotic initiation factor
- SRP
Signal recognition particle
- SSCR
Signal sequence-coding region
- ALREX
Alternative mRNA nuclear export
- Cxcl14
Chemokine ligand 14
- Acc1
Acetyl-CoA carboxylase 1
- SOD1
Cu/Zn Superoxide dismutase 1,
- C9ORF72
Chromosome 9 Open Reading Frame 72
- TARDBP
Transactive response DNA-binding protein (TDP-43)
- FUS
Fused in sarcoma
- DPRs
Dipeptide repeat proteins
- LCCS1
Lethal congenital contracture syndrome 1
- LAAHD
Lethal arthrogryposis with anterior horn cell disease
- Ran
Ras-related nuclear protein
Funding
This work was in part funded by National Institutes of Health Grants GM083165, GM083165-03S1 and EY019492 to P.A.F.
Compliance with ethical standards
Conflict of interest
The author declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The author consents for the publication of this study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
3/28/2019
The original version of this article unfortunately contained the following misspelling and formatting mistakes.
References
- 1.Charcot JM, Joffroy A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol. 1869;2:744–754. [Google Scholar]
- 2.Charcot J. De la sclérose latérale amyotrophique. Prog Med. 1874;2:341–453. [Google Scholar]
- 3.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- 5.Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin. 2015;33(4):855–876. doi: 10.1016/j.ncl.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gizzi M, DiRocco A, Sivak M, Cohen B. Ocular motor function in motor neuron disease. Neurology. 1992;42(5):1037–1046. doi: 10.1212/WNL.42.5.1037. [DOI] [PubMed] [Google Scholar]
- 7.Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133(6):863–885. doi: 10.1007/s00401-017-1708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spiller KJ, Cheung CJ, Restrepo CR, Kwong LK, Stieber AM, Trojanowski JQ, Lee VM. Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J Neurosci. 2016;36(29):7707–7717. doi: 10.1523/JNEUROSCI.1457-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day-Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna-Yasek D, Cady J, Vianney de Jong JM, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A, Consortium FS, Al-Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier-Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, Goldstein DB (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347(6229):1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed]
- 10.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 11.Li HF, Wu ZY. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl Neurodegener. 2016;5:3. doi: 10.1186/s40035-016-0050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 13.Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71(5):1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu YT, Laura M, Hersheson J, Horga A, Jaunmuktane Z, Brandner S, Pittman A, Hughes D, Polke JM, Sweeney MG, Proukakis C, Janssen JC, Auer-Grumbach M, Zuchner S, Shields KG, Reilly MM, Houlden H. Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology. 2014;83(7):612–619. doi: 10.1212/WNL.0000000000000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morais S, Raymond L, Mairey M, Coutinho P, Brandao E, Ribeiro P, Loureiro JL, Sequeiros J, Brice A, Alonso I, Stevanin G. Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur J Hum Genet. 2017;25(11):1217–1228. doi: 10.1038/ejhg.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duis J, Dean S, Applegate C, Harper A, Xiao R, He W, Dollar JD, Sun LR, Waberski MB, Crawford TO, Hamosh A, Stafstrom CE. KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann Neurol. 2016;80(4):633–637. doi: 10.1002/ana.24744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rydzanicz M, Jagla M, Kosinska J, Tomasik T, Sobczak A, Pollak A, Herman-Sucharska I, Walczak A, Kwinta P, Ploski R. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet. 2017;91(5):769–773. doi: 10.1111/cge.12831. [DOI] [PubMed] [Google Scholar]
- 18.Brenner D, Yilmaz R, Muller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P, Ruf W, Neuwirth C, Weber M, Pinto S, Claeys KG, Schrank B, Jordan B, Knehr A, Gunther K, Hubers A, Zeller D, Kubisch C, Jablonka S, Sendtner M, Klopstock T, de Carvalho M, Sperfeld A, Borck G, Volk AE, Dorst J, Weis J, Otto M, Schuster J, Del Tredici K, Braak H, Danzer KM, Freischmidt A, Meitinger T, Strom TM, Ludolph AC, Andersen PM, Weishaupt JH, German ALSnMNDNET Hot-spot KIF5A mutations cause familial ALS. Brain. 2018;141(3):688–697. doi: 10.1093/brain/awx370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, Rivera AM, van Rheenen W, Murphy NA, van Vugt J, Geiger JT, Van der Spek RA, Pliner HA, Shankaracharya, Smith BN, Marangi G, Topp SD, Abramzon Y, Gkazi AS, Eicher JD, Kenna A, Consortium I, Mora G, Calvo A, Mazzini L, Riva N, Mandrioli J, Caponnetto C, Battistini S, Volanti P, La Bella V, Conforti FL, Borghero G, Messina S, Simone IL, Trojsi F, Salvi F, Logullo FO, D’Alfonso S, Corrado L, Capasso M, Ferrucci L, Genomic Translation for ALSCC, Moreno CAM, Kamalakaran S, Goldstein DB, Consortium ALSS, Gitler AD, Harris T, Myers RM, Consortium NA, Phatnani H, Musunuri RL, Evani US, Abhyankar A, Zody MC, Answer ALSF, Kaye J, Finkbeiner S, Wyman SK, LeNail A, Lima L, Fraenkel E, Svendsen CN, Thompson LM, Van Eyk JE, Berry JD, Miller TM, Kolb SJ, Cudkowicz M, Baxi E, Clinical Research in ALS, Related Disorders for Therapeutic Development C, Benatar M, Taylor JP, Rampersaud E, Wu G, Wuu J, Consortium S, Lauria G, Verde F, Fogh I, Tiloca C, Comi GP, Soraru G, Cereda C, French ALSC, Corcia P, Laaksovirta H, Myllykangas L, Jansson L, Valori M, Ealing J, Hamdalla H, Rollinson S, Pickering-Brown S, Orrell RW, Sidle KC, Malaspina A, Hardy J, Singleton AB, Johnson JO, Arepalli S, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Al-Sarraj S, King A, Troakes C, Vance C, de Belleroche J, Baas F, Ten Asbroek A, Munoz-Blanco JL, Hernandez DG, Ding J, Gibbs JR, Scholz SW, Floeter MK, Campbell RH, Landi F, Bowser R, Pulst SM, Ravits JM, MacGowan DJL, Kirby J, Pioro EP, Pamphlett R, Broach J, Gerhard G, Dunckley TL, Brady CB, Kowall NW, Troncoso JC, Le Ber I, Mouzat K, Lumbroso S, Heiman-Patterson TD, Kamel F, Van Den Bosch L, Baloh RH, Strom TM, Meitinger T, Shatunov A, Van Eijk KR, de Carvalho M, Kooyman M, Middelkoop B, Moisse M, McLaughlin RL, Van Es MA, Weber M, Boylan KB, Van Blitterswijk M, Rademakers R, Morrison KE, Basak AN, Mora JS, Drory VE, Shaw PJ, Turner MR, Talbot K, Hardiman O, Williams KL, Fifita JA, Nicholson GA, Blair IP, Rouleau GA, Esteban-Perez J, Garcia-Redondo A, Al-Chalabi A, Project Min EALSSC, Rogaeva E, Zinman L, Ostrow LW, Maragakis NJ, Rothstein JD, Simmons Z, Cooper-Knock J, Brice A, Goutman SA, Feldman EL, Gibson SB, Taroni F, Ratti A, Gellera C, Van Damme P, Robberecht W, Fratta P, Sabatelli M, Lunetta C, Ludolph AC, Andersen PM, Weishaupt JH, Camu W, Trojanowski JQ, Van Deerlin VM, Brown RH, Jr., van den Berg LH, Veldink JH, Harms MB, Glass JD, Stone DJ, Tienari P, Silani V, Chio A, Shaw CE, Traynor BJ, Landers JE (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97(6):1268 e1266–1283 e1266. 10.1016/j.neuron.2018.02.027 [DOI] [PMC free article] [PubMed]
- 20.Rizzo F, Riboldi G, Salani S, Nizzardo M, Simone C, Corti S, Hedlund E. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci. 2014;71(6):999–1015. doi: 10.1007/s00018-013-1480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 22.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312(5778):1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 23.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM, Trojanowski JQ. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7(6):e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9(3):408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- 27.Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586(14):3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350(9071):134–138. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 29.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431(7010):805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 30.Ross CA, Poirier MA. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6(11):891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 31.Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190(5):719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, Liu J, Xu M, Yang Y, Wang C, Zhang D, Bigio EH, Mesulam M, Shen Y, Xu Q, Fushimi K, Wu JY. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol. 2011;18(7):822–830. doi: 10.1038/nsmb.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, Sun H, Boylan KB, Dickson D, Kennedy Z, Lin Z, Zhang YJ, Daughrity L, Jung C, Gao FB, Sapp PC, Horvitz HR, Bosco DA, Brown SP, de Jong P, Petrucelli L, Mueller C, Brown RH., Jr Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88(5):902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L, Lee EB, Lutz CM, Baloh RH. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78(3):426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348(6239):1151–1154. 10.1126/science.aaa9344 [DOI] [PMC free article] [PubMed]
- 37.Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90(3):521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009;8(1):94–109. doi: 10.1016/S1474-4422(08)70293-X. [DOI] [PubMed] [Google Scholar]
- 39.Turner MR, Benatar M. Ensuring continued progress in biomarkers for amyotrophic lateral sclerosis. Muscle Nerve. 2015;51(1):14–18. doi: 10.1002/mus.24470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, Johnston A, Overstreet K, Oh SY, Todd PK, Berry JD, Cudkowicz ME, Boeve BF, Dickson D, Floeter MK, Traynor BJ, Morelli C, Ratti A, Silani V, Rademakers R, Brown RH, Rothstein JD, Boylan KB, Petrucelli L, Disney MD. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83(5):1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Ito H, Wate R, Ohnishi S, Nakano S, Kusaka H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006;112(6):673–680. doi: 10.1007/s00401-006-0130-4. [DOI] [PubMed] [Google Scholar]
- 42.Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2009;68(11):1184–1192. doi: 10.1097/NEN.0b013e3181bc3bec. [DOI] [PubMed] [Google Scholar]
- 43.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagara Y, Tateishi T, Yamasaki R, Hayashi S, Kawamura M, Kikuchi H, Iinuma KM, Tanaka M, Iwaki T, Matsushita T, Ohyagi Y, Kira J. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1alpha in amyotrophic lateral sclerosis. Brain Pathol. 2013;23(5):534–546. doi: 10.1111/bpa.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward ME, Taubes A, Chen R, Miller BL, Sephton CF, Gelfand JM, Minami S, Boscardin J, Martens LH, Seeley WW, Yu G, Herz J, Filiano AJ, Arrant AE, Roberson ED, Kraft TW, Farese RV, Jr, Green A, Gan L. Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J Exp Med. 2014;211(10):1937–1945. doi: 10.1084/jem.20140214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568–583. doi: 10.1002/ana.24469. [DOI] [PubMed] [Google Scholar]
- 47.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351(6269):173–176. doi: 10.1126/science.aad2033. [DOI] [PubMed] [Google Scholar]
- 50.Shang J, Yamashita T, Nakano Y, Morihara R, Li X, Feng T, Liu X, Huang Y, Fukui Y, Hishikawa N, Ohta Y, Abe K. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience. 2017;350:158–168. doi: 10.1016/j.neuroscience.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 51.Zhong Y, Wang J, Henderson MJ, Yang P, Hagen BM, Siddique T, Vogel BE, Deng HX, Fang S. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife. 2017 doi: 10.7554/elife.23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC, Pham JT, Ahmed I, Peng Q, Wadhwa H, Pletnikova O, Troncoso JC, Duan W, Snyder SH, Ranum LPW, Thompson LM, Lloyd TE, Ross CA, Rothstein JD. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94(1):93 e106–107 e106. doi: 10.1016/j.neuron.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG, Cleveland DW, Lagier-Tourenne C. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron. 2017;94(1):48 e44–57 e44. doi: 10.1016/j.neuron.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, Rekhi A, Boudi A, Tziortzouda P, Lee YB, Smith B, Bridi JC, Spinelli G, Dearlove J, Humphrey DM, Gallo JM, Troakes C, Fanto M, Soller M, Rogelj B, Parsons RB, Shaw CE, Hortobagyi T, Hirth F. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-related neurodegeneration. Brain. 2018;141(10):2908–2924. doi: 10.1093/brain/awy241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburgh CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron. 2018;99(5):925 e927–940 e927. doi: 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rush MG, Drivas G, D’Eustachio P. The small nuclear GTPase Ran: how much does it run? Bioessays. 1996;18(2):103–112. doi: 10.1002/bies.950180206. [DOI] [PubMed] [Google Scholar]
- 57.Nachury MV, Weis K. The direction of transport through the nuclear pore can be inverted. Proc Natl Acad Sci USA. 1999;96(17):9622–9627. doi: 10.1073/pnas.96.17.9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akhtar N, Hagan H, Lopilato JE, Corbett AH. Functional analysis of the yeast Ran exchange factor Prp20p: in vivo evidence for the RanGTP gradient model. Mol Genet Genom. 2001;265(5):851–864. doi: 10.1007/s004380100480. [DOI] [PubMed] [Google Scholar]
- 59.Kalab P, Weis K, Heald R. Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science. 2002;295(5564):2452–2456. doi: 10.1126/science.1068798. [DOI] [PubMed] [Google Scholar]
- 60.Kalab P, Pralle A, Isacoff EY, Heald R, Weis K. Analysis of a RanGTP-regulated gradient in mitotic somatic cells. Nature. 2006;440(7084):697–701. doi: 10.1038/nature04589. [DOI] [PubMed] [Google Scholar]
- 61.Bischoff FR, Ponstingl H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature. 1991;354(6348):80–82. doi: 10.1038/354080a0. [DOI] [PubMed] [Google Scholar]
- 62.Bischoff FR, Ponstingl H. Mitotic regulator protein RCC1 is complexed with a nuclear ras-related polypeptide. Proc Natl Acad Sci USA. 1991;88(23):10830–10834. doi: 10.1073/pnas.88.23.10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klebe C, Prinz H, Wittinghofer A, Goody RS. The kinetic mechanism of Ran–nucleotide exchange catalyzed by RCC1. Biochemistry (Mosc) 1995;34(39):12543–12552. doi: 10.1021/bi00039a008. [DOI] [PubMed] [Google Scholar]
- 64.Klebe C, Bischoff FR, Ponstingl H, Wittinghofer A. Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry (Mosc) 1995;34(2):639–647. doi: 10.1021/bi00002a031. [DOI] [PubMed] [Google Scholar]
- 65.Bischoff FR, Klebe C, Kretschmer J, Wittinghofer A, Ponstingl H. RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc Natl Acad Sci USA. 1994;91(7):2587–2591. doi: 10.1073/pnas.91.7.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bischoff FR, Krebber H, Kempf T, Hermes I, Ponstingl H. Human RanGTPase-activating protein RanGAP1 is a homologue of yeast Rna1p involved in mRNA processing and transport. Proc Natl Acad Sci USA. 1995;92(5):1749–1753. doi: 10.1073/pnas.92.5.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Becker J, Melchior F, Gerke V, Bischoff FR, Ponstingl H, Wittinghofer A. RNA1 encodes a GTPase-activating protein specific for Gsp1p, the Ran/TC4 homologue of Saccharomyces cerevisiae. J Biol Chem. 1995;270(20):11860–11865. doi: 10.1074/jbc.270.20.11860. [DOI] [PubMed] [Google Scholar]
- 68.Gorlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996;15(20):5584–5594. doi: 10.1002/j.1460-2075.1996.tb00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, Gorlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997;16(21):6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorlich D, Seewald MJ, Ribbeck K. Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J. 2003;22(5):1088–1100. doi: 10.1093/emboj/cdg113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ribbeck K, Lipowsky G, Kent HM, Stewart M, Gorlich D. NTF2 mediates nuclear import of Ran. EMBO J. 1998;17(22):6587–6598. doi: 10.1093/emboj/17.22.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith A, Brownawell A, Macara IG. Nuclear import of Ran is mediated by the transport factor NTF2. Curr Biol. 1998;8(25):1403–1406. doi: 10.1016/S0960-9822(98)00023-2. [DOI] [PubMed] [Google Scholar]
- 73.Feldherr CM. The nuclear annuli as pathways for nucleocytoplasmic exchanges. J Cell Biol. 1962;14:65–72. doi: 10.1083/jcb.14.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kose S, Imamoto N, Tachibana T, Shimamoto T, Yoneda Y. Ran-unassisted nuclear migration of a 97-kD component of nuclear pore-targeting complex. J Cell Biol. 1997;139(4):841–849. doi: 10.1083/jcb.139.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwoebel ED, Talcott B, Cushman I, Moore MS. Ran-dependent signal-mediated nuclear import does not require GTP hydrolysis by Ran. J Biol Chem. 1998;273(52):35170–35175. doi: 10.1074/jbc.273.52.35170. [DOI] [PubMed] [Google Scholar]
- 76.Nakielny S, Dreyfuss G. Import and export of the nuclear protein import receptor transportin by a mechanism independent of GTP hydrolysis. Curr Biol. 1998;8(2):89–95. doi: 10.1016/S0960-9822(98)70039-9. [DOI] [PubMed] [Google Scholar]
- 77.Ribbeck K, Kutay U, Paraskeva E, Gorlich D. The translocation of transportin-cargo complexes through nuclear pores is independent of both Ran and energy. Curr Biol. 1999;9(1):47–50. doi: 10.1016/S0960-9822(99)80046-3. [DOI] [PubMed] [Google Scholar]
- 78.Paine PL, Moore LC, Horowitz SB. Nuclear envelope permeability. Nature. 1975;254(5496):109–114. doi: 10.1038/254109a0. [DOI] [PubMed] [Google Scholar]
- 79.Paine PL. Nucleocytoplasmic movement of fluorescent tracers microinjected into living salivary gland cells. J Cell Biol. 1975;66(3):652–657. doi: 10.1083/jcb.66.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gorlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 81.Keminer O, Siebrasse JP, Zerf K, Peters R. Optical recording of signal-mediated protein transport through single nuclear pore complexes. Proc Natl Acad Sci USA. 1999;96(21):11842–11847. doi: 10.1073/pnas.96.21.11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ribbeck K, Gorlich D. Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 2001;20(6):1320–1330. doi: 10.1093/emboj/20.6.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naim B, Zbaida D, Dagan S, Kapon R, Reich Z. Cargo surface hydrophobicity is sufficient to overcome the nuclear pore complex selectivity barrier. EMBO J. 2009;28(18):2697–2705. doi: 10.1038/emboj.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohr D, Frey S, Fischer T, Guttler T, Gorlich D. Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009;28(17):2541–2553. doi: 10.1038/emboj.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tu LC, Fu G, Zilman A, Musser SM. Large cargo transport by nuclear pores: implications for the spatial organization of FG-nucleoporins. EMBO J. 2013;32(24):3220–3230. doi: 10.1038/emboj.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Naim B, Brumfeld V, Kapon R, Kiss V, Nevo R, Reich Z. Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J Biol Chem. 2007;282(6):3881–3888. doi: 10.1074/jbc.M608329200. [DOI] [PubMed] [Google Scholar]
- 87.Fiserova J, Richards SA, Wente SR, Goldberg MW. Facilitated transport and diffusion take distinct spatial routes through the nuclear pore complex. J Cell Sci. 2010;123(Pt 16):2773–2780. doi: 10.1242/jcs.070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cardarelli F, Tosti L, Serresi M, Beltram F, Bizzarri R. Fluorescent recovery after photobleaching (FRAP) analysis of nuclear export rates identifies intrinsic features of nucleocytoplasmic transport. J Biol Chem. 2012;287(8):5554–5561. doi: 10.1074/jbc.M111.304899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kimura M, Imamoto N. Biological significance of the importin-beta family-dependent nucleocytoplasmic transport pathways. Traffic. 2014;15(7):727–748. doi: 10.1111/tra.12174. [DOI] [PubMed] [Google Scholar]
- 90.Kimura M, Morinaka Y, Imai K, Kose S, Horton P, Imamoto N. Extensive cargo identification reveals distinct biological roles of the 12 importin pathways. Elife. 2017 doi: 10.7554/elife.21184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aksu M, Pleiner T, Karaca S, Kappert C, Dehne HJ, Seibel K, Urlaub H, Bohnsack MT, Gorlich D. Xpo7 is a broad-spectrum exportin and a nuclear import receptor. J Cell Biol. 2018;217(7):2329–2340. doi: 10.1083/jcb.201712013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Gorlich D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife. 2015 doi: 10.7554/elife.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kutay U, Bischoff FR, Kostka S, Kraft R, Gorlich D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90(6):1061–1071. doi: 10.1016/S0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- 94.Fornerod M, van Deursen J, van Baal S, Reynolds A, Davis D, Murti KG, Fransen J, Grosveld G. The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J. 1997;16(4):807–816. doi: 10.1093/emboj/16.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gorlich D, Dabrowski M, Bischoff FR, Kutay U, Bork P, Hartmann E, Prehn S, Izaurralde E. A novel class of RanGTP binding proteins. J Cell Biol. 1997;138(1):65–80. doi: 10.1083/jcb.138.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vetter IR, Arndt A, Kutay U, Gorlich D, Wittinghofer A (1999) Structural view of the Ran–Importin beta interaction at 2.3 A resolution. Cell 97(5):635–646 [DOI] [PubMed]
- 97.Villa Braslavsky CI, Nowak C, Gorlich D, Wittinghofer A, Kuhlmann J. Different structural and kinetic requirements for the interaction of Ran with the Ran-binding domains from RanBP2 and importin-beta. Biochemistry (Mosc) 2000;39(38):11629–11639. doi: 10.1021/bi001010f. [DOI] [PubMed] [Google Scholar]
- 98.Kutay U, Hartmann E, Treichel N, Calado A, Carmo-Fonseca M, Prehn S, Kraft R, Gorlich D, Bischoff FR. Identification of two novel RanGTP-binding proteins belonging to the importin beta superfamily. J Biol Chem. 2000;275(51):40163–40168. doi: 10.1074/jbc.M006242200. [DOI] [PubMed] [Google Scholar]
- 99.Mingot JM, Bohnsack MT, Jakle U, Gorlich D. Exportin 7 defines a novel general nuclear export pathway. EMBO J. 2004;23(16):3227–3236. doi: 10.1038/sj.emboj.7600338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Radu A, Blobel G, Moore MS. Identification of a protein complex that is required for nuclear protein import and mediates docking of import substrate to distinct nucleoporins. Proc Natl Acad Sci USA. 1995;92(5):1769–1773. doi: 10.1073/pnas.92.5.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bischoff FR, Gorlich D. RanBP1 is crucial for the release of RanGTP from importin beta-related nuclear transport factors. FEBS Lett. 1997;419(2–3):249–254. doi: 10.1016/S0014-5793(97)01467-1. [DOI] [PubMed] [Google Scholar]
- 102.Deane R, Schafer W, Zimmermann HP, Mueller L, Gorlich D, Prehn S, Ponstingl H, Bischoff FR. Ran-binding protein 5 (RanBP5) is related to the nuclear transport factor importin-beta but interacts differently with RanBP1. Mol Cell Biol. 1997;17(9):5087–5096. doi: 10.1128/MCB.17.9.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90(6):1041–1050. doi: 10.1016/S0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- 104.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90(6):1051–1060. doi: 10.1016/S0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 105.Kutay U, Lipowsky G, Izaurralde E, Bischoff FR, Schwarzmaier P, Hartmann E, Gorlich D. Identification of a tRNA-specific nuclear export receptor. Mol Cell. 1998;1(3):359–369. doi: 10.1016/S1097-2765(00)80036-2. [DOI] [PubMed] [Google Scholar]
- 106.Paraskeva E, Izaurralde E, Bischoff FR, Huber J, Kutay U, Hartmann E, Luhrmann R, Gorlich D. CRM1-mediated recycling of snurportin 1 to the cytoplasm. J Cell Biol. 1999;145(2):255–264. doi: 10.1083/jcb.145.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Askjaer P, Bachi A, Wilm M, Bischoff FR, Weeks DL, Ogniewski V, Ohno M, Niehrs C, Kjems J, Mattaj IW, Fornerod M. RanGTP-regulated interactions of CRM1 with nucleoporins and a shuttling DEAD-box helicase. Mol Cell Biol. 1999;19(9):6276–6285. doi: 10.1128/MCB.19.9.6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dong X, Biswas A, Chook YM. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol. 2009;16(5):558–560. doi: 10.1038/nsmb.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390(6657):308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 110.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278(5335):141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- 111.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458(7242):1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17(11):1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 113.Jakel S, Gorlich D. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 1998;17(15):4491–4502. doi: 10.1093/emboj/17.15.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Truant R, Cullen BR. The arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin beta-dependent nuclear localization signals. Mol Cell Biol. 1999;19(2):1210–1217. doi: 10.1128/MCB.19.2.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adam EJ, Adam SA. Identification of cytosolic factors required for nuclear location sequence-mediated binding to the nuclear envelope. J Cell Biol. 1994;125(3):547–555. doi: 10.1083/jcb.125.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gorlich D, Prehn S, Laskey RA, Hartmann E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 1994;79(5):767–778. doi: 10.1016/0092-8674(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 117.Gorlich D, Kostka S, Kraft R, Dingwall C, Laskey RA, Hartmann E, Prehn S. Two different subunits of importin cooperate to recognize nuclear localization signals and bind them to the nuclear envelope. Curr Biol. 1995;5(4):383–392. doi: 10.1016/S0960-9822(95)00079-0. [DOI] [PubMed] [Google Scholar]
- 118.Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem. 2007;282(8):5101–5105. doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jakel S, Albig W, Kutay U, Bischoff FR, Schwamborn K, Doenecke D, Gorlich D. The importin beta/importin 7 heterodimer is a functional nuclear import receptor for histone H1. EMBO J. 1999;18(9):2411–2423. doi: 10.1093/emboj/18.9.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126(3):543–558. doi: 10.1016/j.cell.2006.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Suel KE, Gu H, Chook YM. Modular organization and combinatorial energetics of proline-tyrosine nuclear localization signals. PLoS Biol. 2008;6(6):e137. doi: 10.1371/journal.pbio.0060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang P, Liu GH, Wu K, Qu J, Huang B, Zhang X, Zhou X, Gerace L, Chen C. Repression of classical nuclear export by S-nitrosylation of CRM1. J Cell Sci. 2009;122(Pt 20):3772–3779. doi: 10.1242/jcs.057026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Riviere Y, Blank V, Kourilsky P, Israel A. Processing of the precursor of NF-kappa B by the HIV-1 protease during acute infection. Nature. 1991;350(6319):625–626. doi: 10.1038/350625a0. [DOI] [PubMed] [Google Scholar]
- 124.Li S, Ku CY, Farmer AA, Cong YS, Chen CF, Lee WH. Identification of a novel cytoplasmic protein that specifically binds to nuclear localization signal motifs. J Biol Chem. 1998;273(11):6183–6189. doi: 10.1074/jbc.273.11.6183. [DOI] [PubMed] [Google Scholar]
- 125.Craig E, Zhang ZK, Davies KP, Kalpana GV. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: implications for tumorigenesis. EMBO J. 2002;21(1–2):31–42. doi: 10.1093/emboj/21.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saporita AJ, Zhang Q, Navai N, Dincer Z, Hahn J, Cai X, Wang Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J Biol Chem. 2003;278(43):41998–42005. doi: 10.1074/jbc.M302460200. [DOI] [PubMed] [Google Scholar]
- 127.Fischer U, Schauble N, Schutz S, Altvater M, Chang Y, Faza MB, Panse VG. A non-canonical mechanism for Crm1-export cargo complex assembly. Elife. 2015 doi: 10.7554/elife.05745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cardarelli F, Bizzarri R, Serresi M, Albertazzi L, Beltram F. Probing nuclear localization signal-importin alpha binding equilibria in living cells. J Biol Chem. 2009;284(52):36638–36646. doi: 10.1074/jbc.M109.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kubitscheck U, Grunwald D, Hoekstra A, Rohleder D, Kues T, Siebrasse JP, Peters R. Nuclear transport of single molecules: dwell times at the nuclear pore complex. J Cell Biol. 2005;168(2):233–243. doi: 10.1083/jcb.200411005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mor A, Suliman S, Ben-Yishay R, Yunger S, Brody Y, Shav-Tal Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat Cell Biol. 2010;12(6):543–552. doi: 10.1038/ncb2056. [DOI] [PubMed] [Google Scholar]
- 131.Lim RY, Fahrenkrog B, Koser J, Schwarz-Herion K, Deng J, Aebi U. Nanomechanical basis of selective gating by the nuclear pore complex. Science. 2007;318(5850):640–643. doi: 10.1126/science.1145980. [DOI] [PubMed] [Google Scholar]
- 132.Cardarelli F, Lanzano L, Gratton E. Capturing directed molecular motion in the nuclear pore complex of live cells. Proc Natl Acad Sci USA. 2012;109(25):9863–9868. doi: 10.1073/pnas.1200486109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Koyama M, Matsuura Y. An allosteric mechanism to displace nuclear export cargo from CRM1 and RanGTP by RanBP1. EMBO J. 2010;29(12):2002–2013. doi: 10.1038/emboj.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vetter IR, Nowak C, Nishimoto T, Kuhlmann J, Wittinghofer A. Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature. 1999;398(6722):39–46. doi: 10.1038/17969. [DOI] [PubMed] [Google Scholar]
- 135.Coutavas E, Ren M, Oppenheim JD, D’Eustachio P, Rush MG. Characterization of proteins that interact with the cell-cycle regulatory protein Ran/TC4. Nature. 1993;366(6455):585–587. doi: 10.1038/366585a0. [DOI] [PubMed] [Google Scholar]
- 136.Lounsbury KM, Beddow AL, Macara IG. A family of proteins that stabilize the Ran/TC4 GTPase in its GTP-bound conformation. J Biol Chem. 1994;269(15):11285–11290. [PubMed] [Google Scholar]
- 137.Beddow AL, Richards SA, Orem NR, Macara IG. The Ran/TC4 GTPase-binding domain: identification by expression cloning and characterization of a conserved sequence motif. Proc Natl Acad Sci USA. 1995;92(8):3328–3332. doi: 10.1073/pnas.92.8.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ferreira PA, Hom JT, Pak WL. Retina-specifically expressed novel subtypes of bovine cyclophilin. J Biol Chem. 1995;270(39):23179–23188. doi: 10.1074/jbc.270.39.23179. [DOI] [PubMed] [Google Scholar]
- 139.Wilken N, Senecal JL, Scheer U, Dabauvalle MC. Localization of the Ran-GTP binding protein RanBP2 at the cytoplasmic side of the nuclear pore complex. Eur J Cell Biol. 1995;68(3):211–219. [PubMed] [Google Scholar]
- 140.Wu J, Matunis MJ, Kraemer D, Blobel G, Coutavas E. Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J Biol Chem. 1995;270(23):14209–14213. doi: 10.1074/jbc.270.23.14209. [DOI] [PubMed] [Google Scholar]
- 141.Yokoyama N, Hayashi N, Seki T, Pante N, Ohba T, Nishii K, Kuma K, Hayashida T, Miyata T, Aebi U, et al. A giant nucleopore protein that binds Ran/TC4. Nature. 1995;376(6536):184–188. doi: 10.1038/376184a0. [DOI] [PubMed] [Google Scholar]
- 142.Delphin C, Guan T, Melchior F, Gerace L. RanGTP targets p97 to RanBP2, a filamentous protein localized at the cytoplasmic periphery of the nuclear pore complex. Mol Biol Cell. 1997;8(12):2379–2390. doi: 10.1091/mbc.8.12.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Forler D, Rabut G, Ciccarelli FD, Herold A, Kocher T, Niggeweg R, Bork P, Ellenberg J, Izaurralde E. RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol Cell Biol. 2004;24(3):1155–1167. doi: 10.1128/MCB.24.3.1155-1167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005;15(3):343–351. doi: 10.1101/gr.3266405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Singh BB, Patel HH, Roepman R, Schick D, Ferreira PA. The zinc finger cluster domain of RanBP2 is a specific docking site for the nuclear export factor, exportin-1. J Biol Chem. 1999;274(52):37370–37378. doi: 10.1074/jbc.274.52.37370. [DOI] [PubMed] [Google Scholar]
- 146.Bischoff FR, Krebber H, Smirnova E, Dong W, Ponstingl H. Co-activation of RanGTPase and inhibition of GTP dissociation by Ran-GTP binding protein RanBP1. EMBO J. 1995;14(4):705–715. doi: 10.1002/j.1460-2075.1995.tb07049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seewald MJ, Korner C, Wittinghofer A, Vetter IR. RanGAP mediates GTP hydrolysis without an arginine finger. Nature. 2002;415(6872):662–666. doi: 10.1038/415662a. [DOI] [PubMed] [Google Scholar]
- 148.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88(1):97–107. doi: 10.1016/S0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- 149.Saitoh H, Pu R, Cavenagh M, Dasso M. RanBP2 associates with Ubc9p and a modified form of RanGAP1. Proc Natl Acad Sci USA. 1997;94(8):3736–3741. doi: 10.1073/pnas.94.8.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee GW, Melchior F, Matunis MJ, Mahajan R, Tian Q, Anderson P. Modification of Ran GTPase-activating protein by the small ubiquitin-related modifier SUMO-1 requires Ubc9, an E2-type ubiquitin-conjugating enzyme homologue. J Biol Chem. 1998;273(11):6503–6507. doi: 10.1074/jbc.273.11.6503. [DOI] [PubMed] [Google Scholar]
- 151.Saitoh H, Sparrow DB, Shiomi T, Pu RT, Nishimoto T, Mohun TJ, Dasso M. Ubc9p and the conjugation of SUMO-1 to RanGAP1 and RanBP2. Curr Biol. 1998;8(2):121–124. doi: 10.1016/S0960-9822(98)70044-2. [DOI] [PubMed] [Google Scholar]
- 152.Mueller L, Cordes VC, Bischoff FR, Ponstingl H. Human RanBP3, a group of nuclear RanGTP binding proteins. FEBS Lett. 1998;427(3):330–336. doi: 10.1016/S0014-5793(98)00459-1. [DOI] [PubMed] [Google Scholar]
- 153.Englmeier L, Fornerod M, Bischoff FR, Petosa C, Mattaj IW, Kutay U. RanBP3 influences interactions between CRM1 and its nuclear protein export substrates. EMBO Rep. 2001;2(10):926–932. doi: 10.1093/embo-reports/kve200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lindsay ME, Holaska JM, Welch K, Paschal BM, Macara IG. Ran-binding protein 3 is a cofactor for Crm1-mediated nuclear protein export. J Cell Biol. 2001;153(7):1391–1402. doi: 10.1083/jcb.153.7.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Langer K, Dian C, Rybin V, Muller CW, Petosa C. Insights into the function of the CRM1 cofactor RanBP3 from the structure of its Ran-binding domain. PLoS One. 2011;6(2):e17011. doi: 10.1371/journal.pone.0017011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Arts GJ, Fornerod M, Mattaj IW. Identification of a nuclear export receptor for tRNA. Curr Biol. 1998;8(6):305–314. doi: 10.1016/S0960-9822(98)70130-7. [DOI] [PubMed] [Google Scholar]
- 157.Brownawell AM, Macara IG. Exportin-5, a novel karyopherin, mediates nuclear export of double-stranded RNA binding proteins. J Cell Biol. 2002;156(1):53–64. doi: 10.1083/jcb.200110082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clouse KN, Luo MJ, Zhou Z, Reed R. A Ran-independent pathway for export of spliced mRNA. Nat Cell Biol. 2001;3(1):97–99. doi: 10.1038/35050625. [DOI] [PubMed] [Google Scholar]
- 159.Segref A, Sharma K, Doye V, Hellwig A, Huber J, Luhrmann R, Hurt E. Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 1997;16(11):3256–3271. doi: 10.1093/emboj/16.11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ossareh-Nazari B, Maison C, Black BE, Levesque L, Paschal BM, Dargemont C. RanGTP-binding protein NXT1 facilitates nuclear export of different classes of RNA in vitro. Mol Cell Biol. 2000;20(13):4562–4571. doi: 10.1128/MCB.20.13.4562-4571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Herold A, Klymenko T, Izaurralde E. NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila. RNA. 2001;7(12):1768–1780. [PMC free article] [PubMed] [Google Scholar]
- 162.Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, Hurt E. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417(6886):304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- 163.Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005;19(13):1512–1517. doi: 10.1101/gad.1302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hautbergue GM, Hung ML, Golovanov AP, Lian LY, Wilson SA. Mutually exclusive interactions drive handover of mRNA from export adaptors to TAP. Proc Natl Acad Sci USA. 2008;105(13):5154–5159. doi: 10.1073/pnas.0709167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hautbergue GM, Hung ML, Walsh MJ, Snijders AP, Chang CT, Jones R, Ponting CP, Dickman MJ, Wilson SA. UIF, a New mRNA export adaptor that works together with REF/ALY, requires FACT for recruitment to mRNA. Curr Biol. 2009;19(22):1918–1924. doi: 10.1016/j.cub.2009.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Viphakone N, Hautbergue GM, Walsh M, Chang CT, Holland A, Folco EG, Reed R, Wilson SA. TREX exposes the RNA-binding domain of Nxf1 to enable mRNA export. Nat Commun. 2012;3:1006. doi: 10.1038/ncomms2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, Cheng H. Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res. 2013;41(2):1294–1306. doi: 10.1093/nar/gks1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shi M, Zhang H, Wu X, He Z, Wang L, Yin S, Tian B, Li G, Cheng H. ALYREF mainly binds to the 5’ and the 3’ regions of the mRNA in vivo. Nucleic Acids Res. 2017;45(16):9640–9653. doi: 10.1093/nar/gkx597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Izaurralde E, Lewis J, Gamberi C, Jarmolowski A, McGuigan C, Mattaj IW. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376(6542):709–712. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- 170.Cheng H, Dufu K, Lee CS, Hsu JL, Dias A, Reed R. Human mRNA export machinery recruited to the 5’ end of mRNA. Cell. 2006;127(7):1389–1400. doi: 10.1016/j.cell.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 171.Dias SM, Cerione RA, Wilson KF. Unloading RNAs in the cytoplasm: an “importin” task. Nucleus. 2010;1(2):139–143. doi: 10.4161/nucl.1.2.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Maquat LE, Tarn WY, Isken O. The pioneer round of translation: features and functions. Cell. 2010;142(3):368–374. doi: 10.1016/j.cell.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Le Hir H, Izaurralde E, Maquat LE, Moore MJ. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19(24):6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 2004;18(2):210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Proudfoot N. New perspectives on connecting messenger RNA 3’ end formation to transcription. Curr Opin Cell Biol. 2004;16(3):272–278. doi: 10.1016/j.ceb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 176.Fuke H, Ohno M. Role of poly (A) tail as an identity element for mRNA nuclear export. Nucleic Acids Res. 2008;36(3):1037–1049. doi: 10.1093/nar/gkm1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Taniguchi I, Ohno M. ATP-dependent recruitment of export factor Aly/REF onto intronless mRNAs by RNA helicase UAP56. Mol Cell Biol. 2008;28(2):601–608. doi: 10.1128/MCB.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Flaherty SM, Fortes P, Izaurralde E, Mattaj IW, Gilmartin GM. Participation of the nuclear cap binding complex in pre-mRNA 3’ processing. Proc Natl Acad Sci USA. 1997;94(22):11893–11898. doi: 10.1073/pnas.94.22.11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Visa N, Alzhanova-Ericsson AT, Sun X, Kiseleva E, Bjorkroth B, Wurtz T, Daneholt B. A pre-mRNA-binding protein accompanies the RNA from the gene through the nuclear pores and into polysomes. Cell. 1996;84(2):253–264. doi: 10.1016/S0092-8674(00)80980-0. [DOI] [PubMed] [Google Scholar]
- 180.Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj IW. A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J Cell Biol. 1996;133(1):5–14. doi: 10.1083/jcb.133.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Daneholt B. A look at messenger RNP moving through the nuclear pore. Cell. 1997;88(5):585–588. doi: 10.1016/S0092-8674(00)81900-5. [DOI] [PubMed] [Google Scholar]
- 182.Daneholt B. Assembly and transport of a premessenger RNP particle. Proc Natl Acad Sci USA. 2001;98(13):7012–7017. doi: 10.1073/pnas.111145498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Veith R, Sorkalla T, Baumgart E, Anzt J, Haberlein H, Tyagi S, Siebrasse JP, Kubitscheck U. Balbiani ring mRNPs diffuse through and bind to clusters of large intranuclear molecular structures. Biophys J. 2010;99(8):2676–2685. doi: 10.1016/j.bpj.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fribourg S, Braun IC, Izaurralde E, Conti E. Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol Cell. 2001;8(3):645–656. doi: 10.1016/S1097-2765(01)00348-3. [DOI] [PubMed] [Google Scholar]
- 185.Braun IC, Herold A, Rode M, Izaurralde E. Nuclear export of mRNA by TAP/NXF1 requires two nucleoporin-binding sites but not p15. Mol Cell Biol. 2002;22(15):5405–5418. doi: 10.1128/MCB.22.15.5405-5418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grant RP, Hurt E, Neuhaus D, Stewart M. Structure of the C-terminal FG-nucleoporin binding domain of Tap/NXF1. Nat Struct Biol. 2002;9(4):247–251. doi: 10.1038/nsb773. [DOI] [PubMed] [Google Scholar]
- 187.Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci USA. 1985;82(24):8527–8529. doi: 10.1073/pnas.82.24.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Agutter PS. Models for solid-state transport: messenger RNA movement from nucleus to cytoplasm. Cell Biol Int. 1994;18(9):849–858. doi: 10.1006/cbir.1994.1121. [DOI] [PubMed] [Google Scholar]
- 189.Colon-Ramos DA, Salisbury JL, Sanders MA, Shenoy SM, Singer RH, Garcia-Blanco MA. Asymmetric distribution of nuclear pore complexes and the cytoplasmic localization of beta2-tubulin mRNA in Chlamydomonas reinhardtii. Dev Cell. 2003;4(6):941–952. doi: 10.1016/S1534-5807(03)00163-1. [DOI] [PubMed] [Google Scholar]
- 190.Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell. 2004;117(4):427–439. doi: 10.1016/S0092-8674(04)00448-9. [DOI] [PubMed] [Google Scholar]
- 191.Bridger JM, Kalla C, Wodrich H, Weitz S, King JA, Khazaie K, Krausslich HG, Lichter P. Nuclear RNAs confined to a reticular compartment between chromosome territories. Exp Cell Res. 2005;302(2):180–193. doi: 10.1016/j.yexcr.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 192.Vargas DY, Raj A, Marras SA, Kramer FR, Tyagi S. Mechanism of mRNA transport in the nucleus. Proc Natl Acad Sci USA. 2005;102(47):17008–17013. doi: 10.1073/pnas.0505580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Frey S, Gorlich D. FG/FxFG as well as GLFG repeats form a selective permeability barrier with self-healing properties. EMBO J. 2009;28(17):2554–2567. doi: 10.1038/emboj.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164(6):1162–1171. doi: 10.1016/j.cell.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Schmitt C, von Kobbe C, Bachi A, Pante N, Rodrigues JP, Boscheron C, Rigaut G, Wilm M, Seraphin B, Carmo-Fonseca M, Izaurralde E. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 1999;18(15):4332–4347. doi: 10.1093/emboj/18.15.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hodge CA, Colot HV, Stafford P, Cole CN. Rat8p/Dbp5p is a shuttling transport factor that interacts with Rat7p/Nup159p and Gle1p and suppresses the mRNA export defect of xpo1-1 cells. EMBO J. 1999;18(20):5778–5788. doi: 10.1093/emboj/18.20.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lund MK, Guthrie C. The DEAD-box protein Dbp5p is required to dissociate Mex67p from exported mRNPs at the nuclear rim. Mol Cell. 2005;20(4):645–651. doi: 10.1016/j.molcel.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 198.Napetschnig J, Kassube SA, Debler EW, Wong RW, Blobel G, Hoelz A. Structural and functional analysis of the interaction between the nucleoporin Nup214 and the DEAD-box helicase Ddx19. Proc Natl Acad Sci USA. 2009;106(9):3089–3094. doi: 10.1073/pnas.0813267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.von Moeller H, Basquin C, Conti E. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat Struct Mol Biol. 2009;16(3):247–254. doi: 10.1038/nsmb.1561. [DOI] [PubMed] [Google Scholar]
- 200.Folkmann AW, Collier SE, Zhan X, Aditi Ohi MD, Wente SR. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155(3):582–593. doi: 10.1016/j.cell.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Adams RL, Mason AC, Glass L, Aditi Wente SR. Nup42 and IP6 coordinate Gle1 stimulation of Dbp5/DDX19B for mRNA export in yeast and human cells. Traffic. 2017;18(12):776–790. doi: 10.1111/tra.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lin DH, Correia AR, Cai SW, Huber FM, Jette CA, Hoelz A. Structural and functional analysis of mRNA export regulation by the nuclear pore complex. Nat Commun. 2018;9(1):2319. doi: 10.1038/s41467-018-04459-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol. 2006;8(7):668–676. doi: 10.1038/ncb1424. [DOI] [PubMed] [Google Scholar]
- 204.Tran EJ, Zhou Y, Corbett AH, Wente SR. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA: protein remodeling events. Mol Cell. 2007;28(5):850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 205.Siebrasse JP, Kaminski T, Kubitscheck U. Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy. Proc Natl Acad Sci USA. 2012;109(24):9426–9431. doi: 10.1073/pnas.1201781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Smith C, Lari A, Derrer CP, Ouwehand A, Rossouw A, Huisman M, Dange T, Hopman M, Joseph A, Zenklusen D, Weis K, Grunwald D, Montpetit B. In vivo single-particle imaging of nuclear mRNA export in budding yeast demonstrates an essential role for Mex67p. J Cell Biol. 2015;211(6):1121–1130. doi: 10.1083/jcb.201503135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Grunwald D, Singer RH. In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport. Nature. 2010;467(7315):604–607. doi: 10.1038/nature09438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ma J, Liu Z, Michelotti N, Pitchiaya S, Veerapaneni R, Androsavich JR, Walter NG, Yang W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat Commun. 2013;4:2414. doi: 10.1038/ncomms3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chiu SY, Lejeune F, Ranganathan AC, Maquat LE. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev. 2004;18(7):745–754. doi: 10.1101/gad.1170204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Maquat LE, Hwang J, Sato H, Tang Y. CBP80-promoted mRNP rearrangements during the pioneer round of translation, nonsense-mediated mRNA decay, and thereafter. Cold Spring Harb Symp Quant Biol. 2010;75:127–134. doi: 10.1101/sqb.2010.75.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gross T, Siepmann A, Sturm D, Windgassen M, Scarcelli JJ, Seedorf M, Cole CN, Krebber H. The DEAD-box RNA helicase Dbp5 functions in translation termination. Science. 2007;315(5812):646–649. doi: 10.1126/science.1134641. [DOI] [PubMed] [Google Scholar]
- 212.Bolger TA, Folkmann AW, Tran EJ, Wente SR. The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell. 2008;134(4):624–633. doi: 10.1016/j.cell.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gorlich D, Kraft R, Kostka S, Vogel F, Hartmann E, Laskey RA, Mattaj IW, Izaurralde E. Importin provides a link between nuclear protein import and U snRNA export. Cell. 1996;87(1):21–32. doi: 10.1016/S0092-8674(00)81319-7. [DOI] [PubMed] [Google Scholar]
- 214.Sato H, Maquat LE. Remodeling of the pioneer translation initiation complex involves translation and the karyopherin importin beta. Genes Dev. 2009;23(21):2537–2550. doi: 10.1101/gad.1817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dias SM, Wilson KF, Rojas KS, Ambrosio AL, Cerione RA. The molecular basis for the regulation of the cap-binding complex by the importins. Nat Struct Mol Biol. 2009;16(9):930–937. doi: 10.1038/nsmb.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Usuki F, Yamashita A, Kashima I, Higuchi I, Osame M, Ohno S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol Ther. 2006;14(3):351–360. doi: 10.1016/j.ymthe.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 217.Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20(3):355–367. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Isken O, Maquat LE. The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet. 2008;9(9):699–712. doi: 10.1038/nrg2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Woeller CF, Gaspari M, Isken O, Maquat LE. NMD resulting from encephalomyocarditis virus IRES-directed translation initiation seems to be restricted to CBP80/20-bound mRNA. EMBO Rep. 2008;9(5):446–451. doi: 10.1038/embor.2008.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW. Disassembly of exon junction complexes by PYM. Cell. 2009;137(3):536–548. doi: 10.1016/j.cell.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 221.Wilson KF, Fortes P, Singh US, Ohno M, Mattaj IW, Cerione RA. The nuclear cap-binding complex is a novel target of growth factor receptor-coupled signal transduction. J Biol Chem. 1999;274(7):4166–4173. doi: 10.1074/jbc.274.7.4166. [DOI] [PubMed] [Google Scholar]
- 222.Wilson KF, Wu WJ, Cerione RA. Cdc42 stimulates RNA splicing via the S6 kinase and a novel S6 kinase target, the nuclear cap-binding complex. J Biol Chem. 2000;275(48):37307–37310. doi: 10.1074/jbc.C000482200. [DOI] [PubMed] [Google Scholar]
- 223.Ly TK, Wang J, Pereira R, Rojas KS, Peng X, Feng Q, Cerione RA, Wilson KF. Activation of the Ran GTPase is subject to growth factor regulation and can give rise to cellular transformation. J Biol Chem. 2010;285(8):5815–5826. doi: 10.1074/jbc.M109.071886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Brennan CM, Gallouzi IE, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151(1):1–14. doi: 10.1083/jcb.151.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Yang J, Bogerd HP, Wang PJ, Page DC, Cullen BR. Two closely related human nuclear export factors utilize entirely distinct export pathways. Mol Cell. 2001;8(2):397–406. doi: 10.1016/S1097-2765(01)00303-3. [DOI] [PubMed] [Google Scholar]
- 226.Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol. 2006;175(3):415–426. doi: 10.1083/jcb.200607020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Prechtel AT, Chemnitz J, Schirmer S, Ehlers C, Langbein-Detsch I, Stulke J, Dabauvalle MC, Kehlenbach RH, Hauber J. Expression of CD83 is regulated by HuR via a novel cis-active coding region RNA element. J Biol Chem. 2006;281(16):10912–10925. doi: 10.1074/jbc.M510306200. [DOI] [PubMed] [Google Scholar]
- 228.Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, Pinol-Roma S, Borden KL. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J. 2009;28(8):1087–1098. doi: 10.1038/emboj.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ciufo LF, Brown JD. Nuclear export of yeast signal recognition particle lacking Srp54p by the Xpo1p/Crm1p NES-dependent pathway. Curr Biol. 2000;10(20):1256–1264. doi: 10.1016/S0960-9822(00)00743-0. [DOI] [PubMed] [Google Scholar]
- 230.Grosshans H, Deinert K, Hurt E, Simos G. Biogenesis of the signal recognition particle (SRP) involves import of SRP proteins into the nucleolus, assembly with the SRP-RNA, and Xpo1p-mediated export. J Cell Biol. 2001;153(4):745–762. doi: 10.1083/jcb.153.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Costa EA, Subramanian K, Nunnari J, Weissman JS. Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science. 2018;359(6376):689–692. doi: 10.1126/science.aar3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Alavian CN, Politz JC, Lewandowski LB, Powers CM, Pederson T. Nuclear export of signal recognition particle RNA in mammalian cells. Biochem Biophys Res Commun. 2004;313(2):351–355. doi: 10.1016/j.bbrc.2003.11.126. [DOI] [PubMed] [Google Scholar]
- 233.Takeiwa T, Taniguchi I, Ohno M. Exportin-5 mediates nuclear export of SRP RNA in vertebrates. Genes Cells. 2015;20(4):281–291. doi: 10.1111/gtc.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Mahadevan K, Zhang H, Akef A, Cui XA, Gueroussov S, Cenik C, Roth FP, Palazzo AF. RanBP2/Nup358 potentiates the translation of a subset of mRNAs encoding secretory proteins. PLoS Biol. 2013;11(4):e1001545. doi: 10.1371/journal.pbio.1001545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kassube SA, Stuwe T, Lin DH, Antonuk CD, Napetschnig J, Blobel G, Hoelz A. Crystal structure of the N-terminal domain of Nup358/RanBP2. J Mol Biol. 2012;423(5):752–765. doi: 10.1016/j.jmb.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Palazzo AF, Springer M, Shibata Y, Lee CS, Dias AP, Rapoport TA. The signal sequence coding region promotes nuclear export of mRNA. PLoS Biol. 2007;5(12):e322. doi: 10.1371/journal.pbio.0050322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ferreira PA, Nakayama TA, Pak WL, Travis GH. Cyclophilin-related protein RanBP2 acts as chaperone for red/green opsin. Nature. 1996;383(6601):637–640. doi: 10.1038/383637a0. [DOI] [PubMed] [Google Scholar]
- 238.Ferreira PA, Nakayama TA, Travis GH. Interconversion of red opsin isoforms by the cyclophilin-related chaperone protein Ran-binding protein 2. Proc Natl Acad Sci USA. 1997;94(4):1556–1561. doi: 10.1073/pnas.94.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cho KI, Patil H, Senda E, Wang J, Yi H, Qiu S, Yoon D, Yu M, Orry A, Peachey NS, Ferreira PA. Differential loss of prolyl isomerase or chaperone activity of Ran-binding protein 2 (Ranbp2) unveils distinct physiological roles of its cyclophilin domain in proteostasis. J Biol Chem. 2014;289(8):4600–4625. doi: 10.1074/jbc.M113.538215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Cho KI, Orry A, Park SE, Ferreira PA. Targeting the cyclophilin domain of Ran-binding protein 2 (Ranbp2) with novel small molecules to control the proteostasis of STAT3, hnRNPA2B1 and M-Opsin. ACS Chem Neurosci. 2015;6(8):1476–1485. doi: 10.1021/acschemneuro.5b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Cho KI, Yoon D, Qiu S, Danziger Z, Grill WM, Wetsel WC, Ferreira PA. Loss of Ranbp2 in motoneurons causes disruption of nucleocytoplasmic and chemokine signaling, proteostasis of hnRNPH3 and Mmp28, and development of amyotrophic lateral sclerosis-like syndromes. Dis Model Mech. 2017;10(5):559–579. doi: 10.1242/dmm.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Culjkovic-Kraljacic B, Baguet A, Volpon L, Amri A, Borden KL. The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep. 2012;2(2):207–215. doi: 10.1016/j.celrep.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hamada M, Haeger A, Jeganathan KB, van Ree JH, Malureanu L, Walde S, Joseph J, Kehlenbach RH, van Deursen JM. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J Cell Biol. 2011;194(4):597–612. doi: 10.1083/jcb.201102018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hutten S, Kehlenbach RH. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol Cell Biol. 2006;26(18):6772–6785. doi: 10.1128/MCB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Patil H, Saha A, Senda E, Cho KI, Haque M, Yu M, Qiu S, Yoon D, Hao Y, Peachey NS, Ferreira PA. Selective impairment of a subset of Ran-GTP-binding domains of Ran-binding protein 2 (Ranbp2) suffices to recapitulate the degeneration of the retinal pigment epithelium (RPE) triggered by Ranbp2 ablation. J Biol Chem. 2014;298:29767–29789. doi: 10.1074/jbc.M114.586834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wickramasinghe VO, McMurtrie PI, Mills AD, Takei Y, Penrhyn-Lowe S, Amagase Y, Main S, Marr J, Stewart M, Laskey RA. mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol. 2010;20(1):25–31. doi: 10.1016/j.cub.2009.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Jani D, Lutz S, Hurt E, Laskey RA, Stewart M, Wickramasinghe VO. Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res. 2012;40(10):4562–4573. doi: 10.1093/nar/gks059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Singh SK, Maeda K, Eid MM, Almofty SA, Ono M, Pham P, Goodman MF, Sakaguchi N. GANP regulates recruitment of AID to immunoglobulin variable regions by modulating transcription and nucleosome occupancy. Nat Commun. 2013;4:1830. doi: 10.1038/ncomms2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith DH. Stretch growth of integrated axon tracts: extremes and exploitations. Prog Neurobiol. 2009;89(3):231–239. doi: 10.1016/j.pneurobio.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Anden NE, Hfuxe K, Hamberger B, Hokfelt T. A quantitative study on the nigro-neostriatal dopamine neuron system in the rat. Acta Physiol Scand. 1966;67(3):306–312. doi: 10.1111/j.1748-1716.1966.tb03317.x. [DOI] [PubMed] [Google Scholar]
- 251.Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29(2):444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cai Y, Singh BB, Aslanukov A, Zhao H, Ferreira PA. The docking of kinesins, KIF5B and KIF5C, to Ran-binding protein 2 (RanBP2) is mediated via a novel RanBP2 domain. J Biol Chem. 2001;276(45):41594–41602. doi: 10.1074/jbc.M104514200. [DOI] [PubMed] [Google Scholar]
- 253.Cho KI, Yi H, Desai R, Hand AR, Haas AL, Ferreira PA. RANBP2 is an allosteric activator of the conventional kinesin-1 motor protein, KIF5B, in a minimal cell-free system. EMBO Rep. 2009;10(5):480–486. doi: 10.1038/embor.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Patil H, Cho KI, Lee J, Yang Y, Orry A, Ferreira PA. Kinesin-1 and mitochondrial motility control by discrimination of structurally equivalent but distinct subdomains in Ran-GTP-binding domains of Ran-binding protein 2. Open Biol. 2013;3(3):120183. doi: 10.1098/rsob.120183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Mavlyutov TA, Cai Y, Ferreira PA. Identification of RanBP2- and kinesin-mediated transport pathways with restricted neuronal and subcellular localization. Traffic. 2002;3(9):630–640. doi: 10.1034/j.1600-0854.2002.30905.x. [DOI] [PubMed] [Google Scholar]
- 256.D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136(2):284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43(4):513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 258.Diefenbach RJ, Diefenbach E, Douglas MW, Cunningham AL. The ribosome receptor, p180, interacts with kinesin heavy chain, KIF5B. Biochem Biophys Res Commun. 2004;319(3):987–992. doi: 10.1016/j.bbrc.2004.05.069. [DOI] [PubMed] [Google Scholar]
- 259.Ling SC, Fahrner PS, Greenough WT, Gelfand VI. Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc Natl Acad Sci USA. 2004;101(50):17428–17433. doi: 10.1073/pnas.0408114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Jeong JH, Nam YJ, Kim SY, Kim EG, Jeong J, Kim HK. The transport of Staufen2-containing ribonucleoprotein complexes involves kinesin motor protein and is modulated by mitogen-activated protein kinase pathway. J Neurochem. 2007;102(6):2073–2084. doi: 10.1111/j.1471-4159.2007.04697.x. [DOI] [PubMed] [Google Scholar]
- 261.Palacios IM, Gatfield D, St Johnston D, Izaurralde E. An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature. 2004;427(6976):753–757. doi: 10.1038/nature02351. [DOI] [PubMed] [Google Scholar]
- 262.Shibuya T, Tange TO, Sonenberg N, Moore MJ. eIF4AIII binds spliced mRNA in the exon junction complex and is essential for nonsense-mediated decay. Nat Struct Mol Biol. 2004;11(4):346–351. doi: 10.1038/nsmb750. [DOI] [PubMed] [Google Scholar]
- 263.Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell. 2007;130(1):179–191. doi: 10.1016/j.cell.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 264.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40(6):1095–1104. doi: 10.1016/S0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- 265.Perry RB, Doron-Mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, Twiss JL, Yaron A, Fainzilber M. Subcellular knockout of importin beta1 perturbs axonal retrograde signaling. Neuron. 2012;75(2):294–305. doi: 10.1016/j.neuron.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben-Yaakov K, Hieda M, Yoneda Y, Twiss JL, Fainzilber M. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron. 2008;59(2):241–252. doi: 10.1016/j.neuron.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, Blesch A, Pilpel Y, Twiss JL, Fainzilber M. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012;31(6):1350–1363. doi: 10.1038/emboj.2011.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Twiss JL, Fainzilber M. Ribosomes in axons–scrounging from the neighbors? Trends Cell Biol. 2009;19(5):236–243. doi: 10.1016/j.tcb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 269.Colak D, Ji SJ, Porse BT, Jaffrey SR. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell. 2013;153(6):1252–1265. doi: 10.1016/j.cell.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, Holt CE. Dynamic axonal translation in developing and mature visual circuits. Cell. 2016;166(1):181–192. doi: 10.1016/j.cell.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wong HH, Lin JQ, Strohl F, Roque CG, Cioni JM, Cagnetta R, Turner-Bridger B, Laine RF, Harris WA, Kaminski CF, Holt CE. RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron. 2017;95(4):852 e858–868 e858. doi: 10.1016/j.neuron.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Bellon A, Iyer A, Bridi S, Lee FCY, Ovando-Vazquez C, Corradi E, Longhi S, Roccuzzo M, Strohbuecker S, Naik S, Sarkies P, Miska E, Abreu-Goodger C, Holt CE, Baudet ML. miR-182 Regulates Slit2-mediated axon guidance by modulating the local translation of a specific mRNA. Cell Rep. 2017;18(5):1171–1186. doi: 10.1016/j.celrep.2016.12.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cho KI, Yi H, Yeh A, Tserentsoodol N, Cuadrado L, Searle K, Hao Y, Ferreira PA. Haploinsufficiency of RanBP2 is neuroprotective against light-elicited and age-dependent degeneration of photoreceptor neurons. Cell Death Differ. 2009;16(2):287–297. doi: 10.1038/cdd.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Cho KI, Yi H, Tserentsoodol N, Searle K, Ferreira PA. Neuroprotection resulting from insufficiency of RANBP2 is associated with the modulation of protein and lipid homeostasis of functionally diverse but linked pathways in response to oxidative stress. Dis Model Mech. 2010;3(9–10):595–604. doi: 10.1242/dmm.004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Jiang K, Wright KL, Zhu P, Szego MJ, Bramall AN, Hauswirth WW, Li Q, Egan SE, McInnes RR. STAT3 promotes survival of mutant photoreceptors in inherited photoreceptor degeneration models. Proc Natl Acad Sci USA. 2014;111(52):E5716–E5723. doi: 10.1073/pnas.1411248112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Da Cruz S, Bui A, Saberi S, Lee SK, Stauffer J, McAlonis-Downes M, Schulte D, Pizzo DP, Parone PA, Cleveland DW, Ravits J. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017;134(1):97–111. doi: 10.1007/s00401-017-1688-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18(9):1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A, French C, Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron D TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 282.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276(39):36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- 284.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ayala YM, Pagani F, Baralle FE. TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett. 2006;580(5):1339–1344. doi: 10.1016/j.febslet.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 286.Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(Pt 22):3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 287.Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283(19):13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 289.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18(18):3353–3364. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- 291.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30(2):277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, Moore MJ, Yu G. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286(2):1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Deshaies JE, Shkreta L, Moszczynski AJ, Sidibe H, Semmler S, Fouillen A, Bennett ER, Bekenstein U, Destroismaisons L, Toutant J, Delmotte Q, Volkening K, Stabile S, Aulas A, Khalfallah Y, Soreq H, Nanci A, Strong MJ, Chabot B, Vande Velde C. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain. 2018;141(5):1320–1333. doi: 10.1093/brain/awy062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21(16):3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 299.Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobagyi T, Shaw CE, Rogelj B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133(Pt 6):1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- 300.Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, Miguez R, Dayton RD, Tamir S, Klein RL, Barmada SJ. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8(1):4606. doi: 10.1038/s41598-018-22858-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30(2):639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Miguel L, Frebourg T, Campion D, Lecourtois M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol Dis. 2011;41(2):398–406. doi: 10.1016/j.nbd.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 303.Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep. 2018;8(1):7084. doi: 10.1038/s41598-018-25007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Pinarbasi ES, Cagatay T, Fung HYJ, Li YC, Chook YM, Thomas PJ. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 2018;8(1):7083. doi: 10.1038/s41598-018-25008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang YJ, Petrucelli L, Sattler R, Zarnescu DC, Glass JD, Rossoll W. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–239. doi: 10.1038/s41593-017-0047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 308.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kapeli K, Pratt GA, Vu AQ, Hutt KR, Martinez FJ, Sundararaman B, Batra R, Freese P, Lambert NJ, Huelga SC, Chun SJ, Liang TY, Chang J, Donohue JP, Shiue L, Zhang J, Zhu H, Cambi F, Kasarskis E, Hoon S, Ares M, Jr, Burge CB, Ravits J, Rigo F, Yeo GW. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat Commun. 2016;7:12143. doi: 10.1038/ncomms12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci. 2005;118(Pt 24):5755–5765. doi: 10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- 311.Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15(6):587–593. doi: 10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 312.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Michael WM, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83(3):415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 314.Siomi H, Dreyfuss G. A nuclear localization domain in the hnRNP A1 protein. J Cell Biol. 1995;129(3):551–560. doi: 10.1083/jcb.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G, Fischer U. A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol. 1997;137(1):27–35. doi: 10.1083/jcb.137.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Pollard VW, Michael WM, Nakielny S, Siomi MC, Wang F, Dreyfuss G. A novel receptor-mediated nuclear protein import pathway. Cell. 1996;86(6):985–994. doi: 10.1016/S0092-8674(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 317.Munro TP, Magee RJ, Kidd GJ, Carson JH, Barbarese E, Smith LM, Smith R. Mutational analysis of a heterogeneous nuclear ribonucleoprotein A2 response element for RNA trafficking. J Biol Chem. 1999;274(48):34389–34395. doi: 10.1074/jbc.274.48.34389. [DOI] [PubMed] [Google Scholar]
- 318.Hoek KS, Kidd GJ, Carson JH, Smith R. hnRNP A2 selectively binds the cytoplasmic transport sequence of myelin basic protein mRNA. Biochemistry (Mosc) 1998;37(19):7021–7029. doi: 10.1021/bi9800247. [DOI] [PubMed] [Google Scholar]
- 319.Bekenstein U, Soreq H. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: from structural insights to post-transcriptional regulatory roles. Mol Cell Neurosci. 2013;56:436–446. doi: 10.1016/j.mcn.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 320.Gao Y, Tatavarty V, Korza G, Levin MK, Carson JH. Multiplexed dendritic targeting of alpha calcium calmodulin-dependent protein kinase II, neurogranin, and activity-regulated cytoskeleton-associated protein RNAs by the A2 pathway. Mol Biol Cell. 2008;19(5):2311–2327. doi: 10.1091/mbc.E07-09-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Leal G, Afonso PM, Duarte CB. Neuronal activity induces synaptic delivery of hnRNP A2/B1 by a BDNF-dependent mechanism in cultured hippocampal neurons. PLoS One. 2014;9(10):e108175. doi: 10.1371/journal.pone.0108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K, Gelboin-Burkhart C, Fijany L, Wang HC, Nussbacher JK, Broski SM, Kim HJ, Lardelli R, Sundararaman B, Donohue JP, Javaherian A, Lykke-Andersen J, Finkbeiner S, Bennett CF, Ares M, Jr, Burge CB, Taylor JP, Rigo F, Yeo GW. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron. 2016;92(4):780–795. doi: 10.1016/j.neuron.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, Sanchez-Madrid F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Nousiainen HO, Kestila M, Pakkasjarvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40(2):155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, Szuto A, Levert A, Vidal S, Andre-Guimont C, Camu W, Bouchard JP, Dupre N, Rouleau GA, Wente SR, Dion PA. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24(5):1363–1373. doi: 10.1093/hmg/ddu545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93(7):1147–1158. doi: 10.1016/S0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- 329.Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17(4):2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cho KI, Cai Y, Yi H, Yeh A, Aslanukov A, Ferreira PA. Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic. 2007;8:1722–1735. doi: 10.1111/j.1600-0854.2007.00647.x. [DOI] [PubMed] [Google Scholar]
- 331.Patil H, Yoon D, Bhowmick R, Cai Y, Cho KI, Ferreira PA. Impairments in age-dependent ubiquitin proteostasis and structural integrity of selective neurons by uncoupling Ran GTPase from the Ran-binding domain 3 of Ranbp2 and identification of novel mitochondrial isoforms of ubiquitin-conjugating enzyme E2I (ubc9) and Ranbp2. Small GTPases. 2019;10(2):146–161. doi: 10.1080/21541248.2017.1356432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Ohgomori T, Yamasaki R, Takeuchi H, Kadomatsu K, Kira JI, Jinno S. Differential activation of neuronal and glial STAT3 in the spinal cord of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2017;46(4):2001–2014. doi: 10.1111/ejn.13650. [DOI] [PubMed] [Google Scholar]
- 333.Bonnin E, Cabochette P, Filosa A, Juhlen R, Komatsuzaki S, Hezwani M, Dickmanns A, Martinelli V, Vermeersch M, Supply L, Martins N, Pirenne L, Ravenscroft G, Lombard M, Port S, Spillner C, Janssens S, Roets E, Van Dorpe J, Lammens M, Kehlenbach RH, Ficner R, Laing NG, Hoffmann K, Vanhollebeke B, Fahrenkrog B. Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet. 2018;14(12):e1007845. doi: 10.1371/journal.pgen.1007845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75(5):822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Chen HJ, Anagnostou G, Chai A, Withers J, Morris A, Adhikaree J, Pennetta G, de Belleroche JS. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem. 2010;285(51):40266–40281. doi: 10.1074/jbc.M110.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 337.Del Bo R, Tiloca C, Pensato V, Corrado L, Ratti A, Ticozzi N, Corti S, Castellotti B, Mazzini L, Soraru G, Cereda C, D’Alfonso S, Gellera C, Comi GP, Silani V, Consortium S Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1239–1243. doi: 10.1136/jnnp.2011.242313. [DOI] [PubMed] [Google Scholar]
- 338.Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu ZS, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown RH, Jr, Landers JE. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Yang C, Danielson EW, Qiao T, Metterville J, Brown RH, Jr, Landers JE, Xu Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc Natl Acad Sci USA. 2016;113(41):E6209–E6218. doi: 10.1073/pnas.1605964113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Tran D, Chalhoub A, Schooley A, Zhang W, Ngsee JK. A mutation in VAPB that causes amyotrophic lateral sclerosis also causes a nuclear envelope defect. J Cell Sci. 2012;125(Pt 12):2831–2836. doi: 10.1242/jcs.102111. [DOI] [PubMed] [Google Scholar]
- 341.Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Heon E, Krupin T, Ritch R, Kreutzer D, Crick RP, Sarfarazi M. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 342.De Marco N, Buono M, Troise F, Diez-Roux G. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem. 2006;281(23):16147–16156. doi: 10.1074/jbc.M601467200. [DOI] [PubMed] [Google Scholar]
- 343.Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014;34(24):8083–8097. doi: 10.1523/JNEUROSCI.0543-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Stuven T, Hartmann E, Gorlich D. Exportin 6: a novel nuclear export receptor that is specific for profilin–actin complexes. EMBO J. 2003;22(21):5928–5940. doi: 10.1093/emboj/cdg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Bonner WM. Protein migration into nuclei. I. Frog oocyte nuclei in vivo accumulate microinjected histones, allow entry to small proteins, and exclude large proteins. J Cell Biol. 1975;64(2):421–430. doi: 10.1083/jcb.64.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Feng W, Benko AL, Lee JH, Stanford DR, Hopper AK. Antagonistic effects of NES and NLS motifs determine S. cerevisiae Rna1p subcellular distribution. J Cell Sci. 1999;112(Pt 3):339–347. doi: 10.1242/jcs.112.3.339. [DOI] [PubMed] [Google Scholar]
- 347.Plafker K, Macara IG. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol Cell Biol. 2000;20(10):3510–3521. doi: 10.1128/MCB.20.10.3510-3521.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, Chook YM, Kuhlmann T, Kidd GJ, Shacham S, Casaccia P. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci. 2015;18(4):511–520. doi: 10.1038/nn.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Kosyna FK, Depping R. Controlling the gatekeeper: therapeutic targeting of nuclear transport. Cells. 2018 doi: 10.3390/cells7110221. [DOI] [PMC free article] [PubMed] [Google Scholar]