

Abstract
Background and Purpose
Ulcerative colitis (UC) is an aetiologically refractory inflammatory disease, accompanied by dysfunction of the epithelial barrier and intestinal inflammation. Phosphodiesterase‐4 (PDE4) serves as an intracellular proinflammatory enzyme, hydrolyzing and inactivating cAMP. Though PDE4 inhibitors have been approved for pulmonary and dermatological diseases, the role of PDE4 inhibition in modulating mucosal immunity in the intestine remains ill‐defined. This study was designed to explore whether PDE4 inhibition by apremilast exerts protective effects in dextran sulfate sodium‐induced murine UC.
Experimental Approach
Intestinal inflammation and disease severity were evaluated by morphological, histopathological and biochemical assays, and in vivo imaging. Expression of inflammatory mediators, components of PDE4‐mediated pathways in colon and macrophages were determined using quantitative real‐time PCR, ELISA, Luminex assay, immunostaining, or western blotting, along with siRNA knockdown. Immune cells in mesenteric lymph nodes and colonic lamina propria were analysed by flow cytometry.
Key Results
Apremilast attenuated clinical features of UC, suppressing microscopic colon damage, production of inflammatory mediators, oxidative stresses, and fibrosis. Apremilast also promoted epithelial barrier function and inhibited infiltration of immune cells into inflamed tissues, through decreasing expression of chemokines and chemokine receptors. Furthermore, in UC, PDE4A, PDE4B, and PDE4D were highly expressed in colon. Apremilast not only inhibited PDE4 isoform expression but also activated PKA–CREB and Epac‐Rap1 pathways and subsequently suppressed MAPK, NF‐κB, PI3K–mTOR, and JAK–STAT–SOCS3 activation.
Conclusion and Implications
Inhibition of PDE4 by apremilast protected against UC, by interfering with mucosal immunity. These findings represent a promising strategy for regulating intestinal inflammation.
Abbreviations
- BMDM
bone marrow‐derived macrophages
- CREB
cAMP‐response element binding protein
- DAI
disease activity index
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel disease
- MLN
mesenteric lymph node
- MPO
myeloperoxidase
- TEER
trans‐epithelial electrical resistance
- UC
ulcerative colitis.
What is already known
Clinical trials of the PDE4 inhibitor, apremilast, on UC are ongoing.
What this study adds
This study illustrates the pathological role of PDE4 in intestinal inflammation.
What is the clinical significance
PDE4 inhibition by apremilast represents a potential therapeutic strategy for UC patients.
1. INTRODUCTION
Inflammatory bowel diseases (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), are aetiologically idiopathic, chronic, relapsing, and refractory inflammatory conditions that results from the interactions of gene susceptibility, environmental factors, disturbance of immune homeostasis, and microbiological anomaly in the gastrointestinal tract (Sartor, 2006). In the past decade, UC has grown to become a global health challenge with a prevalence of over 0.3% worldwide (Ng et al., 2018). UC can present in patients with life‐altering syndromes lasting weeks to months, such as diarrhoea, bleeding, abdominal pain, fecal urgency, and severe fever. Recently, in addition to a wide spectrum of therapeutic corticosteroids, immunosuppressants, and biological drugs, inhibitors of phosphodiesterase‐4 (PDE4) have also shown dramatic therapeutic efficacies for the treatment of UC symptoms (Spadaccini, D'Alessio, Peyrin‐Biroulet, & Danese, 2017).
The PDE4 family provides the main PDE activity in a wide range of immune cells and epithelial cells (Chiricozzi et al., 2016). PDE4 is highly specific for cAMP degradation, and elevation of intracellular cAMP by PDE4 inhibition contributes to suppression of cell trafficking and to the release of chemokines and cytokines from inflammatory cells. Lower levels of colon cAMP and up‐regulation of PDE4 expression were observed in IBD, which resulted in abnormal production of cytokines in the inflamed intestine (Banner & Trevethick, 2004; Schafer et al., 2016). Given the suppression of inflammatory cytokines following PDE4 inhibition, compounds such as rolipram, tetomilast, and roflumilast have been applied to experimental models of colitis (El‐Ashmawy, Khedr, El‐Bahrawy, & El‐Adawy, 2018; Hartmann et al., 2000; Ichikawa et al., 2008). Although these inhibitors showed therapeutic effects in acute or chronic colitis, their adverse side‐effects have impeded further clinical application.
In contrast to the inflammation present in CD which is transmural and widespread throughout the entire gastrointestinal tract, inflammation in UC is primarily limited in the mucosal and submucosal layers of the colon (Sartor, 2006). Although the detailed pathogenesis of UC remains undefined, disturbed mucosal immune homeostasis and destruction of epithelial integrity are deemed to be critical to the initiation and perpetuation of UC (Banner & Trevethick, 2004; Xu, Liu, Feng, & Liu, 2014). Pathogens that gain access to lamina propria (LP) following the destruction of epithelial integrity and then expand out of control pose a critical risk factor of UC (Boyapati, Rossi, Satsangi, & Ho, 2016; Cader & Kaser, 2013). Under inflammatory conditions, certain activated immune cells infiltrate into the colonic mucosa, which results in high level of inflammatory cytokines, chemokines and adhesion molecules, along with excessive expression of chemokine receptors and integrin. Macrophages and DCs play a vital role in the aggravation of UC and function conventionally as the antigen‐presenting cells priming naïve T cells and inducing their differentiation into inflammatory phenotypes (Steinbach & Plevy, 2014). Furthermore, there is a substantial increase in the pathological T cells responses, which are responsible for abnormal expression of various tight‐junction proteins and subsequent persistence of mucosal inflammation (de Mattos et al., 2015).
The PDE4 inhibitor apremilast, approved in 2014, represents a breakthrough in effective treatment for patients with psoriasis and psoriatic arthritis (Chiricozzi et al., 2016; Schafer et al., 2010). Apremilast is a well‐tolerated PDE4 inhibitor with mild‐to‐moderate adverse effects. Moreover, apremilast has also been applied clinically in other inflammatory disorders, such as Behcet's syndrome, ankylosing spondylitis, frontal fibrosing alopecia, and discoid lupus erythematosus (Li, Zuo, & Tang, 2018). However, there are no reports of preclinical research on inflammatory colitis, so far. Hence, we aimed to assess the protective effects of apremilast in intestinal inflammation. We have used a murine model of acute colitis induced by dextran sulfate sodium (DSS) to study the effects of apremilast.
2. METHODS
2.1. DSS‐induced colitis and drug treatment
All animal care and experimental procedures were carried out according to the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Bioethics Committee of the Shanghai Institute of Materia Medica. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. Wild‐type male C57BL/6J mice (8 weeks, 22–24 g, RRID:IMSR_JAX:000664) were purchased from Shanghai Lingchang Biotechnology Co., Ltd. (Certificate No.2013‐0018, China). The mice were housed under specific pathogen‐free conditions with a cycle of 12 hr light/12 hr dark, a temperature range of 22 ± 1°C and 55 ± 5% relative humidity. All mice were fed standard laboratory chow and water ad libitum and allowed to acclimatize in our facility for 1 week before any experiments started.
Mice were randomly divided into three groups (normal, vehicle receiving only DSS, and treatment receiving DSS + apremilast) with eight mice per group. UC was induced by administration of DSS (3% in drinking water) for 7 days and then normal drinking water given for another 4 days; this model has been described previously (Chassaing, Aitken, Malleshappa, & Vijay‐Kumar, 2014). Apremilast (50 mg·kg−1) was dissolved in 0.5% carboxymethylcellulose sodium and 0.25% Tween 80 and orally administered once daily. During the treatment, weight loss, stool consistency, and fecal blood, as indicators of disease activity index (DAI), were monitored by three investigators who were blinded to the experimental conditions. The DAI scores were calculated as the sum of the weight loss, stool consistency, and rectal bleeding score, and are shown in Table 1, adapted from Wirtz et al. (2017). On Day 11, mice were anaesthetized with an intraperitoneal injection of 4% chloral hydrate, and the serum samples were collected for biochemical assays, using a HITACHI‐7080 automatic biochemical analyser (Hitachi High Technologies Corporation, Tokyo, Japan). The spleen, mesenteric lymph nodes (MLNs), and colons were collected for further assessment, as described below.
Table 1.
Scoring system for calculating a disease activity index (DAI)
| Score | Weight loss | Stool consistency | Blood |
|---|---|---|---|
| 0 | None | Normal | Negative haemoccult |
| 1 | 1–5% | Soft but still formed | Weakly positive haemoccult |
| 2 | 6–10% | Soft | Positive haemoccult |
| 3 | 11–20% | Very soft; wet | Blood traces in stool visible |
| 4 | >20% | Watery diarrhoea | Gross rectal bleeding |
2.2. Histopathological examination
Colon The length of the colon from the anus to appendix was measured by a ruler. The samples of colon for histological examination were fixed in 10% PBS‐buffered formalin at room temperature and then embedded in paraffin. Sections (5 μm) were cut and stained with haematoxylin and eosin. For colon fibrosis, sections were stained with Masson's trichrome reagents. Histopathological features were observed under a light microscope and scored by three investigators, who were blinded to the experimental conditions, in the Center for Drug Safety Evaluation and Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, which is compliant with Good Laboratory Practice. The scoring was as follows: 0, no evidence of inflammation; 1, low level inflammation with scattered mononuclear cells (1–2 foci); 2, moderate inflammation with multiple foci of mononuclear cells; 3, high level inflammation with increased vascular density and marked wall thickening; and 4, maximal inflammation with transmural leukocyte infiltration and loss of goblet cells.
2.3. Myeloperoxidase, SOD, and MDA measurement
Freshly excised colon was rinsed with PBS, homogenized in tissue lysis buffer, and centrifuged. Individual activities of myeloperoxidase (MPO), SOD, and MDA were determined using an MPO activity assay kit (Nanjing Jiancheng Bioengineering Institute, China), SOD assay kit (Beyotime, Haimen, China), and MDA assay kit (Beyotime) according to the manufacturer's instructions. MPO activity was defined as the quantity of enzyme degrading 1 μmol·ml−1 peroxide at 37°C and expressed as U·mg−1 of colon protein. SOD and MDA activity were expressed as U·mg−1 colonic protein and mol·mg−1 colonic protein.
2.4. In vivo imaging and intestinal permeability measurement
The luminol‐based chemiluminescent probe L‐012 was applied to determine intestinal inflammation. On the day of in vivo imaging, the Living Image software was started and the IVIS Spectrum CT system initialized, according to the manufacturer's instructions (Kielland et al., 2009). Briefly, mice were anaesthetized in a chamber with 1.5–2.0% isoflurane, injected intraperitoneally with 25 mg·kg−1 L‐012 solution, and were placed in a supine position into the anaesthesia manifold in the imaging chamber of the IVIS Spectrum CT bioluminescence imaging system. The bioluminescent images were acquired 1 min after injection using the autoexposure option to automatically regulate acquisition parameters (shutter speed, binning, and aperture of the system) to optimize signal intensity (Wirtz et al., 2017).
For intestinal permeability assay, mice were fasted overnight and FITC‐dextran solution (3–5 kDa, 600 mg·kg−1) were orally dosed with a syringe through a blunt‐ended curved feeding tube. 4‐hr later, mice were anaesthetized and the blood samples were obtained by cardiac puncture. Blood was then centrifuged at 1500× g for 10 min at 4 °C and the fluorescence intensity of FITC in serum was measured at 480‐nm excitation and 520‐nm emission using a microplate reader. Meanwhile, mice were exposed to the IVIS Spectrum CT system, and the fluorescent images were acquired at 480‐nm excitation and 520‐nm emission to determine the retention of FITC‐dextran in the abdominal region (Gupta & Nebreda, 2014).
2.5. Full‐thickness colonic tissue culture
The longitudinal 1‐cm segments of the colon were isolated at the same region using surgical forceps and scissors as described previously (Wirtz et al., 2017). The colonic segments were washed with cold PBS, cut into three or four defined biopsies, and then cultured for 24 hr containing 0.5 ml of sterile cell culture medium at 37°C in a humidified incubator of 5% CO2. The supernatants were collected and cytokine production measured.
2.6. Flow cytometry assay
MLNs and spleen were extracted from the mice using sterile technique and dissected mechanically. Mononuclear splenocyte suspensions were prepared after cell debris, and clumps were removed. Erythrocytes were depleted with ammonium chloride buffer solution. Cells were washed once with fresh medium and were filtered using a 70‐μm filter to obtain a mononuclear cell suspension. Cells from the colonic LP were isolated as described (Uhlig et al., 2006). Colons were cut into small pieces and incubated in RPMI 1640 containing 10% FBS and 5‐mM EDTA for 15 min in a shaking incubator at 37°C for three times to remove epithelial cells. The remaining tissue was digested using RPMI 1640 containing 10% FBS, 0.5 mg·ml−1 Type IV Collagenase, 3 mg·ml−1 Dispase II, and 0.1 mg·ml−1 DNase I for 30 min in a 37°C shaking incubator. LP cells were collected and filtered using a 70‐μm filter to obtain a mononuclear cell suspension.
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Single cell suspensions were washed with PBS and then stained with fixable viability dye eFluor™ 780 (eBioscience, San Diego, CA, USA) for 30 min at 4°C to identify viable cells from the dead cells. Following this procedure, cells were blocked with anti‐CD16/CD32 mAb (Thermo Fisher Scientific Cat# 14‐0161‐86, RRID:AB_467135) and stained with brilliant UV 395‐conjugated CD45 (BD Biosciences Cat# 564279, RRID:AB_2651134), FITC‐conjugated anti‐CD62L (BD Biosciences Cat# 553150, RRID:AB_394665), FITC‐conjugated anti‐γδTCR (Thermo Fisher Scientific Cat# 11‐5711‐82, RRID:AB_465238), FITC‐conjugated anti‐CD4 (BD Biosciences Cat# 553651, RRID:AB_394971), FITC‐conjugated anti‐Gr‐1 (Thermo Fisher Scientific Cat# 11‐5931‐81, RRID:AB_465313), FITC‐conjugated anti‐CD8 (BD Biosciences Cat# 551347, RRID:AB_394159), phycoerythrin (PE)‐conjugated anti‐CD25 (BD Biosciences Cat# 553866, RRID:AB_395101), PE‐conjugated anti‐F4/80 (BD Biosciences Cat# 565410, RRID:AB_2687527), Peridinin‐chlorophyll proteins‐Cyanine5.5 (Percp‐Cy5.5)‐conjugated anti‐CD44 (Thermo Fisher Scientific Cat# 45‐0441‐80, RRID:AB_925747), Percp‐Cy5.5‐conjugated anti‐CD11b (BD Biosciences Cat# 550993, RRID:AB_394002), Percp‐Cy5.5‐conjugated anti‐CD4, allophycocyanin‐conjugated anti‐CD11c (BD Biosciences Cat# 550261, RRID:AB_398460), allophycocyanin‐conjugated anti‐CD4 (BD Biosciences Cat# 553051, RRID:AB_398528), and brilliant violet 421‐conjugated anti‐CD3 (BD Biosciences Cat# 564008, RRID:AB_2732058). For intracellular staining, cells were stained with surface markers, followed by fixation and permeabilization using Foxp3 Staining Buffer set (Thermo Fisher Scientific, MA, USA). Cells were labelled intracellularly with PE‐conjugated anti‐IL‐17A (BD Biosciences Cat# 559502, RRID:AB_397256) and Percp‐Cy5.5‐conjugated anti‐Foxp3 (Thermo Fisher Scientific Cat# 45‐5773‐82, RRID:AB_914351). All immunofluorescent antibodies used in this research were obtained from BD Biosciences (Franklin Lakes, NJ, USA) or Thermo Fisher Scientific. The data were analysed using FlowJo software (RRID:SCR_008520, Tree Star, Ashland, OR, USA).
2.7. Ex vivo proliferation and cytokine production
MLN cells from the three groups were prepared and stimulated with anti‐CD3 antibodies (5 μg·ml−1, Thermo Fisher Scientific Cat# 14‐0031‐81, RRID:AB_467048) and LPS (10 μg·ml−1) respectively. After incubation, the cultures were pulsed with [3H]‐thymidine (0.5 μCi per well) to determine cell proliferation, and the supernatants were collected to determine the cytokine levels.
Polyclonal CD4+ T cells were isolated from MLNs cells using EasySep™ mouse CD4+ T Cell isolation kit (Stemcell, Vancouver, BC, Canada) according to the manufacturer's instructions respectively. To acquire CD4+ T cells, immunomagnetic negative selection was performed for removal with biotinylated antibodies recognizing specific cell surface markers. Unwanted cells (CD8+ cells, B220+ cells, CD11b+ cells, and I‐A+ antigen presenting cells from splenocytes) were separated with an EasySep™ magnet. The purity of the CD4+ T cells was consistently >98% determined by flow cytometry. Purified CD4+ T cells were cultured with medium alone or anti‐CD3 antibodies (5 μg·ml−1) and anti‐CD28 antibodies (2 μg·ml−1, Thermo Fisher Scientific Cat# 14‐0281‐86, RRID:AB_467192). After incubation, the cell cultures were pulsed with [3H]‐thymidine (0.5 μCi per well) to determine CD4+ T cell proliferation, and the supernatants were collected to determine the cytokine levels.
2.8. Cell cultures and treatment
Murine adherent macrophage cell line RAW264.7 cells (ATCC Cat# TIB‐71, RRID:CVCL_0493) and human adherent epithelial cell line Caco‐2 cells (ATCC Cat# HTB‐37, RRID:CVCL_0025) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). RAW264.7 cells were grown in DMEM containing 10% FBS, 2 mmol·L−1 L‐glutamine, 100 U·mL−1 penicillin, and 100 μg·mL−1 streptomycin. Caco‐2 cells were grown in DMEM containing 10% FBS, 2 mmol·L−1 L‐glutamine, 100 U·mL−1 penicillin, and 100 μg·mL−1 streptomycin. The cells were maintained at 37°C in a humidified incubator of 5% CO2. For epithelial barrier function assay, Caco‐2 cells were seeded on collagen‐coated polycarbonate membrane transwell support (Corning Costar, Acton, MA). The cell monolayers were incubated with or without 10 ng·ml−1 TNF‐α and 10 ng·ml−1 IFN‐γ in the absence or presence of different concentrations of apremilast for 72 hr. The electrical resistance of the filter‐grown monolayers was measured by using an EVOM volt ohmmeter (EVOM, Hertfordshire, England) with a pair of STX‐2 chopstick electrodes (WPI, Sarasota, Florida, USA). Then, FITC‐labelled dextran was added to the apical side of the monolayer, and after 2‐hr incubation, FITC‐dextran fluorescence was measured using a microplate reader.
Bone marrow‐derived macrophages (BMDMs) were obtained from the femur and tibia bones of 6‐week old of C57BL/6J mice as previously described (Ying, Cheruku, Bazer, Safe, & Zhou, 2013). BMDMs were cultured for 7 days in Iscove's Modified Dulbecco's Medium containing 10% FBS and 10 ng·ml−1 of mouse CSF. To detach and resuspend mature macrophages after differentiation, 5‐mM EDTA in Ca2+ and Mg2+‐free Hank's balanced buffer were used. The purity of mature macrophages was consistently >98%, defined as the percentage of CD11b+F4/80+ populations by flow cytometry analysis.
BMDMs and RAW264.7 cells were treated with different concentrations of apremilast, with or without LPS. The supernatants were collected for cytokine assays. Meanwhile, cells were centrifuged (1500× g for 10 min at 4 °C) and then assayed by western blot and immunofluorescence for phospho‐cAMP‐response element binding protein (CREB). To investigate the critical role of PKA–CREB in the anti‐inflammation, BMDMs and RAW264.7 cells were incubated with 10‐μM H‐89 (PKA inhibitor) or 10‐μM forskolin (adenylate cyclase activator), and then the cells and supernatants were collected for determining cytokine production and phosphorylation of CREB.
2.9. SiRNA transfection
To knock down PKA expression in BMDMs and RAW264.7 cells, siRNA targeting PKA C‐α (CST, Danvers, MA, USA) was used, following the manufacturer' instructions. Briefly, cells were transfected with siRNA and mixed with Lipofectamine® RNAiMAX Reagent (Thermo Fisher Scientific) in serum‐free opti‐MEM medium. Cells were collected and assayed for PKA C‐α expression after 72 hr of transfection. The percentage of knockdown for PKA C‐α was over 80%.
2.10. cAMP measurements
Briefly, 50–80% confluence of RAW264.7 and BMDMs were treated at the indicated concentrations of apremilast, H89, and forskolin for 30 min. Cells were harvested, lysed by 0.1‐M HCl, and then centrifuged (600× g for 6 min at 4 °C) for supernatant collection. Intracellular cAMP was assayed with the direct cAMP ELISA kit purchased from Enzo Life Sciences (Raamsdonksveer, the Netherlands) according to the manufacturer's instruction. The content of cAMP was expressed as pmol cAMP per mg of total protein, to normalize for protein content.
2.11. Cytokine assay
Quantification of cytokines in serum was performed using the Luminex x‐MAP technology (Luminex Corp, Austen TX, USA). Serum from colitis mice were analysed using a Milliplex multi‐analyte magnetic bead panel obtained from Thermo Fisher Scientific, and all data were collected on a Luminex 200 instrument. All standard curves with four‐parameter logistic fitting generated from the known cytokine concentrations supplied by the manufacturer had R 2 values calculated at or close to 1 and quality controls in the kit performed as expected.
Cytokines in colon homogenates and culture supernatants were determined by using mouse TNF‐α, IFN‐γ, IL‐1β, IL‐2, IL‐6, IL‐10, IL‐12p40, and IL‐17A ELISA kits (BD Pharmingen, San Diego, CA, USA) according to the manufacturer's instructions.
2.12. Immunohistochemistry and immunofluorescence
Formalin‐fixed paraffin‐embedded tissues were sectioned (5 μm) and collected onto coherent glass slides. Tissue sections were dewaxed in xylene and rehydrated through graded alcohol to water. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide before masked antigens were retrieved by 0.01‐M citrate buffer solution. For immunohistochemistry, the sections were blocked with 10% normal horse serum and incubated overnight at 4°C in a humidified environment with antibodies to PDE4D (Proteintech Group Cat# 12918‐1‐AP, RRID:AB_2161097, Rosemont, USA) and phospho‐p65 (Cell Signaling Technology Cat# 3033), phospho‐ERK (Cell Signaling Technology Cat# 4376, RRID:AB_331772, Danvers). Primary labelling was detected using biotinylated horse anti‐rabbit IgG secondary antibody, incubated with streptavidin‐HRP and then signals were detected using diaminobenzidine. For immunofluorescence, the tissue sections were blocked with 10% normal horse serum and incubated overnight at 4°C in a humidified environment with Alexa Fluor 488‐conjugated anti‐E‐cadherin (Cell Signaling Technology Cat# 3199, RRID:AB_10691457), FITC‐conjugated CD11b (Abcam Cat# ab18273, RRID:AB_444372, Cambridge, MA, USA), Alexa Fluor 647‐conjugated F4/80 (Abcam Cat# ab204467), Alexa Fluor 647‐conjugated Ly6G (BioLegend Cat# 127609, RRID:AB_1134162, San Diego, CA, USA), FITC‐conjugated CCR5 (Abcam Cat# ab11466, RRID:AB_2275506), CXCR3 (Proteintech Cat# 26756‐1‐AP, Rosemont, USA), ZO‐1 (Proteintech Cat# 21773‐1‐AP), and Epac1 (Cell Signaling Technology Cat# 4155). Primary labelling for unconjugated fluorescein was detected using FITC‐conjugated secondary antibodies and then counterstained with DAPI and mounted in N‐propyl gallate in glycerol‐PBS. Images were collected on Leica TCS SPS microscope.
2.13. RNA extraction, cDNA synthesis, and qPCR
Total RNA was extracted from colon tissue by using RNAsimple total RNA kit (Tiangen, Beijing, China) and then reverse transcribed by an All‐in‐One cDNA Synthesis SuperMix (Biotool, Houston, TX, USA). Real‐time PCR was performed with SYBR® Green Realtime PCR Master Mix (TOYOBO, Osaka, Japan) on an Applied Biosystems 7500 Fast Real‐Time PCR System (Applied Biosystems, Foster city, CA, USA). The primers used for PCR amplification are listed in Table S1. The fold change in mRNA expression of gene was normalized to β‐actin using the ΔΔCt method.
2.14. Western blotting
Cells and colon samples were lysed with SDS sample buffer, and protein concentration was determined by the Pierce BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein (10–30 μg) were separated by 10% SDS‐PAGE and transferred to a nitrocellulose membrane (Bio‐Rad, Hercules, CA, USA). Non‐specific binding was blocked with 5% BSA, and the membranes were incubated overnight (4°C) with rabbit or mouse primary antibodies (Table S2). Signals were detected with HRP‐conjugated anti‐rabbit IgG (1:20,000) or HRP‐conjugated anti‐mouse IgG (1:10,000) using SuperSigna West Femto Maximum Sensitivity Substrate under visualization in a ChemiDoc™ MP Imaging System (Bio‐Rad).
2.15. Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All the images of western blots and immunohistochemistry (stainings) were quantified using Image‐Pro Plus software (RRID:SCR_007369, Media Cybernetics, Silver Springs, MD, USA). All experimental data are presented as mean ± SEM, and each experiment was performed a minimum of three times. All group data subjected to statistical analysis in the present research have a minimum of n = 5 individuals per group or independent samples according to the power analysis in pharmacology (Curtis et al., 2015). Statistical analyses were evaluated using GraphPad Prism 6.0 software (RRID:SCR_002798, La Jolla, CA, USA). Significant differences between groups were determined using a one‐way ANOVA with Dunnett's multiple comparisons test with no significant variance inhomogeneity (F achieved P < 0.05). P < 0.05 was considered to represent a significant difference between group means.
2.16. Materials
Except where indicated, all the materials and reagents were obtained from Sigma (St Louis, MO, USA). DSS (molecular weight 36–50 kDa) was purchased from MP Biomedicals (Irvine, CA, USA). Apremilast (M.W. = 460.5) was purchased from Selleck (Shanghai, China). The fecal occult blood test kits were obtained from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Anti‐CD3 and CD28 antibodies were purchased from Thermo Fisher Scientific. CCK‐8 (Cell Counting Kit‐8) was purchased from Dojindo (Kumamoto, Japan).
2.17. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017; Alexander et al., 2017a; Alexander et al., 2017b).
3. RESULTS
3.1. Apremilast ameliorated DSS‐induced UC in mice
The DSS‐induced mouse model of colitis is well‐characterized by increased colon ulceration and acute inflammation. Mice with colitis exhibited body weight loss from about Day 5 onward; and diarrhoea and rectal bleeding appeared upon DSS application (Figure 1a). Compared to the vehicle controls, apremilast‐treated colitic mice showed a prominent reduction in body weight loss and DAI scores during the disease progression in a dose‐dependent manner (Figure 1a and Figure S1a,b). Splenomegaly was associated with the colitis and apremilast clearly suppressed the spleen index, shown as the ratio of spleen : body weight (Figure 1b and Figure S1c). In addition, shortening of the colon, caused by inflammation, was an experimental indicator of colitis and apremilast dose‐dependently increased colon length, compared with the mice receiving DSS only (vehicle group; Figure 1c,d and Figure S1d,e). It is worth mentioning that normal mice treated only with apremilast, exhibited no inflammation and showed no symptoms of diarrhoea, splenomegaly, and colon shortening (Figure S1a–e). Considering the therapeutic effects of two doses (50 and 10 mg·kg−1), we used the higher dose of apremilast (50 mg·kg−1) in all subsequent experiments.
Figure 1.
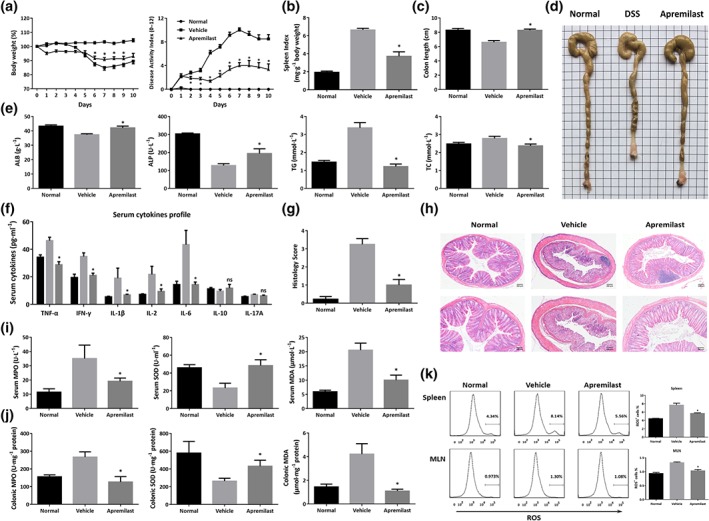
Apremilast ameliorated DSS‐induced inflammation and oxidative stress in colitic mice. Murine colitis was established with 3% DSS for 7 days and drinking water for the next 4 days. (a) Body weight expressed as percentage of initial weight and disease activity index (DAI) mean values assigned based on the criteria described in Table 1. (b) Spleen index calculated by spleen weight (mg)/body weight (g). (c) Colon length. (d) Representative colon images. (e) Serum biochemical indices including ALB, ALP, TG, and TC. (f) Serum cytokine secretion. (g) Histological scores assigned. (h) Representative histological sections of colonic mucosa stained with haematoxylin and eosin (20× and 40× magnification). (i) Serum MPO, SOD, and MDA. (j) Colonic MPO, SOD, and MDA. (k) Flow cytometry analysis and quantification of ROS production in spleen and mesenteric lymph node cells. Data shown are means ± SEM; n = 8 mice per group. *P < 0.05, significantly different from vehicle (DSS only) group
We next determined some variables of blood biochemistry in mice with DSS‐inudced colitis. Due to excessive bleeding and metabolic disturbance, DSS‐treated mice showed decreased serum ALB, ALP, and increased TG and TC. Interestingly, to some extent, apremilast restored the level of these biochemical indices (Figure 1e). Furthermore, assaying serum cytokines, by the Luminex assay, showed that apremilast markedly inhibited the secretion of inflammatory cytokines, such as TNF‐α, IFN‐γ, IL‐1β, IL‐2, and IL‐6, compared with the vehicle group (Figure 1f). The severity of colonic inflammation was further evaluated by histopathological analysis. In the DSS‐treated mice, colon tissue presented obvious mucosal ulceration, loss of crypt, goblet cells and epithelial damage, and neutrophil infiltration (Figure 1h). Colons from the apremilast‐treated group showed markedly less histological damage (Figure 1g,h).
3.2. Apremilast suppressed the oxidative stress responses in mice with DSS colitis
MPO, SOD, and MDA play key roles in oxidative stress responses and inflammation. In mice exposed to DSS only, the levels of MPO and MDA in serum and colonic tissue homogenates were much higher, while the levels of SOD lower than those in normal controls (Figure 1i,j). The abnormal serum and colon levels of MPO, SOD, and MDA were significantly reversed after apremilast treatment (Figure 1i,j). The ROS are known to be involved in oxidative damage and tissue dysfunction. In our models, both in the spleens and MLNs, apremilast decreased DSS‐induced up‐regulation of ROS (Figure 1k), which were mainly derived from CD8+ T cells (Figure S2).
3.3. Apremilast inhibited the inflammatory responses and tissue fibrosis in colon
To evaluate the inflammatory conditions within the gut micro‐environment, we determined the cytokine production in the supernatant of full‐thickness colon tissue cultures. As shown in Figure 2a, inflammatory mediators, such as TNF‐α, IFN‐γ, IL‐6, IL‐12p40, and IL‐17A, were suppressed in the colon cultures of apremilast‐treated mice, in comparison with those treated with DSS only. Correspondingly, the cytokines profile in colonic homogenates showed that the protein levels of TNF‐α, IFN‐γ, IL‐1β, IL‐2, IL‐6, and IL‐17A in apremilast‐treated group were much lower than those in vehicle controls, while IL‐10 was up‐regulated mildly in apremilast‐treated group (Figure 2b). Further analyses of the expression of mRNA for cytokines in colon confirmed the therapeutic effects of apremilast on colon inflammation (Figure 2c). Moreover, apremilast treatment resulted in a reduction of the mRNA levels of iNOS, COX‐2 (Figure 2d), and inflammasome‐associated genes, including NLRP3 and IL‐18, with no effects on ASC and caspase‐1 expression (Figure 2e).
Figure 2.
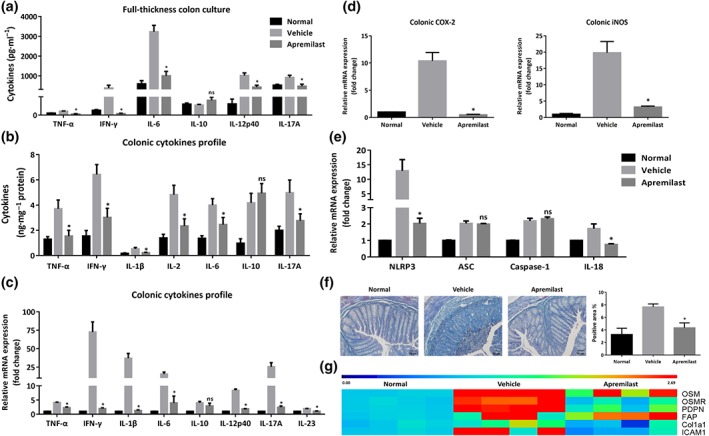
Apremilast suppressed the production of inflammatory mediators and colonic fibrosis in colitic mice. (a) Cytokine production profile in the full‐thickness colon culture. (b) Protein levels of cytokines in the tissue homogenates. (c) The mRNA expression of cytokines in colon. (d) The mRNA expression of iNOS and COX‐2 in colon. (e) The mRNA expression of inflammasome‐related genes in colon. (f) Representative sections and quantification with positive area (%) of colonic mucosa with Masson staining (100× magnification). (g) The mRNA expression of fibrosis‐related genes in colon. Data shown are means ± SEM; n = 8 mice per group. *P < 0.05, significantly different from vehicle (DSS only) group
Intestinal fibrosis is a common complication of IBD, which develops through various immune cells, extracellular matrix, abnormal production of cytokines and collagen deposition (Speca, Giusti, Rieder, & Latella, 2012; Suzuki et al., 2011). During the progression of DSS‐induced colitis, fibrosis has been observed after 6 days of DSS exposure (Suzuki et al., 2011). Histopathological examination of colon fibrosis in our models demonstrated that first signs of fibrotic lesions were observed in the colonic mucosa and submucosa (Figure 2f). Apremilast reduced collagen deposition and suppressed the expression of genes related to fibrosis, including Col1a1, oncostatin M and its receptor, podoplanin, and fibroblast activation protein (Figure 2f,g).
3.4. Apremilast protected the intestinal epithelial barrier function in DSS colitis
The intestinal epithelial integrity serves as the fundamental barrier against exogenous antigens and damage. Therefore, the present study evaluated the effects of apremilast on intestinal barrier function using in vivo imaging of inflammation with L‐012 solution and FITC‐dextran. Severe inflammation appeared in the location of intestinal tract in DSS‐induced colitis mice and apremilast decreased the positive signals following L‐012 injection (Figure 3a). When given orally, FITC‐dextran was mainly discharged through the intestinal and urinary systems and the gut‐transit time is a measure of the movement of FITC‐dextran along the digestive tract (Wang et al., 2015; Woting & Blaut, 2018). Once intestinal injury and inflammation occurred, intestinal retention and blood penetration of FITC‐dextran increased markedly (Figure 3b). The green fluorescent images obtained by IVIS spectrum indicated that apremilast‐treated colitic mice showed much lower retention than DSS‐treated mice (Figure 3b, left). Consistent with these findings, after absorption, distribution, and metabolism, the serum fluorescence intensity of FTIC‐dextran in apremilast‐treated mice was lower than that in DSS‐treated mice (Figure 3b, right and Figure S1f,g). Moreover, there was no change of intestinal permeability in normal mice treated with apremilast (Figure S1f,g). To confirm the involvement of epithelial tight junctions, we further examined the expression of colonic tight junction proteins, such as ZO‐1, E‐cadherin, and occludin, using immunofluorescence, western blot, and quantitative real‐time PCR. Intestinal permeability in mice treated with DSS only (vehicle group) was markedly increased and apremilast protected this epithelial barrier function, reversing the changes in both the protein and mRNA levels of ZO‐1, E‐cadherin, and occludin (Figure 3c,e). Further staining showed that the epithelial integrity in the vehicle group was disrupted, with considerable loss of ZO‐1 and E‐cadherin expression, in situ (Figure 3d). Additionally, apremilast inhibited the expression of MMP2, MMP3, and MMP9 in the colon (Figure 3f).
Figure 3.
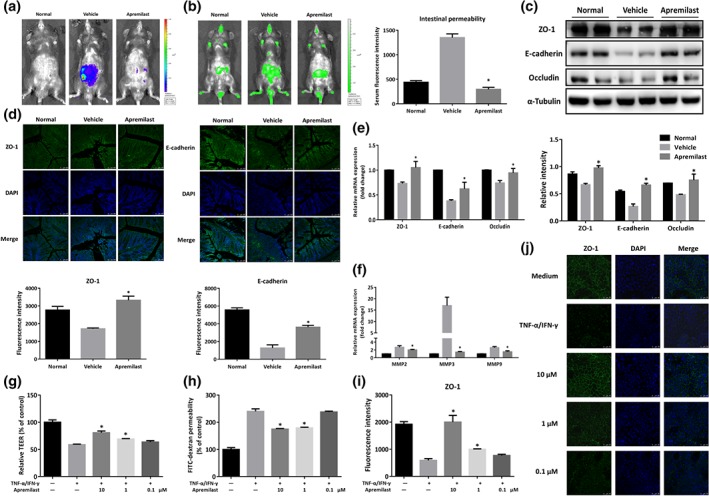
Apremilast protected the intestinal epithelial barrier function and prevented cytokine‐induced epithelial barrier disruption. (a) Bioluminescent imaging with L‐012 sodium was obtained under isoflurane anaesthesia using an IVIS Spectrum CT system. (b) Fluorescence imaging with FITC‐dextran administration (left) and serum fluorescence intensity of FITC‐dextran (right) were measured. (c) The expression of tight junction‐associated proteins (ZO‐1, E‐cadherin, and occludin) detected by western blot and α‐tubulin was used as a loading control. (d) Colonic tissues were immunofluorescently stained for ZO‐1 and E‐cadherin, and the nuclei were stained with DAPI. (e) The mRNA level of tight junction‐associated proteins. (f) The mRNA expression of MMP2, MMP3, and MMP9. (g) Barrier function was measured as TEER in Caco‐2 cell monolayers primed by TNF‐α and IFN‐γ. (h) FITC‐dextran permeability in cytokine‐induced Caco‐2 cells. (i) Quantification of fluorescence intensity of ZO‐1 in Caco‐2 cells. (j) Representative image of immunofluorescent staining for ZO‐1 in Caco‐2 cells. Data shown are means ± SEM. (a–f), n = 8 per group. *P < 0.05, significantly different from vehicle (DSS only) group. (g–j), n = 5. *P < 0.05, significantly different from TNF‐α plus IFN‐γ‐treated group
3.5. Apremilast prevented cytokine‐induced disruption of epithelial integrity and permeability in vitro
To validate the effects on the epithelial barrier, we analysed the protective effects of apremilast against epithelial barrier dysfunction induced by proinflammatory cytokines. TNF‐α and IFN‐γ synergize to reduce the trans‐epithelial electrical resistance (TEER) and to increase the paracellular permeability of Caco‐2 cell monolayers in vitro (Figure 3g,h). Apremilast prevented this decrease of TEER and the increase of paracellular permeability, in a concentration‐dependent manner (Figure 3g,h). Further investigations of ZO‐1 morphology and subcellular distribution showed that the tight junction protein ZO‐1 was localized at the apical cellular junctions and formed a dense reticular structure at the cellular borders (Figure 3i,j). TNF‐α plus IFN‐γ stimulation caused desultory localization and irregular ring structures of membrane, while apremilast markedly attenuated the impairment of ZO‐1 localization in Caco‐2 cell monolayers (Figure 3i,j).
3.6. Apremilast suppressed leukocyte infiltration and expression of chemokines and their receptors
The abnormal interaction and crosstalk between MLNs and gut LP provide the pathogenicity in the initiation and progression of UC (Cader & Kaser, 2013). To gain insights into the pathological role of mucosal immunity in the intestine, the immune cell populations from MLN and colonic LP, were isolated and analysed by flow cytometry (Figure S3). The severity of DSS‐induced colitis was closely associated with an increase in the proportion of leukocytes. As observed both in the MLN (Figure 4a) and LP (Figure 4c), monocytic myeloid cells, macrophages, neutrophils, and dendritic cells were increased in DSS‐treated mice, and these changes were reversed after apremilast treatment. Immune cell infiltration was confirmed by immunofluorescence staining with CD11b, F4/80, and Ly6G, which were mostly scattered across the mucosal layers of colon from normal mice (Figure 4e). In addition, apremilast decreased the percentage of CD44+CD62L− (gated on CD3+CD4+), CD4+IL‐17+ (gated on CD3+) and increased the percentage of CD44−CD62L+ (gated on CD3+CD4+), CD25+Foxp3+ (gated on CD3+CD4+; Figure 4b). Correspondingly, apremilast suppressed the infiltration of γδTCR+, CD4+, and CD8+ T cells (Figure 4d). To further assess the underlying mechanisms, gene expression assays were performed to evaluate the mRNA expression of chemokines and their relevant receptors. Several chemokines (CXCL1, CXCL9, CXCL10, CCL2, CCL3, CCL4, CCL5, CCL20, CCL22, and their receptors (CCR2, CCR4, CCR5, CCR6, CCR9, CXCR2, and CXCR3) were significantly elevated during induction of UC (Figure 4g). Treatment with apremilast inhibited these expression levels, which was consistent with the data from the immunofluorescence assays (Figure 4f,g).
Figure 4.
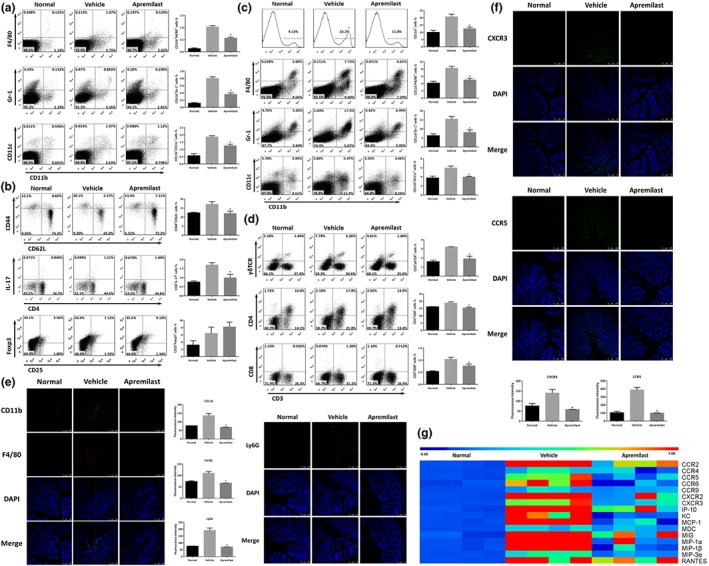
Apremilast regulated the leukocyte populations of mesenteric lymph nodes and lamina propria by suppressing the expression of chemokines and their receptors. (a) The percentage of macrophages (CD11b+F4/80+), neutrophils (CD11b+Gr‐1+), and dendritic cells (CD11b+CD11c+) in MLNs. (b) The percentage of naïve T cells (CD44−CD62L+, gated on CD3+CD4+), effector T cells (CD44+CD62L−, gated on CD3+CD4+), Th17 cells (CD4+IL‐17+, gated on CD3+), and Treg cells (CD25+Foxp3+, gated on CD3+CD4+) in MLNs. (c) The percentage of CD11b+ monocytes, macrophages, neutrophils, and dendritic cells in lamina propria. (d) The percentage of γδTCR+ T cells, CD4+ T cells, and CD8+ T cells in lamina propria. (e) Colonic tissues were immunofluorescence stained with CD11b, F4/80, and Ly6G, and the nuclei were stained with DAPI. (f) Colonic tissues were immunofluorescently stained for CXCR3 and CCR5. (g) The mRNA expression of chemokines and receptors in colon tissue. Data are shown as the representative images under flow cytometry and immunofluorescent staining. The summary data are shown as means ± SEM; n = 8 per group. *P < 0.05, significantly different from vehicle (DSS only) group
3.7. Apremilast inhibited ex vivo proliferation and cytokine production from MLNs
Because apremilast interferes with mucosal immunity in the gut, we cultured mouse MLN cells in the presence or absence of mitogens ex vivo. As expected, the proliferation of CD4+ T cells, purified from MLN cells and after exposure to anti‐CD3 plus CD28 antibodies, was reduced in mice that had received apremilast, (Figure 5a). In parallel with the effects on proliferation, the release of several cytokines (IFN‐γ, IL‐2, IL‐10, and IL‐17A) from CD4+ T cells were also decreased in the apremilast‐treated group (Figure 5b). For whole MLN cells, proliferation was decreased in the apremilast groups in the presence of either anti‐CD3 antibodies (Figure 5c) or LPS (Figure 5e). TCR stimulation led to robust release of cytokines (IFN‐γ, IL‐2, IL‐6, IL‐10, and IL‐17A) from MLN cells in the vehicle group, and apremilast reversed these changes in cytokine release (Figure 5d). Moreover, MLN cells from DSS‐treated mice were strongly activated by LPS induction, with high production of IFN‐γ, IL‐6, IL‐10, and IL‐12p40. However, a relatively low secretion of IFN‐γ, IL‐6, and IL‐12p40, but not of IL‐10, was found in the apremilast group (Figure 5f). Furthermore, the baseline proliferation and cytokine release from CD4+ T cell and whole MLN cells were also reduced, following apremilast treatment (Figure S4).
Figure 5.
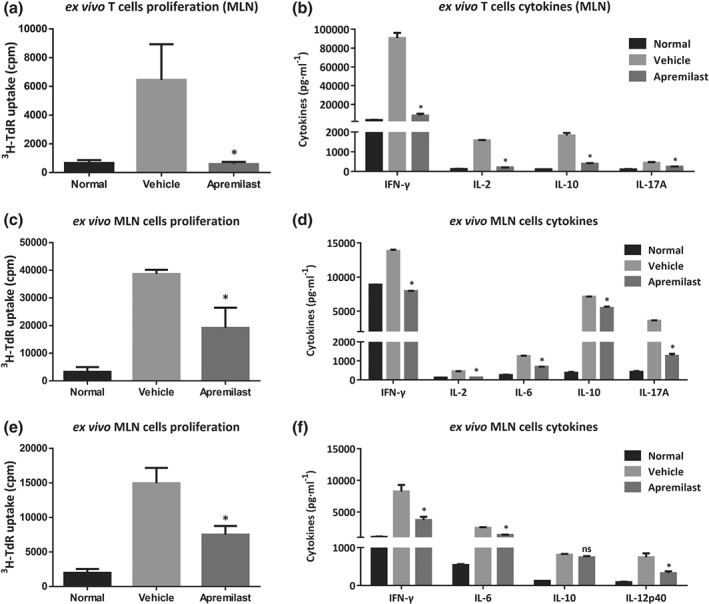
Apremilast inhibited lymphocyte proliferation and cytokine production upon ex vivo stimulation. CD4+ T cells were purified from MLNs and treated with anti‐CD3 plus anti‐CD28 antibodies to determine the CD4+ T cell proliferation (a) and (b) production of cytokines (IFN‐γ, IL‐2, IL‐10, and IL‐17A). (c) The proliferation of MLN cells treated with anti‐CD3 antibodies. and cytokine production (d) were determined. (e) The proliferation of MLN cells treated with LPS and (f) cytokine production. Data shown are means ± SEM; n = 8 mice per group. *P < 0.05, significantly different from vehicle (DSS only) group
3.8. Apremilast down‐regulated the expression of PDE4 isoforms and subsequently activated the PKA–CREB pathway
Previously, gene expression assay of PDE4 isoforms (A, B, C, and D) in peripheral blood mononuclear cells (PBMCs) from patients with CD showed that PDE4C mRNA was preferentially overexpressed, compared with data from normal individuals (Schafer et al., 2016). We first measured the expression of four PDE4 isoforms in the colon tissue from our murine model of UC. As shown in Figure 6a, PDE4A, 4B, and 4D were markedly up‐regulated while PDE4C exhibited little change in DSS‐treated mice. Apremilast decreased both the mRNA and protein level of PDE4 isoforms (Figure 6a,b), which was further supported by immunostaining with PDE4D (Figure 6c). Next, we determined how apremilast affected the downstream signalling mediated by PDE4. In the vehicle (DSS only) group, the expression of exchange protein 1 directly activated by cAMP (Epac1), Epac2, and of phospho‐CREB were decreased and apremilast reversed these decreases. in; meanwhile, apremilast also up‐regulated the protein level of Rap1, without any effects on the expression of CREB and PKA (Figure 6b,d).
Figure 6.
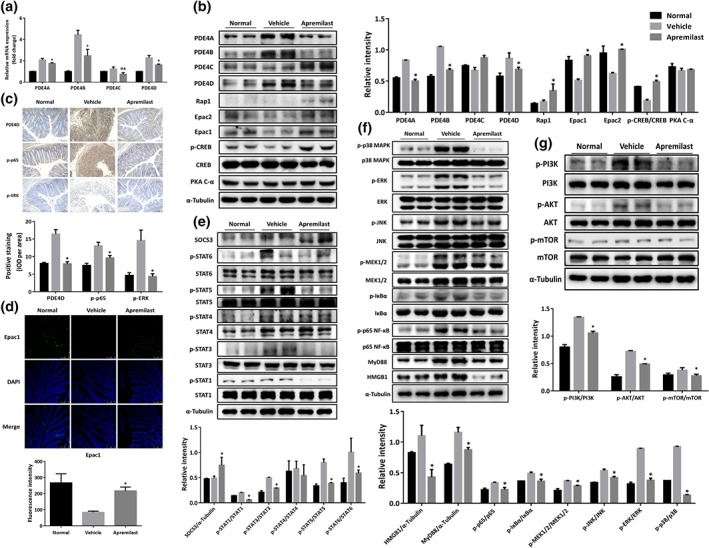
Apremilast inhibited PDE4 expression, activated PKA–CREB and Epac‐Rap1 signalling, and subsequently interfered with JAK–STAT–SOCS3, PI3K–mTOR, NF‐κB, and MAPK pathways. (a) The mRNA expression of the isoforms of PDE4 in colonic tissue. (b) The expression of the isoforms of PDE4 and PKA–CREB signalling‐associated proteins. (c) Immunohistochemical staining for PDE4D, p‐p65, and p‐REK of colonic tissue. (d) Immunofluorescent staining for Epac1 in colonic tissue. Western blot analysis of JAK–STAT–SOCS3 signalling (e), NF‐κB‐ and MAPK‐mediated signalling (f) and PI3K–mTOR signalling (g). Data shown are representative images from western blot assays and from immunohistochemical staining. The summary data are shown as means ± SEM; n = 8 per group. *P < 0.05, significantly different from vehicle (DSS only) group
3.9. Apremilast restrained the activation of NF‐κB‐, MAPK‐, PI3K–AKT–mTOR‐, and JAK–STAT–SOCS‐mediated signalling pathways
PDE4‐mediated PKA–CREB signalling pathways are closely related to MAPK and NF‐κB pathways in many inflammatory conditions (Li et al., 2018). In our model, Western blot assay showed that DSS treatment increased the phosphorylation of p38, ERK, JNK, and MEK1/2 and treatment with apremilast blocked these phosphorylations (Figure 6f). With regard to the NF‐κB pathway, the phosphorylation of IκBα and subunit p65 of NF‐κB were increased markedly upon DSS treatment and these effects were reversed by apremilast treatment (Figure 6f), which were further confirmed in the immunohistochemical analysis (Figure 6c). Apremilast could also decrease the overexpression of MyD88 and HMGB1, induced by DSS (Figure 6f). Moreover, apremilast showed suppressive effects on the phosphorylation of STAT1, STAT3, STAT5, and STAT6 via increasing SOCS3 expression, but only slightly affecting STAT4 phosphorylation (Figure 6e). Strikingly, apremilast also suppressed activation of the PI3K–mTOR pathway, decreasing the phosphorylation of PI3K, AKT, and mTOR (Figure 6g).
3.10. Apremilast suppressed LPS‐stimulated inflammatory responses through the PKA–CREB signalling pathway in macrophages
We additionally corroborated the effect of apremilast on activation of cAMP–PKA–CREB pathway in vitro. In these experiments, we used RAW264.7 cells, a murine macrophage cell line, and BMDMs, with or without LPS challenge. As shown in Figure 7a, apremilast significantly inhibited the production of TNF‐α, whereas it increased production of IL‐10 in a concentration‐dependent manner. In accordance with the results in Figure 6b, apremilast elevated the cellular level of cAMP in BMDMs and RAW264.7 cells (Figure S5) and subsequently increased phosphorylation of CREB with no change on the expression of total CREB protein (Figure 7b). The results were confirmed by immunofluorescent observation of phospho‐CREB (Figure 7c). Although there were some differences between RAW264.7 cells and BMDMs, the cAMP elevation and CREB phosphorylation were blocked in the presence of H89, a specific PKA inhibitor, with no effect on the expression of PKA (Figure 7d–g). In agreement with the results obtained with H89, knock‐down of expression of the catalytic subunit of PKA, using siRNA, provided the same profile of effects (Figure 7e,f and Figure S6). However, apremilast and forskolin, an activator of adenylyl cyclase, showed synergistic effects both in RAW264.7 cells and BMDMs, enhancing the elevation of cellular cAMP and CREB phosphorylation (Figure 7d–g). Further biological assay on TNF‐α release also displayed the antagonistic effects between apremilast and PKA inhibitors and synergistic effects between apremilast and adenylyl cyclase activators (Figure 7h,i).
Figure 7.
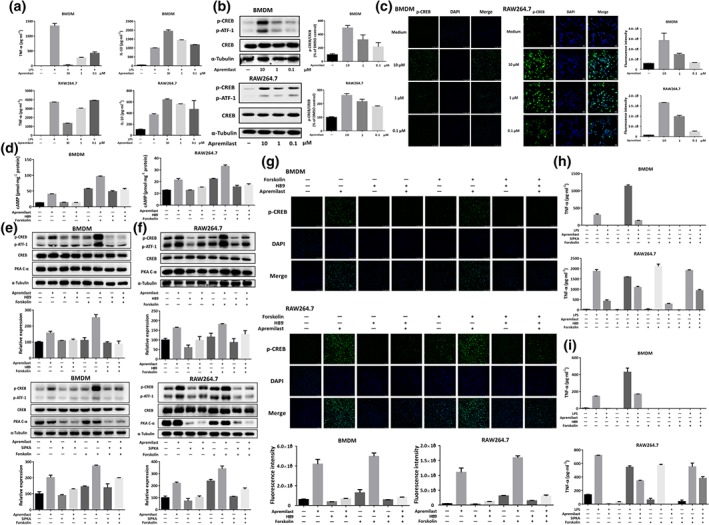
Apremilast suppressed the inflammatory responses in macrophages through PKA–CREB signalling. (a) TNF‐α and IL‐10 production in LPS‐stimulated BMDMs and RAW264.7 cells. (b) The phosphorylation of CREB and ATF‐1, following apremilast treatment, determined by western blot. (c) Immunofluorescent staining for phospho‐CREB upon apremilast treatment. BMDMs and RAW264.7 cells were treated with 10‐μM apremilast, 10‐μM H89 (PKA inhibitor), and 10‐μM Forskolin, cAMP level (d), phosphorylation of CREB and ATF‐1 (upper e, upper f, and g) were measured and TNF‐α production were determined upon LPS stimulation (h). BMDMs and RAW264.7 cells were transfected with PKA siRNA and NC SiRNA; then cells were treated with apremilast, H89, and forskolin. The phosphorylation of CREB and ATF‐1 (lower part of e; lower part of f) and TNF‐α (i) were measured. The summary data are shown as means ± SEM of three independent experiments
4. DISCUSSION
UC has become a worldwide challenging disease that strikes at all ages and leads to lifelong morbidity or even lethality (Ng et al., 2018). The high rate of resistance to therapy for UC generates an important unmet clinical medical needs and, consequently, alternative therapeutic strategies are urgently required. Cytokines are well‐known to mediate intestinal inflammation and targeting cytokine release has been shown to be useful in clinical situations. PDE4 functions as a cellular modulator of cAMP and inhibition of PDE4 is predicted to inhibit a wide array of inflammatory cytokines (Maurice et al., 2014). Over the past decades, various PDE4 inhibitors, including rolipram, roflumilast, and tetomilast, have been introduced as new therapeutic attempts for the treatment of UC. Because of their severe side effects, including headache, vomiting, nausea, and other gastrointestinal problems, further clinical testing of rolipram and tetomilast was abandoned (Spadaccini et al., 2017). Roflumilast, approved for treating asthma and chronic obstructive pulmonary disease, is known to ameliorate the morphological and biochemical alterations in DSS‐induced colitis (El‐Ashmawy et al., 2018). In contrast to roflumilast, another PDE4 inhibitor, apremilast, was approved in 2014 for psoriasis and psoriatic arthritis (Chiricozzi et al., 2016). Recently, a phase 2, multicentre, randomized, double‐blind, placebo‐controlled, parallel‐group study was designed to evaluate the clinical efficacy, safety, and tolerability of apremilast in patients with active UC; whereas no preclinical research on inflammatory colitis has yet been published (Li et al., 2018). The work described here aimed to investigate the efficacy and underlying mechanisms of apremilast in attenuating DSS‐induced murine UC. Our results demonstrated that oral administration of apremilast exerted protective effects by interfering with mucosal immunity (Figure 8). These experiments provide the first evidence of the therapeutic effects of apremilast on intestinal inflammation in vivo, using a murine model.
Figure 8.
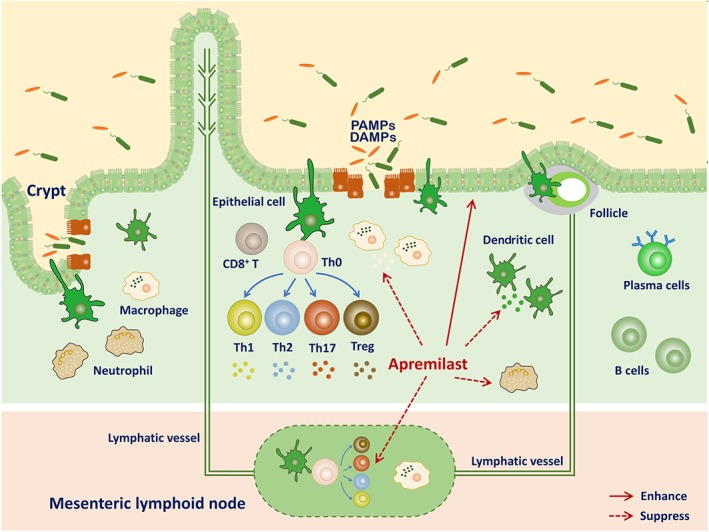
Diagram of how mucosal immunity in the DSS‐induced murine model of UC could be affected by apremilast to attenuate murine UC. PAMPs, pathogen‐associated molecular patterns; DAMPs, damage‐associated molecular patterns
Chemically induced colitis models mimic some key immunological and histopathological features of UC in humans. Oral administration of the sulfated polysaccharide DSS to mice via drinking water results in severe colitis characterized by weight loss, bloody diarrhoea, ulcer formation, and infiltration of inflammatory cells, which are similar to the observations in UC patients. The DSS‐induced colitis model is widely used preclinically due to its reproducibility, simplicity, and controllability (Chassaing et al., 2014). The mechanism by which DSS causes intestinal inflammation is closely associated with damage to the epithelial monolayer and abnormal dissemination of proinflammatory contents into the colon mucosa. In our research, apremilast significantly ameliorated the clinical features of UC, including body weight loss, colon shortening, hematochezia, and diarrhoea. Accompanied by intestinal tract bleeding and severe inflammation, serum biochemical parameters were abnormal with loss of ALB, decreased ALP activity, and dyslipidemia (Vermeire, Van Assche, & Rutgeerts, 2006). The abnormal oxidative stress disrupts the mucosal layer in the gut tract and increases permeability of the barrier contributing to the initiation and exacerbation of UC (Tian, Wang, & Zhang, 2017). We found that apremilast notably alleviated the local and systemic inflammation caused by DSS. Intestinal fibrosis, commonly resulting from epithelial‐to‐mesenchymal and endothelial‐to‐mesenchymal transitions, represents an unavoidable complication of UC and CD (Rieder & Fiocchi, 2008). Thus, in mucosal biopsies from healthy donors and IBD patients, oncostatin M and oncostatin M receptor expression are closely correlated with high expression of Col1a1, fibroblast activation protein, podoplanin, and ICAM1, which display the critical pathology in tissue fibrosis (West et al., 2017). In our DSS‐induced murine UC model, we found these genes were highly expressed in the colon, and inhibition of PDE4 decreased intestinal fibrosis, effects attributed to suppression of these genes.
The intestinal mucosal barrier plays an essential role in protecting homeostasis against the chaotic invasion of many antigens from the external environment (Martini, Krug, Siegmund, Neurath, & Becker, 2017; Turner, 2009). Genome‐wide association studies have identified several UC‐susceptible genes involved in intestinal barrier function, including HNF4, CDH1, and LAMB1 (Oshima & Miwa, 2016). Tight junctions are the constitutive structural component in epithelial cells. Our results from in vivo imaging with FITC‐dextran revealed that apremilast protected the barrier function against DSS‐caused damage. Furthermore, the reduction of tight junction proteins was markedly reversed when mice were treated with apremilast. Besides, increasing studies suggested that TNF‐α driven overexpression of gut MMP activity, especially of MMP3, importantly contributes to the colonic damage, leukocyte accumulation, excessive cytokines production, and fistula formation in UC (Naito & Yoshikawa, 2005). In cultures of LP mononuclear cells, apremilast suppressed TNF‐α and MMP3 production (Gordon et al., 2009).
Although the aetiology and pathogenesis of UC remains to be defined, the integrity and the immune system of the intestine have been shown to maintain tolerance to various inflammatory conditions, in which intestinal epithelial cells, macrophages, dendritic cells, adaptive immune cells, and the newly identified innate lymphoid cells reflect the importance of immune functions in mucosa (Cader & Kaser, 2013). Linked with numerous pathogenetic triggers in UC, the damage appears first in the intestinal epithelial cells, leading to disruption of barrier function (Figure 3). As highlighted in Figure 8, once epithelial integrity is disrupted, immune cells expressing high levels of inflammatory mediators and chemokines receptor infiltrate to mucosal layers (Atreya & Neurath, 2010; Zimmerman, Vongsa, Wendt, & Dwinell, 2008). During the progression of UC, both innate and adaptive immune system are overactivated (Steinbach & Plevy, 2014). In the inflamed tissue, we observed increased numbers of antigen‐presenting cells, neutrophils, γδ‐T cells, and IL‐17‐producing cells, which contributed to intestinal mucosal inflammation (Xu et al., 2014). Moreover, the results from our ex vivo study indicated that the CD4+ T cells purified from MLNs in apremilast‐treated mice exhibited weaker immune activity towards specific antigens than those from DSS‐treated mice (Figure 5). Our research demonstrated that PDE4 inhibition could interfere with mucosal immunity, as shown by inhibition of infiltration by immune cells, chemokines and chemokine receptor expression and reduction of the reactivity of immune cells.
PDE4 acts as a proinflammatory enzymes through the degradation of cAMP, which is well‐established as a potent regulator in both innate and adaptive immune cell functions (Raker, Becker, & Steinbrink, 2016). Previous reports have suggested a difference in gene expression patterns of PDE4 isoforms in PBMC from healthy individuals and from patients with various immune‐mediated or inflammatory diseases including psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis, sarcoidosis, scleroderma, CD, and systemic lupus erythematosus. The isoforms PDE4B and PDE4D were increased at the mRNA level in psoriatic patients whereas PDE4C was preferentially overexpressed in patients with CD (Schafer et al., 2016). However, there is little known about the PDE4 isoforms expressed in UC. We first determined the PDE4 isoforms in the colon tissue of DSS colitis by quantitative real‐time PCR, western blot, and immunohistochemical analyses. Strikingly, among the four isoforms, PDE4D showed the highest level of expression, followed by PDE4A, PDE4B, and PDE4C. Further investigations are required to measure the expression level of PDE4 isoforms in the PBMC or colon biopsies of UC patients. Moreover, DSS treatment in mice led to marked overexpression of PDE4A, 4B, and 4D, but not PDE4C. The interaction of cAMP, PKA, and Epac with PDE4 contributes to the formation of cAMP signalosomes (Maurice et al., 2014). Inhibition of PDE4 could result in accumulation of cellular cAMP and then activate PKA and Epac‐dependent pathways in a spatial and temporal manner (Cheng, Ji, Tsalkova, & Mei, 2008). PKA is composed of two separate subunits, namely, catalytic and regulatory subunits. Upon cAMP elevation, the free catalytic subunit is activated and subsequently affects a wide range of cytoplasmic and nuclear transcriptional factors, including CREB, CREM, and ATF‐1, which contribute to the anti‐inflammation effects in inflammatory conditions (Abdulrahim et al., 2015). This bioactivity was confirmed in our study on RAW264.7 cells and BMDMs using a PKA inhibitor or siRNA to silence the catalytic subunit of PKA. Furthermore, Epac has been identified as another family of cAMP sensor proteins. Epac proteins are highly specific for binding to cAMP and activate the small GTPase Rap1, which plays an important role in the formation of cell–cell junctions and promotes the barrier function of epithelium (Cheng et al., 2008). Consistent with the overexpression of PDE4, our results revealed a reduction of Epac1, Epac2, and phospho‐CREB in DSS‐induced UC and PDE4 inhibition with apremilast markedly down‐regulated the expression of the PDE4 isoforms and increased the expression of Epac1, Epac2, and phospho‐CREB. Meanwhile, colon Rap1 protein was activated in the presence of PDE4 inhibition, which accounted for the protective effects on the epithelial barrier. Alternatively, Epac activation could restrict the actions of inflammatory cytokines IL‐6 by inducing SOCS3 and with negative feedback inhibit JAK–STAT signalling (Parnell, Palmer, & Yarwood, 2015). As we observed, apremilast increased the expression of SOCS3 and inhibited the phosphorylation of STATs. On the other hand, PDE4 inhibition by apremilast indirectly inhibited the MAPK, NF‐κB, and PI3K–mTOR pathways, which are involved in the activation of innate and adaptive immunity (Hernandez‐Florez & Valor, 2016).
In conclusion, the results presented here have demonstrated that oral administration of apremilast exerted protective effects in experimental colitis in mice, through interfering with mucosal immunity, illustrating the pathological role of PDE4 in intestinal inflammation. Accordingly, the effects closely associated with PDE4 inhibition include modulation of the cAMP‐mediated PKA–CREB and Epac‐Rap1 pathways, which subsequently engage the MAPK, NF‐κB, PI3K–mTOR, and JAK–STAT–SOCS3 signalling pathways. Our study indicates the possibility of similar therapeutic effects of PDE4 intervention in UC patients.
AUTHOR CONTRIBUTIONS
H.L., J.Z., and W.T. contributed to the conception and design of this study. H.L., C.F., C.L.F., Y.W., H.M.L., P.L., X.Y., F.Z., Q.Q., and Y.G. performed the experiments. H.L. and W.T. analysed the data and wrote the manuscript. All the authors contributed to the interpretation of data and approved the final draft.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Apremilast dose‐dependently attenuated DSS‐induced ulcerative colitis without inflammation in control mice. (A) The percentage of body weight change during treatment. (B) The score of stool consistency and occult blood and DAI based on the criteria in Table 1. (C) Spleen index calculated by spleen weight (mg)/body weight. (D) Colon length. (E) Representative colon images. (F) Fluorescence imaging with FITC‐dextran administration and (G) serum fluorescence intensity. Data were shown as means±SEM; n = 9 per group. *p < 0.05, significantly different from vehicle group.
Figure S2. Flow cytometry analysis of ROS production from CD3+CD4+ (A), CD3+CD8+ (B), CD3−CD19+ (C) cells in spleen and mesenteric lymph nodes. Colitis was induced with 3% DSS as described in Methods. Splenocytes and mesenteric lymph node cells were isolated and labelled with fluorescent antibodies. Data correspond to experiments conducted on 8 mice.
Figure S3. The gating strategy of flow cytometry. FSC‐A plus SSC‐A was used to identify the total cells for analysis. FSC‐A plus FSC‐W were used to identify the singlets. Subsequently, CD45+FVDint cells were considered as living leukocytes for the further analysis.
Figure S4. Baseline proliferation and cytokines release from CD4+ T cell and whole MLN cells. CD4+ T cells (A, and B), purified from MLNs, and whole MLN cells (C, and D) were cultured to measure the basal proliferation and cytokines levels in accord with Figure 5. Data were shown as means±SEM; n = 8 per group.
Figure S5. Effect of apremilast on cAMP elevation in BMDMs and macrophages. BMDMs and RAW264.7 cells were incubated with the indicated concentrations (10, 1, or 0.1 μM) of apremilast for 30 min. cAMP levels in BMDMs (A) and RAW264.7 cells (B) were detected by ELISA. Data were shown as means±SEM of three independent experiments.
Figure S6. The percentage of knockdown for PKA C‐α.
Table S1. Sequences of primers used for quantitative real‐time PCR
Table S2. Antibodies for immunoblotting
ACKNOWLEDGEMENTS
This work was supported by grants from the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Grant 2018ZX09711002‐006‐011) and the “Personalized Medicines‐Molecular Signature‐based Drug Discovery and Development,” Strategic Priority Research Program of the Chinese Academy of Sciences, Grant XDA12020231. We are kindly grateful for the credible pathological analysis from Center for Drug Safety Evaluation and Research, Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences (CAS) and technical assistance from the Platform of Molecular Imaging and Research, SIMM, and CAS.
Li H, Fan C, Feng C, et al. Inhibition of phosphodiesterase‐4 attenuates murine ulcerative colitis through interference with mucosal immunity. Br J Pharmacol. 2019;176:2209–2226. 10.1111/bph.14667
Contributor Information
Jianping Zuo, Email: jpzuo@simm.ac.cn.
Wei Tang, Email: tangwei@simm.ac.cn.
REFERENCES
- Abdulrahim, H. , Thistleton, S. , Adebajo, A. O. , Shaw, T. , Edwards, C. , & Wells, A. (2015). Apremilast: A PDE4 inhibitor for the treatment of psoriatic arthritis. Expert Opinion on Pharmacotherapy, 16(7), 1099–1108. 10.1517/14656566.2015.1034107 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017a). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017b). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya, R. , & Neurath, M. F. (2010). Chemokines in inflammatory bowel diseases. Digestive Diseases, 28(3), 386–394. 10.1159/000320392 [DOI] [PubMed] [Google Scholar]
- Banner, K. H. , & Trevethick, M. A. (2004). PDE4 inhibition: A novel approach for the treatment of inflammatory bowel disease. Trends in Pharmacological Sciences, 25(8), 430–436. 10.1016/j.tips.2004.06.008 [DOI] [PubMed] [Google Scholar]
- Boyapati, R. K. , Rossi, A. G. , Satsangi, J. , & Ho, G. T. (2016). Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunology, 9(3), 567–582. 10.1038/mi.2016.14 [DOI] [PubMed] [Google Scholar]
- Cader, M. Z. , & Kaser, A. (2013). Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation. Gut, 62(11), 1653–1664. 10.1136/gutjnl-2012-303955 [DOI] [PubMed] [Google Scholar]
- Chassaing, B. , Aitken, J. D. , Malleshappa, M. , & Vijay‐Kumar, M. (2014). Dextran sulfate sodium (DSS)‐induced colitis in mice. Current Protocols in Immunology, 104. Unit 15 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, X. , Ji, Z. , Tsalkova, T. , & Mei, F. (2008). Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochimica et Biophysica Sinica, 40(7), 651–662. 10.1111/j.1745-7270.2008.00438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiricozzi, A. , Caposiena, D. , Garofalo, V. , Cannizzaro, M. V. , Chimenti, S. , & Saraceno, R. (2016). A new therapeutic for the treatment of moderate‐to‐severe plaque psoriasis: apremilast. Expert Review of Clinical Immunology, 12(3), 237–249. 10.1586/1744666X.2016.1134319 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172(14), 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mattos, B. R. , Garcia, M. P. , Nogueira, J. B. , Paiatto, L. N. , Albuquerque, C. G. , Souza, C. L. , … Simioni, P. U. (2015). Inflammatory bowel disease: An overview of immune mechanisms and biological treatments. Mediators of Inflammation, 2015, 493012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Ashmawy, N. E. , Khedr, N. F. , El‐Bahrawy, H. A. , & El‐Adawy, S. A. (2018). Roflumilast, type 4 phosphodiesterase inhibitor, attenuates inflammation in rats with ulcerative colitis via down‐regulation of iNOS and elevation of cAMP. International Immunopharmacology, 56, 36–42. 10.1016/j.intimp.2018.01.004 [DOI] [PubMed] [Google Scholar]
- Gordon, J. N. , Prothero, J. D. , Thornton, C. A. , Pickard, K. M. , Di Sabatino, A. , Goggin, P. M. , … Macdonald, T. T. (2009). CC‐10004 but not thalidomide or lenalidomide inhibits lamina propria mononuclear cell TNF‐α and MMP‐3 production in patients with inflammatory bowel disease. Journal of Crohn's & Colitis, 3(3), 175–182. 10.1016/j.crohns.2009.03.001 [DOI] [PubMed] [Google Scholar]
- Gupta, J. , & Nebreda, A. R. (2014). Analysis of intestinal permeability in mice. Bio‐Protocol, 4(22), e1289. [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, G. , Bidlingmaier, C. , Siegmund, B. , Albrich, S. , Schulze, J. , Tschoep, K. , … Endres, S. (2000). Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. The Journal of Pharmacology and Experimental Therapeutics, 292(1), 22–30. [PubMed] [Google Scholar]
- Hernandez‐Florez, D. , & Valor, L. (2016). Selective phosphodiesterase inhibitors: A new therapeutic option in inflammation and autoimmunity. Reumatologia Clinica, 12(6), 303–306. 10.1016/j.reuma.2016.07.011 [DOI] [PubMed] [Google Scholar]
- Ichikawa, H. , Okamoto, S. , Kamada, N. , Nagamoto, H. , Kitazume, M. T. , Kobayashi, T. , … Hibi, T. (2008). Tetomilast suppressed production of proinflammatory cytokines from human monocytes and ameliorated chronic colitis in IL‐10‐deficient mice. Inflammatory Bowel Diseases, 14(11), 1483–1490. 10.1002/ibd.20524 [DOI] [PubMed] [Google Scholar]
- Kielland, A. , Blom, T. , Nandakumar, K. S. , Holmdahl, R. , Blomhoff, R. , & Carlsen, H. (2009). In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L‐012. Free Radical Biology & Medicine, 47(6), 760–766. 10.1016/j.freeradbiomed.2009.06.013 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Zuo, J. , & Tang, W. (2018). Phosphodiesterase‐4 inhibitors for the treatment of inflammatory diseases. Frontiers in Pharmacology, 9 10.3389/fphar.2018.01048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini, E. , Krug, S. M. , Siegmund, B. , Neurath, M. F. , & Becker, C. (2017). Mend your fences: The epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cellular and Molecular Gastroenterology and Hepatology, 4(1), 33–46. 10.1016/j.jcmgh.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice, D. H. , Ke, H. , Ahmad, F. , Wang, Y. , Chung, J. , & Manganiello, V. C. (2014). Advances in targeting cyclic nucleotide phosphodiesterases. Nature Reviews. Drug Discovery, 13(4), 290–314. 10.1038/nrd4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y. , & Yoshikawa, T. (2005). Role of matrix metalloproteinases in inflammatory bowel disease. Molecular Aspects of Medicine, 26(4–5), 379–390. 10.1016/j.mam.2005.07.009 [DOI] [PubMed] [Google Scholar]
- Ng, S. C. , Shi, H. Y. , Hamidi, N. , Underwood, F. E. , Tang, W. , Benchimol, E. I. , … Kaplan, G. G. (2018). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population‐based studies. Lancet (London, England), 390(10114), 2769–2778. [DOI] [PubMed] [Google Scholar]
- Oshima, T. , & Miwa, H. (2016). Gastrointestinal mucosal barrier function and diseases. Journal of Gastroenterology, 51(8), 768–778. 10.1007/s00535-016-1207-z [DOI] [PubMed] [Google Scholar]
- Parnell, E. , Palmer, T. M. , & Yarwood, S. J. (2015). The future of EPAC‐targeted therapies: Agonism versus antagonism. Trends in Pharmacological Sciences, 36(4), 203–214. 10.1016/j.tips.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raker, V. K. , Becker, C. , & Steinbrink, K. (2016). The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Frontiers in Immunology, 7, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, F. , & Fiocchi, C. (2008). Intestinal fibrosis in inflammatory bowel disease—Current knowledge and future perspectives. Journal of Crohn's & Colitis, 2(4), 279–290. 10.1016/j.crohns.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Sartor, R. B. (2006). Mechanisms of disease: Pathogenesis of Crohn's disease and ulcerative colitis. Nature Clinical Practice. Gastroenterology & Hepatology, 3(7), 390–407. 10.1038/ncpgasthep0528 [DOI] [PubMed] [Google Scholar]
- Schafer, P. H. , Parton, A. , Gandhi, A. K. , Capone, L. , Adams, M. , Wu, L. , … Stirling, D. I. (2010). Apremilast, a cAMP phosphodiesterase‐4 inhibitor, demonstrates anti‐inflammatory activity in vitro and in a model of psoriasis. British Journal of Pharmacology, 159(4), 842–855. 10.1111/j.1476-5381.2009.00559.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, P. H. , Truzzi, F. , Parton, A. , Wu, L. , Kosek, J. , Zhang, L. H. , … Pincelli, C. (2016). Phosphodiesterase 4 in inflammatory diseases: Effects of apremilast in psoriatic blood and in dermal myofibroblasts through the PDE4/CD271 complex. Cellular Signalling, 28(7), 753–763. 10.1016/j.cellsig.2016.01.007 [DOI] [PubMed] [Google Scholar]
- Spadaccini, M. , D'Alessio, S. , Peyrin‐Biroulet, L. , & Danese, S. (2017). PDE4 inhibition and inflammatory bowel disease: A novel therapeutic avenue. International Journal of Molecular Sciences, 18(6). 10.3390/ijms18061276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speca, S. , Giusti, I. , Rieder, F. , & Latella, G. (2012). Cellular and molecular mechanisms of intestinal fibrosis. World Journal of Gastroenterology, 18(28), 3635–3661. 10.3748/wjg.v18.i28.3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach, E. C. , & Plevy, S. E. (2014). The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflammatory Bowel Diseases, 20(1), 166–175. 10.1097/MIB.0b013e3182a69dca [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K. , Sun, X. , Nagata, M. , Kawase, T. , Yamaguchi, H. , Sukumaran, V. , … Asakura, H. (2011). Analysis of intestinal fibrosis in chronic colitis in mice induced by dextran sulfate sodium. Pathology International, 61(4), 228–238. 10.1111/j.1440-1827.2011.02647.x [DOI] [PubMed] [Google Scholar]
- Tian, T. , Wang, Z. , & Zhang, J. (2017). Pathomechanisms of oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxidative Medicine and Cellular Longevity, 2017, 4535194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, J. R. (2009). Intestinal mucosal barrier function in health and disease. Nature Reviews. Immunology, 9(11), 799–809. 10.1038/nri2653 [DOI] [PubMed] [Google Scholar]
- Uhlig, H. H. , Coombes, J. , Mottet, C. , Izcue, A. , Thompson, C. , Fanger, A. , … Powrie, F. (2006). Characterization of Foxp3+CD4+CD25+ and IL‐10‐secreting CD4+CD25+ T cells during cure of colitis. The Journal of Immunology, 177(9), 5852–5860. 10.4049/jimmunol.177.9.5852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeire, S. , Van Assche, G. , & Rutgeerts, P. (2006). Laboratory markers in IBD: Useful, magic, or unnecessary toys? Gut, 55(3), 426–431. 10.1136/gut.2005.069476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Llorente, C. , Hartmann, P. , Yang, A. M. , Chen, P. , & Schnabl, B. (2015). Methods to determine intestinal permeability and bacterial translocation during liver disease. Journal of Immunological Methods, 421, 44–53. 10.1016/j.jim.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, N. R. , Hegazy, A. N. , Owens, B. M. J. , Bullers, S. J. , Linggi, B. , Buonocore, S. , … Powrie, F. (2017). Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor‐neutralizing therapy in patients with inflammatory bowel disease. Nature Medicine, 23(5), 579–589. 10.1038/nm.4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz, S. , Popp, V. , Kindermann, M. , Gerlach, K. , Weigmann, B. , Fichtner‐Feigl, S. , & Neurath, M. F. (2017). Chemically induced mouse models of acute and chronic intestinal inflammation. Nature Protocols, 12(7), 1295–1309. 10.1038/nprot.2017.044 [DOI] [PubMed] [Google Scholar]
- Woting, A. , & Blaut, M. (2018). Small intestinal permeability and gut‐transit time determined with low and high molecular weight fluorescein isothiocyanate‐dextrans in C3H mice. Nutrients, 10(6). 10.3390/nu10060685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. R. , Liu, C. Q. , Feng, B. S. , & Liu, Z. J. (2014). Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World Journal of Gastroenterology, 20(12), 3255–3264. 10.3748/wjg.v20.i12.3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying, W. , Cheruku, P. S. , Bazer, F. W. , Safe, S. H. , & Zhou, B. (2013). Investigation of macrophage polarization using bone marrow derived macrophages. Journal of Visualized Experiments: JoVE, 76 10.3791/50323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, N. P. , Vongsa, R. A. , Wendt, M. K. , & Dwinell, M. B. (2008). Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflammatory Bowel Diseases, 14(7), 1000–1011. 10.1002/ibd.20480 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Apremilast dose‐dependently attenuated DSS‐induced ulcerative colitis without inflammation in control mice. (A) The percentage of body weight change during treatment. (B) The score of stool consistency and occult blood and DAI based on the criteria in Table 1. (C) Spleen index calculated by spleen weight (mg)/body weight. (D) Colon length. (E) Representative colon images. (F) Fluorescence imaging with FITC‐dextran administration and (G) serum fluorescence intensity. Data were shown as means±SEM; n = 9 per group. *p < 0.05, significantly different from vehicle group.
Figure S2. Flow cytometry analysis of ROS production from CD3+CD4+ (A), CD3+CD8+ (B), CD3−CD19+ (C) cells in spleen and mesenteric lymph nodes. Colitis was induced with 3% DSS as described in Methods. Splenocytes and mesenteric lymph node cells were isolated and labelled with fluorescent antibodies. Data correspond to experiments conducted on 8 mice.
Figure S3. The gating strategy of flow cytometry. FSC‐A plus SSC‐A was used to identify the total cells for analysis. FSC‐A plus FSC‐W were used to identify the singlets. Subsequently, CD45+FVDint cells were considered as living leukocytes for the further analysis.
Figure S4. Baseline proliferation and cytokines release from CD4+ T cell and whole MLN cells. CD4+ T cells (A, and B), purified from MLNs, and whole MLN cells (C, and D) were cultured to measure the basal proliferation and cytokines levels in accord with Figure 5. Data were shown as means±SEM; n = 8 per group.
Figure S5. Effect of apremilast on cAMP elevation in BMDMs and macrophages. BMDMs and RAW264.7 cells were incubated with the indicated concentrations (10, 1, or 0.1 μM) of apremilast for 30 min. cAMP levels in BMDMs (A) and RAW264.7 cells (B) were detected by ELISA. Data were shown as means±SEM of three independent experiments.
Figure S6. The percentage of knockdown for PKA C‐α.
Table S1. Sequences of primers used for quantitative real‐time PCR
Table S2. Antibodies for immunoblotting