


Abstract
The Dam DNA methylase of Escherichia coli is required for methyl-directed mismatch repair, regulation of chromosomal DNA replication initiation from oriC (which is DnaA-dependent), and regulation of gene expression. Here, we show that Dam suppresses aberrant oriC-independent chromosomal replication (also called constitutive stable DNA replication, or cSDR). Dam deficiency conferred cSDR and, in presence of additional mutations (Δtus, rpoB*35) that facilitate retrograde replication fork progression, rescued the lethality of ΔdnaA mutants. The DinG helicase was required for rescue of ΔdnaA inviability during cSDR. Viability of ΔdnaA dam derivatives was dependent on the mismatch repair proteins, since such viability was lost upon introduction of deletions in mutS, mutH or mutL; thus generation of double strand ends (DSEs) by MutHLS action appears to be required for cSDR in the dam mutant. On the other hand, another DSE-generating agent phleomycin was unable to rescue ΔdnaA lethality in dam+ derivatives (mutS+ or ΔmutS), but it could do so in the dam ΔmutS strain. These results point to a second role for Dam deficiency in cSDR. We propose that in Dam-deficient strains, there is an increased likelihood of reverse replication restart (towards oriC) following recombinational repair of DSEs on the chromosome.
INTRODUCTION
In the gamma-proteobacteria, adenine bases in the palindromic GATC sequences in DNA are N6-methylated on both strands by the Dam DNA methylase [reviewed in (1–3)]. Discovered more than four decades ago (4,5), the enzyme catalyzes maintenance methylation on the daughter strands of the pair of hemi-methylated duplexes that are generated following semi-conservative replication of DNA, although it can also perform de novo methylation on duplexes in which both strands are unmethylated at these sites.
In Escherichia coli and Salmonella enterica serovar Typhimurium, Dam's action as a maintenance methylase in vivo defines a time window of hemi-methylation following passage of a replication fork when the parental and daughter strands can be distinguished by their GATC methylation status (6). This time window is then exploited by different replication-related processes in bacterial cells. Thus following recognition by MutS of replication errors, the MutH endonuclease cleaves the unmethylated daughter strand at GATC to initiate MutHLS-directed mismatch repair. Second, SeqA binds hemi-methylated GATC sequences to mediate multiple functions including sequestration of the site (oriC) for initiation of chromosomal DNA replication, transcriptional repression of the essential protein DnaA needed for replication initiation at oriC, and cohesion of sister chromatids in the wake of a progressing replication fork. Third, hemi-methylated GATC regions can also serve to control gene transcription and plasmid replication, by modulating the binding of specific proteins to the respective regulatory regions (1–3).
Several phenotypes of dam mutants are related to Dam's role in methyl-directed mismatch repair, and hence are suppressed by mutations in the mutH, -L or -S genes (7,8). The understanding is that, in absence of Dam function, mismatches generated during replication lead to double-strand ends (DSEs) engendered by MutH endonuclease at the unmethylated GATC sequences, and that such DSEs do not occur in the dam mutH/L/S derivatives (8). MutH has been shown in vitro to cleave both strands at unmethylated GATC sites (9). The phenotypes of dam mutants that are mutH/L/S-suppressed (and therefore believed to be consequential to DSE generation) include: synthetic lethalities with mutations in genes for recombinational repair of DSEs (recA, -B, -C, ruvA, -B, -C) and for replication restart (priA, -B, dnaC); sensitivity to 2-aminopurine and genotoxic agents; and hyper-recombination (1–3,8,10,11).
In this work, we have identified a novel function for Dam methylase, which is to control aberrant oriC-independent bacterial replication. In wild-type E. coli, replication of the circular chromosome is achieved by DnaA-mediated initiation at oriC of a pair of replication forks which then proceed bidirectionally to meet in the terminus region [reviewed in (12,13)]. The seven highly transcribed rrn operons are all oriented such as to be co-directional with fork progression on the two replichores. The terminus region is demarcated by the Tus protein-bound TerA and TerC/B sites which act as polar (unidirectional) barriers to fork progression, so that replication forks do not ordinarily progress into the opposite replichore, that is, towards oriC (13–16).
Aberrant oriC-independent chromosomal replication, also referred to as constitutive stable DNA replication (cSDR), has been described in E. coli derivatives harbouring mutations in rnhA, recG or topA, that encode RNase HI, RecG helicase and topoisomerase I, respectively (17–19); most recently, it has also been reported to occur in triple-exo mutant strains, that is, mutants simultaneously deficient for 3′-DNA exonucleases I, VII and SbcCD, or exonucleases I, VII and the 5′-DNA exonuclease RecJ (20). cSDR is critically dependent on the phenomenon of replication restart (17), which in turn is mediated by action of the PriA, PriB, PriC and DnaT proteins in loading of the DnaB replicative helicase at sites of initiation other than oriC (21–24). Increased occurrence of RNA-DNA hybrids or R-loops is believed to be responsible for cSDR initiation in rnhA and topA mutants (17,18), whereas for recG mutants several alternative models have been proposed in this regard: R-loops (17), aberrant replication initiation following fork collisions in the terminus region (25–27), continued replisome progression even after converging forks meet one another (28), and retrograde or reverse replication restart (24,29). A phenomenon related to cSDR is inducible stable DNA replication (iSDR), which is also dependent on replication restart mechanisms but unlike cSDR requires exposure of bacterial cells to DNA damaging agents (17,30).
One of the prominent genetic manifestations of cSDR is the rescue of lethality that is otherwise ordinarily associated with deficiency of DnaA (in other words, cSDR can compensate for loss of oriC-dependent replication in these situations). Such rescue has been demonstrated in rnhA, recG and triple-exo mutants (17,20,25–27). Completion of chromosome duplication by cSDR under these conditions entails that two kinds of obstacles be overcome (13): (i) Tus-bound Ter barriers, and (ii) transcription-replication conflicts, especially at rrn operons. Thus, mutations which help mitigate these impediments—Δtus for the former (13–16), and rpoB*35 encoding an altered RNA polymerase for the latter (31–33)—are either required for (in case of recG or triple-exo mutants) or greatly facilitate (in case of rnhA) the suppression by cSDR of lethality caused by loss of DnaA function (20,25–27).
We now report that Dam deficiency also confers cSDR, and that ΔdnaA inviability is suppressed by Δdam in presence of the Δtus and rpoB*35 mutations. Furthermore, as has been reported earlier for recG, rnhA, topA and the triple-exo mutants (19,20,25–28,34), dam derivatives that are dnaA+ exhibit a prominent ‘mid-terminus’ peak in DNA copy number analysis which is an apparent signature of cSDR. Suppression of ΔdnaA lethality by Δdam was affected by mutH/L/S mutations, suggesting that DSEs are required for cSDR in the Dam-deficient strains. At the same time, our data indicate that Dam deficiency also plays a second role in cSDR, which we propose is in facilitating reverse restart of replication forks on the chromosome.
MATERIALS AND METHODS
Growth media and bacterial strains
The growth media were LB and 0.2% glucose-minimal A (35), the latter supplemented with auxotrophic requirements as appropriate and with Casamino acids at 1% where indicated. Unless otherwise indicated, the growth temperature was 37°C. l-Arabinose (Ara), Xgal, tetracycline (Tet), kanamycin (Kan), ampicillin (Amp), chloramphenicol (Cm) and trimethoprim (Tp) were used at concentrations described previously (36); supplementation with spectinomycin or the various genotoxic agents was at concentrations as mentioned elsewhere in the text. Genotypes of E. coli strains are listed in Supplementary Table S1, with the following knock-out (KanR insertion-deletion) alleles sourced from the collection of Baba et al. (37): dam, tus, mutH, mutL, mutS, seqA, dinG, rnhA, recG, recA, rdgB, sulA, metF and argE.
Plasmids
Previously described plasmids include (brief description for each in parentheses): pCP20 (AmpR CmR, pSC101 derivative carrying Flp recombinase) (38): pBAD18 (AmpR, vector for Ara-induced expression of genes cloned downstream of Para- promoter) (39); pMU575 (TpR, single-copy-number vector with lacZ+) (40); pASKA-dam+ and its corresponding empty vector pCA24N from the ASKA collection (41) (CmR, former expresses Dam methylase); and pAD16 (AmpR, expressing ParB::YGFP protein, that is, a fusion of ParB with a variant green fluorescent protein) (42). The details of construction in this study of plasmids pHYD2388, pHYD4805, and pHYD5701 (pMU575 derivatives carrying S. enterica genes dnaA+, dnaA+ recG+, and recA+, respectively) and of plasmid pHYD4807 (pBAD18 derivative carrying E. coli mutS+) are described in the Supplementary Text.
Genetic methods
Procedures for P1 transduction (43) and transformation (44) were as described. Recombineering of ΔdnaA::Kan was by the method of Datsenko and Wanner (38), as described in more detail in the Supplementary Text. Site-specific deletion of FRT-flanked KanR cassettes was achieved with the aid of plasmid pCP20, as described (38). The polA12 allele was introduced by cotransduction with a linked KanR marker (45); procedures for introduction and testing of alleles rpoB*35 and priA300, and for crossing out of phi80 prophage, are detailed in the Supplementary Text. Presence or absence in strains of phi80 prophage was tested by duplex-PCR with a set of three primers as earlier described (46).
To achieve Ara-induced RNase HI overexpression, a Para-rnhA construct was used that had previously been transferred (36) from a pBAD18-derived plasmid to the λ att site on the chromosome by the method of Boyd et al. (47); the same method was used to construct chromosomal integrations at the λ att site of the Para promoter alone from the pBAD18 vector (as negative control), and of a Para-UvsW construct from the plasmid pHYD2368 that has also been previously described (36).
For scoring rescue of ΔdnaA lethality, cultures of the ΔdnaA derivatives carrying pHYD2388 were grown overnight in Tp-supplemented medium and plated at suitable dilutions on Xgal medium without Tp. Typically for strains in which ΔdnaA was lethal, the proportion of white colonies to total was <0.2%. Rescue of lethality was taken to be robust if (i) white colonies were obtained at >5% of the total, and (ii) these colonies could be maintained indefinitely by sub-culturing on the same medium.
Tn10 stability and pCP20 transformation in dam mutants
dam derivatives carrying btuB::Tn10 yielded spontaneous TetS derivatives at high enough frequency to comprise ∼20% of cells in a culture, indicative of derepression of IS10 transposition in the mutants (48); these strains were therefore routinely tested for their TetR phenotype before use in experiments.
We also discovered in this study that plasmid pCP20, and other pSC101 derivatives, fail to yield transformants in a dam strain but are able to transform a dam seqA mutant; furthermore, plasmid preparations made from the latter now yielded transformants even in the dam strain. These results indicate for the first time that pSC101 also belongs to that category of replicons in which a Dam-methylated template is converted to hemi-methylated DNA in the dam mutant following one round of replication, and is then irreversibly bound by SeqA to stop further replication (49).
Copy number analysis by deep sequencing
In the initial experiments for copy number analysis, genomic DNA was extracted from cultures grown in LB to mid-exponential phase (A600 ≈ 0.4) with the aid of a commercial kit (PureLink; Invitrogen, USA). It is now known that such preparations are depleted of DNA from the rrn operon regions (50,51), and therefore in subsequent experiments we prepared genomic DNA from cultures by phenol-chloroform extraction, as described (44).
Deep sequencing was performed on Illumina HiSeq platforms with a paired-end sequencing strategy, to achieve ∼100-fold coverage for each preparation. Sequence reads (each 100 or 250 bases long) were subjected separately to de novo contig assembly as well as to alignment to reference genomes (either strain MG1655, or MG1655 with phi80 prophage); the aggregate base read counts for each culture (after alignment to MG1655) are given in Supplementary Table S2.
The aligned sequence data were then used for copy number determinations (by a moving-average method), and for mutation detection, as described in the Supplementary Text. The copy number of each genomic region was represented on a semi-log graph as an ‘enrichment ratio’, which is defined as the ratio of (i) the base read count for that region in an exponential-phase culture (normalized to the aggregate of aligned base read counts for that culture) to (ii) the base read count, similarly normalized, for the same region in a stationary-phase culture (where all regions are expected to be equally represented).
cSDR detection by flow cytometry and fluorescence microscopy
For assessment of cSDR by flow cytometry, the method was essentially that described by Martel et al. (52), involving treatment of cultures in exponential phase in LB with 400 μg/ml of the protein synthesis inhibitor spectinomycin for 2 h, followed by addition of the nucleotide analogue EdU (0.08 mM) for 1 h. Cells were fixed with 3.7% paraformaldehyde for 30 min at room temperature, and then permeabilized and treated with Alexa Fluor-488 conjugate as described (52). Flow cytometry was performed on the BD-FACS Aria III platform, and data (for 10 000 cells in each preparation) were analyzed with BD-FACS Diva software (version 6.0).
For parS copy number detection by fluorescence microscopy (42,53), cultures (of strains bearing parS integrated near the chromosomal terminus region and expressing ParB::YGFP constitutively from plasmid pAD16) were grown to early exponential phase (A600 ≈ 0.4) at 37° in 2 ml of glucose-minimal A; one-half of each was treated with spectinomycin at 400 μg/ml for 2 h while the other half was stored on ice for this period. Cells from each of the aliquots were then washed twice in equal volumes of phosphate-buffered saline (PBS) (44), resuspended in 0.5 ml of PBS, and visualized under 60× magnification through a fluorescence microscope (DeltaVision OMX V3, GE Healthcare) at excitation and emission wavelength settings of 488 and 528 nm, respectively. Raw images were processed with the aid of ImageJ software (version 1.52i).
Other methods
The protocols of Sambrook and Russell (44) were followed for DNA manipulations and analysis. Immunoblotting with S9.6 monoclonal antibody for detection of RNA–DNA hybrids was as described (54). For phase contrast microscopy, cells were cultured in glucose-minimal A to mid-exponential phase, washed with PBS and resuspended in PBS; they were then layered atop 1% agarose pads on slides and overlaid with coverslips for viewing under 100× magnification on a Nikon Eclipse 80i microscope, and images were analyzed with the aid of NIS-Elements D3.0 software.
RESULTS
Dam deficiency is associated with cSDR
Given the occurrence of MutHLS-generated DSEs in dam mutants, as well as the similarities that exist between iSDR and cSDR, we examined whether cSDR occurs in Dam-deficient strains. Two different assays were employed, with the common feature being detection of continuing DNA synthesis following complete cessation of oriC-initiated replication. Such cessation was achieved by treatment of cultures for 2 h with the protein synthesis inhibitor spectinomycin (52), since it is known that each round of replication initiation at oriC requires fresh synthesis of DnaA, and that all replication forks which were previously initiated from oriC would have progressed to completion within this period.
In the first assay involving flow cytometry, DNA synthesis in individual cells of cultures was analyzed by fluorescence detection of incorporation of the nucleotide analog EdU before and after spectinomycin addition, as described earlier (52). In the spectinomycin-treated cultures, whereas just 4% of cells in the wild-type strain exhibited new DNA synthesis that exceeded the fluorescence gating threshold of 2 × 103 units, 87% of the Δdam mutant cells did so, indicative of cSDR (Figure 1A). Around 70% of cells in a culture of the wild-type strain exposed to sublethal concentration (2 μg/ml) of the radiomimetic agent phleomycin, which generates DSEs (55,56), also exhibited continued DNA synthesis exceeding the gating threshold following spectinomycin treatment, indicative of iSDR (Figure 1A).
Figure 1.
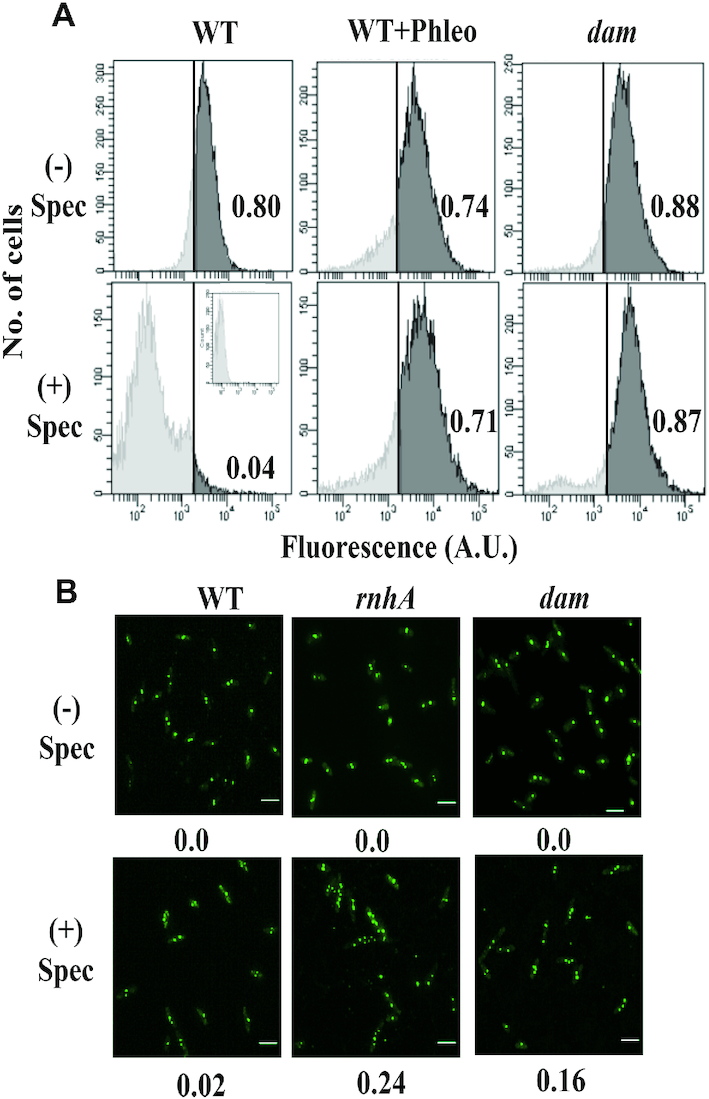
cSDR in dam mutants. (A) Analysis by flow cytometry, as described (52), of DNA synthesis in cultures of (i) parental (WT) strain GJ18609, (ii) WT with phleomycin (WT+Phleo) at 1 μg/ml, and (iii) the isogenic dam mutant GJ16494 (dam), without (–) or with (+) treatment with spectinomycin (Spec). In each panel, the proportion (of the total) of cells in which the fluorescence value exceeded the gating threshold (vertical line) is given at right. Inset within bottom left panel depicts fluorescence in cells exposed to neither EdU nor Alexa Fluor 488 (that is, autofluorescence). (B) Fluorescence microscopy for detection of ParB::YGFP-parS foci in cells from cultures of plasmid pAD16-bearing derivatives of parental (WT) strain GJ15830 and its isogenic rnhA or dam mutants (GJ15831 and GJ15832, respectively) without (–) or with (+) treatment with Spec. Beneath each panel, the proportion of cells with >2 foci is indicated (minimum of 300 cells examined). Scale bar, 3 μm.
For the second assay employing fluorescence microscopy, the number of foci of ParB::YGFP-bound parS was counted in individual cells of strains in which parS had been integrated near the chromosomal terminus region (42,53). The expectation was that normal cells would harbour at most two foci following spectinomycin addition, whereas those exhibiting cSDR would display a substantial proportion with multiple (>2) foci (since DnaA- and oriC-independent replication would lead to additional copies of parS in these cells); this expectation was confirmed with cultures of the wild-type strain and the rnhA mutant, respectively (Figure 1B). Cells of the Δdam mutant also behaved like the rnhA derivative and showed an increased proportion of multiple foci in this assay (Figure 1B). We conclude that Dam deficiency is a new example of a perturbation that confers cSDR in E. coli.
Rescue of ΔdnaA lethality by Dam deficiency in presence of Δtus and rpoB*35 mutations
Based on the findings above, we tested whether cSDR conferred by the dam mutation could restore viability in DnaA-deficient strains. In earlier studies (17,20,25–27,34), the temperature-sensitive dnaA46 mutation had been employed, and hence cSDR-mediated rescue of lethality could only be assessed at 42°. In this work, we chose to use ΔdnaA for the genetic studies on cSDR, and adopted a strategy analogous to those employed by us earlier to study rescue of knockout lethality in other essential E. coli genes such as rne, rho or nusG (36,54,57).
We constructed strain GJ16459/pHYD2388 (as described in Materials and Methods) with ΔdnaA and Δlac mutations on the chromosome, and a single-copy-number TpR shelter plasmid carrying functional copies of lacZ and dnaA from, respectively, E. coli and S. enterica. This strain and its derivatives grow as blue colonies on Xgal-supplemented plates; they also generate spontaneous plasmid-free segregant cells at a frequency of ∼5–15% which, however, can only grow and be subcultured as white colonies on Tp-free Xgal medium if they carry mutation(s) that rescue ΔdnaA lethality. Strain GJ16459 also carries the rpoB*35 and Δtus muations in order to facilitate the progression around the chromosome of cSDR-initiated forks across, respectively, the sites of transcription-replication conflict (especially at rrn operons) and the Ter sequences in the terminus region (20,25–27). We used the mhA and recG mutations as positive controls (17,25–27) to confirm their ability to confer viability in absence of DnaA function (Figure 2, compare panels ii and iii with panel i).
Figure 2.

Perturbations that rescue ΔdnaA lethality. Isogenic ΔdnaA derivatives (also bearing tus rpoB*35 mutations for all but that shown in panel xii) carrying dnaA+ shelter plasmid pHYD2388 (or pHYD4805, in case of recG mutant) and the additional genetic perturbation(s) as indicated on the top of each panel were plated on LB-Xgal plates and incubated for 40 hours at 37°; representative images are shown, and the numbers beneath each of the panels indicate the percentage of white colonies to the total, that is, viable even in absence of dnaA+ shelter plasmid (minimum of 500 colonies counted). Examples of white colonies and white-sectored blue colonies are marked by the white and black arrows, respectively. Strains employed for the different panels were pHYD2388 (or pHYD4805, in case of recG mutant) derivatives of: i, GJ16459; ii, GJ16475; iii, GJ16464; iv, GJ16445; v, GJ16488; vi, GJ18625; vii and viii, GJ16488 with pCA24N and pASKA-dam+, respectively; ix, GJ16469; x, GJ18626; xi, GJ18613; and xii, GJ18628. The medium was supplemented with Ara at 0.2% for panels x and xi.
Two different null alleles of dam (Δdam::Kan, and dam::Tn9) rescued the ΔdnaA lethality in this assay done at 37°C (Figure 2, panels iv–v), which did not occur in the derivative that was tus+ rpoB+ (Figure 2, panel xii). The phenotype was most prominently exhibited on nutrient-rich media [LB (Figure 2), or minimal A supplemented with Casamino acids (Supplementary Figure S1, panels i–iii)] since the combination of dam rpoB*35, even when dnaA+, grows poorly on minimal medium. As expected, the occurrence of white colonies (as an indicator of ΔdnaA viability) was accompanied also by presence of white-sectored blue colonies on the plates (see Figure 2 and Supplementary Figure S4B). Rescue of ΔdnaA lethality was abolished also upon introduction into these derivatives of a plasmid with the cloned dam+ gene but not of the vector alone (Figure 2, compare panels vii and viii), indicating that the phenotype was not being conferred by any additional mutations that may have arisen in the dam strain, which is known to have a mutator phenotype (5,8).
Roles for DinG and PriA in rescue of ΔdnaA lethality by dam
The DinG helicase is required for overcoming the effects of major head-on transcription-replication conflicts in E. coli, as evidenced by the fact that dinG derivatives with one or more of the rrn operons in inverted orientation are unable to grow on rich medium; most interestingly, this growth defect is suppressed by RNase H overexpression, with the implication that growth-inhibitory R-loops are being formed at sites of strong transcription–replication conflict (58). Other recent studies have also pointed to R-loop formation as a contributor to the deleterious effects of head-on collisions on the DNA template between transcription and replication machineries (33,59,60), but the underlying mechanism remains unclear (61). Since retrograde fork progression in cSDR is expected to generate conflicts between the replisome and rrn transcription, we examined the role of DinG on rescue of ΔdnaA lethality in the dam mutant.
Introduction of ΔdinG abolished the rescue of ΔdnaA lethality by the dam mutation (in the derivatives which also carried Δtus and rpoB*35) (Figure 2, panel ix). As with the findings reported earlier for dinG and growth of strains with rrn operon inversions (58), ΔdnaA growth also was restored in the dam ΔdinG derivative upon Ara-induced RNase HI overexpression (Figure 2, compare panels x and xi for derivatives carrying, respectively, Para alone or Para-rnhA). Thus, RNase HI appears to play antagonistic roles in cSDR: its absence (in the rnhA mutant) promotes cSDR ostensibly through R-loop formation, and yet its overexpression also facilitates cSDR in certain situations by overcoming the deleterious consequences of transcription-replication conflict.
The PriA protein, that is crucial for DnaA-independent replication initiation from D-loops and R-loops as well as for restart from intact but inactivated or disintegrated replication forks, has also earlier been shown to be required for cSDR (17). The priA300 mutation, encoding a K230R substitution which abolishes the 3′-5′ DNA helicase activity of the protein, abolishes rescue by recG of lethality associated with DnaA deficiency (25), and that by rnhA in some situations (26). We found in this study that the effect of dam mutation in restoring viability to ΔdnaA tus rpoB*35 is also suppressed upon introduction of the priA300 mutation (Figure 2, panel vi).
Absence of association between SeqA and cSDR
SeqA binds to DNA that is hemi-methylated at GATC sites, and hence the loss of methylation in Dam-deficient strains would lead to an abrogation of SeqA functions (62,63). Given that Δdam conferred viability to ΔdnaA (in presence of Δtus and rpoB*35), we tested the role if any of SeqA on cSDR. ΔseqA by itself was unable to confer viability to the ΔdnaA derivative (Supplementary Figure S1, panel iv), nor was the rescue of ΔdnaA lethality by dam abolished by mutation in seqA (Supplementary Figure S1, panel v).
DSEs in dam mutant contribute to its cSDR phenotype
As mentioned above, Dam deficiency is associated also with occurrence of DSEs in the genome that are generated by action of the proteins MutHLS of methyl-directed mismatch repair (7,8). Rescue of ΔdnaA lethality by dam on LB medium was abolished by mutations in the mutH/L/S genes of mismatch repair (Figure 3A, compare panel i with panels ii–iv). The phenotype was complementable, that is, ΔdnaA viability was restored in the dam mutS mutant carrying mutS+ in trans (Figure 3A, panel v) demonstrating once again that no incidental suppressor mutation had arisen in these derivatives. These results suggest that DSEs are required for conferring robust ΔdnaA viability in the dam strains.
Figure 3.

Roles of dam mutation and DSEs in rescue of ΔdnaA lethality. Isogenic derivatives of ΔdnaA tus rpoB*35 dam::Tn9 (A) or ΔdnaA tus rpoB*35 (B) strains carrying dnaA+ shelter plasmid pHYD2388 and the additional genetic and/or genotoxic perturbations as indicated on top of each panel were plated on LB-Xgal plates and incubated for 40 hours at 37°; representative images are shown, and the percentage of white colonies to total for each panel is indicated as explained in the legend to Figure 2. Strains employed for the different panels were pHYD2388 derivatives of: i, GJ16445; ii, GJ16438; iii, 18615; iv, viii, and xi, GJ16437; v, GJ16437/pHYD4807; vi and ix, GJ16459; and vii and x, GJ16427. Phleomycin (Phleo) supplementation for each of panels ix-xi was at 1 μg/ml, and with Ara for panel v at 0.2%. (C) Spotting assay at serial 10-fold dilutions to examine viability in presence (+) or absence (–) of phleomycin, for which the strains employed were (all tus rpoB*35, relevant genotypes indicated against each): top, GJ16437/pHYD2388; middle, GJ16437; and bottom, GJ16445. The top and middle rows represent blue and white colonies, respectively, from plate shown in Figure 3B, panel xi; and the bottom row represents a white colony from plate shown in Figure 3A, panel i.
Other perturbations that generate DSEs do not rescue ΔdnaA inviability
Since our findings above had implicated DSEs in the Δdam mutant as being required for rescue of ΔdnaA viability, we tested whether other perturbations (DNA-damaging agents, or mutations) that increase DSE frequency would also confer viability in the ΔdnaA Δtus rpoB*35 derivatives. As with dam, the mutations are also associated with a hyper-recombination phenotype and synthetic lethality with mutations affecting recombinational repair or replication restart (64,65).
However, none of the perturbations tested, which included exposure to sublethal concentrations of phleomycin, bleomycin, norfloxacin, ciprofloxacin, 2-aminopurine, or mitomycin C, was able to robustly rescue the ΔdnaA lethality (Supplementary Figure S2, panels vi–xi). Furthermore, mutations in rdgB or polA which are expected to generate nicks in DNA that are converted to DSEs upon arrival of a succeeding replication fork (64–66), did not confer viability to ΔdnaA under these conditions (Supplementary Figure S2, panels iii–iv). A site-specific DSE generated by SbcCD-mediated cleavage of a long palindromic sequence inserted within the lacZ gene (29) was also not proficient in conferring viability to the ΔdnaA tus rpoB*35 cells (Supplementary Figure S2, panel v).
Phleomycin can rescue ΔdnaA lethality in the dam ΔmutS background
Given the observations that DSEs are needed for rescue of ΔdnaA lethality in the dam mutant, but that other perturbations which generate DSEs do not appear to rescue such lethality, we considered the possibility that there is a second property in the dam strain, in addition to DSE formation, that is required for cSDR. This idea was supported by our finding that addition of phleomycin (at 1 μg/ml) could indeed suppress ΔdnaA lethality in a dam ΔmutS derivative which also carried tus and rpoB*35 mutations (Figure 3B, panel xi), under conditions in which there was no suppression in either the dam+ mut+ strain with phleomycin (Figure 3B, panel ix) or the dam ΔmutS strain without the genotoxin (Figure 3B, panel viii). The dam+ ΔmutS strain also was lethal with ΔdnaA, both in absence and presence of phleomycin (Figure 3B, panels vii and x, respectively). The ΔdnaA dam ΔmutS cells remained viable only on medium supplemented with phleomycin and not in its absence (Figure 3C, middle row). Thus, ΔdnaA viability can be conferred by a combination of two perturbations (dam ΔmutS, and phleomycin), neither of which by itself is able to do so.
Copy number analysis of different chromosomal regions by deep sequencing in dam dnaA+ and dam ΔdnaA derivatives
Asynchronously growing cultures of wild-type E. coli display a symmetrical gradient of copy numbers for the different regions around the circular chromosome, with the peak and the trough located near oriC and the terminus, respectively (13). Mutations in rnhA, recG, topA or the three exonucleases (that are associated with cSDR) confer a characteristic ‘mid-terminus peak’ that is superimposed on this gradient in dnaA+ derivatives (19,20,25–27,34).
In similar copy number analysis experiments, the dam dnaA+ derivative also exhibited a prominent mid-terminus peak (between TerA and TerC/B), in comparison with the wild-type strain (Figure 4, compare panels i and ii); see also Supplementary Figure S3A, panels i and ii). The mid-terminus peak was absent in the dam mutS derivative (performed on two replicates which yielded identical profiles, one of which is shown in Figure 4, panel iii). As discussed below, these data provide support to our proposal (13) that the mid-terminus peak is a signature feature of cSDR and that, at least in case of the dam mutant, it is the result of replication forks progressing from stochastically distributed genome-wide supernumerary DNA replication initiation events. In presence of sublethal phleomycin (2 μg/ml), the wild-type strain itself exhibited a mid-terminus peak in the copy number analysis (Supplementary Figure S3A, panel iii).
Figure 4.

Copy number analysis by deep sequencing in dam mutants. From the sequencing reads for the indicated strains (grown to log phase in LB at 37°C), copy number distributions for 1-kb (gray) and overlapping 10-kb (red) intervals have been plotted on semi-log graphs as enrichment ratios across the genome, as described in the text and Supplementary Text. Positions of oriC, TerA and TerC/B are marked. The gap at around 0.3 Mb in each of the distribution plots corresponds to the argF-lac deletion present in the strains. Isogenic strains displayed in the different panels (their relevant genotypes indicated on top of each panel) are: i, GJ13519; ii, GJ18617; iii, GJ18618; iv, GJ16488; v, GJ16494; and vi, GJ18619/pHYD2388. WT, wild-type parent.
It has previously been shown (25) that copy number profiles in the wild-type strain and its tus rpoB*35 derivative are, as expected, identical (with peak at oriC and trough in the TerA-TerC/B interval). Most interestingly, we found that the oriC peak was lost in the dam tus rpoB*35 or dam rpoB*35 derivatives that were dnaA+ (Figure 4, panels iv and v respectively), and that the copy number distributions across the genome were more or less flat in these strains (with only a small peak at the mid-terminus region). As further discussed below, we believe that this pattern is a reflection of the sum of aberrant replication initiation events in any individual dam mutant cell being equal to or exceeding the frequency of initiation from oriC itself.
In the viable ΔdnaA derivative of the dam mutant (carrying the additional tus and rpoB*35 mutations), we confirmed loss of the oriC peak in the copy number analysis by deep sequencing (Figure 4, panel vi). Instead, a terminus-to-oriC gradient was observed [as has been described previously for rnhA (34) and recG (25) mutants in absence of DnaA function], which is also consistent with the model proposed earlier of stochastic genome-wide cSDR initiation events (13).
The genome sequence data from the various strains also allowed us to verify their respective genotype status (such as Δdam, Δtus, rpoB*35, or ΔmutS). These data also served to exclude the possibility that in the ΔdnaA strains (which had been obtained following spontaneous loss of the shelter plasmid carrying the S. enterica dnaA+ gene), viability may have been the consequence of either (i) chromosomal integration of the S. enterica dnaA+ gene from the plasmid by homeologous recombination [which is known to occur when mismatch repair is compromised (67,68)], or (ii) mutation in oriC or any of the candidate genes conferring cSDR such as rnhA, recG, topA, recJ or any of the 3′-DNA exonucleases. Such confirmation was important, given that dam mutants exhibit an elevated frequency of spontaneous mutations (5,8); when compared with the parental strain, only one of the dam cultures (whose data are shown in Supplementary Figure S3A, panel ii) had no sequence changes, whereas each of the other cultures of dam derivatives harboured from 7 up to 18 additional single nucleotide polymorphisms (SNPs).
Rescue of ΔdnaA viability by dam occurs both in presence and absence of phi80 prophage
The DNA sequencing data had also indicated that the parental strain GJ13519 and all its derivatives including the dam and ΔdnaA mutants fortuitously bear the SOS-inducible prophage phi80 (see Supplementary Figure S3B), which is known to spread horizontally across strains in a laboratory environment (46,69,70). The copy number of phi80 genes in the wild-type strain was elevated ∼4-fold upon SOS induction with sublethal phleomycin (Supplementary Figure S3B, compare panels i and iii). It was also increased in one of the two dam mutant cultures (Supplementary Figure S3B, elevated for culture in panel iv but not for that in panel ii); dam mutants have earlier been described to exhibit chronic SOS induction (71,72). In no instance was there copy number elevation of chromosomal genes flanking the prophage attachment sites, suggesting that the prophage was not a source of oriC-independent replication initiation for cSDR or iSDR.
To further test whether the prophage, or its induction, might in any way have contributed to rescue of ΔdnaA lethality by the dam mutation, we prepared derivatives of tus rpoB*35 strains that were free of phi80 prophage and observed identical phenotypes in them (as that described above for the lysogenic strains) of: ΔdnaA dam viability which is abolished by mutS mutation, and ΔdnaA dam mutS viability (but not of ΔdnaA dam+ mutS+) in presence of phleomycin (Supplementary Figure S4B). We conclude that the presence or absence of phi80 prophage is immaterial to exhibition of the cSDR phenotypes associated with Dam deficiency.
Comparisons with cSDR in rnhA and recG mutants
As mentioned above, previous experiments to demonstrate the ability of rnhA or recG mutations to confer growth under DnaA-deficient conditions, have all employed strains carrying the temperature-sensitive dnaA46 allele; hence the derivatives could be tested for growth only at the restrictive temperature (42°). Given our use of a ΔdnaA null allele in this study, we have re-examined some of the cSDR features in rnhA and recG mutants.
The following additional novel properties of rnhA- and recG-mediated rescue of lethality associated with complete deficiency of DnaA were identified in these experiments. (i) It has previously been reported, from the experiments at 42°C, that DnaA-deficient rnhA is viable on minimal medium and that the tus and rpoB*35 mutations are additionally needed for the derivatives to be viable on rich media (26). These results were confirmed in our experiments using ΔdnaA, but we found in addition that ΔdnaA rnhA (without tus or rpoB*35) is viable on LB medium at 30°C (Figure 5A). (ii) The ΔdnaA rnhA derivative was rendered viable on LB at 42°C upon introduction not only of rpoB*35 as earlier reported (26) but also of a sulA mutation (Figure 5B), the latter indicating that ΔdnaA rnhA inviability is only because of SOS-induced hyper-filamentation under these conditions (73). Our microscopy experiments confirmed findings from earlier studies (17) that DnaA-deficient rnhA cells are extremely filamented, and showed additionally that this is reversed by sulA or rpoB*35 mutations (Figure 5C). (iii) As described above for dam ΔdnaA tus rpoB*35, so too the viability of recG ΔdnaA tus rpoB*35, and of rnhA ΔdnaA on LB, was lost upon introduction of the ΔdinG mutation (Figure 5D, compare panels i and iii with, respectively, ii and iv); this loss in the recG derivative was reversed once again with Ara-induced RNase HI overexpression (Figure 5D, compare panels v and vi). (Note that this experiment could not be attempted in the rnhA mutant, because of complementation of the rnhA defect itself under these conditions.) Furthermore, the rnhA ΔdnaA dinG derivative remained viable on glucose-minimal A (data not shown). (iv) In the rnhA ΔdnaA strain carrying tus and rpoB*35 mutations, even RecA was dispensable for viability on glucose-minimal A medium at 30°C, but not so at 42°C nor even on LB at 30°C (Figure 5D, compare panels x–xii). As further discussed below, we interpret these data in terms of the quantitative threshold model introduced above for detecting cSDR-mediated rescue of ΔdnaA lethality, in which facilitatory roles are played by growth on minimal medium at 30° (as opposed to rich medium or 42°C), and by the rpoB*35 and tus mutations.
Figure 5.

cSDR features in rnhA and recG mutants. Spotting at serial 10-fold dilutions, at the indicated temperatures, of suspensions of: (A) GJ16476 (ΔdnaA rnhA) on LB and glucose-minimal A (MM); and (B) in order, GJ16476/pHYD2388, GJ16476, GJ16498 and GJ18612 on LB. (C) Phase contrast microscopy of cultures of strains shown in panel B, after growth at 30°C or 42°C. Scale bar, 3 μm. (D) Plating of isogenic derivatives of ΔdnaA strain carrying dnaA+ shelter plasmid pHYD2388 and the additional genetic perturbation(s) as indicated on top of each panel on (unless otherwise indicated) LB-Xgal at 37°; representative images are shown, and the percentage of white colonies to total for each panel is indicated as explained in the legend to Figure 2. Strains employed for the different panels were pHYD2388 (or pHYD4805, in case of recG mutants) derivatives of: i, GJ16476; ii, GJ18620; iii, GJ16464; iv, GJ16489; v, GJ18621; vi, GJ18622; vii, GJ18623; viii, GJ16490; ix, GJ18627; and x-xii, GJ16499. Plates depicted in panels v–ix were supplemented with 0.2% arabinose. The plate of panel x was incubated at 30°C; while those of panels xi and xii comprised glucose-minimal A medium (MM) incubated at 30°C and 42°C, respectively. (E) Immunoblotting with S9.6 monoclonal antibody of total nucleic acids prepared from cultures of wild-type (WT) strain GJ13519 and its isogenic rnhA, dam, or recG derivatives GJ15833, GJ18617, and GJ17503, respectively.
Relationship between R-loops and cSDR
Finally, we examined the correlation if any between cSDR in the rnhA, recG, or dam mutants on the one hand, and R-loop abundance in them on the other. Kogoma (17) had earlier suggested that R-loops are responsible for cSDR in both rnhA and recG mutants, but their role in the case of recG has subsequently been questioned (25–29). In the dam mutants as well, we presumed that D-loops and not R-loops may be responsible for cSDR. The S9.6 monoclonal antibody (which is specific for RNA-DNA hybrids) has previously been used to demonstrate R-loop accumulation in different E. coli mutants (19,54); through a similar immunoblotting approach, we found that R-loop abundance (in comparison with that in the wild-type strain) is indeed elevated in the rnhA mutant [as has also been reported earlier (19)] but not so in the recG or dam mutants (Figure 5E). In a related observation, ectopic expression of the R-loop helicase UvsW also abolished rescue of ΔdnaA lethality by rnhA but not that by recG or dam mutations (Figure 5D, panels vii–ix).
DISCUSSION
Loss of function mutations that confer cSDR offer insights into mechanisms that otherwise act to prevent aberrant chromosomal replication in bacterial cells. RNase HI, RecG, topoisomerase I, and the single-strand DNA exonucleases have earlier been shown to play such a protective role, and in this work we have identified the Dam DNA methylase as yet another enzyme that is independently required for this purpose. A comparison between the various features of cSDR in the corresponding mutants is given in Supplementary Table S3. Our cSDR studies have also employed the more rigorous ΔdnaA mutation instead of the dnaA46 temperature-sensitive allele that was used in the previous studies.
All the proteins above likely act in different ways to prevent abnormal oriC- and DnaA-independent replication initiation; ΔdnaA viability under these conditions would of course be contingent on the replication forks successfully progressing to completion around the circular chromosome after overcoming both head-on collisions with transcription complexes (especially at rrn operons), and the trapping of forks in the terminus region. As explained below, we believe that a quantitative threshold model can account for many of the findings related to cSDR phenotypes, in both the present and earlier studies.
A quantitative threshold model to link cSDR with rescue of ΔdnaA lethality
Because, as stated above, of the problems faced by ectopically initiated replication forks to progress to completion around the chromosome (13), we propose a quantitative threshold model that links cSDR to the rescue of ΔdnaA lethality. Accordingly, ΔdnaA viability can be achieved either by having sufficiently large numbers of cSDR initiation events, or by fewer events in coordination with features or properties that facilitate the efficient retrograde progression (that is, towards oriC) of replication forks across the chromosome.
In our model, the facilitators for retrograde replication fork progression include the rpoB*35 and Δtus mutations, DinG helicase (and in its absence RNase HI overexpression), growth on minimal medium, and low incubation temperature. The notion that high growth temperatures may be stressful for chromosomal replication might be in accord with the hypothesis of Pogliano and Beckwith (74), which posits a correlation between environmental conditions to which an essential physiological process may be sensitive and conditional phenotypes obtained with mutations affecting that process. The Pogliano-Beckwith model may additionally explain why the sulA mutation is required at 42°C but not at 30°C to allow ΔdnaA rnhA growth on rich medium, since cell division functions may also be considered as sub-optimal at elevated temperatures (75).
Two roles of Δdam in cSDR
At 37°C on LB medium, dam but not dam ΔmutS was able to suppress ΔdnaA lethality, suggesting that increased DSEs are needed for the suppression. On the other hand, sublethal phleomycin could suppress ΔdnaA lethality in the dam ΔmutS but not dam+ derivatives. Alterations of gene expression such as those caused by SOS induction, or by presence of ΔdnaA or rpoB*35 mutations, cannot account for this difference, since they may be expected to have occurred to the same extent in both strains following phleomycin treatment. Therefore, the simplest hypothesis to explain these findings is that increased DSEs (generated either by MutHLS action in Δdam or by phleomycin in dam+) are necessary, but not sufficient, for rescue of ΔdnaA inviability; and that there is a second perturbation imposed by Dam deficiency (which is manifest even in the dam mutS strain) that is also required for the purpose.
In its first role, the dam-generated DSEs are expected to trigger break-induced DNA replication at the lesion sites, via formation of D-loops following the actions of recombinational repair proteins RecA, RecBCD, and RuvABC (17,30,64). The likely second role for Δdam in cSDR is not known, but our results indicate that this role does not involve SeqA. Since DSEs are clearly required for cSDR in the dam mutant, one may presume that the putative second role is in some way connected to replication restart.
It is possible that Dam may modulate replication restart indirectly, by affecting nucleoid structure or through transcriptional regulation of genes involved in the process. On the other hand, we would like to propose that Dam influences replication restart more directly, and that in dam mutants there is an increased likelihood of ‘reverse restart’, so that the newly assembled replisome is directed to proceed towards oriC instead of towards the terminus.
Different models have been put forth earlier for reverse restart-like mechanisms during Red-mediated recombination in phage λ (76) and in E. coli recG (24,29) or recD (77) mutants, any of which could apply in the dam mutant. The feature common to all of them, and which distinguishes them from normal replication restart, is that whereas in the latter there is one terminus-directed replisome that is assembled with the aid of PriABC and DnaT proteins following recombinational repair of a DSE, reverse restart results in a total of three replisomes, one directed towards oriC and two towards the terminus.
One such scheme for the dam mutant is outlined in Figure 6, which is based on the similar proposal earlier for recG (24,29). In this model, in absence of Dam methylation, the actions of PriA (including its helicase activity) in combination with those of PriB-DnaT or PriC lead to aberrant loading of the DnaB replicative helicase on the strand marked in red in panel C of Figure 6, so that the assembled replisome is now oriented for progression towards oriC (Figure 6, panel E). It is noteworthy in this context that the priA300 allele (encoding helicase-deficient PriA) suppresses the rescue, in both dam (this study) and recG (25) mutants, of lethality caused by deficiency of DnaA. The other similarities between these two mutants for cSDR is also brought out in Supplementary Table S3.
Figure 6.

Model for a second role of dam in cSDR. (A) Shown schematically is a MutHLS-generated double strand break (arrowhead) in one of the daughter duplexes immediately behind a replisome that is progressing (ochre arrow) from oriC to Ter on the clockwise replichore in a dam mutant cell. (5′ to 3′ polarity for top strand in each of the duplexes is from left to right.) (B) Following RecBCD-mediated DNA degradation (shown by interrupted lines) from both ends of the break (64), the ori-proximal DSE that remains participates in repair by homologous recombination (green rectangle) with its sister duplex; alternatively (not shown), such recombination can occur also with a ‘cousin’ duplex (50,83). (C) One intermediate in the pathway of recombinational repair is shown, with the pair of mauve arrows marking sites of RuvC-mediated Holliday junction resolution. (D) Disposition of DNA strands following normal replication restart, with DnaB helicase (not shown) translocating from left to right on top strand of the parental duplex, towards Ter. (E) Disposition of DNA strands following reverse restart; PriA-mediated DnaB helicase loading is postulated to have occurred on strand shown in red (this panel, and panel C) with its translocation proceeding from right to left, towards oriC. The step shown by the question mark, in the progression from panel C to panel E, is to indicate that the intermediates in the reverse restart pathway are at present undefined. (F) Disposition of DNA strands and of three replisomes following homologous recombination (at site marked by green rectangle in panel E) and normal restart, similar to pathway depicted in panels B through D.
If our model is correct, it remains to be investigated whether Dam methylation is needed for proper functioning of RecG (24,29), of PriA (78) or of both proteins in avoidance of reverse restart. The role postulated here for Dam in replication restart may also explain a curious finding from phylogenetic analyses that the Dam and PriB proteins have co-evolved, that is, different bacterial clades either possess the two proteins or they have neither (1,3).
Relationship between cSDR and occurrence of the mid-terminus peak in copy number analysis
The occurrence of peaks in chromosomal copy number distribution profiles is most often interpreted as origin sites for replication initiation, which has also been the case for mid-terminus peaks observed for the rnhA, recG, topA and the triple-exo mutants (19,20,25–27,34). On the other hand, we have earlier argued that whereas every origin (of finite strength) will be represented by a peak in the profile, the converse may not necessarily be true, especially for a peak in the mid-terminus region (13).
Thus the alternative model proposed for cSDR (13) suggests a genome-wide distribution of replication origins each of which fire stochastically at a very low frequency, with the mid-terminus peak being generated by algebraic summation of copy numbers from the different subpopulations of cells. A crucial contributory feature in this model is that when a replication fork is arrested at a Tus-bound Ter site for a significant period of time (that is, in the absence of its fusion with another fork coming in the opposite direction), there is marked degradation of DNA proximal to the arrest site; this element of the model is strongly supported by data from a large number of earlier studies on strains with ectopic replication origins or ectopic Ter sites (13,25,27,79,80).
Rudolph and coworkers (51) have also recently shown that for a triple-ori strain (that is, with native oriC and two additional oriC sequences ectopically positioned in the two replichores), the copy number profile comprises not only the three ori-associated peaks but also a mid-terminus peak. For the latter, they have proposed a similar model as discussed above, of copy number summation from different subpopulations in the culture.
Hence the finding in this work of a mid-terminus peak in the Δdam mutant is consistent with the model of genome-wide distribution of stochastically firing replication origins, coupled with DNA degradation proximal to forks arrested at Ter sites (13). We believe it unlikely that the dam mid-terminus peak itself represents a site of replication initiation. The fact that a similar peak is seen also in the wild-type strain grown with sublethal doses of phleomycin (Supplementary Figure S3A, panel iii), suggests that such distributed sites of replication initiation occur under these conditions as well. These results, as well as those from flow cytometry in Figure 1A, point to the close relationship between cSDR and iSDR, as had initially been proposed by Kogoma (17).
Can replication initiation triggered genome-wide by Dam deficiency exceed that from oriC?
It is also remarkable from the copy number analyses that the oriC peak, and bidirectional gradient therefrom, in a dnaA+ dam mutant during log-phase growth is almost completely abolished in derivatives that additionally carry the rpoB*35 or tus rpoB*35 mutations. We propose that the reason for flattening of the gradient in the latter two strains is because the true strength of the stochastically distributed cSDR origins in the dam mutant is revealed only when the problems of fork progression across the chromosome are alleviated (by tus and rpoB*35). Consequently, there is now little difference in copy number distributions between the dnaA+ and ΔdnaA derivatives of the dam tus rpoB*35 strains (compare panels v and vi in Figure 4); and the patterns in both are indeed reminiscent of that in the Haloferax volcanii mutant in which replication initiation was postulated to occur from widely distributed origins following site-specific deletion of its four discrete ori sequences (81).
RecA and cSDR
For cSDR and rescue of ΔdnaA lethality, one can conceive of multiple roles of RecA (17,64,73), including in (i) generation of substrates for aberrant replication initiation, (ii) direct recombinational repair following problems of transcription-replication conflict or of fork breakage at Tus-bound Ter sites, and (iii) SOS induction (of genes such as UvrD that are also involved in taking care of such problems). Earlier work had indicated that cSDR in both rnhA and recG mutants is abolished by recA mutation (17,25,26), and it was suggested that the formation of, respectively, R-loops and D-loops for replication initiation in the two cases required RecA function.
Against this scenario, it is indeed remarkable that our present work has demonstrated rescue of ΔdnaA lethality (at 30°C) even in presence of recA mutation in the rnhA mutant. This finding is consistent with recent models that transcription-associated R-loop formation is RecA-independent (82). The features of the quantitative threshold model may also be important here, in that an absence of RecA may simply reduce the likelihood of successful completion of DnaA-independent replication in the mutants.
DATA AVAILABILITY
The genome sequence data from this work are available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA513325.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jon Beckwith, Olivier Espeli, David Leach, Hirotada Mori and Steve Sandler for strains and plasmids; P Himabindu, Saswat Mohapatra and Prajakta Tathe for assistance with experiments; Aswin Seshasayee and T.V. Reshma for help with deep sequencing and data analysis; and Andrei Kuzminov and the COE team members for advice and discussions.
Author contributions: The major experimental contributions to all aspects of this work were made by N.R. J.K.L. standardized the assays to test for suppression of ΔdnaA lethality, including construction of plasmid pHYD2388 and recombineering of ΔdnaA. S.G. performed the flow cytometry and microscopy experiments for cSDR, assisted by N.R. Some of the sequence data analyses were performed by A.P. J.G. provided overall supervision for the work and drafted the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre of Excellence (COE) project for Microbial Biology – Phase 2; DST-INSPIRE fellowship (NR); CSIR research fellowship (to S.G.); J.C. Bose fellowship and INSA Senior Scientist award (to J.G.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Løbner-Olesen A., Skovgaard O., Marinus M.G.. Dam methylation: coordinating cellular processes. Curr. Opin. Microbiol. 2005; 8:154–160. [DOI] [PubMed] [Google Scholar]
- 2. Wion D., Casadesús J.. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006; 4:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marinus M.G., Løbner-Olesen A.. DNA methylation. EcoSal Plus. 2014; 14:517–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marinus M.G., Morris N.R.. Isolation of deoxyribonucleic acid methylase mutants of Escherichia coli K-12. J. Bacteriol. 1973; 114:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marinus M.G., Morris N.R.. Biological function for 6-methyladenine residues in the DNA of Escherichia coli K-12. J. Mol. Biol. 1974; 85:309–322. [DOI] [PubMed] [Google Scholar]
- 6. Putnam C.D. Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst). 2016; 38:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGraw B.R., Marinus M.G.. Isolation and characterization of Dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol. Gen. Genet. 1980; 178:309–315. [DOI] [PubMed] [Google Scholar]
- 8. Glickman B.W., Radman M.. Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc. Natl. Acad. Sci. U.S.A. 1980; 77:1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Au K.G., Welsh K., Modrich P.. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992; 267:12142–12148. [PubMed] [Google Scholar]
- 10. Marinus M.G. Recombination is essential for viability of an Escherichia coli dam (DNA adenine methyltransferase) mutant. J. Bacteriol. 2000; 182:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harinarayanan R., Gowrishankar J.. A dnaC mutation in Escherichia coli that affects copy number of Co1E1-like plasmids and the PriA-PriB (but not Rep-PriC) pathway of chromosomal replication restart. Genetics. 2004; 166:1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Donnell M., Langston L., Stillman B.. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 2013; 5:a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gowrishankar J. End of the beginning: elongation and termination features of alternative modes of chromosomal replication initiation in bacteria. PLos Genet. 2015; 11:e1004909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neylon C., Kralicek A. V, Hill T.M., Dixon N.E.. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus- Ter complex. Microbiol. Mol. Biol. Rev. 2005; 69:501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duggin I.G., Wake R.G., Bell S.D., Hill T.M.. The replication fork trap and termination of chromosome replication. Mol. Microbiol. 2008; 70:1323–1333. [DOI] [PubMed] [Google Scholar]
- 16. Kaplan D.L., Bastia D.. Mechanisms of polar arrest of a replication fork. Mol. Microbiol. 2009; 72:279–285. [DOI] [PubMed] [Google Scholar]
- 17. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997; 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martel M., Balleydier A., Sauriol A., Drolet M.. Constitutive stable DNA replication in Escherichia coli cells lacking type 1A topoisomerase activity. DNA Repair (Amst). 2015; 35:37–47. [DOI] [PubMed] [Google Scholar]
- 19. Brochu J., Vlachos-Breton É., Sutherland S., Martel M., Drolet M.. Topoisomerases I and III inhibit R-loop formation to prevent unregulated replication in the chromosomal Ter region of Escherichia coli. PLos Genet. 2018; 14:e1007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Midgley-Smith S.L., Dimude J.U., Rudolph C.J.. A role for 3′ exonucleases at the final stages of chromosome duplication in Escherichia coli. Nucleic Acids Res. 2019; 47:1847–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox M.M., Goodman M.F., Kreuzer K.N., Sherratt D.J., Sandler S.J., Marians K.J.. The importance of repairing stalled replication forks. Nature. 2000; 404:37–41. [DOI] [PubMed] [Google Scholar]
- 22. Windgassen T.A., Wessel S.R., Bhattacharyya B., Keck J.L.. Mechanisms of bacterial DNA replication restart. Nucleic Acids Res. 2018; 46:504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michel B., Sandler S.J.. Replication restart in bacteria. J. Bacteriol. 2017; 199:e00102-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michel B., Sinha A.K., Leach D.R.F.. Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 2018; 82:e00013-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rudolph C.J., Upton A.L., Stockum A., Nieduszynski C.A., Lloyd R.G.. Avoiding chromosome pathology when replication forks collide. Nature. 2013; 500:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dimude J.U., Stockum A., Midgley-Smith S.L., Upton A.L., Foster H.A., Khan A., Saunders N.J., Retkute R., Rudolph C.J.. The consequences of replicating in the wrong orientation: bacterial chromosome duplication without an active replication origin. MBio. 2015; 6:e01294-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Midgley-Smith S.L., Dimude J.U., Taylor T., Forrester N.M., Upton L., Lloyd R.G., Rudolph C.J.. Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res. 2018; 46:7701–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wendel B.M., Courcelle C.T., Courcelle J.. Completion of DNA replication in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Azeroglu B., Mawer J.S.P., Cockram C.A., White M.A., Hasan A.M.M., Filatenkova M., Leach D.R.F.. RecG directs DNA synthesis during double-strand break Repair. PLos Genet. 2016; 12:e1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anand R.P., Lovett S.T., Haber J.E.. Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 2013; 5:a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McGlynn P., Lloyd R.G.. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000; 101:35–45. [DOI] [PubMed] [Google Scholar]
- 32. Trautinger B.W., Jaktaji R.P., Rusakova E., Lloyd R.G.. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol. Cell. 2005; 19:247–258. [DOI] [PubMed] [Google Scholar]
- 33. Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E.. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011; 146:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maduike N.Z., Tehranchi A.K., Wang J.D., Kreuzer K.N.. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol. Microbiol. 2014; 91:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller J.H. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. 1992; NY: Cold Spring Harbor Lab Press. [Google Scholar]
- 36. Anupama K., Leela J.K., Gowrishankar J.. Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for Rho-dependent transcription termination. Mol. Microbiol. 2011; 82:1330–1348. [DOI] [PubMed] [Google Scholar]
- 37. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:doi:10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guzman L.M., Belin D., Carson M.J., Beckwith J.. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995; 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andrews A.E., Lawley B., Pittard A.J.. Mutational analysis of repression and activation of the tyrP gene in Escherichia coli. J. Bacteriol. 1991; 173:5068–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H.. Complete set of ORF clones of Escherichia coli ASKA library (A complete Set of E. coli K-12 ORF Archive): unique resources for biological research. DNA Res. 2005; 12:291–299. [DOI] [PubMed] [Google Scholar]
- 42. Galli E., Midonet C., Paly E., Barre F.-X.. Fast growth conditions uncouple the final stages of chromosome segregation and cell division in Escherichia coli. PLos Genet. 2017; 13:e1006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gowrishankar J. Identification of osmoresponsive genes in Escherichia coli: evidence for participation of potassium and proline transport systems in osmoregulation. J. Bacteriol. 1985; 164:434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sambrook J., Russell D.. Molecular Cloning: A Laboratory Manual. 2001; 3rd edn.NY: Cold Spring Harbor Lab Press. [Google Scholar]
- 45. Harinarayanan R., Gowrishankar J.. Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. J. Mol. Biol. 2003; 332:31–46. [DOI] [PubMed] [Google Scholar]
- 46. Harms A., Fino C., Sørensen M.A., Semsey S., Gerdes K.. Prophages and growth dynamics confound experimental results with antibiotic-tolerant persister cells. MBio. 2017; 8:e01964-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boyd D., Weiss D.S., Chen J.C., Beckwith J.. Towards single-copy gene expression systems making gene cloning physiologically relevant: Lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J. Bacteriol. 2000; 182:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roberts D., Hoopes B.C., McClure W.R., Kleckner N.. IS10 transposition is regulated by DNA adenine methylation. Cell. 1985; 43:117–130. [DOI] [PubMed] [Google Scholar]
- 49. del Solar G., Giraldo R., Ruiz-Echevarría M.J., Espinosa M., Díaz-Orejas R.. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998; 62:434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khan S.R., Mahaseth T., Kouzminova E.A., Cronan G.E., Kuzminov A.. Static and dynamic factors limit chromosomal replication complexity in Escherichia coli, avoiding dangers of runaway overreplication. Genetics. 2016; 202:945–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dimude J.U., Stein M., Andrzejewska E.E., Khalifa M.S., Gajdosova A., Retkute R., Skovgaard O., Rudolph C.J.. Origins left, right, and centre: increasing the number of initiation sites in the Escherichia coli chromosome. Genes. 2018; 9:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martel M., Balleydier A., Brochu J., Drolet M.. Drolet M. Detection of oriC independent replication in Escherichia coli Cells. DNA Topoisomerases. Methods in Molecular Biology. 2018; 1703:NY: Humana Press; 131–138. [DOI] [PubMed] [Google Scholar]
- 53. Espeli O., Mercier R., Boccard F.. DNA dynamics vary according to macrodomain topography in the E. coli chromosome. Mol. Microbiol. 2008; 68:1418–1427. [DOI] [PubMed] [Google Scholar]
- 54. Raghunathan N., Kapshikar R.M., Leela J.K., Mallikarjun J., Bouloc P., Gowrishankar J.. Genome-wide relationship between R-loop formation and antisense transcription in Escherichia coli. Nucleic Acids Res. 2018; 46:3400–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sivaramakrishnan P., Sepúlveda L.A., Halliday J.A., Liu J., Núñez M.A.B., Golding I., Rosenberg S.M., Herman C.. The transcription fidelity factor GreA impedes DNA break repair. Nature. 2017; 550:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bhattacharyya S., Soniat M.M., Walker D., Jang S., Finkelstein I.J., Harshey R.M.. Phage Mu Gam protein promotes NHEJ in concert with Escherichia coli ligase. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E11614–E11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leela J.K., Syeda A.H., Anupama K., Gowrishankar J.. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boubakri H., de Septenville A.L., Viguera E., Michel B.. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010; 29:145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lang K.S., Hall A.N., Merrikh C.N., Ragheb M., Tabakh H., Pollock A.J., Woodward J.J., Dreifus J.E., Merrikh H.. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017; 170:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., Mooney R.A., Grass J.A., Sivaramakrishnan P., Herman C., Landick R., Wang J.D.. DksA guards elongating RNA polymerase against ribosome-stalling-induced arrest. Mol. Cell. 2014; 53:766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuzminov A. When DNA topology turns deadly - RNA polymerases dig in their R-Loops to stand their ground: new positive and negative (super)twists in the replication-transcription conflict. Trends Genet. 2018; 34:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu M., Campbell J.L., Boye E., Kleckner N.. SeqA: A negative modulator of replication initiation in E. coli. Cell. 1994; 77:413–426. [DOI] [PubMed] [Google Scholar]
- 63. Løbner-Olesen A., Marinus M.G., Hansen F.G.. Role of SeqA and Dam in Escherichia coli gene expression: a global/microarray analysis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4672–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999; 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kouzminova E.A., Rotman E., Macomber L., Zhang J., Kuzminov A.. RecA-dependent mutants in Escherichia coli reveal strategies to avoid chromosomal fragmentation. Proc. Natl. Acad. Sci. USA. 2004; 101:16262–16267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol. 1995; 16:373–384. [DOI] [PubMed] [Google Scholar]
- 67. Rayssiguier C., Thaler D.S., Radman M.. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989; 342:189–192. [DOI] [PubMed] [Google Scholar]
- 68. Matic I., Rayssiguier C., Radman M.. Interspecies gene exchange in bacteria: The role of SOS and mismatch repair systems in evolution of species. Cell. 1995; 80:507–515. [DOI] [PubMed] [Google Scholar]
- 69. Rotman E., Amado L., Kuzminov A.. Unauthorized horizontal spread in the laboratory environment: The tactics of Lula, a temperate lambdoid bacteriophage of Escherichia coli. PLoS One. 2010; 5:e11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rotman E., Kouzminova E., Plunkett G., Kuzminov A.. Genome of enterobacteriophage lula/phi80 and insights into its ability to spread in the laboratory environment. J. Bacteriol. 2012; 194:6802–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Saisree L., Reddy M., Gowrishankar J.. lon incompatibility associated with mutations causing SOS induction: null uvrD alleles induce an SOS response in Escherichia coli. J. Bacteriol. 2000; 182:3151–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McCool J.D., Long E., Petrosino J.F., Sandler H.A., Rosenberg S.M., Sandler S.J.. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol. Microbiol. 2004; 53:1343–1357. [DOI] [PubMed] [Google Scholar]
- 73. Simmons L.A., Foti J.J., Cohen S.E., Walker G.C.. The SOS regulatory network. EcoSal Plus. 2013; doi:10.1128/ecosalplus.5.4.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pogliano K.J., Beckwith J.. The Cs sec mutants of Escherichia coli reflect the cold sensitivity of protein export itself. Genetics. 1993; 133:763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Blaauwen T. Den, Hamoen L.W., Levin P.A.. The divisome at 25:the road ahead. Curr. Opin. Microbiol. 2017; 36:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Skalka A. Grell R. A replicator's view of recombination (and repair). Mechanisms in Recombination. 1974; Boston, MA: Springer US; 421–432. [Google Scholar]
- 77. White M.A., Azeroglu B., Lopez-Vernaza M.A., Hasan A.M.M., Leach D.R.F.. RecBCD coordinates repair of two ends at a DNA double-strand break, preventing aberrant chromsome replication. Nucleic Acids Res. 2018; 46:6670–6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Windgassen T.A., Leroux M., Satyshur K.A., Sandler S.J., Keck J.L.. Structure-specific DNA replication-fork recognition directs helicase and replication restart activities of the PriA helicase. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:9075–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R.. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015; 520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ivanova D., Taylor T., Smith S.L., Dimude J.U., Upton A.L., Mehrjouy M.M., Skovgaard O., Sherratt D.J., Retkute R., Rudolph C.J.. Shaping the landscape of the Escherichia coli chromosome: replication-transcription encounters in cells with an ectopic replication origin. Nucleic Acids Res. 2015; 43:7865–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hawkins M., Malla S., Blythe M.J., Nieduszynski C.A., Allers T.. Accelerated growth in the absence of DNA replication origins. Nature. 2013; 503:544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gowrishankar J., Leela J.K., Anupama K.. R-loops in bacterial transcription. Transcription. 2013; 4:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kuzminov A. Chromosomal replication complexity: a novel DNA metrics and genome instability factor. PLos Genet. 2016; 12:e1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequence data from this work are available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA513325.