

Abstract
In the course of identifying and cleaving RNA, the RNAi machinery must encounter and contend with the megadalton-sized ribosomes that carry out translation. We investigated this interface by examining the fate of actively translated mRNAs subjected to RNAi in C. elegans. Quantifying RNA levels (RNA-seq) and ongoing translation (Ribo-seq), we found there is a greater fold repression of ongoing translation than expected from loss of RNA alone, observing stronger translation repression relative to RNA repression for multiple, independent double-stranded RNA triggers, and for multiple genes. In animals that lack the RNA helicase SKI complex and the ribosome rescue factor PELOTA, ribosomes stall on the 3′ edges of mRNAs at and upstream of the RNAi trigger. One model to explain these observations is that ribosomes are actively cleared from mRNAs by SKI and PELO during or following mRNA cleavage. Our results expand prior studies that show a role for the SKI RNA helicase complex in removing RNA targets following RNAi in flies and plants, illuminating the widespread role of the nonstop translation surveillance in RNA silencing during RNAi. Our results are also consistent with proposals that RNAi can attack messages during active translation.
Keywords: SKI, PELOTA, RNAi, C. elegans, ribosome, nonstop
INTRODUCTION
Small RNAs regulate gene expression through diverse mechanisms and machineries. Here we studied gene silencing initiated by exogenous double-stranded RNA, termed RNA interference (hereafter RNAi). Work from several labs over the last few decades has led to the following model (for review, see Wilson and Doudna 2013): Double-stranded RNA is chopped up by DICER into short interfering RNAs (siRNAs, ∼20–22 nt) and loaded into an argonaute (e.g., Tabara et al. 1999; Bernstein et al. 2001). The argonaute–siRNA complex finds RNA-bearing complementary sequences which are then cleaved by a ribonuclease (e.g., Liu et al. 2004; Tsai et al. 2015).
Although the above steps are conceptually and functionally sufficient to enforce dsRNA-triggered silencing, many systems add additional levels of surveillance including the amplification of small RNA pools through the action of RNA-directed RNA polymerases (e.g., Schiebel et al. 1998; Cogoni and Macino 1999) and the recruitment of chromatin-modifying components in the vicinity of dsRNA-producing loci (e.g., Wassenegger et al. 1994). Despite these substantial augmentations to the core RNAi machinery, the core machinery remains a common factor in a variety of responses to foreign information and internal signals.
Whatever the mechanism of the initial cleavage event, the resulting RNA fragments are then cleared from the cell. Studies in several systems indicate that the downstream fragment, bearing a free 5′ monophosphate, is degraded by the 5′>3′ exoribonuclease XRN-1, while the upstream fragment, bearing a free 3′ end, is degraded by the SKI complex (hereafter “SKI”) and the 3′>5′ exosome (Orban and Izaurralde 2005; Lima et al. 2016; Hashimoto et al. 2017).
It is unclear if and how RNAi contends with normal gene expression processes, particularly translation. During translation, a ribosome translates an mRNA continuously through its open reading frame until it encounters a stop codon. Cleavage within an open reading frame would be expected to prevent ribosomes from completing translation of the mRNA, since the upstream RNA fragment would lack a stop codon. In other situations where ribosomes translate an mRNA lacking a stop codon, a ribosome rescue mechanism involving the factors PELO/HBS1 is central to the release of ribosomes and subsequent nuclease digestion of the mRNA (Shoemaker et al. 2010; Pisareva et al. 2011). Recent work has also shown that a reduction in PELO or HBS1 stabilizes the upstream RNA fragment during RNAi (Hashimoto et al. 2017; Szádeczky-Kardoss et al. 2018). Confounding a straightforward interpretation of these results, mutants of PELO or HBS1 are inviable in several experimental systems (Castrillon et al. 1993; Adham et al. 2003), requiring interpretation of experiments in which function of these essential factors has been transiently inhibited. Additionally, a recent study argued argonaute-mediated surveillance occurs prior to translation (Seth et al. 2018), further confounding an understanding of the relationship between RNAi and translation.
Here we focus on effects from siRNAs that bear substantial complementary to their mRNA targets, versus that mediated by shorter regions of complementarity characteristic of micro RNAs (miRNAs). Whether and how miRNAs affect target mRNA translation has been the subject of much study and ongoing debate (for reviews, see Bartel 2018; Gebert and MacRae 2018). In many species, the overlap of miRNA- and siRNA-associated machineries would complicate an analysis of siRNAs and translation (e.g., mammalian Ago2 is involved in both miRNA- and siRNA-mediated repression; Hutvágner and Zamore 2002). In C. elegans, the presence of genetically and biochemically distinct argonautes for siRNAs and miRNAs (Tabara et al. 1999; Grishok et al. 2001; Yigit et al. 2006) clearly distinguish siRNA- from miRNA-mediated repression, and affords an experimental system in which translational consequences of siRNAs can be examined.
We set out to specifically test the effect of RNAi on translation of an mRNA in an experimental system where PELO and SKI mutants are viable. By quantifying mRNA levels and ongoing translation, we show that RNAi has an ∼2- to 3-fold greater effect on translation than would be expected based on mRNA levels alone. Using genetic interventions to examine RNAi products in the absence of a functioning nonstop decay pathway, we show that upstream RNA fragments accumulate terminal ribosomes in the absence of PELO and SKI. Our results are consistent with the idea that RNAi can occur cotranslationally, with PELO and SKI required to dismantle the resulting ribosome::RNA complexes.
RESULTS
A system to examine gene expression during exogenous RNAi
We selected C. elegans as a genetically tractable system with a potent and robust exogenous RNAi response against dsRNA. We selected three genes encoding highly expressed transcripts and with a well-defined loss-of-function phenotype (unc-54, unc-15, unc-22) (Brenner 1974; Epstein et al. 1974; Waterston et al. 1977; Benian et al. 1989). To perform RNAi, we fed C. elegans bacteria expressing dsRNA complementary to each of the three genes (Fig. 1A; Timmons and Fire 1998). Upon RNAi of each gene, we saw the expected loss-of-function phenotype, demonstrating a functional response to dsRNA.
FIGURE 1.
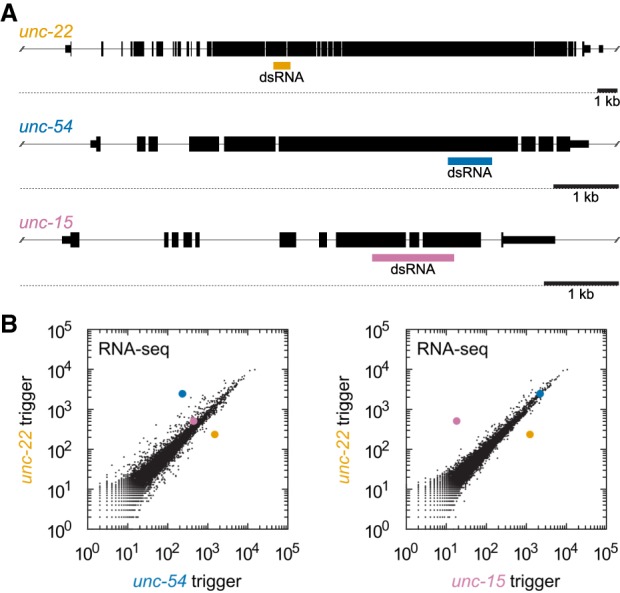
RNAi specifically knocks down transcripts targeted by double-stranded RNA triggers. (A) Diagram of endogenous genes selected as exogenous RNAi targets. Large boxes represent coding regions; narrower boxes represent noncoding regions. Gene diagrams are to scale, with 1 kb scale bar shown. Colored boxes indicate double-stranded RNA (dsRNA) regions expressed in E. coli and fed to the animals to trigger RNAi. (B) Genome-wide RNA-seq of animals after RNAi against unc-22 (y-axis), unc-54 (x-axis, left plot), or unc-15 (x-axis, right plot). Genes are highlighted using the same color scheme as part A. Animals were synchronized and placed on feeding RNAi plates and allowed to develop until the ∼L4 larval stage (∼72 h). Similar knockdown was observed at other developmental stages (Supplemental Fig. S1). Off-diagonal genes other than unc-22, unc-54, and unc-15 are mostly collagens and other periodically expressed transcripts (Supplemental Fig. S2; Hendriks et al. 2014).
To examine mRNA levels upon RNAi, we performed genome-wide RNA-seq on the above samples. We observed an ∼5- to 20-fold knockdown of the target mRNA (unc-54, unc-15, or unc-22) (Table 1; Fig. 1B; Supplemental Fig. S1). Knockdown was specific: (i) unc-54, unc-15, and unc-22 mRNA levels were reduced only when the cognate transcript was targeted by dsRNA and (ii) most other genes’ mRNAs did not change significantly. The handful of genes whose mRNAs did change tended to encode collagens and other transcripts that are periodically expressed during development (Supplemental Fig. S2; Hendriks et al. 2014). At least some of these changes are likely due to secondary effects (e.g., slower development conferred by loss of UNC-54 protein, which is the major myosin in muscle). We note that some gene expression changes (response to elevated dsRNA, commonalities between the various “Unc” phenotypes) would be masked in this analysis as all comparisons were between animals fed dsRNA and exhibiting some form of Unc phenotype. We also failed to detect a significant fold change among myosins paralogous to unc-54 (myo-1, -2, and -3 [Miller et al. 1986]). We conclude that RNAi leads to specific reduction of target mRNA levels for at least unc-15, unc-22, and unc-54.
TABLE 1.
Quantification of fold changes by RNA-seq (with Fig. 1B)
There is a greater loss of ribosomes on RNAi targets than would be expected from RNA loss alone
To analyze translation upon RNAi, we turned to Ribo-seq (ribosome footprint profiling, Ingolia et al. 2009). During Ribo-seq, ribosomes in the act of translation are treated with a nuclease, which digests unbound portions of mRNA, leaving ribosome footprints (Steitz 1969). These footprints can be captured for deep sequencing, yielding information on the number and distribution of ribosomes at the start of the experiment. Ribosomes in the act of translation can protect footprints that are 28–30 nt in size, as well as shorter sizes that report on the occupancy state of the ribosomal A-site (20–24 nt) or ribosomal stalling (15–18 nt) (Steitz 1969; Ingolia et al. 2009; Guydosh and Green 2014; Lareau et al. 2014; Wu et al. 2019). To begin our investigations of translation and RNAi, we initially focused on the 28–30 nt footprint size.
We performed Ribo-seq on the same samples used for RNA-seq and observed an RNAi-dependent and target-specific reduction in the number of Ribo-seq reads on the target mRNAs (Fig. 2A). The loss of Ribo-seq reads was approximately two- to threefold greater than would be expected based on RNA-seq levels alone. We carried out two lines of investigation to scrutinize this effect: (i) We performed a dozen biological replicates and found the effect reproducible (Supplemental Figs. S1, S3). We surveyed animals at four different developmental time points (from early L4 to young adult) after RNAi for each of unc-15, unc-22, and unc-54 by both Ribo-seq and RNA-seq (24 libraries total). (ii) We considered the effects of counting error (Fig. 2B–E). We calculated fold changes in Ribo-seq read counts normalized to RNA-seq read counts. In each case, the mRNA targeted by RNAi exhibited ∼2- to 3-fold fewer counts than would be expected based on RNA-seq alone, compared to mRNAs of a similar expression level. We conclude that RNAi has an ∼2- to 3-fold greater effect on ribosome-protected fragments than mRNA levels.
FIGURE 2.

Repression at the level of translation is greater than expected from RNA loss alone. (A) Genome-wide Ribo-seq of animals after RNAi against unc-22 (y-axis), unc-54 (x-axis, left plot), or unc-15 (x-axis, right plot). Samples are the same as those used for RNA-seq in Figure 1B, and transcripts are highlighted with the same color scheme. Similar knockdown was observed at other developmental stages (Supplemental Fig. S3). Bar graphs on the far right show quantification of fold changes between RNAi and non-RNAi samples for the respective genes’ mRNAs in RNA-seq (with Fig. 1) and Ribo-seq libraries. (B) Equation used to normalize Ribo-seq counts to differences in RNA levels. For each gene in each library, we calculated read counts per million mapped reads. We divided read counts in a Ribo-seq library by read counts from the corresponding RNA-seq library and compared this number between different RNAi conditions. (C) Dot plot showing changes in Ribo-seq read counts (normalized to RNA-seq) between libraries. Y-axis position is determined by equation in part B. A more positive score indicates fewer Ribo-seq read counts normalized to RNA-seq upon RNAi of unc-54; a more negative score indicates fewer Ribo-seq read counts normalized to RNA-seq upon RNAi of unc-22. X-axis position is the total number of read counts across all libraries (2 Ribo-seq and 2 RNA-seq libraries). unc-54, unc-22, and unc-15 are highlighted using the same color scheme as in Figure 1. (D, E) Same as B and C but with unc-22 and unc-15 libraries. A more positive score indicates fewer Ribo-seq read counts normalized to RNA-seq upon RNAi of unc-15; a more negative score indicates fewer Ribo-seq read counts normalized to RNA-seq upon RNAi of unc-22.
In considering models for the difference, we noted recent reports tying translational surveillance factors to orderly recovery after RNAi, nonsense-mediated decay, and other cotranslational mRNA decay events (Lima et al. 2016; Guydosh et al. 2017; Hashimoto et al. 2017; Simms et al. 2017; Szádeczky-Kardoss et al. 2018). We considered the possibility that translational surveillance may add an additional layer of repression during RNAi, accounting for the additional ∼2- to 3-fold effect on ribosome-protected fragments. Our established experimental system was well equipped to investigate one aspect of translational surveillance, namely whether SKI/PELO clear mRNA/ribosomes during ribosome elongation.
Model for SKI/PELO (nonstop) action during RNAi
Previous work shows that when nonstop mRNA decay factors are lost, there are defects in RNA clearance upon RNAi (Orban and Izaurralde 2005; Hashimoto et al. 2017; Szádeczky-Kardoss et al. 2018). During nonstop decay, cells dissociate ribosomes stalled at the 3′ edge of an mRNA lacking a stop codon (for review, see Klauer and van Hoof 2012). The mRNA is endonucleolytically cleaved, then degraded 3′>5′ by the SKI complex and the exosome (van Hoof et al. 2002; Doma and Parker 2006). The ribosome is dissociated by a ribosome rescue factor (pelota/Dom34p) together with Hbs1p and ABCE-1/Rli1p (Shoemaker et al. 2010; Pisareva et al. 2011; Shoemaker and Green 2011). The nascent protein is degraded by listerin and the ribosome quality control complex (Bengtson and Joazeiro 2010). We note that an understanding of the relative order of events in nonstop decay is still emerging; see nonstop decay models from Guydosh et al. (2017) and Simms et al. (2017).
If RNAi were cotranslational, it would result in an upstream RNA fragment bearing ribosomes but lacking a stop codon. Such a complex would be expected to undergo nonstop decay. A model for SKI and PELO action during RNAi based on the existing nonstop decay literature is depicted in Figure 3 (an alternative ordering of events is depicted in Supplemental Fig. S4; Ikeuchi et al. 2016). We set out to test this model explicitly.
FIGURE 3.

Model for SKI and PELO action during RNAi. (Left) Model for ribosome and mRNA clearance after RNA cleavage during RNAi. After RNA cleavage by RISC (yellow), a ribosome elongates to the 3′ edge of the upstream RNA and stalls. An unknown endonuclease (“ribothrypsin,” green scissors) cleaves upstream of the stalled ribosome (Doma and Parker 2006; Ibrahim et al. 2018), and the stalled ribosome is cleared through the action of a specialized ribosome rescue factor (PELO, blue) and ribosome dissociation factor (ABCE-1). The RNA is additionally degraded by SKI and the exosome; the process may repeat itself as the SKI/exosome (orange) chew back the RNA up to another upstream ribosome. The precise ordering of events is not clear; see Supplemental Figure S4 for an alternative ordering (Simms et al. 2017). (Right) Model for cotranslational RNAi in the absence of SKI and PELO. In the absence of PELO, cells are unable to rescue ribosomes after they elongate to the 3′ edge of the upstream RNA. In the absence of SKI, the upstream RNA is less efficiently degraded. Loss of both SKI and PELO would be expected to yield an increase in truncated RNAs and the ribosomes that stall on truncated RNAs.
Ribosomes accumulate on truncated RNA targets of RNAi
We have previously shown that C. elegans’ skih-2 is the ortholog of SKI and C. elegans’ pelo-1 is the ortholog of PELO (Arribere and Fire 2018). Furthermore, the double mutant skih-2 pelo-1 fails to clear ribosomes that stall at mRNA cleavage sites. To test whether SKI and PELO are involved in RNA clearance during RNAi, we performed RNAi in a skih-2 pelo-1 double mutant and performed Ribo-seq and RNA-seq.
Upon RNAi of unc-54, unc-15, or unc-22 in the skih-2 pelo-1 mutant, we observed a loss-of-function phenotype consistent with a functional RNAi response. By RNA-seq, the mRNA levels of cognate transcripts were reduced relative to non-knockdown samples (Fig. 4A; Supplemental Figs. 5A, 6A). Comparing skih-2 pelo-1 double mutant to either wild-type or skih-2 upon RNAi, RNA-seq levels of the target mRNA were elevated, though there was some variability in the extent of knockdown depending on the target mRNA (unc-54, unc-22, or unc-15). We saw a similar effect focusing on 28–30 nt Ribo-seq reads. Taken together, these data are consistent with the idea that the skih-2 pelo-1 strain is competent for RNAi, though knockdown efficiency upon RNAi did not match that of wild type.
FIGURE 4.
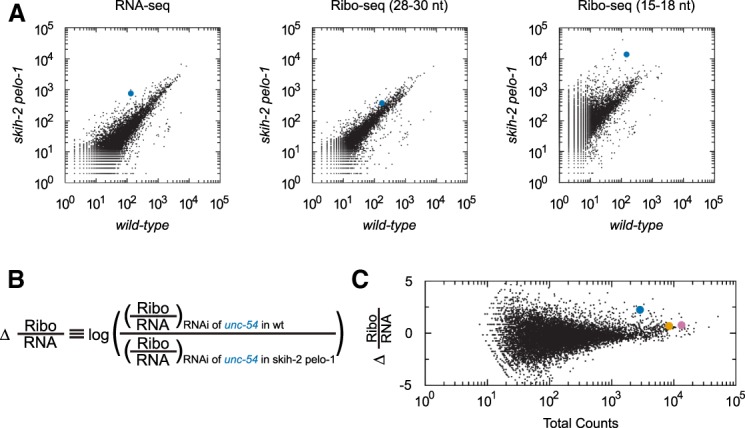
Short Ribo-seq reads accumulate in the skih-2 pelo-1 mutant. (A) Genome-wide RNA-seq, 28–30 nt Ribo-seq, and 15–18 nt Ribo-seq from wild-type (x-axis) and skih-2 pelo-1 (y-axis) knockout upon RNAi of unc-54. Read counts for each library type are displayed as in Figures 1B, 2A. Similar results were observed with RNAi against unc-22 and unc-15 (Supplemental Figs. S5A, S6A). (B) Equation used to normalize Ribo-seq counts to differences in RNA levels. For each gene in each library, we calculated read counts per million mapped reads. We divided read counts in a Ribo-seq library by read counts from the corresponding RNA-seq library and compared this number between different strains. (C) Dot plot showing changes in Ribo-seq (28–30 nt) read counts (normalized to RNA-seq) between libraries. Y-axis position is determined by equation in part B. A more positive score indicates fewer Ribo-seq (28–30 nt) read counts normalized to RNA-seq upon RNAi of unc-54 in the skih-2 pelo-1 strain; a more negative score indicates fewer Ribo-seq (28–30 nt) read counts normalized to RNA-seq upon RNAi of unc-54 in the wild-type strain. X-axis position is the total number of read counts across all libraries (two Ribo-seq and two RNA-seq libraries). unc-54, unc-22, and unc-15 are highlighted using the same color scheme as in Figure 1.
SKI and PELO are required for the clearance of a subpopulation of ribosomes, that is, ribosomes at the 3′ edge of an mRNA, which protect a truncated footprint (15–18 nt) (Guydosh and Green 2014). The exact molecular details of ribosomes that are substrates for SKI and PELO are not yet entirely clear. Recent work in yeasts as well as C. elegans has shown that the bulk of 15–18 nt Ribo-seq reads occur only in the absence of ribosome recycling factors SKI and PELO, consistent with the idea that such reads are indicative of abortive translation structures (Guydosh and Green 2014; Guydosh et al. 2017; Arribere and Fire 2018). Fifteen- to eighteen-nucleotide Ribo-seq reads show lower amounts of phasing than 28–30 nt Ribo-seq reads, but both populations show a highly statistically significant trend toward the zero-frame, consistent with the idea they are derived from translating ribosomes (Supplemental Fig. S7, P-value < 5.73 × 10−22 from χ2 test) (Guydosh and Green 2014; Guydosh et al. 2017; Arribere and Fire 2018). We thus inspected 15–18 nt Ribo-seq reads, using the same samples for the 28–30 nt Ribo-seq libraries, but size-selecting for 15–18 nt instead. We saw an accumulation of 15–18 nt Ribo-seq reads on RNAi target mRNAs in the skih-2 pelo-1 mutant (Fig. 4A; Supplemental Figs. 5A, 6A). The accumulation of 15–18 nt Ribo-seq reads upon RNAi of unc-54 in the skih-2 pelo-1 mutant was higher than comparable libraries from wild type upon unc-54 knockdown, or skih-2 pelo-1 upon unc-22 knockdown. These results are consistent with a model in which SKI and PELO are required for clearance of a subpopulation of ribosomes upon RNAi.
In wild-type animals during RNAi, we observed an extra ∼2- to 3-fold depletion of 28–30 nt Ribo-seq reads relative to RNA-seq (Fig. 2). The skih-2 pelo-1 strain exhibited an even further reduction of this metric, i.e., 28–30 nt Ribo-seq reads were ∼5.5-fold more depleted than RNA-seq when comparing the Ribo-seq/RNA-seq ratio between wild-type and skih-2 pelo-1 upon RNAi of unc-54 (Fig. 4). Similar results were observed with unc-15. These results are consistent with the idea that a population of elongating ribosomes (28–30 nt Ribo-seq reads) is converted to stalled ribosomes (15–18 nt Ribo-seq reads) during RNAi, with the latter population being normally cleared by SKI and PELO. We did not see an additional reduction of the Ribo-seq/RNA-seq ratio in skih-2 pelo-1 animals upon RNAi of the unc-22 gene, though the reasons for this are unclear. One possibility arises from recent work leading to the idea that activity of SKI and PELO depends on collisions between ribosomes (Simms et al. 2017; Juszkiewicz et al. 2018). Under this model, one would expect the density (i.e., ribosomes per stretch of mRNA) to relate to the SKI/PELO sensitivity of ribosome footprints. Consistent with this idea, we note that unc-22 has 10-fold fewer 28–30 nt Ribo-seq reads relative to RNA-seq (compared to unc-54 and unc-15, Figs. 1B, 2A). Thus, it is possible that the lower density of ribosomes on the unc-22 transcript makes this mRNA less prone to SKI/PELO-mediated mRNA decay and ribosomal clearance.
Ribosomes accumulate at and upstream of the region targeted by dsRNA
To illuminate where SKI/PELO-dependent ribosomes occur on an RNAi target, we examined the position-specific distribution of 15–18 nt Ribo-seq fragments (Fig. 5; Supplemental Figs. 5B, 6B). In wild-type and skih-2, we observed a low abundance of 15–18 nt reads distributed throughout the target transcript. In the skih-2 pelo-1 strain, we observed the highest accumulation of reads overlapping the mRNA complementary to the dsRNA trigger. We also observed an accumulation of reads extending upstream of the trigger. This pattern (a high accumulation of 15–18 nt Ribo-seq reads with an upstream accumulation of reads) has been observed for a diverse set of circumstances leading to abortive translation complexes, including nonstop and nonsense-mediated decay (Guydosh et al. 2017; Simms et al. 2017; Arribere and Fire 2018). Our interpretation of this observation is that ribosomes elongate to the 3′ edge of the transcript, stall, and an unknown endonuclease cleaves upstream, thus generating RNA cleavages sites phased by a ribosome footprint length. According to this model, the process of SKI/PELO-mediated decay is expected to generate additional (secondary) cleavages throughout the transcript and upstream of the initial ribosomal stall. We discuss this model and the nature of the stalled ribosomal complexes further in the discussion.
FIGURE 5.

Ribosomes accumulate on target mRNAs at and upstream of a dsRNA trigger in animals that lack SKI and PELO. (A) Location of reads across the unc-54 locus in each strain with each library protocol. unc-54 annotation diagrammed at the bottom, as in Figure 1A, with coding regions in large boxes, noncoding regions in thinner boxes, and intergenic/intronic regions as lines. Location of dsRNA triggers shaded (light gray). Mismatched dsRNA triggers have red stripes. Reads mapping to the dsRNA trigger in RNA-seq were filtered out; see also Materials and Methods “Ribo-seq and RNA-seq analyses.” Antisense reads are displayed below the axis and colored gray. Similar data were obtained with unc-22 and unc-15 (Supplemental Figs. S5B, S6B). (B) The cumulative distribution of 15–18 nt Ribo-seq reads across the spliced transcript (introns removed) for skih-2 pelo-1 libraries with trigger #1 (dotted line), trigger #2 (solid line), and trigger #3 (dashed line) is shown. All reads mapping over a trigger were filtered out, enabling comparisons across the three libraries.
For several systems in which RNA cleavage is coupled to SKI/PELO-mediated decay, the secondary cleavage events are phased by a ribosome footprint length (Guydosh et al. 2017; Simms et al. 2017; Arribere and Fire 2018). We did not detect an obvious phase to the ribosome footprints beyond the 3 nt periodicity from translation (Supplemental Fig. S9). While the initial cleavage associated with nonstop-, nonsense-, or Ire1-mediated decay mechanisms is thought to occur at a discrete site (Guydosh and Green 2017; Guydosh et al. 2017; Arribere and Fire 2018), the initial cleavage during RNAi may occur at any one of a number of siRNA-complementary sites throughout the large region complementary to the dsRNA trigger (hundreds to thousands of bases). According to this model, the initial ribosome stalling pattern would be more diffuse than that observed with nonstop-, nonsense-, or Ire1-mediated decay, and any phasing signal would be lost in a bulk analysis of ribosomal positions. Consistent with this, we failed to detect periodicity in the 15–18 nt Ribo-seq data sets (other than a strong 3 nt periodicity from translation, Supplemental Fig. S9).
A simple hypothesis to explain the high accumulation of 15–18 nt Ribo-seq reads in the skih-2 pelo-1 mutant is that these reads represent ribosomes stalled at abundant cleavage sites of RNAi. To test this hypothesis, we repeated RNAi using two additional triggers that were upstream of (and nonoverlapping with) the initial RNAi trigger used for unc-54 (Fig. 5). In both cases, the highest accumulation of 15–18 nt Ribo-seq reads in the mutant moved with the position of the RNAi trigger, consistent with our expectation.
We wanted to rule out contributions from other RNA species to the 15–18 nt Ribo-seq libraries. Namely, there are at least two known small RNA species produced in a dsRNA trigger-dependent manner on RNAi targets in C. elegans: trigger-derived sense and antisense primary siRNAs, and mRNA-derived antisense secondary siRNAs (Pak and Fire 2007; Sijen et al. 2007). Indeed, in the course of our experiments, we observed a population of antisense RNAs in an RNAi-dependent manner, likely indicating residual siRNA signal (Fig. 5; Supplemental Figs. S5, S6). To deconvolute the relative contributions of trigger-derived and mRNA-derived reads to the signal of short 15–18 nt Ribo-seq reads, we repeated two of the experiments using a mismatched dsRNA trigger. The mismatched dsRNA trigger bears point mutations every 25 nucleotides, enabling in silico deconvolution of trigger-derived and mRNA-derived RNA species. In these experiments, we were able to filter out 18%–23% of reads as being trigger-derived or ambiguous (some 15–18 nt reads do not overlap a mutation). The remaining majority of reads were heavily biased toward the sense-strand (i.e., mRNA-derived), and exhibited properties similar to 15–18 nt Ribo-seq reads from non-RNAi mRNAs (Supplemental Fig. S8, P-value < 1.1 × 10−11 for χ2 test of frame bias). These experiments exhibited the same SKI/PELO-dependent and trigger-dependent accumulations observed in experiments with perfectly matched RNAi triggers, consistent with a model of SKI/PELO-dependent clearance of stalled ribosomes. A statistical analysis also demonstrated significance of the differences in 15–18 nt Ribo-seq read distributions, with the overall distribution of reads differing (P-value < 1.7 × 10−4, KS Test) and median read positions tending more 5′ or 3′ dependent on the RNAi trigger (P-value < 0.0038, Mann–Whitney U test) (P-values computed for all pairwise comparisons between read distributions shown in Fig. 5B, see Materials and Methods).
DISCUSSION
Here we observed a 2- to 3-fold greater repression of ribosome-protected fragments relative to RNA levels upon RNAi. Investigating reasons for the effect, we tested a role for SKI and PELO during RNAi using a system (C. elegans) where SKI and PELO are genetically defined and nonessential. By using mutants of SKI and PELO in an organism where these factors are nonessential, we circumvent the caveats of interpreting effects of transient knockdowns of essential genes. In the absence of SKI and PELO, we observed ribosomes stalled on the upstream fragment during RNAi. This observation is consistent with experiments in which transient knockdown of SKI, PELO, and HBS1 stabilizes the upstream mRNA fragment during exogenous RNAi (Orban and Izaurralde 2005; Hashimoto et al. 2017; Szádeczky-Kardoss et al. 2018). Taken together, these observations are consistent with the idea that RNAi of mRNAs generates stalled ribosome::mRNA complexes, which are dismantled by SKI/PELO-mediated decay.
Our data are consistent with the idea that RNAi can occur concomitantly with translation of an mRNA. There are two models to explain the observed stalled ribosomes over the trigger during RNAi: (i) RNAi occurs coincident with translation elongation, or (ii) RNAi begins prior to ribosome loading, then ribosomes are loaded and elongate to the RNA cleavage site. We favor the former model (i): Given the size of the mRNAs (several kilobases), positions of the RNAi triggers (several kilobases downstream from the start codon), and rate of eukaryotic translation (∼5AA/sec, e.g. [Olofsson et al. 1987]), it is unlikely that a ribosome would have time to elongate from the beginning of the mRNA to the cleavage site without substantial degradation of the RNA fragment from 3′>5′ nucleases. We thus favor the former model, that RNAi can occur cotranslationally.
Our findings are consistent with and build on previous studies showing loss of SKI stabilizes the 5′RNA products of small RNA cleavage (Orban and Izaurralde 2005; Branscheid et al. 2015; Lima et al. 2016; Hashimoto et al. 2017; Szádeczky-Kardoss et al. 2018). We add RNAi to a growing list of cotranslational mRNA decay events where SKI and PELO are required for disassembly of polyribosomes. These decay events include IRE1p-mediated endonucleolytic mRNA cleavage, nonsense-mediated decay, as well as the No-Go/nonstop mRNA decay pathways (Guydosh et al. 2017; Simms et al. 2017; Arribere and Fire 2018). Recent work also indicates ribosome-phased cleavages exist as a notable feature of total cellular mRNA decay (Pelechano et al. 2015; Ibrahim et al. 2018). This literature points to an ancient and conserved solution to the problem of ribosomes bound to an mRNA undergoing decay.
Our data point to SKI and PELO being required for the clearance of a population of abortive translational complexes during RNAi, at least some of which are captured by the 15–18 nt Ribo-seq reads. The population of ribosomes captured by the 15–18 nt Ribo-seq protocol bears clear signatures of translation (Guydosh and Green 2017; Guydosh et al. 2017), and our data recapitulate these features (Arribere and Fire 2018; Supplemental Figs. S7, S9). All these studies have observed clear differences between the 15–18 nt and 28–30 nt Ribo-seq reads, namely, a lower fraction of 15–18 nt Ribo-seq reads in-frame. This point may reflect underlying differing details in the complexes leading to the two populations of fragments and could serve as an impetus for studies of abortive translation-associated structures. The precise nature of the abortive translational complex and the exact molecular role of SKI and PELO in their resolution remains to be determined.
In our system, the initial cleavages from RNAi would be expected to occur over a large area (several hundred nucleotides), generating different ribosomal stall sites on each cleaved mRNA and losing information on the individual relationships between sites of translational distress and precise boundaries of ribosome-protected fragments when viewed in aggregate. We also note that SKI/PELO-mediated ribosome rescue is known to elicit secondary cleavages (by an unknown endonuclease), which would be expected to stall trailing ribosomes. Lastly, ribosomes may stall as a result of collisions with RNAi machinery, possibly even before any mRNA cleavage occurs (Iwakawa and Tomari 2013; Song et al. 2018; Zhang et al. 2018). Our data do not distinguish between ribosomes that stall as a result of an interaction with RNAi components, RNAi cleavage products, or ribosomes that stall in the aftermath. These features of the RNAi system may be a reason to focus studies on the nucleotide-level relationship between translational distress in the absence of SKI/PELO and positioning of 15–18 nt fragments on a more amenable system.
Our results point to the idea that RNAi leads to removal of elongating ribosomes, but it is possible additional interactions between translation and RNAi contribute to the observed effects on ribosomes during RNAi (Fig. 2C,E). For instance, RNAi may reduce the efficiency of translation initiation or differentially target mRNAs that are heavily translated. In any case, our data are consistent with the idea that translation represents an additional level of surveillance in dsRNA-triggered silencing, and that silencing at the translational level contributes to silencing upon RNAi.
MATERIALS AND METHODS
C. elegans strain construction and growth conditions
All strains were derived from “N2” (VC2010) background (Brenner 1974; Thompson et al. 2013) unless otherwise indicated. C. elegans were propagated at 16°C on NGM plates seeded with OP50.
RNA interference and sample preparation
All RNAi experiments were performed at 16°C. RNAi target mRNAs were selected based on their high level of expression, readily detectable phenotype upon RNAi (Unc), and previously reported functional RNAi triggers (Supplemental Table S1, also Kamath et al. 2003).
For a given strain, around 10–15 cm diameter plates with animals were washed off with ∼45 mL N50 (50 mM NaCl). The resulting slurry was added to the top of a 5 mL 5% sucrose cushion in N50 and spun briefly (20″) to separate the worms from bacteria. Animals were washed twice in N50 and then bleached for 7′ (0.5 M NaOH, 20% sodium hypochlorite) or until solution cleared. Eggs were washed three times in M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl), and once in EN50 (1 mM EDTA, 50 mM NaCl). 40,000 eggs were placed on a single 15 cm RNAi feeding plate (NGM with carbenicillin 25 µg/mL and IPTG 232.5 µg/mL), spread the previous day with an RNAi feeding strain ht115(de3) with a plasmid T7-driven dsRNA, described in Supplemental Table S1.
Animals were allowed to develop on RNAi plates until the L4 stage (∼72 h at 16°C, with some deviations, e.g., Supplemental Figs. S1, S3). Animals were visually inspected for the expected Unc phenotypes to ensure successful RNAi: unc-22 (uncontrollable twitching [Brenner 1974]), unc-54 (slow movement but near normal pharyngeal pumping [Epstein et al. 1974]), and unc-15 (similar to unc-54 [Waterston et al. 1977]). Animals were harvested by washing off the plate with ∼15 mL N50 to a 2 mL 5% sucrose cushion in N50. After a brief (20″) centrifugation, the supernatant was aspirated and the pellet was washed twice with N50, then resuspended with about one pellet volume of N50. Pellets were dripped with a Pasteur pipette into 1.5 mL tubes and flash frozen in liquid nitrogen. Pellets were stored at −80°C.
To isolate ribosomes and RNA, samples were ground in liquid nitrogen. A mortar and pestle were prechilled with liquid nitrogen. 200 µl of 1× PLB (20 mM Tris pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 1% Triton) was added to the frozen mortar, followed by the sample. Samples were ground for ∼5′, refilling the mortar with liquid nitrogen as necessary, and the resulting powder was scraped into a 1.5 mL tube on dry ice, later stored at −80°C.
Ribosome footprint isolation
Each ground sample powder was resuspended in 1 mL PLB with cycloheximide (100 µg/mL). Optical density (OD260) of lysates was measured, and 30U of RNase1 (Ambion) was added per OD unit. RNase1 digestion was allowed to proceed at 23°C for 30 min. Samples were layered onto a 10%–60% sucrose gradient and centrifuged at 35,000 rpm in an SW41 rotor (Beckmann) for 4 h and 30 min at 4°C. Monosome peaks were collected by gradient fractionation using Teledyne ISCO's Density Gradient Fractionation System. RNA was isolated from monosome fractions by proteinase K digestion, phenol/chloroform extraction, and ethanol precipitation. Size selection for full length (28–30 nt) or truncated (15–18 nt) ribosome footprints was done on a Urea-TBE 15% polyacrylamide gel using appropriate RNA size standards (AF-MS-24 [28 nt, Stadler and Fire 2011] and AF-JA-267 [17 nt, Arribere and Fire 2018]).
mRNA fragment isolation
Ground sample powder was dissolved in 1 mL TRIzol and RNA harvested per manufacturer's recommendations. Five micrograms of total RNA was depleted of ribosomal RNA using the RiboZero kit per the manufacturer's recommendation (Epicentre/Illumina). Ribosome-subtracted RNA was combined with an equal volume of 2× Alkali Fragmentation Buffer (10 mM Na2CO3, 90 mM NaHCO3, 0.5 mM EDTA, pH ∼9.3) and fragmented for 30′ at 95°C. Fragmented RNA was resolved on a Urea-TBE 15% polyacrylamide gel and 40–60 nt fragments excised and gel purified.
Library construction and sequencing
Libraries for Ribo-seq and RNA-seq were performed essentially as described previously (Arribere and Fire 2018). Size-selected RNA fragments were treated with 10 Units T4 PNK in 1× PNK buffer (NEB) to remove 3′ phosphate. PNK-treated RNA was ligated to 25 pmoles preadenylated 3′ adaptor (IDT, AF-JA-34 /5rApp/NNNNNNAGATCGGAAGAGCACACGTCT/3ddC/) in 20% PEG8000 (NEB), 1× adenylate ligase buffer (3.3 mM DTT, 8.3 mM glycerol, 50 mM HEPES KOH (pH 8.3), 10 mM MgCl2, 10 μg/mL acetylated BSA), and 10 Units T4 RNA Ligase 1 (NEB) for 4 h at 23°C. Excess 3′ adaptor was removed by sequential digestion with 5′ deadenylase (NEB) and RecJ (NEB) in 1× Buffer 2 (NEB). Reverse transcription was performed using AF-JA-126 (IDT/5Phos/AGATCGGAAGAGCGTCGTGT/iSp18/CACTCA/iSp18/GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT) as primer, and superscript II (Invitrogen) in 10 µl. cDNA was size selected and extracted from a Urea-TBE 10% polyacrylamide gel. Gel-purified cDNA was circularized using CircLigase ssDNA Ligase (Epicentre/Illumina). Illumina indexes were added by PCR and amplified for 6, 8, 10, or 12 PCR cycles. Libraries were sequenced on a MiSeq Genome Analyzer for quality control and then sequenced deeper with the NextSeq or HiSeq 4000.
Ribo-seq and RNA-seq analyses
Prior to mapping, Illumina adaptors were trimmed, and reads bearing the same hexamer at their 3′ end were collapsed (to remove PCR biases, based on the strategy of Kivioja et al. 2011). Reads were selected in silico for the expected sizes: RNA-seq (40–60 nt), normal Ribo-seq (28–30 nt), and truncated Ribo-seq (15–18 nt). Reads were mapped using STAR (version 2.5.0a [Dobin et al. 2013]) to the C. elegans’ genome (WBcel235, ENSEMBL release 90) allowing for no mismatches.
In some libraries, we noticed reads mapping to the RNAi trigger region. Trigger-mapping reads were especially abundant in the RNA-seq libraries [which were not poly(A)-selected]. We expect many of the trigger-mapping reads derive from the highly abundant dsRNA trigger used to induce the RNAi response. For RNA-seq libraries, we performed a filter round of mapping to the feeding RNAi plasmids (BJA7 (unc-22), BJA40 (unc-54), BJA77 (unc-15)). To do this, we removed all reads that mapped to the feeding RNAi plasmid, allowing for up to three mismatches. The reads that did not map to the feeding RNAi plasmid were then mapped to the genome, allowing for no mismatches. For the mismatched trigger experiments, reads were mapped to the trigger allowing for no mismatches, and then to the genome, allowing for no mismatches. For Ribo-seq libraries (both 28–30 nt and 15–18 nt sizes), we observed fewer trigger-derived reads. Our Ribo-seq results were robust to whether we removed or included reads overlapping the dsRNA trigger.
Reads were assigned to genes using annotations, ignoring reads that could not be uniquely assigned. Gene counts (e.g., Figs. 1B, 2A, 4A), and read mapping locations (e.g., Fig. 4B), were plotted using custom scripts in python.
We noticed differences between 15- and 18-nt Ribo-seq libraries, including variability in the amount of reads in-frame (Supplemental Fig. S7) and variability in the number of antisense reads (Fig. 4B; Supplemental Figs. S5B, S6B). Because these differences persist outside of the gene targeted by RNAi (where we would not expect a trigger-dependent effect), we interpret them as arising at least in part from day-to-day technical variability (e.g., from differences in RNaseI digestion). Because of this variability, all mutant-control comparisons reported were done between samples grown and libraries prepared in parallel.
Autocorrelation analysis
Ribo-seq data were used to analyze periodicity within 200 nt upstream of each RNAi trigger. For each data set, Pearson and Spearman autocorrelation analysis was performed (Supplemental Fig. S9, only SJA71MS and SJA72MS are shown for simplicity). The Pearson autocorrelation evaluates the linear relationship between two variables and the Spearman autocorrelation evaluates the monotonic (nonparameterized) relationship between two variables. A list of read densities at each position was correlated with itself at offsets ranging from 0 to 50 nt. For each offset, correlation coefficients and P-values were calculated.
Statistical analysis of 15–18 nt Ribo-seq read distributions
We analyzed the statistical significance of differences between 15–18 nt Ribo-seq read distributions in the skih-2 pelo-1 mutant with different unc-54 RNAi triggers. For each of the three samples shown in Figure 5, we identified the positions at which 15–18 nt Ribo-seq occurred across the unc-54 transcript. To test whether these positions differed in their distribution from the other two samples, we performed a Kolmogorov–Smirnov test with the positions of each sample (“ks_2samp” in python). To test whether the positions of one library tended more N or C-terminal, we performed a Mann–Whitney U test with each sample (“mannwhitneyu” in python). All pairwise comparisons were statistically significant at P < 0.00017 and P < 0.0038, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
DATA DEPOSITION
High-throughput sequencing files generated in this study are available at SRA (PRJNA477865).
Supplementary Material
ACKNOWLEDGMENTS
We thank Peter Sarnow and Gabriele Fuchs for use of the ISCO sucrose gradient machine. Thank you to Jana Lim and Anne Brunet for BJA77 (unc-15 RNAi plasmid). We thank Rachel Green and Nick Guydosh for a helpful discussion early on in the project. We thank members of the Arribere and Fire Laboratories for discussions and critical reading of the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.070813.119.
REFERENCES
- Adham IM, Sallam MA, Steding G, Korabiowska M, Brinck U, Hoyer-Fender S, Oh C, Engel W. 2003. Disruption of the pelota gene causes early embryonic lethality and defects in cell cycle progression. Mol Cell Biol 23: 1470–1476. 10.1128/MCB.23.4.1470-1476.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Fire AZ. 2018. Nonsense mRNA suppression via nonstop decay. eLife 7: e33292 10.7554/eLife.33292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. 2010. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467: 470–473. 10.1038/nature09371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benian GM, Kiff JE, Neckelmann N, Moerman DG, Waterston RH. 1989. Sequence of an unusually large protein implicated in regulation of myosin activity in C. elegans. Nature 342: 45–50. 10.1038/342045a0 [DOI] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366. 10.1038/35053110 [DOI] [PubMed] [Google Scholar]
- Branscheid A, Marchais A, Schott G, Lange H, Gagliardi D, Andersen SU, Voinnet O, Brodersen P. 2015. SKI2 mediates degradation of RISC 5′-cleavage fragments and prevents secondary siRNA production from miRNA targets in Arabidopsis. Nucleic Acids Res 43: 10975–10988. 10.1093/nar/gkv1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillon DH, Gönczy P, Alexander S, Rawson R, Eberhart CG, Viswanathan S, DiNardo S, Wasserman SA. 1993. Toward a molecular genetic analysis of spermatogenesis in Drosophila melanogaster: characterization of male-sterile mutants generated by single P element mutagenesis. Genetics 135: 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoni C, Macino G. 1999. Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature 399: 166–169. 10.1038/20215 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564. 10.1038/nature04530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein HF, Waterston RH, Brenner S. 1974. A mutant affecting the heavy chain of myosin in Caenorhabditis elegans. J Mol Biol 90: 291–300. 10.1016/0022-2836(74)90374-X [DOI] [PubMed] [Google Scholar]
- Gebert LFR, MacRae IJ. 2018. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 10.1038/s41580-018-0045-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. 2001. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23–34. 10.1016/S0092-8674(01)00431-7 [DOI] [PubMed] [Google Scholar]
- Guydosh NR, Green R. 2014. Dom34 rescues ribosomes in 3′ untranslated regions. Cell 156: 950–962. 10.1016/j.cell.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, Green R. 2017. Translation of poly(A) tails leads to precise mRNA cleavage. RNA 23: 749–761. 10.1261/rna.060418.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, Kimmig P, Walter P, Green R. 2017. Regulated Ire1-dependent mRNA decay requires no-go mRNA degradation to maintain endoplasmic reticulum homeostasis in S. pombe. eLife 6: e29216 10.7554/eLife.29216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Takahashi M, Sakota E, Nakamura Y. 2017. Nonstop-mRNA decay machinery is involved in the clearance of mRNA 5′-fragments produced by RNAi and NMD in Drosophila melanogaster cells. Biochem Biophys Res Commun 484: 1–7. 10.1016/j.bbrc.2017.01.092 [DOI] [PubMed] [Google Scholar]
- Hendriks G-J, Gaidatzis D, Aeschimann F, Großhans H. 2014. Extensive oscillatory gene expression during C. elegans larval development. Mol Cell 53: 380–392. 10.1016/j.molcel.2013.12.013 [DOI] [PubMed] [Google Scholar]
- Hutvágner G, Zamore PD. 2002. A microRNA in a multiple-turnover RNAi enzyme complex. Science 297: 2056–2060. 10.1126/science.1073827 [DOI] [PubMed] [Google Scholar]
- Ibrahim F, Maragkakis M, Alexiou P, Mourelatos Z. 2018. Ribothrypsis, a novel process of canonical mRNA decay, mediates ribosome-phased mRNA endonucleolysis. Nat Struct Mol Biol 25: 302–310. 10.1038/s41594-018-0042-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Yazaki E, Kudo K, Inada T. 2016. Conserved functions of human Pelota in mRNA quality control of nonstop mRNA. FEBS Lett 590: 3254–3263. 10.1002/1873-3468.12366 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa H, Tomari Y. 2013. Molecular insights into microRNA-mediated translational repression in plants. Mol Cell 52: 591–601. 10.1016/j.molcel.2013.10.033 [DOI] [PubMed] [Google Scholar]
- Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, Hegde RS. 2018. ZNF598 is a quality control sensor of collided ribosomes. Mol Cell 72: 469–481.e7. 10.1016/j.molcel.2018.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. 2011. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9: 72–74. 10.1038/nmeth.1778 [DOI] [PubMed] [Google Scholar]
- Klauer AA, van Hoof A. 2012. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip Rev RNA 3: 649–660. 10.1002/wrna.1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Hite DH, Hogan GJ, Brown PO. 2014. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife 3: e01257 10.7554/eLife.01257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WF, De Hoyos CL, Liang X-H, Crooke ST. 2016. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res 44: 3351–3363. 10.1093/nar/gkw065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J-J, Hammond SM, Joshua-Tor L, Hannon GJ. 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441. 10.1126/science.1102513 [DOI] [PubMed] [Google Scholar]
- Miller DM, Stockdale FE, Karn J. 1986. Immunological identification of the genes encoding the four myosin heavy chain isoforms of Caenorhabditis elegans. Proc Natl Acad Sci 83: 2305–2309. 10.1073/pnas.83.8.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson S-O, Boström K, Carlsson P, Borén J, Wettesten M, Bjursell G, Wiklund O, Bondjers G. 1987. Structure and biosynthesis of apolipoprotein B. Am Heart J 113: 446–452. 10.1016/0002-8703(87)90612-0 [DOI] [PubMed] [Google Scholar]
- Orban TI, Izaurralde E. 2005. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 11: 459–469. 10.1261/rna.7231505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, Fire A. 2007. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315: 241–244. 10.1126/science.1132839 [DOI] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2015. Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161: 1400–1412. 10.1016/j.cell.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CUT, Pestova TV, Pisarev AV. 2011. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J 30: 1804–1817. 10.1038/emboj.2011.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebel W, Pélissier T, Riedel L, Thalmeir S, Schiebel R, Kempe D, Lottspeich F, Sänger HL, Wassenegger M. 1998. Isolation of an RNA-directed RNA polymerase-specific cDNA clone from tomato. Plant Cell 10: 2087–2101. 10.2307/3870786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth M, Shirayama M, Tang W, Shen E-Z, Tu S, Lee H-C, Weng Z, Mello CC. 2018. The coding regions of germline mRNAs confer sensitivity to argonaute regulation in C. elegans. Cell Rep 22: 2254–2264. 10.1016/j.celrep.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R. 2011. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci 108: E1392–E1398. 10.1073/pnas.1113956108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. 2010. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330: 369–372. 10.1126/science.1192430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Steiner FA, Thijssen KL, Plasterk RHA. 2007. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science 315: 244–247. 10.1126/science.1136699 [DOI] [PubMed] [Google Scholar]
- Simms CL, Yan LL, Zaher HS. 2017. Ribosome collision is critical for quality control during no-go decay. Mol Cell 68: 361–373.e5. 10.1016/j.molcel.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Chen H, Sheng G, Wang Y, Lou J. 2018. Argonaute facilitates the lateral diffusion of the guide along its target and prevents the guide from being pushed away by the ribosome. Biochemistry 57: 2179–2183. 10.1021/acs.biochem.8b00213 [DOI] [PubMed] [Google Scholar]
- Stadler M, Fire A. 2011. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17: 2063–2073. 10.1261/rna.02890211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. 1969. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature 224: 957–964. 10.1038/224957a0 [DOI] [PubMed] [Google Scholar]
- Szádeczky-Kardoss I, Csorba T, Auber A, Schamberger A, Nyikó T, Taller J, Orbán TI, Burgyán J, Silhavy D. 2018. The nonstop decay and the RNA silencing systems operate cooperatively in plants. Nucleic Acids Res 46: 4632–4648. 10.1093/nar/gky279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. 1999. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99: 123–132. 10.1016/S0092-8674(00)81644-X [DOI] [PubMed] [Google Scholar]
- Thompson O, Edgley M, Strasbourger P, Flibotte S, Ewing B, Adair R, Au V, Chaudhry I, Fernando L, Hutter H, et al. 2013. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res 23: 1749–1762. 10.1101/gr.157651.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Fire A. 1998. Specific interference by ingested dsRNA. Nature 395: 854 10.1038/27579 [DOI] [PubMed] [Google Scholar]
- Tsai H-Y, Chen C-CG, Conte D Jr, Moresco JJ, Chaves DA, Mitani S, Yates JR III, Tsai M-D, Mello CC. 2015. A ribonuclease coordinates siRNA amplification and mRNA cleavage during RNAi. Cell 160: 407–419. 10.1016/j.cell.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264. 10.1126/science.1067272 [DOI] [PubMed] [Google Scholar]
- Wassenegger M, Heimes S, Riedel L, Sänger HL. 1994. RNA-directed de novo methylation of genomic sequences in plants. Cell 76: 567–576. 10.1016/0092-8674(94)90119-8 [DOI] [PubMed] [Google Scholar]
- Waterston RH, Fishpool RM, Brenner S. 1977. Mutants affecting paramyosin in Caenorhabditis elegans. J Mol Biol 117: 679–697. 10.1016/0022-2836(77)90064-X [DOI] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA. 2013. Molecular mechanisms of RNA interference. Annu Rev Biophys 42: 217–239. 10.1146/annurev-biophys-083012-130404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Zinshteyn B, Wehner KA, Green R. 2019. High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol Cell 73: 959–970. 10.1016/j.molcel.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit E, Batista PJ, Bei Y, Pang KM, Chen C-CG, Tolia NH, Joshua-Tor L, Mitani S, Simard MJ, Mello CC. 2006. Analysis of the C. elegans argonaute family reveals that distinct argonautes act sequentially during RNAi. Cell 127: 747–757. 10.1016/j.cell.2006.09.033 [DOI] [PubMed] [Google Scholar]
- Zhang K, Zhang X, Cai Z, Zhou J, Cao R, Zhao Y, Chen Z, Wang D, Ruan W, Zhao Q, et al. 2018. A novel class of microRNA-recognition elements that function only within open reading frames. Nat Struct Mol Biol 25: 1019–1027. 10.1038/s41594-018-0136-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.