

Modular, tissue-specific hydrogel cross-linkers were designed by the conjugation of various biomolecules to a novel polymer.
Abstract
Synthetic hydrogels are investigated extensively in tissue engineering for their tunable physicochemical properties but are bioinert and lack the tissue-specific cues to produce appropriate biological responses. To introduce tissue-specific biochemical cues to these hydrogels, we have developed a modular hydrogel cross-linker, poly(glycolic acid)–poly(ethylene glycol)–poly(glycolic acid)-di(but-2-yne-1,4-dithiol) (PdBT), that can be functionalized with small peptide-based cues and large macromolecular cues simply by mixing PdBT in water with the appropriate biomolecules at room temperature. Cartilage- and bone-specific PdBT macromers were generated by functionalization with a cartilage-associated hydrophobic N-cadherin peptide, a hydrophilic bone morphogenetic protein peptide, and a cartilage-derived glycosaminoglycan, chondroitin sulfate. These biofunctionalized PdBT macromers can spontaneously cross-link polymers such as poly(N-isopropylacrylamide) to produce rapidly cross-linking, highly swollen, cytocompatible, and hydrolytically degradable hydrogels suitable for mesenchymal stem cell encapsulation. These favorable properties, combined with PdBT’s modular design and ease of functionalization, establish strong potential for its usage in tissue engineering applications.
INTRODUCTION
Polymeric hydrogels are well suited for tissue engineering and other biomedical applications for a number of reasons, including the hydrated environments that they provide for cells and their adjustable physicochemical properties (1, 2). Synthetic polymers used in these hydrogels include thermoresponsive poly(N-isopropylacrylamide) (PNIPAAm), which undergoes physical gelation when elevated above its lower critical solution temperature, as well as derivatives of poly(ethylene glycol) (PEG) and various other polyethers, polyesters, and more (2, 3). These polymers can be cross-linked to form highly organized networks that swell by water uptake to fill the site of a tissue defect and provide a scaffold for cells (3, 4). Chemical cross-linking is necessary to maintain postformation hydrogel integrity, and thermoresponsive polymers such as PNIPAAm must often be cross-linked to prevent the collapse of the hydrogel from chain compaction known as syneresis (3). Hydrogel cross-linkers should, furthermore, be biodegradable by hydrolysis or enzymatic degradation in order for the hydrogel to be replaced by extracellular matrix as tissue regeneration progresses (1). While many hydrogels and synthetic cross-linkers have been developed for tissue engineering, these systems are largely bioinert, and further modification is thus required to produce tissue-specific bioactivity, a critical prerequisite for development of the desired tissue phenotype (1, 3).
Methods of introducing hydrogel bioactivity include the delivery of tissue-specific growth factors and peptides, as well as the usage of bioactive macromolecules such as glycosaminoglycans as hydrogel materials (5, 6). Tissue-specific growth factors such as those from the transforming growth factor–β superfamily, for instance, are often delivered by controlled release from intermediate vessels such as gelatin microparticles to promote bone and cartilage regeneration (7). Alternatively, these tissue-specific biomolecules can directly be conjugated to a hydrogel to produce in situ presentation of bioactive cues and decreased risk of ectopic effects from biomolecule diffusion (8, 9). However, the linker reactions used for biomolecule conjugation, such as 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry and thiol-maleimide chemistry, are susceptible to side reactions with common functional groups found on biomolecules and often involve cytotoxic reagents, which can limit biomolecule selection and compromise biocompatibility (10).
Alkyne-azide click chemistry has emerged as a potent method for achieving biomolecule conjugation due to its high degree of specificity and minimal to no susceptibility of the alkyne or azide moieties to biological side reactions (11, 12). Despite this, the Cu(I) and Ru(II) catalysts commonly used for these reactions are typically inactive under aqueous conditions, mildly reactive with common biomolecule functional groups such as thiols and amines, and cytotoxic, all of which can again restrict biomolecule selection and interfere with tissue engineering applications (11). Thus, the usage of catalyst-free reactions with ring-strained cycloalkynes such as dibenzocyclooctyne (DBCO), photoreactive moieties, and more has been pursued to avoid these issues (13, 14). Nevertheless, these catalyst-free reactions often require highly particular reaction conditions that may not be compatible with biomolecules of varying size, charge/hydrophilicity, and solvent stability, among other conditions. DBCO as an example shows good reactivity via strain-promoted cycloaddition, but the strong hydrophobicity of the multiring structure can limit biomolecule compatibility by necessitating organic cosolvents or extensive chemical modification to be used for biomolecule conjugation (15). For usage in hydrogels, DBCO and other bulky aliphatic rings can also limit polymer design in requiring optimization with hydrophilic components to produce water-soluble polymers (15, 16). Recent studies have suggested that one Ru(II) compound, chloro(pentamethylcyclopentadienyl)(cycloocta-diene)ruthenium(II) [Cp*Ru(cod)Cl], may be able to provide completely aqueous catalysis while being both noncytotoxic and unreactive toward common biological functional groups (17), tackling the major pitfalls of Ru(II) catalyst usage, although studies have been performed so far only using small-molecule reactants and not biologically relevant or polymeric reactants. Ultimately, it is thus of great interest to tissue engineers to develop bioconjugation strategies that are more greatly bioorthogonal, suitable to mild and biocompatible reaction conditions such as room temperature, and useable with a wide range of biomolecules (18).
Using the tools of click chemistry, we have designed a modular and biodegradable hydrogel cross-linking macromer that can be easily functionalized with a variety of tissue-specific biomolecules by a facile mixing process in water, representing a progression from many traditional, bioinert hydrogel cross-linkers in the presentation of modular, tissue-specific biomolecules on the cross-linker (table S1). We here report its conjugation to biologically relevant small peptides and large macromolecules by simply stirring the components in room temperature water in the presence of a Cp*Ru(cod)Cl catalyst. Furthermore, we have established proof of concept of cytocompatible, rapidly cross-linked, and hydrolytically degradable hydrogels generated using both regular and biomolecule-functionalized poly(glycolic acid) (PGA)–PEG–PGA–di(but-2-yne-1,4-dithiol) (PdBT) as cross-linkers for a model poly(N-isopropylacrylamide-co-glycidyl methacrylate) [P(NIPAAm-co-GMA)] system, which is shown to be suitable for mesenchymal stem cell (MSC) encapsulation.
RESULTS
Synthesis and characterization of hydrogel cross-linker
We developed a novel hydrogel cross-linker named PdBT for functionalization with tissue-specific cues. PdBT features modular components including alkyne moieties for bioconjugation, orthogonal sulfhydryl termini for cross-linking, and tunable polyester blocks for hydrolytic degradation (Fig. 1).
Fig. 1. Modular hydrogel cross-linker for click binding of tissue-specific biomolecules.
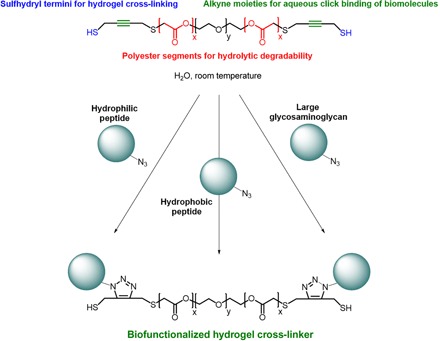
The overall design and modular components of the hydrogel cross-linker PdBT are shown. Biologically relevant peptides and large macromolecules have been conjugated to PdBT to generate a set of biofunctionalized cross-linkers.
We synthesized PdBT using commercially available reagents by the mesylate activation of hydroxyl termini on triblock PGA-PEG-PGA, followed by nucleophilic substitution of mesylate groups using but-2-yne-1,4-dithiol (Fig. 2A). We confirmed the expected chemical structure of PdBT using 1H nuclear magnetic resonance (NMR) (Fig. 3), with additional confirmation by correlating to 1H and 13C NMR spectra of starting materials in fig. S1. We first calibrated 1H NMR peak integrals by setting the left peak “d” to 89.27H in accordance with the expected number of protons on the central PEG chain (Fig. 3). Peak “a” represents PGA repeat units that confer hydrolytic degradability, and peak integration reveals an average of ~7.57 PGA units per PdBT chain that roughly corresponds to the 8 PGA units expected from the monomer feed ratio. The molecular mass of PdBT can be estimated as 1562 Da from NMR analysis of peaks “a” to “g” and gel permeation chromatography (GPC) characterization shows an approximately similar number-average molecular mass (Mn) value of 1381 ± 74 Da and a polydispersity index (PDI) of 1.09 ± 0.03 (n = 3). Peaks “e” and “f” represent protons on the terminal alkyne moieties, which act as sites for the click conjugation of one or two biomolecules per PdBT chain. Peak “g” corresponds to the terminal sulfhydryl groups that are used for nucleophilic cross-linking reactions. In the model hydrogel system described in this paper, we use a spontaneous thiol-epoxy reaction for the cross-linking of P(NIPAAm-co-GMA) by PdBT (Fig. 2).
Fig. 2. Creation of cross-linked, biofunctionalized hydrogels.
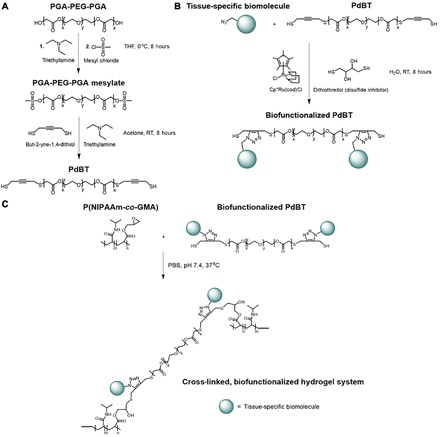
(A) Synthesis of PdBT cross-linker. THF, tetrahydrofuran; RT, room temperature. (B) Generation of biofunctionalized cross-linkers by mixing PdBT with tissue-specific biomolecules in water at room temperature in the presence of catalyst and disulfide inhibitor. (C) Spontaneous chemical cross-linking and physical gelation of a model P(NIPAAm-co-GMA) system induced by mixing the polymer with biofunctionalized PdBT in phosphate-buffered saline (PBS) (pH 7.4) at 37°C.
Fig. 3. Confirmation of PdBT structure using1H NMR.
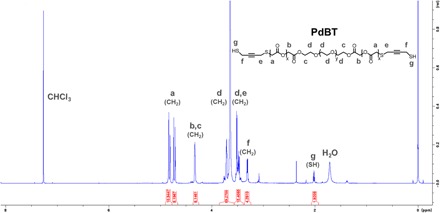
Peaks are identified using the letters a to g. Peak integrals are shown below peaks and are calibrated according to the known molecular mass of the central PEG block (left peak d).
Click functionalization of PdBT
Next, we biofunctionalized PdBT with several bone- and cartilage-specific biomolecules of varying size and hydrophilicity by a facile mixing process in water. PdBT was functionalized with either a hydrophilic bone morphogenetic protein mimetic (BMPm) peptide, a hydrophobic N-cadherin (NC) peptide involved in chondrogenesis, or the cartilage-derived glycosaminoglycan macromolecule, chondroitin sulfate (CS). BMPm and NC peptides with azide-functionalized N termini were synthesized by established solid-phase peptide synthesis procedures (19), while CS was modified with azide moieties as described previously (14). All biofunctionalization reactions of PdBT were conducted at ambient temperature and were performed by mixing of the biomolecule of interest with PdBT at a 2:1 molar ratio for 8 hours in the presence of 0.1 mol equivalent (eq.) Cp*Ru(cod)Cl and 4 mol eq. dithiothreitol (DTT) for catalysis and disulfide inhibition, respectively (Fig. 2B). We then removed impurities and unreacted reagents by 24 hours of dialysis in H2O. CS/PdBT was synthesized in very good yield of 87.1%, while BMPm/PdBT and NC/PdBT were synthesized in fair yields of 65.0 and 58.7%, respectively.
We quantified conversion of azide groups by comparing peak size of the azide-adjacent 1H NMR peaks before and after reaction relative to internal standards, indicating conversions of 83.3% for CS/PdBT, 82.1% for BMPm, and 89.7% for NC/PdBT (Fig. 4, A to C). The high degree of conversion for all biomolecules is thus in agreement with the click nature of this reaction (13).
Fig. 4. Click conjugation of CS, BMPm, and NC biomolecules to PdBT.
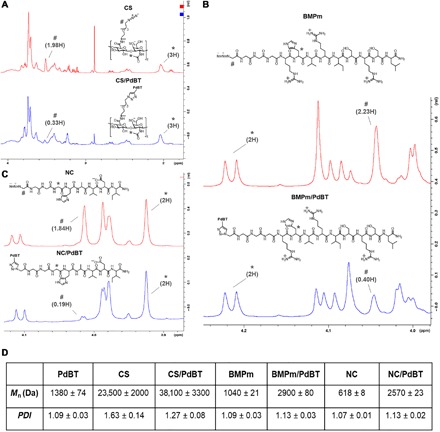
To quantify azide conversion, we compared 1H NMR spectra between unbound biomolecules and the resulting click products for (A) CS, (B) BMPm, and (C) NC, with number sign (#) indicating azide-adjacent peaks, asterisk (*) indicating internal standards used for peak integral calibration, and peak integrals labeled accordingly. Molar conversion, as determined by decrease in azide-adjacent peaks size, was calculated as 83.3% for CS/PdBT, 82.1% for BMPm/PdBT, and 89.7% for NC/PdBT. (D) GPC characterization of unbound biomolecules and their resulting click products, demonstrating increases in Mn after click conjugation to PdBT. Data are reported as means ± SD for a sample size of n = 3.
GPC was used to characterize the molecular mass distributions of unbound biomolecules compared to PdBT-conjugated biomolecules and showed increases in Mn after click conjugation for all three biomolecules (Fig. 4D). For instance, CS and CS/PdBT showed a statistically significant increase in Mn from 23.5 ± 2.0 to 38.1 ± 3.3 kDa after click conjugation, corresponding to chain lengthening of CS produced by the binding of two CS chains to a single PdBT macromer (Fig. 2B). BMPm and NC peptides, similarly, showed statistically significant increases in both Mn and PDI after conjugation that were roughly consistent with the expected molecular mass increase produced by the attachment of two peptides to each PdBT macromer (Fig. 4D). The 1H NMR and GPC data thus indicate that PdBT can be conjugated to hydrophilic and hydrophobic peptides and large biomacromolecules, establishing proof of concept for the bioconjugation of biomolecules with chemically and physically diverse properties.
Hydrogel cross-linking by biofunctionalized PdBT
Next, we established PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT as functioning hydrogel cross-linkers for a model 10% (w/v) P(NIPAAm-co-GMA) hydrogel system (Fig. 2C). PNIPAAm-based gels swell by water uptake when placed in phosphate-buffered saline (PBS) if they are chemically cross-linked but will compact and expel water mass by syneresis instead if they are insufficiently cross-linked, making this hydrogel system a facile model for assessing the performance of PdBT and biofunctionalized PdBT macromers as hydrogel cross-linkers (3, 20). The goal of this study was therefore to identify concentrations of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT that produced a greater swelling ratio at equilibrium, compared to their initial swelling ratio at the point of hydrogel formation, indicating the formation of well-swollen, highly cross-linked hydrogels. For this study, we tested each cross-linking macromer at its maximum soluble concentration and at 1:3 and 2:3 dilutions in PBS (pH 7.4) (Fig. 5A). Concentrations of 3.5% (w/v) PdBT, 4.66% (w/v) CS/PdBT, 10.5% (w/v) BMPm/PdBT, and 3.5% (w/v) NC/PdBT or higher produced a statistically significant degree of postformation swelling (Fig. 5A), representing well–cross-linked systems (20). These minimum concentrations for the formation of well–cross-linked systems were thus used for all further hydrogel characterization. All four hydrogel cross-linking reactions reached completion within ~60 min (Fig. 5B), as shown by differential scanning calorimetry of heat release produced by the thiol-epoxy cross-linking reaction, indicating that biomolecule conjugation did not noticeably interfere with PdBT’s rapid cross-linking kinetics.
Fig. 5. Swelling, reaction kinetics, hydrolytic degradation, and cytocompatibility of PdBT cross-linked hydrogels.
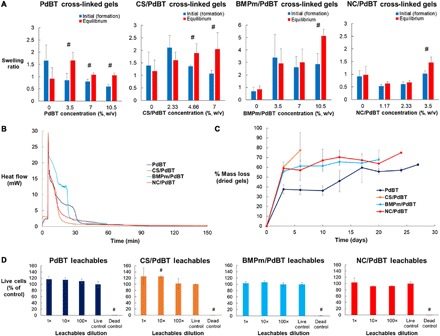
(A) Swelling study performed to assess the cross-linking of 10% (w/v) P(NIPAAm-co-GMA) hydrogels mixed with varying concentrations of PdBT, CS/PdBT, BMPm/PdBT, or NC/PdBT. Data are reported as means ± SD for a sample size of n = 3. Number sign (#) indicates statistical significance of a greater equilibrium swelling ratio than initial swelling ratio at a given concentration, which represents formation of a well–cross-linked system. Representative pictures are shown for 3.5% (w/v) PdBT, 4.66% (w/v) CS/PdBT, 10.5% (w/v) BMPm/PdBT, and 3.5% (w/v) NC/PdBT, which were used for all further studies. (B) Cross-linking kinetics of the aforementioned hydrogel formulations characterized by differential scanning calorimetry of heat flow produced by each cross-linking reaction. Samples were held at 4°C for the first 5 min and then immediately elevated to 37°C to induce gelation and cross-linking. (C) Hydrolytic degradability of PdBT cross-linkers assessed by an accelerated degradation study comparing the degradation kinetics of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT cross-linked hydrogels placed in 0.1 M HCl at 37°C. Data are reported as means ± SD for a sample size of n = 3. (D) Cytocompatibility of cross-linked hydrogels, as demonstrated by a leachable cytotoxicity assay. Data are reported as means ± SD for a sample size of n = 3. Number sign (#) indicates statistical significance from live controls.
To confirm the direct attachment of CS, BMPm, and NC biomolecules to hydrogels, we placed cross-linked hydrogels in PBS (pH 7.4) at 37°C and leached them for 24 hours, followed by quantification of CS, BMPm, and NC content in the leached sol fraction. A dimethylmethylene blue (DMMB) assay was performed for CS, while a Coomassie blue assay was performed for BMPm or NC, using known concentrations of CS/PdBT, BMPm/PdBT, and NC/PdBT as standards. The assays revealed 94.9 ± 1.3% retention of CS, 90.1 ± 1.3% retention of BMPm, and 88.7 ± 0.9% retention of NC by mass (n = 3) in their respective hydrogels (see fig. S2). CS/PdBT may leach less relative to BMPm/PdBT and NC/PdBT due to its substantially higher molecular mass, which would inhibit the diffusion of uncross-linked CS/PdBT out of the hydrogel. Ultimately, nearly all of the conjugated biomolecules are retained inside rather than being eluted in the sol fraction, indicating that these biomolecules are conjugated directly to the PdBT cross-linked systems.
Assessment of biofunctionalized gel degradation
We confirmed the hydrolytic degradability of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT cross-linked hydrogels in a comparative accelerated degradation study using 0.1 M HCl at 37°C. Fabricated hydrogels that were left in acidic solutions and allowed to degrade until the gels at a given time point were soluble in PBS at 4°C, representing the breakdown of chemical cross-linking and the remainder of only thermal gelation effects from P(NIPAAm-co-GMA). Degradation reached completion within 27 days for PdBT, 6 days for CS/PdBT, 20 days for BMPm/PdBT, and 24 days for NC/PdBT (Fig. 5C).
The time spans for degradation of PdBT, BMPm/PdBT, and NC/PdBT are roughly similar, as expected, although degradation occurred more quickly for CS/PdBT, most likely due to increased water and ion uptake from the inclusion of highly charged CS chains. Given that PdBT degradation occurs by the hydrolysis of ester bonds, the degradation kinetics can thus be adjusted by varying the number of PGA units created during polymer synthesis. An accelerated degradation study of PdBT with varying PGA block length, for instance, showed faster degradation kinetics with greater PGA content and a statistically significant higher amount of mass loss at day 3 in particular (fig. S3), most likely due to a greater molar proportion of hydrolysable units causing faster initial hydrolysis of the PGA block. Furthermore, while PEG 1000 was used in synthesizing PdBT in this study, higher molecular mass PEG would likely increase the rate of hydrolysis by increasing the hydrophilicity of the cross-linker.
Leachable cytotoxicity assay
The cytocompatibility of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT cross-linked hydrogels was confirmed in a leachable cytotoxicity assay (20), in which fabricated hydrogels were leached in cell culture media for 24 hours at 37°C, followed by exposure of the media to CRL-1764 fibroblasts at 1×, 10×, and 100× dilutions. As shown in Fig. 5D, no dilutions showed statistical significance from live controls with the exception of the 10× dilution of CS/PdBT gel leachables, which showed a slight increase in cell viability. Given that 1× and 100× dilutions of CS/PdBT showed no effects on cell viability, this outcome is likely due to experimental variability. Hydrogels cross-linked by biofunctionalized PdBT thus demonstrate excellent cytocompatibility, which enables usability for biological applications.
In vitro MSC encapsulation
To demonstrate the suitability of the PdBT cross-linker for stem cell encapsulation, we encapsulated MSCs within either PdBT cross-linked hydrogels or control hydrogels cross-linked by an established polyamidoamine (PAMAM) macromer, which has been previously investigated by our lab and used for MSC encapsulation (3). PdBT and PAMAM were incorporated at 3.5% (w/v) and 7% (w/v), respectively, with the PAMAM concentration representing a minimum concentration for cross-linking based on previous studies from our laboratory (3). MSC-encapsulated hydrogels were cultured in vitro for 7 days, with confirmation of cell viability by measurement of double-stranded DNA content of live cells inside hydrogels with a PicoGreen assay, as well as LIVE/DEAD imaging of cross-sectional slices of the hydrogels. As shown in Fig. 6, control hydrogels demonstrated maintenance of cell viability over 7 days, while PdBT hydrogels displayed an increase in DNA content representing cell proliferation beginning at day 3 and continuing through day 7. These trends were confirmed by LIVE/DEAD imaging, which showed the beginning of visible cell spreading in PdBT cross-linked hydrogels at day 3, followed by substantial cell spreading by day 7. The control hydrogels, on the other hand, displayed only nonspread, spherical cells. The greater cell proliferation seen in PdBT cross-linked gels may be explained by the relatively lower cytotoxicity of unreacted thiol groups on PdBT compared to unreacted amine groups on PAMAM. The strong viability and proliferation of MSCs within PdBT cross-linked hydrogels ultimately establishes its applicability for stem cell encapsulation and the future investigation of tissue-specific differentiation produced by cell-encapsulated and biofunctionalized PdBT hydrogel systems.
Fig. 6. MSC encapsulation in vitro within PdBT cross-linked gels.
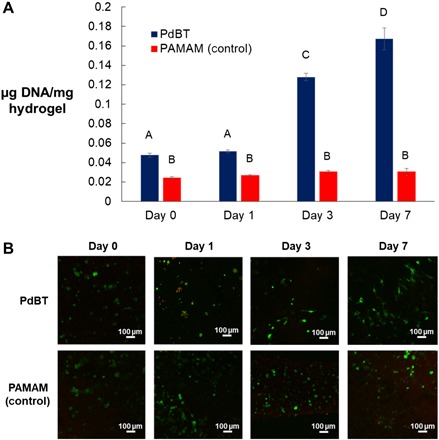
(A) Double-stranded DNA content of live cells normalized to wet hydrogel mass is shown at point of fabrication (0 days) and over the course of 7 days of in vitro culture for hydrogels cross-linked by either PdBT or an established PAMAM cross-linker. Data are reported as means ± SD for a sample size of n = 3. Different letters A to D indicate statistically significant differences between time points. (B) Representative LIVE/DEAD images are shown for cross-sectional slices of hydrogels at each time point. Green and red staining indicates live and dead cells, respectively.
DISCUSSION
It is of longstanding interest for tissue engineers and biomedical scientists to develop bioconjugation methods that are biocompatible, bioorthogonal, and suitable for a variety of biologically relevant molecules (18). Here, modular, tissue-specific hydrogel cross-linkers were developed by a facile and mild click conjugation scheme that is compatible with a diverse set of biomolecules. The successful synthesis of PdBT was first verified by confirming the expected chemical structure and molecular mass using 1H NMR and GPC, respectively. While thiol modification of PEG and PEG copolymers has been performed previously (21), the present modification of PGA-PEG-PGA is unique in its utilization of but-2-yne-1,4-dithiol to simultaneously introduce functional thiol and alkyne moieties, which can then be used with complete orthogonality for cross-linking and biomolecule conjugation, respectively. The bone-specific hydrophilic peptide BMPm, cartilage-specific hydrophobic peptide NC, and cartilage-derived glycosaminoglycan macromolecule CS were conjugated next to PdBT to create tissue-specific cross-linkers in fair to very good yield and high conversion. The mild aqueous conditions, short duration, ambient temperature, and bioorthogonality of this reaction afford notable versatility in terms of biomolecule selection and eliminate any concerns of biomolecule degradation or denaturation associated with the harsher click and nonclick reaction conditions that have been used previously for bioconjugation. While CS, NC, and BMPm represent chondrogenic and osteogenic biomolecules, this click functionalization scheme could feasibly be used for any other peptide or extracellular molecule of interest, addressing the aforementioned need in the field for versatile and biologically compatible conjugation strategies.
Furthermore, we provided proof of concept for the usage of biofunctionalized PdBT macromers as cross-linkers in a model P(NIPAAm-co-GMA) hydrogel system, which produced highly swollen, well–cross-linked systems. We found that all four cross-linkers completed reaction within 60 min, demonstrating that the conjugation of neither small peptides nor large macromolecules interfered with PdBT’s rapid cross-linking kinetics. These fast kinetics are beneficial for biomedical applications in promoting hydrogel stability after formation and limiting the exposure of cells to potentially reactive groups on PdBT (3). It can be inferred that the strong nucleophilicity of the sulfhydryl termini enables these fast reaction kinetics. Furthermore, we confirmed that the conjugated biomolecules CS, BMPm, and NC are incorporated directly into the cross-linked hydrogels at mass retentions of 88.7 to 94.9%, creating in situ presentation of these biological cues directly inside the cross-linked gels. By anchoring these tissue-specific biomolecules to the hydrogel itself, this may mitigate potential issues associated with biomolecule diffusion such as loss of bioactivity or ectopic tissue growth (10).
We then confirmed the hydrolytic degradability of PdBT cross-linked systems under accelerated conditions and confirmed that passive hydrolysis broke down gels for all four cross-linkers. CS/PdBT produced notably quicker degradation than the other cross-linkers, which we attribute to greater water and ion diffusion into CS/PdBT hydrogels resulting from highly charged CS chains. The biophysical properties of the molecule conjugated to PdBT thus affects its degradation kinetics, and one way to modulate this gel life span for one’s intended application can then be to adjust the PGA block size or PEG chain length accordingly. Next, we verified the cytocompatibility of various PdBT cross-linked systems by assessing the contents leached and exposed to fibroblasts, confirming its suitability for tissue engineering and other biomedical applications. While the dialysis and purification procedures should theoretically remove all potentially cytotoxic impurities, the leachable study provides assurance that the end product of PdBT synthesis, biomolecule click conjugation, and hydrogel fabrication is, in fact, cytocompatible. Last, we successfully encapsulated MSCs within PdBT cross-linked hydrogels, demonstrating their viability and proliferation in vitro within a PdBT cross-linked system. By establishing compatibility of PdBT with MSCs, we allow for further studies of stem cell differentiation within biofunctionalized PdBT systems both in vitro and in vivo.
Ultimately, PdBT fulfills several vital requirements of a hydrogel cross-linker—rapid cross-linking, hydrolytic degradability, and cytocompatibility—while maintaining these characteristics when PdBT is conjugated with biomolecules of varying size, charge, and chemical character. PdBT thus represents a biologically friendly cross-linking method for the creation of tissue-specific hydrogels and can be further applied toward various biological systems of interest. One should note, however, that a critical requirement for this functionalization scheme is the introduction of non-native azide groups on any biomolecule of interest. Amine groups are biologically prevalent and, to this benefit, can be converted to azides by an extensive set of strategies ranging from activated ester chemistry to one-pot conversions described in the literature (22, 23). Other predominant biological moieties such as hydroxyl and sulfhydryl groups can also be converted to azides using commercially available linkers or direct functional group conversions (11, 24). Further optimization of the click reaction conditions can help improve yield and reduce the amount of biomolecule needed for the generation of biofunctionalized PdBT macromers. Tuning the pH of the reaction, for instance, may eliminate the need for DTT as a disulfide inhibitor (25), by inhibiting sulfhydryl ion formation, and further simplify the click reaction scheme.
While a model P(NIPAAm-co-GMA) system with a thiol-epoxy cross-linking reaction was used to establish proof of concept in the present study, further investigation will elucidate how PdBT thiol-based cross-linking mechanisms can be used with a diversity of other polymer systems with thiol-reactive groups such as maleimides, alkenes, and mussel-inspired catechols (21, 26, 27). One major implication of the orthogonal design with thiol and alkyne moieties is broad applicability in terms of polymer and biomolecule selection, which can be further expanded in those studies. This is underscored by the finding that the conjugation of chemically diverse biomolecules does not deter cross-linker functionality, as discussed previously. One should also note that the conjugation of biomolecules demonstrated in this study produces covalent anchorage of the tissue-specific biomolecules to the hydrogel matrix, and further investigation is needed to assess the degree of bioactivity of immobilized biochemical cues and the potential effects of biomolecule release to the surrounding environment as a result of the hydrolytic degradation of PdBT. Both of these factors will likely have implications for in vitro and in vivo applications. Additional investigation can then be directed toward the conjugation of growth factors, bioactive dyes, and other biologically relevant molecules to PdBT, as well as the application of biofunctionalized PdBT toward tissue repair and other biological applications.
MATERIALS AND METHODS
Materials
PEG of manufacturer-reported Mn of 1000 Da, glycolide, triethylamine (TEA), mesyl chloride, CS A sodium salt from bovine trachea, EDC hydrochloride, NHS, 2-(N-morpholino)ethanesulfonic acid (MES), 11-azido-3,6,9-trioxaundecan-1-amine (ATA), 2-azidoacetic acid, Cp*Ru(cod)Cl, DTT, and sodium nitrate were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. High-performance liquid chromatography (HPLC)–grade tetrahydrofuran (THF), dichloromethane (DCM), and methanol were also purchased from Sigma-Aldrich and used as is. Ultrapure water was obtained from a Millipore Super-Q water system (Billerica, MA). PBS was prepared using preallocated powder from Sigma-Aldrich and ultrapure water.
PdBT synthesis
PdBT was synthesized from triblock PGA-PEG-PGA in several steps (Fig. 2A). PGA-PEG-PGA is available commercially but, in this case, was synthesized at a molar feed ratio of 4:1 glycolide:PEG, as described in detail elsewhere (28, 29). PGA-PEG-PGA was dissolved in THF and stirred on ice, followed by addition of 10 mol eq. of TEA and 10 mol eq. of mesyl chloride. After stirring on ice for 8 hours, the product was vacuum-filtered to eliminate particulate side products, precipitated in diethyl ether, redissolved in DCM, and washed twice with ultrapure H2O.
The product of this reaction was then dissolved in acetone, followed by addition of 4 mol eq. of but-2-yne-1,4-dithiol (30) and 4 mol eq. of TEA, and then stirred at ambient temperature for 8 hours. The crude product was then precipitated in diethyl ether, redissolved in DCM, and washed twice with ultrapure H2O. DCM was removed by rotoevaporation, and the final product was dried overnight by vacuum.
Preparation of azide-presenting biomolecules
CS, BMPm (“GGGRHVRISRSL”), and NC (“GGGHAVDI”) were used as model biomolecules due to their involvement in the development of native bone and cartilage and their established usage in tissue engineering (5, 31). CS was functionalized with azide groups by attachment of the amine/azide linker ATA, using EDC/NHS chemistry in MES buffer (pH 5), as described in detail elsewhere (13). BMPm and NC were synthesized by 9-fluorenyl methoxycarbonyl–based solid-phase peptide synthesis on a rink amide MBHA (4-methylbenzhydrylamine) low-loading resin, as described elsewhere (19). To introduce N-terminal azide functionalization, 2-azidoacetic acid was added as the final “amino acid” by the same chemistry as regular amino acid addition, followed by peptide cleavage from the resin. Peptide purification was performed by precipitation in cold diethyl ether and dialysis for 24 hours at 500-Da molecular mass cutoff (MWCO), with replacement of H2O every 6 to 8 hours. After dialysis, solutions were flash-frozen in liquid N2 and lyophilized.
Click functionalization of PdBT
PdBT was click-conjugated by alkyne-azide cycloaddition to CS, BMPm, or NC by mixing the biomolecule with PdBT in water at ambient temperature (Fig. 2B). Briefly, the biomolecule of interest was mixed with 0.5 mol eq. of PdBT, 2 mol eq. of DTT for disulfide inhibition, and 0.1 mol eq. of Cp*Ru(cod)Cl in ultrapure H2O, followed by stirring at ambient temperature for 8 hours. Following this, unreacted biomolecules, unmodified PdBT, and impurities were removed by dialysis for 24 hours at either 2-kDa MWCO for BMPm/PdBT and NC/PdBT or 50-kDa MWCO for CS/PdBT, with replacement of H2O every 6 to 8 hours. After dialysis, solutions were flash-frozen in liquid N2 and lyophilized.
1H NMR spectroscopy
NMR was performed using a 600-MHz Bruker spectrometer (Billerica, MA). CDCl3 with 1% (v/v) trimethylsilane and D2O with 1% (w/w) trimethylsilylpropanoic acid from Sigma-Aldrich (St. Louis, MO) were used as NMR solvents. Samples were dissolved at 5 mg/ml in CDCL3 or in D2O in the case of peptide and GAG (glycosaminoglycan) samples, and spectra were processed using Bruker TopSpin software.
GPC determination of molecular mass
Aqueous GPC was used to characterize the molecular mass of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT and compare their molecular mass distributions to those of the unbound biomolecules. Waters Systems (Milford, MA) components in the GPC system included a model 510 HPLC pump, model 410 differential refractometer, model 717 autosampler/injector, and an Ultrahydrogel (6 μm, 6 mm by 40 mm) analytical column. All samples and standards were dissolved at 10 mg/ml and run in 100 mM sodium nitrate at 0.5 ml/min, with filtration of all prepared solutions at 0.2 μm. Mn and PDI were calculated in Waters Empower software by comparison to standard curves (see fig. S4) generated using narrowly dispersed PEG standards of 450, 2500, 10,225, 30,250, 44,000, and 78,300 Da.
Biofunctionalized hydrogel fabrication
P(NIPAAm-co-GMA) was synthesized as previously described to generate a thermoresponsive polymer with pendant epoxy groups for cross-linking (3). Model hydrogels were prepared at 10% (w/v) P(NIPAAm-co-GMA), with varying concentration of PdBT or biofunctionalized PdBT as specified above. To prepare hydrogels, separate solutions of P(NIPAAm-co-GMA) and the chosen cross-linking macromer were prepared twice the intended final concentrations in PBS at pH 7.4 and kept on ice until the point of hydrogel fabrication. At the point of fabrication, the two solutions were prepared at double the intended volume ratio and pipetted at 100 μl per gel construct into cylindrical Teflon molds of 8-mm diameter and 2-mm height (4). The hydrogel-containing molds were then moved to a closed container in a 37°C warm room and given 24 hours to form and cross-link (Fig. 2C), followed by removal from the Teflon molds for characterization.
Assessment of hydrogel cross-linking by postformation swelling
The ability of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT to cross-link P(NIPAAm-co-GMA) was assessed by a study in which fabricated hydrogels were swollen in excess PBS and then assessed for degree of mass swelling, i.e., the ability to resist the natural tendency of P(NIPAAm-co-GMA) to shrink and expel water mass after formation (3). In this study, hydrogels were weighed immediately after fabrication (Wf) and then weighed again after equilibrium swelling (Ws) in 1.5 ml of PBS at pH 7.4 for 24 hours at 37°C. Swollen hydrogels were then flash-frozen in liquid N2 and lyophilized to obtain the dry weight (Wd). From these weights, the initial formation swelling ratio was calculated as , while the equilibrium swelling ratio was calculated as . The minimum PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT concentrations that resulted in a greater equilibrium swelling ratio than the initial formation swelling ratio, indicating sufficiently cross-linked systems, were then used for all further hydrogel characterization.
Differential scanning calorimetry of cross-linking kinetics
A Discovery DSC 250 from TA Instruments (New Castle, DE) was used to monitor cross-linking reaction kinetics (3). Prehydrogel solutions were mixed as described previously and kept on ice, and then, 14 μl of each solution was pipetted into aluminum sample pans to be held at 4°C for 5 min. The sample pans were then immediately elevated to 37°C to induce gelation and cross-linking over a duration of 145 min. The heat release produced by the exothermic thiol-epoxy cross-linking reaction was recorded during the entire 150-min run, and the point at which heat release reached a steady state near 0 was considered the point of reaction completion.
Colorimetric assessment of biomolecule incorporation in hydrogels
Colorimetric assays were purchased from Thermo Fisher Scientific (Waltham, MA) and used according to the manufacturer’s instructions. To quantify the degree of incorporation of biomolecule incorporation in hydrogels, the hydrogels were leached in 1.5 ml of PBS at pH 7.4 for 24 hours at 37°C, and then, the surrounding solution was collected. The liquid solutions containing the hydrogel sol fractions were then quantified for CS, BMPm, or NC concentration using a DMMB kit for sulfated GAGs or a Coomassie blue kit for peptides (32, 33). Following this, the percentage of biomolecule retention in gels was computed by dividing the eluted concentration by the original biomolecule concentration and subtracting from 100%. Standard curves for both assays can be found in the Supplementary Materials.
Assessment of biofunctionalized hydrogel degradation
To confirm the degradability of PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT cross-linked hydrogels via hydrolysis, an accelerated degradation study under acidic conditions was performed according to established protocols (20, 34). Fabricated hydrogels were flash-frozen in liquid N2, lyophilized, and measured for initial dry weight (Wi). Dry gels were then immersed in 4 ml of 0.1 M HCl at 37°C to undergo reswelling and hydrolytic degradation, with replacement of HCl every 3 to 4 days. At time points of interest, the respective hydrogels were flash-frozen in N2 and lyophilized to acquire the degraded dry weight (Wt). The percentage weight loss at each time point was calculated by . Each cross-linking macromer’s degradation study was then concluded once all the lyophilized gels at a given time point could completely solubilize in 4 ml of PBS (pH 7.4) at 4°C, indicating breakdown of chemical cross-linking.
Leachable cytotoxicity assay
The cytocompatibility of hydrogels cross-linked by PdBT, CS/PdBT, BMPm/PdBT, and NC/PdBT was assessed via a leachable cytotoxicity assay (20). Rat2 (CRL-1764) fibroblasts from the American Type Culture Collection (Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotic-antimycotic and used at passage 4 or lower for all experiments. Hydrogels were sterilized for 1 hour under ultraviolet (UV) light immediately after fabrication and then immersed in cell culture media at 1 ml/cm2 of hydrogel surface area for 24 hours at 37°C to leach compounds. Solutions were then prepared from the leachable-containing media at 1×, 10×, and 100× dilutions. Fibroblasts were cultured until 90% confluent at an initial seeding density of 10,000 cells per well in a 96-well plate, followed by replacement of the media in each well with 100 μl of either the 1×, 10×, or 100× leachable solution. For live and dead controls, the media were replaced with fresh media. After 24 hours of incubation under normal cell culture conditions, the dead control was exposed to 100 μl of 70% ethanol for 20 min. Following this, all wells were aspirated, washed twice with 100 μl of PBS, and stained with 100 μl of a 2 μM calcein AM and 4 μM ethidium homodimer-1 solution. After 30 min of incubation at room temperature in the dark, the wells were measured for 494/515 nm (live) fluorescence and 528/617 nm (dead) fluorescence using a BioTek Instruments (Winooski, VT) FLx800 fluorescence plate reader. To calculate percentage of living cells, the live fluorescence value produced by each condition was divided by the average fluorescence value of the live control samples.
MSC harvest and culture
MSCs were harvested by aspiration from the tibia of anesthetized 6-month-old New Zealand White rabbits from Charles River Laboratories (Wilmington, MA) in agreement with protocols approved by the Rice Institutional Animal Care and Use Committee and in accordance with the animal care and use guidelines set forth by the National Institutes of Health, as described previously (35). The MSCs were cultured on T225 flasks in minimum essential medium alpha with 20% (v/v) FBS and 1% (v/v) penicillin-streptomycin inside a humidified incubator at 37°C and 5% CO2 and were used at passage 3.
In vitro MSC encapsulation
All polymer components were UV-sterilized for 3 hours before usage for in vitro experiments. PdBT and P(NIPAAm-co-GMA) were dissolved at triple the intended final concentrations in PBS at pH 7.4 and kept on ice until the point of hydrogel fabrication. At the point of fabrication, the two polymer solutions and an MSC suspension in media were mixed at 1:1:1 volume ratio and then pipetted at 30 μl per gel construct into autoclaved cylindrical Teflon molds of 8-mm diameter and 1-mm height, producing constructs with a final cell density of 15 × 106 cells/ml (36, 37). The hydrogel-containing molds were then placed in a petri dish, moved to a 37°C, 5% CO2 incubator, and given 75 min to form and cross-link. After this, hydrogels were removed from the molds and placed in 1 ml of each of the aforementioned cell culture media, with replacement of media every 2 to 3 days. At time points of interest, hydrogels were soaked in PBS for 15 min, weighed, and processed for either a DNA PicoGreen assay or LIVE/DEAD imaging.
For the DNA PicoGreen assay, hydrogels were placed in 300 μl of filtered ultrapure H2O and then homogenized using a QIAGEN (Hilden, Germany) TissueLyser II at 26 s−1 for 5 min. Double-stranded DNA of live cells was then quantified in each hydrogel sample using an Invitrogen (Eugene, OR) PicoGreen assay, according to kit instructions. For LIVE/DEAD imaging of MSCs within each hydrogel, ~0.5-mm cross-sectional slices were acquired from each PBS-soaked hydrogel using a handheld razor blade and stained with 250 μl of a 2 μM calcein AM and 4 μM ethidium homodimer-1 solution. After 30 min of incubation at room temperature in the dark, representative images of the slices were taken using excitation/emission filters of 494/515 nm (green, live) and 528/617 nm (red, dead) under a 10× objective on an A1-Rsi confocal microscope from Nikon Instruments (Tokyo, Japan).
Statistical analysis
For the swelling study, a one-tailed paired Student’s t test was used to determine whether an increase occurred from initial to equilibrium swelling ratio for each hydrogel formulation. For GPC data, a two-tailed paired Student’s t test was used to identify whether changes in Mn and PDI occurred after click conjugation of biomolecules to PdBT. For the leachable cytotoxicity study, a two-tailed unpaired Student’s t test was used to compare the live control with each experimental condition. PicoGreen data and the supplemental accelerated degradation study were analyzed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test. All tests were performed at α = 0.05.
Supplementary Material
Acknowledgments
We would like to thank J. D. Hartgerink for helpful guidance on peptide synthesis. We also acknowledge the assistance of G. L. Koons with rabbit bone marrow harvest as well as the assistance of E. R. Molina and A. M. Navara with MSC culture. This article reflects the views of the authors and should not be construed to represent FDA’s views or policies. Funding: We acknowledge support by the NIH (R01 AR068073 and P41 EB023833) in the preparation of this work. J.L.G. and V.Y.X. also acknowledge support from the Smalley-Curl Institute Student Training for Advising Research fellowship. H.A.P. acknowledges support from the NSF Graduate Research Fellowship Program. B.T.S. acknowledges support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (F30 AR071258). E.W. acknowledges support from the National Institute of Dental and Craniofacial Research (F31 DE027586). Author contributions: J.L.G. performed data analysis, interpreted the results, and prepared the manuscript and figures. Y.S.K., V.Y.X., P.S.E., and A.G.M. interpreted results and helped prepare the manuscript. B.T.S., E.W., and H.A.P. provided guidance and assistance on rabbit bone marrow harvest. J.L. and H.A.P. helped prepare the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw7396/DC1
Fig. S1. Correlation of PdBT to starting materials on 1H and 13C NMR.
Fig. S2. Measurement of biomolecule incorporation using DMMB and Coomassie blue assays.
Fig. S3. Modulation of PdBT degradation kinetics by PGA block length.
Fig. S4. GPC standard curve.
Table S1. An overview of PdBT compared to several commonly used in situ hydrogel cross-linkers.
REFERENCES AND NOTES
- 1.Drury J. L., Mooney D. J., Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 24, 4337–4351 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Klouda L., Mikos A. G., Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 68, 34–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekenseair A. K., Boere K. W. M., Tzouanas S. N., Vo T. N., Kasper F. K., Mikos A. G., Synthesis and characterization of thermally and chemically gelling injectable hydrogels for tissue engineering. Biomacromolecules 13, 1908–1915 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watson B. M., Kasper F. K., Engel P. S., Mikos A. G., Synthesis and characterization of injectable, biodegradable, phosphate-containing, chemically cross-linkable, thermoresponsive macromers for bone tissue engineering. Biomacromolecules 15, 1788–1796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu X., Li D., Zhou F., Gao C., Biological hydrogel synthesized from hyaluronic acid, gelatin and chondroitin sulfate by click chemistry. Acta Biomater. 7, 1618–1626 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Suzuki Y., Tanihara M., Suzuki K., Saitou A., Sufan W., Nishimura Y., Alginate hydrogel linked with synthetic oligopeptide derived from BMP-2 allows ectopic osteoinduction in vivo. J. Biomed. Mater. Res. A 50, 405–409 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Holland T. A., Tabata Y., Mikos A. G., In vitro release of transforming growth factor-β1 from gelatin microparticles encapsulated in biodegradable, injectable oligo(poly(ethylene glycol) fumarate) hydrogels. J. Control. Release 91, 299–313 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Hubbell J., Matrix-bound growth factors in tissue repair. Swiss Med. Wkly. 136, 387–391 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Vo T. N., Kasper F. K., Mikos A. G., Strategies for controlled delivery of growth factors and cells for bone regeneration. Adv. Drug Deliv. Rev. 64, 1292–1309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reed S., Wu B., Sustained growth factor delivery in tissue engineering applications. Ann. Biomed. Eng. 42, 1528–1536 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Kolb H. C., Finn M. G., Sharpless K. B., Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Zhang L., Chen X., Xue P., Sun H. H. Y., Williams I. D., Sharpless K. B., Fokin V. V., Jia G., Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 127, 15998–15999 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Fan M., Ma Y., Mao J., Zhang Z., Tan H., Cytocompatible in situ forming chitosan/hyaluronan hydrogels via a metal-free click chemistry for soft tissue engineering. Acta Biomater. 20, 60–68 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Truong V. X., Ablett M. P., Gilbert H. T. J., Bowen J., Richardson S. M., Hoyland J. A., Dove A. P., In situ-forming robust chitosan-poly(ethylene glycol) hydrogels prepared by copper-free azide–alkyne click reaction for tissue engineering. Biomater. Sci. 2, 167–175 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Kettenbach K., Ross T. L., A18F-labeled dibenzocyclooctyne (DBCO) derivative for copper-free click labeling of biomolecules. Med. Chem. Commun. 7, 654–657 (2016). [Google Scholar]
- 16.Xu J., Filion T. M., Prifti F., Song J., Cytocompatible poly(ethylene glycol)-co-polycarbonate hydrogels cross-linked by copper-free, strain-promoted click chemistry. Chem. Asian J. 6, 2730–2737 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Destito P., Couceiro J. R., Faustino H., López F., Mascareñas J. L., Ruthenium-catalyzed azide–thioalkyne cycloadditions in aqueous media: A mild, orthogonal, and biocompatible chemical ligation. Angew. Chem. Int. Ed. 56, 10766–10770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahadian S., Sadeghian R. B., Salehi S., Ostrovidov S., Bae H., Ramalingam M., Khademhosseini A., Bioconjugated hydrogels for tissue engineering and regenerative medicine. Bioconjug. Chem. 26, 1984–2001 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Aulisa L., Dong H., Hartgerink J. D., Self-assembly of multidomain peptides: Sequence variation allows control over cross-linking and viscoelasticity. Biomacromolecules 10, 2694–2698 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Vo T. N., Ekenseair A. K., Kasper F. K., Mikos A. G., Synthesis, physicochemical characterization, and cytocompatibility of bioresorbable, dual-gelling injectable hydrogels. Biomacromolecules 15, 132–142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe A. B., Thiol–ene “click” reactions and recent applications in polymer and materials synthesis: A first update. Polym. Chem. 5, 4820–4870 (2014). [Google Scholar]
- 22.Kim N.-H., Le H. T., Yang Y., Byun K. M., Kim T. W., Modified DNA aptamer immobilization via Cu(I)-stabilizing ligand-assisted azide–alkyne cycloaddition for surface plasmon resonance measurement. Bull. Korean Chem. Soc. 36, 2601–2608 (2015). [Google Scholar]
- 23.Goddard-Borger E. D., Stick R. V., An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org. Lett. 9, 3797–3800 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Miller G. P., Kool E. T., Versatile 5′-functionalization of oligonucleotides on solid support: Amines, azides, thiols, and thioethers via phosphorus chemistry. J. Org. Chem. 69, 2404–2410 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Monahan F. J., German J. B., Kinsella J. E., Effect of pH and temperature on protein unfolding and thiol/disulfide interchange reactions during heat-induced gelation of whey proteins. J. Agric. Food Chem. 43, 46–52 (1995). [Google Scholar]
- 26.Lee Y., Chung H. J., Yeo S., Ahn C.-H., Lee H., Messersmith P. B., Park T. G., Thermo-sensitive, injectable, and tissue adhesive sol–gel transition hyaluronic acid/pluronic composite hydrogels prepared from bio-inspired catechol-thiol reaction. Soft Matter 6, 977–983 (2010). [Google Scholar]
- 27.Kuang J., Guo J. L., Messersmith P. B., High ionic strength formation of DOPA-melanin coating for loading and release of cationic antimicrobial compounds. Adv. Mater. Interfaces 1, 1400145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawhney A. S., Pathak C. P., Hubbell J. A., Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(. alpha.-hydroxy acid) diacrylate macromers. Macromolecules 26, 581–587 (1993). [Google Scholar]
- 29.Kaihara S., Matsumura S., Mikos A. G., Fisher J. P., Synthesis of poly(l-lactide) and polyglycolide by ring-opening polymerization. Nat. Protoc. 2, 2767–2771 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Houk J., Whitesides G. M., Structure-reactivity relations for thiol-disulfide interchange. J. Am. Chem. Soc. 109, 6825–6836 (1987). [Google Scholar]
- 31.Bian L., Guvendiren M., Mauck R. L., Burdick J. A., Hydrogels that mimic developmentally relevant matrix and N-cadherin interactions enhance MSC chondrogenesis. Proc. Natl. Acad. Sci. 110, 10117–10122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer J. L., Bertone A. L., McClain H., Assessment of glycosaminoglycan concentration in equine synovial fluid as a marker of joint disease. Can. J. Vet. Res. 59, 205–212 (1995). [PMC free article] [PubMed] [Google Scholar]
- 33.Sapan C. V., Lundblad R. L., Price N. C., Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 29, 99–108 (1999). [PubMed] [Google Scholar]
- 34.Maitland D., Campbell S. B., Chen J., Hoare T., Controlling the resolution and duration of pulsatile release from injectable magnetic ‘plum-pudding’ nanocomposite hydrogels. RSC Adv. 6, 15770–15781 (2016). [Google Scholar]
- 35.Lam J., Lu S., Lee E. J., Trachtenberg J. E., Meretoja V. V., Dahlin R. L., van den Beucken J. J. J. P., Tabata Y., Wong M. E., Jansen J. A., Mikos A. G., Kasper F. K., Osteochondral defect repair using bilayered hydrogels encapsulating both chondrogenically and osteogenically pre-differentiated mesenchymal stem cells in a rabbit model. Osteoarthr. Cartil. 22, 1291–1300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel Z. S., Young S., Tabata Y., Jansen J. A., Wong M. E. K., Mikos A. G., Dual delivery of an angiogenic and an osteogenic growth factor for bone regeneration in a critical size defect model. Bone 43, 931–940 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vo T. N., Shah S. R., Lu S., Tatara A. M., Lee E. J., Roh T. T., Tabata Y., Mikos A. G., Injectable dual-gelling cell-laden composite hydrogels for bone tissue engineering. Biomaterials 83, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hennink W. E., van Nostrum C. F., Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 64, 223–236 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Hoffman A. S., Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 64, 18–23 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Lee C. R., Grodzinsky A. J., Spector M., The effects of cross-linking of collagen-glycosaminoglycan scaffolds on compressive stiffness, chondrocyte-mediated contraction, proliferation and biosynthesis. Biomaterials 22, 3145–3154 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw7396/DC1
Fig. S1. Correlation of PdBT to starting materials on 1H and 13C NMR.
Fig. S2. Measurement of biomolecule incorporation using DMMB and Coomassie blue assays.
Fig. S3. Modulation of PdBT degradation kinetics by PGA block length.
Fig. S4. GPC standard curve.
Table S1. An overview of PdBT compared to several commonly used in situ hydrogel cross-linkers.