

Abstract
Background
Metabolic rewiring allows cancer cells to sustain high proliferation rates. Thus, targeting only the cancer-specific cellular metabolism will safeguard healthy tissues.
Methods
We developed the very efficient FASTCORMICS RNA-seq workflow (rFASTCORMICS) to build 10,005 high-resolution metabolic models from the TCGA dataset to capture metabolic rewiring strategies in cancer cells. Colorectal cancer (CRC) was used as a test case for a repurposing workflow based on rFASTCORMICS.
Findings
Alternative pathways that are not required for proliferation or survival tend to be shut down and, therefore, tumours display cancer-specific essential genes that are significantly enriched for known drug targets. We identified naftifine, ketoconazole, and mimosine as new potential CRC drugs, which were experimentally validated.
Interpretation
The here presented rFASTCORMICS workflow successfully reconstructs a metabolic model based on RNA-seq data and successfully predicted drug targets and drugs not yet indicted for colorectal cancer.
Fund
This study was supported by the University of Luxembourg (IRP grant scheme; R-AGR-0755-12), the Luxembourg National Research Fund (FNR PRIDE PRIDE15/10675146/CANBIO), the Fondation Cancer (Luxembourg), the European Union‘s Horizon2020 research and innovation programme under the Marie Sklodowska- Curie grant agreement No 642295 (MEL-PLEX), and the German Federal Ministry of Education and Research (BMBF) within the project MelanomSensitivity (BMBF/BM/7643621).
Keywords: Metabolic modelling, Cancer, Machine learning, Drug repurposing
Research in context section.
Evidence before this study
High costs in drug development led pharmaceutical companies to invest in drug repurposing, which consists of reusing commercially available and wellassessed drugs with known efficiency and side effects for new indications. Drug repurposing accounted for 20% of the drugs released in 2013.
Metabolic models, in silico knock out strategies, and flux balance analysis (FBA) have successfully been applied to predict cancer drug targets. The in silico essential genes prediction has some main advantage over other technologies, such as CRISPR/Cas9, to be cheap, fast, and easily applied to every cell line and context.
Added value of this study
To build high quality context-specific metabolic models that can be used, among others, for drug target prediction, we developed rFASTCORMICS that builds metabolic models in a matter of seconds based on RNA-seq data. For this study, we reconstructed 10,005 models from the TCGA (The Cancer Genome Atlas) data set that we used to extract gene and reaction signatures for 13 different types of cancers. We also predicted 17 new repurposing drug candidates for colon cancer of which we validated three drugs in vitro on colon cancer cell lines.
Implications of all the available evidence
The drug prediction workflow can easily be adapted for other high proliferative cancer cells and drug targets and drugs can be validated in follow-up studies in vitro and in vivo. Ultimately, the workflow can be used to identify repurposed drugs for different cancer and cancer subtypes.
Alt-text: Unlabelled Box
1. Introduction
The Warburg effect [1] describes the first documented metabolic rewiring event in cancer cells, favouring aerobic glycolysis over oxidative phosphorylation. To compensate for the energetically less efficient glycolysis, cancer cells increase the flux rate through the glycolytic pathway [2], which makes tricarboxylic acid cycle intermediates available to serve as precursors for fatty acids, nucleotides, and amino acids.
Since Otto Warburg's work, other metabolic alterations were reported in different cancer types, such as the up-regulation of the pentose phosphate pathway, the lipid synthesis pathway [3], or a high dependency on glutamine availability to maintain anaplerosis [4]. Metabolic rewiring occurs through mutations of key metabolic enzymes, oncogenes, and tumour suppressors that tightly control metabolic reactions. Further, it affects other hallmarks of cancer such as immune evasion or tissue invasion [5].
By identifying cancer-specific metabolism and the underlying (mutated) genes, new therapies can be developed that reduce the growth of cancer cells without affecting normal cells [6]. Furthermore, focussing on common metabolic alterations, such as glutamine consumption, would allow targeting the whole cancer cell population instead of sub-clones only.
Cancer-specific metabolic networks, extracted from generic reconstructions such as Recon X [[7], [8], [9]] and HMR [10,11], are promising tools to study metabolic rewiring strategies at a cell-wide level [[12], [13], [14]]. Applications to context-specific models include structural analysis (comparing healthy and cancer metabolic networks) to find cancer-specific pathways and in silico gene knock-outs to find potential drug targets [15] or oncometabolites [16]. Moreover, context-specific models allow us to determine cancer-specific flux distributions through random sampling and flux variability analysis [17]. Metabolic modelling approaches were used to successfully integrate cancer patient data and to study the metabolism of cancer cells [16,18]. However, the identification of metabolic rewiring strategies was hindered by the low resolution power and high computational demand of most context-specific model building algorithms [19].
The recently developed FASTCORE family (FASTCORE [20] and FASTCORMICS [21]) can be used in a high-throughput manner offering superior resolution power and accuracy [19]. Because FASTCORMICS builds a model in a matter of seconds to minutes on an ordinary laptop [19,21,22], metabolic models can be used as a routine tool for data analysis and clinical diagnostics.
To systematically study metabolic rewiring and to show that FASTCORMICS can be used to integrate and analyse patient data, we extended FASTCORMICS [21], originally designed for microarray data, to build context-specific models via the integration of RNA-seq data. So far, only two algorithms have been benchmarked by their developers with RNAseq data as input: RegrEx [23] and Lee2012 [24]. Lee 2012 minimizes the error between the flux prediction and the experimental data, which increases the sensitivity of the algorithm to noise. Whereas, RegrEx tries to find a balance between the goodness of the fit of the predicted flux distribution to the measured expression data, (which increases the sensitivity of these algorithms to noise) and the complexity of the model (by reducing the number of features). To address this problem, RegrEx added a L1-norm regularization which in-turn increases the computational demand of the algorithm due to resources extensive cross-validations for parameter tuning.
In this study, 10,005 context-specific models (9264 cancer and 741 control samples, GSE62944 [25] collected by the TCGA Research Network (http://cancergenome.nih.gov/)) were reconstructed using the FASTCORMICS RNA-seq workflow (rFASTCORMICS), a new adaptation of the FASTCORMICS workflow specifically designed for RNA-seq data that prevents the use of arbitrary thresholds. The rFASTCORMICS models showed an enrichment for known essential genes (identified by CRISPR-Cas9 screens in cancer cell lines [26,27]) and approved cancer drug targets among the predicted in silico essential genes.
Three non-cancer drugs (namely naftifine, ketoconazole, and mimosine), targeting the predicted essential genes in colon cancer, were successfully validated in vitro. All three tested drugs significantly reduced the proliferation rates in cancer while having a minor or no effect on the proliferation rate of the control cells.
2. Material and methods
For this study, 10,005 sample-specific and 26 tissue-specific models were reconstructed with rFASTCORMICS using Recon 2. The tissue-specific models consisted of 13 generic cancer and 13 generic control models. rFASTCORMICS is freely available for non-commercial users at:
https://wwwen.uni.lu/research/fstc/life_sciences_research_unit/research_areas/systems_biology/software/rfastcormics. rFASTCORMICS runs under Matlab (MathWorks) and requires a compatible IBM CPLEX solver, the COBRA Toolbox, the Statistics and Machine Learning Toolbox, and the Curve Fitting Toolbox.
2.1. FASTCORMICS RNA-seq (rFASTCORMICS)
For RNA-seq data, arbitrary thresholds between 0.3 and 1 FPKM (Fragments Per Kilobase of transcript per Million mapped reads) are commonly used to segregate between expressed and non-expressed genes. Previous studies [28,29] showed how arbitrary gene expression thresholds affect the model predictions. However, in [30], a novel workflow to identify active genes was proposed; it compares the gene expression levels to the promoter activity signatures using data from the ENCODE project. In this study, the authors showed that the measured signal is composed of 2 partially overlapping Gaussian corresponding to the expressed genes and the noise, respectively. Based on the approach in [30], rFASTCORMICS fits a half-Gaussian curve to the log2-transformed FPKM values of each sample separately and a density plot is drawn for each sample using the ksdensity function in MATLAB (see Fig. 1). The rationale is that the main peak of the signal curve comprises the expressed genes while leaky genes are mostly found in the leftmost side of the curve. The right half of the main peak was mirrored and a Gaussian curve (expression curve) was fitted using the cftool function (see Fig. 1). Then, the Gaussian curve is subtracted from the kernel density estimation of the signal curve to obtain the portion of the signal that was not included in the Gaussian distribution. A second Gaussian curve (inexpression curve) was fitted onto this portion using the cftool function in Matlab.
Fig. 1.
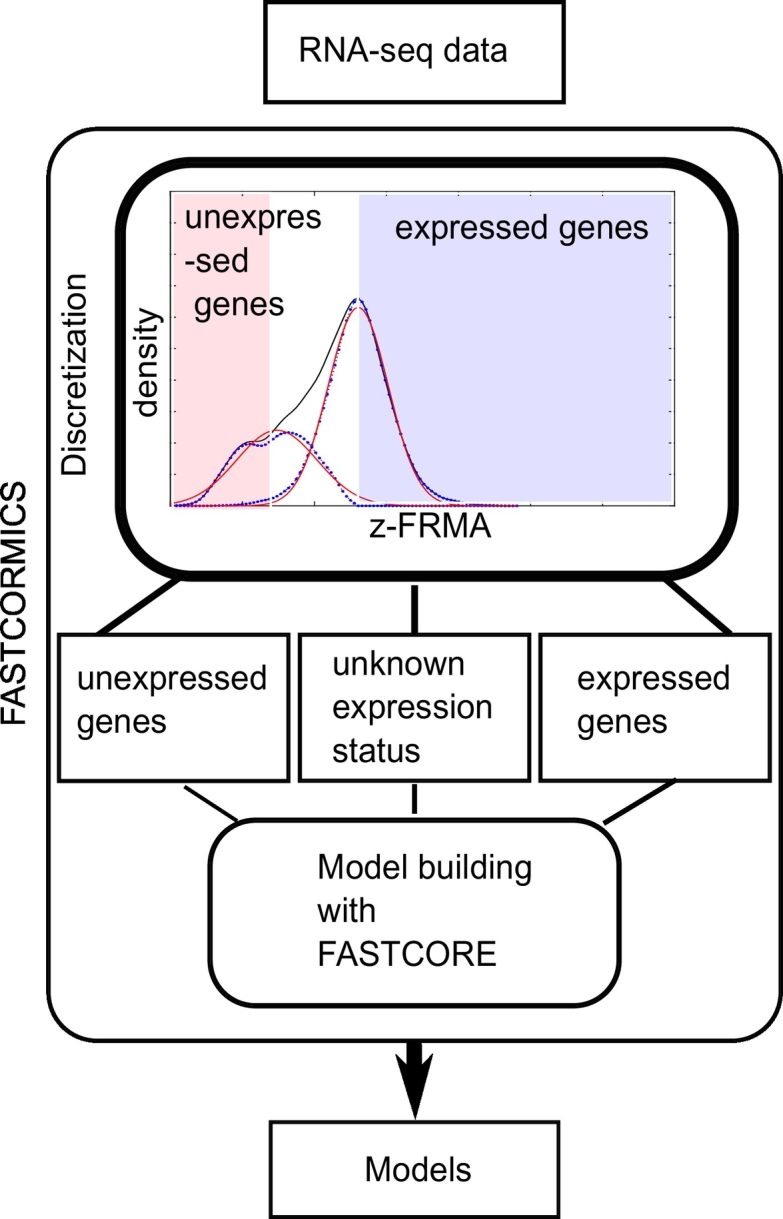
The RNA-seq FASTCORMICS workflow: The workflow uses a discretization step that considers the intensity distribution across all genes to discretize the genes into expressed, undefined expression status, and inexpressed. The discretized values are then mapped to the input model to obtain 3 sets of reactions: core reactions, non-core, and inactive reactions. The bounds of the inactive reactions are set to zero and are removed from the model along with reactions that are no longer able to carry a flux. A modified version of FASTCORE is used to include all the remaining core reactions, with the exception of transporter reactions that are transferred from the core to an unpenalized set (these reactions are not forced in but, as they are not penalized, their inclusion is favoured over non-core reactions). In Recon X models, several hundreds of transporter reactions are controlled by a set of only a few genes. To avoid the unwanted activation of transporter reactions in inactive pathways, transporter reactions are not forced to be active but their activation is not penalized. To obtain consistent models, FASTCORE includes a minimal amount of non-core reactions.
Lastly, the log2-transformed FPKM values were converted into zFPKM values using:
| (1) |
where mu is the log2-transformed FPKM value at the maximum value of the density plot and sigma the standard deviation. Because the best correlation between RNA-seq data and histone marks (at the promoter regions) was found for a zFPKM score of −3 by [30], the maximum of the inexpression curve was chosen as threshold if it is above −3 zFPKM, otherwise −3 is taken. mu (zFPKM score of 0) was selected as expression threshold. If the zFPKM value of a gene is below the inexpression threshold, a score of −1 (not expressed) is assigned; if it is above the expression threshold, a score of 1 (expressed) is assigned; and if it was between the two thresholds a score of 0 (unknown expression status) is assigned (See Fig. 1 and Supplementary Fig. S.1).
Every gene was then mapped to the reactions of Recon 2 via the gene-protein-reaction rules (GPR rules). Similar to FASTCORMICS [21], rFASTCORMICS can be used to reconstruct sample-specific and generic tissue/cell-specific models. For sample-specific models, reactions with a score of −1 are considered as being inexpressed and, therefore, their bounds are set to 0, except for reactions that are required for the biomass function to carry a flux. The score of a reaction is determined by the score of its associated genes through the GPR rules. These Boolean rules define which gene(s) are necessary for a reaction to take place. Depending on the activity of the gene(s) and the Boolean rules (Boolean AND/OR), the associated reaction is active or inactive. E.g. if a reaction is under the control of two genes A and B and the BOOLEAN rule is “Gene A AND Gene B", then both genes have to be expressed for the reaction to be active. In practice, after discretization we obtain a value for each gene 1 (expressed), 0 (unknown) and − 1 (unexpressed). For the previous example, we take the minimum (A,B) as both need to be active to obtain the value associated to the reaction. If A = 1 and B = 1, the reaction is active, if A = 1, and B = 0 then the reaction is tagged as unknown, if A = 1 and B = -1, the reaction is inactive. For OR only one of the genes has to be expressed to get an active reaction. We take the maximum of the discretized gene values accordingly. If A = 1 and B = 1, the reaction is tagged as active, if A = 1 and B = 0 then the reaction is tagged as active as well, if A = 1 and B = -1, the reaction is active as well. If A = 0 and B = -1 then the reaction is tagged as unknown.
For tissue/cell-specific generic models, the bounds of the reactions that are associated with a − 1 in at least 90% of the samples are set to 0.
To eliminate blocked reactions, FASTCC [20] was run and the remaining reactions (not eliminated by FASTCC) with a score of 1 were included in the core set. Note that, for the tissue-specific generic models, a score of 1 has to be found in >90% of the samples. Finally, a modified version of FASTCORE [20] that allows the definition of an unpenalized set is run. Their inclusion is not forced but only favoured over non-core reactions. The unpenalized set contains reactions that tend to be under the control of promiscuous genes such as transporter reactions. The output is a vector that contains the indices of reactions from the input model to be included in the context-specific output model.
Because rFASTCORMICS does not require an optimization or objective function such as biomass production or ATP demand, it can be used to build models of low proliferative cells. rFASTCORMICS needs as input expression data, a generic reconstruction, and a dictionary that maps the expression gene identifiers with the model gene identifiers. Other inputs, such as the medium composition are optional. The medium composition that was used to reconstruct the models can be found in the supplementary Table S.3.
2.2. Cancer and healthy core metabolic genes and reactions
We define the healthy core as genes/reactions that are active in each of the 741 healthy models and the cancer core as genes/reactions that are active in every model in 100 sets of 741 randomly selected cancer models. For non-metabolic genes (genes not present in the human metabolic reconstruction Recon 2), the discretization of rFASTCORMICS was used to determine if a gene/reaction is active. Healthy and cancer core gene/reactions are metabolic (and non-metabolic) genes/reactions that are expressed in every healthy and cancer sample, respectively.
2.3. Reaction and gene signatures
For each reaction and gene, the absolute difference of the expression ratio (number of models that express a gene/reaction divided by the number of models for a given context) between the cancer and the control models for each tissue was computed. The aim is to find genes that tend to have a condition-specific expression pattern. A reverse feature selection approach on the top 100 most different reactions/genes was performed using the rfe function of the caret package [31] in R with the accuracy (AUC score) as metric. Methods such as linear and radial support vector machines and rpart, were tested. For each tissue type, the method and the number of features (gene or reaction signatures) with the best accuracy score were selected.
The aim of the feature selection was to find the best balance between over-fitting (too high number of features) and under-fitting (insufficient number of features) and by such to further reduce the number of reactions/genes that are sufficient to assign a new samples in one of the two conditions. These genes and reactions are more likely to be involved in rewiring strategies and are conserved or eliminated across cancer samples. Secondly, it allows assessing the resolution power of the workflow (defined as the capacity to distinguish between two conditions). The feature selection step starts with a matrix of 100 rows corresponding to the genes/reactions. The number of columns is equal to the number of tissue samples. We used the cancer or control labels as response. After this step, the remaining number of features varies between 4 and 100 genes/reactions depending on the number of genes/reactions that allowed obtaining the best accuracy.
Finally, a cross-validated support vector machine classifier was run, using the Classification Learner toolbox in Matlab, to assess the prediction power of the machine learning model.
2.4. Gene signature enrichment tests
The gene signatures were pooled and a list of 502 unique genes was obtained. An enrichment test has been performed, comparing the 502 unique genes to the 1721 unique metabolic genes of Recon 2 using the hypergeometric test function (hypercdf) in Matlab.
Driver genes. A list of known driver genes for each tissue was retrieved from the driver genes database, driverDBv2 [32]. By default, genes that were called by at least two algorithms in the database are considered driver genes.
Loss-of-function mutations. A list of genes affected by loss-of-function mutations with their corresponding functional annotation was retrieved from the Supplementary Files of [33].
Only gene signatures of tissues also found in the [33] study were considered, corresponding to a set of 362 genes.
Essential genes. Data on essential genes were taken from:
-
(1)
[26] performed CRISPR-Cas9 screens on two chronic myelogenous leukaemia cell lines (KBM7 and K562) and two Burkitt's lymphoma cell lines (Raji and Jiyoye) to identify essential genes. Essential genes were defined by a CRISPR score (CS) that gives information on the effectiveness of the gene inhibition and the reproducibility of the CRISPR-Cas9 screening. A CS-score below 0.1 and a corrected p-value below 0.05 were used as thresholds to consider a gene as essential [26].
-
(2)
[30] who used CRISPR-Cas9 on five TKO cell lines to define core fitness genes (essential genes common to 3–5 cancer cell lines). Two lists with essential genes were obtained; one list which enumerates essential genes in 3 out of 5 cell line and a second list with essential genes in 5 out of 5 cell lines.
Genes with SNPs associated with a high impact on the phenotype. The mutation annotation files (.maf) files containing the impact (high, moderate, and low impact) of each SNP on the phenotype (as predicted by MUSE, MUTECT2, varScan2, and sniper) was downloaded with the GDCqueryMaf function from the TCGAbiolinks package [34] in R.
As stated previously, the number of high impact genes (found in 1 or more of the 13 tissues in the gene signature) was compared to the number of genes found among Recon 2 (See Supplementary Information and Supplementary Figs. S.15 and S.16).
Enrichment in super-enhancers. The curated list of super-enhancers was taken from [35](from Supplementary Table 3) to compute the enrichment analysis in the top 100 highly expressed genes and from [35](from Supplementary Table 5) to compute the enrichment analysis of hypo-methylated super-enhancers in the gene signatures.
2.5. Gene deletion and essential genes
In silico knock-outs were simulated for the 13 cancer and control generic tissue models with two different input models Recon 2.04. To be able to compare the models with an in vitro experiment, the models were constrained for the medium composition with the medium constrain option and the optimization function of the rFASTCORMICS workflow to force the models to produce ATP and biomass.
For each model, in silico gene deletions were performed using the singleGeneDeletion command from the COBRA toolbox, while optimizing for the ATP demand and the biomass reaction separately. Before performing the in silico gene deletion, the maximal flux through the objective function (either ATP demand or biomass) is assessed through optimization. The solution represents the growth rate of the wild type. Then, during the single gene deletion, for each gene, the fluxes of its controlled reactions is set to 0, and the flux through the objective function was maximised. Consequently, for each gene, we obtained a growth rate, measuring how the gene deletion affects the objective function. The growth or production ratio is obtained by dividing the growth rate of the knocked-out model by the growth rate of the wild type model. By comparing the maximal fluxes through the objective function between the wild type (no gene deletion) and the genes were considered essential if their deletion causes a reduction of at most 10% and 50% in the healthy and cancer models, respectively. The deletion (or inhibition) should only minimally affect the healthy cells and mainly target cancer cells, inhibit their growth, or cause them to undergo cell death.
To find possible drug targets in cancer, essential genes between the healthy and cancer models were compared and different lists have been created: genes which are essential in all models (reducing ATP demand/biomass reaction by at least 50%), genes which are essential in cancer (reducing ATP demand/biomass reaction by at least 50%), and genes which are only essential in cancer and not in healthy (reducing growth rate by maximum 10% in healthy and at least 50% in cancer). This resulted in three sub-cases: comparison between ATP demands, biomass reactions, and ATP demand for healthy and biomass reaction for cancer.
For each list, the union and intersection of the essential genes from each tissue was considered, resulting in a total of 14 cases (see Supplementary Table S.5).
2.6. Enrichment analysis for essential genes and cancer drugs
For each list of essential genes, an enrichment analysis for the predicted essential genes and cancer drugs was performed using the hypergeometric test function (hypercdf in Matlab).
Data on essential genes from different cell lines were taken from [26,30] and [36] who determined essential genes in two cell lines (KBM and HAP1) by using extensive gene trapping mutagenesis. Genes with a p-value below 0.05 were defined as essential.
To test for cancer drug enrichment, the DrugBank database [37] was datamined to find approved and inhibiting drugs for each gene present in the genome-scale metabolic reconstruction as well as drugs, which were labelled as antimetabolites and anti-neoplastic agents. Then, the drugs associated to the genes in the different essential gene lists were extracted and compared to 5 different online lists of approved cancer drugs. The online lists (as of April 2017) and the names of the databases can be found in the Supplementary Data (see Supplementary Table S.6).
2.7. Validation
2.7.1. in silico validation
The FASTCORE family was recently benchmarked against several of its most used competitors [38]. The workflow used in this paper was also run with rFASTCORMICS (see Supplementary Fig. S.29). Additionally, models for each tissue were reconstructed using the INIT algorithm [10] from the COBRA toolbox (see supplementary Text and Supplementary Table S.9) to assess the performance of rFASTCORMICS against another model building algorithms.
2.7.2. Compound descriptions
Ketoconazole, a synthetic imidazole based antifungal drug, is known as pan-inhibitor of the PXR nuclear receptor in xenobiotic metabolism (Cyp3A4) in vivo [39]. Ketoconazole increased the intra-tumour drug levels and antitumour activity of fenretinide upon co-treatment [40]. Comparable results, namely effects on the pharmacokinetics of the BCL-2 inhibitor venetoclax, were shown in patients with non-Hodgkin lymphoma [41]. Although ketoconazole reduces cell proliferation in a dose-dependent manner on the HT29 colon cancer cells in vitro [42], its mechanism in CRC is still elusive.
Naftifine, an allylamine derivative, is a synthetic, broad spectrum, antifungal agent. It also shows anti-inflammatory properties such as decreased superoxide production and reduced polymorphonuclear leukocyte chemotaxis and endothelial adhesion [43]. It inhibits squalene epoxidase [44], which catalyses the initial oxygenation step in sterol biosynthesis. As a result, decreased levels of sterols and a corresponding accumulation of squalene is observed in the cells. Recently, SQLE inhibition was proposed as potential therapeutic target for Breast cancer treatment [45]. In vitro, naftifine exhibited anti-tumourigenic effects on lymphoma and myeloma cell lines, whereas no effect was seen on healthy fibroblasts using equal concentrations [46]. The chemical features of naftifine are similar to those of known Wnt inhibitors (cinnarizine, flunnarizine).
Mimosine is a non-protein amino acid similar to tyrosine. Mimosine was shown to have anti-cancer, anti-inflammatory, anti-fibrotic, anti-viral, herbicidal, and insecticidal properties [47]. Recently, it was suggested as promising agent for treatment of malignant gliomas due to the ability to promote ROS generation, resulting in apoptosis in vitro [48]. In SW620 colon adenocarcinoma cells it reversibly inhibits of cell cycle progression [49] and is therefore a potential candidate for CRC treatment (see Supplementary Table S.7 for more details on the action and literature references).
2.7.3. Cell line and culture conditions
The primary tumour cell lines T6 and T18 (stage II and stage IV colon adenocarcinoma) were established from fresh primary colon cancer tissue immediately after surgical resection and fully characterized as previously described in [50]. The cell lines Caco-2 and HT29 both human colon adenocarcinoma and the normal colon cell line CCD-18CO were obtained from the American Type Culture Collection (ATCC, Rockville, USA). All cells as well as the CCD18-CO cell line (normal colonic fibroblast) were cultured in Dulbecco modified Eagle medium (DMEM) F-12 (Lonza), supplied with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS). Cells were regularly split (± twice a week) by using trypsin-EDTA (Lonza) as a detaching agent and they were counted, after Trypan Blue staining, with a Cedex XS cell counting system (Roche).
2.7.4. Compounds preparation
Ketoconazole (Sigma, Ref.: K1003) and naftifine (Sigma, Ref.: N1790) were dissolved in DMSO (Sigma, Ref.: M0253) and stock concentration of 25 mM were prepared. L-mimosine (Sigma) was dissolved in D-PBS, containing 10% NaHCO3 reaching a stock concentration of 50 mM.
2.7.5. Cell proliferation
12,000 colon cancer cells (T6, T18, HT29, Caco-2, or 5,000 colonic fibroblasts (CCD18-CO) were seeded into a 96 well plate in 100 μL of culture medium. After 24 h, new media containing various concentrations of ketoconazole, naftifine, or mimosine was added. The IncuCyte ZOOM system was used to measure cell proliferation over 5 days at 37∘C with 5% CO2. (Essen BioScience).
3. Results and validation
3.1. Performance
The integration of large datasets into metabolic models requires fast model-building algorithms, such as FASTCORMICS [21]. Here, we used rFASTCORMICS to reconstruct 10,005 metabolic models in less than 5 days on a computer with an Intel®Xeon®CPU E3 1241-v3@ 3.50 GHZ processor using Recon 2 [8] and RNA-seq data from the TCGA dataset as input Table 1).
Table 1.
rFASTCORMICS permits the fast reconstruction of models The models were reconstructed using as input the TCGA dataset and Recon 2 for rFASTCORMICS. The average running time per model was around 2.8 min on a computer with an Intel(R)Xeon(R)CPU E3 1241-v3@ 3.50 GHZ processor running in parallel on 4 cores. Only tissues with at least 20 control samples were considered for the analysis.
| Description | Numerics |
|---|---|
| Number of tumour models | 9264 |
| Number of control models | 741 |
| Total number of models | 10,005 |
| Total number of tissue types | 30 |
| Number of tissue considered for analysis | 13 |
| Running time for 10,005 model reconstructions | 112 h |
| Average running time per model reconstruction per core | 161 s |
3.2. rFASTCORMICS captures metabolic variations in cancer
3.2.1. Cancer cells do not lose their metabolic identity
To test if rFASTCORMICS can capture metabolic variations between different contexts, the cosine similarity index and the Jaccard similarity score were calculated, and the models were clustered accordingly (Supplementary Figs. S.2 and S.3, respectively). The cosine similarity index is used to compute the similarity of the models based on the discretized gene intensities of metabolic genes in the model whereas the Jaccard similarity score is used to compute the similarity of the models based on the binarized reaction presence of the models. The clustergrams show that models originating from the same tissue cluster together, regardless of their health state (Supplementary Fig. S.2). Furthermore, considering each tissue individually, a clear difference between cancer subtypes and control models could be observed and, for most tissues, the control models were more homogeneous than the cancer models (Supplementary Fig. S.4 and S.5). Taken together, the Jaccard similarity scores demonstrate that cancer models do not completely lose their metabolic identity but adapt their metabolism to cope with their environment and sustain high proliferation rates.
The reconstructed cancer models contained significantly fewer reactions than the controls but displayed a higher variance in size (number of reactions). Due to the lack of sufficient control samples, only 13 tissue types were considered for further analysis. Among the 13 analysed cancer types, 9 were significantly more compact than their controls (KS-test: p-values between 0.0043 and 8.42 e-36), whereas 3 were significantly larger (KS-test:p-values below 1.59 e-7) and 2 were not significantly different (Supplementary Fig. S.6 a and b and Supplementary Table S.1 and S.2).
To overcome the high intra-variability in cancer and to have a more general overview on the diverse types of cancer, 26 generic tissue-specific models were reconstructed (cancer and control for each of the 13 tissues). We could confirm that cancer models were overall smaller and showed higher variance. Clustering of the generic models also revealed clear differences between cancer and control models (Supplementary Figs. S.7 and S.8). In general, similar tissues cluster together inside the respective clusters (Supplementary Fig. S.8).
3.2.2. Reaction and gene signatures can segregate between cancer and control models
By finding a minimal set of reactions, which can differentiate between cancer and control models, the main player(s) implicated in metabolic rewiring strategies were identified. The reaction signatures were retrieved by a reverse feature selection approach (Supplementary Fig. S.9) (see material and methods for details). The presence or absence of the reactions correctly predicted the labels of the samples through a cross-validation assay with an accuracy >94%, for most tissues (see Supplementary Fig. S.10).
As an example, for liver, the accuracy was 96% with only 19 reactions. Furthermore, many reactions implicated in the consumption of building blocks (nucleotides, amino acids, and cholesterol) appeared in the top 100 most differentially active reactions (reactions that tend to be present in the cancer and absent in the controls or vice-versa), such as the lysine and branched amino acid metabolism which were more often found to be active in the control models (see Supplementary Fig. S.11 for liver models and Supplementary Fig. S.12 for all 13 tissues).
The same approach was used to obtain gene signatures that contained between 4 and 97 genes for all tested tissues (total: 502 genes). Overall, genes from the signatures are specific to a given tissue type and allowed obtaining a prediction accuracy above 94% (see Supplementary Fig. S.13). The gene signatures were enriched for transporters, driver genes, loss-of-function mutations, and genes with SNPs that have a high impact on the phenotype (p-values between 0.02 and 0.0002, see Supplementary Info and Supplementary Figs. S.14, S.15 and S.16).
The metabolic alterations captured by rFASTCORMICS allow us to determine if the sample is cancerous with a high certainty. These alterations are not random but part of a rewiring strategy common to the cancer cells of a tumour.
3.3. The cancer core metabolism is compacter and enriched in essential genes
To discover a common cancer core metabolism, which can be specifically targeted, the expression ratio (number of models that express a gene/reaction divided by the total number of models) of the reactions/genes across every cancer and control model was computed. We separately defined a cancer core metabolism and a healthy core metabolism based on the genes/reactions present in every healthy model and a random set of cancer models of equal size. For the reactions, 211 and 760 reactions have been found to be present in every selected cancer and control model, respectively (see Supplementary Fig. S.17). These results are in accordance with the genes for which the cancer core metabolism was also smaller (65 versus 225 genes, (see Supplementary Fig. S.18).
In addition, we were interested if a smaller size of the cancer core was linked to an enrichment of core fitness genes (essential genes common to several cancer types). Therefore, the core metabolic genes were compared to two CRISPR-Cas9 essential gene screening studies from different cancer cell lines [26,27]. We found an enrichment for essential genes in the cancer metabolic core when compared to all the metabolic genes (hypergeometric test: with p-values between 10e-16 and 0.0309) (see Supplementary Fig. S.19). A weaker enrichment in the healthy core was consistent with the higher number of reactions in the healthy core.
The metabolic rewiring results in a lower pathway presence rate (fraction of active reactions in a pathway) (see Supplementary Figs. S.20, S.21, and S.22 and Supplementary Table S.4) in cancer by loss-of-function mutations in genes controlling alternative metabolic pathways. Notably, N-glycan degradation and synthesis, oxidative phosphorylation, fatty acid, alanine, aspartate, and tetrahydrobiopterin metabolism have a decreased presence rate in the cancer core metabolism whereas the triacylglycerol synthesis and NAD metabolism are increased. Previously [27], showed that core fitness genes had 10-fold higher expression values across the cell lines in the Cancer Cell Line Encyclopedia [51]. In agreement with these findings, cancer core metabolic genes, which are enriched for essential genes, have higher expression values (median = 158) than the healthy core metabolic genes (median = 68) or other metabolic genes (median = 6.5) (KS test p-values were between 1.0e-12 and 0.0146, and between 4.8e-45 and 2.3e-24) (see Supplementary Fig. S.23).
Furthermore, it was shown that genes with high expression values are under high-regulatory control [21], which could also be confirmed for the core metabolic genes with an enrichment for super-enhancers (hypergeometric test p-value = 0.0123).
The cancer and healthy core genes have also been computed for the gene expression data. Here, the data suggests that the cancer core (480 genes in the cancer core against 1725 genes in the controls, the cancer core is almost completely included in the healthy core) is smaller (and mostly included in the healthy core) because of the selection pressure that causes the accumulation of mutations in non-essential genes and the loss of redundant (alternative) pathways. It further shows that core genes, as well as essential genes required for the cell survival, have higher expression values which is guaranteed at least partially by the control of super-enhancers.
3.4. Predicted essential genes are enriched for cancer drugs
Due to the shut-down of branches in different metabolic pathways, cancer cells tend to exhibit more essential genes, which can be used as specific drug targets. Therefore, in silico knock-outs were performed on the 13 cancer and 13 control generic models to assess the effects on ATP and biomass production. Single gene deletion revealed 32 genes that are essential for the biomass production in every cancer model, and 92 essential genes in at least one cancer model, of which 29 and 58 genes did not affect the ATP production in controls, respectively (see Supplementary Fig. S.24 for the workflow).
In general, knock-outs more often affect the biomass reaction (see Supplementary Figs. S.25, S.26, and S.27) and no gene deletion completely eliminated the flux through the ATP demand reaction (minima 0.2173 (PKLR) and 0.2936 (PKM), for controls and cancer, respectively) suggesting that the reactions implicated in the ATP production are maintained by several metabolic pathways able to compensate each other.
To validate gene essentiality, predicted essential genes were compared to different essential gene screenings [26,30,36] and a strong enrichment for each set of genes was found. Out of the 1729 unique metabolic genes, between 128 and 180 genes are considered to be essential by [26,36] (see Supplementary Fig. S.28) and a strong enrichment was found among the different gene lists (Fig. 2).
Fig. 2.

Essential genes predicted by the models are consistent with known cancer drug targets. A strong enrichment in essential genes and cancer drugs was found among the list of predicted essential genes for colorectal cancer. In grey, genes that cause a reduction of the biomass by 50% in either the healthy or the cancer model, in purple, genes that cause a reduction of biomass in the cancer model and in red, genes that are only essential in cancer and not in the healthy model. Metabolic genes are shown in white. The x-axis represents the databases used to extract known cancer drug targets, whereas the “antimetabolites” and “antineoplastic agents” columns have been extracted from the drug categories in the DrugBank. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Because the TCGA dataset contains mainly cancer samples, we were interested if we could find any (already known) cancer drugs that target metabolic genes. To this goal, we retrieved drug data from different resources (see Material and Methods) and tested whether the different lists of predicted essential genes were enriched for anti-metabolites, anti-neoplastic agents, and approved cancer drugs. Highest enrichments in all databases have been found for genes whose deletion affects only the biomass production in cancer without affecting ATP production in the control models (p-values between 0.002 and 6.00E-06) but also in genes whose deletion affects the biomass production regardless of the control models (p-values between 0.02 and 0.0004, see Supplementary Table S.5).
Further analysis of the predicted essential cancer genes revealed gene involvement in the nucleotide metabolism: CAD, DHFR, DHODH, RPIA, RRM1, RRM2, TYMS, and UMPS [52] which are already known targets of antineoplastic agents. Other predicted essential genes in cancer include FASN, KDSR and MVK, for which no anti-cancer drugs yet exist. However, cerulenin and orlistat are both inhibitor of FASN and are currently under investigation [[53], [54], [55]].
3.5. Prediction of drug targets and repurposed drugs for colorectal cancer
To validate the repurposing workflow, based on rFASTCORMICS, which could also be used for any proliferative cancer type, we applied it to colorectal cancer, one of the most common and lethal cancers worldwide. We used our approach to identify, among every potential drug candidate, those drugs that are most likely to kill or reduce the proliferation of cancer cells while having a low toxicity for healthy cells. Predicted drug targets were obtained with the above-described workflow for CRC. 107 genes were predicted to be essential, among which, 17 were already described drug targets and associated to 43 unique drugs of which 17 are not approved for cancer treatment. A brief literature review (see Supplementary Table S.8) was performed for the non-approved drugs to select three drugs, namely naftifine, ketoconazole, and mimosine (See Supplementary Table S.7 for more information). The selected drugs were tested for their effects on cellular proliferation of commercial cell lines as well as on patient-derived primary cultures [50]. In summary, all three drugs reduced the growth rate of the colorectal cancer cells (see Fig. 3 and Supplementary Fig. S.30) but had no or only minimal effects on the healthy control cells. Furthermore, to validate our workflow, we ran the benchmarking workflow proposed by [38] on a rFASTCORMICS liver model and showed an enrichment of reactions associated with genes with high expression level and high confidence levels of gene expression. We also ran the here presented drug repurposing workflow on the models reconstructed with the INIT algorithm that identifies essential genes in order to predict possible drug targets and associated drugs. As already stated in [38], the models reconstructed with INIT are larger, meaning they have more reactions than models reconstructed with members of the FASTCORMICS family (see Supplementary Table S8). Unfortunately, no essential genes could be determined for the models reconstructed with INIT, possibly because of their large model size.
Fig. 3.

Effect of candidate drugs on primary CRC cells (a) and normal fibroblasts (b). 12,000 T18 cells (a) or 5,000 CCD-18-CO cells (b) were seeded in 100 μL of growth medium into each well of a 96 well plate. 24 h after seeding, medium in each well was exchanged and cells were treated with the corresponding drugs at different concentrations. Cell confluence was measured over 5 days. Data show representative experiments of three independent biological replicates per cell line, Data points represent mean confluence +− SD of 6 wells.
4. Discussion and conclusions
The low computational demands of the FASTCORE family [20,21,56] and the here presented rFASTCORMICS [21] allow for the building of metabolic models in a high-throughput manner on an ordinary computer. Having the same characteristics (high resolution, accuracy, and low computational demands) as the original FASTCORMICS workflow, rFASTCORMICS can segregate between cancer and control models by identifying cancer-related rewiring strategies regarding pathways, reactions, and genes. The metabolic alterations common to most cancer cells of a same type, captured by rFASTCORMICS, are not random and are sufficient to distinguish between cancer and controls cells with an accuracy above 94%.
The results showed that cancer models were overall smaller, had a more compact core metabolism, and, among the signatures, genes and reactions were more often absent. This suggests that cancer likely undergoes more loss-of-function mutations causing the inactivation of alternative pathways of the metabolic network as well as an enrichment of essential genes among the cancer metabolic core. Thus, the shut-down of pathways consuming cellular building blocks might confer a selective advantage to hepatocarcinoma cells while enabling their fast proliferation. The selective phenotype hypothesis is further supported by the heterogeneity in model sizes and lower similarity scores between cancer and control models of the same tissue.
Moreover, mutations in the tissue of origin, leading to metabolic alterations and cancer, do not completely change the metabolism of the cancer cells but allow them to sustain high proliferation rates, escape the immune system, or perform angiogenesis while still being similar to the tissue of origin. Taken together, the metabolism of cancer cells depends on the tissue of origin and the driver mutations, an observation which was also made by [57], showing that the dependency of glutamine in a given tumour depends on the tissue of origin and the oncogenic driver(s).
The rFASTCORMICS correctly predicted essential genes and known cancer drug targets with high enrichment scores. The in vitro validation of the three predicted drugs resulted in a reduced proliferation rate of the colorectal cancer cells while having no or only low effects on the control cells. This demonstrated that the models, reconstructed via rFASTCORMICS, mimic the metabolism of both cancer and healthy cells. In the future, the rFASTCORMICS metabolic models can be used for drug repurposing, which consist in finding new indications for already commercialized drugs. More specifically, even though the workflow was used to identify drugs that act alone, the workflow can also be adapted to find genes that are essential only if knocked out together through multiple in silico gene deletions. Hence, one can find drugs combinations that can be used. Further, cancer-type and patient-specific drugs can be determined if the workflow is used together with machine learning approached to identify different patient groups, which are likely to respond to drugs in a different manner.
Furthermore, in this paper, we benchmarked our workflow with different drug and cancer databases. The set of databases and web resources can quite easily be extended. Thus, the workflow could also be combined with other tools that aim to identify drugs with the same target [[58], [59], [60], [61]]. Finally, besides CRISPR-Cas9 screens, the workflow could take advantage of sh-RNA screens or si-RNA screens to validate essential genes. Furthermore, our workflow could be used to identify microRNAs, as described in [[62], [63], [64]], that play an important role in disease progression and target these microRNAs with repurposed drugs. This would require to expand the current genome-scale metabolic reconstructions by including microRNAs and their regulatory action on metabolic genes.”
Funding sources
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska- Curie grant agreement No 642295 (MEL-PLEX) and the Luxembourg National Research Fund (FNR) and the German Federal Ministry of Education and Research (BMBF) within the project MelanomSensitivity (BMBF/BM/7643621). The experimental part was supported by the Fondation Cancer (grant F1R-LSC-PAU-13HY2C). DT is supported by the Fondation du Pélican de Mie and Pierre Hippert-Faber under the aegis of the Fondation de Luxembourg. EL is supported by a CORE (C16/BM/11282028) and a POC grant (PoC18/12554295). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
MPP and TB performed the experiments. MPP and TS created FASTCORMICS RNA-seq and established the machine-learning workflow to obtain the signatures. MPP performed the analysis experiments concerning Fig. 1. TB did the single gene deletion experiments, drug target data-mining, enrichment analysis concerning essential genes and cancer drugs. TB performed the experiments concerning Fig. 2. The supplements were created by TB and MPP. TS and MPP designed the study. MPP, TB, and TS wrote the manuscript. EL and SH established the collection of primary patient material and the isolation of primary colon cancer cell lines. EL and DT planned the validation experiments. DT performed the validation experiments. DT performed the experiments concerning Fig. 3. SH, EL, and DT contributed to the revision of the manuscript draft. DK designed the study and contributed to the revision of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.04.046.
Appendix A. Supplementary data
Supplementary material
References
- 1.Warburg O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften. 1924;12(50):1131–1137. [Google Scholar]
- 2.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 3.Santos C.R., Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 4.DeBerardinis R.J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis N.E., Abdel-Haleem A.M. The evolution of genome-scale models of cancer metabolism. Front Physiol. 2013;4:237. doi: 10.3389/fphys.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duarte N.C., Becker S.A., Jamshidi N., Thiele I., Mo M.L., Vo T.D. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci. 2007;104(6):1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiele I., Swainston N., Fleming R.M.T., Hoppe A., Sahoo S., Aurich M.K. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31(5):419–425. doi: 10.1038/nbt.2488. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swainston N., Smallbone K., Hefzi H., Dobson P.D., Brewer J., Hanscho M. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 2016;12(7):1–7. doi: 10.1007/s11306-016-1051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agren R., Bordel S., Mardinoglu A., Pornputtapong N., Nookaew I., Nielsen J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput Biol. 2012;8(5):e1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardinoglu A., Agren R., Kampf C., Asplund A., Uhlen M., Nielsen J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5 doi: 10.1038/ncomms4083. [DOI] [PubMed] [Google Scholar]
- 12.Resendis-Antonio O., González-Torres C., Jaime-Munoz G., Hernandez-Patino C.E., Salgado-Munoz C.F. Seminars in cancer biology. vol. 30. Elsevier; 2015. Modeling metabolism: A window toward a comprehensive interpretation of networks in cancer; pp. 79–87. [DOI] [PubMed] [Google Scholar]
- 13.Nilsson A., Nielsen J. Genome scale metabolic modeling of cancer. Metab Eng. 2017 Sep;43(Pt B) doi: 10.1016/j.ymben.2016.10.022. (Epub 2016 Nov 4) [DOI] [PubMed] [Google Scholar]
- 14.Hyduke D.R., Lewis N.E., Palsson B.Ø. Analysis of omics data with genome-scale models of metabolism. Mol Biosyst. 2013;9(2):167–174. doi: 10.1039/c2mb25453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folger O., Jerby L., Frezza C., Gottlieb E., Ruppin E., Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7(1):501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agren R., Mardinoglu A., Asplund A., Kampf C., Uhlen M., Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol Syst Biol. 2014;10(3):721. doi: 10.1002/msb.145122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahadevan R., Edwards J.S., Doyle F.J., III Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J. 2002;83(3):1331–1340. doi: 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mardinoglu A., Agren R., Kampf C., Asplund A., Nookaew I., Jacobson P. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol Syst Biol. 2013;9(1):649. doi: 10.1038/msb.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pires Pacheco M., Pfau T., Sauter T. Benchmarking procedures for high-throughput context specific reconstruction algorithms. Front Physiol. 2016;6(410) doi: 10.3389/fphys.2015.00410. http://www.frontiersin.org/systems_biology/10.3389/fphys.2015.00410/abstract Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vlassis N., Pacheco M.P., Sauter T. Fast reconstruction of compact context-specific metabolic network models. PLoS Comput Biol. 2014 Jan;10(1):e1003424. doi: 10.1371/journal.pcbi.1003424. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pacheco M.P., John E., Kaoma T., Heinäniemi M., Nicot N., Vallar L. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genomics. 2015;16(1):809. doi: 10.1186/s12864-015-1984-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Eddy J.A., Price N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst Biol. 2012;6(1):1. doi: 10.1186/1752-0509-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Estévez S.R., Nikoloski Z. Context-specific metabolic model extraction based on regularized least squares optimization. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0131875. e0131875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee D., Smallbone K., Dunn W.B., Murabito E., Winder C.L., Kell D.B. Improving metabolic flux predictions using absolute gene expression data. BMC Syst Biol. 2012;6(1):73. doi: 10.1186/1752-0509-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahman M., Jackson L.K., Johnson W.E., Li D.Y., Bild A.H., Piccolo S.R. Alternative preprocessing of RNA-sequencing data in the Cancer genome atlas leads to improved analysis results. Bioinformatics. 2015;31(22):3666–3672. doi: 10.1093/bioinformatics/btv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J. Identification and characterization of essential genes in the human genome. Science (New York, NY) 2015 nov;350(6264):1096–1101. doi: 10.1088/1751-8113/44/8/085201. http://science.sciencemag.org/content/350/6264/1096.short%5Cnpapers3://publication/doi/10.1126/science.aac7557 http://www.sciencemag.org/cgi/doi/10.1126/science.aac7041 http://arxiv.org/abs/1011.1669 http://www.ncbi.nlm.nih.gov/pubmed/26472758 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4662922 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163(6):1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 28.Opdam S., Richelle A., Kellman B., Li S., Zielinski D.C., Lewis N.E. A systematic evaluation of methods for tailoring genome-scale metabolic models. Cell Syst. 2017;4(3):318–329. doi: 10.1016/j.cels.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machado D., Herrgård M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput Biol. 2014;10(4) doi: 10.1371/journal.pcbi.1003580. e1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hart T., Komori H.K., LaMere S., Podshivalova K., Salomon D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics. 2013;14(1):1–7. doi: 10.1186/1471-2164-14-778. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams C.K., Engelhardt A., Cooper T., Mayer Z., Ziem A., Scrucca L. 2016. Package ‘caret’. [Google Scholar]
- 32.Chung I.F., Chen C.Y., Su S.C., Li C.Y., Wu K.J., Wang H.W. DriverDBv2: a database for human cancer driver gene research. Nucleic Acids Res. 2016;44(D1):D975–D979. doi: 10.1093/nar/gkv1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rykunov D., Beckmann N.D., Li H., Uzilov A., Schadt E.E., Reva B. A new molecular signature method for prediction of driver cancer pathways from transcriptional data. Nucleic Acids Res. 2016;44(11) doi: 10.1093/nar/gkw269. e110–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D. TCGAbiolinks: an R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016 May 5;44(8):e71. doi: 10.1093/nar/gkv1507. (Epub 2015 Dec 23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heyn H., Vidal E., Ferreira H.J., Vizoso M., Sayols S., Gomez A. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17(1):11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blomen V.A., Majek P., Jae L.T., Bigenzahn J.W., Nieuwenhuis J., Staring J. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015 nov;350(6264):1092–1096. doi: 10.1126/science.aac7557. http://www.sciencemag.org/cgi/doi/10.1126/science.aac7557%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/26472760 http://www.sciencemag.org/cgi/doi/10.1126/science.aac7557 Available from: [DOI] [PubMed] [Google Scholar]
- 37.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42(D1):D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pacheco M.P., Pfau T., Sauter T. Benchmarking procedures for high-throughput context specific reconstruction algorithms. Front Physiol. 2016;6:410. doi: 10.3389/fphys.2015.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H., Wang H., Sinz M., Zoeckler M., Staudinger J., Redinbo M. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26(2):258. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Barcons L., Maurer B.J., Kang M.H., Reynolds C.P. P450 inhibitor ketoconazole increased the intratumor drug levels and antitumor activity of fenretinide in human neuroblastoma xenograft models. Int J Cancer. 2017;141(2):405–413. doi: 10.1002/ijc.30706. [DOI] [PubMed] [Google Scholar]
- 41.Agarwal S.K., Salem A.H., Danilov A.V., Hu B., Puvvada S., Gutierrez M. Effect of ketoconazole, a strong CYP3A inhibitor, on the pharmacokinetics of venetoclax, a BCL-2 inhibitor, in patients with non-Hodgkin lymphoma. Br J Clin Pharmacol. 2017;83(4):846–854. doi: 10.1111/bcp.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forgue-Lafitte M.E., Coudray A.M., Fagot D., Mester J. Effects of ketoconazole on the proliferation and cell cycle of human cancer cell lines. Cancer Res. 1992;52(24):6827–6831. [PubMed] [Google Scholar]
- 43.Gupta A.K., Ryder J.E., Cooper E.A. Naftifine: a review. J Cutan Med Surg. 2008;12(2):51–58. doi: 10.2310/7750.2008.06009. [DOI] [PubMed] [Google Scholar]
- 44.Ryder N.S. Specific inhibition of fungal sterol biosynthesis by SF 86-327, a new allylamine antimycotic agent. Antimicrob Agents Chemother. 1985;27(2):252–256. doi: 10.1128/aac.27.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown D.N., Caffa I., Cirmena G., Piras D., Garuti A., Gallo M. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci Rep. 2016;6 doi: 10.1038/srep19435. 19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmeel L.C., Schmeel F.C., Blaum-Feder S., Schmidt-Wolf I.G. In vitro efficacy of naftifine against lymphoma and multiple myeloma. Anticancer Res. 2015;35(11):5921–5926. [PubMed] [Google Scholar]
- 47.Nguyen B.C.Q., Tawata S. The chemistry and biological activities of Mimosine: a review. Phytother Res. 2016;30(8):1230–1242. doi: 10.1002/ptr.5636. [DOI] [PubMed] [Google Scholar]
- 48.Qiao S., Murakami K., Zhao Q., Wang B., Seo H., Yamashita H. Mimosine-induced apoptosis in C6 glioma cells requires the release of mitochondria-derived reactive oxygen species and p38, JNK activation. Neurochem Res. 2012;37(2):417–427. doi: 10.1007/s11064-011-0628-6. [DOI] [PubMed] [Google Scholar]
- 49.Kubens B.S., Niggemann B., Zänker K.S. Prevention of entrance into G2 cell cycle phase by mimosine decreases locomotion of cells from the tumor cell line SW480. Cancer Lett. 2001;162:S39–S47. doi: 10.1016/s0304-3835(00)00651-0. [DOI] [PubMed] [Google Scholar]
- 50.Qureshi-Baig K., Ullmann P., Rodriguez F., Frasquilho S., Nazarov P.V., Haan S. What do we learn from spheroid culture systems? Insights from tumorspheres derived from primary colon cancer tissue. PLoS One. 2016;11(1) doi: 10.1371/journal.pone.0146052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S. The Cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lane A.N., Fan T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015 Feb 27;43(4):2466–2485. doi: 10.1093/nar/gkv047. (Published online 2015 Jan 27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kridel S.J., Axelrod F., Rozenkrantz N., Smith J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64(6):2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 54.Menendez J., Vellon L. Lupu R. Antitumoral actions of the anti-obesity drug orlistat (Xenical™) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene. Ann Oncol. 2005;16(8):1253–1267. doi: 10.1093/annonc/mdi239. [DOI] [PubMed] [Google Scholar]
- 55.Menendez J.A., Vellon L., Mehmi I., Oza B.P., Ropero S., Colomer R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A. 2004;101(29):10715–10720. doi: 10.1073/pnas.0403390101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pacheco M.P., Sauter T. Springer; 2018. The FASTCORE family: For the fast reconstruction of compact context-specific metabolic networks models. Metabolic network reconstruction and modeling; pp. 101–110. [DOI] [PubMed] [Google Scholar]
- 57.Yuneva M.O., Fan T.W., Allen T.D., Higashi R.M., Ferraris D.V., Tsukamoto T. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X., Yan C.C., Zhang X., Zhang X., Dai F., Yin J. Drug–target interaction prediction: databases, web servers and computational models. Brief Bioinform. 2015;17(4):696–712. doi: 10.1093/bib/bbv066. [DOI] [PubMed] [Google Scholar]
- 59.Keiser M.J., Setola V., Irwin J.J., Laggner C., Abbas A.I., Hufeisen S.J. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y., Zeng J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics. 2013;29(13):i126–i134. doi: 10.1093/bioinformatics/btt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X., Liu M.X., Yan G.Y. Drug–target interaction prediction by random walk on the heterogeneous network. Mol Biosyst. 2012;8(7):1970–1978. doi: 10.1039/c2mb00002d. [DOI] [PubMed] [Google Scholar]
- 62.Chen X., Guan N.N., Sun Y.Z., Li J.Q., Qu J. Vol. 16. Brief Bioinform Published online October; 2018. MicroRNA-small molecule association identification: from experimental results to computational models; p. 2018. [DOI] [PubMed] [Google Scholar]
- 63.Qu J., Chen X., Sun Y.Z., Li J.Q., Ming Z. Inferring potential small molecule–miRNA association based on triple layer heterogeneous network. J Chem. 2018;10(1):30. doi: 10.1186/s13321-018-0284-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen X., Sun Y.Z., Zhang D.H., Li J.Q., Yan G.Y., An J.Y. NRDTD: a database for clinically or experimentally supported non-coding RNAs and drug targets associations. Database. 2017:2017. doi: 10.1093/database/bax057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material