

Abstract
Human cryptosporidiosis is caused primarily by two species of apicomplexan parasites, Cryptosporidium parvum and C. hominis. Although infection of cell monolayers with sporozoites does not support the complete parasite life cycle, the in vitro system is used to study the asexual phase of multiplication, which consists of two generations of merogony. To better understand host-parasite interaction and to gain insight into gene regulatory processes driving the complex life cycle of Cryptosporidium parasites, we analyzed the transcriptome of C. parvum in oocysts, sporozoites and infected cell monolayers 2–48 h post-infection. Analysis of RNA-Seq data from replicate oocyst, sporozoite and intracellular samples revealed significant differences between transcriptomes expressed outside and inside the host cell. Compared to the transcriptome found in the host cell, the oocyst transcriptome is less diverse. Biological processes significantly over-represented intracellularly relate to biosynthetic processes. Genes significantly overexpressed in oocysts show evidence of specialized functions not found in other Apicomplexa. A more comprehensive view of gene regulation during the Cryptosporidium life cycle will require the analysis of later time points during the infection, particularly of the poorly studied sexual phase of the life cycle.
Subject terms: Pathogens, Molecular biology
Introduction
Cryptosporidiosis is recognized as one of the most common enteric infections in infants in sub-Saharan Africa and southeast Asia1. The rapid asexual multiplication of the parasite in the intestinal epithelium compromises intestinal function, which can lead to severe diarrhea and have long-term consequences2. There are no effective drugs to control cryptosporidiosis. Drug screening is made difficult by the lack of robust culture methods supporting the entire life cycle.
Commonly used methods to culture Cryptosporidium parasites3 do not support completion of the life cycle, possibly due to inefficient differentiation of gametes and/or deficient fertilization. Oocysts excreted in the feces of naturally or experimentally infected animals can be used to infect cultured epithelial cells. The oocysts release sporozoites which are capable of invading host cells in the intestinal epithelium or in culture. Following invasion, sporozoites transform into trophozoites and divide asexually to generate first-and second-generation meronts in a process known as merogony. Later stages, specifically the sexual phase of the life cycle, do not appear to differentiate consistently in conventional cell monolayers. Progress towards a more stable culture in biphasic and three-dimensional culture systems has been reported4–7.
Compared to better studied Apicomplexa, gene regulation during the Cryptosporidium life cycle has rarely been studied8,9. Reflecting the many technical obstacles to research on these parasites, published Cryptosporidium transcriptome analyses based on RNA-Seq are limited to one study of C. parvum extra- and intracellular gene expression in calf intestinal epithelium and in culture9 and an analysis of the C. parvum life cycle transcriptome in organoids grown from small intestine and lung epithelial cells7. With the goal of improving our understanding of gene regulation in Cryptosporidium parasites, we undertook an RNA-Seq analysis of the Cryptosporidium transcriptome at the oocyst, excysted sporozoite, and intracellular stages. To ensure that the transcriptome features identified in our analysis are of general validity, as opposed to being restricted to a particular cell line, parasite isolate, or experimental condition, we included sequence data originating from infections of different cell lines and different C. parvum isolates. In addition, sporozoite RNA-Seq data generated by Lippuner et al.9 were downloaded from the National Center for Biotechnology Information’s Sequence Read Archive and combined with sequence data generated as part of the present study. We identified functional categories overrepresented in the oocyst transcriptome and found these to be indicative of the specialized functions, like long-term survival and delivery of sporozoites into specific GI tract organs. In contrast, transcriptomic data of intracellular parasite stages indicate that the intracellular transcriptome is tailored for transcription and translation, consistent with rapid asexual multiplication during the initial phase of the infection.
To examine whether sporozoite gene expression responds to environmental stimuli, transcriptomic data were acquired from sporozoites incubated under different conditions. Compared to the difference between intra- and extracellular C. parvum transcriptome, differences in gene expression between oocysts and sporozoites were relatively small.
Methods
Parasites
Figure S1 shows a flowchart of the experimental procedures. Oocyst from three C. parvum isolates were used. First, fecal samples from diarrheic newborn calves raised on a farm in Woodstock, Connecticut were screened for the presence of Cryptosporidium oocysts and one sample with a high number of oocysts (3 × 107 oocysts/ml) was selected. Oocysts from this unnamed isolate were used to infect four replicate monolayers of IPEC-J2 cell monolayers as described10. These monolayers are designated 1M-4M (Table 1). Second, oocysts from isolate TU11411 were used to infect four replicate monolayers of MDBK cells (ATCC CCL-22). TU114 originates from a human infection in Uganda. It has been maintained by serial propagation in immunosuppressed mice since 2003. Third, oocysts of C. parvum isolate MD12 were used to infect a second set of MDBK cell monolayers. MD was originally isolated from a deer and has been maintained by serial propagation in immunosuppressed mice for an unknown number of years. Mouse propagation of C. parvum was approved by and performed in accordance with Tufts University Institutional Animal Care and Use Committee.
Table 1.
Mapping statistics for 37 RNA-Seq datasets.
| ID* | Stage | Sample | Sequences analyzed | Host** | Cryptosporidium parvum | ||
|---|---|---|---|---|---|---|---|
| Mapped sequences | Mapped % | Mapped sequences | Mapped % | ||||
| 15 | Extracellular | Oocysts | 7,437,619 | 9,636 | 0.13 | 6,249,367 | 84.02 |
| 16M | Extracellular | Oocysts | 12,000,000 | 5,449 | 0.05 | 10,503,020 | 87.53 |
| 1 | Extracellular | Sporozoites_2h | 11,525,085 | 5,535 | 0.05 | 10,648,804 | 92.40 |
| 2 | Extracellular | Sporozoites_2h | 9,550,531 | 11,742 | 0.12 | 8,874,905 | 92.93 |
| 3 | Extracellular | Sporozoites_2h | 8,781,223 | 5,287 | 0.06 | 8,332,943 | 94.90 |
| 4 | Extracellular | Sporozoites_2h | 9,791,953 | 7,717 | 0.08 | 9,081,237 | 92.74 |
| 5 | Extracellular | Sporozoites_2h | 8,925,764 | 4,498 | 0.05 | 8,571,776 | 96.03 |
| 6 | Extracellular | Sporozoites_2h | 9,842,080 | 8,410 | 0.09 | 9,023,619 | 91.68 |
| 7 | Extracellular | Sporozoites_0h | 10,346,897 | 2,939 | 0.03 | 9,986,685 | 96.52 |
| 8 | Extracellular | Sporozoites_0h | 7,051,754 | 4,420 | 0.06 | 6,675,995 | 94.67 |
| 9 | Extracellular | Sporozoites_2h | 8,665,564 | 3,466 | 0.04 | 7,969,618 | 91.97 |
| 10 | Extracellular | Sporozoites_2h | 9,217,265 | 10,130 | 0.11 | 8,489,030 | 92.10 |
| 11 | Extracellular | Sporozoites_0h | 10,534,463 | 5,391 | 0.05 | 10,049,403 | 95.40 |
| 12 | Extracellular | Sporozoites_0h | 9,880,483 | 7,078 | 0.07 | 9,417,974 | 95.32 |
| 13 | Extracellular | Sporozoites_2h | 12,067,102 | 5,770 | 0.05 | 11,115,287 | 92.11 |
| 14 | Extracellular | Sporozoites_2h | 8,314,644 | 11,277 | 0.14 | 7,974,108 | 95.90 |
| 1H | Extracellular | Sporozoites_0h | 3,000,000 | 4,074 | 0.14 | 769,974 | 25.70 |
| 2H | Extracellular | Sporozoites_0h | 3,000,000 | 3,696 | 0.12 | 862,410 | 28.70 |
| 23 | Intracellular | infected_MDBK_2h | 17,252,177 | 15,680,504 | 90.89 | 68,676 | 0.39 |
| 24 | Intracellular | infected_MDBK_2h | 19,435,693 | 17,610,681 | 90.61 | 28,243 | 0.12 |
| 25 | Intracellular | infected_MDBK_2h | 19,063,096 | 17,286,415 | 90.68 | 81,325 | 0.43 |
| 26 | Intracellular | infected_MDBK_2h | 19,998,766 | 18,152,880 | 90.77 | 43,074 | 0.22 |
| 28 | Intracellular | infected_MDBK_24h | 20,257,992 | 17,632,556 | 87.04 | 64,235 | 0.32 |
| 29 | Intracellular | infected_MDBK_24h | 21,596,479 | 19,417,394 | 89.91 | 77,453 | 0.36 |
| 30 | Intracellular | infected_MDBK_24h | 18,271,010 | 16,109,550 | 88.17 | 69,870 | 0.38 |
| 31 | Intracellular | infected_MDBK_24h | 17,616,476 | 15,074,419 | 85.57 | 76,016 | 0.43 |
| 1M | Intracellular | infected_IPEC_24h | 7,000,000 | 5,253,821 | 75.10 | 136,862 | 1.96 |
| 2M | Intracellular | infected_IPEC_24h | 47,904,752 | 36,263,897 | 75.70 | 851,774 | 1.78 |
| 3M | Intracellular | infected_IPEC_24h | 7,000,000 | 5,245,701 | 74.93 | 114,070 | 1.63 |
| 4M | Intracellular | infected_IPEC_24h | 7,000,000 | 5,276,146 | 75.37 | 115,771 | 1.65 |
| 38 | Intracellular | infected_MDBK_48h | 16,834,760 | 15,040,175 | 89.34 | 272,638 | 1.62 |
| 39 | Intracellular | infected_MDBK_48h | 16,504,049 | 14,734,815 | 89.28 | 209,349 | 1.27 |
| 40 | Intracellular | infected_MDBK_48h | 15,947,732 | 14,199,861 | 89.04 | 191,336 | 1.20 |
| 41 | Intracellular | infected_MDBK_48h | 18,517,372 | 16,552,679 | 89.39 | 202,993 | 1.10 |
| 42 | Intracellular | infected_MDBK_48h | 20,299,009 | 16,630,978 | 81.93 | 348,552 | 1.72 |
| 18 | Control | uninfected_MDBK | 7,000,000 | 6,258,700 | 89.41 | 17,940 | 0.26 |
| 19M | Control | uninfected IPEC-J2 | 7,000,000 | 5,628,000 | 80.40 | 13,082 | 0.19 |
*Sequences of sample IDs designated with “M” are deposited under accession number PRJEB17685 (Mirhashemi et al., 2018); sequences of samples indicated with “H” are from (Lippuner et al., 2018) (NCBI Sequence Read Archive run # SRR3137248 and SRR3137593). The remaining sequences are deposited under accession numbers PRJEB25665 and PRJEB28268.
**Host: Sporozoites and oocyst sample 15 were mapped to Mus musculus; oocyst sample 16 M, and samples from MDBK cells were mapped to the genome of Bos taurus; sequence reads from IPEC-J2 were mapped to the Sus scrofa genome.
Oocysts were purified from mouse or calf feces on step gradients of 15%/25% (w/v) Nycodenz (Alere Technologies, Oslo, Norway) as described13. To obtain sporozoites, purified oocysts were surface-sterilized with 10% commercial bleach (0.5% sodium hypochlorite) for 10 min on ice. Bleach was removed by precipitating the oocysts and resuspending them in sterile PBS (Fig. S2). Magnetic beads (Dynabeads Protein G, Invitrogen) were reacted with monoclonal antibody 5F10 (a gift from Abhineet Sheoran, Cummings School of Veterinary Medicine). Bead-antibody conjugate was mixed with the oocyst suspension and the suspension incubated in 0.75% taurocholic acid to induce oocyst excystation. Unexcysted oocysts and empty oocyst walls were separated from the sporozoites by capturing the beads and attached oocysts and oocyst walls in a magnetic stand. The unbound sporozoites left in suspension were recovered by centrifugation.
RNA was extracted from sporozoites either immediately after purification, designated in Table 1 as “sporozoite_0h” or after a 2-h incubation at 37 °C/5% CO2 in RPMI-1640 medium supplemented with 10% FBS and 10 μM N-acetylgalactosamine (GalNAc) (Table 1; sporozoites_2h). This treatment was intended to assess whether the effect GalNAc has on sporozoite morphology14 is reflected in the transcriptome.
Animal experiments were performed in accordance with protocols approved by the Tufts University Institutional Animal Care and Use Committee.
Infection of cell monolayers
Monolayers of MDBK cells (Madin Darby Bovine Kidney, ATCC CCL-2215) were infected with C. parvum as described previously16–18. Monolayers of pig epithelial cells (IPEC-J2)19 were grown and infected according to previously described procedures10. MDBK cells were grown to near-confluence in 25 cm2 flasks in Dulbecco’s Modification of Eagle’s Medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum, 1% L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. Confluent or near-confluent cell monolayers were infected with surface-sterilized C. parvum oocysts at a dose of approximately 1 oocyst/cell, equivalent to 1.3 × 105 oocysts per cm2 monolayer or a corresponding dose of 4 sporozoites per cell, taking into consideration that each oocyst contains 4 sporozoites. Following infection, cultures were incubated at 37 °C/5% CO2. Infected cultures were incubated for 2 h, 24 h or 48 h. The experiments were replicated as follows: 4 cultures of MDBK cells incubated for 2 h; 4 cultures of MDBK cells incubated for 24 h, and 5 cultures of MDBK cells incubated for 48 h. RNA-Seq data from 4 infected IPEC-J2 cultures harvested 24 h post-infection10 were also included. Following incubation for 2 h, 24 h or 48 h, monolayers were washed twice with PBS to remove extracellular sporozoites and oocysts. A sample of cells was processed for immunofluorescence20 to confirm the infection. Intracellular development stages were visualized with monoclonal antibody 2E518. For RNA extraction, cells were released from the substrate with a brief incubation in 2 ml Accutase (Millipore Sigma, Burlington, Massachusetts) per 25-cm2 surface.
Molecular biology
RNA was extracted from cells in a Qiacube instrument using an RNeasy kit (Qiagen, Hilden, Germany). RNA from 4 uninfected monolayers of MDBK cells and from uninfected IPEC-J2 cells was extracted in parallel. Prior to extraction, the cells were homogenized in a Minibeadbeater in the presence of 0.5 mm diameter zirconia beads. Extraction of RNA from sporozoites and oocysts was initiated with the same disruption procedure. Guanidine thiocyanate buffer RLT supplemented with 1% β-mercaptoethanol was added to the lysate and the samples transferred to QIAshredder spin columns. RNA was extracted from the filtrate following Qiagen’s RNeasy procedure.
The quality of the RNA was initially assessed by determining the RNA Integrity Number (RIN) using an Agilent Bioanalyzer 2100. An Illumina TruSeq Stranded RNA library kit was used to make strand-specific cDNA libraries from polyA selected RNA from a total of 33 oocyst, sporozoite and infected cell samples plus 4 samples each of uninfected MDBK and IPEC-J2 cells. cDNA libraries were subjected to cluster generation and single-end 100-nucleotide sequencing on an Illumina Hi-Seq 2500 at the Tufts Genomics core facility (tucf.org).
Bioinformatics and statistical analysis
The C. parvum IOWA reference genome and annotation file were downloaded from Cryptosporidium genomics resource cryptodb.org21, release 34, in FASTA and GFF3 format, respectively. The pig (Sus scrofa) references genome (susScr3) and annotation file were downloaded from support.illumina.com/sequencing/sequencing_software/igenome.html. The Bos taurus bosTau8 and Mus musculus mm10 reference genomes provided in the public galaxy instance at usegalaxy.org were used as reference genomes for samples recovered from calf and mouse feces, respectively. To increase the diversity of the samples included in the analysis, RNA-Seq data from two samples of excysted oocysts of C. parvum isolate IPZ:CH-Crypto_K67699 were downloaded from NCBI’s sequence read archive (run # SRR3137248 and SRR3137593). The 75-nt reads were downloaded and randomly subsampled to 3 × 106 reads using program sub.sample in mothur22. Sequence reads in FASTQ format were aligned to the corresponding host genome and to the C. parvum genome using program subread-align23. Alternatively, sequences were mapped in Galaxy24 using HiSat225. Genome-wide values of Fragments Per Kilobase of transcript per Million mapped reads (FPKM) were computed using Cufflinks26 or StringTie27 in Galaxy.
Global differences between FPKM profiles were visualized using Principal Component Analysis (PCA) as implemented in CANOCO28. PCA was applied to a matrix of 35 samples x 3888 C. parvum genes, where each fields of the table contained the FPKM for the corresponding sample and gene. Values were set to 0 if no sequence mapped to a gene. For certain analyses, FPKM values were normalized by gene by converting them to z scores, i.e., subtracting the mean and dividing by the standard deviation. To test for an association between life cycle stage and gene expression, the FPKM data were analyzed using Redundancy Analysis (RDA)28. In this analysis, genes were considered dependent variables, whereas the developmental stage (extracellular or intracellular) represented the independent categorical variable. The percent fit (FitE29) of FPKMs across 35 samples with life cycle stage was calculated using RDA. Shannon diversity was calculated in excel as -Σ pi * ln(pi), where pi is the proportion of gene i FPKM and the sum is over all genes30. Genes differentially expressed in different developmental stages were identified with Linear Discriminant Analysis (LDA) as implemented in program LEfSe31. The Galaxy interface of the program at http://huttenhower.sph.harvard.edu/galaxy/ was used for this analysis.
For rank-abundance analysis, genes were ranked in order of diminishing FPKM. The most abundant gene was assigned rank 1 and the least abundant the highest rank for a particular sample. Because the number of genes detected in RNA-Seq data varied by sample (range: 805–3706), gene ranks were normalized to 100, such that the least abundant gene was at rank 100. Normalized ranks were then calculated by dividing each rank by the total number of genes detected in a sample and multiplying the ratio by 100.
Groups of orthologous proteins were identified using the OrthoMCL database. The database is part of the Eukaryotic Pathogens bioinformatics resource center32.
Results
Gene expression in oocyst and intracellular stages
Sequence volume and statistics of sequences mapping to the C. parvum and to the respective host genome for 37 samples is shown in Table 1. As quality control, oocyst and sporozoite sequence reads were also mapped to the genome of the respective host species (mouse, pig or cow). Similarly, sequence reads from uninfected IPEC-J2 and MDBK cells were mapped to the C. parvum genome. As expected, the proportion of mapped reads in these controls was low, ranging from 0.03% to 0.26%.
PCA was used to visually compare oocyst, sporozoite and intracellular transcriptome (Fig. 1). The intracellular transcriptome was analyzed at 2 h, 24 h and 48 h. The time points were chosen based on the ability of cultured cell monolayers to support growth of C. parvum. PCA was applied to raw FPKM values (Table S1) and to FPKM values normalized by gene. The analysis was duplicated because of the large difference in FPKM values between highly expressed genes and genes expressed at low level; in the analysis based on the raw data, genes with high FPKM are most influential, whereas FPKM normalization by gene gives each gene equal weight. Consistent with differential gene expression during the parasite’s life cycle, PCA clearly discriminated between extracellular parasite stages and intracellular stages, regardless whether FPKM data were normalized or not. The effect of life cycle stage is highly significant based on a randomization of samples with respect to life cycle stages in RDA (pseudo-F = 17.6, p = 0.001). Also apparent in the PCA is the relative similarity between oocyst and sporozoite transcriptome, when compared to the dissimilarity between intracellular and extracellular stages. Incubation of sporozoites post-excystation for 2 h had no apparent effect on the transcriptome profile, regardless whether sporozoites were incubated in PBS or in supplemented culture medium. Significantly, compared to the distance between extracellular and intracellular transcriptome, imported sporozoite RNA-Seq data (Table 1, samples 1 H, 2 H9) were similar to those obtained in this study. Whereas extracellular stages form a relatively compact cluster, FPKM profiles of intracellular stages were found to be more heterogeneous. The heterogeneity among intracellular transcriptomes is consistent with the fact that intracellular development was allowed to progress for 2 h, 24 h or 48 h. The distance between intracellular transcriptomes likely reflects differential gene expression during intracellular development, as implied by the clustering of the 2-h samples. This observation is consistent with previously reported differential gene regulation during merogony33.
Figure 1.

Principal Component Analysis of 35 Cryptosporidium parvum transcriptomes. The analysis is based on raw FPKM values for 3885 annotated C. parvum genes (left) and FPKM values normalized by gene (right). Included in the analysis are two oocyst samples (red circles), 16 sporozoite samples incubated in medium (brown) or PBS (turquoise) for 0 (triangle up) or 2 h (triangle down) as indicated in the key. Infected cells (hexagons) were analyzed at 2 h, 24 h and 48 h post-infection, as indicated by light, medium and dark green symbols, respectively. Crossed triangles indicate samples 1 H and 2 H (Table 1).
We examined whether oocyst excystation is associated with transcriptome remodeling. To visualize differences between oocyst and sporozoite transcriptomes, PCA was applied to the 18 extracellular samples (2 oocyst and 16 sporozoite samples). Excluding the intracellular samples led to a “decompression” of the 18 extracellular samples on PCA plots, enabling a better visualization of sporozoite-oocysts transcriptome differences (Fig. S3). This analysis revealed that the 16 sporozoite samples generated in this laboratory are similar, regardless of the period and condition in which the sporozoites were incubated post-excystation. The two imported sporozoite transcriptomes generated by Lippuner et al. were more distinct, as were the two oocyst transcriptomes. A permutation test showed that in spite of the relatively large difference between the two oocyst samples, sporozoite and oocyst transcriptomes are significantly different (pseudo-F = 3.5; p = 0.02). Together with the PCA of the entire dataset, this analysis indicates that, although extra- and intracellular transcriptomes are clearly distinct, the process of excystation has a detectable impact on the transcriptome. The possible influence of different laboratory procedures used to generate Lippuner’s and our RNA-Seq data on FPKM values revealed in the PCA is discussed below.
Rank abundance plots were used to visualize differences in the diversity (evenness) of the transcriptome across different stages. This analysis showed that gene expression in intracellular parasites was more even (Fig. 2). As in PCA, oocyst and sporozoite transcriptomes were very similar with respect to evenness. Transcriptome diversity was also estimated using the Shannon diversity index. Based on this metric, the intracellular C. parvum transcriptome at 2 h post-infection was the least diverse (mean = 4.19; SD = 0.52, n = 4) (Fig. 3). In contrast, the most diverse C. parvum transcriptome was found in intracellular parasites 48 h post-infection (mean = 6.56; SD = 0.35; n = 5). A Kruskal-Wallis ANOVA on Ranks showed that transcriptome diversity varies significantly among life cycle stages (H = 22.86; 4 d.f.; p < 0.001). Because the dataset we analyzed originates from infected cells and from extracellular stages, the number of sequences mapping to the genome of C. parvum, as expected, varies widely (Table 1). Between intracellular sample #24 with 28,243 of 19,435,693 (0.12%) mapped sequences and sporozoite sample #13 with 11,115,287 of 12,067,102 mapped sequences (92.11%), there is an almost 400-fold difference in the number of sequences mapping to the C. parvum genome. Because the Shannon diversity index is sensitive to sample richness and to population size (in our case the number of mapped sequence reads per sample), we assessed to what extent RNA-Seq Shannon diversity was impacted by sample size. We found little evidence that in our dataset these two variables are correlated (Fig. S4); a linear model fitted to these data shows that sequence abundance only explains 3% of diversity. This observation indicates that the low transcriptome diversity observed in intracellular transcriptomes 2 h post-infection (Fig. 3) is unlikely to be the result of the smaller number of mapped sequences.
Figure 2.
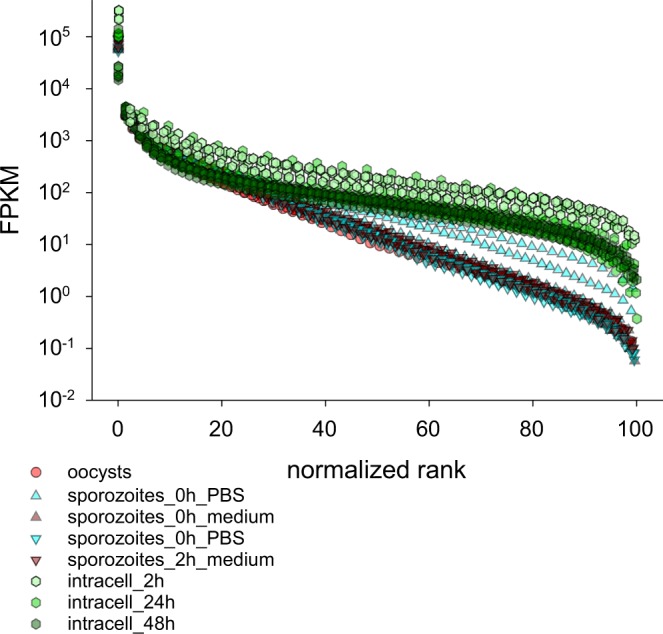
Comparison of normalized rank-abundance plots for 35 C. parvum transcriptomes. Intracellular transcriptomes are more even than transcriptomes from oocysts and sporozoites.
Figure 3.
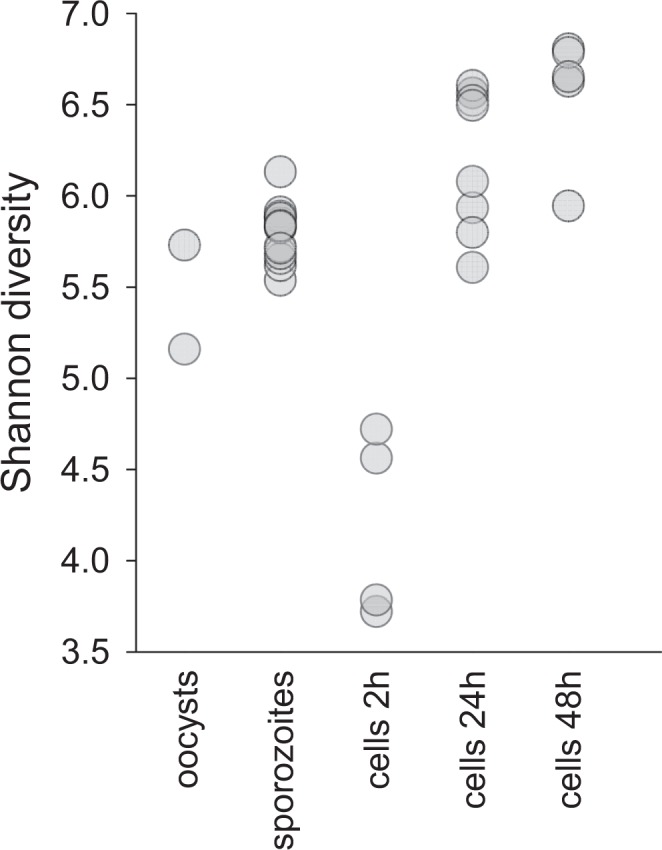
Shannon diversity of 35 C. parvum transcriptomes by life cycle stage. FPKM diversity peaks at 48 h post-infection.
In addition to qualitative properties of the intracellular transcriptome, we also examined the proportion of parasite mRNA in the infected cell transcriptome. As expected from the fact that the parasite multiplies during merogony, as C. parvum intracellular development progresses, the proportion of parasite transcripts increases (Fig. S5). Consistent with our previous analysis10, the parasites transcriptome in cell monolayers does not exceed 2% of the combined host cell – C. parvum transcriptome.
Functional properties of the extra- and intracellular transcriptome
Oocysts and sporozoites are specialized forms of the parasite which have evolved to ensure survival in the environment, delivery of sporozoites to a specific location of the gastro-intestinal (GI) tract34 and invasion of the host cell. We tested whether mRNA encoding specialized functions not commonly found among eukaryotes are important constituents of the extracellular parasite transcriptome by tabulating the number of orthologs of the 50 genes most highly expressed in extracellular and intracellular parasite stages, respectively (Tables S2, S3). The number of orthologous genes in eukaryotic pathogens was not significantly different between extracellular and intracellular transcriptome (extracellular, mean = 11.9 orthologs; intracellular mean = 12.2 orthologs: Mann-Whitney Rank Sum Test, p = 0.230). In contrast, the number of orthologs in the 150 genomes found in the OrthoMCL database35 was almost double for the genes highly transcribed in the host cell (mean = 133.5) as compared to those highly transcribed outside the host cell (mean = 69.7; Mann-Whitney, p < 0.001). This analysis indicates that, in contrast to intracellular developmental stages, oocysts and sporozoites preferentially express genes encoding specialized functions which have few orthologs outside of related protozoa.
We assessed the impact of the transition of non-dividing extracellular life cycle stages to intracellular multiplying stage on functional properties of the parasite transcriptome. Given the importance of ribosomes in the metabolism of dividing cells, the first analysis focused on ribosomal proteins. According to the annotation of the C. parvum IOWA genome found in CryptoDB.org, a total of 55 genes encode ribosomal proteins. Functional enrichment analysis of a smaller number of RNA-Seq datasets from intracellular samples10, (samples 1 M–4 M, Table 1) had already revealed the abundance of mRNA encoding such proteins 24 h post-infection. The analysis of the expanded dataset (n = 35) confirms this observation (Fig. 4).
Figure 4.
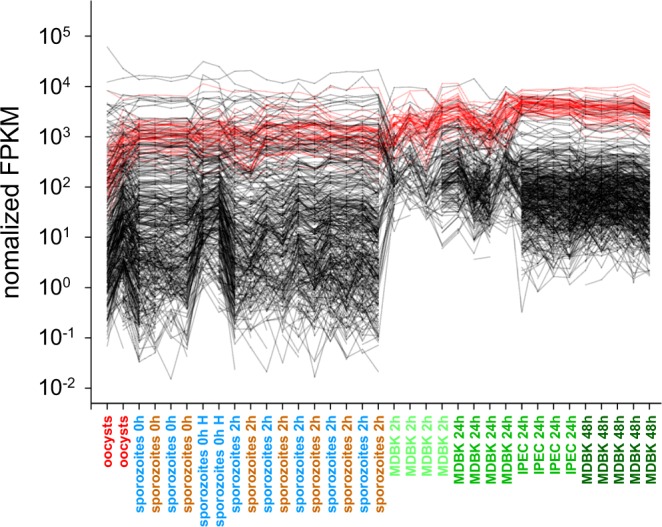
Upregulation of genes encoding ribosomal biosynthesis functions following host cell invasion. Normalized FPKM values of 55 genes encoding ribosomal proteins (red) and 455 randomly chosen genes encoding other functions (black) reveal an upregulation of ribosomal protein expression in relation to other functions. Life cycle stages are ordered on the x axis first in temporal order and second by experiment. The position of replicate samples within each group is arbitrary. The samples are color-coded as in Figs 1 and 2.
The high level of mRNA transcribed from the C. parvum lactate dehydrogenase (LDH) gene cgd7_480, also noticed by Zhang et al.33, was apparent in our dataset. Particularly striking is the fact that LDH mRNA is by far the most abundant transcript in extracellular stages. To assess whether other genes encoding oxidoreductase functions are similarly regulated, we visualized the FPKM values of 40 C. parvum genes with the term “oxidoreductase” in their annotation across the entire RNA-Seq dataset comprising 35 transcriptomes (Fig. 5). The graph shows that other genes in this functional category were also highly expressed in oocysts and sporozoites, but none displayed such and extreme difference between extracellular and intracellular expression as LDH. Using LDA, we also examined the correlation between FPKM and life cycle stage for 21 mRNA transcripts, including LDH, encoding enzymes in the glycolysis pathway (Fig. S6). Unexpectedly, LDH mRNA was clearly unique in the extent of differential regulation among the 21 genes in this metabolic pathway. For instance phosphoglycerate mutase, which is within two metabolic steps of LDH, shows no differential expression in relation to life cycle stages we examined. To ensure that RDA detects co-expression, we ran the same analysis with 7 C. parvum genes encoding Cryptosporidium oocyst wall proteins and found those to be tightly co-regulated (Fig. S6).
Figure 5.
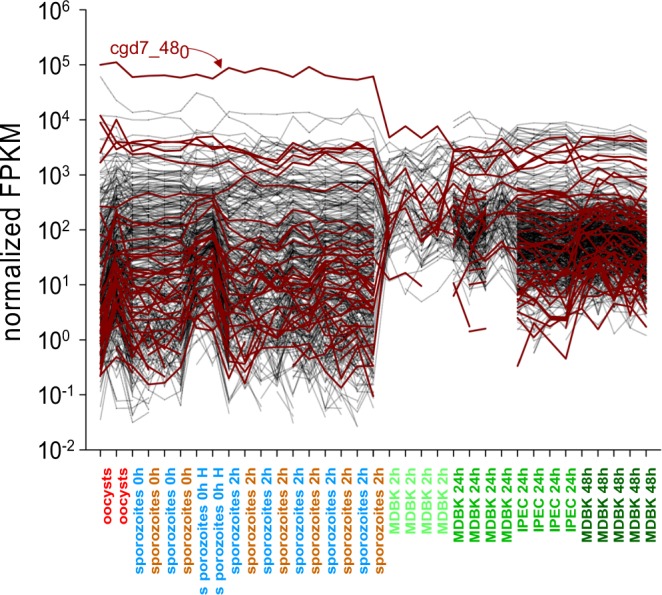
Expression of oxidoreductase functions in oocysts, sporozoites and intracellular developmental stages. Normalized FPKM values of 40 genes encoding oxidoreductase functions (brown) and 460 randomly chosen genes encoding unrelated functions (black) reveal high level of LDH (cgd7_480) transcript in extracellular stages. Life cycle stages are ordered on the x axis in temporal order, then by experiment. The position of samples within each group is arbitrary.
Differential gene expression
To identify genes that are most differentially expressed between extracellular and intracellular life cycle stages, genes were ranked according to the ratio of mean extracellular FPKM/mean intracellular FPKM. Log2 transformed ratios ranged from 5.1 to −16.6. An analysis of enriched biological functions was performed for the 50 genes most overexpressed extracellularly and 50 genes most overexpressed inside the host cell. Gene ontology (GO) enrichment analysis of the former group found no enriched biological function. In contrast, in the 50 genes most overexpressed in the host cell, 10 biological processes were enriched; tetrahydrofolate interconversion (GO:0035999), glycine biosynthetic process from serine (GO:0019264), glycine biosynthetic process (GO:0006545), neurotransmitter biosynthetic process (GO:0042136), L-serine metabolic process (GO:0006563), regulation of neurotransmitter levels (GO:0001505), neurotransmitter metabolic process (GO:0042133), glycine metabolic process (GO:0006544), serine family amino acid biosynthetic process (GO:0009070) and serine family amino acid metabolic process (GO:0009069), (FDR < 0.03). Consistent with the orthology analysis described above suggesting more specialized functions in extracellular stages, there were slightly more uncharacterized genes among the 50 genes most overexpressed in extracellular stages as compared to the 50 genes most overexpressed intracellularly (42% vs 30%). The association between life cycle stage and proportion of uncharacterized genes was, however, statistically not significant (Chi-square 1.6, p = 0.21).
LDA31 was used to identify genes that significantly differ in relative transcript abundance between extracellular and intracellular samples. The analysis identified 641 genes with an LDA score >2; 373 of these were overexpressed in oocyst/sporozoites and 268 in the host cell. GO enrichment analysis found 13 biological processes significantly enriched (FDR ≤ 0.05) in extracellularly overexpressed genes (Table 2), and 28 processes significantly enriched in intracellularly overexpressed genes (Table 3). GO analysis revealed a striking difference between biological processes enriched extra- and intracellularly; in extracellular transcriptomes, transmembrane related processes were particularly frequent (5/13 = 38%) as compared to their occurrence in intracellular transcriptomes (0/28). In contrast, and consistent with the analysis of ribosomal proteins illustrated in Fig. 4, GO terms related to biosynthetic processes were significantly more abundant in intracellular transcriptomes (9/28 = 32%) as compared to extracellular transcriptomes, where this term was not enriched. The association between GO term and life cycle stage was significant by Fisher Exact Test for both processes (transmembrane, p = 0.002; biosynthetic p = 0.038).
Table 2.
GO term enrichment analysis of 373 genes overexpressed in extracellular development stages of Cryptosporidium parvum.
| ID | Name | Fold enrichment | FDR |
|---|---|---|---|
| GO:0042254 | ribosome biogenesis | 6 | 1.18E-08 |
| GO:0022613 | ribonucleoprotein complex biogenesis | 5.78 | 1.364E-08 |
| GO:0044085 | cellular component biogenesis | 4.08 | 4.169E-06 |
| GO:0055085 | transmembrane transport | 3.23 | 2.858E-05 |
| GO:0016072 | rRNA metabolic process | 5.44 | 0.001 |
| GO:0006364 | rRNA processing | 5.44 | 0.001 |
| GO:0071840 | cellular component organization or biogenesis | 2.58 | 0.002 |
| GO:0099132 | ATP hydrol coupled cation transmembr transport | 4.08 | 0.045 |
| GO:0099131 | ATP hydrol coupled ion transmembrane transport | 4.08 | 0.045 |
| GO:0015991 | ATP hydrolysis coupled proton transport | 4.08 | 0.045 |
| GO:0015988 | energy coupled proton transmembr transport, against electrochemical gradient | 4.08 | 0.045 |
| GO:0090662 | ATP hydrolysis coupled transmembrane transport | 4.08 | 0.045 |
| GO:0016070 | RNA metabolic process | 1.79 | 0.046 |
Table 3.
GO term enrichment analysis of 268 genes overexpressed in intracellular development stages of Cryptosporidium parvum.
| ID | Name | Fold enrichment | FDR |
|---|---|---|---|
| GO:0043043 | peptide biosynthetic process | 8.29 | 3.90E-60 |
| GO:0006412 | translation | 8.29 | 3.90E-60 |
| GO:0043604 | amide biosynthetic process | 8.21 | 8.38E-60 |
| GO:0006518 | peptide metabolic process | 8.06 | 6.11E-59 |
| GO:0043603 | cellular amide metabolic process | 7.98 | 1.48E-58 |
| GO:1901566 | organonitrogen compound biosynthetic process | 6.35 | 8.47E-52 |
| GO:0044271 | cellular nitrogen compound biosynthetic process | 5.39 | 4.64E-46 |
| GO:0034645 | cellular macromolecule biosynthetic process | 4.88 | 2.72E-39 |
| GO:0009059 | macromolecule biosynthetic process | 4.85 | 3.94E-39 |
| GO:1901576 | organic substance biosynthetic process | 4.1 | 1.21E-35 |
| GO:0044249 | cellular biosynthetic process | 4.1 | 1.21E-35 |
| GO:0009058 | biosynthetic process | 3.92 | 6.61E-34 |
| GO:0010467 | gene expression | 4.26 | 8.52E-34 |
| GO:0044267 | cellular protein metabolic process | 3.6 | 7.27E-33 |
| GO:1901564 | organonitrogen compound metabolic process | 3.07 | 6.38E-31 |
| GO:0019538 | protein metabolic process | 3.2 | 2.33E-29 |
| GO:0034641 | cellular nitrogen compound metabolic process | 2.98 | 3.47E-24 |
| GO:0044260 | cellular macromolecule metabolic process | 2.67 | 4.78E-23 |
| GO:0006807 | nitrogen compound metabolic process | 2.1 | 1.79E-17 |
| GO:0009987 | cellular process | 1.92 | 2.61E-17 |
| GO:0044237 | cellular metabolic process | 2.09 | 3.92E-17 |
| GO:0044238 | primary metabolic process | 1.99 | 3.68E-16 |
| GO:0071704 | organic substance metabolic process | 1.96 | 6.74E-16 |
| GO:0043170 | macromolecule metabolic process | 2.11 | 7.09E-16 |
| GO:0008152 | metabolic process | 1.82 | 7.34E-15 |
| GO:0008150 | biological process | 1.45 | 1.68E-10 |
| GO:0006414 | translational elongation | 9.99 | 2.75E-05 |
| GO:0006457 | protein folding | 3 | 0.022 |
Discussion
To advance our understanding of gene regulation in the Cryptosporidium life cycle, here we focused on analyzing changes in the transcriptome associated with the transition from non-dividing extracellular stages to intracellular multiplicative forms. In contrast to the differentiation of gametes, meiosis and formation of oocysts, oocyst excystation can readily be studied in vitro, as can the initial phase of merogony in the host cell. Infecting cell monolayers with oocysts or sporozoites generates a reasonably synchronized infection3, facilitating the identification of differential gene expression over time. By including samples from three different studies, we ensure that the results are generally valid, i.e., are not restricted to a specific cell line or C. parvum isolate. We found that transcriptomes of extracellular developmental stages cluster together, regardless of isolate and sequencing strategy. Similarly, parasite transcriptomes 24 h post-infection originating from two different cell lines also were found to cluster (Fig. 1), although not as tightly as sporozoite and oocyst transcriptomes. These results likely reflect, in part, the impact of different laboratory procedures used to generate RNA-Seq data. The position of the sporozoite datapoints from Lippuner’s study in Fig. S3, relative to the datapoints from the present study, illustrates the magnitude of this effect. Lippuner and co-workers used a shorter excystation procedure (20 min vs. 60 min in our laboratory), a difference that may have contributed to their sporozoite transcriptome profiles being slightly different from ours. Validating the approach of incorporating different datasets, the noise introduced by the experimental variables is small compared to the difference between the transcriptome in different life cycle stages.
The global transcriptome analysis (Fig. 1) shows that oocysts and sporozoites express similar transcriptomes. This does not necessarily exclude differential gene expression during and following excystation. The small number of oocyst samples sequenced in this project limits our ability to investigate the transcriptional basis of excystation. In addition, differences between oocyst transcriptomes apparent in Fig. S1 may have been introduced by differences in oocyst age and storage conditions. Indeed, using quantitative PCR, we found that oocyst age impacts the abundance of selected transcripts36. A larger number of oocyst transcriptomes will need to be sequenced to dissect the effect of these experimental variables. The same reasoning applies to the effect of the cell line on C. parvum gene expression. Although our analyses include data from infected bovine and porcine cells, it is unknown to what extent the cell line has impacted C. parvum gene expression, since these samples originated from different experiments. Other variables, such as the degree of confluence of the cell monolayer or the number of infectious oocysts per cell would have to be considered. These analyses require a larger number of samples than analyzed here.
A general limitation of existing methods to culture Cryptosporidium parasites is that the infection is patchy and many host cells remain uninfected. The proportion of infected cells is difficult to measure and to control, precluding an estimation of the true proportion of C. parvum transcripts in the in the infected cell transcriptome. This limitation is reflected in the varying percentage of intracellular sequence reads mapping to the C. parvum genome (Table 1).
Only a small number of published studies have focused on Cryptosporidium transcriptomics. To our knowledge, Lippuner’s study9 is the only publication based on RNA-Seq to characterize the transcriptome of different C. parvum life cycle stages. In light of the inclusion of data from this study into our analyses, we examined the extent to which our data correlated with those previously reported9. Since both studies sequenced sporozoite transcriptomes and the intracellular transcriptome 48 h PI, these stages were included in the comparison. This analysis returned a correlation coefficient (r2) of 0.72. This result has to be evaluated in light of the differences between studies, like laboratory procedures, biological material (cell lines and C. parvum isolate) and sequencing strategy. The relatively high correlation between data from independent studies is encouraging, demonstrating that RNA-Seq is a robust approach to analyzing the complexity of the C. parvum transcriptome, even in the presence of an overwhelming excess of host RNA. This result also argues in favor of combining datasets from different studies as a way of ascertaining the general validity of the results.
The present study confirmed the abundance of LDH mRNA in oocysts previously reported33, and shows that this feature is characteristic of all extracellular stages studied here. What is particularly intriguing about this observation is that among 21 genes encoding enzymes in the glycolytic pathway only LDH mRNA is expressed at such high level. The functional significance of this observation remains to be elucidated.
Conclusions
RNA-Seq analysis of the C. parvum trophozoite/meront transcriptome and of extracellular stages (sporozoites and oocysts) has revealed significant differences in gene expression, both in terms of diversity and function. Whereas in the intracellular transcriptome, functions related to ribosomes and protein synthesis are highly enriched, the oocyst transcriptome does not have a clear functional signature. Genes highly expressed in oocysts appear to fulfill more specialized functions as indicated by a smaller number of orthologs in other eukaryotic genomes.
Supplementary information
Acknowledgements
Monoclonal antibody for immunofluorescence was kindly donated by Abhineet Sheoran, Tufts University. Marzieh Ezzaty Mirhashemi participated in the generation RNA-Seq data. Bruno Cesar Miranda Oliveira assisted with the processing of sequences data. The assistance of Albert Tai and the Tufts Genomics Core Facility staff is gratefully acknowledged. J.M., G.W. and S.T. acknowledge financial support from the National Institute of Allergy and Infections Diseases, NIH (R15 AI122152, U19 AI09776). Funding agencies played no role in the design or execution of this study.
Author Contributions
J.M. and S.T. participated in the design of the study and provided funding. L.S.V.M. performed the bench work. L.S.V.M. and G.W. analyzed the data. G.W. wrote the manuscript with the assistance of L.S.V.M. and K.D.S.B.
Data Availability
Sequence data were deposited in the European Nucleotide Archive under project accession numbers PRJEB25665 and PRJEB28268. Transcriptome data were also deposited with CryptoDB (www.cryptodb.org).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44289-x.
References
- 1.Kotloff KL, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Keusch GT, et al. Implications of acquired environmental enteric dysfunction for growth and stunting in infants and children living in low- and middle-income countries. Food and nutrition bulletin. 2013;34:357–364. doi: 10.1177/156482651303400308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Current WL, Haynes TB. Complete development of Cryptosporidium in cell culture. Science. 1984;224:603–605. doi: 10.1126/science.6710159. [DOI] [PubMed] [Google Scholar]
- 4.Morada Mary, Lee Sangun, Gunther-Cummins Leslie, Weiss Louis M., Widmer Giovanni, Tzipori Saul, Yarlett Nigel. Continuous culture of Cryptosporidium parvum using hollow fiber technology. International Journal for Parasitology. 2016;46(1):21–29. doi: 10.1016/j.ijpara.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 5.DeCicco RePass, M. A. et al. Novel Bioengineered Three-Dimensional Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum. Infect Immun85, 10.1128/IAI.00731-16 (2017). [DOI] [PMC free article] [PubMed]
- 6.Alcantara Warren C, et al. Detection of epithelial-cell injury, and quantification of infection, in the HCT-8 organoid model of cryptosporidiosis. J Infect Dis. 2008;198:143–149. doi: 10.1086/588819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heo I, et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat Microbiol. 2018;3:814–823. doi: 10.1038/s41564-018-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mauzy MJ, Enomoto S, Lancto CA, Abrahamsen MS, Rutherford MS. The Cryptosporidium parvum transcriptome during in vitro development. PLoS One. 2012;7:e31715. doi: 10.1371/journal.pone.0031715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lippuner C, et al. RNA-Seq analysis during the life cycle of Cryptosporidium parvum reveals significant differential gene expression between proliferating stages in the intestine and infectious sporozoites. Int J Parasitol. 2018;48:413–422. doi: 10.1016/j.ijpara.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Mirhashemi ME, et al. Transcriptome analysis of pig intestinal cell monolayers infected with Cryptosporidium parvum asexual stages. Parasites & vectors. 2018;11:176. doi: 10.1186/s13071-018-2754-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widmer G, et al. Comparative genome analysis of two Cryptosporidium parvum isolates with different host range. Infect Genet Evol. 2012;12:1213–1221. doi: 10.1016/j.meegid.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okhuysen PC, et al. Infectivity of a Cryptosporidium parvum isolate of cervine origin for healthy adults and interferon-gamma knockout mice. Journal of Infectious Diseases. 2002;185:1320–1325. doi: 10.1086/340132. [DOI] [PubMed] [Google Scholar]
- 13.Widmer G, Feng X, Tanriverdi S. Genotyping of Cryptosporidium parvum with microsatellite markers. Methods Mol Biol. 2004;268:177–187. doi: 10.1385/1-59259-766-1:177. [DOI] [PubMed] [Google Scholar]
- 14.Edwinson A, Widmer G, McEvoy J. Glycoproteins and Gal-GalNAc cause Cryptosporidium to switch from an invasive sporozoite to a replicative trophozoite. Int J Parasitol. 2016;46:67–74. doi: 10.1016/j.ijpara.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madin SH, Darby NB., Jr. Established kidney cell lines of normal adult bovine and ovine origin. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine. 1958;98:574–576. doi: 10.3181/00379727-98-24111. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths JK, Moore R, Dooley S, Keusch GT, Tzipori S. Cryptosporidium parvum infection of Caco-2 cell monolayers induces an apical monolayer defect, selectively increases transmonolayer permeability, and causes epithelial cell death. Infect Immun. 1994;62:4506–4514. doi: 10.1128/iai.62.10.4506-4514.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Widmer G, Corey EA, Stein B, Griffiths JK, Tzipori S. Host cell apoptosis impairs Cryptosporidium parvum development in vitro. Journal of Parasitology. 2000;86:922–928. doi: 10.1645/0022-3395(2000)086[0922:HCAICP]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 18.Yang YL, et al. Over-expression and localization of a host protein on the membrane of Cryptosporidium parvum infected epithelial cells. Mol Biochem Parasitol. 2009;168:95–101. doi: 10.1016/j.molbiopara.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brosnahan AJ, Brown DR. Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Veterinary microbiology. 2012;156:229–237. doi: 10.1016/j.vetmic.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theodos, C. M., Griffiths, J. K., D’Onfro, J., Fairfield, A. & Tzipori, S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother42 (1998). [DOI] [PMC free article] [PubMed]
- 21.Heiges M, et al. CryptoDB: a Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 2006;34:D419–422. doi: 10.1093/nar/gkj078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braak, C. T. & Šmilauer, P. CANOCO reference manual and CanoDraw for Windows user’s guide: software for canonical community ordination (version 4.5). Microcomputer Power, Ithaca, New York (2002).
- 29.Lepš, J. & Šmilauer, P. Multivariate analysis of ecological data using CANOCO. (Cambridge university press, 2003).
- 30.Magurran, A. E. Measuring biological diversity. (Blackwell Publishing, 2004).
- 31.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aurrecoechea C, et al. EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res. 2017;45:D581–D591. doi: 10.1093/nar/gkw1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Guo F, Zhou H, Zhu G. Transcriptome analysis reveals unique metabolic features in the Cryptosporidium parvum Oocysts associated with environmental survival and stresses. BMC Genomics. 2012;13:647. doi: 10.1186/1471-2164-13-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Widmer G, Klein P, Bonilla R. Adaptation of Cryptosporidium oocysts to different excystation conditions. Parasitology. 2007;134:1583–1588. doi: 10.1017/S0031182007002922. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Widmer G, Orbacz EA, Tzipori S. beta-tubulin mRNA as a marker of Cryptosporidium parvum oocyst viability. Appl Environ Microbiol. 1999;65:1584–1588. doi: 10.1128/aem.65.4.1584-1588.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data were deposited in the European Nucleotide Archive under project accession numbers PRJEB25665 and PRJEB28268. Transcriptome data were also deposited with CryptoDB (www.cryptodb.org).