

Abstract
Hypoglycemic drugs such as metformin increase glucose uptake and utilization by peripheral tissues to maintain glucose homeostasis, and the AMP-activated protein kinase (AMPK) signaling pathway is an important component of this pharmacological activity. Liver kinase B1 (LKB1) acts as a kinase upstream of AMPK and plays an important regulatory role in glucose metabolism. In recent years, as a tumor suppressor, LKB1's antitumor activity has been widely studied, yet its hypoglycemic activity is not clear. Here, using biochemical and cell viability assays, site-directed mutagenesis, immunoblotting, and immunofluorescence staining, we found that a natural product, antroalbol H isolated from the basidiomycete mushroom Antrodiella albocinnamomea, increases cellular glucose uptake in murine L6 myotubes and 3T3-L1 adipocytes. Of note, our results indicated that this effect is related to LKB1-mediated Thr-172 phosphorylation of AMPKα. Furthermore, we observed that antroalbol H induces the phosphorylation of LKB1 specifically at Thr-189 and changes subcellular localization of LKB1. Finally, antroalbol H treatment strikingly promoted glucose transporter type 4 (GLUT4) translocation to the plasma membrane. We conclude that antroalbol H promotes Thr-189 phosphorylation of LKB1, leading to AMPK activation, revealing this residue as a potential target for increasing glucose uptake, and that antroalbol H therefore has potential for managing hyperglycemia.
Keywords: glucose metabolism, liver kinase B1 (LKB1), AMP-activated kinase (AMPK), glucose transporter type 4 (GLUT4), fungi, Basidiomycete, diabetes, energy sensing, glucose homeostasis, sesquiterpene
Introduction
AMP-activated protein kinase (AMPK),4 a major cellular energy sensor that functions in glucose homeostasis, is a heterotrimeric serine-threonine enzyme comprising a catalytic α subunit and two regulatory (β and γ) subunits (1, 2). Phosphorylation of Thr-172 on the α-subunit is the primary factor for the activation of AMPK, and this can be achieved by two signaling pathways: a Ca2+-dependent pathway mediated by Ca2+/calmodulin-dependent protein kinase (CaMKK) and an AMP-dependent pathway mediated by liver kinase B1 (LKB1) (3–6). The activation of the kinases promotes glucose uptake in skeletal muscles and other tissues for systemic energy supply. This biological regulation is closely related to the membrane transport of glucose transporter 4 (GLUT4). It is reported that translocation of GLUT4 from intracellular storage vesicles to the plasma membrane is promoted in 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR)-perfused rat muscle (7). The oral hypoglycemic drug metformin can activate AMPK in the mouse soleus muscle, which enhances glucose uptake (8), linking its effect to AMPK activation. Therefore, the AMPK signaling pathway has been demonstrated as an important target in glucose metabolism, and in recent years, a number of compounds have been reported to activate the AMPK signaling pathway (9–12).
LKB1, known as a tumor-inhibiting factor whose loss-of-function mutations lead to Peutz–Jeghers's syndrome (13), is associated with a variety of tumors, such as sporadic gastric cancer, colorectal cancer, and pancreatic cancer (14–16). LKB1 phosphorylates at least 13 different downstream protein kinases, including AMPK (17). LKB1 is essentially a serine-threonine protein kinase with multiple phosphorylation sites (18). A decade ago, scholars reported that the Ser-307 and Ser-428 residues of LKB1 are required for metformin-enhanced AMPK activation, demonstrating the importance of these two residues in activating AMPK and enhancing glucose metabolism (19–21). Despite this, the effects of the phosphorylation sites of LKB1 in glucose metabolism are still poorly understood.
Higher fungi are a source of a wide range of structurally attractive and biologically active compounds. Basidiomycetes, the largest class of fungi, with more than 1100 genera and over 16,000 species, exhibit anti-tumor, anti-hyperlipidemic, anti-bacterial, and anti-fatigue effects (22–25). In recent years, triterpenoids and sesquiterpenoids isolated from this class of fungi have been reported as potential anti-diabetic compounds (26–30). The isolation of sesquiterpenoids from Antrodiella albocinnamomea allows us to focus more on their anti-diabetic activities.
In this study, we identified a novel compound, antroalbol H, which enhances glucose uptake in myotubes and adipocytes via the LKB1-AMPK pathway by specifically promoting the phosphorylation of the Thr-189 of LKB1, demonstrating the importance of the residue in glucose uptake and metabolism and the potential application of the compound in the regulation of glucose homeostasis in the near future.
Results
Antroalbol H enhances cellular glucose uptake
Differentiated L6 myotubes and 3T3-L1 adipocytes are the two classic cell lines used for the study of glucose uptake and metabolism. Here, we demonstrated that a 24-h incubation of antroalbol H (Fig. 1) dose-dependently enhanced glucose uptake in L6 myotubes (1.71–40 μm) and 3T3-L1 adipocytes (2.5–40 μm) (Fig. 2, A and B). Antroalbol H (10 μm) induced ∼80% of the maximum of the total enhanced glucose uptake in both cells. Then we applied 10 μm antroalbol H to the two cell types and assayed glucose uptake following a series of incubation times. The results showed that the amount of glucose uptake linearly correlated with the incubation time of antroalbol H (6–24 h or 4–24 h; Fig. 2, C and D). Interestingly, as a consequence of enhanced glucose uptake, antroalbol H also induced further production of lactate in a dose-dependent manner in those cells (Figs. 2, E–H). Similarly, lactate production was also enhanced by treatment with antroalbol H in a series of incubation times in these cells (Fig. 2, G and H) that was in parallel with the enhanced glucose uptake (Fig. 2, C and D) without cytotoxicity at higher concentrations (Fig. S1).
Figure 1.

Separation and extraction of antroalbol H.
Figure 2.

Antroalbol H enhances cellular glucose uptake and lactate release. A and B, glucose uptake assay with different concentrations of antroalbol H in L6 myotubes and 3T3-L1 adipocytes. Cells were incubated with 0.1% DMSO (Con), 100 nm insulin (Ins), or incremental concentrations of antroalbol H (AH) for 24 h. The results are expressed as -fold change relative to the control values. C and D, glucose uptake assay for different treatment times of AH in those cells. L6 myotubes and 3T3-L1 adipocytes were incubated with 10 μm AH for a series of incubation periods up to 24 h. E–H, the concentrations of lactate were assayed in cells parallel to the glucose uptake assay. Results are expressed as mean ± S.D. (error bars) (n = 6). *, p < 0.05; **, p < 0.01 compared with control.
Antroalbol H promotes phosphorylation of AMPK at the Thr-172 residue rather than Akt
To determine the signaling pathways associated with antroalbol H–induced cellular glucose uptake, Akt, the classical key protein kinase in insulin signaling, was studied first. In general, insulin strongly stimulates phosphorylation of the Ser-473 and Thr-308 residues of Akt in the L6 myotubes and 3T3-L1 adipocytes. Antroalbol H, however, had no effects on these two phosphorylation residues in both of the cells after 15-min, 4-h, or 24-h incubations (Fig. 3), indicating that the mechanism involved in antroalbol H–induced glucose uptake is independent of the insulin signaling pathway.
Figure 3.

Antroalbol H does not affect Akt. A–D, immunoblots of p-AktS473, p-AktT308, t-Akt, and β-actin in cells stimulated by AH for a short time. L6 myotubes or 3T3-L1 adipocytes were incubated with 100 nm insulin or 10 μm AH for 15 min or 4 h. E and F, the same assay was performed in L6 myotubes or 3T3-L1 adipocytes when cells were incubated with 100 nm insulin or 2.5–20 μm AH for 24 h. The band intensity of the phosphorylation protein was normalized to that of total protein in the right panel (the same as below). The values are expressed as -fold change relative to the value from the DMSO-treated cells (Con). Results are represented as means ± S.E. (error bars) (n = 3). **, p < 0.01 compared with control.
It has been previously reported that insulin-independent glucose uptake is mediated through AMPK (31). Activation of AMPK is achieved through phosphorylation of Thr-172 of the AMPKα; therefore, it is necessary to determine the effect of antroalbol H on the Thr-172 residue. The results showed that by antroalbol H treatments (5–20 μm) for 24 h, the phosphorylation levels of AMPKα on the Thr-172 residue were dose-dependently enhanced in 3T3-L1 adipocytes (1.52-fold the control level by 20 μm antroalbol H) and L6 myotubes (4.88-fold the control by 20 μm antroalbol H). Acetyl-CoA carboxylase (ACC) is a direct substrate of AMPKα, and its phosphorylation level is regulated by AMPKα. We found that the phosphorylation of ACC was also up-regulated in the antroalbol H treatment groups (Fig. 4, A and B). In addition, the phosphorylation of AMPK was time-dependently enhanced by treatment with antroalbol H (10 μm) in 3T3-L1 adipocytes and L6 myotubes (Fig. 4, C and D).
Figure 4.

Antroalbol H promotes AMPK phosphorylation. A and B, immunoblots of p-AMPKT172, t-AMPK, p-ACCS79, t-ACC, and β-actin in L6 myotubes and 3T3-L1 adipocytes. Cells were incubated with 100 nm insulin or 1.25–20 μm AH for 24 h. C and D, immunoblots of p-AMPKT172, t-AMPK, and β-actin in L6 myotubes and 3T3-L1 adipocytes when cells were treated with 10 μm AH at the indicated hours. All values are expressed as -fold change relative to the value from the DMSO-treated cells (Con or 0 h). Results are presented as mean ± S.E. (error bars) (n = 3). *, p < 0.05; **, p < 0.01 compared with control.
Antroalbol H induces cellular glucose uptake through AMPK
To further elucidate how AMPK controls the effect of antroalbol H on glucose uptake, AICAR (AMPK activator) was applied together with antroalbol H for the estimation of AMPK phosphorylation at the Thr-172 residue and glucose uptake. Consistent with the additive effect of phosphorylation of AMPK, the combination treatment caused a tendency of additive effects as compared with antroalbol H or AICAR alone in L6 myotubes (Fig. 5, A and B) and 3T3-L1 adipocytes (Fig. 5, C and D).
Figure 5.

Antroalbol H induces cellular glucose uptake through AMPK. A, immunoblots of p-AMPKT172, t-AMPK, and β-actin in L6 myotubes. L6 myotubes were incubated with 5 μm AH and/or 500 μm AICAR for 24 h, with values expressed as -fold change relative to the value from the DMSO-treated cells. B, glucose uptake assay parallel to the above Western blotting experiments. C and D, the same immunoblots and glucose uptake assays were performed in 3T3-L1 adipocytes. E and H, the same immunoblots and glucose uptake assays co-incubated with compound C in those cells. L6 myotubes and 3T3-L1 adipocytes were preincubated with 10 μm compound C for 30 min followed by treatment with or without 10 μm AH for 24 h. I and J, immunoblots and glucose uptake assays after siRNA in L6 myotubes. L6 myotubes were transfected with scramble siRNA (siNC) or siAMPKα for 24 h, followed by incubation with 0.1% DMSO or 10 μm AH for 24 h. K and L, the same assays were performed using 2 mm metformin (Met) in L6 myotubes. All immunoblotting values are expressed as -fold change relative to the value from the DMSO-treated cells (Con). Results are expressed as mean ± S.E. (n = 3) or mean ± S.D. (n = 6) (error bars). *, p < 0.05; **, p < 0.01 compared with appropriate control.
On the other hand, we treated cells by a combination of antroalbol H with compound C, an AMPK inhibitor, and estimated the phosphorylation of AMPK and glucose uptake. As predicted, as compared with antroalbol H alone in both 3T3-L1 adipocytes and L6 myotubes, this approach induced the reduction of AMPK phosphorylation and glucose uptake (Fig. 5, E–H), indicating that compound C eliminated the effects of antroalbol H on AMPK phosphorylation and glucose uptake.
Furthermore, we knocked down the catalytic α subunit of AMPK by siRNA, which resulted in a decrease in both AMPK phosphorylation and glucose uptake (Fig. 5, I and J). A similar effect could also be observed in the metformin-treated group as the control (Fig. 5, K and L), which is in line with previous reports involving skeletal muscle in subjects with type 2 diabetes (32).
LKB1 deficiency abolishes antroalbol H–induced phosphorylation of AMPK and glucose uptake
CaMKK and LKB1 are the two major upstream kinases of AMPK that can directly stimulate phosphorylate of AMPK at the Thr-172 residue (6). We aimed to determine which of the kinases might play a role in antroalbol H–induced AMPK phosphorylation and glucose uptake, and experiments related to CaMKK were first performed. Our data showed that there was no increase in the phosphorylation of CaMKII, downstream kinase of CaMKK, treated by series doses of antroalbol H for 24 h or 10 μm antroalbol H for 2 h in L6 myotubes (Fig. S2, A and B). Antroalbol H did not change the cytosolic Ca2+ level in L6 myotubes and 3T3-L1 adipocytes, whereas ionomycin, the Ca2+ ionophore, was capable of increasing the cellular Ca2+ levels as the control (Fig. S2, C and D). Those results indicated that the CaMKII and Ca2+ were not involved in the antroalbol H–induced AMPK phosphorylation and glucose uptake.
As another upstream factor of AMPK, we next elucidated the role of LKB1 in antroalbol H–induced effects by RNAi of LKB1 in L6 myotubes. Surprisingly, the down-expression of LKB1 resulted in abolished effects on antroalbol H–induced phosphorylation of AMPK and glucose uptake (Fig. 6, A and B). As a control, metformin also exhibited similar effects of antroalbol H–induced AMPK phosphorylation and glucose uptake (Fig. 6, C and D). Moreover, we elucidated the effects of AICAR on phosphorylation of AMPK in HeLa cells, which naturally lack LKB1 protein expression. As we expected, AICAR was able to significantly increase AMPK phosphorylation in the cells (2.67-fold the control level), but antroalbol H treatment did not change the phosphorylation of AMPK in these LKB1-deficient cells (Fig. 6E). Together, these findings strongly demonstrated that LKB1 is required for antroalbol H–induced AMPK phosphorylation and the phosphorylation-related glucose uptake.
Figure 6.

LKB1 deficiency abolishes antroalbol H–induced phosphorylation of AMPK and glucose uptake. A, immunoblots of p-AMPKT172, t-AMPK, and β-actin after siRNA in L6 myotubes. L6 myotubes were transfected with 100 nm scramble siRNA (siNC) and 100 nm siLKB1 RNA (siLKB1) for 24 h, followed by incubation with 0.1% DMSO or 10 μm AH for 24 h. B, glucose uptake assay parallel to the above Western blotting experiments. C and D, the same immunoblotting and glucose uptake assays were performed using metformin in L6 myotubes. E, immunoblots of p-AMPKT172, t-AMPK, and β-actin in HeLa cells. HeLa cells were incubated with DMSO, 500 μm AICAR, or 10 μm AH for 24 h. All immunoblotting results are expressed as -fold change relative to the DMSO-treated cells (Con). Results are presented as mean ± S.E. (n = 3) or mean ± S.D. (n = 6) (error bars). *, p < 0.05; **, p < 0.01 compared with the appropriate control.
Antroalbol H–induced phosphorylation of AMPK and glucose uptake depends on the Thr-189 residue of LKB1
After elucidating the dependence of antroalbol H on LKB1, we next set out to examine the effects of antroalbol H on activating LKB1. Our data showed that a significant and dose-dependent increase of LKB1 phosphorylation at Thr-189 resides was observed by treatment with a series of doses of antroalbol H (2.5–20 μm) for 24 h in L6 myotubes (Fig. 7A), whereas phosphorylation of LKB1 at other residues (Ser-307 and Ser-428) was not altered by the same treatment (Fig. 7B).
Figure 7.

Antroalbol H promotes phosphorylation and nuclear translocation of LKB1. A, immunoblots of p-LKB1T189, t-LKB1, and β-actin in L6 myotubes. L6 myotubes were treated with 0–20 μm AH for 24 h. B, immunoblots of p-LKB1S307, p-LKB1S428, t-LKB1, and β-actin in L6 myotubes. L6 myotubes were treated with AH as above. C, immunoblots of p-AMPKT172, t-AMPK, and β-actin after point mutation in HeLa cells. HeLa cells were transfected with T189A, S307A, and LKB1 plasmid for 24 h and then treated with AH for 8 h followed by immunoblot analysis. D, the same assay using 2 mm metformin in HeLa cells. Results are expressed as mean ± S.E. (error bars) (n = 3). *, p < 0.05; **, p < 0.01 compared with the appropriate control. E and F, immunofluorescence staining of LKB1 in cells. L6 cells and 3T3-L1 adipocytes were treated with 0.1% DMSO, 2 mm metformin, or 10 μm AH for 24 h prior to immunofluorescence staining. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm.
Furthermore, if the antroalbol H–induced phosphorylation of AMPK is LKB1-dependent, we assumed that overexpressing LKB1 would rescue the antroalbol H–induced phosphorylation of AMPK in LKB1-deficient HeLa cells. Therefore, we constructed LKB1 mutants at the antroalbol H–activated Thr-189 (LKB1-T189A) or non-activated Ser-307 residue (LKB1-S307A) and then expressed and co-treated with or without antroalbol H in HeLa cells. As we predicted, antroalbol H induced enhanced phosphorylation of AMPK at the WT LKB1– or LKB1-S307A–overexpressing cells, whereas the phosphorylation of AMPK was not changed by antroalbol H in the LKB1-T189A–expressing group, strongly indicating that the Thr-189 residue is required for antroalbol H–induced activations of LKB1, AMPK, and glucose uptake as a consequence in the cells. Interestingly, there was an absence of activation effect in the LKB1-S307A–expressing cells treated with metformin, but not for the LKB1-T189A–expressing group (Fig. 7, C and D), indicating the distinct molecular targets involved in glucose uptake for antroalbol H and metformin. Furthermore, we visualized subcellular localization of LKB1 in L6 myotubes and 3T3-L1 adipocytes. We found that LKB1 predominantly was located within the nucleus in a basal state, and the diffused distribution was intensified by antroalbol H or metformin (Fig. 7E), revealing the nucleocytoplasmic transport of LKB1 as a consequence of LKB1 phosphorylation by antroalbol H or metformin.
Antroalbol H enhances the translocation of GLUT4 to the plasma membrane
We addressed the fact that antroalbol H is capable of enhancing glucose uptake via the LKB1-AMPK signaling pathway. It has been reported that activation of AMPK promotes translocation of GLUT4 to the plasma membrane and then functions in glucose uptake (33), so we next elucidated the effect of antroalbol H on GLUT4 translocation to the plasma membrane. The membrane fraction results demonstrated that antroalbol H facilitated endogenous GLUT4 translocation from the cytosol to the plasma membrane (52.5 ± 2.1%), which was similar to that of insulin treatment (49.3 ± 0.5%) as compared with the control (35.9 ± 1.9%) (Fig. 8A). Furthermore, we exogenously expressed HA-GLUT4-mOrange2 in 3T3-L1 adipocytes and quantified the amount of the GLUT4 on the plasma membrane. The results of immunofluorescence staining of GLUT4 showed that antroalbol H enhanced the distribution of GLUT4 on the plasma membrane (Fig. 8, B and C).
Figure 8.

Antroalbol H increases GLUT4 translocation to the plasma membrane. A, GLUT4 protein content assay in 3T3-L1 adipocyte subcellular fractions. 3T3-L1 adipocytes were incubated with 0.1% DMSO, 100 nm insulin, or 20 μm AH for 24 h. The subcellular fractions were prepared, and the plasma membrane (PM) and light microsome (LM) pellet fractions were immunoblotted with anti-GLUT4, N-cadherin, or β-actin antibody. The data are expressed as a percentage of the total value of light microsome and plasma membrane in the right panel. Results are expressed as mean ± S.E. (n = 3). **, p < 0.01 compared with the appropriate control. B and C, exogenous GLUT4 fluorescent staining assay in 3T3-L1 adipocytes. After electroporation of the plasmid, 3T3-L1 adipocytes were treated with 0.1% DMSO, 100 μm insulin, or 10 μm AH for 24 h. Cells were fixed in 3.7% paraformaldehyde for 8 min and then incubated with anti-HA primary antibody and Cy3 secondary antibody and finally analyzed by fluorescence microscopy (Nikon, Japan). The values were the ratio of surface (S, green) and total (T, red) GLUT4. Results are presented as mean ± S.D. (n = 6). **, p < 0.01 compared with the control.
Discussion
Skeletal muscle and adipose tissues are important organs/tissues of glucose uptake, and glucose uptake by these cells is the first rate-limiting step in glucose metabolism in mammalian organisms (34, 35). In the present study, we report that a new nor-sesquiterpene, antroalbol H, isolated from fermentation of A. albocinnamomea, was able to enhance glucose uptake in basal L6 myotubes and 3T3-L1 adipocytes through LKB1-AMPK signaling.
Insulin is the hormone that lowers blood glucose in the body, and it works through a series of protein signaling cascades. Among these protein members, Akt is considered to be a characteristic protein, which is phosphorylated at Ser-473 and Thr-308 residues when the insulin signaling pathway is activated. In this study, antroalbol H showed no effects on the phosphorylation of residues both in short (15 min and 4 h) and long (24 h) treatments in L6 myotubes and 3T3-L1 adipocytes, indicating that its mechanism of glucose metabolism is independent of this pathway under this environment.
AMPK-mediated glucose uptake is insulin-independent. It can be confirmed in the skeletal muscle of type 2 diabetes patients that AMPK-mediated glucose uptake worked normally, whereas insulin-induced glucose uptake is impaired (31). Antroalbol H induced phosphorylation of AMPKα on the Thr-172 residue in parallel to the performance on glucose uptake, linking antroalbol H's role in promoting glucose uptake to the AMPK pathway. As an important kinase downstream of AMPK, ACC can be phosphorylated by AMPK signaling pathway activators. In the antroalbol H–treated cells, phosphorylation of the Ser-79 residue of ACC was up-regulated, demonstrating that antroalbol H activates AMPK. This association was further confirmed in a subsequent experiment involving the manipulation of AMPK activity by activators, inhibitors, and siRNA. We also found that phosphorylation of AMPKβ on the Ser-182 residue in antroalbol H–treated cells was not changed (data not shown), thus eliminating the possibility that antroalbol H promotes cellular glucose uptake by AMPKβ.
AMPK is activated by phosphorylation at the Thr-172 residue within the catalytic α-subunit, catalyzed by upstream AMPK kinases, such as LKB1 and CaMKK (3–5). CaMKII, a kinase downstream of CaMKK, is the major signaling kinases implicated by many studies in regulating contraction-stimulated glucose uptake through GLUTs (36, 37), which is activated by phosphorylation of the Thr-286 residue of CaMKII after a rapid rise in cytosolic Ca2+. In this study, neither Ca2+ signal nor the phosphorylation of Thr-286 was changed in L6 myotubes by antroalbol H treatment, indicating that the molecular basis for the promotion of glucose uptake by antroalbol H is CaMKK-independent and distinct from that of muscle contraction. By contrast, the mechanism of AMPK action by nootkatone, a sesquiterpene constituent of grapefruit with a similar structure to antroalbol H, seems to be the same as the mechanism of muscle contraction on glucose uptake (38).
Interestingly, antroalbol H induced phosphorylation of LKB1 at the Thr-189 residue and altered nucleocytoplasmic shuttling of LKB1 both in L6 myoblasts and myotubes (Fig. 7). Phosphorylation and subcellular localization are two main mechanisms of LKB1 activation, although the physiological relevance of its phosphorylation remains controversial (39). It has been reported that phosphorylation of LKB1 at Ser-307 regulates the nuclear transport of LKB1 into the cytosol and is required for metformin-enhanced AMPK activation in various cells (18, 19). Our results with metformin are consistent with these reports by mutation of Ser-307 in LKB1-deficient HeLa cells. Under the same circumstances, however, mutation of Thr-189 blocks AMPK activation in response to antroalbol H, revealing the distinct mechanism of antroalbol H and metformin on Thr-189 or Ser-307 residues of LKB1 and highlighted that the Thr-189 residue serves as potential target for the LKB1-AMPK pathway in glucose uptake and metabolism. The Thr-189 residue has been reported by scholars as the autophosphorylation site of LKB1 (40), and its natural activator has not been reported so far. Therefore, antroalbol H may be a useful tool for the study of the mechanism of how the Thr-189 residue affects in LKB1-dependent AMPK activity.
AMPK activation contributes to GLUT4 glucose transport in adipocytes and skeletal muscle (41, 42). Our results showed that, similar to insulin, antroalbol H promotes translocation of GLUT4, whereas the expression of GLUT4 was not changed (Fig. S3). Therefore, as a consequence of activation of the LKB1-AMPK pathway, it is inferred that antroalbol H improves glucose uptake, at least in part, by inducing GLUT4 translocation to the plasma membrane.
Taken together, the mechanism of antroalbol H on glucose uptake specifically involves the activation of LKB1 phosphorylation at the Thr-189 residue (Fig. 9). Thr-189, an important phosphorylation site of LKB1, may be a new potential target for regulation of glucose homeostasis through the LKB1-AMPK pathway. Antroalbol H can be a useful tool for the study of the LKB1-AMPK signaling pathway and has great potential for the treatment of hyperglycemia.
Figure 9.
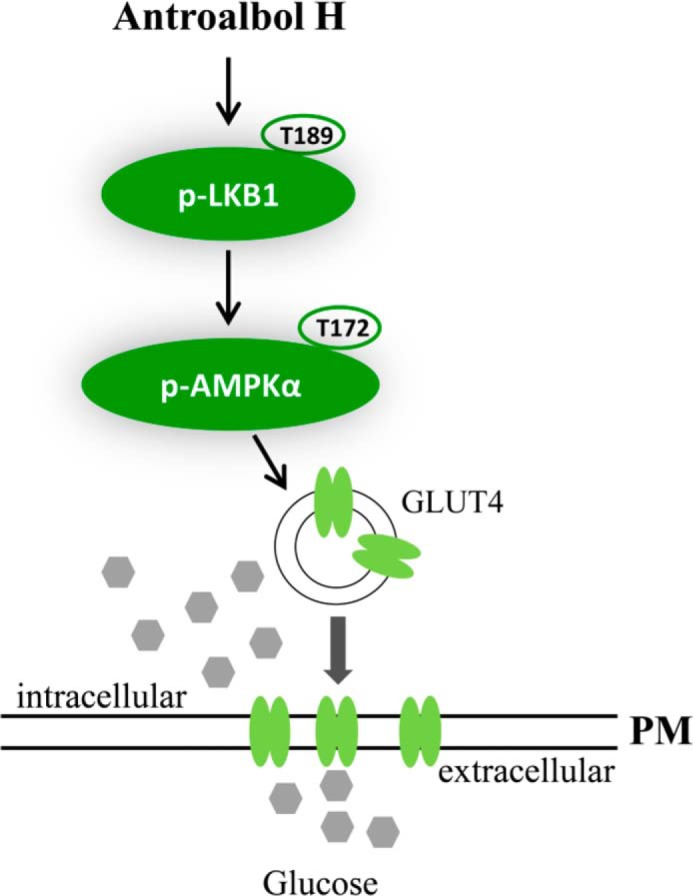
Summary of the role of antroalbol H in the AMPK signaling pathway.
Experimental procedures
The cell culture medium was purchased from Biological Industries (Kibbutz Beit Haemek, Israel). Insulin was purchased from Roche (Basel, Switzerland), and Compound C was purchased from MedChemExpress. All other chemicals and organic solvents were of the highest grade and were obtained from Sigma-Aldrich. The glucose assay kits were purchased from Shanghai Rongsheng Bioengineering Institute (Shanghai, China). The lactate assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Plasmid (pcDNA3-FLAG-LKB1) was prepared by Addgene (Cambridge, MA). Specific antibodies and siRNA sequences are indicated in the supporting information.
Preparation of antroalbol H
The A. albocinnamomea strain was cultured from the tissue of its fruiting bodies collected from the Changbai Mountain Nature Reserve (Antu County, Jilin Province, China), in August 2009. Authentication of A. albocinnamomea was performed by Prof. Yucheng Dai (Beijing Forestry University). The voucher specimen (number CGBWSHF00182) was deposited in the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences. The culture medium consisted of 5% glucose, 0.15% peptone, 0.5% yeast powder, 0.05% KH2PO4, and 0.05% MgSO4. Fermentation was carried out on a shaker at 24 °C and 150 rpm for 26 days. The whole-culture broth of A. albocinnamomea (20 liters) was initially filtered, and the filtrate was extracted three times using EtOAc. The organic layer was concentrated using reduced pressure to give an oily residue (7.9 g). The residue was subjected to reversed-phase C18 eluting with MeOH-H2O from 0:100 to 100:0, to obtain four subfractions, A–D. The fraction B (1.2 g) was separated by Sephadex LH-20 (CHCl3/MeOH, 1:1) to obtain three subfractions (B1–B3). Fraction B3 (200 mg) was separated by reversed-phase C18 eluting with MeOH-H2O from 0:100 to 100:0 to obtain nine subfractions, B3a–B3i. The fraction B3c was separated by repeated silica gel CC and Sephadex LH-20 (CHCl3/MeOH, 1:1) and Me2CO treatment to obtain antroalbol H (25.9 mg). The optical rotations were determined on a Horiba SEPA-300 polarimeter. IR spectra were recorded on a Bruker Tensor 27 spectrometer with KBr pellets. One- and two-dimensional NMR spectra were performed on a Bruker AM-400 spectrometer with tetramethylsilane as the internal standard. Mass spectra were recorded using a VG Auto Spec-3000 or an APIQSTAR TOF spectrometer. The NMR data and spectrum are provided as supporting information. Antroalbol H: Colorless oil, [α]D24 + 49.9 (c 1.42, MeOH), and IR (KBr) νmax of 3443, 2965, 1722, 1689, 1631, 1548, 1529, 1461, 1424, 1401, 1381, 1272, 1163, 1114, 1082, and 1054 cm−1, 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz) data, see Table S1; ESIMS (positive) m/z 255 [M + H]+, HREIMS (positive) m/z 254.1522 (calculated for C14H22O4, 254.1518). Spectral data is shown in the supporting information (Fig. S4–S12).
Cell culture
3T3-L1, HeLa, and L6 myoblast cells were purchased from the American Type Culture Collection (Manassas, VA). The cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) or calf serum (for 3T3-L1 cells), 100 units/ml penicillin, and 100 mg/ml streptomycin in 10-cm diameter dishes in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. The cells were maintained in continuous passages by trypsinization of subconfluent cultures and supplied with fresh medium every 48 h. For differentiation, L6 myoblasts were transferred to Dulbecco's modified Eagle's medium with 2% FBS in tissue culture plates for 5–6 days. 3T3-L1 cells were exposed to 0.5 mm 3-isobutyl-1-methylxanthine, 1 mm dexamethasone, 1 mm rosiglitazone, and 1 mg/ml insulin for 3 days; 1 mg/ml insulin (only) was administered the day after the 3-day treatment period.
Glucose and lactate measurements
The cells were serum-starved for 4 h in 96-well plates and then incubated with insulin, rosiglitazone, AICAR, and/or antroalbol H for the indicated times. To inhibit the signal pathways, L6 myotubes were preincubated with 10 μm compound C for 30 min. Finally, the supernatant of cultured cells was collected and used in the glucose and lactate assay by a commercially available kit. The quantified values were normalized using the results of the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay.
Cell viability assay
Cell viability was evaluated using the MTS assay. After treatment, the cells were incubated with 100 ml of fresh culture medium containing 20% (v/v) MTS for 4 h. The color intensity, which reflects cell viability, was measured at 492 nm using a microplate reader (PerkinElmer Life Sciences 2104 multilabel reader).
Western blot analysis
Following treatment, the cells were homogenized in lysis buffer containing 20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and 1 mm phenylmethylsulfonyl fluoride. The resulting lysates were centrifuged at 14,000 × g for 20 min at 4 °C. The supernatants were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and probed with specific antibodies (Table S2). The immunoblots were quantified using MetaMorph software. Information related to antibodies is provided in the supporting information.
Subcellular fractionation for the 3T3-L1 adipocytes
The 3T3-L1 adipocytes were cultured in 3.5-cm dishes, and they were lysed after treatment with insulin or antroalbol H for 24 h. The subcellular proteins were fractionated by the membrane protein and cytoplasmic protein extraction kit (P0033, Beyotime Biotechnology) according to the instructions. Proteins (20 μg/sample) were separated by SDS-PAGE. N-cadherin and β-actin were assayed as a membrane or cytoplasm loading control, respectively.
Transfection of siRNAs
L6 myotubes were transfected with specific AMPKα and LKB1 siRNAs (siRNA, 100 nmol/liter each) for 48 h using jetPRIME reagent (PolyplusTransfection, Illkirch, France). siRNA nucleotide sequences are listed in the supporting information (Table S3).
Immunofluorescence staining
At the end of treatment, the cells grown on the coverslips were fixed with 3.7% paraformaldehyde solution in PBS for 8 min. After permeabilization with 0.2% Triton X-100 buffer for 20 min, the cells were incubated with 1% FBS for blocking at 37 °C for 60 min, followed by incubation with primary and appropriate secondary antibodies. The nuclei were counterstained with DAPI. The coverslips were mounted with glycerin containing paraphenylenediamine. Images were obtained using a confocal microscope (Olympus, Japan).
Mutagenesis
The Thr-189 or Ser-307 residues of human LKB1 were mutated to Ala using the QuikChange II site-directed mutagenesis kit (Agilent Technologies), according to the manufacturer's instructions. The mutations were confirmed by DNA sequencing. Point mutation plasmid and pcDNA3-FLAG-LKB1 plasmid were transfected into the HeLa cells using the jetPRIME reagent.
Electrotransfection
3T3-L1 adipocytes where suspended at a concentration of 5 × 106 cells/ml in the electroporation medium (242 mm sucrose, 5.5 mm Na2HPO4, 3 mm NaH2PO4, and 1.7 mm MgCl2, pH 7.1). The cell suspension (400 μl) and 14 μg of HA-GLUT4-orange plasmid were mixed and put into a 2-mm electroporation cuvette (Eppendorf, Hamburg, Germany). The cell/plasmid suspension was treated with electric pulses (400 V, 80 μs) and then incubated at room temperature for 10 min. Finally, 100 μl of the treated suspension was taken and transferred into a 24-well plate for the exogenous GLUT4 fluorescent staining assay.
Statistical analysis
The data were analyzed using GraphPad Prism version 5.0 (La Jolla, CA). Multiple comparisons were performed by one-way analysis of variance followed by Student's t-test. A value of p < 0.05 was considered statistically significant. Data are represented as mean ± S.E. or mean ± S.D.
Author contributions
F. W., X. Y., Y. L., Z. L., J. H., and J. L. formal analysis; F. W. and X. Y. investigation; F. W., X. Y., and Y. L. writing-original draft; X. Y., Y. L., Z. L., and Y. X. resources; Y. X. and J. H. data curation; Y. X. and J. H. software; J. H. and J. L. validation; J. L. and W. X. project administration.
Supplementary Material
Acknowledgments
We thank Prof. Xianghai Cai (Kunming Institute of Botany, Chinese Academy of Sciences) and Ling Li (Kunming Medical University) for helpful advice, and we thank Prof. Yan Li (Kunming Institute of Botany, Chinese Academy of Sciences) for provision of reagents.
This work was supported by National Key R&D Program of China Grant 2017YFC1700906 (to Fei Li), National Natural Science Foundation of China Grant 81561148013 (to J. L.), and Yunnan Provincial Science and Technology Department of China Grants 2017FA044 and 2013HA023 (to W. X.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1–S12.
- AMPK
- AMP-activated protein kinase
- LKB1
- liver kinase B1
- CaMKK
- Ca2+/calmodulin-dependent protein kinase kinase
- GLUT4
- glucose transport 4
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- FBS
- fetal bovine serum
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)- 2-(4-sulfophenyl)-2H-tetrazolium.
References
- 1. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 10.1038/nrm2249 [DOI] [PubMed] [Google Scholar]
- 2. Hardie D. G., Ross F. A., and Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carling D., Sanders M. J., and Woods A. (2008) The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. (Lond.) 32, 55–59 10.1038/sj.ijo.0803765 [DOI] [PubMed] [Google Scholar]
- 4. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., and Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 10.1016/j.cmet.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 5. Xiao B., Heath R., Saiu P., Leiper F. C., Leone P., Jing C., Walker P. A., Haire L., Eccleston J. F., Davis C. T., Martin S. R., Carling D., and Gamblin S. J. (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 10.1038/nature06161 [DOI] [PubMed] [Google Scholar]
- 6. Zhang B. B., Zhou G., and Li C. (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9, 407–416 10.1016/j.cmet.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 7. Ha J., Guan K. L., and Kim J. (2015) AMPK and autophagy in glucose/glycogen metabolism. Mol. Aspects Med. 46, 46–62 10.1016/j.mam.2015.08.002 [DOI] [PubMed] [Google Scholar]
- 8. Kristensen J. M., Treebak J. T., Schjerling P., Goodyear L., and Wojtaszewski J. F. (2014) Two weeks of metformin treatment induces AMPK-dependent enhancement of insulin-stimulated glucose uptake in mouse soleus muscle. Am. J. Physiol. Endocrinol. Metab. 306, E1099–E1109 10.1152/ajpendo.00417.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arha D., Ramakrishna E., Gupta A. P., Rai A. K., Sharma A., Ahmad I., Riyazuddin M., Gayen J. R., Maurya R., and Tamrakar A. K. (2018) Isoalantolactone derivative promotes glucose utilization in skeletal muscle cells and increases energy expenditure in db/db mice via activating AMPK-dependent signaling. Mol. Cell. Endocrinol. 460, 134–151 10.1016/j.mce.2017.07.015 [DOI] [PubMed] [Google Scholar]
- 10. Han J., Yang N., Zhang F., Zhang C., Liang F., Xie W., and Chen W. (2015) Rhizoma Anemarrhenae extract ameliorates hyperglycemia and insulin resistance via activation of AMP-activated protein kinase in diabetic rodents. J. Ethnopharmacol. 172, 368–376 10.1016/j.jep.2015.05.016 [DOI] [PubMed] [Google Scholar]
- 11. Han J., Yi J., Liang F., Jiang B., Xiao Y., Gao S., Yang N., Hu H., Xie W. F., and Chen W. (2015) X-3, a mangiferin derivative, stimulates AMP-activated protein kinase and reduces hyperglycemia and obesity in db/db mice. Mol. Cell. Endocrinol. 405, 63–73 10.1016/j.mce.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 12. Adachi Y., Kanbayashi Y., Harata I., Ubagai R., Takimoto T., Suzuki K., Miwa T., and Noguchi Y. (2014) Petasin activates AMP-activated protein kinase and modulates glucose metabolism. J. Nat. Prod. 77, 1262–1269 10.1021/np400867m [DOI] [PubMed] [Google Scholar]
- 13. Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., et al. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184–187 10.1038/34432 [DOI] [PubMed] [Google Scholar]
- 14. Park W. S., Moon Y. W., Yang Y. M., Kim Y. S., Kim Y. D., Fuller B. G., Vortmeyer A. O., Fogt F., Lubensky I. A., and Zhuang Z. (1998) Mutations of the STK11 gene in sporadic gastric carcinoma. Int. J. Oncol. 13, 601–604 [PubMed] [Google Scholar]
- 15. Dong S. M., Kim K. M., Kim S. Y., Shin M. S., Na E. Y., Lee S. H., Park W. S., Yoo N. J., Jang J. J., Yoon C. Y., Kim J. W., Kim S. Y., Yang Y. M., Kim S. H., Kim C. S., and Lee J. Y. (1998) Frequent somatic mutations in serine/threonine kinase 11/Peutz-Jeghers syndrome gene in left-sided colon cancer. Cancer Res. 58, 3787–3790 [PubMed] [Google Scholar]
- 16. Avizienyte E., Loukola A., Roth S., Hemminki A., Tarkkanen M., Salovaara R., Arola J., Bützow R., Husgafvel-Pursiainen K., Kokkola A., Järvinen H., and Aaltonen L. A. (1999) LKB1 somatic mutations in sporadic tumors. Am. J. Pathol. 154, 677–681 10.1016/S0002-9440(10)65314-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkelä T. P., Hardie D. G., and Alessi D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 10.1038/sj.emboj.7600110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alessi D. R., Sakamoto K., and Bayascas J. R. (2006) LKB1-dependent signaling pathways. Annu. Rev. Biochem. 75, 137–163 10.1146/annurev.biochem.75.103004.142702 [DOI] [PubMed] [Google Scholar]
- 19. Song P., Xie Z., Wu Y., Xu J., Dong Y., and Zou M. H. (2008) Protein kinase C ζ-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J. Biol. Chem. 283, 12446–12455 10.1074/jbc.M708208200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Xie Z., Dong Y., Scholz R., Neumann D., and Zou M. H. (2008) Phosphorylation of LKB1 at serine 428 by protein kinase C-ζ is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117, 952–962 10.1161/CIRCULATIONAHA.107.744490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xie Z., Dong Y., Zhang J., Scholz R., Neumann D., and Zou M. H. (2009) Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 29, 3582–3596 10.1128/MCB.01417-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Umeo S. H., Souza G. P., Rapachi P. M., Garcia D. M., Paccola-Meirelles L. D., Valle J. S., Colauto N. B., and Linde G. A. (2015) Screening of basidiomycetes in submerged cultivation based on antioxidant activity. Genet. Mol. Res. 14, 9907–9914 10.4238/2015.August.19.25 [DOI] [PubMed] [Google Scholar]
- 23. Al-Fatimi M., Schröder G., Kreisel H., and Lindequist U. (2013) Biological activities of selected basidiomycetes from Yemen. Pharmazie 68, 221–226 [PubMed] [Google Scholar]
- 24. Ćilerdžić J. L., Sofrenić I. V., Tešević V. V., Brčeski I. D., Duletić-Laušević S. N., Vukojević J. B., Stajić M. M. (2018) Neuroprotective potential and chemical profile of alternatively cultivated Ganoderma lucidum Basidiocarps. Chem. Biodivers. 15, e1800036 10.1002/cbdv.201800036 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Li L., An S., Zhang Y., Feng S., Zhao L., Teng L., and Wang D. (2017) Antifatigue effects of Antrodia cinnamomea cultured mycelium via modulation of oxidative stress signaling in a mouse model. Biomed. Res. Int. 2017, 9374026 10.1155/2017/9374026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tao Q. Q., Ma K., Bao L., Wang K., Han J. J., Wang W. Z., Zhang J. X., Huang C. Y., and Liu H. W. (2016) Sesquiterpenoids with PTP1B inhibitory activity and cytotoxicity from the edible mushroom Pleurotus citrinopileatus. Planta Med. 82, 639–644 10.1055/s-0041-111629 [DOI] [PubMed] [Google Scholar]
- 27. Kim J. H., Park Y. K., Kim J. E., Lee S. P., Kim B. C., and Jang B. C. (2013) Crude extract of Ceriporia lacerata has a protective effect on dexamethasone-induced cytotoxicity in INS-1 cells via the modulation of PI3K/PKB activity. Int. J. Mol. Med. 32, 179–186 10.3892/ijmm.2013.1364 [DOI] [PubMed] [Google Scholar]
- 28. Thyagarajan-Sahu A., Lane B., and Sliva D. (2011) ReishiMax, mushroom based dietary supplement, inhibits adipocyte differentiation, stimulates glucose uptake and activates AMPK. BMC Complement. Altern. Med. 11, 74 10.1186/1472-6882-11-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuo Y. H., Lin C. H., and Shih C. C. (2015) Antidiabetic and antihyperlipidemic properties of a triterpenoid compound, dehydroeburicoic acid, from Antrodia camphorata in vitro and in streptozotocin-induced mice. J. Agric. Food Chem. 63, 10140–10151 10.1021/acs.jafc.5b04400 [DOI] [PubMed] [Google Scholar]
- 30. Xiao C., Wu Q., Zhang J., Xie Y., Cai W., and Tan J. (2017) Antidiabetic activity of Ganoderma lucidum polysaccharides F31 downregulated hepatic glucose regulatory enzymes in diabetic mice. J. Ethnopharmacol. 196, 47–57 10.1016/j.jep.2016.11.044 [DOI] [PubMed] [Google Scholar]
- 31. Koistinen H. A., Galuska D., Chibalin A. V., Yang J., Zierath J. R., Holman G. D., and Wallberg-Henriksson H. (2003) 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 52, 1066–1072 10.2337/diabetes.52.5.1066 [DOI] [PubMed] [Google Scholar]
- 32. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., and Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin, Science 310, 1642–1646 10.1126/science.1120781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalamidas S. A., and Kotoulas O. B. (1999) The degradation of glycogen in the lysosomes of newborn rat hepatocytes: glycogen-, maltose- and isomaltose-hydrolyzing acid α glucosidase activities in liver. Histol. Histopathol. 14, 23–30 10.14670/HH-14.23 [DOI] [PubMed] [Google Scholar]
- 34. Ismail-Beigi F. (1993) Metabolic regulation of glucose transport. J. Membr. Biol. 135, 1–10 [DOI] [PubMed] [Google Scholar]
- 35. Ren J. M., Marshall B. A., Gulve E. A., Gao J., Johnson D. W., Holloszy J. O., and Mueckler M. (1993) Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J. Biol. Chem. 268, 16113–16115 [PubMed] [Google Scholar]
- 36. Holloszy J. O. (2003) A forty-year memoir of research on the regulation of glucose transport into muscle. Am. J. Physiol. Endocrinol. Metab. 284, E453–E467 10.1152/ajpendo.00463.2002 [DOI] [PubMed] [Google Scholar]
- 37. Li Q., Zhu X., Ishikura S., Zhang D., Gao J., Sun Y., Contreras-Ferrat A., Foley K. P., Lavandero S., Yao Z., Bilan P. J., Klip A., and Niu W. (2014) Ca2+ signals promote GLUT4 exocytosis and reduce its endocytosis in muscle cells. Am. J. Physiol. Endocrinol. Metab. 307, E209–E224 10.1152/ajpendo.00045.2014 [DOI] [PubMed] [Google Scholar]
- 38. Murase T., Misawa K., Haramizu S., Minegishi Y., and Hase T. (2010) Nootkatone, a characteristic constituent of grapefruit, stimulates energy metabolism and prevents diet-induced obesity by activating AMPK. Am. J. Physiol. Endocrinol Metab. 299, E266–E275 10.1152/ajpendo.00774.2009 [DOI] [PubMed] [Google Scholar]
- 39. Fogarty S., and Hardie D. G. (2009) C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J. Biol. Chem. 284, 77–84 10.1074/jbc.M806152200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Collins Q. F., Liu H. Y., Pi J., Liu Z., Quon M. J., and Cao W. (2007) Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5′-AMP-activated protein kinase. J. Biol. Chem. 282, 30143–30149 10.1074/jbc.M702390200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russell R. R. 3rd, Bergeron R., Shulman G. I., and Young L. H. (1999) Translocation of myocardial GLUT4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. 277, H643–H649 10.1152/ajpheart.1999.277.2.H643 [DOI] [PubMed] [Google Scholar]
- 42. Kurth-Kraczek E. J., Hirshman M. F., Goodyear L. J., and Winder W. W. (1999) 5′AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48, 1667–1671 10.2337/diabetes.48.8.1667 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.