

Abstract
The zoonotic transmission of hantaviruses from their rodent hosts to humans in North and South America is associated with a severe and frequently fatal respiratory disease, hantavirus pulmonary syndrome (HPS)1,2. No specific antiviral treatments for HPS are available, and no molecular determinants of in vivo susceptibility to hantavirus infection and HPS are known. Here we identify the human asthma-associated gene protocadherin-1 (PCDH1)3–6 as an essential determinant of entry and infection in pulmonary endothelial cells by two hantaviruses that cause HPS, Andes virus (ANDV) and Sin Nombre virus (SNV). In vitro, we show that the surface glycoproteins of ANDV and SNV directly recognize the outermost extracellular repeat domain of PCDH1—a member of the cadherin superfamily7,8—to exploit PCDH1 for entry. In vivo, genetic ablation of PCDH1 renders Syrian golden hamsters highly resistant to a usually lethal ANDV challenge. Targeting PCDH1 could provide strategies to reduce infection and disease caused by New World hantaviruses.
Reporting summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Hantaviruses systemically infect and replicate in endothelial cells, and the nonlytic dysregulation of these cells is thought to underlie the changes in vascular permeability that are a hallmark of the viral disease in humans2,9. αvβ3 integrins have been identified as in vitro determinants of hantavirus infection10, and viral subversion of β3-integrin signalling in endothelial cells has been proposed to compromise vascular integrity9,10. Gene-complementation experiments have yielded other receptor candidates, including [β2 integrin11 and numerous components of the complement system12,13. However, the roles of these host factors in animal models of HPS or in humans remain undefined. Therefore, the identities of host molecules that mediate hantavirus infection in vivo and influence pathogenesis so far remain unknown.
To systematically uncover host factors for hantavirus entry, we14 and others15 previously used a recombinant vesicular stomatitis virus bearing the ANDV Gn/Gc glycoproteins (rVSV-ANDV Gn/Gc) to perform a loss-of-function genetic screen in HAP1 haploid human cells (Extended Data Fig. 1a). These screens identified several genes involved in the sterol regulatory element binding protein (SREBP) pathway as determinants of viral entry in endothelial cells and showed that membrane cholesterol has a key role in hantavirus membrane fusion14,16.
To extract hantavirus-receptor candidates from our dataset, we filtered our hits for genes that encode known plasma-membrane proteins17, and found a single gene, PCDH1—which encodes a cadherin-superfamily protein7,8, protocadherin-1—with proposed roles in human airway function and disease3–6 (Extended Data Fig. 1a and Supplementary Table 1). PCDH1 was not a hit in any other published haploid screens for pathogen host factors18,19, suggesting that it has a specific role in hantavirus entry. To evaluate this hypothesis, we used CRISPR-Cas9 genome engineering to generate cell clones deficient for PCDH1 in two human cell lines—HAP1 haploid cells (Fig. 1a, b and Extended Data Fig. 1b, c) and U2OS osteosarcoma cells (Fig. 2b and Extended Data Fig. 1d–f). PCDH1-knockout cells were poorly susceptible to infection by rVSVs bearing Gn/Gc glycoproteins from ANDV, SNV, Maporal virus (MPRLV) and Prospect Hill virus (PHV), all of which belong to the New World hantavirus clade. However, PCDH1-knockout cells remained susceptible to rVSVs bearing the VSV glycoprotein G or Gn/Gc from Hantaan virus (HTNV) and Seoul virus (SEOV)—hantaviruses in the Old World hantavirus clade that are not associated with HPS. The susceptibility of the PCDHl-knockout cell lines to Gn/Gc-dependent entry could be restored by expression of human PCDH1 complementary DNA (Figs. 1a, b, 2b and Extended Data Fig. 1). Infections with authentic hantaviruses corroborated and extended our observations: ANDV and SNV required PCDH1 for infection, whereas HTNV did not (Figs. 1b, 2b). Thus, PCDH1 mediates Gn/Gc-dependent cell entry and infection by four New World hantaviruses—including two that are associated with HPS (ANDV and SNV)—but not by two Old World hantaviruses that are associated with haemorrhagic fever with renal syndrome.
Fig. 1 |. A haploid genetic screen identifies PCDH1 as a host factor for ANDV and SNV entry and infection.
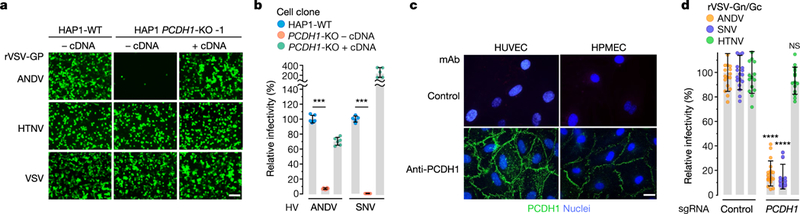
a, b, Relative infectivity of rVSVs bearing the indicated viral glycoproteins. Wild-type (WT) and PCDH1-knockout (KO) HAP1 cell lines lacking (-cDNA) or expressing (+cDNA) WT human PCDH1 cDNA were exposed to rVSVs expressing hantavirus glycoproteins (rVSV-GPs) (a) or to hantaviruses (HVs) (b).a, Infected cells positive for enhanced green fluorescent protein (eGFP; pseudocoloured green) were detected by fluorescence microscopy. Representative images are shown. Scale bar, 100 μm. b, Hantavirus-infected cells were detected and enumerated by immunofluorescence microscopy. Averages ± s.d. from three experiments are shown in b; n = 6 (ANDV); n = 5 (SNV); WT versus PCDH1-KO cells, two-way ANOVA with Tukey’s test, ***P< 0.001 (n indicates the number of biologically independent samples). c, Expression of PCDH1 in HUVECs and HPMECs was detected by immunostaining with PCDHl-specific monoclonal antibody (mAb) 3305 or negative control antibody (see Extended Data Fig. 4d) and visualized by immunofluorescence microscopy. Scale bar, 20 μm. Experiments were performed three times with similar results. d, HPMECs transduced to co-express the endonuclease Cas9 and control or single-guide RNAs (sgRNAs) targeting PCDH1 were exposed to rVSVs. The results are averages ± s.d. from five experiments; n = 16 for ANDV; n = 18 for SNV; n = 14 for HTNV. PCDH1 sgRNA versus control sgRNA, two-way ANOVA with Sidak’s test; NS, P > 0.05; ****P < 0.0001.
Fig. 2 |. The first extracellular cadherin domain of PCDH1 is required for New World hantavirus entry and infection.
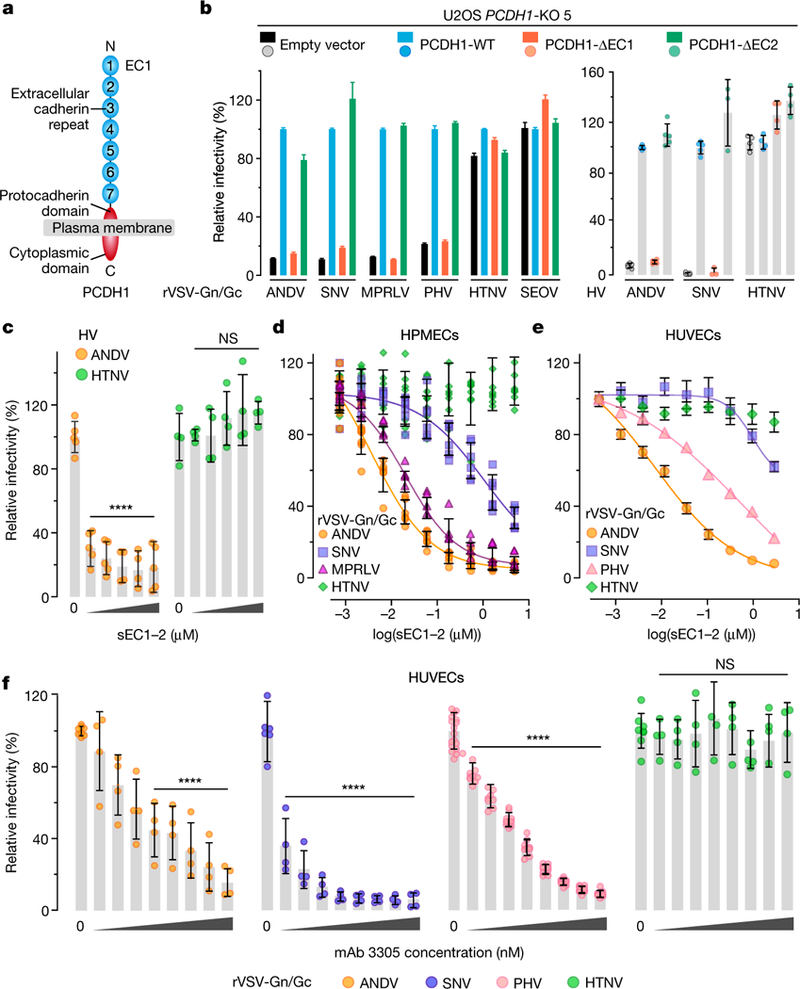
a, Organization of PCDH1. b, U2OS PCDH1-KO cell lines complemented with the indicated PCDH1 proteins were exposed to rVSVs or hantaviruses. Left, averages ± s.e.m.: five experiments, n = 25 for ANDV and HTNV; five experiments, n = 15 for SNV except full-length PCDH1 (four experiments, n = 12)); four experiments, n = 24 for MPRLV and SEOV; three experiments, n = 10 for PHV. Right, averages ± s.d.: three experiments, n = 5 for ANDV and SNV, except SNV ΔEC1 and ΔEC2 (two experiments, n = 3)); two experiments, n = 4 for HTNV. c, Capacity of sEC1–2 (0–2.2 μM) to block authentic hantavirus infection. Viruses were preincubated with sEC1–2, and then allowed to infect WT U2OS cells. Averages ± s.d.: two experiments, n = 4 or 5 for ANDV; two experiments, n = 4 for HTNV. Untreated versus sEC1–2-treated, two-way ANOVA with Dunnett’s test; NS, P > 0.05; ****P < 0.0001. d, e, Capacity of soluble, truncated PCDH1 (sEC1–2, 0–5 μM) to block viral entry. rVSVs were preincubated with sEC1–2, and then allowed to infect HPMECs (d) and HUVECs (e). d, Averages ± s.d.: three experiments, n = 8 for ANDV and HTNV; n = 6 for SNV; n = 4 for MPRLV. e, Averages ± s.e.m.: six experiments, n = 15 or 16 for ANDV and HTNV; three experiments, n = 5 or 6 for SNV; four experiments, n = 8 for PHV. f, Capacity of PCDH1 EC1-specific mAb 3305 to block viral entry. HUVECs were preincubated with mAb 3305 (0–680 nM), and then exposed to rVSVs. Averages ± s.d.: three experiments, n = 4 for ANDV, SNV and HTNV); four experiments, n = 9 for PHV. Untreated versus antibody-treated by two-way ANOVA with Dunnett’s test: NS, P > 0.05; ****P < 0.0001.
Endothelial cells, including those of the lung microvasculature, are major targets of hantavirus infection in vivo2,20. Consistent with this, previous work showed that PCDH1 is expressed in human airway epithelial and endothelial cells3,6. Here we found abundant cell-surface expression of PCDH1 in both human primary umbilical vein endothelial cells (HUVECs) and human primary pulmonary microvascular endothelial cells (HPMECs) (Fig. 1c). Further, depletion of PCDH1 in HPMECs by CRISPR–Cas9 engineering selectively reduced infection by rVSVs that bear ANDV and SNV Gn/Gc glycoproteins (Fig. 1d), which suggests that PCDH1 has a critical role in ANDV and SNV Gn/Gc-dependent viral entry into primary endothelial cells.
PCDH1 is a type I membrane protein with seven extracellular cadherin-repeat (EC) domains and a protocadherin-specific cytoplasmic domain7,8,21 (Fig. 2a). To identify PCDH1 sequences that are required for hantavirus entry, we generated constructs that lack (Δ) the first or second EC domains (EC1 (human PCDH1 amino-acid residues 61–172) and EC2 (residues 173–284)) and tested their ability to complement U2OS PCDH1-knockout cells (Fig. 2). Although both proteins were expressed and localized to the plasma membrane (Extended Data Fig. 2), only PCDH1-ΔEC1 failed to restore infection (Fig. 2b), suggesting a role for EC1 in hantavirus entry. Reasoning that EC1 may interact with hantavirus Gn/Gc, we next expressed and purified recombinant, soluble forms of EC1-EC2 (sEC1–2) and EC1 (sEC1) (Extended Data Fig. 3a–c) and evaluated their effects on viral infection. Preincubation with sEC1–2 or sEC1 selectively inhibited entry and infection by New World hantaviruses (Fig. 2c–e and Extended Data Fig. 3d).
To directly target PCDH1 EC1, we developed and characterized a panel of monoclonal antibodies against purified PCDH1 EC domains (Extended Data Fig. 4a–c) and confirmed that they could decorate cell surfaces in a PCDH1-EC1-specific manner (Extended Data Fig. 4d). Preincubation of HUVECs (Fig. 2f) and HPMECs (Extended Data Fig. 5) with EC1-specific antibody 3305 specifically inhibited Gn/Gc-dependent entry by New World viruses. Taken together, these findings suggest that an interaction between viral Gn/Gc glycoproteins and the first extracellular cadherin-repeat domain of PCDH1, EC1, drives entry by four New World hantaviruses.
We used four distinct approaches to evaluate the hypothesis that New World hantavirus Gn/Gc glycoproteins directly recognize and engage PCDH1 EC1. First, we infected cells with rVSVs bearing ANDV and HTNV Gn/Gc and incubated the resulting Gn/Gc-displaying cells with sEC1–2 (Extended Data Figs. 6, 7a, b). Only ANDV Gn/Gc-expressing cells could be decorated with sEC1–2, and sEC1–2 capture was sensitive to the EC1-specific antibody 3305 (Extended Data Fig. 7c). Second, we incubated sEC1–2 immobilized on ELISA plates with pre-titrated amounts of viral particles bearing Gn/Gc. sEC1–2 could selectively capture ANDV and SNV rVSV Gn/Gc from solution (Fig. 3a), in a manner that was sensitive to both antibody 3305 (Fig. 3b) and convalescent sera from two Chilean survivors of HPS22 (Fig. 3c), providing evidence that the interaction is specific to both PCDH1 and Gn/Gc. Third, we incubated biotinylated rVSV Gn/Gc particles with purified sEC1–2 and recovered the particles with streptavidin beads. rVSV-ANDV Gn/Gc co-precipitated substantially higher levels of sEC1–2 compared with the negative control (beads alone), whereas rVSV-HTNV Gn/Gc did not (Extended Data Fig. 7d). Fourth, purified, recombinant virus-like particles (VLPs)23 constituted with Gn/Gc from MPRLV (Extended Data Fig. 8a, b)—but not the Old World hantavirus Puumala virus (PUUV)—could recognize sEC1–2 in the capture ELISA described above (Fig. 3d), and MPRLV VLPs bound to sEC1–2 with an equilibrium dissociation constant (Kd) of around 7 nM, as determined by biolayer interferometry (BLI) (Fig. 3e, f and Extended Data Fig. 8c). Finally, we assessed the capacity of the interaction-blocking reagents sEC1–2 and antibody 3305 to interfere with viral attachment to the cell surface (Fig. 3g, h). Both sEC1–2 (Fig. 3g) and the antibody (Fig. 3h) specifically blocked attachment of rVSV-ANDV Gn/Gc particles to HUVECs. Together, these findings provide evidence that New World hantavirus glycoproteins directly recognize the EC1 domain of PCDH1 with nanomolar affinity and exploit this interaction to mediate hantavirus attachment to the cell surface.
Fig. 3 |. PCDH1 mediates ANDV and SNV attachment to cells by binding directly to the viral glycoproteins.
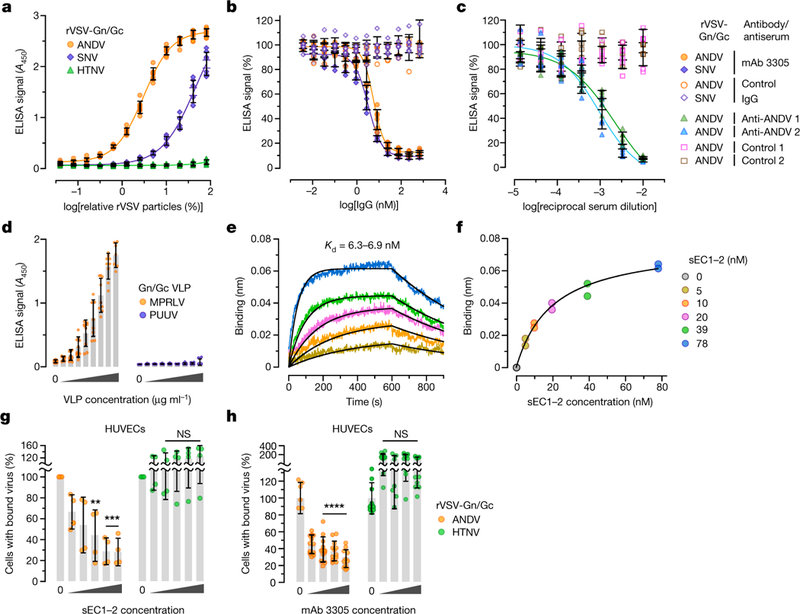
a, Biotinylated rVSVs were added to sEC1–2-coated plates and rVSV capture was measured by ELISA. Averages ± s.d.: four experiments, n = 8. b, The capacity of PCDH1-specific mAb 3305 to block rVSV-Gn/Gc capture by sEC1–2 was measured by ELISA as in a. Immobilized sEC1–2 was incubated with a control IgG or with mAb 3305 before addition of biotinylated rVSVs. Averages ± s.d.: three experiments, n = 6. c, The capacity of Gn/Gc-reactive convalescent sera from two Chilean survivors of HPS to block binding between sEC1–2 and rVSV-Gn/Gc was measured by ELISA as in a. Biotinylated rVSV-ANDV Gn/Gc was incubated with serial dilutions of antisera and then added to sEC1–2-coated plates. Averages ± s.d.: three experiments, n = 6. d,The capacity of sEC1–2 to capture purified, Strep-tagged MPRLV or PUUV VLPs (0–170 μml–1) was measured by ELISA. Averages ± s.d.: three experiments, n = 7 for PUUV, n = 7 or 9 for MPRLV. e, Sensorgrams of sEC1–2 binding to MPRLV VLPs by biolayer interferometry. Experimental curves (coloured traces) were fit using a 1/1 binding model (black traces) to derive equilibrium dissociation constant (Kd) values. f, Response curve for steady-state analysis. Coloured dots correspond to the coloured curves in e. Results from two independent experiments are shown in e and f. g, Capacity of sEC1–2 to block viral attachment to cells. rVSVs bearing ANDV or HTNV Gn/Gc and labelled with functional-component spacer diacyl lipid (FSL)–fluorescein were preincubated with sEC1–2 (0–1.6 μM), and then exposed to HUVECs at 4 °C for 1 hour. Cells with bound viral particles were enumerated by flow cytometry. Averages ± s.d.: four experiments, n = 4. Untreated versus sEC1–2-treated, two-way ANOVA with Dunnett’s test; NS, P > 0.05; **P < 0.01; ***P < 0.001. h, Capacity of mAb 3305 to block viral attachment to cells. HUVECs were preincubated with mAb 3305 (0–68 nM) at 4 °C, and then exposed to DiD lipophilic-dye-labelled rVSVs bearing ANDV or HTNV Gn/Gc at 4 °C. We obtained 6–12 images per coverslip; virus-bound cells were analysed with Volocity software. Averages ± s.d.: three experiments, n = 12. Untreated versus antibody-treated, two-way ANOVA with Dunnett’s test; NS, P > 0.05; ****P < 0.0001.
Finally, we sought to evaluate the importance of PCDH1 to hantavirus infection and disease pathogenesis in the Syrian golden hamster (Mesocricetus auratus)—the gold-standard rodent model for lethal HPS24. The human and Syrian hamster PCDH1 orthologues are highly conserved in amino-acid sequence (with more than 97% sequence identity in EC1), suggesting that interactions of PCDH1 with both hantavirus Gn/Gc and antibody 3305 are likely to be preserved in hamster cells. Indeed, sEC1–2 and sEC1—but not sEC2—selectively blocked rVSV-ANDV Gn/Gc infection of hamster primary lung endothelial cells, as did the EC1-specific monoclonal antibody, which confirms that Gn/Gc–PCDH1 recognition drives ANDV entry into endothelial cells from this species (Extended Data Fig. 9a, b).
Next, we used CRISPR-Cas9 genome engineering to generate hamsters carrying a germline-transmitted, single-nucleotide deletion in PCDH1 (Extended Data Fig. 10a–c). This frameshift mutation causes multiple premature stop codons in PCDH1 and its genetic inactivation (knockout), as confirmed by immunoblotting of isolated lung tissue from animals homozygous for the knockout allele (Extended Data Fig. 10d). PCDH1-knockout hamsters were viable and fertile, and morphological and histopathological analysis revealed no lesions in the lungs (Fig. 4c, left) or in other sampled organs, suggesting that PCDH1 loss is not associated with tissue malformation or injury. We then subjected wild-type and PCDH1-knockout hamsters to intranasal challenge with ANDV (Fig. 4a). Wild-type hamsters largely succumbed to ANDV infection, as previously shown25,26. By contrast, PCDH1-knockout hamsters largely survived ANDV challenge, indicating that the loss of PCDH1 is highly protective (Fig. 4a). Measurement of viral titres in sera collected from challenged animals on day 14—near the onset of lethality—indicated reduced levels of viraemia in knockout animals (Extended Data Fig. 10e). Histopathological analysis of lungs isolated from challenged wild-type animals (Fig. 4b) revealed severe interstitial pneumonitis, perivascular inflammation associated with macrophage and neutrophil infiltration, oedema, haemorrhage, and fibrin deposition. By contrast, knockout animals had only mild inflammation with thickening of alveolar septae and occasional macrophage and neutrophil infiltration (Fig. 4b). Furthermore, lung tissue collected from wild-type but not knockout animals at 18 days post-infection showed extensive staining for viral antigen in endothelial and alveolar septal cells (Fig. 4c). Concordantly, we measured substantially higher levels of viral RNA in lung homogenates from wild-type animals relative to their knockout counterparts (Fig. 4c). Blocking the hantavirus–PCDH1 interaction both in vitro and in vivo did not fully prevent viral attachment, infection and histopathology, which suggests the existence of PCDH1-independent entry pathways. Nevertheless, our findings establish a role for PCDH1 in ANDV infection in vivo and in the development of disease in the Syrian hamster model.
Fig. 4 |. PCDH1 is required for lethal ANDV pulmonary infection in Syrian hamsters.
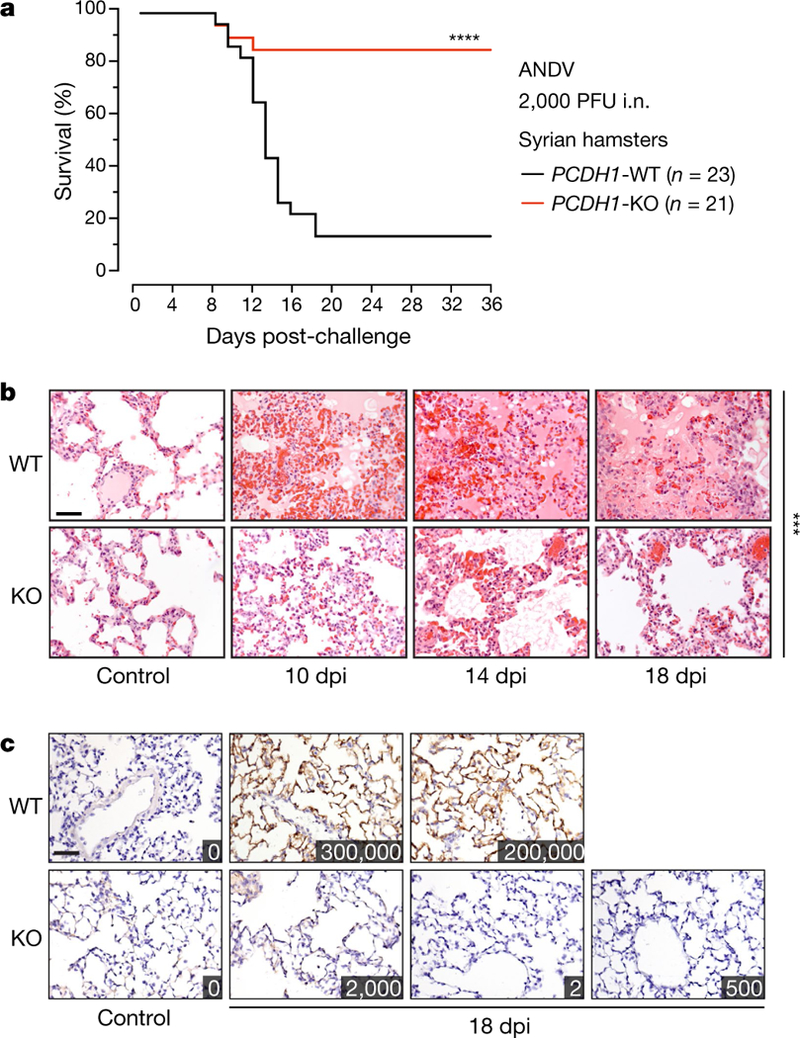
a, Wild-type hamsters (two experiments, n = 23) and knockout hamsters (two experiments, n = 21) were exposed to ANDV (target dose 2,000 plaque-forming units (PFU); actual dose 1,500 PFU) via the intranasal (i.n.) route, and mortality was monitored for 36 days. PCDH1-KO versus PCDH1-WT, two-sided Mantel-Cox test; ****P < 0.0001. b, c, Representative images of lung sections from control and ANDV-challenged Syrian hamsters were stained with haematoxylin and eosin (b) or with a mAb targeting the ANDV nucleoprotein (c). Insets in c show viral loads in lung tissue (in units of viral genome equivalents per μg of total RNA). Experiments were performed twice with similar results. Scale bar, 100 μm (b, c). WT versus KO (n = 3) at 18 days postinfection (dpi), two-way ANOVA with Dunnett’s test; ***P < 0.001.
Herein we have used global gene disruption in human cells to discover a protein from the cadherin superfamily, PCDH1, as a critical host factor for cell entry and infection by multiple New World hantaviruses, including ANDV and SNV—the major aetiological agents of HPS in the Americas. Pharmacological disruption of the interaction between the viral glycoproteins and PCDH1 might afford the development of anti-HPS therapeutics.
METHODS
Cells.
Human U2OS osteosarcoma cells, 293T human embryonic kidney fibroblast cells and Freestyle-293F suspension-adapted HEK-293 cells were obtained from ATCC. U2OS and 293T lines were cultured in modified McCoy’s 5A medium and high-glucose Dulbecco’s modified Eagle medium (DMEM), respectively (Thermo Fisher), supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals), 1% GlutaMAX (Thermo Fisher) and 1% penicillin-streptomycin (Thermo Fisher). HAP1 cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher), supplemented as above. HUVECs (Lonza) were cultured in endothelial growth medium (EGM) supplemented with EGM-SingleQuots (Lonza). HPMECs (Promocell) were cultured in MV2 endothelial cell growth medium (Promocell). Syrian hamster primary lung microvascular endothelial cells (Cell Biologics) were cultured in complete endothelial cell medium (Cell Biologics). All cell lines were authenticated by their respective manufacturers or vendors, and no additional authentication was performed. Cell lines from ATCC were authenticated by human short-tandem-repeat analysis. All cell lines were routinely screened for mycoplasma contamination (once monthly) and found to be negative. Adherent cell lines were maintained in a humidified 37 °C, 5% CO2 incubator. Shake flasks of 293F cells were cultured in Gibco FreeStyle 293 expression medium (Thermo Fisher) at 37 °C and 8% CO2.
Generation of PCDH1-knockout cell lines.
A CRISPR sgRNA was designed to target the PCDH1 gene (NCBI Gene identification number 5,097), nucleotides 9,776–9,798 (target sequence 5′-GTTTGAGCGGCCCTCCTATGAGG-3′, with the protospacer acceptor motif (PAM) sequence underlined), in exon 2 of the PCDH1 isoform 1 precursor messenger RNA (GenBank accession number NM_002587.). This sgRNA sequence was cloned into Addgene vector 41,824 (gRNA cloning vector; a gift from G. Church27) and used in conjunction with a Cas9-encoding plasmid (Addgene plasmid 41,815; a gift from G. Church27, Harvard Medical School and Massachusetts Institute of Technology) to generate HAP1 and U2OS PCDH1-KO single-cell clones, as described14. The PCDH1 genotypes of these cell lines were verified by Sanger sequencing of cloned polymerase chain reaction (PCR) amplicons flanking the sgRNA target sequence, as described14, except that the PCDH1-specific primers 5′-CCTTAGGGGTCACGGGAAAC-3′ and 5′-GACAACACACCCAACTTCGC-3′ were used for PCR amplification of genomic DNA. HAP1 and U2OS PCDH1-KO clones both contained a single nucleotide insertion (T) after PCDH1 nucleotide 9,792 (PCDH1 amino-acid residue Y285). The resulting frame-shifted transcript is predicted to encode a truncated 285-amino-acid polypeptide.
CRISPR–Cas9 engineering of HPMECs.
The human PCDH1-specific sgRNA sequence mentioned above was cloned into Addgene vector 52,961 (lentiCRISPR v2; a gift from F. Zhang28, Massachusetts Institute of Technology and Broad Institute) and was used to produce lentiviruses following triple transfection of Addgene vector 12,260 (psPAX2; a gift from D. Trono, École Polytechnique Fédérale de Lausanne) and a plasmid expressing VSV glycoprotein G in 293FT cells. The empty vector was used as a negative control. Lentiviral supernatants were filtered and applied to monolayers of HPMECs. At 5–7 days post-transduction, cells were exposed to rVSVs bearing hantavirus Gn/Gc, and infection was monitored by fluorescence microscopy.
rVSVs and infections.
rVSVs expressing eGFP and bearing Gn/Gc from ANDV14, SNV14, HTNV14, SEOV (GenBank accession number M34882.1) and PHV (GenBank accession number X55129.1) were engineered, rescued and propagated as described14,29. Cells were exposed to rVSVs at a multiplicity of infection (MOI) of 0.02–0.2 infectious units (IU) per cell (see below for specific MOIs and times post-infection used in each experiment), and viral infectivity was measured by automated enumeration of eGFP-positive cells from captured images using a custom analysis pipeline in CellProfiler30 or directly from multiwell plates using a CellInsight CX5 automated fluorescence microscope and onboard HCS Studio software (Thermo Fisher). Throughout this paper, MOIs for rVSVs are based on the calculations for the particular cell lines on which the experiments were performed.
For Figs. 1, 2, infection MOIs, times post-infection at which viral infectivity was measured, and the percentage of infected cells that corresponds to 100% relative infectivity are as follows. Values of these parameters for the Extended Data Figs. are presented in each figure legend. Figure 1a: cells were infected with rVSVs at a MOI of 0.02 IU per cell and scored for infection at 20 hours post-infection (hpi). Figure 1d: cells were infected with rVSVs at a MOI of 0.2 IU per cell and scored for infection at 9 hpi; 100% relative infectivity corresponds to 10–20% infected cells. Figure 2b (left): cells were exposed to rVSVs at a MOI of 0.02 IU per cell and scored for infection at 20–24 h; 100% relative infectivity corresponds to 20–40% infected cells for rVSVs (but 8–12% for rVSV-PHV). Figure 2d–f: cells were exposed to rVSVs at a MOI of 0.2 IU per cell and scored for infection at 9 hpi; 100% relative infectivity corresponds to 15–20% infected cells.
For soluble PCDH1 and PCDH1-specific-antibody experiments, pretitrated amounts of rVSV particles (0.1–0.2 IU per cell; see above and the next section for specific MOIs for rVSVs and authentic hantaviruses, respectively) were incubated with increasing concentrations of test reagent at room temperature or 37 °C, respectively, for 1 h before addition to cell monolayers in 96-well plates. Viral infectivities were measured as above.
Authentic hantaviruses and infections.
ANDV strain Chile-9717869, SNV strain CC107 and HTNV strain 76–118 were propagated in Vero E6 cells as described31–33. Hantavirus infections were performed, and infected cells were immunostained for viral antigen, as described14. In brief, HAP1 and U2OS cells were exposed to virus at a MOI of 0.5–3 PFU per cell (ANDV and HTNV) or fluorescent focus-forming units (FFU) per cell (SNV) (see below for specific MOIs), and viral infectivity was determined by immunostaining of formalin-fixed cells at 72 h post-infection (see below for specific times) with rabbit polyclonal sera specific for ANDV, HTNV or SNV nucleoproteins (NR-9673, NR-9674 and NR-12152 (all from BEI Resources), respectively). Images were acquired at 20 fields per well with a ×20 objective on an Operetta high-content imaging device (PerkinElmer). Images were analysed with a customized scheme built from imageanalysis functions present in Harmony software, and the percentage of infected cells was determined using the analysis functions. MOI values for authentic hantaviruses are based on the titres estimated from infections of Vero cell monolayers.
Infection MOIs, times post-infection at which viral infectivity was measured, and the percentage of infected cells that corresponds to 100% relative infectivity in each figure panel are as follows. Figure 1b: cells were exposed to authentic hantaviruses at a MOI of 1 PFU (ANDV and HTNV) or 1 FFU (SNV) per cell and scored for infection at 72 hpi; 100% relative infectivity corresponds to 10–20% infected cells. Figure 2b (right): cells were exposed to authentic hantaviruses at a MOI of 1 PFU per cell (ANDV and HTNV) or 3 FFU per cell (SNV) and scored for infection at 72 hpi; 100% relative infectivity corresponds to 10–20% infected cells. Figure 2c, cells were exposed to authentic hantaviruses at a MOI of 1 PFU per cell; cells were scored for infection at 72 hpi; 100% relative infectivity corresponds to 10–20% infected cells.
Truncated and soluble PCDH1 variants.
A cDNA encoding human PCDH1 (isoform 1, GenBank accession number NM_002587) was synthesized in frame with Myc and Flag epitope tags at the carboxyl terminus (Epoch Biolabs) and cloned into the pBABE-puro retroviral vector34. PCDH1 constructs engineered to lack the first or second extracellular cadherin repeats (ΔEC1, Δ61–172 amino-acid residues; ΔEC2, Δ173–284 residues) were also cloned into this vector. Constructs encoding soluble (secreted) PCDH1 variants (GenBank accession number NM_002587) were generated by cloning the following sequences into the pcDNA3.1 mammalian expression vector (Thermo Fisher): EC1 (residues 1–172), EC2 (residues 1–60, 172–284), and EC1–EC2 (residues 1–284), each in frame with a C-terminal GSG linker, followed by Myc, Flag and decahistidine tags (Extended Data Fig. 3 a, b), or with a GSG linker followed by a decahistidine tag alone (Extended Data Fig. 3c). Each construct also retained the endogenous PCDH1 amino-terminal signal sequence (residues 1–60). Sequences of all the plasmids were verified by Sanger sequencing.
Stable cell populations expressing PCDH1 variants.
HAP1 and U2OS PCDH1-KO cells ectopically expressing the above PCDH1 variants were generated by transduction with pBABE-puro vectors. Retroviruses packaging the transgenes were produced by triple transfection in 293T cells14, and target cells were directly exposed to sterile-filtered (0.45 μm) retrovirus-laden supernatants in the presence of polybrene (6 μg ml–1). Transduced cell populations were selected with puromycin (2 μg ml–1), and transgene expression was confirmed by immunostaining.
Detection and measurement of cellsurface PCDH1 expression.
U2OS cells were seeded on six-well plates, chilled on ice and incubated for 1 h with PCDH1 EC7-specific monoclonal antibody 3677 or human IgG (negative control) diluted to 5 μg ml–1 in cold U2OS medium containing 1% HEPES. Cells were washed extensively with cold Hank’s balanced salt solution (HBSS), scraped off the plate, fixed with 4% paraformaldehyde and stained with anti-human IgG1 conjugated to Alexa Fluor-488 for 1 h. Cells were washed with phosphate-buffered saline (PBS) and analysed by flow cytometry.
Expression and purification of secreted PCDH1 variants.
Secreted PCDH1 variants cloned into pcDNA3.1 (see above) were expressed in 293F cells in shake flasks by transient transfection with linear polyethyleneimine (Polysciences), as described35, and purified by nickel-chelation chromatography. In brief, clarified cell supernatants were stirred overnight at 4 °C with nickel-NTA resin (2–3 ml packed resin per 600 ml supernatant). Beads were then retrieved, washed with PBS containing 50 mM imidazole, and eluted with PBS containing 250 mM imidazole. The eluted protein was buffer-exchanged with PBS, concentrated, and stored in aliquots at –20 °C. Purity of the secreted PCDH1 variants was determined by size-exclusion chromatography with multi-angle light scattering (SEC-MALS) and SDS–PAGE, stained with Bio-Safe Coomassie G-250 Stain (BioRad) and imaged on a ChemiDoc Touch Imaging System (BioRad).
Isolation and characterization of anti-PCDH1 antibodies.
The panel of PCDH1-specific mAbs was isolated from a phage-displayed synthetic human antigen-binding fragment (Fab) library (Library F)36. Binding selections, phage ELISAs and Fab protein purification were performed as described37. In brief, phage particles displaying the Fabs from Library F were cycled through rounds of binding selections with purified PCDH1–Fc fusion proteins (either full-length extracellular domain (EC1–7; residues 58–851) or EC2–7 (residues 190–851)) immobilized on 96-well Maxisorp Immunoplates (Thermo Fisher) (Extended Data Fig. 4). After four rounds of selection, phages were produced from individual clones grown in a 96-well format and phage ELISAs were performed to detect specific binding clones. Clones with positive binding were subjected to DNA sequencing. The DNAs encoding for variable heavy-and light-chain domains of the positive binders were cloned into vectors designed for the production of Fabs or light chain (human Kappa) or heavy chain (human IgG1). Fabs were expressed from bacterial cells and IgGs from Expi293F cells (Thermo Fisher). Fab and IgG proteins were affinity-purified on protein A affinity columns (GE Healthcare) as described37.
Generation of anti-Gn/Gc monoclonal antibody.
To detect native Gn/Gc in virus particles or at the cell surface, hybridoma cell clones were prepared from splenocytes of Balb/c mice (12 weeks old) following four immunizations with 20 μg of ANDV virus-like particles (VLPs)23 in complete and incomplete Freund’s adjuvant (Sigma-Aldrich) using standard techniques. Clones were screened for reactivity to ANDV VLPs by ELISA and positive clones were further subcloned. 293FT cells (Thermo Fisher) expressing ANDV38 or HTNV39 Gn/Gc were used to select and characterize the 1E11/D3 hybridoma clone by flow cytometry using an AlexaFluor-488 conjugated goat anti-mouse secondary antibody (Thermo Fisher). For total protein expression, cells were fixed and permeabilized with Triton X-100 before staining with the antibody. For analysis of cell-surface expression, cells were stained with primary antibody before fixing and no permeabilization step was involved (Extended Data Fig. 6).
Cell-based assays for rVSV-Gn/Gc-PCDH1 binding.
To visualize the binding of sEC1–2 to cells expressing hantavirus Gn/Gc, we infected U2OS cells with rVSVs bearing ANDV or HTNV Gn/Gc. At 12–14 h post-infection, cells were washed with cold PBS, and blocked with cold 10% FBS in PBS at 4 °C for 30 min. Cells were then stained for surface expression of viral Gn/Gc with mouse mAb anti-hantavirus Gn/Gc 1E11/D3 followed by anti-mouse Alexa Fluor 568 (Thermo Fisher) for 1 h each at 4 °C. In parallel, rVSV-ANDV/HTNV Gn/Gc-infected cells were incubated with purified Flagtagged sEC1–2 (5 μM for 1 h at 4 °C, and then washed extensively with PBS. To visualize bound sEC1–2, we fixed cells with 4% formaldehyde (Sigma-Aldrich), and stained them with anti-Flag M2 mouse mAb (Sigma-Aldrich) and secondary antibody anti-mouse Alexa Fluor 568 (Thermo Fisher) for 1 h each at 4 °C. To test the ability of mAb 3305 to block sEC1–2 binding to hantavirus Gn/Gc expressing cells, we preincubated 200 nM sEC1–2 with either control human IgG or mAb 3305 (5–500 nM) for 1 h at 4 °C and performed immunostaining for sEC1–2 as described above. Cells were visualized by fluorescence microscopy.
Immunofluorescence microscopy and co-immunostaining.
For visualization of PCDH1, U2OS or primary endothelial cells plated on gelatin or fibronectin-coated glass coverslips were chilled on ice for 5 min and stained with control (hIgG ctrl) or PCDH1-specific mAb (5 μg ml–1) for 1 h at 4 °C in cell-growth medium containing 10% FBS. Cells were then washed with cold PBS, fixed with 4% paraformaldehyde for 5 min on ice, and then stained with anti-human IgG secondary antibodies conjugated to Alexa Fluor-555 or Alexa Fluor-488 (Thermo Fisher) for 1 h each at room temperature. For immunostaining with an anti-Flag antibody, paraformaldehyde-fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 10% FBS in PBS at room temperature for 30 min. Cells were then stained with anti-Flag M2 mouse mAb (Sigma-Aldrich) and an anti-mouse IgG secondary antibody conjugated to Alexa Fluor-568 (Thermo Fisher), for 1 h each at 4 °C. Coverslips were mounted in Prolong with DAPI (Thermo Fisher), and cells were imaged with a Zeiss Axio Observer inverted microscope under a ×63 objective.
rVSV-Gn/Gc-PCDH1 binding and competition ELISAs.
The capacity of rVSV-Gn/Gc to recognize sEC1–2 was determined by capture ELISA (Fig. 3a). In brief, high-protein-binding 96-well ELISA plates (Corning) were coated with purified sEC1–2 (100 ng per well) overnight at 4 °C and blocked with 5% nonfat dry milk in PBS (PBS-milk). The membranes of rVSV particles normalized for protein concentration were labelled with a short-chain phospholipid probe, functional-component spacer diacyl lipid conjugated to biotin (FSL–biotin; Sigma-Aldrich), as described14, and then pretitrated for biotin content by streptavidin ELISA. The sEC1–2-coated plates were incubated with dilutions of biotinylated viral particles in PBS-milk for 1 h at 37 °C. Bound rVSVs were detected by incubation with a streptavidin—horseradish peroxidase (HRP) conjugate.
The capacity of PCDH1-specific mAb 3305 to inhibit virus–PCDH1 binding in the capture ELISA was determined as above, except that, first, the sEC1–2 coated plate was incubated with the indicated dilutions of each mAb before addition of viral particles; and second, a single concentration of viral particles, normalized for binding to the sEC1–2-coated plate, was then added to the plates.
The capacities of ANDV Gn/Gc-reactive convalescent sera (serial numbers 703,328 and 703,329)22 and two ANDV-seronegative human serum controls to inhibit virus–PCDHl binding in the capture ELISA was determined as above, except that rVSV-Gn/Gc particles at a single concentration normalized for binding to sEC1–2 were incubated with the indicated dilutions of each serum before their addition to sEC1–2-coated plates. Human sera were provided by H. Galeno, Instituto de Salud Pública de Chile.
Co-precipitation assay for rVSV-Gn/Gc-PCDH1 association.
The capacity of rVSV-Gn/Gc to recognize Flag-tagged sEC1–2 was determined by co-precipitation. FSL-biotin-labelled rVSV particles bearing ANDV or HTNV Gn/Gc were immobilized on streptavidin-coated magnetic beads (Spherotech) by incubation for 1 h at room temperature. Coated beads were then blocked with 5% nonfat dry milk in PBS for 30 min and washed twice with PBS. Beads were incubated with purified sEC1–2 (37 μg ml–1) for 1 h at room temp and washed five times with PBS. Captured proteins were eluted by heating the beads at 98 °C for 5 min with Laemmli sample buffer, subjected to SDS–PAGE and immunoblotting using an anti-Flag M2 mAb–HRP conjugate (Sigma-Aldrich) for sEC1–2 detection and mAb 23H1240, followed by an anti-mouse IgG secondary antibody-HRP conjugate (Santa Cruz) to detect the matrix protein M in rVSV particles.
Preparation and characterization of MPRLV and PUUV VLPs.
The DNA fragments encoding MPRLV Gn/Gc (residues 21 to 1138) and PUUV Gn/Gc (residues 20 to 1148) were amplified by PCR and fused in-frame to a double streptavidin tag (at the N-terminus of Gn). HEK293 cell lines expressing the VLPs were established after selection of individual clones in the presence of geneticin at 0.5 mg ml–1. The cells were then grown in DMEM supplemented with 0.2 mg ml–1 geneticin for five days and the VLP-containing supernatant was collected, clarified at 500g for 15 min, concentrated to 50 ml, and supplemented with 10 μg ml–1 avidin and 0.1 M Tris-HCl (pH 8.0) before being passed through a 0.2-μm pore filter. The VLPs were then purified by streptactin-affinity chromatography and the eluate was concentrated and stored at –80 °C.
For visualization by electron microscopy, MPRLV VLPs (80–100 ng μl–1) were spotted on glow-discharged carbon grids, negatively stained with 2% uranyl acetate, analysed with a Tecnai G2 Bio-Twin electron microscope (FEI) and imaged with an Eagle camera (FEI).
Measurement of Gn/Gc-PCDH1 interaction by biolayer interferometry.
The OctetRed 384 system (ForteBio, Pall LLC) was used to determine kinetic binding properties. Aminopropylsilane (APS) sensors were used to load VLPs (20 μg ml–1) in PBS (pH 7.4) for 900 s until saturation was reached at 6 nM, washed for 60 s with PBS and quenched for 600 s in PBS–bovine serum albumin (BSA) (0.2 mg ml–1). Subsequently, association of sEC1–2 was monitored for 600 s in PBS-BSA over the indicated concentration range, and dissociation was monitored for 300 s in PBS-BSA. Independent experiments were performed with different sensors to account for potential experimental artefacts resulting from intersensor variability. Non-specific interactions were monitored by applying the identical experimental set-up to empty APS sensors (PBS, pH 7.4 during the loading step). Global data fitting to a 1:1 binding model was used to estimate values for the kon (association-rate constant), koff (dissociation-rate constant) and Kd (equilibrium dissociation constant). The steady-state equilibrium concentration curve was fitted using a one-site-specific binding fit in Graphpad Prism.
Assay for sEC1–2-mediated inhibition of virus-cell attachment.
HUVECs were seeded on six-well plates and chilled on ice. rVSVs bearing ANDV or HTNV Gn/Gc (1 μg total protein) were labelled with a lipophilic dye, FSL–fluorescein (Sigma-Aldrich; 5 μg ml–1) as described14. Labelled viruses were preincubated with sEC1–2 (0–30 μg ml–1; 0–1.1 μM) for 1 h at 37 °C, and then allowed to attach to cells at a MOI of 1.5 IU per cell by centrifugation (2,500 r.p.m. for 60 min at 4 °C) in HBSS (Corning). Cells were then placed on ice, washed extensively with cold HBSS to remove unbound virus, and fixed with 4% paraformaldehyde. Cell-surface-bound virus was analysed by flow cytometry as described14.
Assay for mAb-3305-mediated inhibition of virus–cell attachment.
HUVECs, seeded on 24-well plates the previous day, were chilled on ice and incubated with mAb 3305 diluted in HUVEC medium at concentrations of 0–10 μg ml–1 (in serial three-fold dilutions) for 30 min on ice. rVSVs bearing ANDV or HTNV Gn/Gc (0.375 μg) were labelled with a lipophilic dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine 4-chlorobenzenesulfonate (DiD; 0.2–0.4 μM), by mixing and incubating the dye with the virus at 37 °C for 1 h. Labelled viruses were added to the cells at an MOI of 0.4 IU per cell and allowed to attach by centrifugation (1,500g for 20 min at 4 °C). Cells were placed on ice and washed extensively with cold HBSS (Corning) to remove unbound virus, collected from the plate, then fixed with 4% paraformaldehyde. Coverslips were mounted in Prolong with DAPI and imaged the same day by immunofluorescence microscopy. Virus and cell nuclei were detected and enumerated using Volocity (Perkin-Elmer) software’s ‘Find Objects’ module. A total of330–500 cells was analysed.
Animal welfare statement.
Breeding, CRISPR–Cas9 genome engineering and challenge studies with Syrian golden hamsters were conducted under Institutional Animal Care and Use Committee (IACUC)-approved protocols in compliance with the Animal Welfare Act, Public Health Service Policy, and other applicable federal statutes and regulations related to animals and experiments involving animals. The facilities where this research was conducted (Utah State University and US Army Medical Research Institute of Infectious Diseases (USAMRIID)) are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), and adhere to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011.
Generation of a PCDH1-KO Syrian hamster model.
A portion of exon 2 of PCDH1 was PCR-amplified from Syrian golden hamsters housed at Utah State University with primers MA-PCDH1–6723F (5′-TGCCTGTCGTTTTACCCACC-3′) and MA-PCDH1–7908R (5′-GGGAAAAGGGAGCTTCCCAC-3′) and subjected to Sanger sequencing. A panel of candidate sgRNAs was designed and assembled by overlapping PCR to generate human U6-promoter-driven sgRNA expression cassettes and screened for genome-editing efficiency in BHK21 baby hamster kidney cells stably expressing spCas9. The best candidate sgRNA (target sequence 5′-GGTAGTATACAAGGTGCCAGAGG-3′; PAM sequence is underlined), targeting exon 2 of the hamster PCDH1 gene (nucleotides 7,063–7,085; GenBank accession number NW_004801621), was used for in vivo gene editing. This gRNA sequence was in vitro transcribed using the MEGAscript T7 Transcription kit (Thermo Fisher). Cas9 (PNA Bio; 2 μg) was incubated with sgRNA (1 μg) at room temperature for 15 min to generate sgRNA-Cas9 ribonucleoprotein (RNP) complexes, and then diluted to a concentration of 100 ng μl–1 Cas9 and 50 ng μl–1 sgRNA with 10 mM RNase-free TE buffer for pronuclear injections. Two-to three-month-old female Syrian hamsters (body weight 110–135 g) were superovulated by an intraperitoneal injection of pregnant mare’s serum gonadotropin (PMSG; Sigma-Aldrich; 10–20 IU) at 09:00 on the day of post-oestrus discharge. The females were mated to fertile males at 19:00 on day 4 of the oestrous cycle and were humanely euthanized approximately 19 h later for zygote isolation. Zygotes were flushed from oviducts with warmed and equilibrated HECM-9 medium supplemented with 0.5 mg ml–1 human serum albumin. Zygotes were then washed twice with HECM-9, transferred into 20-μl drops of HECM-9 covered by mineral oil in groups (of about 20) in a culture dish, and cultured at 37.5 °C under 10% CO2, 5% O2 and 85% N2.
Cas9–sgRNA RNPs were injected into the zygote pronuclei in a dark room, and red filters were used on the microscope light source. A group of 15–20 hamster zygotes was transferred to a 100-μl HECM-9 drop in the microinjection dish, and 1–2 pl of Cas9–sgRNA RNP complex was injected into the male pronucleus of each zygote. After injection, zygotes were washed twice with equilibrated HECM-9 medium and incubated in HECM-9 medium covered by mineral oil for at least 30 min before embryo transfer. Embryos with normal morphology were bilaterally transferred into the oviducts of pseudopregnant recipients (10–15 embryos per oviduct) that were prepared by mating with vasectomized males at the same time that zygote donors were mated.
Genomic DNA was isolated from the pups at the age of 2 weeks, and a 595-base-pair (bp) product flanking the sgRNA target site was PCR-amplified by using primers MA-PCDH1–6723F (5′-TGCCTGTCGTTTTACCCACC-3′) and MA-PCDH1–7317R (5′-GCCATTCTGCACGAGGTCTGT-3′) and subjected to a T7 endonuclease I assay (NEB) to detect indels. Amplicons from pups bearing indels were topoisomerase (TOPO)-cloned and Sanger-sequenced to identify a founder female animal carrying a single-nucleotide (C) deletion at nucleotide 7,080 of the PCDH1 gene. This deletion results in a frameshift at amino-acid position 58, leading to production of a truncated, 84-amino-acid polypeptide. Because it destroys a unique BanI restriction-enzyme site, a restriction-fragment-length polymorphism (RFLP) was used to genotype pups: the 595-bp wild-type allele can be digested by BanI into 343-bp and 252-bp products, whereas the edited allele is indigestible (see Extended Data Fig. 10a, b). Loss of PCDH1 expression was confirmed by immunoblotting of lung homogenates from age-matched wild-type and PCDH1-knockout hamsters with mAb 3305 (Extended Data Fig. 10d).
Hamster challenge studies.
Wild-type (Envigo) or PCDH1-knockout (Utah State University), 8–10-week-old male and female Syrian hamsters anaesthetized with isoflurane inhalation were exposed to a target dose of 2,000 PFU (actual dose determined by back-titration = 1,500 PFU) of ANDV strain Chile-9717869 diluted in PBS via the intranasal route, with 50 μl of the virus preparation delivered by pipette. Animals were observed daily for clinical signs of disease, morbidity and mortality. Moribund animals—described as being unresponsive or presenting with severe respiratory distress—were humanely euthanized on the basis of IACUC-approved criteria. Animal numbers were chosen so that the survival studies were adequately powered. Study personnel checking the animals were not blinded to treatment group; however, personnel were not privy to the details of specific treatments.
Histopathology and immunochemistry.
Age-matched wild-type and PCDH1-knockout hamsters (one animal each) were humanely euthanized, and various tissues—including lungs, heart, liver, spleen, kidneys, urinary bladder, brain, spinal cord, intestinal tract, testis and epididymis—were collected and preserved in 10% neutral-buffered formalin for 48 h at room temperature. Tissue were paraffin-embedded for sectioning (5–8 μm) and processed for haematoxylin-eosin staining for histopathological examination at the Utah Veterinary Diagnostic Laboratory. The presence of any pathological lesions was scored on a scale of 0–4, 0 indicating no lesions and 4 indicating severe lesions. No substantial microscopic lesions attributable to PCDH1 loss were detected by the veterinary pathologist.
For histopathology of ANDV-challenged wild-type and PCDH1-knockout hamsters, lungs fixed in buffered formalin for 30 days, received from USAMRIID, were paraffinembedded and 5-μm-thin sections were cut and stained with haematoxylin and eosin using standard procedures at the Histopathology and Comparative Pathology facility at Albert Einstein College of Medicine. Blinded slides were then scored on a scale of 1–4 for histopathological lesions by a veterinary pathologist using a previously described scoring metric26.
Lung sections were also immunostained for viral antigen (ANDV nucleoprotein) as described33. Briefly, deparaffinized and rehydrated tissue sections were subjected to antigen retrieval with 10 mM sodium citrate for 20 min in a steamer. Sections were then treated with 0.3% hydrogen peroxide for 10 min to block endogenous peroxidase activity and stained with a 1/1,000 dilution of an ANDV nucleoprotein-specific monoclonal antibody (clone 1A8F6, Austral Biologicals) for 1 h at room temperature, followed by ImmPress HRP anti-mouse IgG (peroxidase) polymer detection kit (Vector Labs) for 30 min at room temperature. After developing an immunohistochemical signal with the DAB peroxidase (HRP) substrate kit (Vector Labs), sections were counterstained with haematoxylin. Blinded slides were imaged by a veterinary pathologist.
Quantitative RT–PCR of hantavirus genomic RNA.
RNA was purified from 0.25 ml of 10% lung homogenates or serum following inactivation with 0.75 ml of TRIzol LS reagent (Invitrogen), in accordance with the manufacturer’s instructions. RNA concentrations were determined in a NanoDrop spectrophotometer (Thermo Fisher) and normalized to 40 μg pl–1 in nuclease-free water. ANDV-specific quantitative reverse transcriptase (RT)–PCR reactions were completed with 250 ng of total RNA per 20-μl reaction, as described41. A standard curve of amplicon fluorescence intensity versus ANDV infectious titre (PFU per ml) was generated with serial 100-fold dilutions of RNA purified (as described above) from ANDV stock virus with a known infectious titre, and results were reported as genome equivalents per μg of total RNA.
Statistics and reproducibility.
The n number associated with each dataset in the figures indicates the number of biologically independent samples. The number of independent experiments and the measures of central tendency and dispersion used in each case are indicated in the figure legends. The testing level (alpha) was 0.05 for all statistical tests. All analyses were carried out in GraphPad Prism. No statistical methods were used to predetermine sample size, except in Fig. 4a which used a power calculation (80% power, 5% type I error) to see a fourfold effect in survival. The experiments were not randomized.
Extended Data
Extended Data Fig. 1 |. PCDH1 is required for entry of New World hantaviruses into HAP1 and U2OS cells.
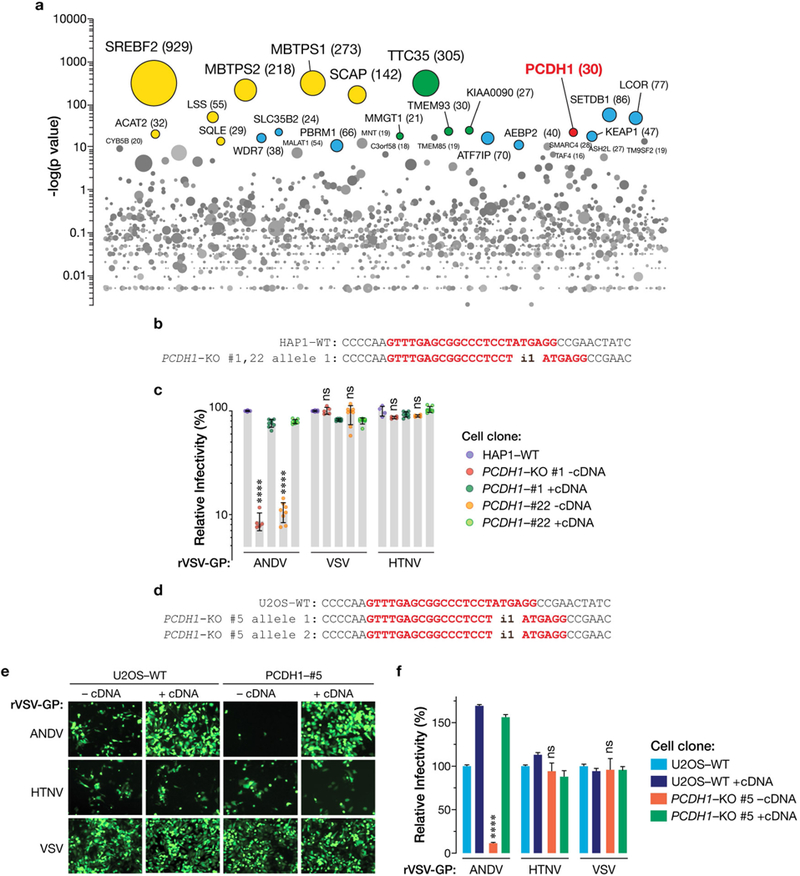
a, Genes enriched for genetrap insertions in the rVSV-ANDV Gn/Gc-selected population versus unselected control cells. The size of each bubble reflects the number of independent gene-trap insertions observed. Candidate genes are associated with cholesterol metabolism (yellow), the endoplasmic reticulum membrane complex network (green), and transcription (blue). PCDH1 (red) is a singleton hit. b, Single-cell HAP1 clones deficient for PCDH1 were generated by CRISPR–Cas9 genome engineering. The sequences of PCDH1-knockout alleles in clones 1 and 22 are shown. The sgRNA target sequence is highlighted in red. i1, insertion of a single nucleotide in PCDH1. c, WT and HAP1 PCDH-KO cell lines were exposed to rVSVs bearing the indicated viral glycoproteins at a MOI of 0.02 IU per cell. ‘+cDNA’ indicates complementation of PCDH1-KO cells with PCDH1. Cells were scored for infection at 20 hpi. One hundred per cent relative infection corresponds to 20–30% infected cells. Averages ± s.d. are shown: HAP1-WT, three experiments, n = 6; PCDH1-KO#1-cDNA, two experiments, n = 5; PCDH1-KO#1+cDNA, three experiments, n = 8; PCDH1-KO#22-cDNA, three experiments, n = 8, except for HTNV Gn/Gc, for which two experiments, n = 4; PCDH1- KO#1+cDNA, three experiments, n = 8; PCDH1-KO#22+cDNA, three experiments, n = 8. d, Single-cell U2OS clones deficient for PCDH1 were generated by CRISPR–Cas9 genome engineering. The sequences of PCDH1-knockout alleles in clone 5 are shown. The sgRNA target sequence is highlighted in red. i1, insertion of a single nucleotide in PCDH1. e,f, WT and U2OS PCDH1-KO cell lines were exposed to the indicated rVSVs at a MOI of 0.02 IU per cell. Cells were scored for infection at 20 hpi. eGFP-positive infected cells (pseudocoloured green) were detected by fluorescence microscopy (e) and enumerated by automated counting (f). One hundred per cent relative infection corresponds to 10–20% infected cells. Averages ± s.e.m. are shown, three experiments, n = 13. In c, f, wild type versus PCDH1-KO by two-way ANOVA with Tukey’s (c) or Sidak’s (f) tests: NS, P > 0.05; ****, P < 0.0001.
Extended Data Fig. 2 |. Expression and plasma-membrane localization of PCDH1 variants lacking domains EC1 or EC2 of PCDH1.
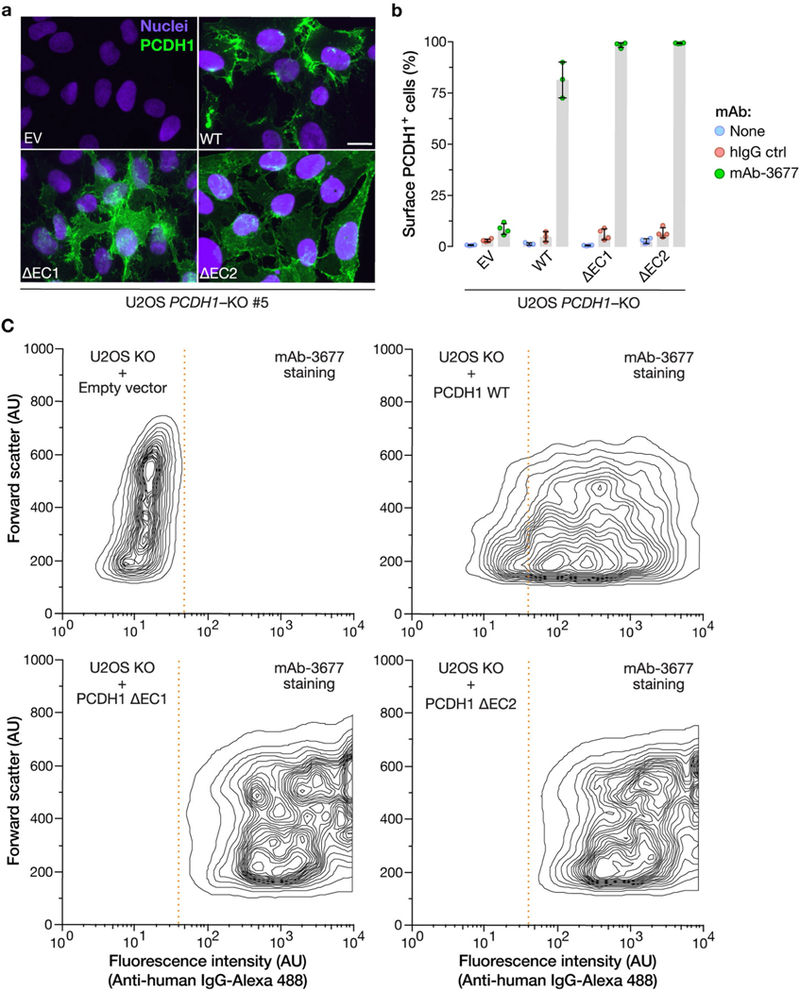
a, U2OS PCDH1-KO cell lines complemented with the indicated PCDH1 proteins were immunostained with an anti-Flag antibody and visualized by fluorescence microscopy. EV, empty vector. Scale bar, 20 μm. b, c, Live cells from a were stained with the PCDHl-EC7-specific mAb 3677 at 4 °C to detect cell-surface PCDH1 and visualized by flow cytometry. Cells were gated on PCDH1 immunofluorescence intensity (dotted red lines in c) to determine the percentage of cells with surface expression of each PCDH1 protein. Averages ± s.d. are shown in b: two experiments, n = 4, except in the case of WT, for which n = 3. c, Representative flow plots from b. Experiments were performed three times with similar results.
Extended Data Fig. 3 |. Expression, purification and characterization of soluble PCDH1 proteins.
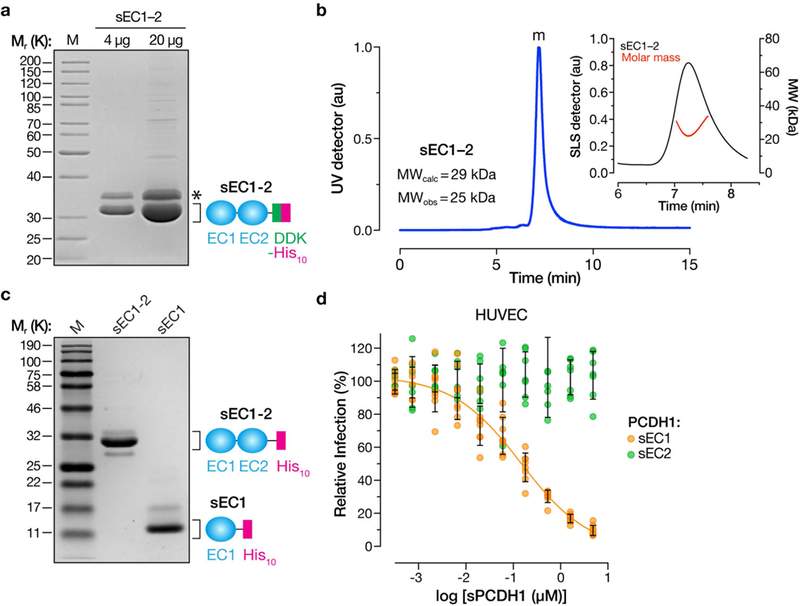
a, Purified sEC1–2 bearing C-terminal Flag and decahistidine (His10) epitope tags was resolved on an SDS–polyacrylamide gel and visualized by Coomassie blue staining. Asterisk, minor component of unknown origin. Mr, relative molecular weight (K denotes × 1,000); M, monomer peak. The experiment was performed three times with similar results. b, SEC-MALS analysis of purified sEC1–2 from a. Absorbance (arbitrary units, au) was monitored at 280 nm; m, monomer peak. Calculated (MWcalc) and observed (MWobs) molecular-weight estimates from MALS are shown in the inset. The experiment was performed twice with similar results. c, Generation of purified sEC1, comprising the first extracellular cadherin domain of PCDH1, and bearing a C-terminal His10 epitope tag. sEC1–2 is shown for comparison. The experiment was performed three times with similar results. d, Capacity of sEC1 and sEC2 to block hantavirus glycoprotein-dependent entry. rVSVs bearing ANDV Gn/Gc were preincubated with sEC1 or sEC2 (0–5 μM, in serial threefold dilutions), and then allowed to infect HUVECs at a MOI of 0.2 IU per cell. Cells were scored for infection at 9 hpi. One hundred per cent relative infection corresponds to 10–20% infected cells. Averages ± s.d.: three experiments, n = 8 for sEC1; n = 7 for sEC2.
Extended Data Fig. 4 |. Isolation of PCDHl-specific monoclonal antibodies.
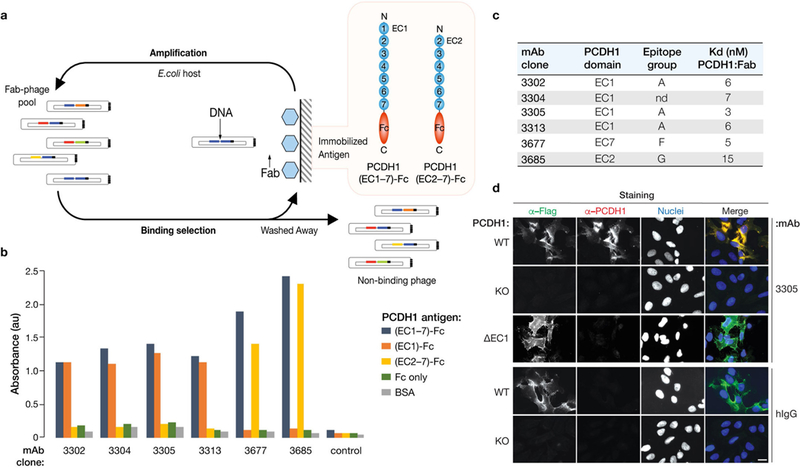
a, Flow chart showing isolation of PCDH1-specific mAbs from Library F by phage display. b, Capacity of selected mAb clones to recognize recombinant fusion proteins comprising PCDH1 ectodomains (containing or lacking EC1) and the Fc antibody domain. BSA, bovine serum albumin. Data from a representative ELISA experiment are shown. Experiments were performed three times with similar results. c, Kinetic binding analysis of selected Fabs to PCDH1(ectodomain)–Fc fusion proteins by surface plasmon resonance. nd, not determined; Kd, equilibrium dissociation constant. d, mAb 3305 recognizes the first extracellular cadherin (EC1) domain of PCDH1. U2OS PCDH1-KO cells, uncomplemented (KO) or complemented either with full-length PCDH1 (WT) or with a PCDH1 variant lacking the EC1 domain (ΔEC1), were co-immunostained with anti-Flag (α-Flag) mAb and anti-PCDH1 mAb 3305, or with anti-Flag and negative control antibodies (hIgG). Cells were visualized by fluorescence microscopy. Scale bar, 20 μm. Data from a representative experiment are shown. Experiments were performed three times with similar results.
Extended Data Fig. 5 |. PCDH1 ECl-specific mAb 3305 blocks ANDV and SNV entry into primary HPMECs.
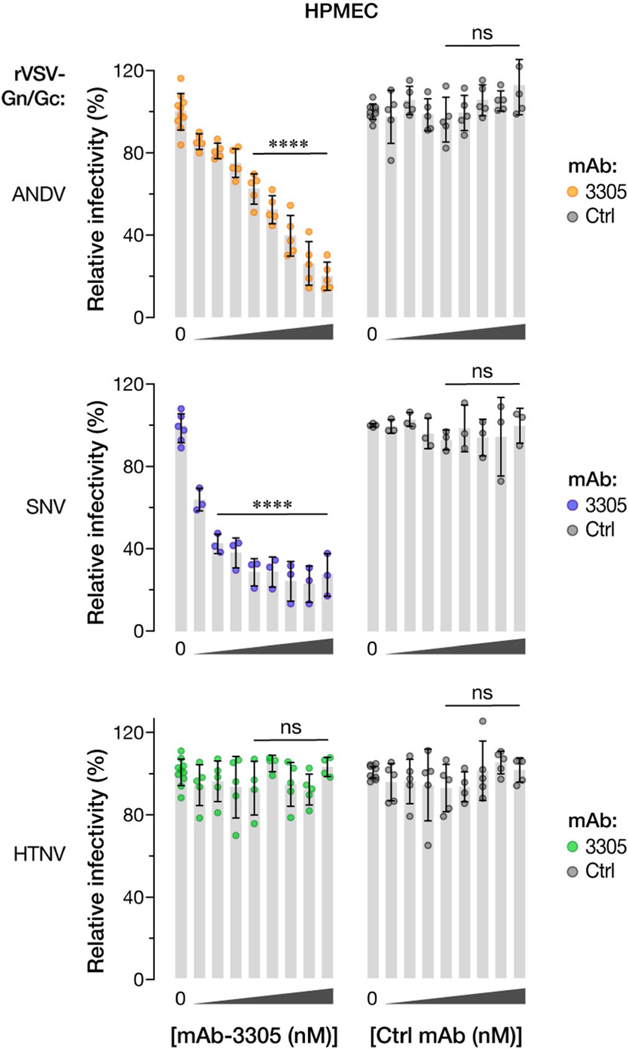
HPMECs were preincubated with mAb 3305 or with human IgG control (Ctrl) (0–100 μg ml–1, 0–680 nM, in serial threefold dilutions), and then exposed to the indicated rVSVs Gn/Gc glycoproteins at a MOI of 0.2 IU per cell. Infected cells were scored at 9 hpi. One hundred per cent relative infectivity corresponds to 15–20% infected cells. Averages ± s.d.: three experiments; no-mAb samples (‘0’), n = 10 for ANDV and HTNV, n = 3 for SNV; mAb-treated samples, n = 5 for ANDV and HTNV, n = 3 for SNV. mAb 3305 versus control mAb, two-way ANO VA with Dunnett’s test: ns, P > 0.05; ****P < 0.0001.
Extended Data Fig. 6 |. The monoclonal antibody 1E11/D3 recognizes HTNV and ANDV Gn/Gc.
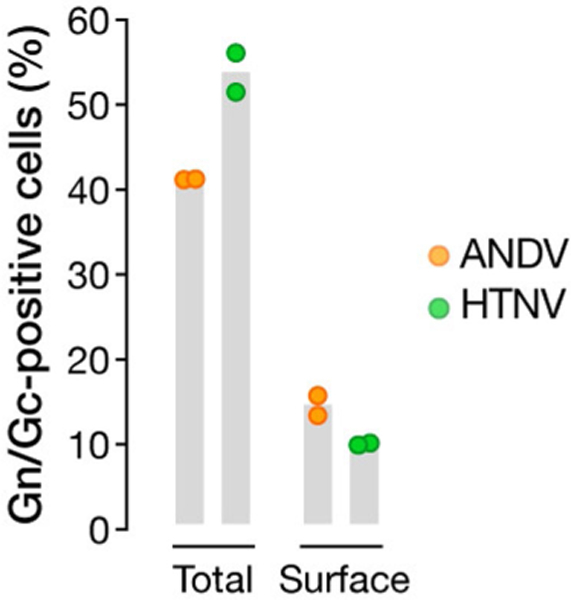
293FT cells transfected with plasmids encoding ANDV or HTNV Gn/Gc were analysed by flow cytometry using 1E11/D3 mAb for total (after permeabilization) and surface (without permeabilization) expression of Gn/Gc. Emptyvector-transfected cells were used as a negative control for gating. Results are from a representative experiment with n = 2. Experiments were performed five times for ANDV and twice for HTNV with similar results.
Extended Data Fig. 7 |. PCDH1 mediates viral entry by direct binding to hantavirus glycoproteins.
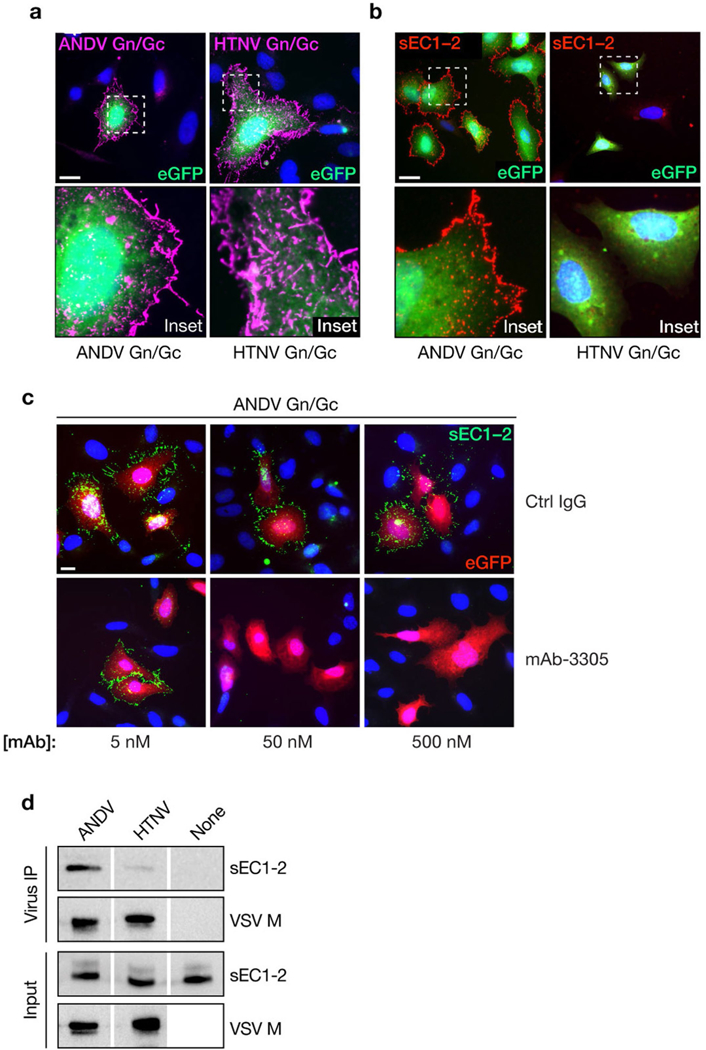
Capacity of hantavirus glycoproteins expressed at the cell surface to capture Flag-tagged sEC1–2 from solution. a, rVSVs bearing ANDV or HTNV Gn/Gc were allowed to infect U2OS cells, and cell-surface expression of Gn/Gc was detected by immunofluorescence microscopy using mAb 1E11/D3. b, Cells expressing Gn/Gc were then exposed to sEC1–2 (200 nM), and sEC1–2 binding to the cell surface was detected by immunofluorescence microscopy using an anti-Flag mAb. c, The capacity of PCDH1-specific mAb 3305 to block binding of sEC1–2 to Gn/Gc-expressing cells was determined as in b. sEC1–2 (50 nM) was preincubated with the indicated amounts of a control IgG or mAb 3305, and then exposed to Gn/Gc-expressing cells. In a-c, representative images are shown, and experiments were performed three times with similar results. Scale bars, 20 μm. d, Biotinylated rVSVs bearing ANDV or HTNV Gn/Gc were incubated with sEC1–2 and then captured with streptavidin magnetic beads. Co-precipitated viral particles and sEC1–2 (‘Virus IP’) and a fraction of the input material (‘Input’) were detected by immunoblotting with mAb 23H12, specific for the VSV M matrix protein, and an anti-Flag mAb, respectively. ‘None’ indicates control precipitation of sEC1–2 in the absence of viral particles. Representative images are shown. Experiments were performed three times with similar results.
Extended Data Fig. 8 |. Expression and purification of MPRLV VLPs and their interaction with soluble PCDH1.
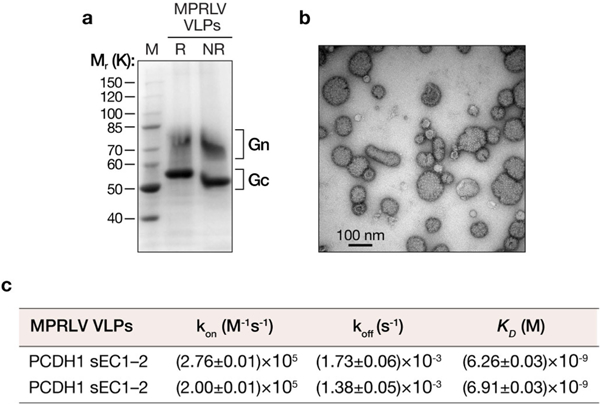
a, Purified MPRLV VLPs (8 μg protein) bearing an N-terminal StrepTag were resolved on an SDS–polyacrylamide gel and visualized by Coomassie blue staining. R, reducing conditions; NR, nonreducing conditions. b, MPRLV VLPs were negatively stained with uranyl acetate and visualized by electron microscopy. c, Kinetic constants for MPRLV VLP-sEC1–2 interaction were determined by biolayer interferometry. kon, association-rate constant; koff, dissociation rate constant. Values ± 95% confidence intervals derived from curve fits are shown for two independent experiments. These experiments were performed three times with similar results.
Extended Data Fig. 9 |. PCDH1-EC1 is required for hantavirus Gn/ Gc-dependent entry and infection in primary Syrian hamster lung endothelial cells.
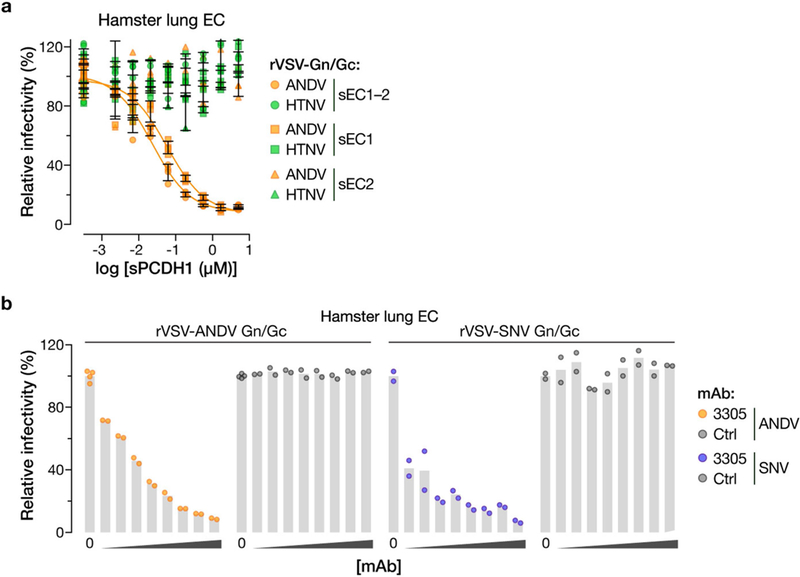
a, Capacity of soluble PCDH1 (sPCDHl) variants (sEC1–2, sEC1 or sEC2) to block hantavirus glycoprotein-dependent entry. rVSVs bearing ANDV or HTNV Gn/Gc glycoproteins were preincubated with the indicated amounts of the indicated sPCDHl variants (0–5 μM, in serial threefold dilutions) at room temperature for 1 h, and then allowed to infect Syrian hamster lung endothelial cells (ECs). One hundred per cent relative infectivity corresponds to 15–20% infected cells. Averages ± s.d.: two experiments, n = 4. b, Capacity of PCDH1-EC1-specific mAb 3305 to block hantavirus glycoprotein-dependent entry. Syrian hamster lung ECs were preincubated with mAb 3305 or control human IgG (0–100 μg ml–1, 0–680 nM, in serial threefold dilutions), and then exposed to rVSVs bearing ANDV or SNV Gn/Gc at an MOI of 0.2 IU per cell. Cells were scored for infection at 9 hpi. One hundred per cent relative infectivity corresponds to 15–20% infected cells. Two experiments, n = 2, except in the case of no mAb (‘0’) for which n = 4.
Extended Data Fig. 10 |. Generation of a PCDH1-KO knockout Syrian hamster by CRISPR-Cas9 genome engineering.
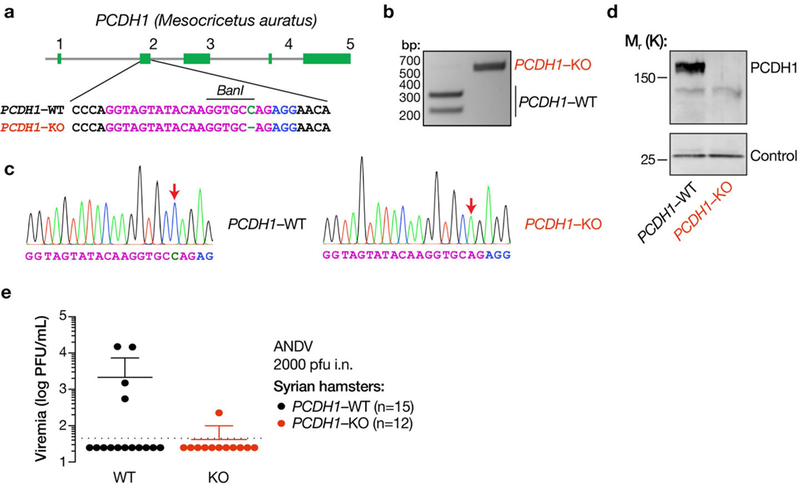
a, Organization of the PCDH1 gene of the Syrian hamster (M. auratus). The sequence in exon 2 of PCDH1 that was targeted by an sgRNA is shown in magenta. Knockout animals bear two PCDH1 alleles that have been edited to lack a single nucleotide, highlighted in green. The sgRNA PAM is highlighted in blue. b, A PCR–RFLP strategy, based on loss of digestion by the restriction endonuclease BanI, was used to detect genome editing and genotype animals. c, PCR–RFLP results were confirmed by Sanger DNA sequencing of PCR amplicons from WT and genome-edited animals. Sequencing traces are shown. Sequence features are highlighted as in a. Red arrows denote the site of gene editing in the PCDH1-KO allele. Experiments were performed twice with similar results. d, Lung tissue isolated from WT and PCDH1-KO hamsters was solubilized, normalized by protein content, and subjected to SDS–PAGE. PCDH1 was detected by immunoblotting with EC1-specific mAb 3305. Control, nonspecific loading control. Experiments were performed three times with similar results. e, Viral loads in the sera of WT and PCDH1-KO hamsters at 14 dpi. The limit of detection is shown as a dotted line. Experiments in b-e were performed twice with similar results.
Supplementary Material
Acknowledgements
See Supplementary Information for grants supporting this work. We thank A. Beck and the Einstein Histopathology Core for histopathology support; S. Garforth and the Einstein Macromolecular Therapeutics Development Facility for assistance with SEC-MALS; G. Pehau-Arnaudet, Institut Pasteur Paris, for electron microscopy of MPRLV VLPs; H. Galeno, Instituto de Salud Pública de Chile, for convalescent sera from Chilean patients with hantavirus; and M. Evans for feedback and discussions. Opinions, conclusions, interpretations and recommendations are those of the authors and are not necessarily endorsed by the US Army. The mention of trade names or commercial products does not constitute endorsement or recommendation for use by Department of the Army or Department of Defense.
Footnotes
Reviewer information Nature thanks B. Lee, G. Schönrich and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Competing interests R.K.J., L.TJ., K.C. and TR.B. are named inventors on US patent application US20170173141A1, covering methods to treat hantavirus infections by targeting PCDH1 (the entry factor described here), the application being assigned to Albert Einstein College of Medicine. TR.B. is co-founder and Scientific Advisory Board member of Haplogen GmbH, and co-founder and managing director of Scenic Biotech BV. F.A.R. is a consultant for Flagship Pioneering.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41586-018-0702-1.
Extended data is available for this paper at https://doi.org/10.1038/s41586-018-0702-1.
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-018-0702-1.
Reprints and permissions information is available at http://www.nature.com/ reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.MacNeil A, Nichol ST & Spiropoulou CF Hantavirus pulmonary syndrome. Virus Res. 162, 138–147 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Zaki SR et al. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 146, 552–579 (1995). [PMC free article] [PubMed] [Google Scholar]
- 3.Kozu Y et al. Protocadherin-1 is a glucocorticoid-responsive critical regulator of airway epithelial barrier function. BMC Pulm. Med. 15, 80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mortensen LJ, Kreiner-Moller E, Hakonarson H, Bønnelykke K & Bisgaard H The PCDH1 gene and asthma in early childhood. Eur. Respir. J. 43, 792–800 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Toncheva AA et al. Genetic variants in protocadherin-1, bronchial hyper-responsiveness, and asthma subphenotypes in German children. Pediatr. Allergy Immunol. 23, 636–641 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Koppelman GH et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am. J. Respir. Crit. Care Med. 180, 929–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gul IS, Hulpiau P, Saeys Y & van Roy F Evolution and diversity of cadherins and catenins. Exp. Cell Res. 358, 3–9 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Sotomayor M, Gaudet R & Corey DP Sorting out a promiscuous superfamily: towards cadherin connectomics. Trends Cell Biol. 24, 524–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackow ER & Gavrilovskaya IN Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 102, 1030–1041 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH & Mackow ER (33 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl Acad. Sci. USA 95, 7074–7079 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raftery MJ et al. (β2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 211, 1485–1497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buranda T et al. Recognition of decay accelerating factor and αvβ3 by inactivated hantaviruses: toward the development of high-throughput screening flow cytometry assays. Anal. Biochem. 402, 151–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krautkrämer E & Zeier M Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55). J. Virol. 82, 4257–4264 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinfelter LM et al. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. MBio 6, e00801–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen J et al. The major cellular sterol regulatory pathway is required for Andes virus infection. PLoS Pathog. 10, e1003911 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tischler ND, Gonzalez A, Perez-Acle T, Rosemblatt M & Valenzuela PD Hantavirus Gc glycoprotein: evidence for a class II fusion protein. J. Gen. Virol. 86, 2937–2947 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Uhlén M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Pillay S & Carette JE Hunting viral receptors using haploid cells. Annu. Rev. Virol. 2, 219–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staring J, Raaben M & Brummelkamp TR Viral escape from endosomes and host detection at a glance. J. Cell Sci. 131, jcs216259 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Nolte KB et al. Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum. Pathol. 26, 110–120 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Sano K et al. Protocadherins: a large family of cadherin-related molecules in central nervous system. EMBO J. 12, 2249–2256 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tischler ND, Galeno H, Rosemblatt M & Valenzuela PD Human and rodent humoral immune responses to Andes virus structural proteins. Virology 334, 319–326 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Acuña R et al. Hantavirus Gn and Gc glycoproteins self-assemble into virus-like particles. J. Virol. 88, 2344–2348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safronetz D, Ebihara H, Feldmann H & Hooper JW The Syrian hamster model of hantavirus pulmonary syndrome. Antiviral Res. 95, 282–292 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hooper JW, Ferro AM & Wahl-Jensen V Immune serum produced by DNA vaccination protects hamsters against lethal respiratory challenge with Andes virus. J. Virol. 82, 1332–1338 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Safronetz D et al. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog. 7, e1002426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong AC, Sandesara RG, Mulherkar N, Whelan SP & Chandran K A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J. Virol. 84, 163–175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamentsky L et al. Improved structure, function and compatibility for CellProfller: modular high-throughput image analysis software. Bioinformatics 27, 1179–1180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hooper JW, Larsen T, Custer DM & Schmaljohn CS A lethal disease model for hantavirus pulmonary syndrome. Virology 289, 6–14 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Lee HW, Lee PW & Johnson KM Isolation of the etiologic agent of Korean Hemorrhagic fever. J. Infect. Dis. 137, 298–308 (1978). [DOI] [PubMed] [Google Scholar]
- 33.Schmaljohn AL et al. Isolation and initial characterization of a newfound hantavirus from California. Virology 206, 963–972 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Morgenstern JP & Land H Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helperfree packaging cell line. Nucleic Acids Res. 18, 3587–3596 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wec AZ et al. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 354, 350–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Persson H et al. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol. 425, 803–811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hornsby M et al. A high through-put platform for recombinant antibodies to folded proteins. Mol. Cell. Proteomics 14, 2833–2847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cifuentes-Muñoz N, Darlix JL & Tischler ND Development of a lentiviral vector system to study the role of the Andes virus glycoproteins. Virus Res. 153, 29–35 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Hooper JW, Custer DM, Thompson E & Schmaljohn CS DNA vaccination with the Hantaan virus M gene protects hamsters against three of four HFRS hantaviruses and elicits a high-titer neutralizing antibody response in Rhesus monkeys. J. Virol. 75, 8469–8477 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lefrancois L & Lyles DS The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121, 157–167 (1982). [PubMed] [Google Scholar]
- 41.Trombley AR et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am. J. Trop. Med. Hyg. 82, 954–960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.