

Abstract
Motivation
The emergence of single-cell RNA-sequencing has enabled analyses that leverage transitioning cell states to reconstruct pseudotemporal trajectories. Multidimensional data sparsity, zero inflation and technical variation necessitate the selection of high-quality features that feed downstream analyses. Despite the development of numerous algorithms for the unsupervised selection of biologically relevant features, their differential performance remains largely unaddressed.
Results
We implemented the neighborhood variance ratio (NVR) feature selection approach as a Python package with substantial improvements in performance. In comparing NVR with multiple unsupervised algorithms such as dpFeature, we observed striking differences in features selected. We present evidence that quantifiable dataset properties have observable and predictable effects on the performance of these algorithms.
Availability and implementation
pyNVR is freely available at https://github.com/KenLauLab/NVR.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Complex tissue systems consist of heterogeneous cell populations, and single-cell RNA-sequencing (scRNA-seq) is capable of extracting transcriptomic information while preserving this complexity. Each cell and its respective state become a high-dimensional data point (Tang et al., 2009). These data encompass nuanced transitional cell states, and through pseudotemporal trajectory reconstruction, these transitional states can be ordered to describe developmental dynamics (Trapnell et al., 2014). Computational techniques such as density-dependent k-Nearest Neighbors (k-NN) network traversal, minimum spanning trees and reverse graph embedding are examples of trajectory reconstruction approaches utilizing high-dimensional data (Herring et al., 2018a,b; Qiu et al., 2011, 2017).
However, these algorithmic approaches perform best when provided with high-quality features, which are often confounded by artifacts such as gene dropouts, resulting from incomplete transcriptomic sampling, and stochasticity, arising from the amplification of single-cell scale reaction materials (Kim and Marioni, 2013). Detectable cell-to-cell variation can also originate from stochastic gene expression, where an underlying level of randomness is captured at the time of sample processing (Elowitz, 2002; Raj and van Oudenaarden, 2008). These sources of variation necessitate machine learning strategies for the selection of biologically meaningful features (Herring et al., 2018a). Unsupervised feature selection algorithms such as neighborhood variance ratio (NVR), dpFeature (dpF), FindVariableGenes (FVG) and PCA-Based Feature Extraction (PCAFE) are distinct strategies for achieving this goal in the context of pseudotemporal analysis (Butler et al., 2018; Qiu et al., 2017; Taguchi, 2018; Welch et al., 2016). Although feature selection is essential, the assumptions and performance of these algorithms have not been systematically evaluated, confounding the applicability of these methods to different datasets. Here, we examine an underlying characteristic of high-dimensional data that interacts with these algorithms with a focus on NVR and dpFeature.
2 Software description
2.1 Implementation
pyNVR (Welch et al., 2016) was implemented in Python 2.7 with 3.7 compatibility. It is available through Github with installation instructions for multiple operating systems and tutorials with example datasets (https://github.com/KenLauLab/NVR). This package works as an unsupervised feature selection pipeline on quality controlled scRNA-seq data. scRNA-seq count or FPKM data act as inputs into this pipeline. The pseudocode for NVR is documented (Supplementary Material S1.1). We benchmarked our Python implementation of NVR against a previous R implementation across multiple machines.
2.2 Subject datasets
Datasets used in this study can be found on the NCBI GEO repository as GSE102698, GSE52529 and GSE60781 (Herring et al., 2018b; Schlitzer et al., 2015; Trapnell et al., 2014). These data were generated using different platforms and biological contexts (Supplementary Table S1).
2.3 Comparative performance
To compare the performance of NVR and dpFeature (Supplementary Material S1.2) in a controlled manner, we examined GSE102698, a dataset generated from the colonic epithelium, and sampled the data in two ways. First, we sampled the dataset to different cell numbers. We surmised that relationships within high-density subspaces would be more robust to data sampling, thus affecting how extensively their metric space neighborhoods are defined. Second, we directly controlled the distribution of the data by imposing closeness centrality thresholds on cell sampling (Supplementary Material S1.3, Supplementary Fig. S1). Closeness centrality, as defined by the normalized sum of the length of the shortest paths between a given node and all other nodes, is calculated on a density-based k-NN graph (Lever et al., 2017; Pearson, 1901). Nodes with low closeness represent disjointed cells comprising separable cell subpopulations, while those with high closeness occupy more central and inseparable positions within the graph. As a correlative output, we calculated gene set similarities by taking their Jaccard indices, describing divergence between algorithm performance (Levandowsky and Winter, 1971) (Supplementary Material S1.4). This workflow is further described in Supplementary Figure S2.
2.4 Pseudotemporal analysis and gene ontology term enrichment
Given that cell closeness is a controllable parameter affecting data distribution, we further investigated its effects on downstream analyses. We performed pseudotemporal analysis on GSE102698 using feature-selected gene sets with p-Creode (Herring et al., 2018b) (Supplementary Material S1.5). To better understand the gene sets each algorithm selected for, we performed gene ontology (GO) term enrichment analysis using WebGestalt as described by the Zhang group (Supplementary Material S1.6) (Wang et al., 2017).
3 Results
We noted significant improvements in speed, with an average 14-fold decrease in runtime, when comparing our Python implementation against the R implementation (Fig. 1A, Supplementary Table S2). In applying NVR and dpFeature to distinct scRNA-seq datasets, we observed striking inconsistencies in the genes selected (Fig. 1B, Supplementary Table S1). We observed significant, positive linear relationships (Supplementary Table S3) between gene set Jaccard index, cell number (≤ 2.2e-16, Fig. 1C), and cell closeness sampling thresholds (P = 1.227e-06, Fig. 1D). We observed another performance divergence in examining their robustness, as defined by the resistance to performance decay given decreasing input data quality (Supplementary Fig. S3). Given these gene sets, we also observed distinct differences in GO term enrichment (Supplementary Fig. S4, Supplementary Table S4). Downstream p-Creode trajectory reconstruction using low closeness dpFeature gene sets resulted in the underrepresentation of stem-like developmental projections (Fig. 1E). Additionally, we analyzed two more methods in parallel, findVariableGenes and PCAFE (Supplementary Material S1.7–S1.8), and observed significant differences in performance (Supplementary Figs S5–S7).
Fig. 1.
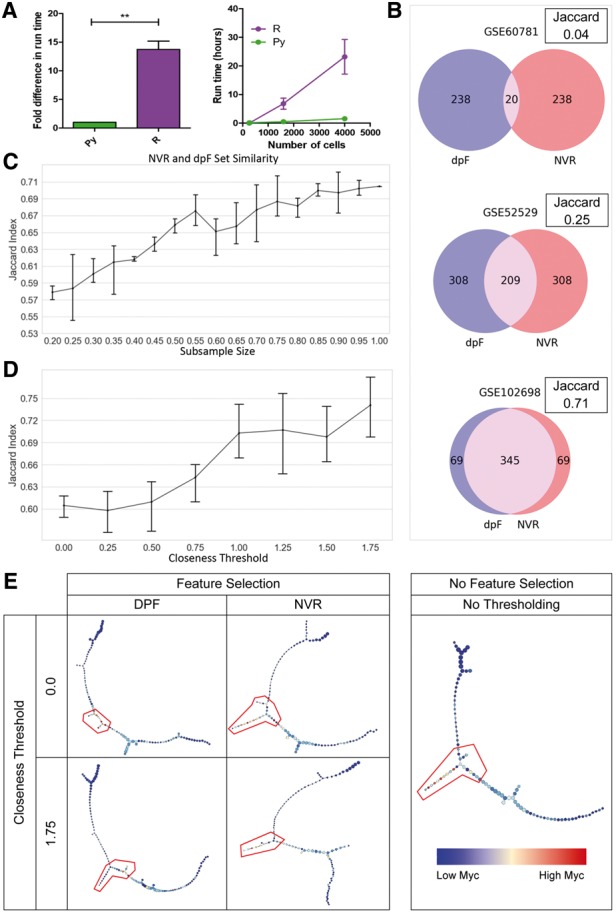
Evaluation of pyNVR performance. (A) Fold difference in runtime between the Python and R implementations of NVR. (B) Gene set similarity given different datasets. (C) Gene set similarity and its relationship with cell number. (D) Gene set similarity and its relationship with closeness threshold-imposed sampling. (E) Representative p-Creode graphs generated using genes selected from closeness-thresholded samples. Heatmap overlay and gating depicts Myc and putative stem-like cell states, respectively
4 Discussion
We reasoned that the algorithms examined were differentially impacted by data distributions, through examinations of cell number and closeness centrality. NVR and dpFeature rely on calculating metric space neighbors, in the context of graphs and t-Distributed Stochastic Neighbor Embedding (t-SNE) embeddings, respectively (Qiu et al., 2017; van der Maaten and Hinton, 2008; Welch et al., 2016). Both cell number and closeness affect the calculation of neighborhood-defining radial distances. NVR uses these distances to generate a k-NN graph. t-SNE, instead, uses these distances to calculate a probabilistic representation of neighborhoods. Beyond neighborhood representations, the algorithms also have unique selection criteria.
5 Conclusion
We created an accessible and significantly faster implementation of NVR feature selection. We present evidence suggesting that the performance of different unsupervised feature selection algorithms diverge based on dataset properties such as cell number and closeness. Downstream pseudotemporal or gene ontological analyses are demonstrably affected by the feature selection algorithm used.
Supplementary Material
Acknowledgements
The authors would like to thank the Vanderbilt Department of Biomedical Informatics for access to their compute servers.
Funding
K.S.L is funded by NIDDK [R01DK103831] and NCI [U2CCA233291]. B.C. is funded by a training grant from NLM [T32LM012412] and C.A.H. is funded by a pre-doctoral F31 from NIGMS [F31GM120940] and a training grant from NICHD [T32HD007502].
Conflict of Interest: none declared.
References
- Butler A., et al. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol., 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M.B. (2002) Stochastic gene expression in a single cell. Science, 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Herring C.A., et al. (2018a) Single-cell computational strategies for lineage reconstruction in tissue systems. Cell. Mol. Gastroenterol. Hepatol., 5, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring C.A., et al. (2018b) Unsupervised trajectory analysis of single-cell RNA-seq and imaging data reveals alternative tuft cell origins in the gut. Cell Syst., 6, 37–51.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Marioni J.C. (2013) Inferring the kinetics of stochastic gene expression from single-cell RNA-sequencing data. Genome Biol., 14, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levandowsky M., Winter D. (1971) Distance between sets. Nature, 234, 34–35. [Google Scholar]
- Lever J., et al. (2017) Points of significance: principal component analysis. Nat. Methods, 14, 641–642. [Google Scholar]
- Pearson K. (1901) LIII. On lines and planes of closest fit to systems of points in space. London, Edinburgh, Dublin Philos. Mag. J. Sci., 2, 559–572. [Google Scholar]
- Qiu P., et al. (2011) Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol., 29, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., et al. (2017) Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods, 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., van Oudenaarden A. (2008) Nature, nurture, or chance: stochastic gene expression and its consequences. Cell, 135, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A., et al. (2015) Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol., 16, 718–728. [DOI] [PubMed] [Google Scholar]
- Taguchi Y. (2018) Principal component analysis-based unsupervised feature extraction applied to single-cell gene expression analysis In: Huang D. et al. (eds), ICIC 2018: Intelligent Computing Theories and Application. Cham, Springer, 816–826. [Google Scholar]
- Tang F., et al. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods, 6, 377–382. [DOI] [PubMed] [Google Scholar]
- Trapnell C., et al. (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol., 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L.J.P., Hinton G.E. (2008) Visualizing high-dimensional data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Wang J., et al. (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res., 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.D., et al. (2016) SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol., 17, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.