

Abstract
Diet influences dopamine transmission in motor- and reward-related basal ganglia circuitry. In part, this reflects diet-dependent regulation of circulating and brain insulin levels. Activation of striatal insulin receptors amplifies axonal dopamine release in brain slices, and regulates food preference in vivo. The effect of insulin on dopamine release is indirect, and requires striatal cholinergic interneurons that express insulin receptors. However, insulin also acts directly on dopamine axons to increase dopamine uptake by promoting dopamine transporter (DAT) surface expression, counteracting enhanced dopamine release. Here we determined the functional consequences of acute insulin exposure and chronic diet-induced changes in insulin on DAT activity after evoked dopamine release in striatal slices from adult ad-libitum fed (AL) rats and mice, and food-restricted (FR) or high-fat/high-sugar obesogenic (OB) diet rats. Uptake kinetics were assessed by fitting evoked dopamine transients to the Michaelis-Menten equation and extracting Cpeak and Vmax. Insulin (30 nM) increased both parameters in the caudate putamen and nucleus accumbens core of AL rats in an insulin receptor- and PI3-kinase-dependent manner. A pure effect of insulin on uptake was unmasked using mice lacking striatal acetylcholine, in which increased Vmax caused a decrease in Cpeak. Diet also influenced Vmax, which was lower in FR versus AL. The effects of insulin on Cpeak and Vmax were amplified by FR but blunted by OB, consistent with opposite consequences of these diets on insulin levels and insulin receptor sensitivity. Overall, these data reveal acute and chronic effects of insulin and diet on dopamine release and uptake that will influence brain reward pathways.
Keywords: rats, mice, Michaelis-Menten, cholinergic interneurons, nAChRs, obesity
Graphical Abstract
Insulin and diet have marked effects on dopamine (DA) transmission in the brain. Insulin increases evoked striatal DA release indirectly via cholinergic interneurons (ChIs) but enhances DA transporter activity and DA uptake directly by activating insulin receptors coupled to the PI3K / Akt pathway. Food restriction (FR) or obesogenic (OB) diets that produce lower or higher plasma insulin respectively decrease DA release and uptake, relative to an ad-libitum (AL) diet. Moreover, insulin-induced increases in DA release and uptake are enhanced with FR but blunted with OB diets.
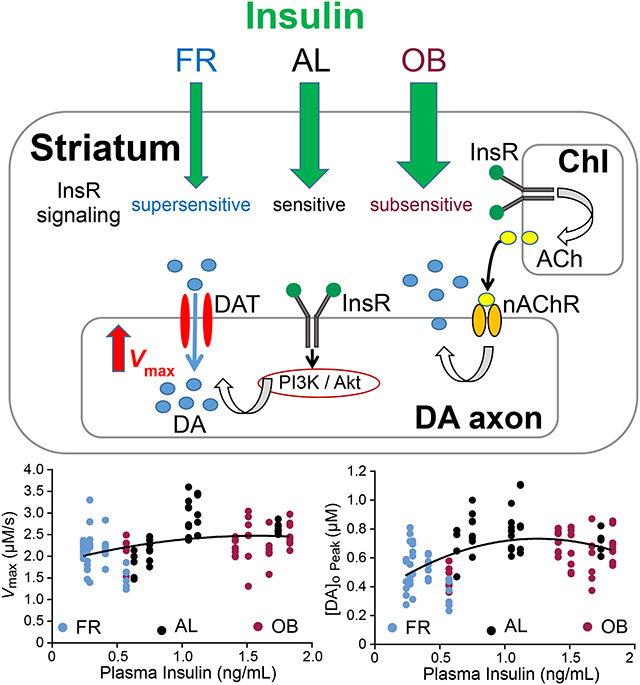
Introduction
Eating disorders, whether consumption of too little or too much, can have serious negative consequences on health, and become cycles that are difficult to break. There is increasing evidence that chronic alterations in diet can have marked effects on the reward-related circuitry of the brain (Carr, 2007; Davis et al., 2008; Carr, 2011; Stice et al., 2013; Ferrario et al., 2016) that can influence decisions on what, when, and how much we eat. Indeed, peak evoked extracellular dopamine (DA) concentration ([DA]o) detected by fast-scan cyclic voltammetry (FSCV) in striatal slices is decreased by either food restriction (FR) or a high-fat/high-sugar obesogenic (OB) diet versus ad-libitum (AL) feeding (Stouffer et al., 2015). Among other factors, the pancreatic hormone insulin has been shown to regulate DA release and uptake in reward-related pathways (Mebel et al., 2012; Labouebe et al., 2013; Stouffer et al., 2015; Fordahl et al., 2017). Insulin receptors (InsRs) are expressed in midbrain DA neurons (Figlewicz et al., 2003), and mediate the effects of insulin on DA neurons in the ventral tegmental area (VTA) to decrease feeding behavior (Mebel et al., 2012; Labouebe et al., 2013). In addition, physiological concentrations of insulin (i.e., 10 to 30 nM) that are typically reached in the CSF and brain after feeding (Strubbe et al., 1988; Banks and Kastin, 1998; Woods et al., 2003; Banks, 2004) enhance evoked [DA]o throughout the rat striatal complex with the greatest effect in the ventral striatum (Stouffer et al., 2015; Fordahl et al., 2017), which also has the highest levels of InsR expression (Werther et al., 1987; Schulingkamp et al., 2000).
The mechanism by which insulin enhances evoked increases in [DA]o in the striatum is indirect through InsRs on striatal cholinergic interneurons (ChIs); InsR activation increases ChI excitability and subsequently boosts DA release induced by nAChRs on DA axons (Stouffer et al., 2015). Moreover, the sensitivity of evoked [DA]o to insulin is markedly enhanced with FR, in which plasma insulin levels are lower than in AL, whereas the response to insulin is absent in slices from rats on an OB diet in which plasma insulin levels are higher than in AL (Stouffer et al., 2015). Thus, chronic diet-dependent changes in circulating insulin levels appear to alter acute insulin signaling in both motor and reward related pathways of the brain, although other nutritive factors including glucose and leptin that are also chronically altered with feeding state could play a role. Importantly, intact insulin signaling in the ventral striatum is required for the development of flavor preference, which has implications for food choice (Stouffer et al., 2015; Woods et al., 2016).
As well as enhancing striatal DA release, insulin also increases dopamine transporter (DAT) trafficking to the plasma membrane via a PI3-kinase (PI3K) and Akt signaling pathway, thereby increasing overall DAT activity and DA uptake (Carvelli et al., 2002; Garcia et al., 2005; Schoffelmeer et al., 2011; Mebel et al., 2012; Jones et al., 2017). However, little is known about how opposite changes in circulating plasma insulin levels with FR versus OB diets in vivo influence striatal DAT activity in intact, ex vivo striatal slices and DAT responsiveness to acute insulin exposure. Elucidating these interactions is important because of the role of insulin in feeding behavior (Stouffer et al., 2015; Woods et al., 2016), and also because the DAT is instrumental in mediating the effects of psychostimulant drugs (e.g., Sulzer, 2011).
To assess DAT activity, we fitted the initial portion of the falling phase of evoked increases in [DA]o to the integrated form of the Michaelis-Menten equation to extract Vmax (maximal uptake velocity, which is proportional to the number of functional DATs), as well as the accurate peak [DA]o (Cpeak) for each record (see Li et al., 2010). The primary data set analyzed were from Stouffer et al. (2015), in which data were presented mainly as measured peak evoked [DA]o normalized to the appropriate control for each condition. Here we focused on the effects of insulin and diet on DA uptake, and report unpublished Vmax values and computed peak evoked [DA]o extracted from the original data set, using Michaelis-Menten analysis. For comparison, we have also illustrated the basic effect of insulin on Vmax under control conditions using data that were previously reported in table form (Stouffer et al., 2015). Overall, this new analysis allowed us to assess the role of the PI3-kinase/Akt signaling pathway and feeding state on DAT-dependent uptaketo complement our previous studies of the effects of insulin on DA release via activation of ChIs.. Lastly, we compared DAT activity between AL-fed rats and mice, and assessed the influence of insulin on DA uptake in the absence of striatal acetylcholine (ACh).
Methods
Animals:
All animal procedures followed NIH guidelines and were approved by the NYU Langone Health Animal Care and Use Committee. Animals were kept on a 12 h light:dark cycle, with lights on from 06:00 to 18:00 local time. Male Sprague-Dawley Rats (Taconic) were normally housed in pairs but were singly housed for comparison of the effects of AL, FR and OB diets. Generation of mice lacking choline acetyltransferease (ChAT) and thus ACh in the forebrain (Nkx2.1cre;ChATflox/flox) or non-mutant littermate controls (Nkx2.1Cre-;ChATflox/flox or Nkx2.1cre;ChATflox/+) was as described previously (Patel et al., 2012); male mice were examined and were housed in groups of five or fewer.
Diets:
As described previously (Stouffer et al., 2015), adult rats (8–10 weeks old) were assigned to AL, FR or OB diet regimens such that initial body weights were similar across groups. AL rats received both food (chow) and water ad-libitum. Rats on the FR diet received titrated amounts of chow to maintain body weight at 80% of initial weight but had free access to water. Rats on the OB diet were allowed free access to a moderately high-fat/high-sugar chocolate flavored liquid (Ensure), as well as chow and water. The data used for this study were taken from a subset of rats from each diet group for which plasma insulin values were available (n = 5 rats per group) (Stouffer et al., 2015). Diets for these rats lasted a total of 21–32 days. FR rats exhibited significantly lower final body weights versus AL (one-way ANOVA, F2,12 = 109.4, with Dunnett’s post hoc vs. AL; P = 0.0001) fed rats, whereas OB rats had higher final body weights than AL (one-way ANOVA, F2,12 = 109.4 with Dunnett’s post hoc vs. AL; P = 0.008) fed rats (FR = 308 ± 3 g; AL = 456 ± 6 g; OB = 505 ± 15 g).
Plasma insulin:
At the time of decapitation for slice preparation (see below), trunk blood was collected in EDTA-containing tubes and centrifuged at 1500 × g for 15 min. The plasma supernatant was then stored at −80 °C until processing with an ALPCO Rat Insulin ELISA kit.
DA recordings:
Procedures for preparing ex vivo brain slices were as described in Patel et al. (2013) and Stouffer et al. (2015). All animals were deeply anesthetized with sodium pentobarbital (SleepAway) (50 mg/kg, i.p.) before decapitation and preparation of coronal striatal slices (400 μm thickness for rats, 300 μm for mice); these procedures were initiated between 08:00 and 10:00 each experimental day. Slices were allowed to recover at room temperature for exactly one hour in a HEPES-buffered aCSF containing (in mM): NaCl (120); KCl (5); NaHCO3 (20); HEPES acid (6.7); HEPES sodium salt (3.3); MgSO4 (2); glucose (10); CaCl2 (2); equilibrated with 95% O2/5% CO2; two slices were then transferred to a submersion recording chamber (Warner Instruments) superfused with a bicarbonate-buffered aCSF containing (in mM): NaCl (124); KCl (3.7); NaHCO3 (26); MgSO4 (1.3); KH2PO4 (1.3); glucose (10); CaCl2 (2.4); and bovine serum albumin (0.1 mg/mL) equilibrated with 95% O2/5% CO2, and maintained at 32 °C. Slices were allowed to acclimatize to this environment for 30 min before recordings were initiated. [DA]o evoked by local single-pulse electrical stimulation (0.1 ms duration) was monitored with 7 μm diameter carbon fiber electrodes and FSCV using a Millar Voltammeter scanning from −700 mV to +1300 mV to −700 mV at a rate of 800 V/s every 100 ms (Patel et al., 2013; Patel, 2016). Initial recordings were made in three to five sites per region in the caudate putamen (CPu) and nucleus accumbens (NAc) core in each of two slices over 20 to 30 mins as an index of slice viability and to compare differences in DA release and uptake in feeding groups closer to the time of sacrifice. Immediately after this sampling, superfusion with either aCSF alone, aCSF plus insulin, aCSF plus test drug, or insulin plus test drug was begun; after 60 min, recording of evoked release in three to five sites per region was initiated in the continued presence of the superfusing solution. Responses were quantified by post-experiment calibration of carbon fiber electrodes with 1 μM DA in aCSF in the recording chamber at 32 °C. Release and uptake in the presence of insulin were compared to those obtained in the time-matched control experiments with aCSF alone or the test drug alone.
Cpeak and Vmax analysis:
The falling phase of single-pulse evoked DA transients detected by FSCV is governed by Michaelis-Menten uptake kinetics (Wightman et al. 1988; Wightman and Zimmerman, 1990; Wu et al., 2001). To evaluate changes in DAT activity we used a MATLAB® script in which an integrated form of the Michaelis-Menten equation was solved and then fitted to the initial segment of the falling phase of single-pulse evoked [DA]o curves (Figure 1) to extract Vmax (maximal uptake velocity) as described previously (Li et al., 2010). The value for Km (which is inversely related to the affinity of the DAT for DA) was fixed at values obtained by prior FSCV studies which were 0.2 μM for rats (Wu et al., 2001) or 0.9 μM for mice (Schmitz et al., 2001). Previous studies have determined that Km is similar across striatal subregions in a given species (Wu et al., 2001). Importantly, Km does not change with either FR or OB diets and is unaffected by insulin (Zhen et al., 2006; Jones et al., 2017). We also assumed that Km was unaltered in ChAT-KO mice or with exogenous application of drugs used in these studies. During kinetic analysis, we found that the tail of some computed [DA]o curves fell more rapidly than seen in the corresponding experimental record; this suggested that additional factors, including DA adsorption to the electrode, might play a role in prolonging the time course of experimental curves. To avoid this complication, we restricted the curve fitting to the initial portion of the falling phase (either three points for Km = 0.2 μM or four points for Km = 0.9 μM) after the peak (see Figure 1). It should also be noted that unlike some methods for evaluating Vmax from FSCV data (e.g. Wu et al., 2001), our method is not restricted to [DA]o much greater than Km.
Figure 1. Derivation of Cpeak and Vmax from MM-analysis of evoked [DA]o transients detected using FSCV.

(A) Representative evoked [DA]o plot showing the experimental data point for peak [DA]o (green cross), and the three data points used for curve fitting (red circles) to extract peak [DA]o (Cpeak) and Vmax in the CPu of an AL fed rat. (B) Fit of representative data to Michaelis-Menten (MM) equation with a fixed Km of 0.2 μM gave Cpeak = 1.25 μM (blue circle), Vmax = 3.27 μM/s and R2 = 0.997.
A value for Cpeak was also determined from the integrated Michaelis-Menten equation (Li et al., 2010) because peak concentration is difficult to capture faithfully with 100 ms sampling intervals. These concentration values have not been reported previously. Goodness-of-fit and coefficient of determination (R2) values were estimated; R2 values < 0.95 were rejected from the data sets reports here.
Drugs:
Salts for all buffers as well as insulin (from porcine pancreas), LY294002, and DA were purchased from Sigma Aldrich (St. Louis, MO, USA). Membrane permeable HNMPA (hydroxyl-2-naphthalenylmethylphosphonic acid trisacetoxymethyl ester) was from Enzo Life Sciences (Farmingdale, NY, USA), and picropodophyllotoxin (PPP) was from Tocris Bioscience (Bio-Techne Cooperation, Minneapolis, MN, USA).
Statistics:
Statistical analyses were performed using GraphPad Prism version 7.0. for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com. Data are expressed as means ± SEM or as individual measurements with means ± SEM, where n denotes the number of DA recording sites from three to five rats or mice per group. Significance of differences was assessed by unpaired Student’s t-tests, with or without Welch correction for unequal variance if appropriate, for comparison of two groups, or by one-way ANOVA, as indicated; significance was consider to be P < 0.05..
Results
Insulin increases DA release and DAT activity
Using Michaelis-Menten kinetics, we calculated Cpeak and Vmax for DA release and uptake from evoked DA release transients monitored by FSCV. In the striatum of AL rats, average Cpeak for single-pulse evoked [DA]o obtained in the absence and presence of insulin showed significant differences (one-way ANOVA, F3,226, = 33.15, P < 0.0001) as did Vmax (one-way ANOVA, F3,226, = 73.47, P < 0.0001). Cpeak was significantly higher in the CPu than in the NAc core, as was Vmax (Figure 2). These data reflect the well documented decreasing dorsoventral gradient of DA axon innervation and DAT density (Ciliax et al. 1995; Gonzalez-Hernandez et al., 2004; Marshall et al. 1990). Consistent with our previous report of increased evoked [DA]o with insulin (Stouffer et al., 2015), the present Michaelis-Menten analysis shows a significant increase (unpaired two-tailed t-tests) in Cpeak with insulin (30 nM) in both CPu (P = 0.0003) and NAc (P < 0.0001) (Figure 2A,B), despite the corresponding increase in Vmax in both regions (CPu, P = 0.0368; NAc core, P = 0.0020) (Figure 2C) previously published in Table 1 of Stouffer et al., 2015. Thus, the influence of insulin on DAT mediated DA uptake, which alone would be expected to decrease evoked [DA]o, is outcompeted by enhanced DA release. Notably, this physiological concentration of insulin appears to be optimal for modulation of DA release and DA uptake since higher concentrations of insulin are less effective at augmenting FSCV-detected peak [DA]o or Vmax (Stouffer et al., 2015).
Figure 2. Insulin enhances DA release and DAT activity in AL.

(A) Average single-pulse (1p) evoked [DA]o transients recorded in CPu and NAc core in the absence and presence of insulin (30 nM). Error bars have been omitted for clarity. (B) Insulin (Ins) significantly enhanced Cpeak derived from fitting individual [DA]o records to extract Michaelis-Menten parameters in both CPu and NAc core. (C) Insulin also increased average Vmax for DA uptake in both striatal regions. Data are means ± s.e.m. In CPu, n = 58 recording sites from 9 rats for control and n = 39 recording sites from 5 rats for insulin. In NAc core, n = 73 sites from 11 rats for control and n = 60 sites from 9 rats for insulin. One-way-ANOVA with Bonferroni’s multiple comparisons test of selected pairs was used to compare CPu versus NAc core under control conditions. Unpaired-two-tailed t-tests were used to compare each striatal subregion with and without insulin. ####p < 0.0001 versus CPu; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus respective control. Data in (C) are taken from Table 1, Stouffer et al., (2015).
The effect of insulin on DAT activity and DA release requires InsRs and PI3-kinase
Our previous study demonstrated that the effect of insulin on evoked increases in [DA]o occurred via InsRs rather than signaling through insulin-like growth factor receptors (IGF-1Rs) (Stouffer et al., 2015). It is also important to establish whether the effect of insulin on Vmax is mediated by InsRs, as well. Application of HNMPA (5 μM), an intracellular inhibitor of InsRs, had no effect on either Vmax or Cpeak (Figure 3). However, this InsR inhibitor blocked the effect of insulin on Vmax and Cpeak in both the CPu (unpaired one-tailed t-tests, P = 0.2685 for Vmax and P = 0.1098 for Cpeak) and NAc core (unpaired one-tailed t-tests, P = 0.3192 for Vmax and P = 0.4903 for Cpeak) (Figure 3). By contrast, insulin-induced increases in Vmax (unpaired one-tailed t-tests, P = 0.0378 for CPu and P = 0.0080 for NAc core) and Cpeak (unpaired one-tailed t-tests, P = 0.0008 for CPu and P = 0.0001 for NAc core) persisted in the presence of a selective IGF-1R inhibitor, PPP (1 μM) (Figure 3). Interestingly, PPP alone significantly decreased Cpeak (CPu: one-way ANOVA, F6,205, = 3.69, P = 0.0017, with Bonferroni’s post hoc, P = 0.0033; NAc core one-way ANOVA, F6,255, = 5.092, P = 0.0017, with Bonferroni’s post hoc, P = 0.0178), but not Vmax in both striatal subregions (Figure 3), implying that tonic activity at IGF-1Rs, presumably on ChIs, may play a part in regulating DA release.
Figure 3. Insulin enhances DA release and DAT activity via InsRs and PI3-kinase.

(A) In both the CPu and NAc core of AL rats the average insulin-induced enhancement of Cpeak was prevented by the insulin receptor (InsR) antagonist HNMPA (5 μM) and the PI3-kinase inhibitor LY29002 (1 μM) but not by the insulin-like growth factor-1 receptor (IGF-1R) antagonist PPP (1 μM). (B) In both CPu and NAc core the insulin-induced increases in Vmax were prevented by HNMPA but not by PPP. Insulin caused a decrease in Vmax in the presence of LY29002. Data are means ± s.e.m. For control n numbers see Figure 2. For HNMPA, CPu n = 31 recording sites, NAc core n = 38 sites (4 rats). For HNMPA + Ins CPu, n = 30 sites, NAc, n = 33 sites (4 rats). For PPP, CPu, n = 22 sites, NAc core n = 29 sites (3 rats). For PPP + Ins, CPu n = 23 sites, NAc core n = 29 sites (3 rats). For LY29002 or LY29002 + Ins, CPu n = 24 sites, NAc core n = 30 sites (3 rats). One-way-ANOVA with Bonferroni’s multiple comparisons test of selected pairs was used to compare each drug alone versus control. Unpaired one-tailed t-tests were used to compare each drug with and without Ins. n.s. = not significant (P > 0.05); #P < 0.05; ## P < 0.01 versus control; *P < 0.05; **P < 0.01;***P < 0.001.
The enhancement of DAT activity induced by insulin is in keeping with the established role of insulin in promoting surface expression of monoamine transporters, including DAT (Carvelli et al., 2002; Garcia et al., 2005; Jones et al., 2017). This has been shown to involve activation of PI3-kinase and the Akt pathway (Carvelli et al., 2002; Garcia et al., 2005; Robertson et al., 2010). Inhibiting PI3-kinase activity with LY249002 (1 μM) completely prevented insulin-induced increases in Vmax and Cpeak in both the CPu and NAc, without changing either parameter when applied alone (Figure 3). In fact, Vmax was significantly lower when insulin was applied in the presence of LY249002 (unpaired one-tailed t-tests, P = 0.0114) (Figure 3), suggesting that insulin may also activate another as yet unknown mechanism/pathway that opposes DAT insertion and is only revealed when the PI3-kinase/Akt pathway is blocked.
Insulin’s effect on DA release but not DAT activity is via ChIs
Our previous study revealed that the effect of insulin on evoked DA release was indirect by activating InsRs on striatal ChIs, increasing ChI excitability, and enhancing facilitation of DA release via nAChRs on DA axons (Stouffer et al., 2015). Of course, DA axons, as well as ChIs express InsRs (Stouffer et al., 2015), but whether the effect of insulin on DA uptake reflects a direct action or some indirect response to cholinergic activation has not been established. Because pharmacological nAChR agents can alter Km and potentially confound data interpretation (Middleton et al., 2007), we examined the effect of insulin on Vmax and Cpeak in mice that lack the ACh synthesizing enzyme ChAT in the forebrain (ChAT-KO) and therefore lack ACh release from otherwise intact ChIs (Patel et al., 2012). Our analysis assumed no change in Km for the DAT in ChAT-KO mice.
Interestingly, Vmax was slightly lower (one-way ANOVA, F3,107, = 12.26, with Bonferroni’s post hoc, P = 0.0410) in the CPu of ChAT-KO mice versus control mice (see Figure 4). Activation of nAChRs by nicotine has been shown to enhance DAT function, without increasing DAT surface expression (Middleton et al., 2007). Consequently, lower Vmax in KO mice could reflect loss of enhanced DA uptake by tonic nAChR activation that occurs in control mice. Importantly, the enhancing effect of insulin on both Cpeak and Vmax seen in AL rat CPu and NAc core was also seen in these regions in AL mice (Figure 4). Although the effect of insulin on Vmax persisted in ChAT-KO mice (unpaired one-tailed t-tests, P = 0.0035 for CPu; P < 0.0001 for NAc core), its enhancing effect on evoked DA release was lost in both CPu and NAc core (Figure 4). In fact, Cpeak decreased significantly in the presence of insulin in both regions from ChAT-KO mice (unpaired one-tailed t-tests, P = 0.0003 for CPu; P = 0.0179 for NAc core), revealing the true consequence of insulin on increasing DAT activity when cholinergic transmission is absent (Figure 4). Thus, these data demonstrate that modulation of DAT activity by insulin is direct, in contrast to insulin regulation of DA release which is indirect via ChIs.
Figure 4. Insulin-enhanced DA release requires ACh, but increased DAT activity does not.

(A) In both CPu and NAc core insulin (Ins; 30 nM) increased average Cpeak in ChAT control (ChAT-con) mice, but decreased Cpeak in ChAT-KO mice. (B) In both CPu and NAc core insulin increased Vmax in ChAT-con and ChAT-KO mice. Data are means ± s.e.m. For ChAT-con CPu, n = 22 recording sites, NAc core n = 28 sites (3 mice). For ChAT-con + Ins, CPu, n = 25 sites, NAc core n = 27 sites (3 mice). For ChAT-KO CPu, n = 25 sites, NAc core n = 28 sites (3 mice). For ChAT-KO + Ins CPu, n = 23 sites, NAc core n = 28 sites (3 mice). One-way-ANOVA with Bonferroni’s multiple comparisons test of selected pairs was used to compare ChAT-con versus ChAT-KO mice under control conditions. Unpaired one-tailed t-tests were used to compare each genotype with and without insulin. n.s. = not significant; #P < 0.05 versus ChAT-con; *P < 0.05; **P < 0.01;***P < 0.001; ****P < 0.001 versus respective control.
Striatal DA release and uptake kinetics in mice versus rats
We also took advantage of our FSCV data from AL mice and AL rats to compare Cpeak and Vmax in CPu and NAc core between these species obtained using the same methods. We found that Cpeak was significantly greater in both striatal subregions from mice than from rats (one-way ANOVA, F3,177, = 32.4, with Bonferroni’s post hoc; CPu, P < 0.0001, 1.63 ± 0.09 μM, n = 22 sites from 3 mice vs. 1.25 ± 0.03 μM, n = 58 recording sites from 9 rats; NAc core, P = 0.005, 1.14 ± 0.07 μM, n = 28 recording sites from 3 mice vs. 0.91 ± 0.03 μM, n = 73 recording sites from 11 rats). Moreover, Vmax values extracted from these same records were also significantly higher in mouse versus rat in both CPu and NAc core (one-way ANOVA, F3,177, = 164.0 with Bonferroni’s post hoc, P < 0.0001 for both; CPu, 5.67 ± 0.24 μM/s vs. 3.25 ± 0.08 μM/s; NAc core, 3.93 ± 0.13 μM/s, vs. 2.06 ± 0.07 μM/s). These data indicate greater DA release, as well as greater DAT activity in mice than in rats. In contrast, Calipari et al. (2012), also using FSCV, found no difference in Vmax between rat and mouse, although lower Vmax in monkey than in rodents. However this analysis used a single fixed value for Km of 0.16 μM for all species. In the present study, Cpeak and Vmax were derived using species-dependent values of Km. It is known that calculated Km values can differ according to the methods used to obtain them as well as the tissue preparations examined (see Nicholson, 1995). The values used here were taken from two separate studies of evoked DA release transients using FSCV (0.2 μM for rats from Wu et al., 2001 and 0.9 μM for mice from Schmitz et al., 2001). These values correlate extremely well with Km values for DA inhibition of [3H]-DA uptake obtained in cell lines expressing rat DAT (Km = 0.32 μM; Giros et al., 1991) or mouse DAT (Km = 0.93 μM; Brüss et al., 1999). Other studies in the literature find differences between Km in rat versus human or bovine DAT (Lee et al., 1996), possibly due to differences in N-linked glycosylation sites (Patel et al., 1993), offering further support for the value of using species-dependent values for Km.
FR and OB diets decrease DA release and DAT activity
We reported previously that single-pulse evoked [DA]o is significantly lower throughout the striatum of rats on either a FR or OB diet versus AL feeding, with no significant changes in DA tissue content among these diet groups (Stouffer et al., 2015). Given that plasma insulin is positively correlated with body adiposity (Strubbe, 1988; Stouffer et al., 2015; Woods et al., 2016) and that brain insulin, which mirrors plasma insulin, modulates surface DAT expression, we hypothesized that diet might influence evoked [DA]o via changes in DAT activity. As expected, average plasma insulin in FR rats was significantly lower than ALs (one-way ANOVA, F2,12 = 9.518, with Dunnett’s post hoc vs. AL; P = 0.0252), whereas plasma insulin with OB diet rats was generally higher, albeit not significantly (P = 0.3099), than ALs (FR: 0.36 ± 0.06; AL: 1.06 ± 0.19; OB: 1.40 ± 0.22 ng/mL, n = 5 rats per group). To assess the influence of diet and circulating insulin levels on DA release and uptake, FSCV recordings were made as soon as possible after preparation of brain slices (typically 90 min). It should be noted that Cpeak in these early measurements are slightly lower than those made in fresh sites in slices that were in the recording chamber at 32oC for ~150 min for insulin and drug studies (compare Cpeak values in Figure 5 with those in Figures 2 and 6). Higher levels in Cpeak possibly reflect on-going DA synthesis in the recording chamber during prolonged incubation. In vivo, on-going DA neuron activity promotes DA release and thereby a DA tone at striatal D2 DA autoreceptors (Benoit-Marand et al., 2001). By contrast, DA axons in striatal slices are no longer connected to DA neurons, so that on-going DA release is minimal, and slices lack a DA tone at D2 receptors (Patel et al., 1992). Thus, minimal release coupled with lack of D2 feedback to dampen the rate of DA synthesis by tyrosine hydroxylase (Vulto & Fowler, 1986) would increase DA content. Although other factors could be involved, this is the most likely explanation for higher evoked [DA]o with increasing incubation time.
Figure 5. DA release and DAT-mediated uptake are decreased by FR and OB diets.

(A) Inverted-U relationship between individual Cpeak values (upper panel) or Vmax (lower panel) in CPu and NAc core with plasma insulin levels from rats on a food restricted (FR), ad libitum (AL) or obesogenic (OB) diet. (B) In both CPu and NAc core average Cpeak (upper panel) was lower with FR and OB diets versus and AL diet. Average Vmax (lower panel) in CPu was lower with FR and OB diets and was also lower with FR versus an AL diet in NAc. Data are either individual values (A) or means ± s.e.m (B). For FR CPu, n = 36 recording sites, NAc core n = 40 sites (5 rats). For AL, CPu, n = 35 sites, NAc, n = 41 sites (5 rats). For OB, CPu, n = 38 sites, NAc core n = 40 sites (5 rats). A second order polynomial curve fit (Graphpad Prism) was used for Cpeak or Vmax versus plasma insulin data. One-way-ANOVA with Bonferroni’s multiple comparisons test of selected pairs was used to compare average Cpeak or Vmax for each feeding group to AL; n.s. = not significant (P > 0.05); **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure 6. Effects of insulin on DA release and DAT activity are enhanced by FR diet, but blunted by OB.

(A) In both CPu and NAc core insulin (Ins, 30 nM) increased average Cpeak in slices from FR but not from OB rats. (B) In both the CPu and NAc core insulin increased Vmax in FR rats but failed to increase Vmax in the CPu or NAc core of OB fed rats. Data are means ± s.e.m. For FR, CPu n = 39 recording sites, NAc core n = 45 sites (5 rats). For FR + Ins, CPu n = 25 sites, NAc core n = 30 sites (3 rats). For OB, CPu n = 23 sites, NAc core n = 28 sites (3 rats). For OB + Ins CPu, n = 24 sites, NAc core n = 30 sites (3 rats). One-way-ANOVA with Bonferroni’s multiple comparisons test of selected pairs was used to compare each diet group with and without Ins; n.s. = not significant (P > 0.05); ****P < 0.0001.
We found that the relationship between plasma insulin and Cpeak or Vmax values derived for individual evoked DA release transients in both the CPu and NAc core was an inverted U-shape that could be fitted to a second order polynomial (quadratic) (Figure 5A). Thus, with either low plasma insulin concentrations in FR rats or higher plasma insulin concentrations in OB rats, Cpeak and Vmax were lower than in AL-fed rats. Indeed, in both the CPu and NAc, average Cpeak was significantly lower in rats on the FR diet compared to AL-fed rats (CPu: one-way ANOVA, F2,106, = 28.03, with Bonferroni’s post hoc, P < 0.0001; NAc core: one-way ANOVA, F2,118, = 12.94, with Bonferroni’s post hoc, P < 0.0001) and was also significantly lower in OB rats versus AL (CPu: one-way ANOVA, F2,106, = 28.03, with Bonferroni’s post hoc, P = 0.0001; NAc core: one-way ANOVA, F2,118, = 12.94, with Bonferroni’s post hoc, P = 0.0065) (Figure 5B). Notably, average Vmax values were generally lower in CPu (CPu: one-way ANOVA, F2,106, = 15.12, P < 0.0001) and NAc core (NAc core: one-way ANOVA, F2,118, = 6.505, P = 0.0021) in both feeding groups (Figure 5B). Regardless of the cause, this observation eliminates enhanced DA uptake as a contributing factor to lower Cpeak with either the FR or OB diet.
The effect of insulin on DAT activity is amplified in FR but blunted with OB diets
We next evaluated the effect of exogenous insulin application on peak evoked [DA]o and DAT-mediated uptake in these diet groups to test the hypothesis that InsR-dependent regulation of the DAT is chronically under-stimulated in FR rats but chronically over-stimulated and consequently insensitive in OB rats. In FR rats, insulin increased Cpeak and Vmax in both the CPu (Cpeak: one-way ANOVA, F3,107, = 38.08 with Bonferroni’s post hoc, P < 0.0001; Vmax: one-way ANOVA, F3,107, = 40.27 with Bonferroni’s post hoc, P < 0.0001) and NAc core (Cpeak: one-way ANOVA, F3,129, = 9.593 with Bonferroni’s post hoc, P < 0.0001; Vmax: one-way ANOVA, F3,129 = 10.6 with Bonferroni’s post hoc, P < 0.0001) (Figure 6A,B). Indeed, the effect of 30 nM insulin on both Cpeak and Vmax in FR rats was greater than in AL rats (e.g., in CPu ~60% increase in Cpeak in FR vs. ~20% in AL; and ~50% increase in Vmax in FR vs. ~16% in AL). In contrast, the enhancing effects of insulin on Cpeak and Vmax were completely absent in OB rats in both the CPu (Cpeak: one-way ANOVA, F3,107, = 38.08 with Bonferroni’s post hoc, P > 0.9999; Vmax: one-way ANOVA, F3,107, = 40.27 with Bonferroni’s post hoc, P = 0.2015) and NAc core (Cpeak: one-way ANOVA, F3,129, = 9.593 with Bonferroni’s post hoc, P > 0.9999; Vmax: one-way ANOVA, F3,129 = 10.6 with Bonferroni’s post hoc, P = 0.3480) (Figure 6A,B). Together, these data show that insulin-dependent regulation of DA uptake, as well as DA release (Stouffer et al., 2015), is amplified by FR, but lost with an OB diet, reflecting consequences of dynamic changes in the sensitivity of InsR signaling pathways.
Discussion
We recently reported that exogenous insulin amplifies single-pulse evoked [DA]o by increasing ChI excitability and nAChR-dependent facilitation of DA release (Stouffer et al., 2015). However, evoked [DA]o transients reflect a net balance between DA release and DA uptake. Given that insulin can promote DAT trafficking to the plasma membrane (Carvelli et al., 2002; Garcia et al., 2005; Schoffelmeer et al., 2011; Mebel et al., 2012; Jones et al., 2017), we hypothesized that insulin-dependent regulation of [DA]o also includes changes in DAT-mediated DA uptake, and that dietary changes that chronically alter endogenous circulating, and therefore brain, insulin levels, should also affect DAT function and activity.
Our approach of fitting the initial part of the falling phase of evoked [DA]o to Michaelis-Menten kinetics allowed us to extract Cpeak for DA release, as well as Vmax for DA uptake. This analysis assumes that a relatively large volume of tissue surrounding the recording carbon-fiber microelectrode is at the same [DA]o so that diffusion does not contribute to DA clearance, as in previous models (Wightman et al. 1988; Wightman and Zimmerman, 1990; Wu et al., 2001). The goodness of fit for the data presented here support the validity of this assumption; this is beneficial as the involvement of a diffusional component would require a much more complicated analysis (Nicholson, 1995). It should be noted that the Cpeak values reported here are typically higher than peak evoked [DA]o obtained experimentally with FSCV, presumably because the 100 ms sampling interval used and imperfect response time of carbon-fiber microelectrodes prevent capturing true peak concentration. On the other hand, our reported Vmax values should not be viewed as absolute Vmax for the DAT, as this varies with experimental preparations and approaches, but rather as an index of DAT activity for the comparisons of conditions and diet groups evaluated using the same protocols.
Insulin enhances DAT Vmax independently of effects on ChIs and ACh
Physiological concentrations of insulin that amplify evoked DA transients also enhance Vmax for DA uptake (Stouffer et al., 2015). We show here that this effect occurs through activation of InsRs rather than IGF-1Rs that also bind insulin. Inhibition of PI3-kinase had no effect on peak [DA]o or Vmax alone, yet prevented insulin-induced increases in DAT function. These findings are consistent with PI3-kinase dependent trafficking of DAT to the surface of DA axons (Carvelli et al., 2002; Garcia et al., 2005; Schoffelmeer et al., 2011; Mebel et al., 2012; Jones et al., 2017) as the likely mechanism for increased DA uptake with insulin.
Activation of striatal ChIs powerfully regulates DA release, with ChI stimulation able to trigger nAChR-dependent DA release in the absence of action-potential dependent stimulation of DA axons (Threlfell et al., 2012; Cachope et al., 2012). Moreover, pharmacological antagonism of nAChRs by application of nicotine at desensitizing concentrations (Rice & Cragg, 2003; Zhang & Sulzer, 2003; Koranda et al., 2014), or genetic deletion of striatal ACh synthesis (Patel et al., 2012) leads to enhanced frequency responsiveness of axonal DA release, thus enhancing signal-to-background DA signaling. Insulin acts at InsRs on ChIs to increase striatal ChI excitability, and thereby enhance evoked striatal DA release in an ACh and nAChR-dependent manner; a striking demonstration of this dependence is the complete absence of an effect of insulin on evoked DA release in striatal slices from ChAT-KO mice (Stouffer et al., 2015). Here, we demonstrate that ChAT-KO striatum still responds to insulin, but through a direct effect on DAT function in DA axons. Indeed, a significant proportion of striatal InsRs are on DA axons (Stouffer et al., 2015). In the absence of cholinergic transmission, the analyses reported here reveal a significant insulin-dependent decrease in net evoked [DA]o resulting from enhanced DA uptake. Thus, the effects of insulin on ChI-facilitated DA release will normally dominate over effects on DAT activity. However, in the absence of ChI activation, enhanced uptake could dominate, which would also enhance signal-to-background for striatal DA transmission.
FR and OB diets similarly decrease baseline evoked DA release and DAT activity, but oppositely affect sensitivity to insulin
Insulin levels in both plasma and CSF increase quickly after peripheral glucose elevation, with increased insulin detected in striatum in less than 5 min of an increase in plasma insulin elevation (Strubbe et al., 1988; Banks et al., 1997; Banks and Kastin, 1998), albeit with brain levels ~25% lower than in plasma (Gray et al., 2014). Are the brain levels functional? The answer is yes; Woods et al. (2016) recently demonstrated that InsRs and Akt-pathways in the NAc can be stimulated within 7 minutes of glucose ingestion. Moreover, insulin in the NAc also influence food choices, as indicated by loss of development of preference for the flavor of a glucose-containing solution when paired with NAc infusion of antibodies to insulin which disrupts normal InsR signaling (Stouffer et al., 2015). These rapid effects in a key brain reward center add a new dimension to the established role of insulin in the hypothalamus, where it provides a satiety signal to end further food consumption (Baskin et al., 1999; Porte et al., 2002).
Given that circulating insulin levels correlate with body adiposity (Strubbe et al. 1988; Stouffer et al., 2015; Woods et al., 2016), an opposing change in InsR sensitivity provides the most likely explanation for the enhanced insulin sensitivity of striatal DA release with FR and loss of responsiveness with OB (Stouffer et al.. 2015). In support of this, using the same diets, Woods et al. (2016) showed enhanced glucose-induced InsR phosphorylation in the NAc with FR relative to AL, with the absence of InsR phosphorylation in OB rats. Data reported here extend these previous results by showing that FR and OB diets decrease baseline striatal DA release and DA uptake. The seemingly paradoxical observation that dramatically different diets have the same negative effect on Cpeak and Vmax can also be explained by chronic, diet-induced changes in striatal InsR sensitivity. As already discussed, acute elevation of physiological levels of insulin leads to increased ChI excitability and nAChR-dependent DA release, as well as an increase in DAT surface expression and Vmax. In FR, the opposite conditions prevail, with chronically decreased insulin levels predicted to decrease ChI excitability and Cpeak, and well as decrease surface DAT and Vmax, as seen (Figure 5). Analogously, loss of striatal InsR sensitivity and insulin responsiveness with an OB diet (Stouffer et al., 2015; Woods et al. 2016) should also decrease ChI excitability and DA release, as well as impair surface DAT expression and lower Vmax from loss of tonic effects of insulin on these parameters. Although absolute values for Vmax differ among experimental paradigms, the 25–30% decreases reported for CPu and NAc with these diets parallel decreases obtained under non-depolarizing conditions using synaptosomes and Bmax (maximal binding) for surface DAT expression with either shorter food deprivation (Patterson et al. 1998) or the same FR and OB diets examined here (Zhen et al., 2006; Jones et al., 2017). Notably, a lower Vmax was also seen in FSCV studies in striatal slices from mice on a high-fat only diet (normal sugar), although the decrease was <20% from control, and not accompanied by a change in peak evoked [DA]o (Fordahl & Jones, 2017). Whether these slight differences from the present results reflect diet or species differences has not been addressed. In this light, mice with global KO of brain InsRs show the opposite effect on DA uptake monitored in striatal slices with amperometry, with a 40% narrowing of the width of evoked [DA]o transients versus control suggesting enhanced uptake (Kleinridders et al., 2015). However, these mice were 14 months old at the time of study, with other consequences of InsR KO, including increased monoamine oxidase activity and oxidative stress that could indirectly alter DA transmission (Kleinridders et al., 2015).
We show here that the effect of acute exogenous insulin on DAT Vmax, like the previously reported effects on DA release and InsR signaling in striatal slices (Stouffer et al., 2015; Woods et al., 2016), is amplified by FR and blunted by OB. These data mirror the influence of these diets on DAT-mediated 3[H]-DA uptake in striatal synaptosomes (Jones et al., 2017). The enhancing effect of insulin on DA release and uptake in mouse striatal slices is also lost with a chronic high-fat diet (Fordahl & Jones, 2017). Together, these results support the notion that diet-regulated levels of brain insulin set the tone for acute InsR-pathway mediated DA signaling. Loss of insulin sensitivity with a high-fat diet appears to be a consequence of impaired efficacy of downstream InsR-substrates, rather than a decrease in the number of InsRs per se, as it can be reversed by inhibiting tyrosine phosphatase 1B, which preserves phosphorylation at tyrosine residues on InsR-substrates (Fordahl and Jones, 2017). Similarly, restoring phosphorylation of the downstream target Akt selectively in DA neurons by striatal viral-injection can also rescue impairments in DAT surface expression and function (Speed et al., 2011).
Implications for reward signaling and conclusions
Reward prediction and reward presentation information is encoded in the basal ganglia by phasic increases in midbrain DA neuronal activity that correlates with phasic striatal DA release transients (Schultz, 1998; Matsumoto & Hikosaka, 2009; Sombers et al., 2009). Further regulation of striatal DA signaling typically occurs via local factors including locally released transmitters and neuromodulators (Rice et al., 2011; Sulzer et al., 2016). Here, we demonstrate that evoked increases in [DA]o are shaped by systemic changes in a circulating neuromodulator that crosses the blood-brain barrier, namely insulin. Altered responsiveness of striatal DAT function following phasic DA release with chronic changes in insulin signaling in different feeding states could have implications for behavioral responses to both natural rewards and the rewarding effects of abused drugs, particularly psychostimulants that preferentially target the DAT. In general, hypoinsulinemic FR diets tend to enhance the rewarding effects of drugs (Carroll and Meisch, 1984; Pothos et al. 1995; Bell et al. 1997). For example, the FR regime used here decreases brain reward thresholds and basal locomotor activity but increases self-administration of psychostimulant drugs, as well as enhancing cocaine conditioned place preference (Cabeza de Vaca and Carr, 1998, Carr, 2007; Carr, 2011; Zheng et al, 2012). By contrast, hyperinsulinemic OB diets dampen brain reward systems (Davis et al., 2008; Geiger et al., 2009; Speed et al., 2011; Hryhorczuk et al., 2016) and lead to increased sensitization to psychostimulants (McGuire et al., 2011; Baladi et al., 2012; Fordahl et al., 2016). Importantly, responsiveness to food preference is also altered with feeding state, with FR rats displaying enhanced flavor-nutrient preference versus AL rats, but hyperinsulinemic OB rats losing their ability to acquire significant flavor-nutrient preference (Woods et al., 2016). Given that DA operates as a subsecond modulator for food seeking (Roitman et al., 2004), incorrect valuation of nutrients by altered insulin-induce DA signaling could exacerbate maladaptive eating patterns such as those seen with anorexia nervosa or with obesity. Indeed, animals on a high-fat diet have increased motivation for food consumption (Speed et al,, 2011; Narayanaswami et al., 2013).
Overall, the data presented here reveal that acute and chronic insulin dynamically regulate striatal DA release and uptake, which could contribute to the influence of diet and insulin on regulation of the rewarding effects of food and drugs.
Acknowledgment
We are grateful to Dr. Robert P. Machold at NYU School of Medicine for initially providing ChAT KO mice, and consulting as we established our own colony.
Funding
These studies were supported by NIH grants DA033811 (M.E.R., K.D.C., and M.E.A. Reith), DA03956 (K.D.C.), and a NARSAD Independent Investigator Award (K.D.C.).
Abbreviations
- ACh
acetylcholine
- AL
ad libitum
- ChAT
choline acetyltrasferase
- DA
dopamine
- [DA]o
extracellular dopamine concentration
- DAT
dopamine transporter
- FR
food restricted
- HNMPA
hydroxyl-2-naphthalenylmethylphosphonic acid trisacetoxymethyl ester
- InsR
insulin receptor
- KO
knockout
- nAChR
nicotinic acetylcholine receptor
- OB
obesogenic
- PPP
picropodophyllotoxin;Vmax, maximum uptake velocity
- Cpeak
extrapolated peak concentration
Footnotes
Conflict of Interest Statement: The authors declare no conflict of interest.
Data accessibility: The authors confirm that all average data underlying the findings have been provided within the article. Raw data will be made available following publication in correspondence with the corresponding author.
References
- Baladi MG, Koek W, Aumann M, Velasco F, & France CP (2012) Eating high fat chow enhances the locomotor-stimulating effects of cocaine in adolescent and adult female rats. Psychopharmacology (Berl.), 222, 447–457. [DOI] [PubMed] [Google Scholar]
- Banks WA (2004) The source of cerebral insulin. Eur. J. Pharmacol, 490, 5–12. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB, Huang W & Kastin AJ (1997) Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides 18, 1423–1429. [DOI] [PubMed] [Google Scholar]
- Banks WA & Kastin AJ (1998) Differential permeability of the blood-brain barrier to two pancreatic peptides: Insulin and amylin. Peptides 19, 883–889. [DOI] [PubMed] [Google Scholar]
- Baskin DG, Figlewicz Lattemann D, Seeley RJ, Woods SC, Porte D Jr. & Schwartz MW (1999) Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brai. Res 848, 114–123. [DOI] [PubMed] [Google Scholar]
- Bell SM, Stewart RB, Thompson SC & Meisch RA (1997) Food-deprivation increases cocaine-induced conditioned place preference and locomotor activity in rats. Psychopharmacology, 131, 1–8. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E,& Gonon F (2001) Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 21, 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüss M, Wieland A & Bönisch H (1999) Molecular cloning and functional expression of the mouse dopamine transporter. J. Neural. Transm 106, 657–662. [DOI] [PubMed] [Google Scholar]
- Cabeza de Vaca S & Carr KD (1998) Food restriction enhances the central rewarding effect of abused drugs. J. Neurosci, 18, 7502–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM & Cheer JF (2012) Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Reports, 2, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Huggins KN, Mathews TA & Jones SR (2012) Conserved dorsal-ventral gradient of dopamine release and uptake rate in mice, rats and rhesus macaques. Neurochem. Int 61, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr KD (2007) Chronic food restriction: enhancing effects on drug reward and striatal cell signaling. Physiol. Behav, 91, 459–472. [DOI] [PubMed] [Google Scholar]
- Carr KD (2011) Food scarcity, neuroadaptations, and the pathogenic potential of dieting in an unnatural ecology: binge eating and drug abuse. Physiol. Behav, 104, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll ME & Meisch RA (1984) Increased drug-reinforced behavior due to food deprivation. Adv. Behav. Pharmacol, 4, 47–88. [Google Scholar]
- Carvelli L, Morón JA, Kahlig KM, Ferrer JV, Sen N, Lechleiter JD, Leeb-Lundberg LMF, Merrill G, Lafer EM, Ballou LM, Shippenberg TS, Javitch JA, Lin RZ, & Galli A (2002) PI3-kinase regulation of dopamine uptake. J. Neurochem, 81, 859–869. [DOI] [PubMed] [Google Scholar]
- Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, & Levey AI (1995) The dopamine transporter: immunochemical characterization and localization in brain. J. Neurosci, 15, 1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JF, Tracy AL, Schurdak JD, Tschöp MH, Lipton JW, Clegg DJ, & Benoit SC (2008) Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav. Neurosci, 122, 1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Labouèbe G, Liu S, Nieh EH, Routh VH, Xu S, O’Connor EC (2016) Homeostasis meets motivation in the battle to control food intake. J. Neurosci, 36, 11469–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Evans SB, Murphy J, Hoen M & Baskin DG (2003) Expression of receptors for insulin and leptin in ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res, 964,107–115. [DOI] [PubMed] [Google Scholar]
- Fordahl SC, Locke JL, & Jones SR (2016) High fat diet augments amphetamine sensitization in mice: Role of feeding pattern, obesity, and dopamine terminal changes. Neuropharmacology 109, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordahl SC & Jones SR (2017) High-fat-diet-induced deficits in dopamine terminal function are reversed by restoring insulin signaling. ACS Chem. Neurosci, 8, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BG Wei Y, Moron JA, Lin RZ, Javitch JA & Galli A (2005) Akt is essential for insulin modulation of amphetamine-induced human dopamine transporter cell-surface redistribution. Mol. Pharmacol, 68, 102–109. [DOI] [PubMed] [Google Scholar]
- Giros B, El Mestikawy S, Bertrand L & Caron MG (1991) Cloning and functional characterization of a cocaine-sensitive dopamine transporter. FEBS, 295, 149–154. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez T, Barroso-Chinea P, De La Cruz Muros I, Del Mar Perez-Delgado M & Rodriguez M (2004) Expression of dopamine and vesicular monoamine transporters and differential vulnerability of mesostriatal dopaminergic neurons. J. Comp. Neurol, 479, 198–215. [DOI] [PubMed] [Google Scholar]
- Gray SM, Meijer RI & Barrett EJ (2014) Insulin regulates brain function, but how does it get there? Diabetes, 63, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG & Pothos EN (2009) Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience, 159, 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryhorczuk C, Florea M, Rodaros D, Poirier I, Daneault C, Des Rosiers C, Arvanitogiannis A, Alquier T & Fulton S (2016) Dampened mesolimbic dopamine function and signaling by saturated but not monounsaturated dietary lipids. Neuropsychopharmacology, 41, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KT, Woods C, Zhen J, Antonio T, Carr KD & Reith MEA (2017) Effects of diet and insulin on dopamine-transporter activity and expression in rat caudate-putamen, nucleus accumbens, and midbrain. J. Neurochem 140, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, Pothos EN & Kahn CR (2015) Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. U.S.A, 112, 3463–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koranda JL, Cone JJ, McGehee DS, Roitman MF, Beeler JA and Zhuang X (2014) Nicotinic receptors regulate the dynamic range of dopamine release in vivo. J. Neurophysiol, 111, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouebe G, Liu S, Dias C, Zou H, Wong JC Karunakaran S, Clee SM, Phillips AG, Boutrel B & Borgland SL (2013) Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci, 16, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Rhee J, Koh J-K & Lee Y-S (1996) Species differences in functions of dopamine transporter: paucity of MPP+ uptake and cocaine and cocaine binding in bovine dopamine transporter. Neurosci. Lett, 214, 199–201. [DOI] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME and Yue Z (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci 30, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JF, O’Dell SJ, Navarrete R & Rosenstein AJ (1990) Dopamine high-affinity transport site topography in rat brain: major differences between dorsal and ventral striatum. Neuroscience, 37, 11–21. [DOI] [PubMed] [Google Scholar]
- Matsumoto M & Hikosaka O (2009) Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature, 459, 837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire BA, Baladi MG & France CP (2011) Eating high-fat chow enhances sensitization to the effects of methamphetamine on locomotion in rats. Eur. J. Pharmacol, 658, 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebel DM, Wong JC, Dong YJ & Borgland SL (2012) Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur. J. Neurosci, 36, 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton LS, Apparsundaram S, King-Pospisil KA & Dwoskin LP (2007) Nicotine increases dopamine transporter function in rat striatum through a trafficking-independent mechanism. Eur. J. Pharmacol, 554, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswami V, Thompson AC, Cassis LA, Bardo MT & Dwoskin LP (2013) Diet-induced obesity: dopamine transporter function, impulsivity and motivation. Int. J. Obes 37, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C (1995) Interaction between diffusion and Michaelis-Menten uptake of dopamine after iontophoresis in striatum. Biophys. J, 68, 1699–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, Trout SJ & Kruk ZL (1992) Regional differences in evoked dopamine efflux in brain slices of rat anterior and posterior caudate putamen. Naunyn Schmiedebergs Arch. Pharmacol, 346, 267–276. [DOI] [PubMed] [Google Scholar]
- Patel A, Uhl G & Kuhar MJ (1993) Species differences in dopamine transporters: postmortem changes and gylcosylation differences. J. Neurochem, 61, 496–500. [DOI] [PubMed] [Google Scholar]
- Patel JC, Rossignol E, Rice ME, & Machold RP (2012) Opposing regulation of striatal dopamine release and exploratory motor behavior by forebrain and brainstem cholinergic inputs. Nat. Commun, 3, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, & Rice ME (2013) Monitoring axonal and somatodendritic dopamine release using fast-scan cyclic voltammetry in brain slices. Methods Mol. Biol, 96, 243–273. [DOI] [PubMed] [Google Scholar]
- Patel JC (2016) Voltammetry: Electrochemical detection of neurotransmitters in the brain In: Encyclopedia of Life Sciences (eLS). John Wiley & Sons, Ltd: Chichester. [Google Scholar]
- Patterson TA, Brot MD, Zavosh A, Schenk JO, Szot P & Figlewicz DP (1998) Food deprivation decreases mRNA and activity of the rat dopamine transporter. Neuroendocrinology 68, 11–20. [DOI] [PubMed] [Google Scholar]
- Porte D Jr., Baskin GJ & Schwartz MW (2002) Leptin and insulin action in the central nervous system. Nutr. Rev 60, S20–29; discussion S68–84, 85–87. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Creese I & Hoebel BG (1995) Restricted eating with weight loss selectively decreases extracellular dopamine in the nucleus accumbens and alters dopamine response to amphetamine, morphine, and food intake. J. Neurosci 15, 6640–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, & Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nature Neuroscience, 7, 583–584. [DOI] [PubMed] [Google Scholar]
- Rice ME, Patel JC & Cragg SJ (2011) Dopamine release in the basal ganglia. Neurosci, 198, 112–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SD, Matthies HJ, Sathananthan V, Christianson NSB, Kennedy JP, Lindsley CW, Daws LC & Galli A (2010). Insulin reveals Akt signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine homeostasis. J. Neurosci, 30, 11305–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PE, Wightman RM & Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J. Neurosci, 24, 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffelmeer AN, Drukarch B, De Vries TJ, Hoenboom F, Schetters D & Pattij T (2011) Insulin modulates cocaine-sensitive monoamine transporter function and impulsive behavior. J. Neurosci, 31, 1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F & Sulzer D (2001) Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J. Neurosci, 21, 5916–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulingkamp RJ, Pagano TC, Hung D & Raffa RB (2000) Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci. Biobehav. Rev, 24, 855–872. [DOI] [PubMed] [Google Scholar]
- Schultz W (1998) Predictive reward signal of dopamine neurons. J. Neurophysiol, 80, 1–27 [DOI] [PubMed] [Google Scholar]
- Sombers LA, Beyene M, Carelli RM & Wightman RM (2009) Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J. Neurosci,, 29, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed N, Saunders C, Davis AR, Owens WA, Matthies HJG, Saadat S, Kennedy JP, Vaughan RA, Neve RL, Lindsley CW, Russo SJ, Daws LC, Niswender KD & Galli A (2011) Impaired striatal Akt signaling disrupts dopamine homeostasis and increases feeding. PLoS One, 6, e25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stice E, Figlewicz DP, Gosnell BA, Levine AS & Pratt WE (2013) The contribution of brain reward circuits to the obesity epidemic. Neurosci. Biobehav. Rev, 37, 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, Machold RP, Jones KT, de Vaca SC, Reith ME, Carr KD & Rice ME (2015). Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun, 6, 8543. doi: 10.1038/ncomms9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubbe JH, Porte D Jr. & Woods SC (1988) Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in the rat. Physiol. Behav, 44, 205–208. [DOI] [PubMed] [Google Scholar]
- Sulzer D (2011) How addictive drugs disrupt presynaptic neurotransmission. Neuron, 69, 628–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Cragg SJ & Rice ME (2016) Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia, 6, 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ (2012) Striatal dopamine release is triggered by synchronous activity in cholinergic interneurons. Neuron 75, 58–64. [DOI] [PubMed] [Google Scholar]
- Vulto AG & Fowler CJ (1986) The effect of the dopamine agonist pergolide on tyrosine hydroxylase activity in rat striatal and limbic miniprisms in vitro: a model for the dopamine autoreceptor? Naunyn Schmiedebergs Arch. Pharmacol, 333, 349–533. [DOI] [PubMed] [Google Scholar]
- Werther GA, Hogg A, Oldfeld BJ, McKinley MJ, Figdor R, Allen AM & Mendelsohn FAO (1987) Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology, 121, 1562–1570. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG & May LJ (1988). Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience, 25, 513–523. [DOI] [PubMed] [Google Scholar]
- Wightman RM & Zimmerman JB (1990) Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res. Rev, 15, 135–144. [DOI] [PubMed] [Google Scholar]
- Woods CA, Guttman ZR, Huang D, Kolaric RA, Rabinowitsch AI, Jones KT, Cabeza de Vaca S, Sclafani A & Carr KD (2016) Insulin receptor activation in the nucleus accumbens reflects nutritive value of a recently ingested meal. Physiol. Behav, 159, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Baskin DG & Schwartz MW (2003) Insulin and the blood brain barrier. Curr. Pharm. Des, 9, 795–800. [DOI] [PubMed] [Google Scholar]
- Wu Q, Reith MEA, Wightman RM, Kawagoe KT and Garris PA (2001) Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J. Neurosci. Meth, 112, 119–133. [DOI] [PubMed] [Google Scholar]
- Zhang H & Sulzer D (2004) Frequency-dependent modulation of dopamine release by nicotine. Nature Neurosci. 7, 581–582. [DOI] [PubMed] [Google Scholar]
- Zhen J, Reith MEA & Carr KD (2006) Chronic food restriction and dopamine transporter function in rat striatum. Brain Res, 1082, 98–101. [DOI] [PubMed] [Google Scholar]
- Zheng D, Cabeza de Vaca S & Carr KD (2012) Food restriction increases acquisition, persistence and drug prime-induced expression of a cocaine-conditioned place preference in rats. Pharmacol. Biochem. Behav, 100, 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]