

SUMMARY
Antibiotics can induce mutations that cause antibiotic resistance. Yet, despite their importance, mechanisms of antibiotic-promoted mutagenesis remain elusive. We report that the fluoroquinolone antibiotic ciprofloxacin (cipro) induces mutations by triggering transient differentiation of a mutant-generating cell subpopulation, using reactive oxygen species (ROS). Cipro-induced DNA breaks activate the Escherichia coli SOS DNA-damage response and error-prone DNA polymerases in all cells. However, mutagenesis is limited to a cell subpopulation in which electron transfer together with SOS induce ROS, which activate the sigma-S (σS) general- stress response, allowing mutagenic DNA-break repair. When sorted, this small σS-response- “on” subpopulation produces most antibiotic cross-resistant mutants. An FDA-approved drug prevents σS induction specifically inhibiting antibiotic-promoted mutagenesis. Further, SOS-inhibited cell division, causing multi-chromosome cells, promotes mutagenesis. The data support a model in which within-cell chromosome cooperation together with development of a “gambler” cell subpopulation promote resistance evolution without risking most cells.
Keywords: antibiotic resistance, ciprofloxacin, error-prone DNA polymerases, Escherichia coli, evolution, fluoroquinolones, reactive oxygen species (ROS), RpoS (σS) stress response, SOS response, stress-induced mutagenesis
In Brief
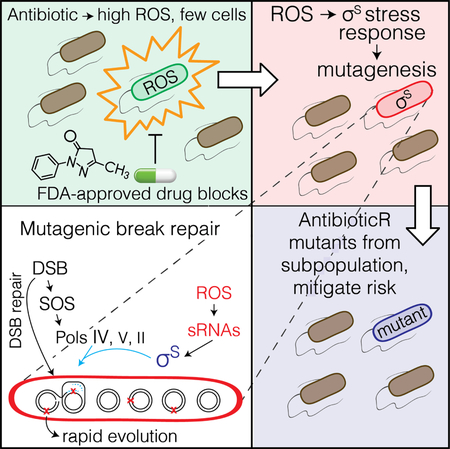
Bacteria exposed to antibiotic acquire reactive oxygen in a transient “gambler” cell subpopulation that harbors general stress response-induced mutagenic DNA break repair, evolves resistance to new antibiotics, and is inhibited by an FDA approved drug acting to inhibit evolvability.
Graphical Abstract
INTRODUCTION
Antibiotic resistance is a world health threat and occurs both by uptake of resistance genes from other bacteria, and mutation of resident genes. New mutations underpin resistance to diverse antibiotics and dominate the World Health Organization’s “priority pathogens” (Magrini, 2017). Historically, resistance has been addressed with new antibiotics. A complementary approach could be to discover, then inhibit molecular mechanisms that drive evolution of resistance (Al Mamun et al., 2012; Cirz et al., 2005; Rosenberg and Queitsch, 2014), with successes in fungi (Cowen and Lindquist, 2005; Shekhar-Guturja et al., 2016), but none yet for bacteria. Antimicrobials both select resistant mutants and can induce their formation (Cirz et al., 2005; Gutierrez et al., 2013; Kohanski et al., 2010). Although their mechanisms of growth arrest are detailed, how antibiotics induce new mutations is poorly understood.
Fluoroquinolone antibiotics inhibit bacterial type-II topoisomerases and kill cells via DNA double-strand breaks (DSBs) (Drlica, 1999). Resistance, including to ciprofloxacin (cipro), occurs mostly by de novo mutation. Cipro exposure at so-called “sub-inhibitory” concentrations (below minimal inhibitory concentration, MIC), which occurs in ecosystems and during therapies, both induces and selects cipro resistance (Cirz et al., 2005). A fluoroquinolone also induced resistance mutations to antibiotics not yet encountered (Kohanski et al., 2010)—antibiotic “cross” resistance. The mutagenesis required reactive oxygen species (ROS) induced by the drug (Kohanski et al., 2010), as does the antibiotic (killing) activity at higher MIC doses (Kohanski et al., 2007). ROS promote killing by oxidizing DNA bases, which cause more lethal DNA breaks during repair (Foti et al., 2012), but whether this underlies the ROS mutagenic activity is unknown.
Here we show that low, sub-inhibitory doses of cipro induce transient differentiation of a small cell subpopulation with high ROS and σS general stress-response activity, that generates cross-resistant mutants: a “gambler” subpopulation. We show that in gamblers, ROS activate the σS response, which allows mutagenic repair of cipro-triggered DSBs—a novel signaling/differentiating role of ROS in mutagenesis. We also find a requirement for SOS- induced inhibition of cell division, causing multiple chromosomes per cell. The findings imply a highly regulated, transient differentiation process and support a model in which within-cell chromosome cooperation together with development of a transient gambler subpopulation drive evolution of resistance to new antibiotics without risk to most cells.
RESULTS
Cipro-Induced Mutagenesis
We developed two assays for cipro-induced mutagenesis without cipro selection of the mutants (Figure 1A). In both, strains are grown in liquid, each with cipro at its minimum antibiotic concentration (MAC, final cfu 10% of no-drug cultures) (Lorian and De Freitas, 1979). These are “low-dose” and “sub-inhibitory” relative to MICs (cfu ≤10−4 of untreated). Table S1 shows MACs and MICs for all strains assayed (wild-type MAC, 8.5ng/mL). Cells are then removed from cipro and plated selectively for colonies resistant to rifampicin (RifR) or ampicillin (AmpR) antibiotics (Figure 1A), and mutation rates estimated (Methods). RifR arises by specific base-substitutions in the rpoB gene (Figure S1A), and AmpR by ampD null mutations in engineered Escherichia coli (Petrosino et al., 2002) (Figures S1B and C, Methods). Strikingly, cipro increased RifR and AmpR mutation rates 26- and 18-fold above no-cipro rates (Figure 1B, all mutation rates Table S2). The RifR or AmpR mutants are not selected in sub-inhibitory cipro, and are at a slight but significant disadvantage (Figure 1C), implying that mutation not selection of the mutants is elevated by MAC cipro. Additional controls show negligible cell death in the low-dose cipro (Figure S1D, other controls Figure S2).
Figure 1. Cipro-Induced Mutagenesis via Cipro-Induced ROS and Mutagenic Break Repair.

(A) Assays for base substitution (RifR) and null mutations (AmpR). Per Methods, with MAC cipro.
(B) MAC cipro induces RifR and AmpR mutagenesis, sequences Figure S1A–C. Mean ± 95% confidence interval (CI), ≥ 3 independent experiments. *Differ from no cipro, p<0.001, two-tailed Student’s t-test.
(C) Competition experiments: neither RifR nor AmpR mutants is selected in MAC cipro. Initial mixtures 50% mutant cfu. rpoB and ampD mutants are <50% after growth, AmpR p = 0.0098; RifR p = 0.0014, 1 sample t-test, indicating growth disadvantage. Means ± SEM, 3 independent experiments.
(D) ROS scavenger (thiourea, TU), or preventer (2,2′-bipyridine, BP) reduces mutation rates (additional controls Figure S2). Fold induction of mutation rate, raw rates Table S2. Means ± 95% CI, ≥3 experiments. *Different from medium, p<0.001, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data.
(E) Starvation-stress-induced mutagenic break repair (MBR), reviewed (Al Mamun et al., 2012; Fitzgerald et al., 2017), Results.
(F) Cipro-induced mutation requires MBR-pathway proteins. Mutants grown at their respective MACs, Table S1. Means ± 95% CIs, ≥ 4 experiments. *Different from wild-type (WT), p<0.001, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data; n.s. not significant. Epistasis analyses, Figure S1E, raw rates Table S2.
(G) Cipro induces DSBs dose-dependently. Log phase, 8.5ng/ml MAC. DSBs as fluorescent foci of phage Mu GamGFP (Shee et al., 2013). Representative images. Scale bars, 10μm. Mean ± SEM, 3 experiments.
(H) Reparable DSBs are required for cipro-induced mutagenesis. DSB-trapping GamGFP inhibits DSB repair (Shee et al., 2013) and cipro-induced mutagenesis. Means ± 95% CIs, ≥3 experiments. *Different from no GamGFP, p<0.01, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data.
(I) Cipro binding to its target type-II topoisomerases is required for induction of mutagenesis. gyrA* parC* encode functional gyrase and topoisomerase IV that are not bound by cipro. Means ± 95% CIs, 3 experiments. *Different, p<0.001, two-tailed Student’s t-test of natural-log transformed data.
See also Figures S1, S2, and S3, S4, and Tables S1, S2, and S4.
ROS-dependent Mutagenesis is σS-dependent Mutagenic Break Repair
The cipro-induced mutagenesis requires ROS, and is inhibited by ROS scavenging/preventing agents thiourea (TU) and 2,2′-bipyridine (BP) (Figure 1D, Table S2). The following indicate that the ROS instigate a σS-licensed mutagenic DNA break-repair (MBR) mechanism triggered by cipro-induced DSBs.
MBR is regulated mutagenesis during repair of DSBs, requiring the SOS and σS responses (Figure 1E) (Fitzgerald et al., 2017), causing mutations when cells are maladapted to their environments: when stressed. Spontaneous DSBs induce the SOS DNA-damage response and are repaired by homology-directed DSB repair (HR repair, Figure 1E). SOS transcriptionally upregulates error-prone DNA polymerases (Pols) IV, V and II; but repair synthesis is non-mutagenic unless the σS response is also induced (Ponder et al., 2005; Shee et al., 2011) (Figure 1E). σS, by unknown means, allows formation or persistence of errors made by Pols IV, V and II in DSB repair causing mutations (Fitzgerald et al., 2017; Ponder et al., 2005; Shee et al., 2011) near DSBs (Shee et al., 2012).
Most cipro-induced ampD and rpoB mutagenesis requires MBR proteins (Figure 1F, raw rates Table S2): RecA, RecB, and RuvC (DSB-repair), SOS- and σS-response activators, and SOS-upregulated DNA Pols IV, V, and II, implying a MBR-like mechanism. SOS non-inducible (lexAInd) or ΔrpoS (σS) strains (Table S1) showed 87%±3% and 70%±9% decreases (AmpR and RifR combined, mean ± 95% CI). Thus, two stress responses and repair are required—SOS is not sufficient. Figure 1F, Table S2). Double SOS-, σS-defective mutants show no further reduction (Figure S1E), implying action in the same pathway, as do ROS and σS (Figures S1F, S1D, S2); neither cell death nor no-drug mutation rates differ between strains (Table S2). Thus, cipro-induced ROS-dependent mutagenesis occurs by the σS-dependent MBR pathway.
The mutagenesis also requires reparable DSBs. MAC cipro induced DSBs, quantified as fluorescent foci of GamGFP DSB-end-specific binding protein (Shee et al., 2013), 28±9 times above spontaneous levels (mean ± SEM Figures 1G, S3A, S4A). GamGFP binds DSB ends preventing HR repair (Shee et al., 2013), and also inhibited cipro induction of mutagenesis (Figure 1H, Table S2), indicating that reparable DSBs are required. RecBCD, interacts specifically with DSB ends (Kuzminov, 1999), and its requirement (Figure 1F, recB) also implies the necessity of DSBs, supporting a MBR mechanism.
Special functional gyrase- and topo IV-mutant proteins that are not bound by cipro (Khodursky et al., 1995) block induction of mutagenesis (Figure 1I; Table S2), implicating cipro- induced DSBs, and making “off-target” effects unlikely. Further, σE and R-loop-promoting proteins promote starvation-stress-induced MBR by promoting spontaneous DSBs (Gibson et al., 2010; Wimberly et al., 2013) (Figure 1E), and are not required for cipro-induced MBR (Figure S1G), implying an MBR mechanism with the DSBs not from spontaneous sources, rather from cipro action on topoisomerases.
ROS Differentiate a Cell Subpopulation, Activate σS Response
We surveyed single log-phase cells for ROS and stress-response induction by flow cytometry. At time “0” cipro is added in early log-phase, and all log-specific work is at 16h unless stated otherwise. SOS reporter PsulAmCherry at a non-genic chromosomal site (Nehring et al., 2016; Pennington and Rosenberg, 2007) revealed population-wide dose-dependent SOS induction (Figure 2A), with 208±26 times more SOS-positive cells at the 8.5ng/mL mutagenic MAC than without drug. Auto-fluorescence (Renggli et al., 2013) is negligible (Figures S4B–D).
Figure 2. ROS Form in Minority Cell Subpopulation, Activate σS Response and MBR.

(A-C) Cells analyzed in log-phase growth (16h). The MAC cipro is 8.5ng/mL (WT), for all strains, Table S1.
(A) Dose-dependent activation of the SOS response by cipro. Flow-cytometry with chromosomal SOS reporter PsulAmCherry. SOS-positive cells, right of the gate shown (black bars, Methods). Afu, arbitrary fluorescence units. Means ± SEM, 3 experiments. *Different from no cipro, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(B) MAC cipro induces high ROS in a 20%±3% cell subpopulation in log phase. Flow cytometry, ROS dye dihydrorhodamine 123 (DHR). ROS-high cells, within the gate (black bar). Means ± SEM, 3 experiments. *Different from no cipro, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(C) MAC cipro induces high σS-response activity in 27%±3% cell subpopulation in log phase. σS-response reporter yiaG-yfp. σS-high cells, within the gate (black bar, Methods). Note: smaller σS-high subpopulations of ~10% of cells in stationary phase (E below, Figures 3 and 4). Means ± range, 2 experiments. *Different from no cipro, p<0.01, one-way ANOVA with Tukey’s post- hoc test.
(D) Cipro induction of mutagenesis occurs maximally at the 8.5ng/mL MAC. Means ± range, ≥2 experiments.
(E) ROS are required for cipro-induced σS response. ROS scavenger TU removes the σS-high cell subpopulation in log (16h) and stationary phase (24h), MAC cipro. Means ± SEM, 3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(F) ROS are required for MAC-cipro-induced σS increase. TU reduces σS accumulation in log (16h) and stationary phase (24h). Representative western blot and quantification. Means ± range, 2 experiments. *Different from no cipro, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(G) Engineered σS production substitutes for ROS, allowing mutagenesis in TU-treated cells. Means ± 95% CIs, ≥3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test; n.s., not significant.
(H) Cipro induces DSBs ROS independently, unaffected by TU or BP. GamGFP (DSB) foci log-phase (16h) with or without MAC cipro. Representative images. Scale bar, 5 μm. Means ± SEM, ≥ 3 experiments. *Different from no cipro, p<0.001, one-way ANOVA with Tukey’s post-hoc test. (I) SOS induction is independent of ROS, unaffected by TU or BP. Per A. Means ± SEM, ≥ 3 experiments, one-way ANOVA with Tukey’s post-hoc test.
(J) Summary: cipro-induced ROS induce the σS response, which allows mutagenic break repair (MBR) and mutations. Not shown: the ROS and σS response occur in minority cells subpopulation(s).
See also Figures S1, S2, S3, S4, and S5, and Tables S1, S2, and S3
Surprisingly, only discreet cell subpopulation(s) showed strong ROS or σS induction. ROS, detected with dihydrorhodamine 123 dye (DHR, Figure 2B) in log phase, appear in a distinct 20%±3% cell subpopulation (mean ± SEM), and high σS activity [yiaG-yfp fluorescence (Al Mamun et al., 2012)] in a discreet 27%±3% of the cells (Figures 2C and S3B). Both ROS- or σS-high subpopulations arose above a threshold, with the 8.5ng/mL MAC dose most inducing (Figure 2B, C), then declined at higher doses (Figure 2B, C) mimicking the dose response of mutagenesis (Figure 2D). MAC doses are used for all following work (e.g., Figures 3 and 4). ROS- and σS-high subpopulation sizes change throughout growth, peaking in log phase (16h) and declining to near 10% at stationary phase (24h, 48h, Figures 2E, S3C), when mutagenesis is assayed (time survey Table S3, graphs Figure 4A). Antibiotic growth inhibition requires ROS (Kohanski et al., 2007), and also occurred above an 8.5ng/ml threshold (Figure S3D). The discreet ~20% subpopulation(s) of the log-phase cells have very high ROS or σS-activity (Figure 2B, C).
Figure 3. σS-response-high Gambler Cell Subpopulation Generates Mutants, Is Inhibited by FDA-approved Drug.

(A) Most cross-resistant mutants are produced by the minority σS high-activity cell subpopulation. MAC cipro-treated cells (24h) with σS-response or lac reporters FACS sorted and assayed for mutagenesis. ≥88% of cipro-induced mutants arose in 13% of cells (text). Means ± 95% CI, 3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test; n.s., not significant.
(B) High catalase activity in σS-high cells confirms σS-high status. HPII, the σS-regulated catalase. Means ± SEM, 3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(C) FDA-approved antioxidant drug edaravone reduces the σS-response-activated cell subpopulation. Flow-cytometry of stationary-phase (24h) cells. Means ± range, 2 experiments. *Different from medium, p<0.001, one-way ANOVA with Tukey’s post-hoc test.
(D) Edaravone decreases ROS-high subpopulation. Per Figure 2E,F,G. Flow-cytometry of log-phase (16h) cells, MAC cipro, stained with DHR ROS dye. Different from medium, p<0.001, one-way ANOVA with Tukey’s post-hoc test.
(E) Edaravone inhibits cipro-induced mutagenesis. Means ± range, 2 experiments. *Different from no-drug, p<0.001, one-way ANOVA with Tukey’s post-hoc test.
(F, G) Edaravone does not affect (F) MAC cipro induction of GamGFP (DSB) foci, log phase (16h), or (G) the SOS response. Means ± range, 2 experiments, one-way ANOVA with Tukey’s post-hoc test.
(H) Edaravone does not reduce high-dose cipro antibiotic killing activity. Log-phase cells grown with or without 1.5μg/mL cipro. Means ± range, 2 experiments.
(I) Summary: the σS high-activity cell subpopulation generates most resistant mutants: a gambler cell subpopulation. FDA-approved drug edaravone inhibits mutagenesis, reduces ROS and σS-high gambler subpopulations. Ovals, E. coli cells.
See also Figures S2, S4, S5, S6, and S7, and Tables S1 and S2.
Figure 4. ROS-high Subpopulation Cells Become σS-High Cells.

(A) ROS-high cells become many of the σS high cells. Flow cytometry time course of cells with MAC cipro, with and without ROS reducers BP, TU, or edaravone (edar). ROS-high cells precede σS-high cells, and double-positive cells (upper right quadrants 8–48h) indicate that many σS-high cells arise from ROS-high cells. Representative of 3 experiments.
(B) Live ROS-high cells, detected by PoxyRgfp or PsodAgfp oxidative-stress-response reporters. MAC cipro time course. Double-positive cells indicate that many σS-high cells had high ROS. Representative of 2 experiments.
(C) Most or all σS-high red cells arise from oxidative response-activated green cells. Live-cell time-lapse deconvolution microscopy imaging of cells carrying PsodAgfp and σS-response reporter yiaG-mCherry grown with MAC (8.5ng/mL) cipro for 8 hours, were imaged over 12 additional hours. Essentially all σS-high cells at 24h arose from cells that were ROS-high at 9–19h (>99%). Also, most (54%) but not all (28%) ROS-high cells at 19h became σS-high at 24h, and some 19h ROS-high cells lose their ROS by 24h (19%). Scale bar, 10μM. Mean ± range, 2 experiments tracking ≥ 250 cells.
ROS scavenging/preventing agents TU or BP blocked σS-response induction, removing the σS-high subpopulation (Figures 2E and S3E), reduced accumulation of σS-protein (Figure 2F), or of a σS-β-galactosidase reporter (Figure S3F). Thus, ROS are required for induction of the σS response, as for mutagenesis (Figure 1D, additional controls Figure S5A and B).
Moreover, engineered σS upregulation fully substituted for ROS in mutagenesis (Figures 2G, S5C,D), implying that the main ROS role in cipro-induced MBR (Figure 1D) is σS induction. ROS and σS also act in the same mutation pathway (Figure S1F). Cells with active but cipro-non-binding GyrA* and ParC* proteins (Khodursky et al., 1995) showed no induction of SOS, ROS, or σS responses (Figure S3G–I), indicating that the events that lead to SOS, ROS, and σS induction begin with cipro interaction with its targets.
ROS promote antibiotic (growth-inhibitory) activity (Table S1) by creating DNA breaks via oxidized guanine (8-oxo-dG) in DNA (Foti et al., 2012; Kohanski et al., 2007). In contrast, reduction of cellular ROS with TU or BP, though inhibitory to the MBR mutagenesis (Figure 1D), did not reduce MAC-cipro induction of DSBs, quantified as GamGFP foci (Figures 2H and S3J), nor SOS (Figure 2I). Also, 8-oxo-dG incorporation appears not to underlie ROS action in the mutagenesis in that ROS-mediated 8-oxo-dG-signature mutations [G·C➝T·A and A·T➝C·G, (Schaaper and Dunn, 1987)] are less frequent in cipro-induced than spontaneous forward mutations (Figure S1C). Thus, ROS drive mutagenesis other than by DNA damage, SOS, or misincorporation opposite 8-oxo-dG.
The data show that cipro action on topoisomerases leads to induction of high ROS in a discreet cell subpopulation (Figure 2B); the ROS activate σS in a subpopulation (Figure 2C,E–F); and σS activation is how ROS promote cipro-induced MBR (Figure 2G,J). This constitutes a novel role for ROS in mutagenesis—signaling induction of the σS stress response—unlike those in antibiotic activity or starvation-stress-induced MBR (Moore et al., 2017).
σS-active Gambler Cell Subpopulation Generates Mutants
Fluorescence-activated cells sorting (FACS) revealed that the small σS-high subpopulation produces most cipro-induced mutants (Figure 3). We sorted σS high- and low-activity in 24h stationary cells, when 13%±1% of cells are σS- high, to at least 97% enrichment (Figures S6A–C). Remarkably, whereas unsorted and mock-sorted cells show (mean) 25±4-fold induction of RifR mutant frequencies (Figure 3A), the sorted σS-high cells displayed 410±70-fold induction—16±3-times higher than unsorted or mock-sorted cells (Figures 3A,B, S6D,E, and S7A). Though untreated cells have higher σS-activity at 24h than during log phase (4h and 24h, Figure S5G) the activity is much less than in cipro-treated σS-high cells (Figures 4A and S5E). Moreover, the σS-low subpopulation, 87%±1% of cells, showed 8±2-times fewer mutants than unsorted or mock-sorted cells (Figure 3A), indicating that few if any mutants arise in the majority subpopulation. To estimate the contribution of each subpopulation to yields of mutants: because σS-low cells display only a 3±1-fold increase in RifR mutants (Figure 3A), we can conclude that the σS-low cells produced about 12% of the mutants (3-fold increase / 25-fold increase in un/mock-sorted = 12%, Figure 3A). Thus, at least 88% of RifR mutant yield originates in the σS-high cells.
σS-high cells could have higher levels, or better survival, of mutagenesis. Either way, most mutants, and so most evolvability, arises from the them. Death is similar in σS-high and - low cells (Figure S7B) suggesting more mutagenesis, not obviously better survival. Both high σS activity and mutability are transient. The RifR mutants recovered are neither σS-high (Figure S5F) nor heritably “mutator” (Figure S7C). Greater mutant production does not result indirectly from high fluorescence (possible high metabolic activity): see Placcfp (Figures 3A, S4E and S2A,B).
Thus, a small, transiently differentiated σS-high subpopulation is transiently hypermutable and produces most cipro-induced Rif (cross)-resistant mutants, suggesting a potential “bet-hedging” developmental strategy (Norman et al., 2015; Veening et al., 2008) that may allow evolution while only some cells risk mutagenesis; we call these gamblers.
FDA-approved Drug Inhibits Evolvability
The gambler subpopulation could be a therapeutic target for inhibition of cipro-induced mutagenesis to antibiotic resistance, cross resistance and immune evasion. We found that the ROS-reducing drug edaravone, indicated for ALS and cerebral infarction (Watanabe et al., 2018), inhibits cipro-induced mutagenesis but not its antibiotic activity. At concentrations used clinically (100μM) (Parikh et al., 2016), edaravone inhibited appearance of σS-high cells (Figure 3C), σS-fusion protein (Figure S3F), ROS-high cells (Figure 3D), and most (82%±1% of) RifR mutagenesis (Figure 3E). Edaravone did not affect cipro induction of DSBs (Figure 3F), SOS (Figure 3G), cell growth (Figure S2A), colony formation (Figures S2B), or negative-control β-gal activity (Figure S5B), implying specific inhibition of σS induction (Figure 3I). Importantly, edaravone did not reduce high-dose cipro killing (Figure 3H), showing that it can reduce mutagenesis without altering cipro utility as an antibiotic. These data serve as a proof-of-concept for small-molecule inhibitors that could be administered with antibiotics to reduce resistance evolution by impeding differentiation of gamblers, without harming antibiotic activity.
ROS-high Cells Become Gamblers via sRNAs
We explored how gamblers are differentiated (Figure 3I), following single cells over time with flow cytometry, and found that ROS (DHR dye, green) appear 4 hours after cipro, before σS activity (red fluorescence reporter) is detectable (Figure 4A, Methods). Then, double-positive ROS (green) σS-active (red) cells develop between 8 and 16 hours (Figures 4A and S5G), showing that at least some σS-high cells begin as ROS-high cells. At 24h, when cells were harvested for sorting/mutagenesis assays (Figure 1A,B), many double-positive ROS-/σS-high cells were present (upper right quadrant, Figure 4A 24h), as were some σS-high single-positive cells (lower right quadrant, Figure 4A 24h). We used live-cell imaging with fluorescence-reporter genes (green) for two different oxidative stress responses, in cells that also carry the red σS-response reporter, to follow single live cells over time from their burst of ROS to σS-response induction. The reporters are transcriptional GFP fusions for oxyR (peroxide) and sodA (superoxide) responses, and both show double-positive and some σS-single-positive cells with flow cytometry at 24 hours (Figure 4B).
Time-lapse microscopy showed that essentially all red σS-active gambler cells arose from oxidative-stress-response-activated green cells (sodA reporter, > 99%, Figure 4C, Movie S1). Some of the σS-high cells showed reduced ROS after σS induction (Figure 4C, Movie S1), suggesting amelioration of high ROS levels by the σS response.
We investigated how ROS activate the σS response (Figure 5). σS is regulated at multiple levels including upregulation by small RNAs (sRNAs) ArcZ, RprA, and DsrA which promote σS translation assisted by Hfq RNA chaperone (Battesti et al., 2011). Hfq, DsrA and ArcZ, but not RprA, are required for induction of σS protein (Figure 5A), differentiation of the gambler subpopulation (Figure 5B), and mutagenesis Figure 5C,D), with no further decrease in the double or triple mutants, implying action of Hfq and the sRNAs in the same pathway (Figure 5A–C). Further, Hfq can be substituted by production of σS from a plasmid, which restored 86%±10% of mutagenesis to Δhfq cells (Figure 5D, S2A,B, Table S2), implying that Hfq promotes mutagenesis mostly or wholly by promoting σS induction, presumably via ArcZ and DsrA sRNAs.
Figure 5. ROS induce transcription of sRNAs that upregulate σS general stress response.

(A) sRNAs DsrA and ArcZ and the Hfq RNA chaperone are required for cipro-induction of σS protein. Stationary phase (24h) with MAC cipro. RssB facilitates degradation of σS protein. The increase of σS levels in ΔrssB cells without cipro, but not with, implies reduced σS degradation when cipro-induced ROS upregulate σS. Means ± SEM, 3 experiments. *Different from WT with cipro (top half) or WT without cipro (bottom half), p<0.01, one-way ANOVA with Tukey’s post- hoc test.
(B) DsrA, ArcZ, and Hfq allow cipro induction of σS activity, stationary-phase (24h). Representative flow cytometry histograms show loss of σS-high cells in dsrA, arcZ and hfq null mutants. Means ± SEM, 3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test; n.s. not significant.
(C) DsrA and ArcZ required for cipro-induced mutagenesis and act in the same pathway. Means ± range, ≥2 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(D) Artificial upregulation of σS substitutes for Hfq in mutagenesis indicating that Hfq promotes mutagenesis by σS upregulation. Means ± 95% CIs, ≥3 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(E) Cipro-induced ROS induce the dsrA and arcZ promoters. β-galactosidase activity, PdsrAlacZ and ParcZlacZ reporters in log (16h) and stationary phase (24h), ± ROS reducers TU, BP, or edaravone. Means ± range, 2 experiments. *p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(F) Summary: Cipro-induced ROS in subpopulation cells induce transcription of DsrA and ArcZ sRNAs which, with the Hfq RNA chaperone, upregulate σS in the ROS-high cells (Figure 4).
See also Figures S2, S5, and Tables S1 and S2.
Moreover, cipro induced dsrA and arcZ transcriptional lacZ fusions, which showed 2.3±0.3- and 53±3-fold induction in log (16h), and 7.4±0.4- and 48±1.3-fold in stationary phase (24h) (Figure 5E), ROS-dependently, reduced by TU, BP, and edaravone (Figure 5E). Thus, cipro-induced ROS increase transcription of DsrA and ArcZ sRNAs, which, with Hfq, allow creation of the σS-high gambler subpopulation. The smaller stationary-phase increase of σS activity in the main population (Figure S5G no cipro 4h and 24h) did not require these sRNAs (Figure 5B, middle histogram, compare arcZ dsrA with rpoS), suggesting that they specifically allow cipro-induction of the ROS-promoted σS response. Although DsrA and ArcZ are necessary for ROS upregulation of σS and mutagenesis (Figure 5A–C), they may not be sufficient; ROS might promote σS induction via other mechanisms. The sRNAs are required for and their levels correlate with differentiation of ROS-high subpopulation cells into σS-active gamblers (Figure 5F).
One way that the σS response is kept “off” in unstressed cells is via RssB, which delivers σS to the ClpXP protease for degradation. Using a rpoS-lacZ reporter (Zhou and Gottesman, 2006), deletion of rssB increased σS in untreated, but not in cipro-treated cells (Figure 5A), implying that detectable RssB-mediated σS degradation occurs without but not with cipro treatment. Similarly, ΔrssB did not increase cipro-induced mutagenesis (Figure 5C), suggesting that cipro induction of σS already includes downregulation or saturation of RssB-mediated degradation.
ROS Induced via SOS Response and Ubiquinone
The ROS-induction pathway is only partly characterized (Figure 3I) (Dwyer et al., 2015). We found that mutagenesis was reduced in cells lacking UbiD (biosynthesis of ubiquinone), but not other components of the electron-transfer-chain (ETC) shown to promote σS activity during starvation-stress-induced MBR: NuoC (ubiquinone oxidoreductase I, an ETC “complex I” subunit), and CyoD (a subunit of cytochrome bo’ oxidase, an ETC “complex II” subunit) (Al Mamun et al., 2012) (Figures 6A and S2A,B). UbiD/ubiquinone appear to act upstream of σS-response induction in mutagenesis, in that artificial production of σS substituted for UbiD, restoring most or all mutagenesis (87%±16%) to ΔubiD cells (Figure 6B). We found reduction of σS accumulation, σS activity, ROS, but not SOS activation in ΔubiD (ubinquinone-deficient) cells (Figure 6C–F), indicating that ubiquinone, and by implication, electron transfer, are required for cipro induction of the σS response, and act downstream of (after) SOS induction and before (upstream of and by promoting) ROS induction.
Figure 6. ROS Induction Requires SOS and Ubiquinone (Electron Transfer).

(A) Ubiquinone promotes cipro-induced mutagenesis. Mutant MACs, Table S1. Means ± CIs, ≥ 3 experiments. *Different from wild-type (WT), p<0.01, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data.
(B) Artificial upregulation of σS substitutes for UbiD in mutagenesis, implying that UbiD promotes mutagenesis by upregulation of σS. Means ± 95% CIs, ≥ 3 experiments. *Different from WT, p<0.01, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data; n.s. not significant.
(C) Cipro-induced σS-β-galactosidase activity, reflecting σS accumulation, is promoted by UbiD. MAC cipro-grown 24h stationary-phase cells. Means ± range, 2 experiments. *Different from WT, p<0.01, two-tailed Student’s t-test.
(D) Cipro-induced σS activity requires UbiD. Flow-cytometry shows loss of the σS-high subpopulation in ubiD-null cells. MAC cipro-grown 24h stationary-phase cells. Means ± SEM, 2 experiments. *Different from WT, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(E) Cipro induction of ROS requires UbiD. ROS-positive cells in log phase (16h) MAC cipro, seen with DHR dye. Means ± SEM, 3 experiments. *Different from WT, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(F) Cipro induction of the SOS response does not require UbiD. SOS activity in stationary-phase (24h) MAC cipro-grown cells. Means ± range, 2 experiments, two-tailed Student’s t-test.
(G) UbiD promotes cipro induction of the H2O2 responsive katG-lacZ fusion. MAC cipro log-phase (16h) cells. Means ± range, 2 experiments. *p<0.01, two-tailed Student’s t-test
(H) The SOS response is required for cipro induction of the ROS-high cell subpopulation. SOS non-inducible lexAInd− and recB cells, which are defective in SOS induction by DSBs. Cells grown in low-dose MAC cipro and assayed in log phase (16h). Means ± range of 2 independent experiments. *Different from WT at p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(I) The SOS response is required for cipro induction of the σS-high subpopulation. SOS non inducible lexAInd− cells, and recB cells at 24h MAC cipro (stationary phase). Means ± range, 2 experiments. *Different from WT, p<0.01, one-way ANOVA with Tukey’s post-hoc test.
(J) Model for cipro-induction of ROS via the SOS response and ubiquinone. Cipro-induced DSBs activate the SOS response in all cells. SOS slows aerobic respiration (Swenson and Schenley, 1974), which promotes autoxidation of ubiquinone (Gonzalez-Flecha and Demple, 1995; Skulachev, 1998), we suggest in a cell subpopulation that becomes the σS-high subpopulation (Figure 5).
Ubiquinone functions in the aerobic ETC, mediating oxido-reduction cycles for ATP production (Meganathan and Kwon, 2009). The ΔubiD cells showed severely reduced ROS in cipro with 8%±4% ROS-high cells in log phase compared with 32%±9% in WT (Figure 6E), and reduced katG-lacZ activity, a reporter activated by H2O2 (Table S5, Figure 6G). We can infer that SOS acts upstream of, or in parallel with, ubiquinone in ROS induction (Figure 6H), not downstream of ubiquinone, which is not needed for SOS induction (Figure 6F). In assays without cipro, SOS inhibited aerobic respiration (Swenson and Schenley, 1974), and slowed respiration-promoted autoxidation of quinols leading to superoxide (Gonzalez-Flecha and Demple, 1995; Skulachev, 1998). These data without cipro and ours with cipro support a model in which SOS activation may inhibit the ETC leading to ROS (Figure 6J). Though necessary for the cascade to σS induction (Figure 6H,I), SOS seems not to be sufficient in that most SOS-induced cells do not display high ROS or σS (Figure 2A–C). One possibility is that the cipro-induced SOS response might inhibit/slow aerobic respiration in only a cell subpopulation, allowing autoxidation of ubiquinone to produce high ROS levels in those cells (Figure 6J).
Multi-Chromosome Cells Allow Evolvability
In MAC cipro E. coli forms long, multi-chromosome cell “filaments” that “bud off” small, normal- length daughter cells that produce cipro-resistant mutants efficiently (Bos et al., 2015), suggesting that multiple chromosomes might promote adaptation. Recombination or allele sharing might mitigate deleterious effects of multiple mutations (Bos et al., 2015), and/or increase repair probability, allowing survival.
We reduced the multiple chromosomes and cell length by a significant half by knock-out of the SulA SOS-induced cell-division inhibitor (Figure 7), which promotes filamentation (Huisman and D’Ari, 1981). Reducing filamentation, seen by counting TetR-mCherry-marked chromosomes as foci (Figure 7A–F) (Joshi et al., 2013), and cell size by microscopy (Figure 7D,E, filament definition Methods), reduced mutagenesis (Figure 7G).
Figure 7. Multi-chromosome Bacterial Cells Promote Cipro-induced Mutagenesis.

(A) Scheme for labeling chromosomes as red fluorescent foci using a chromosomal tetO array bound by Tet-repressor-mCherry (TetR-mCherry) in a replication-origin (oriC)-proximal site. Red circle, plasmid that produces TetR-mCherry. Multiple TetR-mCherry foci represent approximate number of ori-proximal chromosomal equivalents (Joshi et al., 2013).
(B) More than 33% of log-phase MAC cipro-grown cells carry multiple chromosomes: ≥4 per cell, per A, 1919 cells counted. Representative images of DAPI-stained WT cells. Scale bar, 5μm.
(C) Fewer than 1% of log-phase cells grown without cipro have ≥4 chromosomes per cell. Per (A), 3915 cells counted. Representative images. Scale bar, 5μm.
(D-F) Cipro induction of the multi-chromosome state requires SulA. Scatterplots of microscopically determined distributions of cell lengths (μM) and chromosome (TetR-mCherry) foci, with and without MAC cipro. Data from 3 experiments.
(D) Cipro induction of multi-chromosome cells.
(E, F) SulA is required for the cipro-induction of long, multi-chromosome cells. 98% of untreated cells show ≤4 chromosomes per cell, more than half dependent on SulA. nc, no cipro. Means ± SEM, 3 experiments. *Different from WT at p<0.001, one-way ANOVA with Tukey’s post-hoc test.
(G) The SulA-dependent multi-chromosome state promotes cipro-induced mutagenesis. SulA and RuvC act in the same MBR pathway (are epistatic). Data also presented as fold induction of mutation rate per chromosome per generation (green bars). sulA cells still show less mutagenesis (p<0.05). σS-high cells’ induction of mutagenesis per chromosome per generation (yellow bar) exceeds WT using either mutations per chromosome or per cell per generation (p<0.01). Means ± 95% CIs, ≥ 4 experiments. *Different from WT, p<0.05, one-way ANOVA with Tukey’s post-hoc test of natural-log transformed data; n.s. not significant.
(H) Mathematical model shows that multi-chromosome filaments have a large advantage for adaptation and survival at high mutation rates. Left panel, expected relative cfu of all surviving cells (adapted and not-adapted) as a function of the number of deleterious mutations accumulated. Middle panel, the expected relative cfu of adapted cells as function of the number of deleterious mutations accumulated. Right panel, formation of multi-chromosome filaments can increase the surviving population size when selection is harsh. The fold-increase of surviving population size due to filamentation as function of the fold increase in mutation rate due to filamentation, for several selection parameters. s – selection coefficient of the major stress (e.g., antibiotics). Model description and parameters: Methods.
(I) Model: mechanism of cipro-induced transient differentiation of an evolvable gambler cell subpopulation that allows stress-responsive MBR without risk to most cells. Left to right: cipro-binding to type-II topoisomerases causes DSBs that activate the SOS response throughout the cell population. SOS upregulates error-prone DNA polymerases and SulA, which inhibits cell division causing multi-chromosome cells. SOS also slows aerobic respiration, we suggest, in a cell subpopulation, which generates ROS promoted by autoxidation of ETC component ubiquinone. The ROS activate transcription of σS-upregulating sRNAs DsrA and ArcZ, which, with Hfq RNA chaperone, promote translation of σS protein, thus activating the general stress response in the cell subpopulation, and allowing MBR in those cells—a transient hypermutable state in gambler cells (red cells). The multi-chromosome state promotes survival and adaptation of highly mutated cells by amelioration (complementation and reassortment) of deleterious recessive mutant phenotypes generated.
See also Figures S2, S3, S4, and S7 and Tables S1, S2, and S4.
Because multiple chromosomes per cfu in cipro might increase apparent mutation rate—mutations per cfu per generation—without increasing mutagenesis per chromosome, we counted marked chromosomes (Figure 7A) and calculated cipro-induced mutation rates per chromosome per generation. Mirroring the per-cell rates (Figure 1F), WT per-chromosome mutation rate is induced 7±1.5-fold by cipro (Figure 7G; mean Amp and Rif), whereas, the per chromosome rates are not induced in mutants that lack σS (0.73±0.03), recA (0.29±0.11), recB (1.0±0.19), ruvC (0.41±0.07), Pol IV (0.64±0.05), Pol II (1.0±0.4), Pol V (0.83±0.35), Pols II, IV and V (0.83±0.17) or are SOS-non-inducible lexAInd− (1.2±0.15) (raw rates, fold inductions, p values Table S4). ΔsulA cells show significant reduction of per-chromosome mutation rate by cipro: 4.0±0.5-fold compared with 7±1.5-fold in WT (Figure 7G, Table S4), implying that mutagenesis itself is promoted by SulA/multi-chromosome cells. Per-cell mutation rate in ruvC cells is not reduced further by ΔsulA (Figure 7G, Table S4), implying that SulA promotes RuvC-dependent (MBR).
We also allowed cipro-treated cells to resolve their filaments to small cells by 4–6 hours growth without cipro after their 18–19h in cipro, and mutation rate per cell per generation did not differ from the standard 24-hr cipro assay (Figure S7D). Thus, neither filamentation nor mutation rate calculation method alters conclusions drawn here. Previously SulA was required for about half of starvation-induced MBR (McKenzie et al., 2000), though whether the starving (not dividing) cells filamented was not examined, making interpretations tentative.
SulA is required for formation of the ROS-high and most of the σS-high gambler cells (Figures S7E, F), and not via promoting HR, shown with HR-defective, SOS-proficient ΔruvC cells (Figure S7G). Overproduction of σS did not substitute for SulA (Figures S7H), indicating a possible SulA role in MBR in addition to promoting σS. Alternatively, the optimal intensity of σS- high cells for mutagenesis is not known, and the profile of σS activity levels produced by overproduction (Figures S5D) might be altered by ΔsulA and insufficient for MBR in ΔsulA cells. Also, SOS, which is fully required for ROS/σS-active gambler formation (Figure 6I), appears to promote ROS/σS activity both SulA-dependently and -independently.
We formulated a mathematical model to test possible benefits of multi-chromosome filaments for rapid adaptation (Methods), per Bos et al. (2015). Results of the model (Figure 7H) show that increasing filament mutation rate increases the probability of both adaptation and survival of a chromosome relative to non-filamented cells, supporting cooperation accelerating complex adaptations (Obolski et al., 2017). The advantage of multiple chromosomes increases with increasing selection coefficient (Figure 7H, e.g., lethality of a drug in cells under selection for resistance). This model shows that multiple chromosomes could facilitate adaptation by mutagenesis (model, Figure 7I).
DISCUSSION
Our findings (Figure 7I) unite quinolone-induced mutagenesis with σS-dependent stress-induced mutagenesis, defined as mutation-producing mechanisms upregulated by stress responses (Fitzgerald et al., 2017). Coupling mutagenesis to stress responses generates mutants preferentially when cells or organisms are maladapted to their environments—when stressed—potentially accelerating adaptation (Fitzgerald et al., 2017; Ram and Hadany, 2012).
ROS Regulate Mutagenesis
We found a novel regulatory and differentiating mutagenic role of ROS (Figure 7I). ROS can promote mutagenesis by direct mechanisms, including oxidation of guanines to 8-oxo-dG, which pairs with A, causing G-to-T and T-to-G (A-to-C) mutations (Schaaper and Dunn, 1987), and are repaired causing DSBs (Foti et al., 2012), neither of which is observed (Figures S1B,C, 2H), counter-indicating both possible mechanisms. Rather, the ROS mutagenic role can be substituted by production of σS, which activates the general/starvation stress response (Figures 2G) and allows MBR, showing σS activation to be the main ROS role. The ROS induce transcription of ArcZ and DsrA sRNAs (Figure 5E), which, assisted by Hfq RNA chaperone (Figure 5A–F), promote translation of σS protein (Battesti et al., 2011), which allows MBR mutagenesis (Figures 1E–I, 3A and 7I). This differs from a mutagenic role of Hfq with another sRNA via downregulation of translation of a mismatch-repair protein (Chen and Gottesman, 2017). Induction of ROS by cipro precedes σS-response activation (Figure 4), and the ROS-high cells become σS-high cells (Figure 4C, movie S1) that generate mutants (Figure 3A). These data highlight the centrality of stress-response-control of mutagenesis, and that ROS are signaling molecules in this regulation.
Mutagenesis in Transiently Differentiated Gamblers
Cipro-induced ROS lead to high σS activity in a 10–25% cell subpopulation (Figures 2B,C,E and 4) that is transiently mutable (Figure S7B), and produces most of the mutants (Figure 3A,C). Transient differentiation in subpopulations is a potential evolutionary “bet-hedging” strategy, in which some cells risk a phenotype that may be advantageous or not depending on the environment (Norman et al., 2015; Veening et al., 2008). “Persisters” tolerate lethal drugs but reduce proliferation (Balaban et al., 2004); competence for natural transformation (Chen and Dubnau, 2004), sporulation (Norman et al., 2015), and even programmed cell death (Amitai et al., 2009; Gonzalez-Pastor et al., 2003), are hypothesized or demonstrated (Gonzalez-Pastor et al., 2003) to aid siblings of the sacrificed bacteria. Limitation of mutagenesis to a subpopulation appears to embed environmentally tuned mutagenesis within a “bet hedging” strategy (Norman et al., 2015; Torkelson et al., 1997; Veening et al., 2008). Though transiently mutable subpopulations have been hypothesized (Hall, 1990; Ninio, 1991), supported by genetic evidence (Torkelson et al., 1997), and cells with stress responses linked to mutagenesis of unknown mechanism (Woo et al., 2018), our data provide the first isolation (Figure 3) of a hypermutable cell subpopulation in the act of mutagenesis, and show the defining, differentiating inputs: ROS and the general stress response (Figures 3 and 4), as well as the mutagenesis mechanism: MBR (Figure 7I)—all novel mechanisms of potential promotion of the ability to evolve. Unlike “persisters,” these cells take the risk of inducing mutations, which can lead to heritable resistance to never-before-encountered antibiotics. They are “gamblers.”
Drugging Evolvability
The FDA-approved drug edaravone behaved as an “anti-evolvability” drug by removing the ROS- then σS-high gambler subpopulation, without reducing the antibiotic power of cipro (Figure 3C–H), providing a promising proof-of-concept. Other ROS-promoted mutagenesis mechanisms may involve upregulation of the σS response and so be similarly susceptible. σS promotes MBR (Lombardo et al., 2004; Ponder et al., 2005; Shee et al., 2011), downregulates mismatch repair activity (Gutierrez et al., 2013), activates transposition (Ilves et al., 2001), and possibly other mechanisms. Stress-response regulators, such as σS, are non-redundant hubs in the MBR network (Al Mamun et al., 2012), making them attractive targets for drugs to slow evolution of pathogen resistance and immune evasion (Al Mamun et al., 2012; Fitzgerald et al., 2017; Rosenberg and Queitsch, 2014).
Multichromosome Cells Promote Evolvability
Multiple chromosomes may aid mutagenesis by providing more repair partners for MBR, and/or promoting adaptation by cooperation (Obolski et al., 2017): sharing of alleles (recombination) and/or gene products (while compensatory mutations occur), masking deleterious phenotypes (Figure 7H, I). Cell “filaments” may be biomarkers of rapid evolution. Bacillus subtilis undergoes natural transformation activated by the Com stress response, which also upregulates mutagenesis (Sung and Yasbin, 2002), thus engaging recombination with mutagenesis (Lenhart et al., 2012). E. coli is incapable of natural transformation, but may achieve the mutate-and- recombine (or share) strategy via multiple sibling chromosomes within one cell, rather than exogenous sibling DNA. In addition to targeting stress-response regulators as an anti- evolvability drug strategy [(Al Mamun et al., 2012; Fitzgerald et al., 2017; Rosenberg and Queitsch, 2014) and Figure 3C–H], dividing (and conquering) the multiple chromosomes might also reduce evolvability as a therapeutic strategy.
STAR★Methods
CONTACT FOR REAGENTS AND RESOURCE SHARING
The corresponding author, Susan M. Rosenberg (smr@bcm.edu), is the contact for reagents and resource sharing.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli (strain MG1655) and isogenic derivatives were used for all experiments.
METHODS DETAILS
Strains, Media, and Growth
E. coli strains used are shown in the Table S5, and the specific strains used in each figure listed in the following section. Bacteria were grown in LBH rich medium (Torkelson et al., 1997) at 37°C with aeration, and additives where indicated a t the following concentrations: ciprofloxacin (cipro, 1–64 ng/mL, Table S1), ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), kanamycin (50 μg/ml), tetracycline (10 μg/ml), rifampicin (110 μg/ml), and sodium citrate (20 mM).
Bacterial Strains Used in Each Figure
Figure 1: (B) MG1655, SMR5223. (C) MG1655, SMR5223, SMR24603, SMR23099. (D) MG1655, SMR5223. (F) SMR5223, SMR11641, SMR11642, SMR5226, SMR21948, SMR23928, SMR11640, SMR23957, SMR23925, SMR23962, MG1655, SMR20479, SMR21338, SMR20475, SMR20467, SMR20477, SMR21321, SMR23974, SMR23930, SMR23982. (G) SMR14334; (H) SMR14334. (I) MG1655, SMR24600. Figure 2: (A) SMR24100, SMR24156. (B) MG1655. (C) SMR24096, SMR24134. (D) MG1655. (E) SMR24096, SMR24134. (F) MG1655, SMR20479. (G) SMR24450, SMR24451. (H) SMR14334. (I) SMR24100, SMR24156. Figure 3: (A) SMR24096, SMR14471. (B) SMR24096, SMR24134, SMR14471. (C) SMR24096, SMR24134. (D) MG1655. (E) MG1655. (F) SMR14334. (G) SMR24100, SMR24156. (H) MG1655. Figure 4: (A) SMR24268. (B) SMR24852, SMR24853, SMR24854. (C) SMR24854. Figure 5: (A) SG30013, SG30018, SMR24524, SMR24516, SMR24520, SMR24542, BA701, BA709, SMR24546. (B) SMR24096, SMR24692, SMR24688, SMR24694, SMR24695, SMR24436, SMR24134. (C) SMR24096, SMR24690, SMR24688, SMR24694, SMR24118. (D) SMR24450, SMR24451, SMR24452, SMR24453. (E) CH2046, PM1450. Figure 6: (A) MG1655, SMR24682, SMR24678, SMR24680, SMR5223, SMR24676, SMR24672, SMR24674. (B) SMR24450, SMR24451, SMR24684, SMR24686. (C) SG30013, SMR24539. (D) SMR24096, SMR24134, SMR24725. (E) SMR24100, SMR24156, SMR24705. (F) MG1655, SMR24682. (G) SMR24462, SMR24466. (H) MG1655, SMR21338, SMR20467. (I) SMR24096, SMR24561, SMR24563, SMR24134. Figure 7: (B) SMR24700. (C) SMR24700. (D) SMR24700. (E) SMR24347. (F) SMR24700, SMR24347. (G) SMR24096, MG1655, SMR21774, SMR23984, SMR23985, SMR5223, SMR21772, SMR239990, SMR23991. Figure S1: (D) MG1655, SMR20479, SMR21338, SMR20475, SMR20467, SMR20477, SMR21321, SMR23982. (E) MG1655, SMR20479, SMR21338, SMR24004, SMR5223, SMR11641, SMR11642, SMR24002. (F) SMR24096, SMR24134. (G) MG1655, SMR21938, SMR21940, SMR21946, SMR5223, SMR21911, SMR21913, SMR21919. Figure S2: (A) MG1655, SMR5223, SMR24603, SMR24604, SMR24606, SMR24608, SMR24612, SMR24620, SMR24626, SMR24627, SMR24629, SMR24631, SMR24635, SMR24643, SMR24649, SMR24650, SMR24652, SMR24654, SMR24658, SMR24666, SMR24707, SMR24708, SMR24709, SMR24711, SMR24712, SMR24714, SMR23097, SMR23099, SMR23100, SMR23101, SMR23102, SMR23104, SMR23107, SMR23113, SMR23120, SMR20479, SMR11641, SMR20475, SMR5226, SMR20467, SMR21948, SMR20477, SMR23928, SMR21321, SMR11640, SMR24682, SMR24600, SMR24450, SMR24451, SMR24452, SMR24453, SMR24096, SMR14471. (B) MG1655, SMR816, SMR24603, SMR24604, SMR24606, SMR24608, SMR24612, SMR24620, SMR24626, SMR24627, SMR24629, SMR24631, SMR24635, SMR24643, SMR24649, SMR24650, SMR24652, SMR24654, SMR24658, SMR24666, SMR24707, SMR24708, SMR24709, SMR24711, SMR24712, SMR24714, SMR23077, SMR23079, SMR5880, SMR23081, SMR23087, SMR20479, SMR11641, SMR20475, SMR5226, SMR20467, SMR21948, SMR20477, SMR23928, SMR21321, SMR11640, SMR24682, SMR24600, SMR24450, SMR24451, SMR24452, SMR24453, SMR24096, SMR14471. Figure S3: (A) SMR14334. (B) SMR24096, SMR24134. (C) MG1655, SMR20479. (D) MG1655. (E) SMR24096, SMR24134. (F) MG1655. (G) SMR24096, SMR24134. (H) SG30013. (I) SMR14334. (J) SMR24100, SMR24156, SMR24422. (K) MG1655, SMR24600. (L) SMR24096, SMR24134, SMR24439. Figure S4: (A) SMR14333, SMR14334. (B) MG1655, SMR24100. (C) SMR24096, MG1655. (D) MG1655. (E) MG1655, SMR14471. Figure S5: (A) SMR14471. (B) MG1655. (C) SMR24450, SMR24451. (D) SMR24134, SMR25222, SMR25223. (E) SMR24096, SMR24134. (F) SMR24096, SMR24134. (G) MG1655, SMR24268, SMR24312. Figure S6: (A) SMR24096, SMR14471. (B) SMR24096, SMR14471. (C) SMR24096, SMR14471. (D) SMR24096, SMR14471. (E) SMR24096, SMR14471. Figure S7: (A) SMR24096, SMR14471, SMR24134. (B) MG1655, SMR24024. (C) SMR24268. (D) MG1655. (E) MG1655, SMR21774. (F) SMR24096, SMR24134, SMR24430. (G) MG1655, SMR23984. (H) SMR24450, SMR25224, SMR24451, SMR25225.
Assays for Ciprofloxacin-induced Mutagenesis
Strains and selections.
Assays for rifampicin-resistant (RifR) mutants were performed in the wild-type (WT) E. coli MG1655 strain, and its isogenic derivates, and select base substitutions in the rpoB gene encoding a subunit of RNA polymerase (see Figure S1A). To assay ampicillin resistant (AmpR) mutants, we used engineered E. coli strains developed previously (Petrosino et al., 2002) that mutate to AmpR similarly to most clinically relevant Enterobacterial pathogens. The engineered E. coli carry a chromosomal cassette of the divergently transcribed resistance by upregulation of the E. cloacae ampC (b-lactamase) gene (Petrosino et al., 2002). The cassette allows E. coli to mimic most Enterobacteria, which have ampR ampC with their intervening promoters. E. coli (and Shigella) differ by having an apparent deletion (relative to most Enterobacteria) that fuses the E. coli ampC gene to a constitutive low-activity promoter.
Cipro concentrations used at MAC and in dose-response experiments.
Saturated overnight LBH cultures, started each from a single colony, were diluted 1:4×106 into 25 ml in a 250ml flask in fresh LBH broth and incubated at 37°C with shaking for 3–3.5 h, then diluted 1:3 into fresh LBH broth (“no-cipro” controls) or into LBH with cipro at a final “sub-inhibitory” MAC, which causes a final cfu titer of 10% of the titer observed in the no-cipro control. Each strain’s MAC was used in mutagenesis assays (fluctuation tests, below). These concentrations were determined individually for each experimental strain, and are shown in Table S1. For dose- response fluctuation tests, the final cipro concentrations were 1, 2, 4, 8.5, 10, 12 and 14 ng/ml.
Fluctuation test protocol.
For all fluctuation tests, between 10 and 60 independent saturated overnight cultures per strain were assayed as above. From the diluted cultures (above), between 10 and 60 1-ml aliquots were dispensed into 96-deep-well plates or 14-ml tubes as the start of each independent culture, which gives between 104 and 105 cfu per well/tube. The tubes/plates were incubated at 37°C with shaking. These cultures are independent because no mutants are present at these low numbers of cells. The time of cipro addition to the early log (3–3.5hr) cultures is called time 0. After 24h (RifR) or 48h incubation (AmpR), samples were plated onto LBH agar for determination of total viable cfu titers or selective LBH-agar plates containing rifampicin (110 μg/ml) or ampicillin (100 μg/ml) to select mutants resistant to each drug. LBH-agar plate cfu were counted after 16–24h growth at 37°C. Ampicillin-agar plate cfu were counted after 20–24h growth at 37°C. Rifampicin-agar plate cfu were counted after 44–60h growth at 37°C.
Total and resistant cfu were counted, and mutation rates (mutations per cell per generation, or mutations per chromosome per generation) estimated with the MSS-MLE algorithm using the FALCOR calculator (www.mitochondria.org/protocols/FALCOR.html#interface). The raw mutation rates (Table S2) and their fold induction by cipro-induced were determined as the ratio of the mutation rates of the treated divided by the untreated control samples. Raw rates and fold-induction values for all strains assayed are given in Table S2.
For fluctuation tests performed with addition of reagents that reduce ROS, the final concentrations were 100 mM for thiourea, 0.25 mM for 2,2’-bipyridine, and 100 μM for edaravone. Ten aliquots of log-phase cultures were diluted 1:3 and dispensed into 14-mL tubes with and without chemicals that reduce reactive oxygen and with and without MAC cipro and then grown at 37° shaking for 24h (RifR) or 48h (Am pR) as for the mutagenesis assays.
Gam and GamGFP.
For assays in which GamGFP was produced to trap double-strand breaks (DSBs) (Shee et al., 2013), GamGFP was induced from the chromosome using 10 and 20 ng/mL doxycycline in LBH liquid or in plates, as used for determining cfu/ml. Doxycycline was added to cells at their initial 1:4×106 dilution and cultures were grown for 3–3.5h and then diluted 1:3 into fresh LBH and fresh LBH with cipro and dispensed into 10 14-mL tubes and grown for 24h (RifR) as described above. Previously, we found that GamGFP production stops the divisions of cells that obtain a GamGFP focus—a DSB the repair of which is blocked by GamGFP (Shee et al., 2013). Because we do not expect rates of spontaneous DSB formation to differ in RifR mutants from their RifS parents, the killing effect of Gam on cells with DSBs is not expected to affect measurements of RifR mutagenesis when Gam is produced.
Plasmids for σS artificial upregulation and the empty-vector control were obtained from the mobile plasmid collection (see Table S5), and were induced with 30 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) at the initial 1:4×106 cell dilution from saturated cultures into fresh medium, and cultures were grown for 3–3.5h (37° sha king), then diluted 1:3 into fresh LBH and fresh LBH with cipro, dispensed into 10 14-mL tubes and grown at 37° shaking for 24h (RifR) from the addition of cipro, as described above. IPTG was not present in the plates used to determine RifR or total cfu/ml. σS production was confirmed by flow cytometry using the yiaG- yfp σS-response reporter (see below Flow Cytometric Assays for σS- and SOS-Response- Regulated Promoter Activity).
Reconstruction Experiments
Reconstruction experiments were performed to verify that differences in cipro-induced mutant cfu titers observed between wild-type and various mutant strains were not caused by differences in colony-formation efficiency or speed under exact reconstructions of selection conditions: selective plates with varying amounts of isogenic sensitive neighbor cells. From two replicate cultures for each strain, about 100 cfu of ampicillin-resistant ampRC ΔampD cells or rifampicin-resistant rpoB A1687C, rpoB Δ1593–1598, rpoB A1547T mutant cells of each experimental strain genotype were mixed with ~109 or ~108 isogenic sensitive neighbor cells and plated onto ampicillin or rifampicin selective plates, respectively, and their numbers and speed of forming colonies scored. These platings reconstruct the experimental conditions in which mutant cells form colonies scored in our Assays for Ciprofloxacin-induced Mutagenesis. Resistant mutants were also plated alone for reference. We quantified cfu observed after 24 h (ampicillin) or 48 h (rifampicin) at 37°C. Numbers of independent experiments for each given in the Figure S2B legend.
Competition Experiments
Cultures of sensitive and resistant mutants of each experimental strain genotype were mixed at a 50:50 ratio and grown per fluctuation tests, then plated at the end of the growth period on selective rifampicin or ampicillin medium and non-selectively, to obtain the final ratios of sensitive and resistant cfu after growth in competition. Pure cultures were also established as controls. These experiments showed that neither RifR not AmpR mutants is selected (wins the competition ending over 50% of cfu), and both are actually significantly counter-selected relative to their sensitive parent strains (Figures 1C and S2A). These data indicate that all of our estimates of the induction of mutagenesis to RifR and AmpR are underestimates. Numbers of independent experiments for each given in the figure legends.
Flow Cytometric Assays for σS- and SOS-Response-Regulated Promoter Activity
Quantification of cells that have induced their σS or SOS responses, and how much they have, were achieved using engineered chromosomal fluorescence reporter genes and flow cytometry, per (Nehring et al., 2016; Pennington and Rosenberg, 2007) for SOS, and per (Al Mamun et al., 2012) for σS-response activation. We used the yiaG-yfp σS-response reporter (Al Mamun et al., 2012) and the Δattλ::PsulAmCherry SOS reporter (Nehring et al., 2016) modified from (Pennington and Rosenberg, 2007) in strains grown under fluctuation-test conditions as described for Assays for Ciprofloxacin-induced Mutagenesis, with or without cipro, at indicated concentration(s), and harvested the cells in late log phase or stationary phase. For quantification, flow cytometry “gates” were calibrated, for SOS, using the negative-control SOS-off lexA(Ind−), and SOS-response proficient cells, per (Pennington and Rosenberg, 2007) as the dividing place between peaks of the bimodal distribution of SOS-proficient cells at which most cells diverge from the spontaneously SOS-induced fluorescent cell subpopulation, usually at between 0.5% and 1% of cells cultured in LBH broth. With this gate, ~10−4 of SOS-non-inducible recA or lexAInd− cells cross the gate, scoring as “SOS-positive,” per (Pennington and Rosenberg, 2007). For the σS response, gates for σS-high activity cells were set to the point at which fewer than 0.5% of cells with cipro but without the reporter gene were positive. At this gate fewer than 10−3 of ΔrpoS cells, which are σS-response deficient, cross the gate and would be scored as positive. For all, the percent of the population that scored as positive is reported.
Fluorescence-Activated Cell Sorting
Cell sorting was performed using a FACS Aria II cell sorter (BD Biosciences, San Jose, CA) with a 70-μm nozzle. E. coli cells were identified using forward and side scatter parameters, and these were sorted using sterile 1X phosphate buffered saline (PBS) as sheath fluid. After treatment with MAC cipro for 24 hours (RifR) (identical to Assays for Ciprofloxacin-induced Mutagenesis, above), yellow fluorescent protein-positive (σS activity, yiaG-yfp) and non- fluorescent cells were sorted into 14 mL conical tubes (20–30×106 negative cells and 3–8×106 positive cells) and plated on LB agar with and without rifampicin to determine cfu/mL (per Assays for Ciprofloxacin-induced Mutagenesis, above). These data were used to calculate RifR mutant frequencies in the sorted σS-high, σS-low, unsorted, and mock-sorted populations, the last being cells run through the machine and all cells collected. For Figure 3A, the fold induction of RifR mutant frequency among sorted cells, the cipro-treated mutant frequencies from σS-high, σS-low, unsorted, and mock-sorted populations were divided by the mutant frequency of unsorted cells grown without cipro in the same experiments. Cells grown without cipro do not have a distinct σS-high population—all are somewhat higher than in log-phase cells and not nearly as high as the σS-high subpopulation cells after cipro treatment (see Figure S5E and G). Control sorts for fluorescence from Placcfp, a negative control for metabolically active cells, and mutagenesis assays, were performed similarly in parallel with the experimental sorts.
HPII Catalase Activity
HPII (σS-dependent catalase) activity was measured as described (Iwase et al., 2013). The viable cell titers (cfu/mL) of cells growing in LBH broth were determined at appropriate time points in log or stationary phase. HPI catalase was inactivated by heating 100 μL culture aliquots at 55°C for 15 min. After inactivation, 100 μL 30% H2O2 and 1% Triton-X 100 (Sigma) were added. After an additional 15 min incubation, the height of bubble formation was measured in millimeters. The millimeters of bubbles were then normalized to cfu/mL of cells. Controls in ΔrpoS cells demonstrated that these assays report on σS-response-dependent catalase activity.
Microscopy and Quantification of GamGFP (DSB) and TetR-mCherry (Chromosome) Foci
Saturated overnight LBH cultures of cells carrying the chromosomal inducible GamGFP cassette were diluted 1:4×106 into 25ml LBH broth in 250mL flasks and grown for 3 h. These were then diluted 1:3 into LBH with or without cipro (1–8.5 ng/ml). GamGFP, a DNA DSB-specific binding protein that traps DSBs and inhibits their repair (Shee et al., 2013), was induced in late log phase using 40 ng/mL of doxycycline. After 2 h of induction, cells were fixed with 1% paraformaldehyde and placed at 4°C until microscopy images were taken. For chromosome quantification, saturated overnight LBH cultures of cells containing the inducible TetR-mCherry plasmids and the tetO chromosomal array were diluted 1:4×106 into 25ml in 250mL flasks and grown for 3 h. These were then diluted 1:3 into LBH with or without cipro (MACs). The TetR-mCherry protein binds to the chromosomal tetO array labeling oriC-proximal chromosomal units as red foci, and was induced in late log-phase using 2 μM of sodium salicylate. After 4h of induction, cells were fixed with 1% paraformaldehyde and placed at 4°C until microscopy images were taken. Images were obtained with an inverted DeltaVision Core Image Restoration Microscope (GE Healthcare) with a 100X UPlan S Apochromat (numerical aperture, 1.4) objective lens (Olympus) and a CoolSNAP HQ2 camera (Photometrics). Captured images for analysis were chosen randomly. The images were taken with Z-stacks (0.15- m intervals) and then deconvoluted (DeltaVision SoftWoRx software) to visualize the whole cell for precise and accurate quantification of foci per (Xia et al., 2019). For each experiment, >400 cells were counted using ImageJ software (NIH) with visual inspection from each independent experiment. Only foci that overlapped with DAPI DNA stain were quantified (≥99% of all foci).
Live Cell Deconvolution Microscopy
Cells were grown as for Assays for Ciprofloxacin-induced Mutagenesis. At 8 hours after the addition of ciprofloxacin (8.5 ng/mL), 4 μL of culture were plated onto 35mm glass bottom cell culture plates. An agar pad containing spent medium from replicate cultures (8.5 ng/mL cipro in cells grown for 8h) was placed on top of the cells, and a glass cover slip placed over the agar pad and sealed with silicon grease to limit evaporation. Images were taken every 1–2 hours for 12 hours with an inverted DeltaVision Core Image Restoration Microscope (GE Healthcare) with a 100X UPlan S Apochromat (numerical aperture, 1.4) objective lens (Olympus) and a CoolSNAP HQ2 camera (Photometrics). Captured images for analysis were randomly chosen. The images were taken with Z-stacks (0.15- m intervals) and then deconvoluted (DeltaVision SoftWoRx software) to visualize the whole cell. For each experiment, >250 cells were followed to track the activation of the GFP (PsodAgfp oxidative stress response) and mCherry (σS activity) using ImageJ software (NIH) with visual inspection from each independent experiment.
rpoB and ampD Sequencing
A sole RifR or AmpR colony was isolated from each of 24 cipro-treated or 24 control independent cultures and the rpoB or ampD gene sequenced. RifR rpoB mutations occur mostly within two mutation clusters (Reynolds, 2000), and all isolated mutants contained mutations within one of these two sites of clustering (or rarely both sites). ampD loss of function mutations confer ampicillin resistance in engineered E. coli that carry the Enterobacter cloacae ampRC genes in the chromosome, per (Petrosino et al., 2002) and Assays for Ciprofloxacin-induced Mutagenesis, Strains and selections. The rpoB cluster I and II sites were amplified, as described (Reynolds, 2000), STAR METHODS KEY RESOURCES TABLE for primers. The ampD gene was amplified using primers described in STAR METHODS KEY RESOURCES TABLE. PCR fragments were subjected to Sanger sequencing (GeneWIZ, Massachusetts) to identify insertions, deletions, and/or base substitutions.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals and Recombinant proteins | ||
| ciprofloxacin | MP Biomedicals | Cat# 199020 |
| rifampicin | Research Products International | Cat# 13292–46-1 |
| ampicillin | Sigma-Aldrich | Cat# A9518 |
| doxycycline | Alfa Aesar | Cat# J60422 |
| thiourea | Sigma-Aldrich | Cat# T8656 |
| 2,2’ bipyridyl | Sigma-Aldrich | Cat# D216305 |
| edaravone | Sigma-Aldrich | Cat# M70800 |
| isopropyl β-D-1- thiogalactopyranoside | Research Products International | Cat# 156000–5 |
| 2-Nitrophenyl β-D-galactopyranoside | Sigma-Aldrich | Cat# N1127 |
| dihydrorhodamine | Life Technologies | Cat# D632 |
| sodium salicylate | Sigma-Aldrich | Cat# 54–21-7 |
| Sytox Blue dead cell stain | Life Technolgies | Cat# S34857 |
| E. coli Sigma S antibody | Neoclone | Cat# WP009 |
| E. coli RpoB antibody | BioLegend | Cat# 663903 |
| Experimental Models | Escherichia coli K12 strains Table S5 | |
| Sequence-Based Reagents | ||
| primers | ||
|
rpoB cluster I - FWD REV |
GAC AGA TGG GTC GAC TTG TCA G AGG TGG TCG ATA TCA TCG ACT T |
|
| rpoB cluster I - Sequencing | GAA GGC ACC GTA AAA GAC AT | |
|
rpoB cluster II - FWD REV |
TCG AAG GTT CCG GTA TCC TGA G GGA TAC ATC TCG TCT TCG TTA AC |
|
| rpoB cluster II - Sequencing | CGT GTA GAG CGT GCG GTG AAA | |
|
ampD - FWD REV |
GTC GGG TGT CAG GGT TAT AG CGC TTC AAG ACG ATG ATC AAG |
|
| ampD - Sequencing | ATA AGG TAG AAA CAT GCT ACT CT | |
|
yiaG-mCherry SH – FWD REV |
CCCGGCATTAAGTAAGCAGTTGATGGAATAGACTTTTATCATG GTTTCCAAGGGCGAGGA GCGGGTGATGCAACAATTATTTTTCATATTTATGATTAATGTG TAGGCTGGAGCTGCTTC |
Western Analyses of σS Protein Levels
Proteins were separated by SDS-PAGE and transferred to 200 polyvinylidine (PVDF) membranes (Amersham Biosciences), blocked with 2% blocking buffer, and probed with polyclonal mouse anti-σS antibody (1:700 dilution) (Neoclone). Polyclonal mouse anti-RNA polymerase subunit beta (RpoB) was used to detect RpoB as a loading control in Figure 2F (1:1000) (BioLegend). Goat anti-mouse antibody conjugated to Cy5 fluorescent dye (1:5000 dilution) (Amersham Biosciences) was used to detect the primary antibody-bound σS or RpoB protein. Fluorescence was quantified using a Typhoon scanner, with a PMT of 500 and 670BP 30Cy5emission filter, and the bands quantified using ImageJ software (NIH). Quantifications from two or three separate western blots for σS are reported, each with band intensities normalized to the values from isogenic wild-type cells with no cipro treatment run in parallel, and the means ±SEM shown.
Beta-galactosidase Assays
Cells were grown as for Assays for Ciprofloxacin-induced Mutagenesis to equivalent ODs and frozen at −20°C until assays were carried out. Determination of the β-galactosidase activity of the ParcZ-lacZ, PdsrA-lacZ, rpoS-lacZ, and katG-lacZ fusions was accomplished using the standard assay described by JH Miller, as previously (Gibson et al., 2010), except that the assays were carried out in 96-well plates to ease sample processing.
Flow Cytometric Detection of Intracellular ROS or GFP and σS Activity in Single Cells
Cells were grown in the absence or presence of MAC cipro to early-, late-log, and stationary phase as for Assays for Ciprofloxacin-induced Mutagenesis. The ROS measurement protocol was modified from Gutierrez et al. (Gutierrez et al., 2013). Cells were incubated with ROS-staining dye dihyrdorhodamine 123 (DHR) (Invitrogen) for 30 min at 37°C in PBS. After washing twice with PBS buffer, flow cytometry analyses were performed immediately. Gates for ROS-positive cells were set so that <0.5% of cells treated with cipro without DHR dye were positive. For experiments in which ROS or GFP and σS activity were measured, cells were grown in the absence or presence of MAC cipro as for Assays for Ciprofloxacin-induced Mutagenesis (above), then harvested serially from cultures at 4, 8, 12, 16, 24, and 48 hours for ROS detection using DHR or at 12, 16, and 24 hours for ROS detection using the PoxyRgfp or PsodAgfp transcriptional fusions (Table S5). For ROS detection using DHR, cells containing σS-activity reporter yiaG-mCherry were collected and ROS detected as green fluorescence, and σS activity as red fluorescence. For ROS detection using PoxyRgfp and PsodAgfp, cells containing both σS-activity reporter yiaG-mCherry and plasmids carrying the PoxyRgfp or PsodAgfp reporters, or a promoterless gfp parental plasmid Pvector-gfp, were maintained with 35μg/mL kanamycin, and used to detect both GFP and red fluorescence. Single color and no-fluorescence controls were also collected at time points for spectral compensation. For the PoxyRgfp or PsodAgfp transcriptional fusions, gates were drawn so that the promoterless-gfp vector Pvector-gfp had <0.5% GFP-positive cells. σS high-activity-cell gates were drawn so that <0.5% of cells without cipro were positive, and MAC cipro-treated wild-type cells without the chromosomal σS-response reporter had fewer than 0.5% scored as positive.
Controls for Appearance of ROS-high Subpopulation Before σS-high Subpopulation
In Figure 4A, the ROS-high cell subpopulation is apparent hours before the σS-high cell subpopulation, with ROS detected by DHR dye and σS activity by mCherry fluorescence from a gene the transcription of which requires σS. We can be sure that the appearance of ROS before σS activity is not the result of the lag between induction of transcription and appearance of a translated fluorescent protein because the same result is obtained when ROS and σS activity are both detected by fluorescence reporters each of which requires transcription and translation Figure 4B. Additionally the lag between induction and appearance of flow-cytometry-detectable fluorescent protein is under 15 minutes (Pennington and Rosenberg, 2007), much less than the lag between ROS-high and σS-high cells (Figure 4).
Experimental Definition of Cipro-induced Multi-chromosome Cell Filaments
Without cipro only 1% ± 0.7% of exponential (16h post-cipro) wild-type cells have four or more chromosome copies (Figure 7C), so we defined a multi-chromosome cell as those with ≥4 chromosome copies. With cipro, 33% ± 2% of wild-type cells have ≥4 chromosome copies (Figure 7B). By contrast, ΔsulA cells show much reduced cell length and chromosome content (Figure 7D–F).
Mathematical Modeling of Cipro-induced Multi-chromosome Cell Filaments
In our model, a population of microbes is exposed to severe external stress (e.g., antibiotics), and two strategies are available: either growing into “filament” cells, that can contain multiple DNA copies, or reproducing individually. We consider a case in which resistance to the external stress can be acquired by a single mutation, with baseline rate μ, and deleterious mutations occur at many other loci, with the number of deleterious mutations per replication following a Poisson distribution with average λ. We assume that during the external stress the basic mutation rates of all cells (both μ and λ) increase A-Fold, and mutation rates in filament cells are further increased B-fold relative to non-filament cells.
We denote by s and δ the selection coefficients against the external stress and each deleterious mutation, respectively. We denote by Ia the level of adaptation to the external stress, where . The fitness (modeled here as the probability to replicate) of an individual that possess n deleterious mutations is thus . In the filament population, we assume that DNA copies in the same cell filament share gene products, and that deleterious mutations are recessive. Once a genome copy within the filament acquires the beneficial mutation that confers resistance to the major stress, it buds out of the filament, and begins to duplicate regularly (in proportion to the number of deleterious mutations it possesses).
We follow the two strategies for k replication cycles, starting from a population that doesn’t carry any deleterious mutations nor is adapted to the external stress. In the filament population the cells duplicate their genome without dividing and have up to 2k DNA copies. Because the populations begin without any deleterious mutations, we neglect filaments in which all DNA copies share the same deleterious mutation. Therefore, the fitness of DNA copies in the filament population is affected only by the external stress, while in the non-filament population the fitness of each DNA copy (or cell) is affected both by the external stress and by the number of deleterious mutations it carries. After k replication cycles the filaments divide to cells, each containing a single DNA copy. We then compare the population size and fitness, the proportion of adapted individuals, and the distribution of deleterious mutations, between the filament population and the non-filament population.
Parameter values in Figure 7H: λ = 0.003, μ = 6·10−7, δ = 0.03, A = 100, k = 4. In left and middle panels we use B = 4 and s =0.9, whereas in the right panel B is the value on the x-axis. The value B = 4 is derived from empirical results presented in Figure 7G, in which the we see that during antibiotic stress the mutation rate of cells that do filament (WT) have a fold- increase of ~4 relative to non-filamented cells.
The model tests the effect of filaments on evolvability, where mutation serves as the variation mechanism. However, if chromosomes in filaments also experience recombination, then the system corresponds to the case of Fitness-Associate Recombination (FAR) (Hadany and Beker, 2003b) – the less fit chromosomes experience higher recombination rate then the fitter ones. Previous work has shown that this mode of recombination results in increased mean fitness and improved adaptability (Hadany and Beker, 2003a).
Parameters:
Beneficial mutation rate ~Ber(μ)
Deleterious mutation rate ~Poisson(λ)
A – stress-induced increase in mutation rate
B – filament cells fold increase in mutation rate relative to non-filament cells
s - selection coefficient of the antibiotic
δ - selection coefficient of each deleterious mutation (multiplicative model)
k - number of replication cycles
Measurement of High-Dose Cipro Antibiotic Activity
Cells were grown to log phase OD600 ~0.5, then cipro (1.5 μg/mL) with or without edaravone (100μM) was added, and cells were harvested 0.75, 1.25, 2.25, and 3 hours later to determine cfu/mL. Cells were washed twice with PBS and then assayed for viable cfu.
Nalidixic-Acid Test for Heritable Hypermutability
Tests for heritable mutator phenotype were as described (Torkelson et al., 1997). Ten independent cipro-induced RifR mutant isolates, each with a different mutation, were grown in parallel with control wild-type (non-mutator) and mutS mismatch repair-defective (mutator) strains each in duplicate independent cultures. 100μL of each saturated overnight culture was spread onto an LBH agar plate. After 10 minutes, dry nalidixic acid powder was spotted onto each plate using a capillary tube. The plates were incubated for 24 hours at 37°C, after which the number of microcolonies in the zones of inhibition were counted, and compared with the positive (mutS) and negative (isogenic wild-type) controls.
Flow-Cytometric Detection of Dead Cells
Cells were grown in the presence of MAC cipro per Assays for Ciprofloxacin-induced Mutagenesis (above), and harvested serially from cultures at log phase (4 and 12 hours) and stationary phase (24 hours) for dead cell detection using SYTOX blue dead cell stain. Cells were stained according to manufactures recommendation. Cells were incubated with SYTOX blue dye (1μM) for 30 minutes at room temperature and flow cytometry analyses were performed immediately. As a positive control, cells were incubated in 95% ethanol for 10 minutes before staining. Positive gates for dead cells were set so that <0.2% of undyed cipro- treated cells were positive, at which 90% ± 5% of the SYTOX-blue dyed positive-control ethanol-treated cells were positive.
Statistics
Statistics were performed in Microsoft Excel or GraphPad PRISM. For comparisons of two groups, a two-tailed Students t-test was used if data were normally distributed and homoscedastic. For comparisons of 3 or more groups, ANOVA with Tukey post-hoc test was used if data were normally distributed and homoscedastic, otherwise a Kruskal-Wallis non-parametric test was used. For mutation rates and ratios, which are not normally distributed, natural-logarithm transformed data were used to calculate 95% confidence intervals (CIs) as well as performing statistical significance tests. 95% CIs appear in bar graphs as error bars that are not symmetrical above and below the top of the bar.
Supplementary Material
Movie S1. Example of formation of σS-high red cells arising from oxidative response-activated green cells, Related to Figure 4
Table S4. Mutation Rates per Cell per Generation and per Chromosome per Generation
Table S5. Escherichia coli Strains and Plasmids
Table S2. Mutation Rates and Fold Induction of Mutation Rate by Cipro in RifR andz AmpR Fluctuation Tests
Table S3. Percent of ROS-high, σS-high, and Double-Positive Cells over 48 Hours in Cipro, and Mutation Rates at 16 and 24 Hours
Highlights.
Antibiotic-induced mutable cell subpopulation generates resistant mutants
Mitigates risk to most cells; reactive oxygen → σS stress response → gamblers
FDA-approved drug blocks σS response and mutagenesis: anti-evolvability drug
Multiple chromosomes needed: chromosome cooperation can allow rapid adaptation
ACKNOWLEDGEMENTS
We thank S Gottesman, J Imlay, I Matic, and L Zechiedrich for E. coli strains, N Majdalani and S Kozmin for advice, S Henikoff for helpful conversation, and KM Miller and Meng Wang for improving the manuscript. Supported by NIH Grants R35-GM122598 (SMR), R01-GM088653 (CH), R01-GM102679 (DB), R01-GM106373 (PJH), Israeli Science Fund ISF 1568/13 (LH), and the Baylor College of Medicine (BCM) Integrated Microscopy Core funded by NIH (DK56338, and CA125123), the Dan L Duncan Comprehensive Cancer Center, postdoctoral fellowships RP160283 and 132206-PF-18–035–01–DMC (DMF) from the Cancer Prevention and Research Institute of Texas BCM Cancer Training Program and the American Cancer Society; the John S. Dunn Gulf Coast Consortium for Chemical Genomics; and the BCM Cytometry and Cell Sorting Core (NIH: P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of JM Sederstrom.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Al Mamun AAM, Lombardo M-J, Shee C, Lisewski AM, Gonzalez C, Lin D, Nehring RB, Saint-Ruf C, Gibson JL, Frisch RL, et al. (2012). Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai S, Kolodkin-Gal I, Hananya-Meltabashi M, Sacher A, and Engelberg-Kulka H (2009). Escherichia coli MazF leads to the simultaneous selective synthesis of both “death proteins” and “survival proteins”. PLoS Genet 5, e1000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, and Leibler S (2004). Bacterial persistence as a phenotypic switch. Science 305, 1622–1625. [DOI] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, and Gottesman S (2011). The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65, 189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos J, Zhang Q, Vyawahare S, Rogers E, Rosenberg SM, and Austin RH (2015). Emergence of antibiotic resistance from multinucleated bacterial filaments. Proc Natl Acad Sci USA 112, 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, and Dubnau D (2004). DNA uptake during bacterial transformation. Nat Rev Microbiol 2, 241–249. [DOI] [PubMed] [Google Scholar]
- Chen J, and Gottesman S (2017). Hfq links translation repression to stress-induced mutagenesis in E. coli. Genes Dev 31, 1382–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, and Romesberg FE (2005). Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3, e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE, and Lindquist S (2005). Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 309, 2185–2189. [DOI] [PubMed] [Google Scholar]
- Drlica K (1999). Mechanism of fluoroquinolone action. Curr Opin Microbiol 2, 504–508. [DOI] [PubMed] [Google Scholar]
- Dwyer DJ, Collins JJ, and Walker GC (2015). Unraveling the physiological complexities of antibiotic lethality. Annu Rev Pharmacol Toxicol 55, 313–332. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DM, Hastings PJ, and Rosenberg SM (2017). Stress-induced mutagenesis: implications in cancer and drug resistance. Annu Rev Cancer Biol 1, 119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Devadoss B, Winkler JA, Collins JJ, and Walker GC (2012). Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenoy A, and Bonhoeffer S (2018). Death and population dynamics affect mutation rate estimates and evolvability under stress in bacteria. PLoS Biol 16, e2005056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JL, Lombardo M-J, Thornton PC, Hu KH, Galhardo RS, Beadle B, Habib A, Magner DB, Frost LS, Herman C, et al. (2010). The sigma(E) stress response is required for stress-induced mutation and amplification in Escherichia coli. Mol Microbiol 77, 415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Flecha B, and Demple B (1995). Metabolic sources of hydrogen peroxide in aerobically growing Escherichia coli. J Biol Chem 270, 13681–13687. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Pastor JE, Hobbs EC, and Losick R (2003). Cannibalism by sporulating bacteria. Science 301, 510–513. [DOI] [PubMed] [Google Scholar]
- Gutierrez A, Laureti L, Crussard S, Abida H, Rodríguez-Rojas A, Blázquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, et al. (2013). β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4, 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadany L, and Beker T (2003a). Fitness-associated recombination on rugged adaptive landscapes. J Evol Biol 16, 862–870. [DOI] [PubMed] [Google Scholar]
- Hadany L, and Beker T (2003b). On the evolutionary advantage of fitness-associated recombination. Genetics 165, 2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG (1990). Spontaneous point mutations that occur more often when advantageous than when neutral. Genetics 126, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman O, and D’Ari R (1981). An inducible DNA replication-cell division coupling mechanism in E. coli. Nature 290, 797–799. [DOI] [PubMed] [Google Scholar]
- Ilves H, Horak R, and Kivisaar M (2001). Involvement of sigma(S) in starvation-induced transposition of Pseudomonas putida transposon Tn4652. J Bacteriol 183, 5445–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase T, Tajima A, Sugimoto S, Okuda K-I, Hironaka I, Kamata Y, Takada K, and Mizunoe Y (2013). A simple assay for measuring catalase activity: a visual approach. Sci Rep 3, 3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi MC, Magnan D, Montminy TP, Lies M, Stepankiw N, and Bates D (2013). Regulation of sister chromosome cohesion by the replication fork tracking protein SeqA. PLoS Genet 9, e1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodursky AB, Zechiedrich EL, and Cozzarelli NR (1995). Topoisomerase IV is a target of quinolones in Escherichia coli. Proc Natl Acad Sci USA 92, 11801–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, DePristo MA, and Collins JJ (2010). Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, and Collins JJ (2007). A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810. [DOI] [PubMed] [Google Scholar]
- Kuzminov A (1999). Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63, 751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart JS, Schroeder JW, Walsh BW, and Simmons LA (2012). DNA repair and genome maintenance in Bacillus subtilis. Microbiol Mol Biol Rev 76, 530–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo M-J, Aponyi I, and Rosenberg SM (2004). General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics 166, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorian V, and De Freitas CC (1979). Minimal antibiotic concentrations of aminoglycosides and beta-lactam antibiotics for some gram-negative bacilli and gram-positive cocci. J Infect Dis 139, 599–603. [DOI] [PubMed] [Google Scholar]
- Magrini ETN (2017). Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics In Essential medicines and health products (World Health Organization: World Health Organization; ), pp. 1–7. [Google Scholar]
- McKenzie GJ, Harris RS, Lee PL, and Rosenberg SM (2000). The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA 97, 6646–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meganathan R, and Kwon O (2009). Biosynthesis of Menaquinone (Vitamin K2) and Ubiquinone (Coenzyme Q). EcoSal Plus 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JM, Correa R, Rosenberg SM, and Hastings PJ (2017). Persistent damaged bases in DNA allow mutagenic break repair in Escherichia coli. PLoS Genet 13, e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehring RB, Gu F, Lin HY, Gibson JL, Blythe MJ, Wilson R, Bravo Nunez MA, Hastings PJ, Louis EJ, Frisch RL, et al. (2016). An ultra-dense library resource for rapid deconvolution of mutations that cause phenotypes in Escherichia coli. Nucleic Acids Res 44, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio J (1991). Transient mutators: a semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics 129, 957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman TM, Lord ND, Paulsson J, and Losick R (2015). Stochastic switching of cell fate in microbes. Annu Rev Microbiol 69, 381–403. [DOI] [PubMed] [Google Scholar]
- Obolski U, Ram Y, and Hadany L (2017). Evolution on rugged adaptive landscapes. Rep Prog Phys 81, 012602. [DOI] [PubMed] [Google Scholar]
- Parikh A, Kathawala K, Tan CC, Garg S, and Zhou XF (2016). Development of a novel oral delivery system of edaravone for enhancing bioavailability. Int J Pharm 515, 490–500. [DOI] [PubMed] [Google Scholar]
- Pennington JM, and Rosenberg SM (2007). Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39, 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino JF, Pendleton AR, Weiner JH, and Rosenberg SM (2002). Chromosomal system for studying AmpC-mediated beta-lactam resistance mutation in Escherichia coli. Antimicrob Agents Chemother 46, 1535–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder RG, Fonville NC, and Rosenberg SM (2005). A switch from high-fidelity to error- prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19, 791–804. [DOI] [PubMed] [Google Scholar]
- Ram Y, and Hadany L (2012). The evolution of stress-induced hypermutation in asexual populations. Evolution 66, 2315–2328. [DOI] [PubMed] [Google Scholar]
- Renggli S, Keck W, Jenal U, and Ritz D (2013). Role of autofluorescence in flow cytometric analysis of Escherichia coli treated with bactericidal antibiotics. J Bacteriol 195, 4067–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MG (2000). Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156, 1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM, and Queitsch C (2014). Combating evolution to fight disease. Science 343, 1088–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper RM, and Dunn RL (1987). Escherichia coli mutT mutator effect during in vitro DNA synthesis. J Biol Chem 262, 16267–16270. [PubMed] [Google Scholar]
- Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu L-Y, Magnan D, Halliday JA, Frisch RL, Gibson JL, et al. (2013). Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife 2, e01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee C, Gibson JL, Darrow MC, Gonzalez C, and Rosenberg SM (2011). Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci USA 108, 13659–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee C, Gibson JL, and Rosenberg SM (2012). Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli. Cell Rep 2, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T, Gunaherath GM, Wijeratne EM, Lambert JP, Averette AF, Lee SC, Kim T, Bahn YS, Tripodi F, Ammar R, et al. (2016). Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat Chem Biol 12, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP (1998). Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta 1363, 100–124. [DOI] [PubMed] [Google Scholar]
- Sung HM, and Yasbin RE (2002). Adaptive, or stationary-phase, mutagenesis, a component of bacterial differentiation in Bacillus subtilis. J Bacteriol 184, 5641–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson PA, and Schenley RL (1974). Respiration, growth and viability of repair-deficient mutants of Escherichia coli after ultraviolet irradiation. Int J Radiat Biol Relat Stud Phys Chem Med 25, 51–60. [DOI] [PubMed] [Google Scholar]
- Torkelson J, Harris RS, Lombardo MJ, Nagendran J, Thulin C, and Rosenberg SM (1997). Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J 16, 3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JW, Smits WK, and Kuipers OP (2008). Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol 62, 193–210. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Tanaka M, Yuki S, Hirai M, and Yamamoto Y (2018). How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J Clin Biochem Nutr 62, 20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly H, Shee C, Thornton PC, Sivaramakrishnan P, Rosenberg SM, and Hastings PJ (2013). R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat Commun 4, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo AC, Faure L, Dapa T, and Matic I (2018). Heterogeneity of spontaneous DNA replication errors in single isogenic Escherichia coli cells. Sci Adv 4, eaat1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Chiu LY, Nehring RB, Bravo Nunez MA, Mei Q, Perez M, Zhai Y, Fitzgerald DM, Pribis JP, Wang Y, et al. (2019). Bacteria-to-Human Protein Networks Reveal Origins of Endogenous DNA Damage. Cell 176, 127–143 e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, and Gottesman S (2006). Modes of regulation of RpoS by H-NS. J Bacteriol 188, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Example of formation of σS-high red cells arising from oxidative response-activated green cells, Related to Figure 4
Table S4. Mutation Rates per Cell per Generation and per Chromosome per Generation
Table S5. Escherichia coli Strains and Plasmids
Table S2. Mutation Rates and Fold Induction of Mutation Rate by Cipro in RifR andz AmpR Fluctuation Tests
Table S3. Percent of ROS-high, σS-high, and Double-Positive Cells over 48 Hours in Cipro, and Mutation Rates at 16 and 24 Hours