

Abstract
The trkB family of transmembrane proteins serves as receptors for BDNF and NT-4/5. The family is composed of a tyrosine kinase-containing isoform as well as several alternatively spliced “truncated receptors” with identical extracellular ligand-binding domains but very small intracellular domains. The two best-characterized truncated trkB receptors, designated as trkB.T1 and trkB.T2, contain intracellular domains of only 23 and 21 amino acids, respectively. Although it is known that the tyrosine kinase isoform (trkB.FL) is capable of initiating BDNF and NT-4/5-induced signal transduction, the functional role or roles of the truncated receptors remain enigmatic. At the same time, the potential importance of the truncated receptors in the development, maintenance, and regeneration of the nervous system has been highlighted by recent developmental and injury paradigm investigations. Here we have used trkB cDNA transfected cell lines to demonstrate that both trkB.T1 and trkB.T2 are capable of mediating BDNF-induced signal transduction. More specifically, BDNF activation of either trkB.T1 or trkB.T2 increases the rate of acidic metabolite release from the cell, a common physiological consequence of many signaling pathways. Further, these trkB.T1- and trkB.T2-mediated changes occur with kinetics distinct from changes mediated by trkB.FL, suggesting the participation of at least some unique rate-limiting component or components. Mutational analysis demonstrates that the isoform-specific sequences within the intracellular domains of each receptor are essential for signaling capability. Finally, inhibitor studies suggest that kinases are likely to be involved in the trkB.T1 and trkB.T2 signaling pathways.
Keywords: trkB, BDNF, NT4/5, neurotrophin, truncated receptors, signal transduction
The neurotrophin family of neurotrophic factors, composed of NGF, BDNF, NT3, NT4/5 and NT6, is essential for the proper development and maintenance of the nervous system. Classically known to serve as target-derived trophic factors regulating naturally occurring neuronal cell death during development, recent work has shown that neurotrophins also act during neurogenesis, neuronal differentiation, and pathfinding, as well as influencing synaptic efficiency (for review, see Davies, 1994, 1996; Snider, 1994).
The trk family of transmembrane receptors plays fundamental roles in mediating neurotrophin action (Kaplan et al., 1991a,b; Klein et al., 1991a; Jing et al., 1992; Meakin et al., 1992). Alternative RNA splicing generates three well characterized trkB isoforms that bind BDNF and NT-4/5 (see Fig. 1). The trkB.FL isoform is a receptor tyrosine kinase with an extracellular ligand-binding domain, a single transmembrane domain, and a typical tyrosine kinase-containing intracellular domain (Berkemeier et al., 1991; Klein et al., 1991b,1992; Soppet et al., 1991; Squinto et al., 1991). Two other trkB isoforms (trkB.T1 and trkB.T2) possess the same extracellular and transmembrane domains as trkB.FL but with “truncated” intracellular domains (23 and 21 amino acids, respectively) lacking the kinase domain (Klein, 1990; Middlemas et al., 1991). The first 12 intracellular amino acids of both trkB.T1 and trkB.T2 are common with trkB.FL. The remaining 11 and 9 residues, respectively, are isoform-specific and lack obvious homology to any known protein motifs.
Fig. 1.

Schematic diagram of wild-type and carboxy end-truncated trkB receptors used in this study.
Although trkB.FL is capable of mediating ligand-induced signal transduction via a tyrosine kinase-activated cascade (Berkemeier et al., 1991; Klein et al., 1991b, 1992; Soppet et al., 1991; Squinto et al., 1991), little is known about truncated trkB receptor function. However, several observations suggest an important role or roles. First, the intracellular domain of trkB.T1 is 100% conserved among humans, mice, rats, and felines (Klein et al., 1990; Middlemas et al., 1991; Shelton et al., 1995; B. Hinkle, V. Makrides, M. Radeke, S. Feinstein, unpublished data). Second, although initial studies suggested that trkB.T1 is expressed solely in non-neuronal cells, more recent work reveals high levels of trkB.T1 expression in both neurons and glia, including mature motor neurons, developing cerebellar granule neurons (Armanini et al., 1995), and trigeminal neurons (Ninkina et al., 1996). Third, although trkB.FL is the predominant trkB isoform expressed during embryogenesis, trkB.T1 is the major isoform expressed in adult brain (Escandón et al., 1993, 1994; Allendoerfer et al., 1994; Armanini et al., 1995). Fourth, trkB.T1 expression is induced markedly at the time of axon arrival at targets (Escandón et al., 1993, 1994; Allendoerfer et al., 1994; Armanini et al., 1995; Ninkina et al., 1996) and in hippocampal glia after lesion of the fimbria–fornix (Beck et al., 1993). Although much less is known about trkB.T2, recent in situ hybridization studies show trkB.T2 mRNA expression in brain neurons (Armanini et al., 1995).
Several models have been proposed for truncated trkB action. Recent heterologous expression studies examining coexpression of truncated and full-length trkB cDNAs in Xenopus laevis embryos (Eide et al., 1996) and sympathetic neurons (Ninkina et al., 1996) have shown that coexpression of truncated trkB receptors with trkB.FL reduced the ability of trkB.FL to induce particular BDNF-dependent events in a truncated trkB:trkB.FL ratio-dependent manner. Taken together with recent work demonstrating that these receptors are coexpressed in at least some neurons, the suggestion is that the truncated receptors can act as naturally occurring dominant negative elements in cells coexpressing trkB.FL. Another model suggests that truncated receptors might act as “sponges” to soak up excess ligand and act as a barrier to diffusion (Biffo et al., 1995). Yet another model suggests that they might serve to bind ligand reversibly in the extracellular space, thereby maintaining an elevated local concentration of ligand for use by nearby trkB.FL receptors (Beck et al., 1993).
On the other hand, it is difficult to reconcile any of these relatively passive roles with either the extraordinarily high degree of intracellular domain sequence conservation or the fact that multiple truncated isoforms even exist, neither of which would be predicted by any of these models. Might the truncated trkB receptors serve additional functions?
In this study we have tested an alternative hypothesis, that is, that the truncated trkB isoforms trkB.T1 and trkB.T2 are capable of mediating ligand-dependent signal transduction. Previous investigations designed to test this hypothesis have used responses characteristic of known signaling pathways to assay for signaling capability, such as ligand-mediated induction of immediate early genes, calcium efflux, mitogenesis, neurite outgrowth, or survival. Using these assays, investigators found no evidence to support a ligand-dependent signaling role for trkB.T1 or trkB.T2 (Biffo et al., 1995; Roback et al., 1995;Eide et al., 1996; Ninkuna et al., 1996). In contrast, we hypothesize that the truncated trkB receptor isoforms signal via a presently unknown signaling pathway. To test this version of the hypothesis, we have used an extremely general yet well established physiological assay for signal transduction–ligand-inducible release of acidic metabolites. With the use of this assay, our data demonstrate that both trkB.T1 and trkB.T2 are capable of mediating ligand-dependent signal transduction and, further, that their short isoform-specific intracellular sequences are required for that capability.
MATERIALS AND METHODS
Generation of trkB cDNA transfected cell lines. All trkB cDNAs were of rat origin except for the trkB.T1 cDNA, which was isolated from a human cerebellum cDNA library. However, the intracellular domains of trkB.T1 are conserved 100% among human, rat, mouse, and feline, and the extracellular domains are very highly conserved (Klein et al., 1990; Middlemas et al., 1991; Shelton et al., 1995; our unpublished data). The trkB.FL and trkB.T2 cDNAs were generous gifts from D. Middlemas and T. Hunter (The Salk Institute La Jolla, CA; Middlemas et al., 1991). Mutant cDNAs encoding trkBΔ435 and trkBΔ423 were generated by PCR, using rat trkB.FL cDNA as a template. The upstream primer in both cases was 5′ GGACCCGCCATGGCGCGGCT 3′. The trkBΔ435 downstream primer was 5′ CTATCAACCTTTCATGCCAAACTTGG 3′, and the trkBΔ423 downstream primer was 5′ CTATCACTTGAGCAGAAGCAGCATC 3′. Both downstream primers encode two in-frame stop codons at the desired site of truncation. Both mutant cDNAs were sequenced in their entirety on both strands to ensure that no spurious mutations had occurred during PCR amplification. cDNAs encoding trkB.FL, trkB.T1, trkBΔ435, and trkBΔ423 were ligated into pBJ5 (Elliot et al., 1990) and then introduced into Ltk−cells by calcium phosphate cotransfection along with pCHtk (Radeke et al., 1987). Stable clones were selected with hypoxanthine-aminopterin-thymidine (HAT)-containing media. The cDNA encoding trkB.T2 was ligated into pBABEpuro (Morgenstern and Land, 1990) and introduced into Ltk− cells by calcium phosphate transfection (Radeke et al., 1987). Stable clones were selected with puromycin-containing media. To control for any possible effect of the puromycin selection on cell signaling capability, we subsequently transfected stable transfectants selected in HAT with pBABEpuro vector lacking any cDNA insert and clonally selected them for resistance to puromycin.
Routine cell culture maintenance was in DMEM supplemented with 5% horse serum (Gemini Bio-Products, Calabasas, CA) and 5% supplemented calf serum (Gemini Bio-Products).
Cell staining with biotinylated BDNF. BDNF was biotinylated commercially by Vector Laboratories (Burlingame, CA) and was a generous gift from Geoff Lewis and Steve Fisher (UCSB). Cells were plated on poly-d-lysine-coated coverslips and then incubated with 20 ng/ml biotin-BDNF in HEPES–Kreb’s–Ringer’s solution (HKR) containing (in mm): 10 HEPES, 137 NaCl, 4.7 KCl, 2.6 CaCl2, 1.2 KH2PO4, and 1.2 MgSO4, pH 7.4, plus 0.02% azide and 0.1% BSA for 1 hr at 4°C. Cells were washed twice with HKR at 4°C and fixed with 4% paraformaldehyde in 100 mm Na cacodylate, pH 7, for 20 min, again at 4°C. Next, cells were rinsed three times with HKR and once with Tris saline (20 mm Tris-HCl and 150 mmNaCl, pH 7.4). Bound biotin-BDNF was detected with streptavidin–Cy3 (Jackson Laboratories, Bar Harbor, ME) on a Bio-Rad (Richmond, CA) 1024 laser scanning confocal microscope.
BDNF Cross-linking. Cells were grown to 50–75% confluency on 150 mm tissue culture dishes. Cells were washed twice with 10 mm HEPES and 150 mm NaCl, pH 7.4, and harvested in 10 mm HEPES, 150 mm NaCl, and 1 mm EDTA, pH 7.4. The optical density of the cell suspension at 600 nm was determined, cells were pelleted at 500 ×g for 5 min at 4°C, and then the cells were resuspended in 4°C cross-linking buffer containing (in mm): 10 HEPES, 150 NaCl, 1 MgCl2, 1 CaCl2, and 0.1% BSA) at an A600 per ml = 3. Next, 500 μl of the resulting cell suspension was added to 500 μl of 20 nm125I-BDNF (prepared as described in Radeke and Feinstein, 1991) in cross-linking buffer and incubated at 4°C for 60 min, either plus or minus 10 μg/ml unlabeled BDNF. Then cells were pelleted at 500 × g for 5 min at 4°C and resuspended in 500 μl of cross-linking buffer lacking BSA; an equal volume of 8 mm EDAC [1,ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride] (Sigma, St. Louis, MO) in cross-linking buffer was added. After a 10 min incubation at 4°C, cells were washed three times in cross-linking buffer lacking BSA and then lysed in 500 μl of lysis buffer (1% Triton X-100, 20 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1 mm PMSF, 2.5 μg/ml pepstatin, and 2.5 μg/ml leupeptin, pH 7.4) at 37°C. Immediately after lysis, the lysate was chilled on ice and then centrifuged at 10,000 × g for 10 min at 4°C. The resulting supernatant was incubated with 20 μl of wheat germ agglutinin (WGA)–agarose slurry (Sigma) for 60 min at 4°C. The WGA–agarose was washed three times at 4°C with lysis buffer, and bound proteins were eluted with 2× gel sample buffer plus 5% β-mercaptoethanol. Samples were fractionated on a 7% SDS-polyacrylamide gel and detected by fluorography.
c-fos Immunoblotting. Subconfluent log-phase cells were cultured in serum-free DMEM containing 0.1% endotoxin-free BSA for 16–18 hr, treated with 50 ng/ml BDNF or NGF (as indicated) for an additional 1 hr, and then lysed in 1% Triton, 20 mmTris-HCl, 150 mm NaCl, 1 mm EDTA, 2.5 μg/ml leupeptin, 2.5 μg/ml pepstatin, and 1 mm PMSF, pH 7.4, at 4°C for 10 min. Nuclei were isolated by low-speed centrifugation (500 × g, 5 min, 4°C) and resuspended in 2× protein sample buffer; the DNA was sheared mechanically with a 25 gauge needle. Nuclear protein (20 μg), as determined by the Amido–Schwartz assay of Schaffner and Weissmann (1973), was fractionated on a reducing 7.5% SDS-polyacrylamide gel and transferred to nitrocellulose. Nonspecific antibody binding was blocked by incubating the membrane in 5% Blotto (5% nonfat milk, 0.1% Tween-20, 50 mm Tris, and 150 mm NaCl, pH 7.4). c-fos protein was detected with 0.1 μg/ml SC-52, an affinity-purified rabbit anti-fos polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibody was visualized by HRP-conjugated goat anti-rabbit IgG (Bio-Rad), followed by enhanced chemiluminescence (Pierce, Rockford, IL).
Microphysiometric assays. Cells were harvested at ∼75% confluency and seeded onto 3 μm polycarbonate microporous transwells (Molecular Devices, Sunnyvale, CA) at a density of 5 × 105 cells/ml. Then cells were serum-starved for 16–18 hr in DMEM containing 0.1% endotoxin-free BSA and 10,000 U/ml each of penicillin and streptomycin (Life Technologies, Gaithersburg, MD). After serum starvation, the transwells were transferred to flow chambers of a Cytosensor microphysiometer (Molecular Devices), as described in Baxter et al. (1992). Cells were perfused with low phosphate-buffered RPMI-1640 medium with a pump cycle of 150 sec, composed of a flow-on period (100 μl/min for 110 sec) followed by a flow-off period (40 sec). Extracellular acidification rate was determined from the slope of a linear least-squares fit during the central 30 sec of each flow-off period. Acidification rate data were normalized mathematically to 100% at the data point just before BDNF exposure. BDNF was used at 50 ng/ml.
RESULTS
Transfected cells expressing trkB.T1 or trkB.T2 fail to induce c-fos in response to BDNF administration
To test the hypothesis that the truncated trkB isoforms trkB.T1 and trkB.T2 are capable of ligand-dependent signal transduction, we generated stably transfected clonal L-cell lines expressing trkB.FL, trkB.T1, or trkB.T2. Two sets of analyses, one microscopic and one biochemical, were performed to assess trkB receptor surface expression on each of these cell lines. First, cells on coverslips were treated with biotinylated BDNF, fixed, stained with streptavidin–Cy3, and viewed by confocal microscopy. Figure 2 presents a projection series of 0.5 μm optical sections of nonpermeabilized representative cells for each of the transfected cell lines and nontransfected control cells, providing a pseudo-three-dimensional view of the trkB expression on each cell surface. The images show that transfected cells, but not the nontransfected host cell line, exhibit BDNF binding sites on their surfaces. Addition of excess unlabeled BDNF resulted in a complete loss of labeling (data not shown). Although there appears to be a concentration of receptors at endfeet-like structures, rotation of the images reveals that this results from extensive membrane ruffling associated with these structures. Additionally, the distribution pattern of receptors on the cell surfaces is indistinguishable among the different transfected cell lines.
Fig. 2.
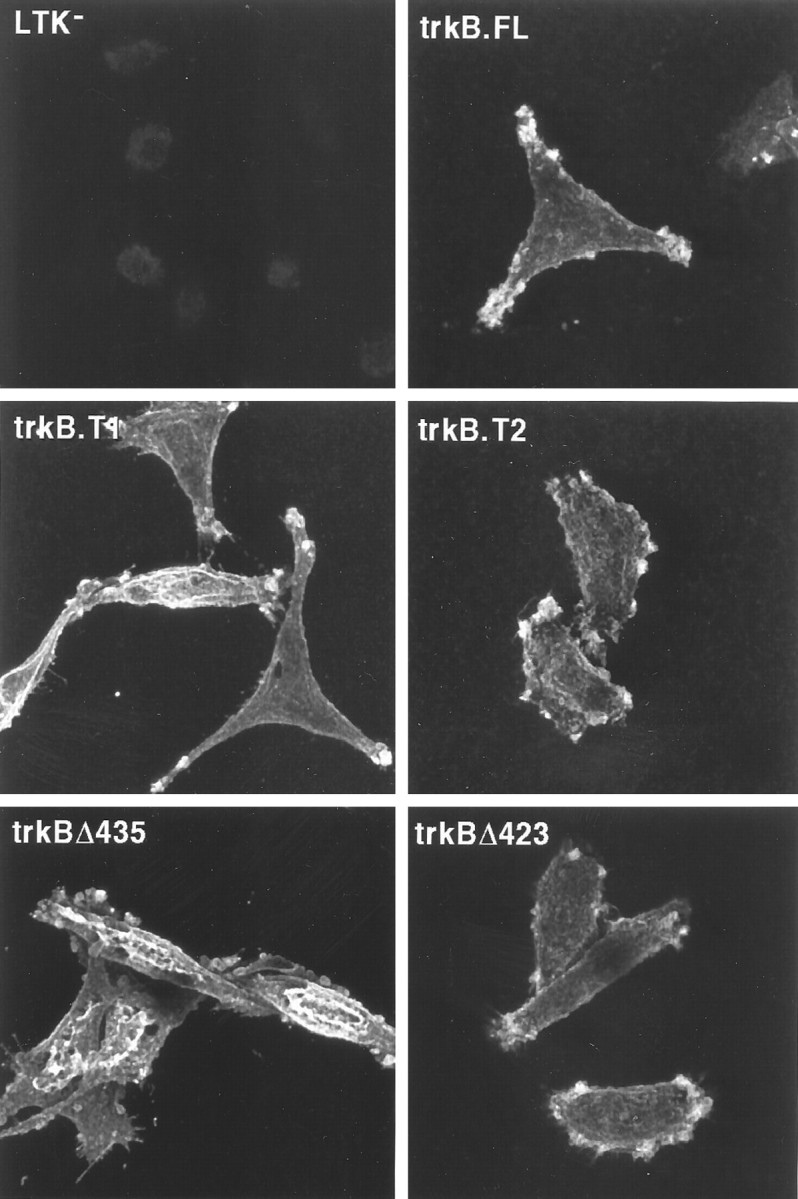
Distribution of BDNF binding on the plasma membranes of wild-type (trkB.FL, trkB.T1, trkB.T2) and mutant (trkBΔ435 and trkBΔ423) trkB receptors in clonal, stably transfected L-cells: confocal microscopy. Transfected and nontransfected control cells were treated on coverslips with biotinylated BDNF, fixed, treated with streptavidin–Cy3, and viewed by laser scanning confocal microscopy. A Z-series of 0.5 μm optical sections encompassing the entire thickness of the cells was acquired; images presented here are a projection of all of the optical sections. Laser strength was 3% for trkB.T1, trkBΔ435, and trkBΔ423 transfected cells, and 10% for trkB.T2 and trkB.FLtransfected cells and for nontransfected control cells.
Second, cells were exposed to 125I-BDNF for 1 hr at 4°C and then treated with the covalent cross-linker EDAC. Then cells were lysed in a Triton X-100 buffer, and the lysate was enriched for trkB receptors by WGA affinity chromatography. This WGA eluate was fractionated by SDS-PAGE, and proteins covalently coupled to125I-BDNF were visualized by fluorography. As seen in Figure 3, each transfected cell line expresses specific radiolabeled complexes corresponding to the correct size for each trkB isoform–125I-BDNF complex. Simultaneous addition of an excess of unlabeled BDNF eliminated formation of the radiolabeled complex (data not shown). In addition, both immunoblotting and BDNF binding studies confirmed that each of these cell lines expresses trkB receptors (data not shown).
Fig. 3.

Biochemical characterization of cell-surface BDNF receptors on cells expressing wild-type (trkB.FL, trkB.T1, trkB.T2) and mutant (trkBΔ435 andtrkBΔ423) trkB receptors in clonal, stably transfected L-cells: 125I-BDNF cross-linking. Transfected and nontransfected control cells were treated with 125I-BDNF, cross-linked with EDAC, and lysed with Triton X-100 containing buffer. Then the lysate was enriched for trkB receptors by wheat germ agglutinin chromatography, fractionated by SDS-PAGE, and visualized by fluorography. Exposure times were 8 hr for trkB.T1 andtrkBΔ435 transfected cells, 3 d fortrkBΔ423 transfected cells, and 2 weeks fortrkB.T2 and trkB.FL transfected cells and for nontransfected control cells. (The x-ray film was not preflashed, which exaggerates the differences between stronger and weaker signals.)
As an initial test of signaling capability, we analyzed each transfected cell line and nontransfected control cells for their ability to induce a common immediate early gene product, c-fos, in response to BDNF administration. Consistent with expectations based on previous trkB.FL studies (Marsh et al., 1993; Nakagawara et al., 1994) and other receptor tyrosine kinases (Cochran et al., 1984; Kruijer et al., 1984, 1985; Muller et al., 1984; Bravo et al., 1985; Greenberg et al., 1985; Milbrandt, 1986; Stumpo and Blackshear, 1986), trkB.FL transfectants treated with BDNF exhibited a rapid induction of c-fos protein expression comparable to that exhibited by NGF-treated PC12 cells (Fig. 4). Nontransfected control cells failed to induce c-fos expression. Most importantly, no induction of c-fos was observed with either trkB.T1 or trkB.T2 transfected cells treated with BDNF. We also examined another common receptor tyrosine kinase-mediated response, the induction of ornithine decarboxylase activity (Greene et al., 1978; Guroff et al., 1981; Feinstein et al., 1985). Again, trkB.FL transfectants, but not trkB.T1 or trkB.T2 transfectants, responded to BDNF treatment (data not shown).
Fig. 4.

trkB.FL transfectants, but nottrkB.T1, trkB.T2, trkBΔ435, ortrkBΔ423 transfectants, induce c-fos expression in response to BDNF administration. Cells were treated with 50 ng/ml recombinant BDNF for the indicated amounts of time; nuclei were isolated and immunoblotted with an anti-c-fos probe (Santa Cruz Biotechnology SC-52). The arrow marks the migration of the 62 kDa c-fos protein. PC12 cells were treated with 50 ng/ml NGF instead of BDNF, because they express trkA, but not trkB, receptors.
BDNF signal transduction by trkB.T1 and trkB.T2: microphysiometric assays
Because the intracellular domains of trkB.T1 and trkB.T2 bear no sequence resemblance to a tyrosine kinase domain, it is not surprising that they were unable to mediate characteristic tyrosine kinase signaling events. In fact, the intracellular domains of trkB.T1 and trkB.T2 are not reminiscent of any sequence motif known to be involved in signaling. Therefore, we next sought a means to test the ability of each transfectant to mediate ligand-induced signal transduction that was not dependent on previous knowledge of the specific signal transduction mechanism or endpoint. For this purpose, we used a Cytosensor microphysiometer, an instrument based on a pH-sensitive, light-addressable potentiometric sensor (LAPS; Owicki et al., 1990;McConnell et al., 1992; Owicki and Parce, 1992). This instrument detects ligand-induced receptor activation and downstream signaling mechanisms by monitoring the rate of extracellular media acidification. This acidification is a consequence of the energy-producing metabolic pathways, glycolysis and aerobic respiration, which produce protons via formation and release of the acidic byproducts lactic acid and CO2, respectively. To a first approximation, the extracellular acidification rate represents the sum of cellular glycolytic and respiratory activity and is therefore a measure of cellular metabolic activity (Owicki and Parce, 1992). In addition, in some instances it is likely that activation of specific H+antiporters contributes to the change in the rate of media acidification (Wada et al., 1993; Baxter et al., 1994). Most important for this investigation, receptor activation has been shown to alter the rate at which cells release acidic metabolites in a large number of signaling pathways. Indeed, microphysiometry has been used in studies of CNTF signaling in SH-SY5Y cells (Johnson et al., 1994), kainic acid effects on hippocampal cells (Raley-Susman et al., 1992), NGF:trkA interactions (Pitchford et al., 1995), D1 and D2 dopamine receptor action (Neve et al., 1992; Bouvier et al., 1993), m1 and m3 muscarinic receptor action (Baxter et al., 1994), ACh action on the Na+/K+ ATPase (Miller et al., 1993), PKC action (Omary et al., 1992), cAMP effects on the H+/K+ATPase (Baxter et al., 1992; Thibodeau et al., 1994), glucocorticoid action (Redish et al., 1993), the role of PKC ε on granulocyte-macrophage–colony-stimulating factor (GM–CSF) action (Wada et al., 1993), angiotensin II action (Dickinson et al., 1994), and CC-chemokine receptor action (Samson et al., 1996). The microphysiometer also has been used recently to examine the interactions between synthetic peptides and cognate receptors in an effort to design effective competitive inhibitors (Renschler et al., 1995). Thus, microphysiometry has become an extremely valuable tool to address important questions in many different signaling systems.
The microphysiometer readily detected BDNF-induced signal transduction by trkB.FL receptors (Fig. 5), revealing a kinetic profile essentially identical to NGF stimulation of trkA and other ligand–receptor tyrosine kinase signaling systems (Parce et al., 1989;Baxter et al., 1992; McConnell et al.; 1992; Pitchford et al., 1995). Nontransfected control cells did not respond to BDNF. The most intriguing results were that BDNF treatment of both the trkB.T1 and trkB.T2 transfected cells induced significant changes in their respective rates of extracellular acidification. All of these BDNF effects were concentration-dependent and saturable (data not shown). Finally, NT4/5, another ligand known to bind to the trkB receptors (Berkemeier et al., 1991), produced similar responses as BDNF (data not shown).
Fig. 5.

trkB.T1 and trkB.T2are both capable of mediating BDNF-dependent signal transduction requiring their respective intracellular domains. Transfected cell lines were treated with 50 ng/ml BDNF at time t = 0, and the rate of extracellular media acidification was monitored with a Cytosensor microphysiometer. Data are presented as the percentage of increase in acidification rate over initial basal activity before treatment with BDNF.
These data make several points. First, the data demonstrate that trkB.T1 and trkB.T2 are capable of mediating ligand-induced changes in cell physiology, i.e., signal transduction. Moreover, in contrast to the rapid burst and plateau kinetics of the trkB.FL receptors and other tyrosine kinases, the trkB.T1 and trkB.T2 receptors exhibited more gradual kinetics in their rates of media acidification. These observations suggest that at least some features of the trkB.T1 and trkB.T2 signaling pathways must be distinct from those used by trkB.FL.
The intracellular domains of trkB.T1 and trkB.T2 are necessary for their signaling capabilities
To begin assessing the molecular basis of trkB.T1 and trkB.T2 signaling, we next investigated the necessity of the intracellular domain of each receptor for signaling capability. We generated two clonal stably transfected cell lines expressing trkB deletion mutants, as schematized in Figure 1. Both possess the entire extracellular and transmembrane domains of trkB. However, trkBΔ435 possesses only the first 12 intracellular amino acids (common to all trkB isoforms), whereas trkBΔ423 contains only a single lysine on the intracellular domain. Microscopic (Fig. 2) and biochemical cross-linking (Fig. 3) analyses confirmed expression of these mutant trkB receptors on the plasma membranes, as did ligand-binding assays (data not shown). Microphysiometric analyses of both the trkBΔ423 and trkBΔ435 cell lines demonstrated that these deletion mutants were unable to signal in response to BDNF (Fig. 4). Thus, the unique carboxyl tails of trkB.T1 and trkB.T2, only 11 and 9 amino acids long, respectively, are necessary for signal transduction by these receptors.
Phosphorylation is likely to be involved in the trkB.T1 and trkB.T2 signal transduction mechanisms
To investigate further the signaling mechanism or mechanisms used by trkB.T1 and trkB.T2, we tested their signaling abilities in the presence of the kinase inhibitors K252a and staurosporine. K252a is known to inhibit signaling by the tyrosine kinase isoforms of the trk family of receptors (Berg et al., 1992; Ohmichi et al., 1992). Staurosporine is a more general kinase inhibitor (Tapley et al., 1992;Roback et al., 1995). As seen in Figure6A and B, staurosporine completely blocks signaling by both trkB.T1 and trkB.T2. On the other hand, K252a diminishes and delays, but does not eliminate, signaling by both receptors. In both cases the responses were dose-dependent with respect to inhibitor concentration (data not shown). Further, parallel experiments with the trkB.FL transfectants demonstrated that K252a and staurosporine decreased tyrosine phosphorylation of trkB.FL by ∼90 and ∼99%, respectively, at the concentrations used for the data presented in Figure 5 (data not shown). These are relatively high doses that are likely to inhibit multiple kinases. K252a or staurosporine alone had no effect on the rate of acidic metabolite release (data not shown). Taken together, these data suggest that protein kinases are likely to be involved at some point in the trkB.T1 and trkB.T2 signaling pathways.
Fig. 6.

Effects of K252a and staurosporine on trkB.T1 and trkB.T2 signaling capability. A, TrkB.T1 transfected cell lines were pretreated for 20 min with either no inhibitor, 100 nm K252a, or 200 nm staurosporine and then treated simultaneously with the same inhibitor plus 50 ng/ml BDNF at time t = 0 and analyzed as in Figure 5.B, TrkB.T2 transfected cell lines were pretreated for 20 min with either no inhibitor, 100 nm K252a, or 200 nm staurosporine and then treated simultaneously with the same inhibitor plus 50 ng/ml BDNF at time t = 0 and analyzed as in Figure 5. All exposures to BDNF or BDNF plus inhibitor were for the duration of the experiment. For both the staurosporine and K252a experiments, these are relatively high concentrations that likely inhibit multiple kinases.
DISCUSSION
The hypothesis tested in this study proposes that the trkB.T1 and trkB.T2 receptors are capable of mediating ligand-dependent signal transduction by a presently unknown molecular mechanism. The rationale for the hypothesis is as follows. First, current models for truncated trkB function (action as a dominant negative modulating trkB.FL effects, action as a barrier to diffusion, or action as a molecular sponge to soak up excess ligand) require nothing functionally of the intracellular region other than to tether the molecule in the cell membrane. On the other hand, the facts that (1) the intracellular domain of trkB.T1 exhibits 100% sequence conservation across four mammalian species and (2) multiple truncated isoforms exist suggest that their intracellular domains are likely to have more demanding functions required of them beyond simply serving as molecular anchors. Second, studies to date have failed to detect any ligand-dependent signaling capabilities for the truncated receptors with the use of commonly observed signaling endpoints, such as calcium fluxes, immediate early gene inductions, or survival.
With the use of microphysiometry as an assay, our data suggest that both trkB.T1 and trkB.T2 are capable of mediating ligand-induced changes in cell physiology. Further, we have shown that the isoform-specific intracellular sequences are necessary for this response. The simplest interpretation is that the truncated trkB receptors are capable of mediating ligand-induced signal transduction.
Alternatively, it is possible that the microphysiometric signals observed in the trkB.T1 and trkB.T2 transfected cells represent the energy consumption caused by receptor internalization. This explanation would suggest that the energy required for receptor internalization alone accounts for the microphysiometric response of trkB.T1 and trkB.T2 transfected cells, whereas the lack of response in trkBΔ423 and trkBΔ435 cells results from their possible inability to internalize. However, calculations by Owicki and Parce (1992) suggest that the amount of ATP consumed by receptor-activated signaling mechanisms, including internalization, is small relative to the metabolic responses detected by the microphysiometer. Thus, although energy consumed in the actual signaling and internalization processes themselves may account for a small fraction of the observed responses, it is unlikely to be a major factor influencing the overall level of acidic metabolite release. Rather, the long-term changes observed in cellular metabolism likely reflect more global changes in cellular function, such as those associated with differentiation or long-term maintenance of a cell type.
Another noteworthy point is that different transfected cell lines express somewhat different levels of their respective trkB protein. However, we do not believe that this affects the interpretation of the data. Indeed, the fact that cell lines expressing either relatively higher or lower levels of receptor are capable of responding to BDNF (trkB.T1 has higher levels of expression relative to trkB.T2 and trkB.FL cells) whereas other cells expressing either higher or lower levels of receptor fail to respond (trkBΔ435 expresses higher levels of receptor similar to trkB.T1 cells, but trkBΔ423 is expressed at lower levels more similar to trkB.T2 and trkB.FL cells) suggests that expression levels over the range present in these cells is not a significant factor.
Although the intracellular domains are quite small, there is ample precedent for signaling by other transmembrane proteins with small intracellular domains. For example, integrins and some cytokine receptors have small intracellular domains that lack any known enzymatic activity and still promote ligand-induced signal transduction by using a host of signaling elements (McLean et al., 1990; Clarke and Brugge, 1995; Schwartz et al., 1995; Taniguchi, 1995). β-1,4-Galactosyltransferase, a transmembrane protein found on the surface of mammalian sperm that possesses only 24 intracellular amino acids, nonetheless activates a G-protein complex when sperm interact with egg surface proteins (Gong et al., 1995).
It is important to note that signaling capability is not mutually exclusive of other functions, such as action as a dominant negative effector (Eide et al., 1996; Ninkina et al., 1996). In fact, superimposing these functions may provide a cell with unique capabilities. For example, trkB.FL is the predominant trkB isoform expressed early in development, whereas later, trkB.T1 levels become elevated. At the time of transition, cells coexpressing both isoforms might experience a gradually increasing dominant negative effect of trkB.T1 receptors on trkB.FL signaling pathway via formation of trkB.FL:trkB.T1 heterodimers as trkB.T1 levels increase relative to trkB.FL levels. In parallel, the trkB.T1 signaling pathway or pathways would be expected to activate gradually via trkB.T1 homodimer formation.
Mechanistically, ligand-induced dimerization of trkB.T1 or trkB.T2 would be expected to bring together two intracellular domains plus any associated proteins. This dimerized unit then could serve as an activation complex for initiating a signaling cascade. Included within the first 12 intracellular amino acids common to all three known trkB isoforms (as well as trkA and trkC) is the amino acid sequence KFG, which has been shown in trkA to be essential for activation of a ras-independent NGF signaling pathway and phosphorylation of SNT (Peng et al., 1995). This sequence may be involved in signaling by trkB.T1 and trkB.T2. In addition, although the intracellular domain of trkB.T1 lacks any tyrosines or threonines, it does have a serine residue. TrkB.T2 has all three residues. Phosphorylation of the receptors themselves is therefore a possible component of the signaling pathway. Alternatively, the phosphorylation dependence of the signaling pathways suggested by the staurosporine and K252a experiments might occur at a step further downstream in the cascade.
That trkB.T1 and trkB.T2 receptors are capable of ligand-mediated signaling may have broad functional implications for both neuronal and glial cells. Because trkB.T1 becomes the predominant trkB isoform as development proceeds and its expression is induced strongly after injury, it is possible that trkB.T1 signaling may influence or regulate aspects of neural maturation and/or response to injury. Although trkB.T2 expression has not been examined yet in these contexts, recent work has revealed that it is found primarily in neurons (Armanini et al., 1995). On the basis of the data presented here, a role in neurotrophin signaling now must be considered in developing models for the action of both trkB.T1 and trkB.T2. These data also raise the possibility that at least some of the numerous trkC isoforms lacking tyrosine kinase domains may be capable of signaling (Lamballe et al., 1993; Tsoulfas et al., 1993; Valenzuela et al., 1993). From a larger evolutionary point of view, generating multiple receptor isoforms with distinct signaling capabilities from a single gene provides functional versatility in a very efficient manner. For example, five different PACAP (pituitary adenylyl cyclase-activating polypeptide) receptors, each with distinct signaling patterns, arise from a single gene by virtue of alternative splicing of exons encoding different regions of the intracellular domain of the protein (Spengler et al., 1993).
The next steps in the process of understanding trkB.T1 and trkB.T2 action and function will be to identify molecular components involved in these intracellular signaling pathways and to gain a more detailed understanding of the developmental and cell type-specific expression of each isoform. In addition, mouse knock-out experiments in which trkB.FL is expressed, but each truncated isoform in turn is deleted, might be especially valuable in elucidating their respective functions.
Footnotes
This project was supported by a fellowship to B.H. in memory of Natalie Holland by Fight for Sight, New York Research Division of Prevent Blindness America, a Beckman Fellowship to R.C.K., an Advanced Research Projects Agency contract to Molecular Devices Corporation (MDA972-92-C-0005), and Grant EY10739 from National Institutes of Health to S.C.F. The University of California Santa Barbara (UCSB) Advanced Instrumentation Center is part of the Materials Research Laboratory Central Facilities, supported by the National Science Foundation under Award DMR-9123048. We thank David Middlemas and Tony Hunter for trkB.FL and trkB.T2 cDNA clones. We are grateful to Andy Welcher and his colleagues at Amgen, Thousand Oaks, CA, for providing recombinant BDNF and NT4/5 and to Geoff Lewis and Steve Fisher for providing biotinylated BDNF. We thank Kathy Foltz for excellent comments on this manuscript, Geoff Lewis and Brian Matsumoto for help with confocal microscopy, and Maura Jess and the Neuroscience Research Institute Computer Laboratory for assistance with computer graphics. We are also grateful to the UCSB Advanced Instrumentation Center for synthetic oligonucleotides.
Correspondence should be addressed to Dr. Stuart C. Feinstein, Neuroscience Research Institute, University of California, Santa Barbara, CA 93106.
REFERENCES
- 1.Allendoerfer KL, Cabelli RJ, Escandón E, Kaplan DR, Nikolics K, Shatz CJ. Regulation of neurotrophin receptors during the maturation of the mammalian visual system. J Neurosci. 1994;14:1795–1811. doi: 10.1523/JNEUROSCI.14-03-01795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armanini MP, McMahon SB, Sutherland J, Shelton DL, Philips HS. Truncated and catalytic isoforms of trkB are co-expressed in neurons of rat and mouse CNS. Eur J Neurosci. 1995;7:1403–1409. doi: 10.1111/j.1460-9568.1995.tb01132.x. [DOI] [PubMed] [Google Scholar]
- 3.Baxter GT, Miller DL, Kuo RC, Wada HG, Owicki JC. PKC epsilon is involved in granulocyte-macrophage colony-stimulating factor signal transduction: evidence from microphysiometry and antisense oligonucleotide experiments. Biochemistry. 1992;31:10950–10954. doi: 10.1021/bi00160a002. [DOI] [PubMed] [Google Scholar]
- 4.Baxter GT, Young ML, Miller DL, Owicki JC. Using microphysiometry to study the pharmacology of exogenously expressed m1 and m3 muscarinic receptors. Life Sci. 1994;55:573–583. doi: 10.1016/0024-3205(94)00483-8. [DOI] [PubMed] [Google Scholar]
- 5.Beck KD, Lamballe F, Klein R, Barbacid M, Schauwecker PE, McNeill TH, Finch CE, Hefti F, Day JR. Induction of noncatalytic TrkB neurotrophin receptors during axonal sprouting in the adult hippocampus. J Neurosci. 1993;13:4001–4014. doi: 10.1523/JNEUROSCI.13-09-04001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg MM, Sternberg DW, Parada LF, Chao MV. K252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- 7.Berkemeier LR, Winslow JW, Kaplan DR, Nikolics K, Goeddel DV, Rosenthal A. Neurotrophin-5: a novel neurotrophic factor that activates trk and trkB. Neuron. 1991;7:857–866. doi: 10.1016/0896-6273(91)90287-a. [DOI] [PubMed] [Google Scholar]
- 8.Biffo S, Offenhäuser N, Carter BD, Barde YA. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development (Camb) 1995;121:2461–2470. doi: 10.1242/dev.121.8.2461. [DOI] [PubMed] [Google Scholar]
- 9.Bouvier C, Salon JA, Johnson RA, Civelli O. Dopaminergic activity measured in D1- and D2-transfected fibroblasts by silicon microphysiometry. J Recept Res. 1993;13:559–571. doi: 10.3109/10799899309073679. [DOI] [PubMed] [Google Scholar]
- 10.Bravo R, Burckhardt J, Curran T, Muller R. Stimulation and inhibition of growth by EGF in different A431 cell clones is accompanied by the rapid induction of c-fos and c-myc proto-oncogenes. EMBO J. 1985;4:1193–1197. doi: 10.1002/j.1460-2075.1985.tb03759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 12.Cochran BH, Zullo J, Verma IM, Stiles CD. Expression of the c-fos gene and of a fos-related gene is stimulated by platelet-derived growth factor. Science. 1984;226:1080–1082. doi: 10.1126/science.6093261. [DOI] [PubMed] [Google Scholar]
- 13.Davies AM. The role of neurotrophins in the developing nervous system. J Neurobiol. 1994;25:1334–1348. doi: 10.1002/neu.480251103. [DOI] [PubMed] [Google Scholar]
- 14.Davis AM. The neurotrophic hypothesis: where does it stand? Philos Trans R Soc Lond [Biol] 1996;351:389–393. doi: 10.1098/rstb.1996.0033. [DOI] [PubMed] [Google Scholar]
- 15.Dickinson KE, Cohen RB, Skwish S, Delaney CL, Serafino RP, Poss MA, Gu Z, Ryono DE, Moreland S, Powell JR. BMS-180560, an insurmountable inhibitor of angiotensin II-stimulated responses: comparison with losartan and EXP3174. Br J Pharmacol. 1994;113:179–189. doi: 10.1111/j.1476-5381.1994.tb16191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliot JF, Albrecht GR, Gilladoga A, Handunnetti SM, Neequaye J, Lallinger G, Minjas JN, Howard RJ. Genes for Plasmodium falciparum surface antigens cloned by expression in COS cells. Proc Natl Acad Sci USA. 1990;87:6363–6367. doi: 10.1073/pnas.87.16.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Escandón E, Burton LE, Szonyi E, Nikolics K. Characterization of neurotrophin receptors by affinity crosslinking. J Neurosci Res. 1993;34:601–613. doi: 10.1002/jnr.490340602. [DOI] [PubMed] [Google Scholar]
- 19.Escandón E, Soppet D, Rosenthal A, Mendoza-Ramirez JL, Szonyi E, Burton LE, Henderson CE, Parada LF, Nikolics K. Regulation of neurotrophin receptor expression during embryonic and postnatal development. J Neurosci. 1994;14:2054–2068. doi: 10.1523/JNEUROSCI.14-04-02054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feinstein SC, Dana S, McConlogue S, Shooter EM, Coffino PC. Nerve growth factor rapidly induces ornithine decarboxylase mRNA in PC12 cells. Proc Natl Acad Sci USA. 1985;82:5761–5765. doi: 10.1073/pnas.82.17.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong X, Dubois DH, Miller DJ, Shru BD. Activation of a G-protein complex by aggregation of β-1,4-galactosyltransferase on the surface of sperm. Science. 1995;269:1718–1721. doi: 10.1126/science.7569899. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg ME, Greene LA, Ziff EB. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- 23.Greene LA, McGuire JC. Induction of ornithine decarboxylase by nerve growth factor dissociated from effects on survival and neurite outgrowth. Nature. 1978;276:191–194. doi: 10.1038/276191a0. [DOI] [PubMed] [Google Scholar]
- 24.Guroff G, Dickens G, End D. The induction of ornithine decarboxylase by nerve growth factor and epidermal growth factor in PC12 cells. J Neurochem. 1981;37:342–349. doi: 10.1111/j.1471-4159.1981.tb00461.x. [DOI] [PubMed] [Google Scholar]
- 25.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9:1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RM, McNeeley PA, DeMoor K, Stewart GR, Glaeser BS, Pitchford S. Recombinant human ciliary neurotrophic factor stimulates the metabolic activity of SH-SY5Y cells as measured by a cytosensor microphysiometer. Brain Res. 1994;646:327–331. doi: 10.1016/0006-8993(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991a;252:554–557. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 29.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990a;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 30.Klein R, Jing S, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991a;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 31.Klein R, Nanduri V, Jing S, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991b;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein R, Lamballe F, Bryant S, Barbacid M. The trkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron. 1992;8:947–956. doi: 10.1016/0896-6273(92)90209-v. [DOI] [PubMed] [Google Scholar]
- 33.Kruijer W, Cooper JA, Hunter T, Verma IM. Platelet-derived growth factor induces rapid but transient expression of the c-fos gene and protein. Nature. 1984;312:711–716. doi: 10.1038/312711a0. [DOI] [PubMed] [Google Scholar]
- 34.Kruijer W, Schubert D, Verma IM. Induction of the proto-oncogene fos by nerve growth factor. Proc Natl Acad Sci USA. 1985;82:7330–7334. doi: 10.1073/pnas.82.21.7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamballe F, Tapley P, Barbacid M. trkC encodes multiple neurotrophin-3 receptors with distinct biological properties and substrate specificities. EMBO J. 1993;12:3083–3094. doi: 10.1002/j.1460-2075.1993.tb05977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh HN, Scholz WK, Lamballe F, Klein R, Nanduri V, Barbacid M, Palfrey HC. Signal transduction events mediated by the BDNF receptor gp145trkB in primary hippocampal pyramidal cell culture. J Neurosci. 1993;13:4281–4292. doi: 10.1523/JNEUROSCI.13-10-04281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McConnell HM, Owicki JC, Parce JW, Miller DL, Baxter GT, Wada HG, Pitchford S. The cytosensor microphysiometer: biological applications of silicon technology. Science. 1992;257:1906–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- 38.McLean JW, Vestal DJ, Cheresh DA, Bodary SC. cDNA sequence of the human integrin b5 subunit. J Biol Chem. 1990;265:17126–17131. [PubMed] [Google Scholar]
- 39.Meakin SO, Suter U, Drinkwater CC, Welcher AW, Shooter EM. The rat trk proto-oncogene product exhibits properties characteristic of the slow nerve growth factor receptor. Proc Natl Acad Sci USA. 1992;89:2374–2378. doi: 10.1073/pnas.89.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milbrandt J. Nerve growth factor rapidly induces c-fos mRNA in PC12 rat pheochromocytoma cells. Proc Natl Acad Sci USA. 1986;83:4789–4793. doi: 10.1073/pnas.83.13.4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller DL, Olson JC, Parce JW, Owicki JC. Cholinergic stimulation of the Na+/K+ adenosine triphosphatase as revealed by microphysiometry. Biophys J. 1993;64:813–823. doi: 10.1016/S0006-3495(93)81442-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller R, Bravo R, Burckhardt J, Curran T. Induction of c-fos gene and protein by growth factors precedes activation of c-myc. Nature. 1984;312:716–720. doi: 10.1038/312716a0. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawara A, Azar G, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neve KA, Kozlowski MR, Rosser MP. Dopamine D2 receptor stimulation of Na+/H+ exchange assessed by quantification of extracellular acidification. J Biol Chem. 1992;267:25748–25753. [PubMed] [Google Scholar]
- 47.Ninkina N, Adu J, Fischer A, Pinon LGP, Buchman VL, Davies AM. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996;15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- 48.Ohmichi M, Decker SJ, Pang L, Saltiel AR. Inhibition of the cellular actions of nerve growth factor by staurosporine and K252A results from the attenuation of the activity of the trk tyrosine kinase. Biochemistry. 1992;31:4034–4039. doi: 10.1021/bi00131a019. [DOI] [PubMed] [Google Scholar]
- 49.Omary MB, Baxter GT, Chou CF, Riopel CL, Lin WY, Strulovici B. PKC epsilon-related kinase associates with and phosphorylates cytokeratin 8 and 18. J Cell Biol. 1992;117:583–593. doi: 10.1083/jcb.117.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owicki JC, Parce JW. Biosensors based on the energy metabolism of living cells: the physical chemistry and cell biology of extracellular acidification. Biosens Bioelectron. 1992;7:255–272. doi: 10.1016/0956-5663(92)87004-9. [DOI] [PubMed] [Google Scholar]
- 51.Owicki JC, Parce JW, Kercso KM, Sigal GB, Muir VC, Venter JC, Fraser CM, McConnell HM. Continuous monitoring of receptor-mediated changes in the metabolic rates of living cells. Proc Natl Acad Sci USA. 1990;87:4007–4011. doi: 10.1073/pnas.87.10.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parce JW, Owicki JC, Kercso KM, Sigal GB, Wada HG, Muir VC, Bousse LJ, Ross KL, Sikic BI, McConnell HM. Detection of cell-affecting agents with a silicon biosensor. Science. 1989;246:243–247. doi: 10.1126/science.2799384. [DOI] [PubMed] [Google Scholar]
- 53.Peng X, Greene LA, Kaplan DR, Stephens RM. Deletion of a conserved juxtamembrane sequence in trk abolishes NGF-promoted neuritogenesis. Neuron. 1995;15:395–406. doi: 10.1016/0896-6273(95)90043-8. [DOI] [PubMed] [Google Scholar]
- 54.Pitchford S, De Moor K, Glaeser BS. Nerve growth factor stimulates rapid metabolic responses in PC12 cells. Am J Physiol. 1995;268:C936–C943. doi: 10.1152/ajpcell.1995.268.4.C936. [DOI] [PubMed] [Google Scholar]
- 55.Radeke MJ, Feinstein SC. Analytical purification of the slow, high affinity NGF receptor: identification of a novel 135 kd polypeptide. Neuron. 1991;7:141–150. doi: 10.1016/0896-6273(91)90082-b. [DOI] [PubMed] [Google Scholar]
- 56.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 57.Raley-Susman KM, Miller KR, Owicki JC, Sapolsky RM. Effects of excitotoxin exposure on metabolic rate of primary hippocampal cultures: application of silicon microphysiometry to neurobiology. J Neurosci. 1992;12:773–780. doi: 10.1523/JNEUROSCI.12-03-00773.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Redish DM, Raley-Susman KM, Sapolsky RM. Inhibition of acidification rate in cultured fibroblasts by glucocorticoids. Application of silicon microphysiometry to endocrinology. Horm Metab Res. 1993;25:264–267. doi: 10.1055/s-2007-1002093. [DOI] [PubMed] [Google Scholar]
- 59.Renschler MF, Wada HG, Fok KS, Levy R. B-lymphoma cells are activated by peptide ligands of the antigen-binding receptor or by anti-idiotypic antibody to induce extracellular acidification. Cancer Res. 1995;55:5642–5647. [PubMed] [Google Scholar]
- 60.Roback JD, Marsh HN, Downen M, Palfrey HC, Wainer BH. BDNF-activated signal transduction in rat cortical glial cells. Eur J Neurosci. 1995;7:849–862. doi: 10.1111/j.1460-9568.1995.tb01072.x. [DOI] [PubMed] [Google Scholar]
- 61.Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996;35:3362–3367. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- 62.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 63.Schaffner W, Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem. 1973;56:502–514. doi: 10.1016/0003-2697(73)90217-0. [DOI] [PubMed] [Google Scholar]
- 64.Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Philips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 66.Soppet D, Escandón E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LW, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parada LF. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 67.Spengler E, Waeber C, Pantoloni C, Holsboer F, Bockaert J, Seeburg PH, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- 68.Squinto SP, Still TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 69.Stumpo DJ, Blackshear PJ. Insulin and growth factor effects on c-fos expression in normal and protein kinase C-deficient 3T3–L1 fibroblasts and adipocytes. Proc Natl Acad Sci USA. 1986;83:9453–9457. doi: 10.1073/pnas.83.24.9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taniguchi T. Cytokine signaling through nonreceptor protein tyrosine kinases. Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- 71.Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- 72.Thibodeau A, Kuo RC, Crothers JM, Jr, Yao X, Owicki JC, Forte JG. Direct measurement of extracellular proton flux from isolated gastric glands. Am J Physiol. 1994;267:C1473–C1482. doi: 10.1152/ajpcell.1994.267.5.C1473. [DOI] [PubMed] [Google Scholar]
- 73.Tsoulfas P, Soppet D, Escandón E, Tessarollo L, Mendoza-Ramirez JL, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- 74.Valenzuela DM, Maisonpierre PC, Glass DJ, Rojas E, Nunez L, Kong Y, Gies DR, Stitt TN, Ip NY, Yancopoulos GD. Alternative forms of rat trkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- 75.Wada HG, Indelicato SR, Meyer L, Kitamura T, Miyajima A, Kirk G, Muir VC, Parce JW. GM-CSF triggers a rapid, glucose-dependent extracellular acidification by TF-1 cells: evidence for sodium/proton antiporter and PKC-mediated activation of acid production. J Cell Physiol. 1993;154:129–138. doi: 10.1002/jcp.1041540116. [DOI] [PubMed] [Google Scholar]