

Abstract
Mitochondrial ribosomes are functionally specialized for the synthesis of several essential inner membrane proteins of the respiratory chain. Although remarkable progress has been made toward understanding the structure of mitoribosomes, the pathways and factors that facilitate their biogenesis remain largely unknown. The long unstructured domains of unassembled ribosomal proteins are highly prone to misfolding and often require dedicated chaperones to prevent aggregation. To date, chaperones that ensure safe delivery to the assembling ribosome have not been identified in the mitochondrion. In this study, a respiratory synthetic lethality screen revealed a role for an evolutionarily conserved mitochondrial matrix protein called Mam33 in Saccharomyces cerevisiae mitoribosome biogenesis. We found that the absence of Mam33 results in misassembled, aggregated ribosomes and a respiratory lethal phenotype in combination with other ribosome-assembly mutants. Using sucrose gradient sedimentation, native affinity purifications, in vitro binding assays, and SILAC-based quantitative proteomics, we found that Mam33 does not associate with the mature mitoribosome, but directly binds a subset of unassembled large subunit proteins. Based on these data, we propose that Mam33 binds specific mitoribosomal proteins to ensure proper assembly.
Keywords: mitochondria, chaperone, ribosome assembly, RNA helicase, mitochondrial respiratory chain complex, complement C1q-binding protein (C1QBP), COX1, protein misfolding, synthetic lethality
Introduction
Proper mitochondrial function requires the coordinated expression of two genomes. Although nuclear genes encode for roughly 99% of the mitochondrial proteome, the mitochondrial genome encodes several essential inner membrane proteins of the respiratory chain (1–4). Mitochondrial gene transcription, translation, and membrane insertion machinery are coupled and colocalized adjacent to the matrix surface of the inner membrane (3). Expression is predominantly controlled by a set of gene-specific translational activators, which cooperate to deliver specific mRNA transcripts to the mitoribosomes (5).
Mitochondrial ribosomes are highly specialized for translating the small set of highly hydrophobic membrane proteins encoded by mtDNA. Recent high-resolution structural analyses of human and yeast mitoribosomes have revealed major structural and functional differences from bacterial and eukaryotic ribosomes (6–12). These differences include: 1) the 5S rRNA of the large subunit (mtLSU)2 has been lost, resulting in a remodeled central protuberance (CP) region. 2) Mitoribosomes have incorporated additional proteins resulting in an increased protein to rRNA ratio. 3) Hydrophobic residues line the walls of the polypeptide tunnel exit (PTE), which presumably slows protein synthesis to facilitate cotranslational membrane insertion and assembly.
Recent knowledge has emphasized the biomedical importance of understanding the structure and assembly of mitoribosomes. Importantly, mutations in mitoribosomal proteins (MRPs), rRNAs, and assembly factors are responsible for a growing number of severe human disorders. These include a heterogeneous group of infantile multisystemic OXPHOS diseases such as sensorineural hearing loss, hypertrophic cardiomyopathy, Leigh's syndrome, and encephalomyopathy (13). In addition, mitoribosomes and their accessory proteins are increasingly being recognized as cancer biomarkers and targets for cancer therapy (14, 15). Furthermore, mitoribosomes are the unintended target of clinically important antibiotics used to treat bacterial infections, which can produce deleterious side effects (16, 17). For these reasons, it is important to understand how mitochondrial ribosomes are assembled.
Mitoribosome biogenesis is a remarkably complex process that requires coordinated expression of mtDNA-encoded rRNAs with nuclear-encoded chaperones, assembly factors, and MRPs. Although recent studies have provided a general framework for the order in which MRPs are assembled in the mtLSU, the details remain largely unknown (18, 19). Ten assembly factors have been reported thus far in Saccharomyces cerevisiae and can be classified as rRNA-modifying enzymes, MRP-processing enzymes, GTPases, and RNA helicases (13–15, 20). Surprisingly, proteins that bind and chaperone newly-imported MRPs during assembly have not been reported. This class of proteins is important because the long unstructured domains and high basic amino acid content of MRPs makes them particularly vulnerable to misfolding and aggregation.
Here, we report that an evolutionarily conserved mitochondrial acidic matrix protein called Mam33 chaperones newly-imported MRPs prior to their incorporation into the assembling ribosome. The broad clinical significance of this finding is emphasized by the ubiquitous nature of the mammalian ortholog p32/HABP1/gC1qR and its implications in cancer progression and mitochondrial disease (21). p32 is a multicompartmental protein with established roles in immunological response, cell cycle regulation, apoptosis, nuclear transcription, splicing, sperm-oocyte interactions, and respiratory chain biogenesis. Importantly, overexpression of p32 has been detected in nearly all tissue-specific forms of cancer and is correlated with poor prognosis in patients.
Despite its diverse cellular functions, p32 primarily localizes to the mitochondrial matrix in most cell types, and mutations or changes in its expression ultimately result in mitochondrial dysfunction (22–25). In normal fibroblasts, p32 overexpression results in ROS generation, increased Ca2+ uptake, loss of membrane potential, and the subsequent induction of apoptosis (26–28). In contrast, homozygous disruption of the p32 ortholog in mice leads to mid-gestation lethality (25). In human patients, bi-allelic mutations cause severe multisystemic defects in mitochondrial energy metabolism (25, 29). Given that p32 has been the subject of more than 150 reports and the accumulating evidence for its involvement in human disease, it is surprising that its precise mitochondrial function remains unknown.
Studies in mammals suggest that the function of p32 is conserved in other eukaryotes. Crystal structures show that the mammalian, fungal, and protozoan orthologs all form a homotrimeric doughnut-shaped protein, with a peculiar acidic face (30–32). The introduction of p32 can restore the respiratory growth impairment in yeast cells lacking Mam33 (23). Furthermore, the mtDNA instability and reduced mitochondrial gene expression observed in mice and human patients with disrupted p32 are reminiscent of yeast mitochondrial translation mutants (25, 29). We chose to study this protein in yeast because it is the only model organism with established techniques to genetically manipulate mtDNA and does not require a functional respiratory chain for viability.
We previously reported that Mam33 promotes the translation of mtDNA-encoded Cox1, the core catalytic subunit that nucleates the assembly of cytochrome c oxidase (33). Mam33 is not required for exponential-phase respiratory growth, but rather to efficiently adapt from fermentation to respiration, suggesting that a second factor compensates for Mam33 activity during respiration in yeast. In our present study, we sought to find proteins functionally redundant with Mam33 during respiration. Interestingly, the proteins identified were also necessary for Cox1 translation, and suggested a role for Mam33 in mitoribosome assembly.
Results
Point mutations in genes necessary for mitochondrial translation are respiratory synthetic lethal with mam33Δ
We previously reported that in the absence of Mam33, Cox1 translation is impaired, and cells poorly adapt to respiratory conditions because they lack basal fermentative levels of Cox1 (33). Given that these cells still respire normally, we reasoned that this fermentation-specific phenotype could be explained by a redundant pathway that functions during respiration. To identify the components of this pathway, we utilized the phenomenon of respiratory synthetic lethality. This approach assumes that if an alternate pathway compensates for Mam33 during respiration, then mam33Δ cells bearing a mutation in this second pathway will fail to respire. Additionally, mutations that are respiratory lethal in combination with mam33Δ might reveal factors that regulate the same process as Mam33.
To identify respiratory synthetic lethal mutants, chemically mutagenized clones of mam33Δ cells were screened for the inability to respire following the loss of a plasmid containing WT MAM33 using an ADE3/ade2/ade3-based colony-sectoring assay (34–36). Three recessive mutants that fulfilled the criteria of being respiratory synthetically lethal with mam33Δ were divided into two complementation groups, and the corresponding WT genes were cloned from a genomic library. From this screen, we identified three alleles in two different genes that are respiratory synthetic lethal in combination with mam33Δ: mrh4-1, mrpl4-1, and mrpl4-2 (Fig. 1).
Figure 1.

Point mutations in genes necessary for mitochondrial translation are respiratory synthetic lethal with mam33Δ. A, Mrh4 and its human ortholog DDX28 contain conserved sequence motifs typical of DEAD-box RNA helicases. A single amino acid substitution in an RNA-binding domain produces a respiratory synthetic lethal phenotype in mam33Δ cells (shown in red). B, MrpL4 contains a conserved sequence domain of the MrpL47 superfamily. The two amino acid substitutions obtained from the mam33Δ respiratory synthetic lethal screen (shown in red). C, mutations in MRH4 and MRPL4 are respiratory synthetic lethal in combination with mam33Δ. Cells were streaked onto SC plates containing fermentable (glucose) or respiratory (glycerol + ethanol) carbon sources and incubated at 30 °C for 2 or 4 days, respectively.
Interestingly, Mrh4 and MrpL4 are directly associated with the mitochondrial translation machinery. Mrh4 is a mitochondrial DEAD-box RNA helicase that is essential for mtLSU assembly (37, 38). This protein has several conserved domains and motifs that are typical of DEAD-box RNA helicases: a DEAD-box motif (Asp-Glu-Ala-Asp), an ATP- binding domain, and four RNA-binding motifs (39, 40). The mrh4-1 allele identified in this study encodes a protein with a single amino acid substitution (P209L) within the first RNA-binding motif (Fig. 1A). The second protein, MrpL4 (uL29), is a mtLSU PTE protein that is positioned in close proximity to the co-translational membrane insertion machinery (6, 8, 41–43). The mrpl4-1 and mrpl4-2 alleles identified in this study result in E260K and P67L amino acid substitutions, respectively (Fig. 1B).
Given that cells require Mam33 to adapt efficiently from fermentative to respiratory carbon sources (33), we asked whether cells harboring any of the respiratory synthetic lethal alleles alone also had difficulty adapting to respiratory media. To test this possibility, serial dilutions of fermenting exponential phase cells were spotted onto plates containing glucose or nonfermentable carbon sources. Although the mrpl4-2 mutant formed colonies indistinguishable from WT on respiratory medium, the mrh4-1 mutant adapted slower than mam33Δ cells (Fig. S1A). None of the mutations compromised the reverse shift, from respiratory to fermentative growth. Increased adaptation time for a cell culture population could result from some cells being unable to adapt, all cells adapting slowly, or a combination of both. To assay cell viability following adaptation, fermenting exponential-phase cells were spread for single colonies on plates containing either glucose or nonfermentable carbon sources, and the number of colonies was compared. We found that only about 3% of mrh4-1 cells were able to adapt compared with WT cells (Fig. S1B). This result demonstrates that mrh4-1 mutants have a more severe adaptation defect than previously observed for mam33Δ cells (33).
Mrh4 is necessary for Cox1 translation during fermentative growth
Cox1 translation is impaired in mam33Δ cells during fermentative conditions (33), we speculated this might account for the synthetic respiration defect observed in mam33Δ mrh4-1 double mutants. To test this possibility, mitochondrial translation was monitored in vivo by pulse-labeling cells with [35S]methionine and cysteine while blocking cytosolic protein synthesis with cycloheximide. In S. cerevisiae, the mitoribosomes synthesize eight proteins including three cytochrome c oxidase subunits (Cox1, Cox2, Cox3), one apocytochrome b subunit (Cob), three F1F0-ATP synthase subunits (Atp6, Atp8, Atp9), and a ribosomal small subunit protein (Var1) (2, 3). The mam33Δ mrh4-1 allele combination led to mtDNA instability (ρ−) and as a result, no mitochondrial translation products were produced (Fig. 2A). This phenotype is common in mutants defective in mitochondrial translation and has previously been observed in mrh4Δ cells (37, 44). However, the mtDNA remained stable in cells containing the mrh4–1 allele alone (84% ρ+) and the translational profile of these cells resembled that observed for the mam33Δ strain. Cox1 synthesis was strongly diminished during fermentation, whereas Cox2 and Cox3 were reduced to a lesser extent, suggesting an indirect effect. In contrast, respiring cells produced all three Cox proteins at WT levels (Fig. 2B). Given that mam33Δ mrh4-1 cells are unable to respire, this latter result is consistent with the idea that Mam33 and Mrh4 are functionally related.
Figure 2.

Mrh4 is necessary for Cox1 translation during fermentative growth. A–C, mitochondrial translation profiles of cells grown to mid-log in galactose (A and C) or lactate (B) and pulse-labeled with [35S]methionine and cysteine in the presence of cycloheximide. Proteins were resolved by SDS-PAGE and visualized by autoradiography. Protein molecular masses are indicated in kilodaltons on the right. Atp8 and Atp9 run well below the smallest protein marker (11 kDa), whose position is not indicated. D, qRT-PCR analysis of total COX1 transcript levels and specific COX1 splicing intermediates in cells fermenting galactose. The qRT-PCR amplicons examined are indicated in the diagram above the graph. Each target was normalized to ACT1 and WT was set equal to 1 (dashed line). Bars represent the average fold-change of three biological replicates (black dots) and error bars represent S.E. E, growth phenotypes of strains carrying an ARG8m reporter under control of the COX1 or COX3 UTRs. Cells grown to mid-log in rich glucose medium were washed twice with dH2O and 1/8 dilutions were spotted onto media containing (+Arg) or lacking (−Arg) arginine. Relevant nuclear and mitochondrial genotypes are indicated on the left and right side of the panel, respectively.
We next asked whether the reduction of Cox1 protein synthesis in mrh4-1 mutants results from transcription, mRNA stability, or splicing defects. Total transcript levels were measured by qRT-PCR using primers within COX1 exon 4, a region present in all intermediate and mature transcripts. COX1 mRNA levels in the mrh4-1 mutant were comparable with WT, demonstrating that reduced mRNA levels are not responsible for the Cox1 translation defect (Fig. 2D). Because COX1 contains up to seven introns, we next considered the possibility that reduced Cox1 translation in mrh4-1 cells results from splicing defects. Mature transcript levels were measured by qRT-PCR using primers spanning COX1 exon junctions. If splicing were impaired, large introns would remain within the cDNA amplicon, preventing PCR amplification. The relative abundance of spliced transcripts was normal in mrh4-1 cells indicating efficient splicing (Fig. 2D). As a second approach to monitor splicing, Cox1 translation was evaluated in a strain carrying intronless mtDNA. Although Cox1 synthesis was mildly restored in this strain, band intensity was still well below WT levels (Fig. 2C). Although this result suggests that MRH4 is required for optimal COX1 intron splicing, the defect is likely indirect due to impaired translation. This interpretation is consistent with the observation that splicing of the first two COX1 introns is most sensitive to a translation block (45).
Because the insertion of Cox1 into the mitochondrial inner membrane nucleates the assembly of cytochrome c oxidase, Cox1-specific translation defects can destabilize other complex subunits (46, 47). To distinguish between translation and protein stability in mrh4-1 cells, we utilized mitochondrial reporter strains in which ARG8, which encodes a mitochondrial protein required for arginine biosynthesis, is placed under the control of the COX1 or COX3 UTRs (Fig. 2E). These gene constructs support growth on minimal glucose medium lacking arginine. Introduction of the mrh4-1 mutation produced an Arg− growth phenotype in cox1ΔARG8m cells, indicating that protein synthesis is impaired in these cells. We also observed a mild growth defect in the cox3ΔARG8m background, consistent with the weak Cox3 in vivo translation defect (Fig. 2A). These results indicate that mrh4-1 cells have a specific defect in Cox1 synthesis, but not stability.
The mrpl4 mutations intensify the Cox1 translation defect in mam33Δ cells
The two mrpl4 alleles that produced a respiration-deficient phenotype in mam33Δ cells were characterized as described for the mrh4-1 mutant. Interestingly, the mrpl4 mutations alone did not impair mitochondrial translation during either fermentative or respiratory growth (Figs. 3, A and C). However, Cox1 translation was completely abolished in fermenting mrpl4 mam33Δ double mutants. This was not due to defects in either COX1 mRNA transcription or stability, as judged by qRT-PCR using primers within exon 4 (Fig. 3B). We next measured spliced COX1 transcripts using primers spanning exon junctions. Although a ∼60% decrease was observed in splicing of the first three introns, translation was not restored in the strain carrying intronless mtDNA (Fig. 3D). These results indicate that the decrease in protein synthesis cannot be attributed to impaired splicing.
Figure 3.

The mrpl4 mutations intensify the Cox1 translation defect in mam33Δ cells. A, mitochondrial translation profiles of cells grown to mid-log in galactose and pulse-labeled with [35S]methionine and cysteine in the presence of cycloheximide. Proteins were resolved by SDS-PAGE and visualized by autoradiography. Protein molecular masses are indicated in kilodaltons on the right. Atp8 and Atp9 run well below the smallest protein marker (11 kDa), whose position is not indicated. B, qRT-PCR analysis of total COX1 transcript levels and specific COX1 splicing intermediates in cells fermenting galactose. The qRT-PCR amplicons examined are indicated in the diagram above the graph. Each target was normalized to ACT1 and WT was set equal to 1 (dashed line). Bars represent the average fold-change of three biological replicates (black dots) and error bars represent S.E. C–E, experiment performed as described in A, except that cells in C, were grown in lactate. The WT (MRPL4) lane in C is duplicated from Fig. 2B (labeled MRH4). In both figures, WT and mutant cells were grown identically, simultaneously labeled, and loaded on the same gel. The WT sample was loaded between the mrpl4 and mrh4 mutant samples to serve as a control for both experiments. Protein molecular masses are indicated in kilodaltons on the right. Atp8 and Atp9 run well below the smallest protein marker (11 kDa), whose position is not indicated. F, growth phenotypes of strains carrying an ARG8m reporter under control of the COX1 or COX3 UTRs. Cells grown to mid-log in rich glucose medium were washed twice with dH2O and 1/8 dilutions were spotted onto media containing (+Arg) or lacking (−Arg) arginine. Plates were incubated at 30 °C for the time indicated below each panel.
Since MrpL4 is located at the PTE in close proximity to the membrane insertion machinery, we tested whether decreased Cox1 translation results from a membrane insertion or assembly defect (6, 8, 41–43). In a negative-feedback mechanism that coordinates Cox1 protein synthesis with assembly, the translational activator Mss51 binds the nascent Cox1 polypeptide and is released at a point late in cytochrome c oxidase assembly (48–56). If assembly is compromised, Mss51 remains sequestered with the misassembled complex and is unable to reinitiate Cox1 synthesis. This negative-feedback mechanism can be bypassed by using a truncated version of Cox1 (Cox1ΔC15) that is unable to bind Mss51 (54). This allele leaves Mss51 constitutively available to activate Cox1 synthesis, which eliminates feedback in various Cox1 assembly mutants. In the mrpl4 mam33Δ double mutants, the Cox1ΔC15 allele did not restore Cox1 synthesis, whereas translation in the pet100Δ assembly mutant control was significantly restored (Fig. 3E). This result indicates that the Cox1 translation defect in the mrpl4 mam33Δ double mutant does not indirectly result from complex assembly defects.
To determine whether the translation defect in mrpl4 mam33Δ double mutant results from protein instability, translation was measured using the cox1Δ::ARG8m and cox3Δ::ARG8m reporters (Fig. 3F). Because mrpl4-1 mam33Δ double mutants displayed significant genome instability (40% ρ+), this experiment was only performed with the mrpl4-2 allele (98% ρ+; Fig. 3A). Furthermore, because the mam33Δ mutation alone produces an Arg− phenotype, incubation at 30 °C was extended to better distinguish between the single and double mutants. Following extended incubation, mam33Δ cells grew slowly on Arg− medium, whereas the mrpl4-2 mam33Δ double mutant maintained an Arg− growth phenotype. In contrast, expression of the cox3Δ::ARG8m reporter was unaffected by the mrpl4-2 mutation.
Taken together, it appears that there is a specific defect in the translation of Cox1 in mrpl4 mam33Δ double mutants. This phenotype was more severe than in mam33Δ single mutants, which may explain the synthetic respiratory defects.
Mam33 interacts with a subset of unassembled mtLSU proteins, but not mature mitoribosomes
Given that Mam33 is necessary for optimal mitochondrial translation and functionally interacts with two proteins directly involved in protein synthesis, we explored the possibility that Mam33 might associate with mitoribosomes. To answer this question, we assessed whether Mam33 and mitoribosomes cosediment in sucrose gradients. WT mitochondria were solubilized with the nonionic detergent n-dodecyl β-d-maltoside (DDM) in the presence of low (25 mm KOAc) or high (300 mm KOAc) salt concentrations to favor either monosomes or individual subunits, respectively. Mam33 remained in the top fractions of the gradient under both salt conditions and did not sediment with either the monosome or the individual subunits (Fig. 4A). Although sucrose gradient sedimentation analysis has successfully been used to detect physical interactions with the ribosome, this result does not conclusively demonstrate that Mam33 does not bind the mature ribosome as actively translating ribosomes are disrupted upon solubilization (37, 57, 58).
Figure 4.

Mam33 interacts with a subset of unassembled mtLSU proteins, but not mature mitoribosomes. A, sucrose gradient sedimentation analysis of Mam33 and ribosomal subunits in mitochondrial extracts prepared from WT cells grown in rich galactose medium. Mitochondria were solubilized under the indicated magnesium and salt conditions, and gradients were centrifuged for 2 (high salt) or 3 (low salt) h. Twelve equal fractions were analyzed by Western blotting using the indicated antibodies. Protein molecular masses are indicated in kilodaltons on the left. B, mitochondria isolated from respiring (Gly, Lac) or fermenting (Gal) cells expressing either WT (−) or 8×His-tagged (+) Mam33 were solubilized and 8×His-Mam33 was purified by metal affinity chromatography. Eluted proteins were resolved by SDS-PAGE and visualized with silver stain. Indicated bands were excised and sent for identification by MS. Protein molecular masses are indicated in kilodaltons on the left. C, mitochondria isolated from fermenting (Gal) cells expressing either WT (−) or 3×Myc-tagged (+) Mam33 were solubilized and 3×Myc-Mam33 was purified by using anti-myc–agarose beads. Eluted proteins were resolved by SDS-PAGE and visualized with silver stain. Protein molecular masses are indicated in kilodaltons on the left. D, experiment performed as described in B, comparing ρ+ and ρo cells. E, list of identified protein bands from B to D. Also, see Table S6. F, Mam33 copurifies with proteins in specific regions of the ribosome. Crystal structure of the yeast mtLSU. Proteins that copurified with Mam33 are colored, whereas all others are shown in gray. Also, see Fig. S3.
Because Mam33 appeared to function independently of the mitoribosome, a native affinity purification strategy was used to identify other interacting proteins. To facilitate Mam33 purification, an 8×His tag was inserted at the N terminus of the mature protein. The tagged protein was functional and expressed at native levels (Fig. S2). Mitochondria isolated from both respiring and fermenting cells were solubilized and 8×His-Mam33 was purified by metal affinity chromatography using the high-stringency conditions employed to assess potential mitoribosome interactions. Several proteins consistently copurified with 8×His-Mam33 but not the untagged control (Fig. 4B). To demonstrate that copurification was not due to interaction with the 8×His epitope tag, the experiment was repeated using 3×Myc-Mam33. Although this fusion protein was less active based on Cox1 steady-state protein levels, the anti-Myc pulldown produced a comparable set of copurifying proteins (Fig. 4C and Fig. S2). In addition, because 3×Myc- and 8×His-Mam33 migrate at different molecular weights, proteins that comigrated with either tagged protein could be resolved. Copurifying proteins present in at least three biological replicates were excised and identified by LC-MS/MS (Fig. 4E). Given that Mam33 does not sediment with the mitoribosome, we were surprised to find that 16 of the 46 mtLSU proteins copurified with Mam33. Interestingly, these proteins were generally localized to two distinct regions of the large subunit, the CP and PTE (Fig. 4F and Fig. S4). Other less abundant interacting proteins were predominantly associated directly or indirectly with the translation machinery. Together with the cosedimentation experiments, these protein interactions suggest that Mam33 associates with individual mtLSU proteins, but not the fully assembled large subunit.
Based on these protein interactions, we speculated that Mam33 might play a role in mitoribosome biogenesis and bind unassembled MRPs prior to their assembly. To test this possibility, we repeated the 8×His-Mam33 pulldown experiment with ρo cells, which lack mtDNA. These cells lack assembled mitoribosomes because both rRNAs are encoded by the mtDNA. Consistent with our hypothesis, we found that Mam33 still interacts with MRPs in the absence of assembled ribosomes (Fig. 4D).
Mam33 directly interacts with individual mtLSU proteins in vitro
To determine which MRPs directly interact with Mam33, candidate proteins were individually tested for their ability to copurify with Mam33. Each MRP tested was translated in either rabbit reticulocyte lysates or wheat germ extracts supplemented with [35S]methionine and recombinant 6×His-Mam33 (Fig. 5A). Full-length radiolabeled polypeptides were obtained for all MRPs except MrpL40 (uL24), which was synthesized as several truncated products (Fig. 5B). Following expression, 6×His-Mam33 was affinity purified from each reaction and copurifying MRPs were visualized by autoradiography and quantified by scintillation counting (Fig. 5, C and D). Mam33 directly interacted with RmL2 (uL2), Mrp7 (bL27), MrpL10 (uL15), MrpL27 (mL41), MrpL32 (bL32), and MrpL49 (bL21). Although this experiment does not definitively exclude the remaining MRPs as binding partners, it demonstrates that Mam33 is capable of directly binding individual unassembled MRPs.
Figure 5.

Mam33 directly interacts with individual mtLSU proteins in vitro. A, workflow: candidate ribosomal proteins from the 8×His-Mam33 affinity purification (Fig. 4) were expressed from a SP6 promoter in either rabbit reticulocyte lysates or wheat germ extracts containing [35S]methionine and recombinant 6×His-Mam33. Following nickel purification, radiolabeled proteins were visualized by autoradiography and quantified by scintillation counting. B, full-length candidate proteins were expressed in the cell-free transcription/translation reaction in the presence of recombinant 6×His-Mam33. Proteins from 1/100 of each reaction were resolved by SDS-PAGE and radiolabeled products were visualized by autoradiography. Predicted molecular weights based on amino acid sequence are shown directly below each lane. Protein molecular masses are indicated in kilodaltons on the left (first two panels) and right (third panel). C, several proteins synthesized in cell-free lysates copurify with recombinant 6×His-Mam33. Percent copurified was calculated as cpm in the bead elution divided by cpm in a TCA precipitate of the total transcription-translation reaction. Bars represent the average of at least three biological replicates minus negative control reactions lacking 6×His-Mam33 performed in parallel. Error bars represent S.E. and two or three asterisks indicate p values smaller than 0.01 or 0.001, respectively (see “Experimental procedures”), for more information on statistical analysis). Raw data are provided in Table S7. D, a fraction of the total reaction and bead eluate from the nickel-agarose beads were visualized by SDS-PAGE and autoradiography for the six interacting proteins (Mrp7, MrpL10, RmL2, MrpL27, MrpL32, MrpL49) and one protein that did not interact (MrpL4). Protein molecular masses are indicated in kilodaltons on the left and right of the upper and lower panel sets.
Mam33, MrpL4, and Mrh4 are necessary for proper assembly of the mtLSU
Given that Mam33 physically interacts with specific unassembled MRPs, we hypothesized that it might be necessary for mitoribosome biogenesis. Likewise, this process might also be disrupted in the mrpl4 and mrh4 respiratory synthetic lethal mutants identified in this study. To assess mitoribosome integrity, we analyzed the sedimentation patterns of MRPs in sucrose gradients, using either low (100 mm KOAc) or high (300 mm KOAc) salt conditions. Although low salt conditions were useful for visualizing general assembly defects and aggregation, stringent salt conditions were ideal for visualizing the specific defects described below because they favored dissociation of the ribosome into its individual subunits, partially disrupted protein/rRNA aggregates, and destabilized weak interactions to reduce smearing through the gradient. Fractions of the gradient were analyzed for rRNA content and a representative set of MRPs by qRT-PCR and immunoblotting, respectively (Fig. 6, A and B). In WT extracts, the small and large subunits were confined to the middle of the gradient with distinct peaks at fractions 6 and 7, respectively. The mrpl4-2 mutant exhibited similar sedimentation patterns, consistent with its normal translation phenotype (Fig. 3A). In contrast, MRPs in mam33Δ, mrh4-1, and mrpl4-2 mam33Δ mutants were found not only in the middle of the gradient, but also in fractions of low density and the highest density. This additional distribution likely reflects free or partially assembled MRPs (top fractions) and aggregates (bottom fraction). In the mrh4-1 single mutant and mrpl4-2 mam33Δ double mutants, the mtLSU shifted to fraction 6, indicating that many components were missing from the large subunit. In mam33Δ single mutants and mrpl4-2 mam33Δ double mutants, a large percentage of 21S rRNA was found at the bottom of the high salt gradients, suggesting the aggregation of not just MRPs, but the assembling large subunit. In summary, these results demonstrate that ribosome assembly is severely impaired in these mutants.
Figure 6.
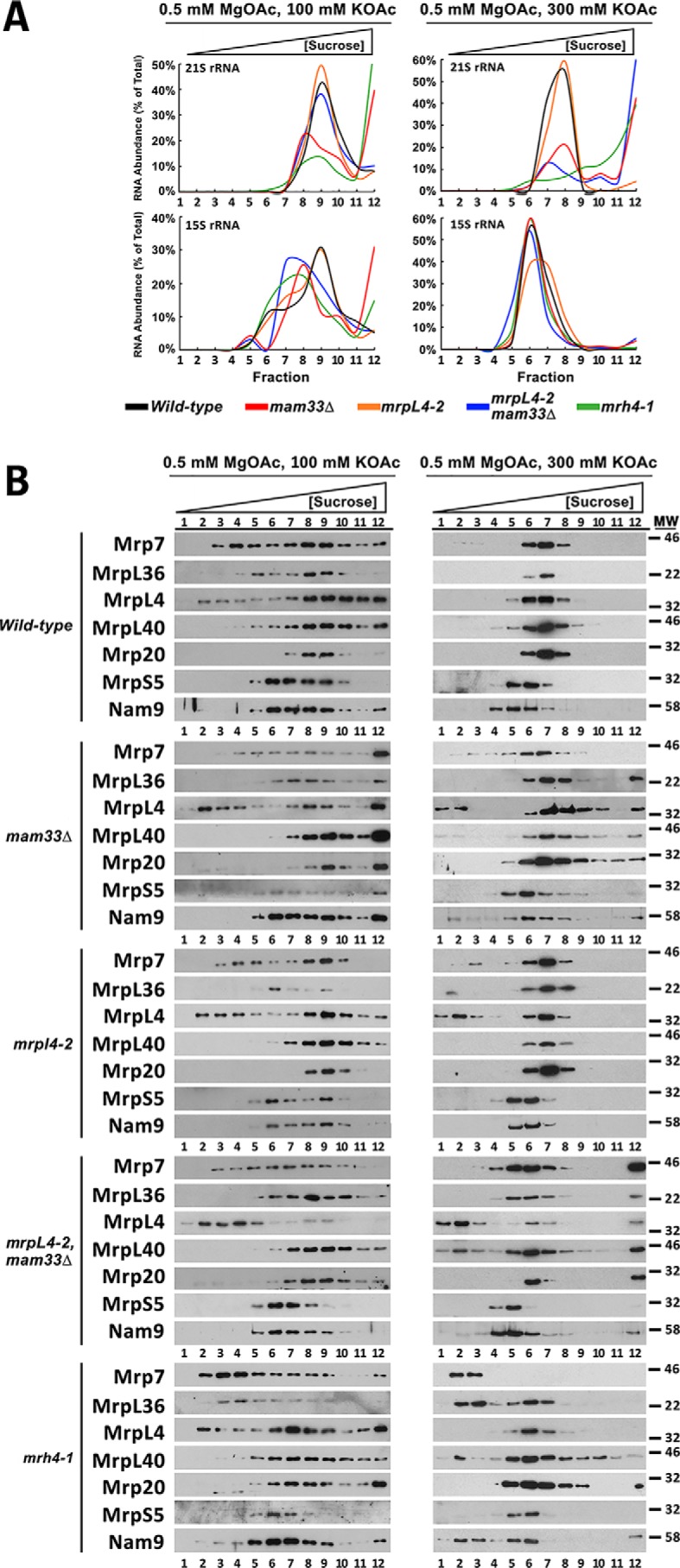
Mam33, MrpL4, and Mrh4 are necessary for proper assembly of the mtLSU. A, sucrose gradient sedimentation analyses of the 15S and 21S rRNAs in mam33Δ and respiratory synthetic lethal mutants. Mitochondria isolated from cells grown in rich galactose medium were solubilized under the magnesium and salt conditions stated, loaded on a linear 10–40% sucrose gradient, and centrifuged for 2 (high salt) or 3 (low salt) h. RNA purified from 12 equal gradient fractions was analyzed by qRT-PCR using primers within the 15S or 21S rRNAs. Curves represent the amount of target rRNA in each gradient fraction divided by the sum target rRNA in all fractions. The raw data used to create each curve is provided in Table S8. B, proteins in each gradient fraction described in A were resolved by SDS-PAGE and mitoribosomal proteins were detected by Western blotting using antibodies recognizing a representative set of large (Mrp7, MrpL36, MrpL4, MrpL40, Mrp20) and small (MrpS5, Nam9) subunit mitoribosomal proteins. Protein molecular masses are indicated in kilodaltons on the right.
The sucrose gradient experiments were also used to compare the assembly of MrpL4 P67L into the mtLSU and determine whether Mam33 impacts its insertion (Fig. 6B). Under both salt conditions, a moderate percentage of MrpL4 shifted to the top of the gradient in mam33Δ and mrpl4-2 cells. However, a significant portion of the protein remained with the assembled large subunit, consistent with their respiratory-competent phenotypes. The amount of MrpL4 P67L dissociated from the large subunit increased substantially in the mam33 mrpl4-2 double mutant. Taken together with the physical interaction data, these results suggest that MrpL4 P67L is incorrectly assembled into the ribosome. The absence of Mam33 exacerbates this phenotype suggesting a mechanism for the respiratory lethal interaction.
Mam33 has a minimal effect on MRP steady-state levels
Given its role in preventing misfolding and aggregation, we next asked if Mam33 is also required to maintain MRP steady-state levels. Although most MRPs remained stable, Mrp20 (uL23) was moderately decreased in mam33Δ and mrpl4-2 single mutants, and nearly abolished in the double mutant (Fig. 7). Because antibodies are not available for most MRPs, we used a MS approach to detect differences in their abundances. Mitochondria were prepared from fermenting WT and mam33Δ cells grown in parallel, except that WT proteins were labeled with heavy isotopes of arginine and lysine. The use of these isotopes did not noticeably alter growth rate. Light (mam33Δ) and heavy (WT) samples were mixed at a 1:1 ratio and analyzed using SILAC-based quantitative proteomic analysis. Consistent with our Western blot analysis, most large and small subunit ribosomal proteins remained stable, whereas Mrp20 was decreased (62% of WT; Tables S5, S9, and S10). Of the proteins that Mam33 directly binds, only MrpL27 was destabilized in mam33Δ cells (mL41; 48% of WT). In addition, MrpL13 (mL50; 69% of WT) and MrpL38 (uL14; 46% of WT) were decreased. Although MrpL13 does not appear to physically interact with Mam33, it interacts with Mrp20. It remains unclear whether Mam33 directly binds MrpL38, because it may have been missed in our copurification experiments due to its small size (<15 kDa). Together, these results indicate that Mam33 is necessary to prevent misfolding and aggregation but has a minimal effect on MRP steady-state levels.
Figure 7.
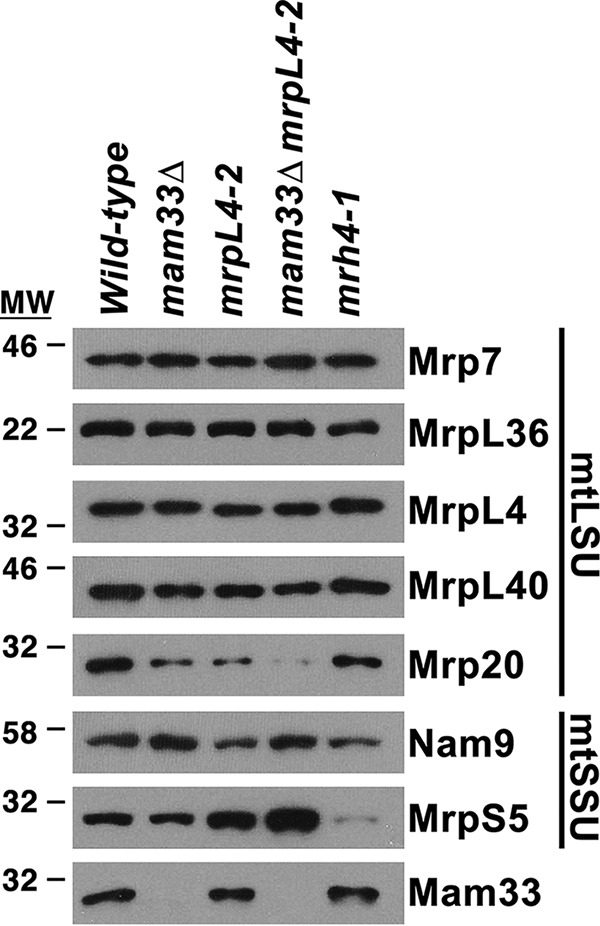
Mam33 has a minimal effect on MRP steady-state levels. Mitochondrial proteins (5 μg) from cells grown in galactose were resolved by SDS-PAGE and detected by Western blotting using the antibodies indicated on the right side of the panel. Protein molecular masses are indicated in kilodaltons on the left.
Discussion
We recently identified an evolutionarily conserved protein called Mam33 that promotes the translation of Cox1 in yeast mitochondria (33). In this study, we screened for genes that functionally compensate for Mam33 during respiration and identified mutants with Cox1-specific translation defects and misassembled, aggregated mitochondrial ribosomes. Interestingly, Mam33 does not associate with the assembled ribosome, but rather binds a subset of large subunit MRPs prior to their incorporation. Furthermore, Mam33 is necessary for proper assembly of the large subunit. Based on these data, we propose that Mam33 is a mitoribosome assembly factor.
In our working model, Mam33 binds newly-imported mtLSU proteins and facilitates assembly into the large subunit (Fig. 8). Given Mam33's peripheral inner membrane location, we propose that it interacts with its target during or immediately after import into the mitochondria (24). This association may serve to maintain the extended unstructured regions of these proteins until they can be incorporated into the large subunit and stabilized by adjacent MRPs and rRNA. Likewise, the acidic face of Mam33 may mask the basic regions that are prevalent in MRPs. Another interesting possibility is that the polypeptide passes through the central channel of the Mam33 trimer. Depending on the degree of blockage by the inner loops, which may undergo conformational changes, the opening would have a diameter sufficient to accommodate a polypeptide chain (31). It remains unclear whether Mam33 accompanies its MRPs to the assembling ribosome or mediates insertion into large subunit subassemblies.
Figure 8.

Working model for the role of Mam33 in mitoribosome biogenesis. A, structure and surface charge distribution of the Mam33 trimer (PDB 3QV0 (31)). There are three Mam33 molecules in an asymmetric unit that form a bowl- or doughnut-shaped trimer. The central channel has a diameter of ∼15 Å. However, the central loops that partially cover the channel reduce the diameter to about 4 Å. Electrostatic surface potential reveals an unusual asymmetric charge distribution on the two sides of the trimer (center and right panels). Positively and negatively charged electric potential are indicated in blue and red, respectively. B, schematic model of the role of Mam33 as a chaperone of newly-synthesized MRP's. We propose that the Mam33 trimer physically interacts with a specific subset of MRPs during or immediately after import into the mitochondria. This association may serve to maintain the extended unstructured regions of these proteins until they can be incorporated into the large subunit and stabilized by adjacent MRPs and rRNA. Likewise, the acidic face of Mam33 may stabilize the basic regions, prevalent in MRPs. Alternatively, the polypeptide may pass through the central channel of the Mam33 trimer, depending on the degree of blockage by the inner loops. Finally, it is unclear whether Mam33 accompanies its targets to the assembling ribosome or mediates insertion into large subunit subassemblies. In the absence of Mam33 (lower pathway), the unstructured or highly basic regions misfold and aggregate with other MRPs and the 21S rRNA
Our Mam33 physical interaction data suggests a mechanism responsible for the respiratory synthetic lethal interaction between MRPL4 and MAM33. A small percentage of Mrpl4 does not sediment with the mature large subunit in either respiratory-competent single mutant (Fig. 6B). This defect was exacerbated in the mam33Δ mrpL4-2 double mutant, explaining the respiratory-deficient phenotype. Why does the absence of Mam33 further impair MrpL4 P67L assembly? We have shown that Mam33 directly binds MrpL27, which forms extensive physical contacts with MrpL4 (Fig. S3) (6, 8). Although MrpL4 can assemble into the ribosome in the absence of Mam33, albeit less efficiently, MrpL4 P67L cannot be incorporated. This result suggests that MrpL27 is not properly incorporated in mam33Δ mutants. The decreased steady-state levels of MrpL27 in mam33Δ cells are consistent with this hypothesis (Table S5). Furthermore, the copurification of PTE proteins surrounding MrpL4 (MrpL40, MrpL27, MrpL13, Mhr1, and MrpL3) with Mam33 indicates an association with this assembling subcomplex (Fig. 4).
The model that Mam33 chaperones MRPs during their assembly is supported by the genetic relationship between MAM33 and MRH4. The complete absence of Mrh4 results in partially assembled ribosomes that are missing MrpL16 (uL16) and MrpL39 (bL33), two proteins present at the interface between the CP and the rest of the ribosome (Fig. S3B) (37). In our mrh4-1 mutant, the CP region partially dissociated from the large subunit. Interestingly, Mam33 copurified with several CP proteins (Mrp7, MrpL7, MrpL17, MrpL35, MrpL36), indicating that it associates with this subassembly. Mam33 directly interacted with Mrp7, which has a long N-terminal extension reaching the ribosome center that appears to serve as a structural anchor for the CP. Together, these observations suggest that the MrpL16, MrpL39, and Mrp7 anchors may not be properly incorporated in the mam33Δ mrh4-1 double mutant, causing the CP region to completely dissociate from the ribosome.
Several lines of evidence confirm that the Mam33-mtLSU protein interactions reported in this study are physiologically significant. First, they are consistent with genetic data including respiratory lethal interactions with MRH4 and MRPL4, mutant translation phenotypes, and mtLSU assembly defects. Second, the interactions between Mam33 and positively-charged ribosomal proteins were detected using stringent salt conditions to prevent nonspecific electrostatic interactions. Direct binding was observed in highly concentrated lysates containing a wide variety of proteins to compete for binding. Moreover, interactions were only identified with a small subset of the numerous basic proteins tested. Third, Mam33 was previously detected in binding CP proteins in a ribosome assembly mutant using tagged ribosomal proteins, instead of tagged Mam33 (59). This observation confirms that endogenous Mam33 binds unassembled ribosomal proteins. Finally, Mam33 expression correlates highly with MRPs (60). Taken together, these data support our model that Mam33 aids in mitoribosome assembly, acting as a chaperone for unassembled MRPs.
It is intriguing that the ribosome assembly mutants in this study produce Cox1-specific defects rather than an overall impairment of translation. Several mechanisms could account for this phenotype. First, Mam33, MrpL4, and Mrh4 may also function as Cox1 translational activators. Second, the ribosome defects in these mutants may trigger regulatory signaling pathways that inhibit Cox1 translation. Synthesis of Cox1 appears to be under the most stringent regulation to prevent ROS formation. For instance, mechanisms for sensing its own assembly status and heme biosynthesis have been reported (48–56). Third, Mam33, MrpL4, and Mrh4 may be necessary for a subset of ribosomes specialized for the synthesis of Cox1. Mitoribosomes have evolved to synthesize the unique subset of proteins encoded by the mtDNA, but the question remains whether different populations exist that further specialize for particular proteins. For example, misassembled ribosomes may be missing a component dispensable for protein synthesis in general, but necessary for binding a Cox1-specific translational activator.
Why is Mam33 dispensable for respiration in yeast cells? Given that the mammalian ortholog is essential for fetal development and specific mutations cause severe defects in mitochondrial energy metabolism, we predict that a second protein may compensate in yeast (22, 23, 25). Although the respiratory synthetic lethal screen was designed to identify functionally redundant genes, it remains unclear whether MrpL4 or Mrh4 perform the same function as Mam33 in mitoribosome assembly.
Seven chaperones have been identified in the nucleus and cytosol of eukaryotic cells that accompany and stabilize unassembled ribosomal proteins prior to their incorporation into the ribosome (61, 62). Interestingly, the doughnut-shaped structure and asymmetric charge distribution of one chaperone (Sqt1) is reminiscent of Mam33 and its orthologs (63). The acidic face stabilizes Rpl10 (uL16) by directly binding a positively charged region at the N terminus of the protein (64–66). Although Sqt1 binds a different ribosomal protein, those chaperoned by Mam33 also contain positively charged regions at their N terminus (Fig. S4). Future experiments aim to define the precise regions that Mam33 recognizes and to determine specifically when it binds and releases mitoribosomal proteins.
Experimental procedures
Strains, media, and genetic methods
Yeast strains used in this study are listed in Table S1. All strains are congenic with either D273-10B or W303, except MHY1934 and MHY1935, which are derived CH1304 and CH1305. Cells were grown at 30 °C in rich medium (YP) containing 1% yeast extract and 2% peptone or minimal medium containing 0.17% yeast nitrogen base, 0.5% ammonium sulfate, and the appropriate amino acids, unless otherwise indicated. Media contained 2% glucose, galactose, lactate, ethanol or 3% glycerol, as indicated. The rich or minimal “D” medium used for the respiratory synthetic lethal screen contained 3% glycerol, 2% ethanol, and 0.2% glucose. The mrh4-1 (c626t), mrpl4-2 (c200t), and mrpl4-1 (g778a) respiratory synthetic lethal point mutations were re-engineered by site-directed mutagenesis and integrated into the chromosome using the yeast integrative vectors pRS306 (mrh4-1) or YIp5 (mrpl4-2 and mrpl4-1) (67–69). Mam33 was tagged with 8×His, 1×Myc, or 3×Myc (70) immediately downstream of the mitochondrial targeting signal (MTS). Recombinant 6×His- and GST-Mam33 fusion constructs were created by inserting MAM33ΔMTS into the EcoRI site of pRSETA (pMHY448; Invitrogen) and pGEX-4T-1 (pMHY446; Amersham Biosciences), respectively. The sequences of these fusion proteins are provided in Table S4. mtDNA stability was measured by growing cells to exponential phase using conditions identical to the corresponding experiment, spreading cells for single colonies on YPD plates, and crossing at least 50 colonies to the ρo tester strains. Percent of ρ+ represents the fraction of the resulting diploids that became respiratory competent. Strains were made ρo by maintaining exponential phase growth for 36 h in rich glucose medium containing 10 μg/ml of ethidium bromide. Cells unable to respire were stained with 4′,6-diamidino-2-phenylindole and visualized using fluorescence microscopy to confirm the complete loss of mtDNA.
MAM33 respiratory synthetic lethality screen
In this strategy, mam33Δ, ade2−, ade3− cells harboring a plasmid containing WT MAM33, a URA3 selectable marker, and an ADE3 color marker formed red colonies, whereas those that lost the plasmid turned white. When grown on complete respiratory media, the red colonies contained white sectors, these genes are not required for respiration. Following chemical mutagenesis, red nonsectoring colonies that required the MAM33 plasmid for respiration were further analyzed as described below.
To create the parental strain for the screen, the MAM33 ORF in CH1305 (35) was replaced with a KANMX module from the yeast knockout collection (71) and transformed with pMHY606 to generate MHY1935. Plasmid pMHY606 (2 μm URA3 ADE3 MAM33) was generated by subcloning a 2-kb BamHI-XhoI fragment containing MAM33 from pMHY437 (CEN URA3 MAM33) into BamHI-SalI–linearized pPS709 (72).
MHY1935 was grown in minimal glucose medium (−ura) to a density of ∼2 × 107 cells/ml, then mutagenized with EMS to 50% killing as described previously (36). Following mutagenesis, cells were plated on rich D medium and incubated until colonies developed a red color. Of the 225,000 colonies screened, 359 red nonsectoring colonies were isolated and restreaked onto fresh rich D plates. Three candidates satisfied all the criteria for synthetic lethality with mam33Δ on respiratory medium (36, 72)
The dominance or recessiveness of each mutant allele was tested by mating with the unmutagenized parental strain of the opposite mating type (MHY1934) as previously described (36). On this basis, all mutations were judged recessive. Each resulting diploid was subsequently sporulated under conditions to maintain plasmid pMHY606 and tetrads were scored for their sectoring phenotypes. All mutants displayed 2:2 segregation of the sectoring phenotype, indicating that the respiratory synthetic lethality resulted from a mutation in a single gene. Three respiratory synthetic lethal mutants were placed into two complementation groups. Each mutant was also tested for heat (36 °C) and cold (15 °C) sensitive phenotypes, but none showed conditional growth phenotypes linked to the nonsectoring.
Genes complementing the respiratory synthetic lethal mutations were identified by transforming one candidate from each complementation group, carrying only pMHY607 (CEN LEU2 ADE3 MAM33), with a YCp50-based genomic library containing a URA3 selectable marker as previously described (36, 73). Following selection for library plasmids on minimal D medium lacking uracil, library plasmids were isolated from white colonies, which no longer required pMHY607 to respire. The specific chromosomal regions contained within each library plasmid were determined using upstream and downstream vector primers that flanked the insert. Sequence obtained from each end was subsequently compared with the yeast genome database. Because each library clone insert contained more than one gene, smaller regions were subcloned and tested individually.
RNA isolation and qRT-PCR
For RNA extraction, cells were grown to exponential phase in rich galactose medium and RNA was isolated using RiboZol RNA extraction reagent (VWR) according to the manufacturer's instructions. RNA from sucrose gradient fractions was extracted using the Direct-zol RNA Miniprep Kit (Zymo Research). cDNA synthesis and subsequent qRT-PCR was performed using the One-Step Real-time PCR system (Applied Biosystems) and the Luna qRT-PCR kit (New England Biolabs) according to the manufacturer's recommendations. Reactions were run in triplicate and represent the average of three biological replicates. Primer sequences are provided in Table S2.
Purification of mitochondria
Exponential-phase yeast cells (OD600 = 1.0–1.5) harvested by centrifugation at 2,000 × g for 5 min were washed with water, resuspended in reducing buffer (2 ml/g cells; 100 mm Tris-SO4, pH 9.4, and 10 mm DTT), and incubated with gentle shaking for 40 min at 30 °C. Cells were washed once with 1.2 m sorbitol, resuspended in zymolyase buffer (6.7 ml/g of cells; 1.2 m sorbitol, 20 mm potassium Pi, pH 7.4, and zymolyase 100T (Amsbio; 1 mg/g cells)), and incubated with gentle shaking for 40 min at 30 °C. Spheroplasts were harvested by centrifugation at 1,500 × g for 5 min, gently resuspended in regeneration buffer (growth media containing 1.2 m sorbitol; 6.7 ml/g of cells), and incubated for 30 min at 30 °C with gentle shaking. All of the subsequent steps and centrifugations were performed on ice or at 4 °C, respectively. Regenerated spheroplasts were harvested by centrifugation at 1,500 × g for 5 min, resuspended in SE buffer (8.5% sucrose, 10 mm MOPS-KOH, pH 7.4, 1 mm EDTA), and homogenized by 10 strokes in a glass douncer. The homogenate was centrifuged at 2,000 × g for 5 min and the supernatant, containing mitochondria, was transferred to a new tube. The pellet was rehomogenized and the supernatants were combined. Homogenization was repeated until the cell debris was clear and colorless (approximately four times). The combined supernatants were centrifuged at 2,000 × g for 10 min to pellet any remaining cell debris, and mitochondria were subsequently harvested by centrifugation at 12,000 × g for 20 min. Crude mitochondria were further purified in two consecutive sucrose gradients and protein concentrations were determined as previously described (74, 75).
Analysis of mitochondrial proteins
In vivo radiolabeling of mitochondrial translation products and Western blotting were performed as previously described (33, 76, 77). Western blot analysis of proteins in sucrose gradients was performed by adding an equal volume of 2× SDS-loading buffer directly to each gradient fraction and probing the resulting blots with the indicated antibodies. The anti-Mam33 polyclonal antibody was obtained from Covance Inc. (Denver, PA) by immunizing rabbits with recombinant full-length GST-Mam33 purified from Escherichia coli. The anti-Cox1 mouse mAb (11D8B7) was obtained from Abcam. The following antibodies were gifts and used at the indicated dilutions: anti-Mrp7 mouse mAb (1:1,000) from Tom Fox at Cornell University (78); anti-Mrp20 mouse mAb (1:8,000) from Carolyn Suzuki at Rutgers University (79); anti-MrpL36 (1:60,000), anti-MrpL4 (1:80,000), anti-MrpL40 (1:150,000), and anti-MrpS5 (1:12,000) rabbit polyclonal antibodies from Martin Ott at Stockholm University (43, 80, 81); anti-Aco1 (1:200,000) rabbit polyclonal antibody from Ophry Pines at The Hebrew University of Jerusalem (82); and anti-Nam9 (1:100,000) rabbit polyclonal antibody from Godfrey Getz at The University of Chicago (83). Blots were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies (Cell Signaling Technologies) and signals were detected using WesternBright Sirius ECL (Advansta).
Purification of tagged proteins
To find proteins that copurify with 8×His-Mam33, mitochondria (5 mg of protein) were gently resuspended in 1.5 ml of solubilization buffer (1% DDM, 30 mm imidazole, 300 mm KOAc, 5 mm MgOAc, 0.8 mm EDTA, 10 mm Tris-HCl, pH 7.4, 5% glycerol, 1 mm spermidine, 5 mm β-mercaptoethanol (β-ME), 1 mm PMSF, and 2.5 μg/ml of pepstatin, leupeptin, antipain, and chymostatin (PLAC)) and incubated for 15 min with gentle rocking (all steps performed on ice or at 4 °C). Clarified lysates obtained by centrifugation at 16,000 × g for 10 min were incubated with gentle rocking for 90 min with Ni-NTA–agarose beads (GoldBio, catalog number H-320-35; 12 μl of slurry pre-equilibrated with solubilization buffer). The beads were washed five times for 15 min with 1.2 ml of wash buffer (solubilization buffer containing 0.1% DDM and 70 mm imidazole), and purified proteins were eluted with 30 μl of elution buffer (0.1% DDM, 400 mm imidazole, 100 mm NaOAc, 10 mm Tris-HCl, pH 7.4, 5% glycerol, 5 mm β-ME, 0.5 mm PMSF, and 2.5 μg/ml of PLAC). Eluates were diluted with 4× SDS-loading buffer and analyzed by SDS-PAGE (76). Proteins were visualized using a MS-compatible silver stain (Thermo Scientific, catalog number 24600). Bands of interest were excised and proteins were identified by NanoLC-ESI-MS/MS (ProtTech Inc., Phoenixville, PA).
Affinity purification of 3×Myc-Mam33 was performed exactly as described for 8×His-Mam33, except that mitochondrial extracts were incubated with anti-myc–agarose beads (Thermo Scientific, catalog number 20168) and proteins were eluted with SDS-loading buffer without reducing agent. Samples were adjusted to 5% β-ME prior to SDS-PAGE analysis.
Recombinant 6×His-Mam33 was purified from E. coli BL21(DE3) cells containing pMHY448. Cells were grown to exponential phase (OD600 = 0.6) at 27 °C in Luria broth supplemented with 50 μg/ml of carbenicillin and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h. The remainder of the purification was performed at 4 °C or on ice. Cells harvested by centrifugation were resuspended in lysis buffer (10 mm imidazole, 300 mm NaCl, 40 mm Tris-HCl, pH 8, 5% glycerol, 5 mm β-ME, 1 mg/ml lysozyme, 1 mm PMSF, 2.5 μg/ml of PLAC; 2 ml per 50 ml of culture), incubated on ice for 30 min, and lysed by sonication with six 10-s pulses at a medium setting on an ultrasonicator (Branson Sonifier 150). The resulting lysate was supplemented with 5 μg/ml of DNase I (Sigma) and 10 μg/ml of RNase A (Sigma) and incubated on ice for 15 min. The clarified lysate obtained by 30 min centrifugation at 10,000 × g was incubated with gentle rocking for 60 min with Ni-NTA–agarose beads (200 μl slurry). Following five 15-min washes with 1.2 ml of wash buffer (60 mm imidazole, 300 mm NaCl, 40 mm Tris-HCl, pH 8, 5% glycerol, 5 mm β-ME, 1 mm PMSF), 6×His-Mam33 was eluted with 800 μl of elution buffer (wash buffer with 300 mm imidazole). The eluate was concentrated in 100 mm NaCl, 40 mm Tris-HCl, pH 8, 5% glycerol using a Amicon Ultra 50,000 NMWL filter (Millipore).
Purification of recombinant GST-Mam33 was performed as previously described (36) except that expression was induced at 25 °C for 3 h. GST-Mam33 was eluted in 20 mm GSH, 100 mm Tris-HCl, pH 8, 120 mm NaCl and subsequently dialyzed and stored in PBS.
Sucrose gradient sedimentation
Mitochondria (2 mg of protein) were resuspended in 300 μl of solubilization buffer (1% DDM, 25–300 mm KOAc, 10 mm Tris-HCl, pH 7.4, 0.5 mm MgOAc, 5% glycerol, 5 mm β-ME, 1 mm PMSF, and 2.5 μg/ml of PLAC) and incubated for 15 min with gentle rocking (all steps performed on ice or at 4 °C). Clarified lysates obtained by centrifugation at 16,000 × g for 10 min were layered on a linear 3.6 ml of 10–40% sucrose gradient with the same buffer conditions except less DDM (0.1%). Following centrifugation at 370,000 × g (Beckman SW 60 Ti rotor; spin time indicated for each experiment), gradients were collected in 12 equal fractions. Proteins and mt-rRNAs were analyzed by Western blotting or qRT-PCR as described above.
Cell-free protein expression
To synthesize candidate proteins in vitro, we used the TnT (rabbit reticulocyte lysate or wheat germ extract) SP6/T7 Coupled Transcription/Translation system (Promega, catalog numbers L5020 and L5030), in the presence of l-[35S]methionine (PerkinElmer, EasyTag catalog number NEG709A). Each gene was expressed from the pTnT vector (Promega, catalog number L5610), containing a T7/SP6 promoter, poly(A)30 tail, and T7 terminator. Primers were designed to amplify each gene with an upstream XhoI or MluI restriction site, a start codon with a Kozak consensus sequence (CCACC-ATG-GCG), the mature coding region, and a downstream NotI restriction site. The resulting proteins began with Met-Ala and terminated at their natural stop codons. This expression cassette is shown in Fig. 5A and the primers used for amplification are listed in Table S3. Coding regions were amplified from W303 chromosomal DNA using either Q5 or Vent High-Fidelity DNA polymerases (New England Biolabs). PCR products were subsequently digested with either XhoI/Not1 or MluI/NotI, and ligated into the pTnT vector cut with the same enzymes. Full-length expression of Mrp7, MrpL4, MrpL40, and MrpL36 was optimal in the rabbit reticulocyte system, whereas all others were ideal in wheat germ extracts. SP6 was superior to T7 for the expression of all constructs.
20 μl of TnT reactions were performed according to the manufacturer's protocol with the addition of 1 μg of purified recombinant 6×His-Mam33. One-fifth of each reaction was set aside to determine percent incorporation (as described by manufacturer), for analysis by SDS-PAGE and autoradiography. 6×His-Mam33 was purified by metal affinity chromatography as follows. The remainder of the reaction was diluted with 200 μl of binding buffer (150 mm KOAc, 0.5 mm MgOAc, 0.05% DDM, 20 mm Tris-HCl, pH 7.4, 5% glycerol, 10 mm imidazole, 5 mm β-ME, 1 mm PMSF) and incubated with Ni-NTA beads (5 μl of bead slurry) for 1 h with rocking (all steps performed on ice or at 4 °C). Beads were subjected to five 10-min washes with 500 μl of solubilization buffer containing 50 mm imidazole. 6×His-Mam33 was eluted with 30 μl of elution buffer (100 mm NaCl, 20 mm Tris-HCl, pH 7.4, 5% glycerol, 400 mm imidazole, 5 mm β-ME, 0.5 mm PMSF). For each eluate, 2 μl was used for scintillation counting and 7.5 μl was analyzed by SDS-PAGE and autoradiography.
SILAC-based quantitative proteomic analysis
Mitochondria prepared from mam33Δ cells (MHY2021) were grown to mid-log (OD600 = 1.2) in minimal galactose medium containing all amino acids. WT cells (NB40–36a) were grown under identical conditions, except arginine and lysine were substituted with heavy isotopes (l-arginine-HCl (13C6), l-lysine-2HCl (13C6); Thermo Scientific). SILAC-based quantitative proteomic analysis was performed by Creative Proteomics (Shirley, NY). Briefly, light (mam33Δ) and heavy (WT) purified mitochondria were mixed at a 1:1 ratio, lysates were transferred to a Microcon device YM-10 (Millipore), centrifuged at 12,000 × g at 4 °C for 10 min, and washed twice with 200 μl of 50 mm ammonium bicarbonate. The resulting sample was reduced with 10 mm DTT at 56 °C for 1 h and then alkylated with 20 mm indole-3-acetic acid at room temperature in the dark for 1 h. The device was centrifuged at 12,000 × g at 4 °C for 10 min, washed once with 50 mm ammonium bicarbonate, recentrifuged, and then 100 μl of 50 mm ammonium bicarbonate was added to the protein concentrate. Free trypsin (Promega) was added a ratio of 1:50 to the protein solution, and the solution was incubated at 37 °C overnight. The device was centrifuged at 12,000 × g at 4 °C for 10 min and washed twice with 100 μl of 50 mm ammonium bicarbonate. The extracted peptides were lyophilized to near dryness and subsequently resuspended in 2–20 μl of 0.1% formic acid before Nano LC-MS/MS analysis.
Nano LC was performed using an Ultimate 3000 nano UHPLC (Thermo Fisher Scientific, USA) with a 100 μm × 10 cm in-house made nanocolumn packed with a reversed-phase ReproSil-Pur C18-AQ resin (3 μm, 120 Å, Dr. Maisch GmbH, Germany). Mass spectrometry was performed using a Q Exactive HF mass spectrometer (Thermo Fisher Scientific, USA). Raw MS files were analyzed and searched against the S. cerevisiae protein database based on the species of the samples using Proteome Discoverer 2.0. The parameters were set as follows: the protein modifications were carbamidomethylation (C) (fixed), oxidation (M) (variable); the enzyme specificity was set to trypsin; the maximum missed cleavages were set to 2; the precursor ion mass tolerance was set to 10 ppm, and MS/MS tolerance was 0.6 Da. Only high confident identified peptides were chosen for downstream protein identification analysis. Percentages in Tables S5, S9, and S10 represent the abundances of light samples (mam33Δ) relative to medium samples (WT). Note that although these data only represent one biological replicate, it was generally consistent with other data in the paper and therefore included for qualitative purposes.
Statistical analysis
All experiments were performed at least in triplicate unless otherwise indicated. Error bars represent mean ± S.E. The statistical significance in Fig. 5C was determined using an unpaired one-tailed Student's t test with Microsoft Excel. Significant difference in means is indicated as: **, p < 0.01 and ***, p < 0.001.
Software
Crystal structure images were generated using PyMOL.
Author contributions
G. A. H. and M. F. H. conceptualization; G. A. H. and M. F. H. formal analysis; G. A. H. and M. F. H. investigation; G. A. H. visualization; G. A. H. methodology; G. A. H. and M. F. H. writing-original draft; G. A. H. and M. F. H. writing-review and editing; M. F. H. resources; M. F. H. supervision; M. F. H. funding acquisition; G. A. H. designed and performed experiments, analyzed the results, and made the figures. The manuscript was written by M. F. H. and G. A. H.; M. F. H. initiated the study, designed experiments, and analyzed results. The manuscript was written by M. F. H. and G. A. H.
Supplementary Material
Acknowledgments
We thank Kyle Hubble and Dr. Michael Anikin for insightful discussions and comments on the manuscript. We are grateful to Thomas D. Fox, Carolyn Suzuki, Martin Ott, Xochitl Perez-Martinez, Ophry Pines, Godfrey Getz, and Alexander Tzagoloff for strains and antisera.
This work was supported by NIGMS National Institutes of Health Grant GM117483 (to M. F. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Tables S1–S10.
- mtLSU
- mitochondrial large subunit
- CP
- central protuberance
- PTE
- polypeptide tunnel exit
- qRT
- quantitative RT
- DDM
- n-dodecyl β-d-maltoside
- SILAC
- stable isotope labeling by amino acids in cell culture
- MTS
- mitochondrial targeting signal
- GST
- glutathione S-transferase
- β-ME
- β-mercaptoethanol
- PMSF
- phenylmethylsulfonyl fluoride
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. de Zamaroczy M., and Bernardi G. (1986) The primary structure of the mitochondrial genome of Saccharomyces cerevisiae: a review. Gene 47, 155–177 10.1016/0378-1119(86)90060-0 [DOI] [PubMed] [Google Scholar]
- 2. Foury F., Roganti T., Lecrenier N., and Purnelle B. (1998) The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 440, 325–331 10.1016/S0014-5793(98)01467-7 [DOI] [PubMed] [Google Scholar]
- 3. Fox T. D. (2012) Mitochondrial protein synthesis, import, and assembly. Genetics 192, 1203–1234 10.1534/genetics.112.141267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tzagoloff A., and Myers A. M. (1986) Genetics of mitochondrial biogenesis. Annu. Rev. Biochem. 55, 249–285 10.1146/annurev.bi.55.070186.001341 [DOI] [PubMed] [Google Scholar]
- 5. Herrmann J. M., Woellhaf M. W., and Bonnefoy N. (2013) Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim. Biophys. Acta 1833, 286–294 10.1016/j.bbamcr.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 6. Amunts A., Brown A., Bai X. C., Llácer J. L., Hussain T., Emsley P., Long F., Murshudov G., Scheres S. H. W., and Ramakrishnan V. (2014) Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 10.1126/science.1249410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amunts A., Brown A., Toots J., Scheres S. H. W., and Ramakrishnan V. (2015) Ribosome: the structure of the human mitochondrial ribosome. Science 348, 95–98 10.1126/science.aaa1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai N., Brown A., Amunts A., and Ramakrishnan V. (2017) The structure of the yeast mitochondrial ribosome. Science 355, 528–531 10.1126/science.aal2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greber B. J., Bieri P., Leibundgut M., Leitner A., Aebersold R., Boehringer D., and Ban N. (2015) Ribosome: the complete structure of the 55S mammalian mitochondrial ribosome. Science 348, 303–308 10.1126/science.aaa3872 [DOI] [PubMed] [Google Scholar]
- 10. Greber B. J., Boehringer D., Leibundgut M., Bieri P., Leitner A., Schmitz N., Aebersold R., and Ban N. (2014) The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–286 10.1038/nature13895 [DOI] [PubMed] [Google Scholar]
- 11. Greber B. J., Boehringer D., Leitner A., Bieri P., Voigts-Hoffmann F., Erzberger J. P., Leibundgut M., Aebersold R., and Ban N. (2014) Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–519 10.1038/nature12890 [DOI] [PubMed] [Google Scholar]
- 12. Brown A., Amunts A., Bai X. C., Sugimoto Y., Edwards P. C., Murshudov G., Scheres S. H. W., and Ramakrishnan V. (2014) Structure of the large ribosomal subunit from human mitochondria. Science 346, 718–722 10.1126/science.1258026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Silva D., Tu Y. T., Amunts A., Fontanesi F., and Barrientos A. (2015) Mitochondrial ribosome assembly in health and disease. Cell Cycle 14, 2226–2250 10.1080/15384101.2015.1053672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H. J., Maiti P., and Barrientos A. (2017) Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 47, 67–81 10.1016/j.semcancer.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sotgia F., and Lisanti M. P. (2017) Mitochondrial biomarkers predict tumor progression and poor overall survival in gastric cancers: companion diagnostics for personalized medicine. Oncotarget 8, 67117–67128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalghatgi S., Spina C. S., Costello J. C., Liesa M., Morones-Ramirez J. R., Slomovic S., Molina A., Shirihai O. S., and Collins J. J. (2013) Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 5, 192ra185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singh R., Sripada L., and Singh R. (2014) Side effects of antibiotics during bacterial infection: mitochondria, the main target in host cell. Mitochondrion 16, 50–54 10.1016/j.mito.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 18. Bogenhagen D. F., Ostermeyer-Fay A. G., Haley J. D., and Garcia-Diaz M. (2018) Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep. 22, 1935–1944 10.1016/j.celrep.2018.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng R., Smith E., and Barrientos A. (2018) Yeast mitoribosome large subunit assembly proceeds by hierarchical incorporation of protein clusters and modules on the inner membrane. Cell Metab. 27, 645–656.e7 10.1016/j.cmet.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Silva D., Poliquin S., Zeng R., Zamudio-Ochoa A., Marrero N., Perez-Martinez X., Fontanesi F., and Barrientos A. (2017) The DEAD-box helicase Mss116 plays distinct roles in mitochondrial ribogenesis and mRNA-specific translation. Nucleic Acids Res. 45, 6628–6643 10.1093/nar/gkx426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saha P., and Datta K. (2018) Multi-functional, multicompartmental hyaluronan-binding protein 1 (HABP1/p32/gC1qR): implication in cancer progression and metastasis. Oncotarget 9, 10784–10807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fogal V., Richardson A. D., Karmali P. P., Scheffler I. E., Smith J. W., and Ruoslahti E. (2010) Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 30, 1303–1318 10.1128/MCB.01101-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muta T., Kang D., Kitajima S., Fujiwara T., and Hamasaki N. (1997) p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. 272, 24363–24370 10.1074/jbc.272.39.24363 [DOI] [PubMed] [Google Scholar]
- 24. Seytter T., Lottspeich F., Neupert W., and Schwarz E. (1998) Mam33p, an oligomeric, acidic protein in the mitochondrial matrix of Saccharomyces cerevisiae is related to the human complement receptor gC1q-R. Yeast 14, 303–310 [DOI] [PubMed] [Google Scholar]
- 25. Yagi M., Uchiumi T., Takazaki S., Okuno B., Nomura M., Yoshida S., Kanki T., and Kang D. (2012) p32/gC1qR is indispensable for fetal development and mitochondrial translation: importance of its RNA-binding ability. Nucleic Acids Res. 40, 9717–9737 10.1093/nar/gks774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chowdhury A. R., Ghosh I., and Datta K. (2008) Excessive reactive oxygen species induces apoptosis in fibroblasts: role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR). Exp. Cell Res. 314, 651–667 10.1016/j.yexcr.2007.10.033 [DOI] [PubMed] [Google Scholar]
- 27. Meenakshi J., Anupama, Goswami S. K., and Datta K. (2003) Constitutive expression of hyaluronan binding protein 1 (HABP1/p32/gC1qR) in normal fibroblast cells perturbs its growth characteristics and induces apoptosis. Biochem. Biophys. Res. Commun. 300, 686–693 10.1016/S0006-291X(02)02788-2 [DOI] [PubMed] [Google Scholar]
- 28. Saha P., Chowdhury A. R., Dutta S., Chatterjee S., Ghosh I., and Datta K. (2013) Autophagic vacuolation induced by excess ROS generation in HABP1/p32/gC1qR overexpressing fibroblasts and its reversal by polymeric hyaluronan. PLoS ONE 8, e78131 10.1371/journal.pone.0078131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feichtinger R. G., Oláhová M., Kishita Y., Garone C., Kremer L. S., Yagi M., Uchiumi T., Jourdain A. A., Thompson K., D'Souza A. R., Kopajtich R., Alston C. L., Koch J., Sperl W., Mastantuono E., et al. (2017) Biallelic C1QBP mutations cause severe neonatal-, childhood-, or later-onset cardiomyopathy associated with combined respiratory-chain deficiencies. Am. J. Hum. Genet. 101, 525–538 10.1016/j.ajhg.2017.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang J., Zhang Y., Krainer A. R., and Xu R. M. (1999) Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl. Acad. Sci. U.S.A. 96, 3572–3577 10.1073/pnas.96.7.3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pu Y.-G., Jiang Y.-L., Ye X.-D., Ma X.-X., Guo P.-C., Lian F.-M., Teng Y.-B., Chen Y., and Zhou C.-Z. (2011) Crystal structures and putative interface of Saccharomyces cerevisiae mitochondrial matrix proteins Mmf1 and Mam33. 175, 469–474 [DOI] [PubMed] [Google Scholar]
- 32. Sprehe M., Fisk J. C., McEvoy S. M., Read L. K., and Schumacher M. A. (2010) Structure of the Trypanosoma brucei p22 protein, a cytochrome oxidase subunit II-specific RNA-editing accessory factor. J. Biol. Chem. 285, 18899–18908 10.1074/jbc.M109.066597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roloff G. A., and Henry M. F. (2015) Mam33 promotes cytochrome c oxidase subunit I translation in Saccharomyces cerevisiae mitochondria. Mol. Biol. Cell 26, 2885–2894 10.1091/mbc.E15-04-0222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bender A., and Pringle J. R. (1991) Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 1295–1305 10.1128/MCB.11.3.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kranz J. E., and Holm C. (1990) Cloning by function: an alternative approach for identifying yeast homologs of genes from other organisms. Proc. Natl. Acad. Sci. U.S.A. 87, 6629–6633 10.1073/pnas.87.17.6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Henry M. F., and Silver P. A. (1996) A novel methyltransferase (Hmt1p) modifies poly(A)+-RNA-binding proteins. Mol. Cell. Biol. 16, 3668–3678 10.1128/MCB.16.7.3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Silva D., Fontanesi F., and Barrientos A. (2013) The DEAD box protein Mrh4 functions in the assembly of the mitochondrial large ribosomal subunit. Cell Metab. 18, 712–725 10.1016/j.cmet.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmidt U., Lehmann K., and Stahl U. (2002) A novel mitochondrial DEAD box protein (Mrh4) required for maintenance of mtDNA in Saccharomyces cerevisiae. FEMS Yeast Res. 2, 267–276 10.1016/S1567-1356(02)00109-5 [DOI] [PubMed] [Google Scholar]
- 39. Linder P., Lasko P. F., Ashburner M., Leroy P., Nielsen P. J., Nishi K., Schnier J., and Slonimski P. P. (1989) Birth of the D-E-A-D box. Nature 337, 121–122 10.1038/337121a0 [DOI] [PubMed] [Google Scholar]
- 40. Schmid S. R., and Linder P. (1992) D-E-A-D protein family of putative RNA helicases. Mol. Microbiol. 6, 283–291 10.1111/j.1365-2958.1992.tb01470.x [DOI] [PubMed] [Google Scholar]
- 41. Graack H. R., Grohmann L., and Choli T. (1988) Mitochondrial ribosomes of yeast: isolation of individual proteins and N-terminal sequencing. FEBS Lett. 242, 4–8 10.1016/0014-5793(88)80975-X [DOI] [PubMed] [Google Scholar]
- 42. Graack H. R., Grohmann L., Kitakawa M., and Goldschmidt-Reisin S. (1995) Gene MRP-L4, encoding mitochondrial ribosomal protein YmL4, is indispensable for proper non-respiratory cell functions in yeast. Gene 152, 107–112 10.1016/0378-1119(94)00633-4 [DOI] [PubMed] [Google Scholar]
- 43. Gruschke S., Gröne K., Heublein M., Hölz S., Israel L., Imhof A., Herrmann J. M., and Ott M. (2010) Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J. Biol. Chem. 285, 19022–19028 10.1074/jbc.M110.113837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Myers A. M., Pape L. K., and Tzagoloff A. (1985) Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae. EMBO J. 4, 2087–2092 10.1002/j.1460-2075.1985.tb03896.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Decoster E., Simon M., Hatat D., and Faye G. (1990) The MSS51 gene product is required for the translation of the COX1 mRNA in yeast mitochondria. Mol. Gen. Genet. 224, 111–118 [DOI] [PubMed] [Google Scholar]
- 46. Carr H. S., and Winge D. R. (2003) Assembly of cytochrome c oxidase within the mitochondrion. Acc. Chem. Res. 36, 309–316 10.1021/ar0200807 [DOI] [PubMed] [Google Scholar]
- 47. Soto I. C., Fontanesi F., Liu J., and Barrientos A. (2012) Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 1817, 883–897 10.1016/j.bbabio.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barrientos A., Zambrano A., and Tzagoloff A. (2004) Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 23, 3472–3482 10.1038/sj.emboj.7600358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fontanesi F., Clemente P., and Barrientos A. (2011) Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 286, 555–566 10.1074/jbc.M110.188805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mick D. U., Wagner K., van der Laan M., Frazier A. E., Perschil I., Pawlas M., Meyer H. E., Warscheid B., and Rehling P. (2007) Shy1 couples Cox1 translational regulation to cytochrome c oxidase assembly. EMBO J. 26, 4347–4358 10.1038/sj.emboj.7601862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perez-Martinez X., Broadley S. A., and Fox T. D. (2003) Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 22, 5951–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perez-Martinez X., Butler C. A., Shingu-Vazquez M., and Fox T. D. (2009) Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol. Biol. Cell 20, 4371–4380 10.1091/mbc.e09-06-0522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pierrel F., Bestwick M. L., Cobine P. A., Khalimonchuk O., Cricco J. A., and Winge D. R. (2007) Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 26, 4335–4346 10.1038/sj.emboj.7601861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shingú-Vazquez M., Camacho-Villasana Y., Sandoval-Romero L., Butler C. A., Fox T. D., and Pérez-Martínez X. (2010) The carboxyl-terminal end of Cox1 is required for feedback assembly regulation of Cox1 synthesis in Saccharomyces cerevisiae mitochondria. J. Biol. Chem. 285, 34382–34389 10.1074/jbc.M110.161976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soto I. C., Fontanesi F., Myers R. S., Hamel P., and Barrientos A. (2012) A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab. 16, 801–813 10.1016/j.cmet.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zambrano A., Fontanesi F., Solans A., de Oliveira R. L., Fox T. D., Tzagoloff A., and Barrientos A. (2007) Aberrant translation of cytochrome c oxidase subunit 1 mRNA species in the absence of Mss51p in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 18, 523–535 10.1091/mbc.e06-09-0803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jia L., Kaur J., and Stuart R. A. (2009) Mapping of the Saccharomyces cerevisiae Oxa1-mitochondrial ribosome interface and identification of MrpL40, a ribosomal protein in close proximity to Oxa1 and critical for oxidative phosphorylation complex assembly. Eukaryot. Cell 8, 1792–1802 10.1128/EC.00219-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Szyrach G., Ott M., Bonnefoy N., Neupert W., and Herrmann J. M. (2003) Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J. 22, 6448–6457 10.1093/emboj/cdg623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Box J. M., Kaur J., and Stuart R. A. (2017) MrpL35, a mitospecific component of mitoribosomes, plays a key role in cytochrome c oxidase assembly. Mol. Biol. Cell 28, 3489–3499 10.1091/mbc.e17-04-0239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hibbs M. A., Hess D. C., Myers C. L., Huttenhower C., Li K., and Troyanskaya O. G. (2007) Exploring the functional landscape of gene expression: directed search of large microarray compendia. Bioinformatics 23, 2692–2699 10.1093/bioinformatics/btm403 [DOI] [PubMed] [Google Scholar]
- 61. Pillet B., Mitterer V., Kressler D., and Pertschy B. (2017) Hold on to your friends: dedicated chaperones of ribosomal proteins: dedicated chaperones mediate the safe transfer of ribosomal proteins to their site of pre-ribosome incorporation. Bioessays 39, 1–12 10.1002/bies.201670013 [DOI] [PubMed] [Google Scholar]
- 62. Ting Y. H., Lu T. J., Johnson A. W., Shie J. T., Chen B. R., Kumar S. S., and Lo K. Y. (2017) Bcp1 is the nuclear chaperone of Rpl23 in Saccharomyces cerevisiae. J. Biol. Chem. 292, 585–596 10.1074/jbc.M116.747634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frénois F., Legrand P., and Fribourg S. (2016) Sqt1p is an eight-bladed WD40 protein. Acta Crystallogr. F Struct. Biol. Commun. 72, 59–64 10.1107/S2053230X15024097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eisinger D. P., Dick F. A., Denke E., and Trumpower B. L. (1997) SQT1, which encodes an essential WD domain protein of Saccharomyces cerevisiae, suppresses dominant-negative mutations of the ribosomal protein gene QSR1. Mol. Cell. Biol. 17, 5146–5155 10.1128/MCB.17.9.5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pausch P., Singh U., Ahmed Y. L., Pillet B., Murat G., Altegoer F., Stier G., Thoms M., Hurt E., Sinning I., Bange G., and Kressler D. (2015) Co-translational capturing of nascent ribosomal proteins by their dedicated chaperones. Nat. Commun. 6, 7494 10.1038/ncomms8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. West M., Hedges J. B., Chen A., and Johnson A. W. (2005) Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol. Cell. Biol. 25, 3802–3813 10.1128/MCB.25.9.3802-3813.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gietz R. D., Schiestl R. H., Willems A. R., and Woods R. A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 10.1002/yea.320110408 [DOI] [PubMed] [Google Scholar]
- 68. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Struhl K., Stinchcomb D. T., Scherer S., and Davis R. W. (1979) High-frequency transformation of yeast: autonomous replication of hybrid DNA molecules. Proc. Natl. Acad. Sci. U.S.A. 76, 1035–1039 10.1073/pnas.76.3.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schneider B. L., Seufert W., Steiner B., Yang Q. H., and Futcher A. B. (1995) Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast 11, 1265–1274 10.1002/yea.320111306 [DOI] [PubMed] [Google Scholar]
- 71. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 10.1038/nature00935 [DOI] [PubMed] [Google Scholar]
- 72. Koepp D. M., Wong D. H., Corbett A. H., and Silver P. A. (1996) Dynamic localization of the nuclear import receptor and its interactions with transport factors. J. Cell Biol. 133, 1163–1176 10.1083/jcb.133.6.1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rose M. D., Novick P., Thomas J. H., Botstein D., and Fink G. R. (1987) A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene 60, 237–243 10.1016/0378-1119(87)90232-0 [DOI] [PubMed] [Google Scholar]
- 74. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 75. Meisinger C., Sommer T., and Pfanner N. (2000) Purification of Saccharomcyes cerevisiae mitochondria devoid of microsomal and cytosolic contaminations. Anal. Biochem. 287, 339–342 10.1006/abio.2000.4868 [DOI] [PubMed] [Google Scholar]
- 76. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 77. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fearon K., and Mason T. L. (1988) Structure and regulation of a nuclear gene in Saccharomyces cerevisiae that specifies MRP7, a protein of the large subunit of the mitochondrial ribosome. Mol. Cell. Biol. 8, 3636–3646 10.1128/MCB.8.9.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fearon K., and Mason T. L. (1992) Structure and function of MRP20 and MRP49, the nuclear genes for two proteins of the 54 S subunit of the yeast mitochondrial ribosome. J. Biol. Chem. 267, 5162–5170 [PubMed] [Google Scholar]
- 80. Kehrein K., Schilling R., Möller-Hergt B. V., Wurm C. A., Jakobs S., Lamkemeyer T., Langer T., and Ott M. (2015) Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep. 10, 843–853 10.1016/j.celrep.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 81. Prestele M., Vogel F., Reichert A. S., Herrmann J. M., and Ott M. (2009) Mrpl36 is important for generation of assembly competent proteins during mitochondrial translation. Mol. Biol. Cell 20, 2615–2625 10.1091/mbc.e08-12-1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Regev-Rudzki N., Karniely S., Ben-Haim N. N., and Pines O. (2005) Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol. Biol. Cell 16, 4163–4171 10.1091/mbc.e04-11-1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Biswas T. K., and Getz G. S. (1999) The single amino acid changes in the yeast mitochondrial S4 ribosomal protein cause temperature-sensitive defect in the accumulation of mitochondrial 15S rRNA. Biochemistry 38, 13042–13054 10.1021/bi990058u [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.