

Abstract
Isoniazid (INH) is the cornerstone of tuberculosis (TB) chemotherapy, used for both treatment and prophylaxis of TB. The antimycobacterial activity of INH was discovered in 1952, and almost as soon as its activity was published, the first INH-resistant Mycobacterium tuberculosis strains were reported. INH and its structural analog and second-line anti-TB drug ethionamide (ETH) are pro-drugs. INH is activated by the catalase-peroxidase KatG, while ETH is activated by the monooxygenase EthA. The resulting active species reacts with NAD+ to form an INH-NAD or ETH-NAD adduct, which inhibits the enoyl ACP reductase InhA, leading to mycolic acid biosynthesis inhibition and mycobacterial cell death. The major mechanism of INH resistance is mutation in katG, encoding the activator of INH. One specific KatG variant, S315T, is found in 94% of INH-resistant clinical isolates. The second mechanism of INH resistance is a mutation in the promoter region of inhA (c-15t), which results in inhA overexpression and leads to titration of the drug. Mutations in the inhA open reading frame and promoter region are also the major mechanism of resistance to ETH, found more often in ETH-resistant clinical isolates than mutations in the activator of ETH. Other mechanisms of resistance to INH and ETH include expression changes of the drugs’ activators, redox alteration, drug inactivation, and efflux pump activation. In this article, we describe each known mechanism of resistance to INH and ETH and its importance in M. tuberculosis clinical isolates.
THE EMERGENCE OF DRUG RESISTANCE
Tuberculosis (TB) chemotherapy started in the 1930s with the discovery by Domagk and colleagues of the anti-TB activity of sulfonamides. Since these compounds were very toxic and highly insoluble, analogs were synthesized, leading to the discovery of Tibione (thiacetazone, Fig. 1), a highly effective thiosemicarbazone against Mycobacterium tuberculosis (1, 2). In parallel, the natural product streptomycin (SM), discovered by Schatz and Waksman, showed activity against M. tuberculosis (3) and was used successfully to treat TB patients. Two new anti-TB drugs were discovered soon after: para-aminosalicylic acid (PAS) in 1946 (4) and isonicotinic acid hydrazine (isoniazid, INH) in 1952 (5, 6). Each drug had activity against M. tuberculosis; however, drug-resistant mutants emerged rapidly during clinical trials (7–9). To prevent drug resistance, in 1959, SM, PAS, and INH were combined to form the first successful multidrug, biphasic chemotherapy for TB (10). This combination treatment was so impressive that Selman Waksman wrote, “the ancient foe of man, known as consumption, the great white plague, tuberculosis, or by whatever other name, is on the way to being reduced to a minor ailment of man. The future appears bright indeed, and the complete eradication of the disease is in sight” (185, p. 217). Nevertheless, the treatment was long and expensive, and patients often dropped out prior to completing chemotherapy. In 1984, a new short-course treatment was established that showed improved efficacy and patient compliance; the drug regimen consisted of two months on INH, rifampicin (RIF), pyrazinamide (PZA), and ethambutol, followed by four months on INH and RIF only.
FIGURE 1.
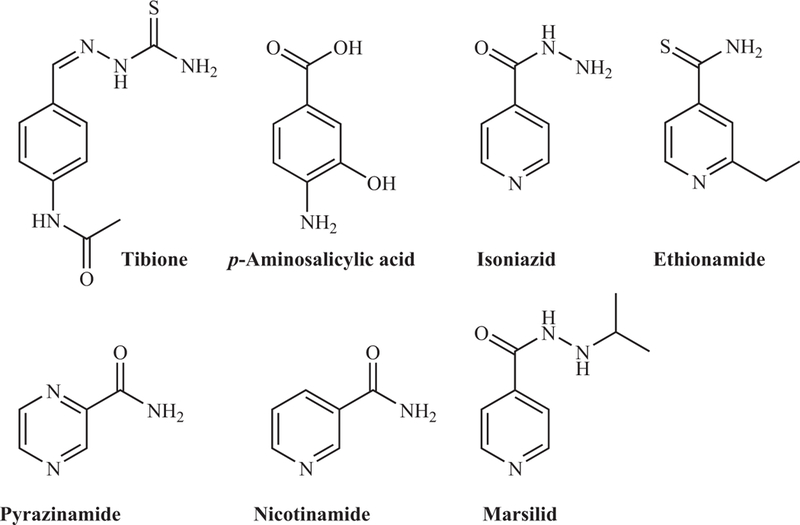
Early synthetic antituberculosis drugs. doi:10.1128/microbiolspec.MGM2-0014-2013.f1
Despite this successful chemotherapy, the rate of multidrug-resistant (MDR) M. tuberculosis strains, defined as strains resistant to INH and RIF, started to increase as early as 1985. Nowadays, the World Health Organization (WHO) estimates that 3.7% of new TB cases and 20% of previously treated TB cases are caused by MDR-TB (11). The highest incidence of MDR-TB (up to 76%) is found in Russia and the former Soviet republics. Extensively drug-resistant (XDR)-TB, defined as TB strains resistant to INH, RIF, fluoroquinolones, and one second-line injectable drug, is found in up to 9% of MDR-TB cases and has been reported in at least 84 countries so far. Furthermore, strains of M. tuberculosis that are resistant to up to 10 TB drugs, referred to as totally drug-resistant (TDR-TB), have been isolated in Europe, Africa, India, and Iran (12). One factor in the emergence and rapid spread of drug resistance is the paucity of rapid diagnostics. While new tools are available to quickly assess drug resistance, these methods are based on known drug resistance mechanisms. Understanding all these mechanisms is key to improving diagnosis and eradicating drug-resistant M. tuberculosis. In this article, we will discuss the mechanisms that M. tuberculosis developed to become resistant to the first-line anti-TB drug INH and its analog, the second-line anti-TB drug ethionamide (ETH).
MODE OF ACTION OF INH AND ETH
INH
The antimycobacterial activity of INH was published and commercialized simultaneously by three independent pharmaceutical companies: Bayer (Neoteben) (13), Hoffman-La Roche (Rimifon) (6), and Squibb Institute for Medical Research (Nydrazid) (5). INH had been first synthesized 40 years earlier and reported in a doctoral thesis; therefore, none of the pharmaceutical companies could patent the discovery. Fox (6) described the discovery of INH as an attempt to combine the anti-TB activity of nicotinamide, which had been found to arrest M. tuberculosis growth in guinea pigs (14), and thiosemicarbazones. By replacing the benzene ring of Tibione by the pyridine ring found in nicotinamide (Fig. 1), meta- and para-pyridylaldehyde thiosemicarbazones were synthesized. All the intermediates in the synthesis of these pyridine thiosemicarbazones were tested for activity against TB, and among them, one intermediate, INH, had antimycobacterial activity far superior to any compound at the time (6). Notably, another analog synthesized, Marsilid (1-isonicotinoyl-2-isopropylhydrazine) (Fig. 1), also showed good anti-TB activity. The use of Marsilid as a TB drug was soon discontinued because it induced euphoria in TB patients. However, Marsilid did go on to become the first antidepressant. Interestingly, the earlier synthetic compounds active to some degree against M. tuberculosis share a similar chemical skeleton (Fig. 1), although their modes of action are different.
The mechanism of action of INH has been the subject of intensive research and controversies since its discovery in 1952. From 1953 to 1980, multiple modes of action for INH were proposed: INH was thought to interfere with cell division (15), pyridoxal-dependent metabolic pathway(s) (16), lipid biosynthesis (17), fatty acid biosynthesis (18), nucleic acid biosynthesis (19), glycerol conversion to hexose phosphate (20), NAD+ biosynthesis (21), and NADH dehydrogenase activity (22–23). A major breakthrough in understanding the mechanism of action of INH came with the study by Winder and Collins (186) in which they showed that INH inhibited synthesis of mycolic acids, long-chain α-alkyl β-hydroxy fatty acids that are a crucial component of the mycobacterial cell wall. The effect of INH on mycolic acids has been subsequently confirmed by numerous researchers. Among them, Takayama and coworkers were the first to demonstrate that inhibition of mycolic acid biosynthesis by INH correlated with cell death (24), accumulation of long-chain fatty acids (25), and inhibition of C24 and C26 monounsaturated fatty acid biosyntheses (26). Takayama and colleagues concluded that the mode of action of INH involved a desaturase required for the biosynthesis of these unsaturated fatty acids (26).
INH, like other TB drugs (ETH, PZA, isoxyl, thiacetazone) is a pro-drug. The catalase peroxidase KatG (encoded by Rv1908c) activates INH to form a hypothetical isonicotinoyl anion or radical (27–30). This entity reacts with NAD+ to yield an INH-NAD adduct, which binds to the active site of the NADH-dependent enoyl-ACP reductase InhA (Rv1484) (Fig. 2) (31). This enzyme reduces monounsaturated acyl-ACP to acyl-ACP (32, 33) and is part of the fatty acid synthase type II (FASII) (34). FASII elongates fatty acids up to 56 carbons long where they are derivatized and coupled to a C24-C26 fatty acid generated by FASI to form mycolic acids. The INH-NAD adduct binds to and inhibits InhA (28, 35, 36), leading to disruption of mycolic acid biosynthesis and cell death (37, 38).
FIGURE 2.

Mechanism of action of INH and ETH. INH and ETH are activated by the catalase peroxidase KatG and monooxygenase EthA, respectively, to form a reactive species that binds to NAD+. The resulting adducts, INH-NAD or ETH-NAD, inhibit the enoyl-ACP reductase InhA of the FASII system, resulting in mycolic acid biosynthesis inhibition. doi:10.1128/microbiolspec.MGM2-0014-2013.f2
ETH
2-Ethylthioisonicotinamide, ETH (Fig. 1), is a structural analog of INH. ETH was first synthesized in 1956 by a French team trying to improve on the antimycobacterial properties of thioisonicotinamide (39). Grumbach and coworkers found that ETH was more active than SM but less so than INH against M. tuberculosis. However, ETH was also active against INH-, PAS-, and SM-resistant M. tuberculosis strains. ETH was shown to be efficacious in combination with PZA and with or without INH in a clinical trial to treat TB patients infected with INH- and SM-resistant strains (40, 41). Nowadays, ETH is a second-line drug, mostly used to treat MDR-TB cases in South Africa.
Similar to INH (42, 43), M. tuberculosis treated with ETH loses its acid fastness (39) and its ability to synthesize mycolic acids (44). ETH is also a pro-drug, activated by the NADPH-specific flavin adenine dinucleotide-containing monooxygenase EthA (also called EtaA, encoded by Rv3854c) (45–48). Once activated, the mode of action of ETH is very similar to INH (Fig. 2). The active form of ETH reacts with NAD+ to yield an ETH-NAD adduct (49), which inhibits InhA, leading to mycolic acid biosynthesis inhibition. Interestingly, while KatG only activates INH, EthA activates two other second-line anti-TB drugs: Tibione (thiacetazone) and isoxyl (46, 50).
MECHANISMS OF RESISTANCE TO INH AND ETH
Drug resistance in mycobacteria is due to the acquisition of mutations or efflux pump activation, not due to the acquisition of resistance plasmids or transposons, common resistance mechanisms in other bacterial species. The main mechanisms of resistance to INH and ETH can be divided into two categories. First, prevention of the activation of INH and ETH can be obtained by mutating the activators of the drugs katG and ethA, respectively, or by mutating regulators of their expression. For example, the katG(S315T) mutation is found in up to 94% of INH-resistant M. tuberculosis clinical isolates. Second, the inhibition of InhA by the INH-NAD or ETH-NAD adduct can be overcome by mutations in inhA or its promoter region. Other mechanisms of resistance exist such as drug inactivators, redox alteration, and efflux pumps. We will first describe the mechanisms of coresistance to INH and ETH and then the mechanisms of resistance specific to each drug.
Common Mechanisms of Resistance to INH and ETH
Clinical isolates coresistant to INH and ETH were isolated from TB patients who had received INH but had never been treated with ETH (51–53). This conundrum led to the hypothesis that INH and ETH shared a common mechanism of resistance, a hypothesis that could not be tested until a plasmid transformation system was developed for mycobacteria (54, 55). It would take another three decades to discover that INH and ETH target the same enzyme in M. tuberculosis: the enoyl-ACP reductase InhA (56). Other mechanisms of coresistance to INH and ETH have been discovered. These mechanisms along with resistance mechanisms due to inhA mutations are listed below.
Alteration of InhA, the target of INH and ETH
The target of INH and ETH was discovered by isolating a Mycobacterium smegmatis mutant coresistant to INH and ETH (56). A genomic DNA cosmid library of this M. smegmatis mutant and of drug-susceptible Myco-bacterium avium, Mycobacterium bovis, M. smegmatis, and M. tuberculosis strains was constructed and transformed into M. smegmatis. A single open reading frame (ORF) was found to be sufficient to confer coresistance to INH and ETH in M. smegmatis and was named inhA. These experiments demonstrated that a single amino acid mutation in inhA(S94A) or overexpression of inhA conferred coresistance to INH and ETH in mycobacteria. To prove that inhA inactivation was sufficient to lead to death in a manner similar to INH action, a temperature-sensitive mutant in inhA was isolated (37). Heat-inactivation of InhA(V238F) mimicked the effects of INH in M. tuberculosis described by Takayama and colleagues: inhibition of mycolic acid biosynthesis (24), alteration of the bacterium morphology (57), accumulation of long-chain fatty acids (25), and cell death (24), demonstrating that inhibition of InhA alone reproduced the biochemical characteristics of INH treatment of M. tuberculosis. Moreover, the S94A mutation identified in the INH- and ETH-resistant M. smegmatis mutant was transferred into wild-type M. tuberculosis by specialized linkage transduction, and the resulting strain was at least five times more resistant to INH and ETH than wild-type M. tuberculosis (38). This combined set of data confirms that InhA is the main target and the main mechanism of coresistance to INH and ETH.
The mechanism by which the S94A mutation leads to resistance to INH and ETH has been well studied. InhA is an NADH-dependent enoyl-ACP reductase, and the binding of the enoyl substrate to InhA is not disturbed by the S94A mutation; however, the mutation results in a 5-fold increase in the KM for the InhA cofactor NADH (32). On the other hand, the ability of the INH-NAD adduct to inhibit InhA(S94A) is markedly reduced, because the IC50 and Ki are 17 and 30 times higher, respectively, for the mutated protein. Comparison of the crystal structures of InhA(S94A) to wild-type InhA revealed the loss of a water molecule and disruption of a hydrogen bonding network in the mutated protein, which was enough to reduce the binding of the INH-NAD adduct (38). Others disputed this conclusion and hypothesized that InhA interacts with FASII enzymes and that this interaction is perturbed by the S94A mutation, resulting in INH resistance (58). Overexpression of inhA is also a common factor of INH and ETH resistance in clinical isolates. The c-15t base pair change in the inhA regulatory region increases inhA mRNA levels by 20-fold, resulting in the overexpression of InhA. This leads to a titration of INH or ETH and consequently an eight-fold increase in INH and ETH MICs in M. tuberculosis (38).
Numerous point mutations in inhA and its promoter region have been identified in INH- and ETH-resistant M. tuberculosis clinical isolates (Tables 1 and 2); however, no base pair insertions or deletions have been observed. Mutations in the inhA promoter and ORF regions are associated with low-level resistance to INH, even when strains contained mutations in both regions (MIC <1 mg/liter) (59). The c-15t mutation in the promoter region of inhA is found in up to 35% of INH-resistant and 55% of ETH-resistant clinical isolates, but never in INH- or ETH-sensitive strains. This mutation was also overly represented in XDR-TB cases in South Africa. In a study on clinical isolates from the Western Cape Province, South Africa, the c-15t mutation was present in 30% of strains mono-resistant to INH, 48% of MDR-TB, and 85% of XDR-TB (60), suggesting that this mutation could be a marker for XDR-TB. Interestingly, in a survey of the Eastern Cape Province clinical isolates, the c-17t inhA promoter mutation was the predominant genetic modification present in 83% of XDR-TB cases. The combined inhA promoter mutations (at positions −8, −15, and −17) were found in 92% of the XDR-TB cases versus 62% of the MDR-TB cases in the Eastern Cape Province (60).
TABLE 1.
Identified mutations in genes other than katG in INH-resistant M. tuberculosis strainsa
| oxyR’-ahpC intergenic region | furA-katG intergenic region | Ndh |
| t-89a (130) | c-1 ins (102) | R13C (67) |
| g-88a (130) | g-7a (96) | V18A* (66–67) |
| c-81t (131) | a-10c (96) | T110A (68) |
| g-74a (132) | g-12a (96) | R268H (66, 68) |
| c-72t (131) | G313R* (116) | |
| g-67a (66) | fabG-inhA regulatory region | |
| g-66a (133) | g-147t (88) | iniB |
| atgt-54 ins (92) | a-113c (134) | t198ins (135) |
| c-54t (132) | g-102a (134) | a211del (135) |
| c-52t (136) | a-92t (69) | 222 12bp del (88) |
| g-51a (131) | g-67c (137) | N88S* (116) |
| t-49g (137) | g-47c,a (134, 138) | G192* (135) |
| g-48a (135, 139) | c-34t (138) | H481Q* (116) |
| g-46a* (102, 140) | t-24g (141) | |
| g-46 del* (66) | g-22c (142) | iniA |
| c-45t (84) | g-17t* (66, 135, 139, 143) | P3A (88) |
| t-44a (99) | a-16g (84) | nt282–286 del (88) |
| t-40c (144) | c-15t (66, 88, 135, 139, 140, 143) | H481Q* (88, 135) |
| c-39t (66, 135, 139, 140, 144) | t-12a (145) | R537H (88, 135) |
| t-34c,a (102, 140) | a-11t (66) | |
| t34 del (102) | t-8a*,g,c (66, 88, 140–142) | iniC |
| g-32a (140) | t-5a (146) | t79ins (135) |
| c-30t (66, 144) | c-4a (140) | a98ins (135) |
| c-20t* (102) | A5P (137) | W83G (88) |
| c-15t (66, 135, 139, 144) | V14L (137) | |
| c-12t (66, 135, 139, 140) | T21A (66) | Rv0340 |
| c-10a,g,t (66, 135, 139, 140) | T143* (135) | |
| g-9a* (66, 135, 139, 140, 144) | inhA ORF | G149* (135) |
| g-6a (66, 140, 144) | M1L (147) | V163I (88, 135) |
| a-4g (140) | K8N (148) | |
| I16T (149) | nat | |
| ahpC ORF | I21T,V (66, 88, 135, 139, 149) | G67R* (88) |
| P2S (69) | I25T (136) | Y177H (150) |
| L3K (140) | I47T* (66,149) | G207R* (88, 103) |
| L4R (130) | V78A (149) | |
| T5I (66) | S94A (59, 66,144) | Rv1592c |
| F10I (144) | I95P (151) | P42L (88) |
| D33N (84) | A190S (152) | V430A (88) |
| D73H* (66) | I194T* (66, 88) | |
| E76K (153) | R202G (133) | fadE24 |
| L191K (66) | E217D (133) | -64 2 bp ins (88) |
| T241M (142) | a-23c* (88) | |
| oxyR’ | T253A* (152) | |
| G12a (130) | D256N (152) | Rv1772 |
| g18a (69) | I258T,V (135, 152) | T4A (88) |
| g27t (69) | Y259H (152) | |
| c28a (69) | efpA | |
| c37t (99) | kasA | T15R* (116) |
| bp67 ins ggcg (99) | D66N* (66, 109) | I73T* (135) |
| g325t (99) | M77I* (88, 135) | Q513R (116) |
| a331c (99) | R121K (69) L245R (135) | E520V (135) |
| furA | G269S* (66, 69, 88, 109, 135,139) | fabD |
| S5P (88, 135) | G312S* (66, 69, 109, 135) | S275N* (88) |
| c34 del (96) | S341* (135) | A199T* (88) |
| A14V (96) | G387D (69) | |
| A46V* (116) | F413L (109) | accD6 |
| L68F (94) | D229G* (88) | |
| C97Y (94) | srmR homolog | |
| D3G (88) | fbpC | |
| M323T* (88) | G158S* (88) |
Asterisk (*), found in INH-resistant and/or INH-sensitive strains.
TABLE 2.
Identified mutations in ETH-resistant M. tuberculosis strainsa
| ethA | ethA | ethA |
| M1R (62) | A234D (154) | C403G (62) |
| I9T (154) | t703 del (61) | R404L (154) |
| G11A (62) | Q246STOP (61) | G413D (61) |
| g32 del (154) | A248D (154) | c1254 del (154) |
| A20 ins (62) | Y250ST0P (154) | g1268 del (154) |
| a65 del (46) | cg754 ins (62) | c1290 del (61, 154) |
| H22P (62) | Q254P (154) | gc1322,1323 del (61) |
| Y32D (154) | Q254ST0P (154) | T453I (154) |
| a110 del (61, 154) | g768 del (61)* | Y461H (62) |
| G43S (61), C (46) | S266R (62) | R463D (61) |
| T44N (154) | Q269ST0P (62) | a1391 ins (62) |
| D49A (155) | Q271ST0P (154) | |
| Y50C (154) | L272P (62) | fabG-inhA regulatory region |
| P51L (46) | P288R (154) | g-17t (61) |
| D55A (61) | Q291ST0P (154) | c-15t (38, 61, 156) |
| D58A (46) | R292ST0P (154) | |
| T61M (62) | C294F (154) | inhA ORF |
| Y84D (46) | F302L (154) | I21T(61) |
| 1bp271 del (46) | T324 ins (154) | S94A (61) |
| cg282–283 del (154) | L328M (155) | I95P (151) |
| g337 del (154) | S329L (62) | |
| a338 ins (61) | L333R (155) | mshA |
| a342 del (154) | I338S (61)* | N111S* (62, 70) |
| G124D (62) | T342K (46) | Q128STOPa(70) |
| G139S (154) | d1029 del (154) | V171G(62) |
| Y140 STOP (154) | N345K (155) (154) | A187V(62) |
| Q165P (62) | A352P (154) | R273Cb (70) |
| W167STOP (154) | g1054 del (154) | G299Cb (70) |
| T186K (46) | P378L (154) | Q331STOPb (70) |
| g593 del (154) | A381P (46) | G356Db (Z0) |
| c613 del (155) | t1152 del (154) | E361Ab (70) |
| gt638–639 del (155) | G385D (61) | |
| D219G (154) | Y386C (62) | ethR |
| E223K (61) | S390F (154) | A95T (62) |
| g673 ins (154) | W391ST0P (154) | F110L (62) |
| tc675 ins (62) | T392A (61) | |
| T232A (155) | L397R (155) |
Asterisk (*), also found in ETH-sensitive strains.
Mutations found in in vitro cultures.
inhA is an essential gene; therefore, mutations in the coding region of inhA are rare. About 15 mutations in inhA have been identified in INH-resistant clinical isolates, although two of them were also found in INH-sensitive strains (Table 1). I21T, S94A, and I95P are the only amino acid changes found in both INH- and ETH-resistant clinical isolates. The first mutation identified in inhA in M. smegmatis, resulting in the S94A variant, has since been found in INH-resistant M. tuberculosis clinical isolates with no other mutation present in katG or the fabG-inhA intergenic region (59, 61). This confirms that the S94A mutation is sufficient to confer INH and ETH resistance in M. tuberculosis clinical isolates.
While mutations in the inhA promoter region can represent up to 35% of the INH-resistant cases, and mutations in the inhA gene are rare in INH-resistant clinical isolates, this is the predominant region where mutations are found in ETH-resistant clinical isolates. In one study (62), 62% of the ETH-resistant clinical isolates had mutations in inhA (gene and/or promoter region), while 47% had mutations in ethA. The inhA promoter mutation c-15t was therefore proposed as a marker for ETH resistance (63).
Alteration of redox potential
M. smegmatis and M. bovis BCG mutants coresistant to INH and ETH were isolated in in vitro experiments from nonmutagenized independent cultures. The mutants had mutations in ndh (Rv1854c), a gene encoding a type II NADH dehydrogenase, which oxidizes NADH into NAD+. In M. smegmatis, the ndh mutants contained single base pair changes resulting in amino acid changes and a pleitropic phenotype: INH resistance, ETH resistance, temperature sensitivity, and for some mutants, Ser/Gly auxotrophy (64, 65). In M. bovis BCG, the mutants had either single base pair changes or insertions (65). The ndh mutants lost up to 95% of their Ndh activity compared to wild type and had higher levels of NADH, while their NAD+ levels were similar to wild type. An increase in NADH concentration was shown to prevent the binding of the INH-NAD adduct to InhA by acting as a competitive inhibitor, leading to INH resistance (65).
In M. tuberculosis clinical isolates, ndh mutations have been found in both INH-sensitive and INH-resistant strains at a very low rate (66) (Table 1). However, two studies from Singapore and Brazil identified ndh mutations in 8 to 10% of INH-resistant M. tuberculosis clinical isolates and found no ndh mutations in their INH-sensitive clinical isolate strains (67, 68). The R13C mutation was found in a strain containing the katG (S315T) mutation, so the contribution to the INH resistance of this ndh mutation is uncertain (67). The ndh (T110A) mutation was only associated with an ahpC mutation resulting in the (T5I) variant (68). However, in that study, katG was only partially sequenced, and inhA was not (68, 69), so the INH resistance in that isolate may or may not be due to the ndh(T110A) mutation. The R268H mutation is the only mutation so far to have been identified in two independent studies and only in INH-resistant strains, but it was associated with katG, inhA, or ahpC mutations (66).
M. tuberculosis has an additional NADH dehydrogenase named ndhA (Rv0392c). Therefore, mutations in one NADH dehydrogenase such as ndh might not alter the redox balance in M. tuberculosis and might not lead to INH and/or ETH resistance as long as the second NADH dehydrogenase is functional. ndh and ndhA are also present in M. bovis BCG, but M. bovis ndhA codes for a single amino acid change (V241A) relative to M. tuberculosis, which might explain why INH- and ETH-resistant ndh mutants were isolated in that strain. For that reason, ndh mutations might not be a mechanism of resistance to INH and ETH in M. tuberculosis unless both NADH dehydrogenase genes are mutated.
Alteration in mycothiol biosynthesis
Mutations in ndh in INH-resistant M. bovis BCG mutants were isolated in vitro by plating nonmuta-genized, independent M. bovis BCG cultures on plates containing both INH and ETH to avoid mutants carrying mutations in the activator of INH or ETH. When the same experiment was repeated in M. tuberculosis H37Rv or the virulent M. bovis Ravenel strain, ndh mutants were not obtained. Instead, all of the INH- and ETH-resistant M. tuberculosis mutants had mutations in mshA (Rv0486) (70), a gene encoding a glycosyl transferase involved in the biosynthesis of mycothiol (N-acetylcysteine glucosamine inositol, MSH), while the M. bovis mutants coresistant to INH and ETH carried mutations either in mshA or in mshC (Rv2130c) encoding the cysteine ligase of the mycothiol biosynthesis (71). Five enzymes are required to synthesize mycothiol: MshA, MshA2, MshB (Rv1170), MshC, and MshD (Rv0819) (72). Mycothiol is the major thiol and the main reducing and detoxifying agent in mycobacteria (72), yet the role of mycothiol during infection is ambiguous since mycothiol-deficient M. tuberculosis strains do not have a growth defect in vivo (70). Eight in vitro M. tuberculosis mshA mutants were isolated containing a single base pair modification in mshA, resulting in amino acid changes, stop codons, and frameshifts, all of which caused a drastic decrease in mycothiol levels (from 83 to 99.9%). The mutants had different levels of resistance to INH (2-fold to >10-fold) and ETH (4- to 8-fold increase). Complementation with wild-type M. tuberculosis mshA restored ETH susceptibility but not INH susceptibility in all the mutants. Interestingly, deletion of mshA in M. tuberculosis led to a strain that did not produce mycothiol and was highly resistant to ETH but fully sensitive to INH (70). This suggests that mycothiol is mostly involved in ETH resistance and might play a role in ETH activation. The observed INH resistance in M. tuberculosis mshA point mutants and INH susceptibility in M. tuberculosis AmshA strains might also indicate that mshA is required for INH resistance in mycothiol-deficient strains.
In M. smegmatis, a 4- to 8-fold increase in INH and ETH resistance was obtained when mshA or mshC was deleted, while deletion of mshB resulted in a strain resistant only to ETH, and deletion of mshD had no effect on INH or ETH resistance (73). M. smegmatis mycothiol mutants obtained from chemical mutagenesis or transposon insertion had slightly different INH and ETH resistance patterns (74). The role of mycothiol deficiency in INH resistance might be species-dependent. So far, we can only conclude that mutations in mshA will result in high-level ETH resistance and at most low-level INH resistance in M. tuberculosis.
In a highly INH- and ETH-resistant clinical isolate, a double mutation in mshA (V171G, A187V) was found (62). That strain had the katG(S315T) mutation to account for the INH resistance but no other mutation to explain its resistance to ETH. However, the mshA (A187V) mutation is present in wild-type M. tuberculosis Beijing strain. Other mshA mutations have also been found in drug-sensitive mycobacterial strains. The N111S mutation is present in M. tuberculosis Erdman and Haarlem strains (70, 75), while the M. bovis Ravenel and M. bovis ATCC19210 strains carry a g316a base pair change in mshA, resulting in a G106R amino acid change (71, 75). Mycothiol genes such as mshA and mshC should be added to the list of candidates responsible for ETH resistance in clinical isolates, although correlation between mshA mutations and ETH resistance should be carefully analyzed since mutations in mshA might not lead to drug resistance in M. tuberculosis.
Degradation of the INH-NAD or ETH-NAD adduct
In a recent study, Wang and colleagues suggested that the NADH pyrophosphatase NudC (Rv3199c), an enzyme from the NAD+ recycling pathway, could hydrolyze the INH-NAD or ETH-NAD adduct, leading to INH and ETH resistance (76). M. smegmatis and M. bovis BCG NudC are functional enzymes, while M. tuberculosis H37Rv NudC has a point mutation (Q237P) that renders the enzyme inactive. The authors demonstrated that NudC from M. smegmatis and M. bovis BCG could release the adenosine mono-phosphate group from the INH-NAD and ETH-NAD adducts and that overexpression of M. smegmatis or M. bovis BCG nudC resulted in at least a 10-fold increased resistance to INH and ETH, while deletion of nudC rendered the strains more sensitive to the drugs. A small portion of M. tuberculosis clinical isolates (2%) were found to have the glutamine residue at position 237, suggesting that in these M. tuberculosis strains, NudC might be capable of hydrolyzing the adducts. However, no transfer of mutation was performed to prove that this mutation is sufficient to confer INH and ETH resistance in M. tuberculosis. The role of nudC in INH and/or ETH resistance in M. tuberculosis clinical isolates remains to be determined.
Mechanisms of Resistance Specific to INH
Alterations in KatG, the activator of INH
The first mutants isolated in in vitro cultures that were highly resistant to INH had the characteristic of being catalase-negative and avirulent in guinea pigs (77, 78). Winder hypothesized that the loss of catalase activity might imply that INH was activated by a catalase to yield some highly reactive species (79). The relationship between INH resistance and the catalase-negative phenotype was elucidated many years later when a highly INH-resistant strain, BHI, a mutant of M. smegmatis mc2155 (55), was complemented with an M. tuberculosis library (30). INH susceptibility was restored in BHI by the introduction of a single gene, katG (Rv1908c), encoding a catalase-peroxidase. Furthermore, the authors also found katG deletion or mutations in INH-resistant M. tuberculosis clinical isolates and demonstrated that transformation of these INH-resistant isolates with a wild-type copy of katG restored INH susceptibility (30, 80).
In INH-resistant M. tuberculosis clinical isolates, more than 300 mutations in katG have been identified throughout the ORF (Table 3). Complete deletion of the gene has been found in clinical isolates, including the first INH-resistant M. tuberculosis mutant identified in the Zhang study (30); it has subsequently been identified in other studies (Table 3). Point mutations causing a single amino acid substitution or premature termination, frameshift mutations after addition or deletion of bases, and partial or complete deletion of the gene have been identified. The incidence of katG mutations differs between geographical regions but represents at least 30% and up to 95% of INH-resistant clinical isolates.
TABLE 3.
Identified katG mutations in INH-resistant M. tuberculosis strainsa
| Complete deletion (30, 88, 134, 141, 157) | E217G,del (84, 158) | W397Y (88) STOP (66) |
| Partial deletion (138, 159–161) | N218K (153) | A409D,R,T,V (142,152, 153, 162) |
| V1A (84) L (92) | Q224E (163) | Y413H (133) |
| P2S (163) | Y229F (164) | K414N (165) |
| a17 ins (139) | V230A (153) | R418Q (166) |
| c30 del (160) | P232R,S (139, 167) | D419A,Y,E, H (59,139) |
| T11A (160) | G234E (102) R (153) | M420T (167) |
| T12P (160) | N236T (153) | A424E V (163) |
| S17N (163) | A243S (163) | G428R (168) |
| G19D (163) | A245V* (146) | P429S (163) |
| a98ins (160) | R249C (159) | c1297 ins, c1305 del (167) |
| N35D (160) | T251M (92) | Q434stop (88) |
| g109del (160) | F252L (84) | t1311 ins (139) |
| W38stop (66) | R254L (139) | W438R (88) |
| L48Q (146) | M257I,T (143, 146) | a1329 ins (139) |
| a149 del (102) | N258S (163) | c1339 del (139) |
| A61T (160) | E261Q (142) | L449F (169) |
| c185 ins (160) | T262R (84, 141) | S457I (165) |
| D63E (88, 141) | A264T,V* (152,163) | K459ST0P (168) |
| A65T (163) | H270Q (170) | R463L* (85, 141, 162,171) |
| A65 or cccc ins (84,157) | T271P (172) | W477stop (84) |
| A66P (163) | G273C,S (139, 173) | R484S (102) |
| I71N (84) | T275A,P, (84, 159, 162,171) | G485V (84) |
| D72G (153) K (66) | G279D, A* (59, 139) | K488N (157) |
| D73N (160) | P280H,L*,P (92, 93,152) | R489S (167) |
| D74Y, G (153) | A281V (163) | G490S (102, 139) |
| M84I (153) | G285C,D,V (116, 145, 152) | G491C (135, 143) |
| T85P (92) | E289D (153) | N493H (142) |
| Q88R (157) | A291P,V (152, 174) | G494D (59) |
| W90R (139) STOP (88, 135, 163) | L293V (139) | R496L (168) |
| W91R (88) | Q295K,P,STOP (130, 146, 157) | P501A (157) |
| D94A (84) | M296V (145) | W505S (175), R (163) |
| G96C (153) | Q297V (146) | D511del (84) |
| H97R (139) | G299S (139) C (153) A (133) | D513del (84) |
| G99E (84) | W300D,G,C,ST0P (69, 84, 138, 157) | R515C (66, 84,135) |
| R104L (141), Q (92) | gc900 ins (138) | L521del (84) |
| M105I (93) | S302R (139, 163) | Q525P (84) |
| A106V (92) | Y304S (165) | N529D (88) |
| W107R (92) | T308P (176) | D542H (167) |
| W107ST0P (102) | G309C,S,V (177–179) | L546P (172) |
| H108E,Q (84, 141) | D311E (180) G (153) Y (179) | A550D (163) |
| A109V (139) | A312G,V (152) | E553K (59) |
| A110V (59, 84) | S315T,N,I,R,G,L (59, 66, 84, 88, 135, 139, 141–144, 157) | c1667del (66) |
| D117A (143) | G316D, S* (66) | F567S (84) |
| G120del (84) | I317L (153) | D573N (92) |
| G121C (92) V (157) | E318G,V (152, 180) | A574V (84) E (66) |
| A122del (84) | W321G (66) R (153) | L587I,M,P* (66, 84) |
| G123E (167) | T322A,I (136, 179) | P589T (163) |
| g371 del (167) | T324A,P (146, 179) | G593D (84) |
| G125C (153) | T326D (181) | R595stop (59) |
| H125 ins (171) | K327I (178) | E607K (92) |
| M126I (163) | W328L,C,F,S,G (84, 155, 172,182,183) | M609I (163) |
| Q127E,P (153, 167) | D329A,C (138, 145) | L617del (84) |
| R128P (66) E (174) | S331C (180) | ac1849 ins (157) |
| F129S (116) | I334T (84) | L619P (84) |
| N133T (167) | I335T (84, 141) V (184) | G629S (88, 141) |
| N138S,H*,T (61, 66, 84, 88, 178) | L336R,P (88) | R632C (167) |
| A139P (84) | Y337F,C (163) | L634F (84) |
| S140A,N (84) | W341S,G (59,139) | A636E (92) |
| L141F (59) | T344A,P (152,175) | L653P (92) |
| D142A (84) | K345T (92) | F658V (59) |
| K143T (92) | P347L* (152) | L662R (139) |
| A144V (139) | A350S,T (84, 141) | G685R (135) |
| R146W (93) | Q352ST0P (155) | D695A,G (162,172) |
| L148A,P (84, 170) | A361D (133) | G699Q (102) |
| Y155C (92) S (157) | T363A (152) | S700P (84,157) |
| Y155C (92) S (157) | T363A (152) | S700P (84, 157) |
| S160L (84) | P365S* (152) | V708P (146) |
| cc478–479 del (167) | F368L* (152) | V710A (84) |
| A162T (92) | G372 ins (88) | c139 del (139) |
| G169A (163) | S374P* (152) | A713P (66) |
| A172T (66, 84) V (153) | L378P (133) | A714P (84) |
| M176I,T (153, 167) | A379T (142) V (133) | A716P (141) |
| T180C (84) K (66) | T380I (153) | Q717P (66) |
| G186V,H (153) | D381G (141) | V725A (146) |
| W191R, STOP (102, 172) | S383P (167) | A726T (139) |
| g572 del (153) | L384R (92) | A727D (159) |
| WE191–192 del (167) | D387H (167) | W728C (92) |
| E195K (61) | P388L,S (152) | D735 del (92) |
| W198stop (84) | T390I (153) | D735A (66, 84), N (92) |
| K200E (139), STOP (84) | L390 ins (146) | |
| W204R | I393N (84) |
Asterisk (*), found in INH-resistant and/or INH-sensitive strains.
The most frequent katG mutation is at codon S315, where each base (AGC) can be mutated to produce a Thr, Asn, Arg, Ile, Gly, or Leu residue. The S315T mutation can be found in up to 94% of the INH-resistant clinical isolates (81). Two independent biochemical analyses reported that KatG(S315T) has catalase-peroxidase activities, yet its ability to oxidize INH was significantly reduced (82, 83). Biochemical analyses of other KatG mutants showed a wide range of catalase-peroxidase and INH oxidase activities (Table 4). Nevertheless, there is a link between INH oxidase activity and INH resistance. M. tuberculosis strains with KatG proteins deficient in INH oxidase activity were highly resistant to INH, while katG mutants with INH oxidase activity close to wild-type levels had only 2- to 4-fold increases in the MIC for INH. However, the level of INH resistance might not be defined by location of a mutation: S315T, W341G, G494D, and R595STOP variants are highly resistant to INH (84), while L141F, E553K, and F658V variants are associated with low-level INH resistance (84). A very common polymorphism, R463L, is often found associated with other katG mutations and is more likely to be present in INH-sensitive strains than in INH-resistant strains (85).
TABLE 4.
| Variant |
|||
|---|---|---|---|
| Activity | Similar to wt | Partial | None |
| Catalase peroxidase | A110V, A139P, A245V, S315N, S315T, R463L, L587M, L619P, L634F, D735A | L48Q | L141F, T275P, Q295P, G297V, T324P, L587P |
| INH oxidase | L48Q, A110V, A245V, R463L | A139P, Q127E, N133T, L141F, P232S, Q295P, G297V, T324P, S383P, D387H, D419H, M420T, R489S, L634F, D735A | M176T, S315 (N,R,T), D542H, L619P, R632C |
The high-level resistance to INH associated with katG (S315T) (MIC >1 mg/liter) was reported to be specific to the Ser→Thr amino acid change (59, 86). Brossier and colleagues found katG(S315N) only in low-level INH-resistant clinical isolates (62). Curiously, KatG(S315N) had been shown to prevent the formation of the INH-NAD adduct, suggesting that INH cannot be activated by KatG(S315N), which conflicts with the above mentioned results (87). In a different study, a clinical isolate carrying katG(S315N) and inhA(c-15t), which is associated with low-level resistance to INH (see above), had an MIC for INH of >256 mg/liter (88). This was the same level of resistance found in clinical isolates where katG was missing or the mutation resulted in early termination of the protein (Q434STOP) (88). Furthermore, the INH-resistant clinical isolate containing only the inhA promoter mutation had an MIC of 0.19 mg/liter, confirming the low-level INH resistance associated with this mutation. This suggests that katG(S315N) might be associated with a high level of INH resistance in clinical isolates.
One of the first studies on the isolation of INH-resistant M. tuberculosis mutants in vitro found that the mutants could be classified into catalase-negative and highly INH-resistant or catalase-positive and weakly INH-resistant (MIC <10 mg/liter) (89). Catalase-negative mutants were unable to grow in guinea pigs and rabbits, while catalase-positive mutants grew relatively well in vivo (90). The relationship between catalase activity and fitness of an INH-resistant strain in vivo has been investigated. Pym and colleagues demonstrated that the INH-resistant M. tuberculosis KatG(T275P) variant had no detectable catalase peroxidase activity and was highly attenuated in a mouse model of infection, while M. tuberculosis carrying the KatG(S315T) variant was found to be fully virulent in mice, to have no bacterial fitness cost, and to be fully transmissible (91). Consequently, the S315T variant is more often found in MDR-TB patients than in INH mono-resistant clinical isolates (86) and might be related to the higher transmission capabilities of this particular strain (92). A recent study of TB patients showed that those infected with non-katG INH-resistant strains (such as inhA[c-15t]) were more likely to exhibit sputum conversion after 1 month than those infected with katG mutant strains (93). Transmissibility, virulence, and response to chemotherapy seem to be affected by katG mutations.
Alterations of katG expression
katG is cotranscribed with its negative regulator furA (Rv1909c), a gene encoding a ferric uptake regulation protein (94, 95). Deletion of furA in M. tuberculosis results in overexpression of katG and hypersusceptibility to INH (94). Mutations have been identified in furA as well as in the 38-bp region between furA and katG (Table 1). To assess the role of these mutations, Ando and colleagues constructed isogenic strains containing the mutations found in the intergenic region (g-12a, a- 10c, g-7a) or in the furA coding sequence (A14V). katG expression in the g-12a strain (intergenic region) was only slightly lower than wild-type levels, and the A14V variant had no effect on KatG levels (96); however, the strains with the a-10c or g-7a mutation exhibited an 80% reduction in katG expression. This reduction was associated with a decrease in INH oxidase activity and a 2- to 4-fold increase in INH resistance, suggesting that the furA-katG intergenic region should be examined in low-level INH-resistant M. tuberculosis clinical strains.
Transcriptional analyses indicate that katG is also regulated by the sigma factor sigl (Rv1189)(97). M. tuberculosis ΔsigI had decreased catalase capabilities and was more resistant to INH than wild-type M. tuberculosis in vitro and in vivo. Furthermore, overexpression of sigI increased the susceptibility of M. tuberculosis to INH by 2-fold. Mutations in sigI could therefore be another factor that modulates katG expression and induces low-level INH resistance in M. tuberculosis clinical isolates.
Compensatory mutations
The activity of INH against mycobacterial species is very specific. Other bacteria such as Escherichia coli and Salmonella typhimurium are not inhibited by a high dose of INH (500 mg/liter or higher; the MIC for M. tuberculosis is 0.05 mg/liter). Yet when E. coli and S. typhimurium have a deficient oxidative stress response regulator, encoded by oxyR, the strains become more sensitive to INH (MIC <50 mg/liter) (98). M. tuberculosis oxyR’ is nonfunctional, because the coding region contains multiple frameshifts and deletions. Downstream of oxyR’ is ahpC (Rv2428), which encodes an alkyl hydroperoxide reductase. Up to 29% of INH-resistant clinical isolates contain mutations in the oxyR’-ahpC region (99). Mutations in the oxyR’-ahpC intergenic region such as g-9a and c-15t have been shown to increase the expression of ahpC by 9- and 18-fold, respectively (100). This increase in ahpC expression is thought to compensate for the loss of KatG activity occurring in INH-resistant strains, which would render the strains more susceptible to hydrogen and organic peroxides (101) and to prevent further oxidative damage. However, most variant KatG enzymes, and in particular, KatG (S315T), are competent catalase-peroxidases, meaning that the organism is not deficient in its ability to detoxify peroxides or other compounds. With the present knowledge, the role of ahpC in INH resistance is a matter of debate. Baker et al. reported that mutations in the oxyR’-ahpC region did not contribute to INH resistance since these mutations could be found in 20% of their INH-resistant clinical isolates but also in 8% of their INH-sensitive isolates (102).
Detoxification of INH
In humans, INH is acetylated by the arylamine N-acetyltransferase NAT2. The rate of this detoxification reaction varies between individuals, leading to the classification of TB patients as rapid or slow inactivators depending on their NAT activity. An enzyme similar to NAT is expressed in M. tuberculosis (103). When M. tuberculosis nat was overexpressed in M. smegmatis, the resulting strain was more resistant to INH, with an MIC 3-fold higher than wild-type M. smegmatis, suggesting that the mycobacterial arylamine N- acetyltransferase can acetylate and inactivate INH (104). Mutations in nat (nhoA/Rv3566c) have been identified in INH-resistant clinical isolates (Table 1); G67R and G207R variants were found in INH-resistant and INH-sensitive clinical isolates. Biochemical analyses revealed that the KM for INH N-acetylation of Nat (G207R) was 10 times that of wild-type Nat (103), indicating that the variant Nat protein is mostly unable to acetylate and therefore inactivate INH. Thus, there is no obvious association between nat mutations and INH resistance. Nevertheless, deletion of nat in M. bovis BCG affected the biosynthesis of mycolic acids, glycolipids, and complex lipids as well as survival in mouse macrophages, indicating that nat might modulate other factors involved in INH resistance (105).
Genes induced upon INH treatment
When M. tuberculosis comes in contact with INH, numerous genes are upregulated, as first evidenced using a method that employed differential expression using customized amplification libraries (DECAL) (106). The availability of the M. tuberculosis genome sequence (107) led to the development of microarrays that have been used to explore the response of M. tuberculosis to INH and other drug treatments (108). Transcriptional analysis of INH-treated M. tuberculosis revealed that M. tuberculosis upregulated a set of genes encoding proteins involved in fatty acid biosynthesis (fabD, acpM, kasA, kasB, accD6; Rv2243–2247), trehalose dimycolyl transfer (fbpC, Rv0129c), fatty acid degradation (fadE23, fadE24; Rv3139–3140), peroxidase activity (ahpC), transport (iniB, iniA; Rv0341–0342), efflux pump (efpA, Rv2846c), and unknown functions (Rv1592c, Rv1772) (106,108). Since genes that respond to drug treatment could be implicated in the mechanisms of resistance to the drug, Ramaswany and colleagues sequenced all the genes induced by INH except kasB, fadE23, and acpM in 38 INH-resistant and 86 INH-sensitive clinical isolates (88). The kasA operon is composed of fabD, acpM, kasA, kasB, and accD6. Rv2242, an srmR homolog, is located just upstream of the kasA operon and was added to this study. Mutations in kasA, fadE24, Rv1592c, and Rv2242 were found in INH-resistant strains carrying katG mutations or in INH-sensitive strains. No mutations in fbpC, fabD, accD6, or efpA were found in INH-resistant clinical isolates. One T4A mutation was found in Rv1772 in a low-level INH-resistant clinical isolate, with no mutation in the other 19 genes sequenced. This mutation was not found in the INH-sensitive isolates. Another study found a mutation in efpA resulting in a variant (E520V) found in INH-resistant strains only but associated with katG and oxyR ‘-ahpC mutations. In conclusion, most of the mutations identified in these genes (except for Rv1772) in INH-resistant clinical isolates were found either in INH-sensitive strains or in combination with other mutations known to confer INH resistance. Their roles in INH resistance cannot be assessed at this point.
KasA is a beta-ketoacyl-ACP synthase that condenses an elongating fatty acyl-ACP with malonyl-ACP and is the first enzyme in the FASII system. KasA was once considered a more likely target of INH than InhA based on the fact that INH treatment of M. tuberculosis resulted in the inhibition of mycolic acid biosynthesis and accumulation of the long-chain fatty acid hexacosanoic acid. The accumulation of fatty acids correlated better with the inhibition of a beta-ketoacyl-ACP synthase (KasA) than with the inhibition of an enoyl-ACP reductase (InhA), which should result in the accumulation of enoyl products (unsaturated fatty acids) (109). This biochemical red herring, however, did not consider the work of Takayama and coworkers, who demonstrated that a short exposure (5 min) of M. tuberculosis to INH resulted in the accumulation of unsaturated fatty acids, while longer INH exposures led to accumulation of hexacosanoic acid due to the total shutdown of the FASII system (26). Mdluli et al. demonstrated that in INH-treated M. tuberculosis, KasA formed a complex with INH and the acyl carrier protein AcpM and identified four variants in KasA (D66N, G269S, G312S, F413L) in INH-resistant clinical isolates (109). The authors thus concluded that KasA was the main target of INH. This conclusion was disproved by the demonstration that the complex between KasA, INH, and AcpM was formed only when InhA was inhibited (110) and by the discovery of the KasA variants D66N, G269S, and G312S in INH-sensitive strains (66). Other kasA mutations have been identified in INH-resistant M. tuberculosis clinical isolates; however, these mutations are usually associated with katG mutations.
The INH-inducible gene iniA was shown to confer tolerance to INH when overexpressed in M. bovis BCG but not in M. tuberculosis, while deletion of iniA rendered M. tuberculosis more susceptible to INH (111). iniA is in an operon with the INH-inducible genes iniB and iniC. Immediately upstream of this operon is Rv0340, which is transcribed in the same direction. This operon is induced specifically by drugs inhibiting mycobacterial cell wall biosynthesis such as INH and ethambutol (112). The iniBAC operon encodes a membrane transporter, but it was shown not to transport INH (111). All the mutations in this cluster (Table 1) were only present in INH-resistant clinical isolates; however, they were always associated with mutations in katG and/or inhA. Hence, mutations in the iniBAC operon may only have a minor role in INH resistance.
Interestingly, the target of INH and ETH, inhA, is not upregulated upon INH or ETH treatment of M. tuberculosis, which may be an important determinant of successful drug targets.
Overexpression of NAD+/NADP+-binding enzymes
The INH-sensitive E. coli oxyR mutant described above was also used to identify other molecular determinants of INH resistance in M. tuberculosis. The E. coli mutant was transformed with an M. tuberculosis genomic plasmid library and screened for clones that became INH-resistant. Three genes, glf (ceoA), ceoB, and ceoC, restored INH resistance in E. coli. Each gene contains an NAD+ binding motif. Only overexpression of M. tuberculosis glf (Rv3809c), an NAD+- and flavin adenine dinucleotide-dependent UDP galactopyranose mutase, led to low-level INH resistance in BCG. However, a binding experiment with radioactive INH showed that Glf did not bind to INH. The authors concluded that upregulation of NAD+-binding proteins might play a role in INH resistance by either reducing the levels of NAD+ available for the formation of the INH-NAD adduct or by binding an unknown derivative of INH (113).
The finding that the NADPH-dependent β-ketoacyl-ACP reductase FabG (Rv1483), part of the FASII system, was inhibited in vitro by an INH-NADP adduct similarly to InhA (114) led others to investigate whether additional enzymes could be inhibited by this INH- NADP adduct. The enzymatic activity of the M. tuberculosis dihydrofolate reductase DHFR (Rv2763c), the target of the broad-spectrum antibiotic trimethoprim, was shown to be inhibited by the 4R-INH-NADP adduct with subnanomolar affinity (115). Overexpression of M. tuberculosis dfrA in M. smegmatis increased INH resistance by 2-fold at 30°C (115). However, overexpression of M. tuberculosis dfrA in M. tuberculosis did not increase resistance to INH (116), and the sequences of 127 INH-resistant clinical isolates revealed no mutation in dfrA, suggesting that DHFR is not a marker for INH resistance in M. tuberculosis (117). Affinity chromatography identified 16 other proteins that could bind to the INH-NAD or INH-NADP adduct (118), but overexpression of the M. tuberculosis genes encoding these proteins did not confer resistance to INH or ETH in M. smegmatis (116).
Overexpression of efflux pumps
Active export of drugs from cells by efflux pumps was first described in E. coli for tetracycline (119). In mycobacteria, low-level resistance to tetracycline and other aminoglycosides has been attributed to efflux pumps (120, 121). iniA, a gene induced by INH treatment (106), was shown to be a component of an MDR-like efflux pump. Overexpression of iniA conferred tolerance to INH, while deletion of iniA increased the susceptibility of mycobacteria to INH (111). However, IniA does not pump INH out of the cells.
A microarray study of M. tuberculosis clinical isolates resistant to INH, RIF, SM, ethambutol, and ofloxacin revealed that upon INH treatment, expression of several predicted efflux pump genes was upregulated: Rv1819c, Rv2459, Rv2846, Rv3065, and Rv3728 (122). Furthermore, administration of an efflux pump inhibitor decreased the MIC for INH in these strains by at least 4-fold, suggesting that these efflux pumps may play a role in drug resistance in M. tuberculosis.
Rv1217c-1218c, an operon encoding an ATP binding cassette transporter, was shown by RT-qPCR to be overexpressed in MDR- and XDR-TB strains; the overexpression of Rv1218C was associated with increased INH resistance (123). However, deletion of Rv1218c did not affect the MIC of INH in M. tuberculosis (124), nor did deletion of other efflux pumps such as Rv1877, mmr, and mmpL7 (125,126), although overexpression of mmpL7 conferred resistance to INH (127). Conversely, deletion of lfrA led to a 2-fold decrease in the INH MIC (126). Unexpectedly, deletion of efpA, which encodes an efflux pump that is specifically induced upon INH treatment of M. tuberculosis, resulted in increased resistance to INH and RIF in M. smegmatis (126).
In light of these studies, the role of efflux pumps in INH resistance in M. tuberculosis needs to be further evaluated in order to assess their importance in the resistance mechanisms.
Mechanisms of Resistance to ETH Only
Alterations of EthA, the activator of ETH
The mechanisms of resistance to ETH are mutations in genes encoding its activator (ethA), its target (inhA), or the ethA regulator (ethR). So far, 85 ethA mutations have been identified, although some were also found in drug-susceptible or partially ETH-resistant M. tuberculosis strains (Table 2). Mutations have been identified throughout the length of the coding region. Approximately two-thirds of the nucleotide changes are missense mutations that result in amino acid changes, while the remaining mutations are insertions, deletions, or nonsense mutations. Unlike the katG(S315T) variant, which can be present in up to 94% of the INH-resistant clinical isolates, no dominant ethA mutation occurs in ETH-resistant clinical isolates. Morlock and colleagues (61) hypothesized that the lack of cluster or dominant ethA mutations in ETH-resistant clinical isolates could be attributed to the presence of numerous monooxygenase homologs in M. tuberculosis that could protect the cells against a loss of EthA activity.
In a study by Brossier and colleagues (62), 47 ETH-resistant clinical isolates were analyzed for mutations in ethA, ethR, inhA, ndh, or mshA. Of these, 22 clinical isolates (47%) had mutations in ethA, while 29 strains (62%) had a mutation in inhA (promoter region/gene). On average, the proportion of inhA mutations in ETH-resistant clinical isolates is 68%, suggesting that this is the main mechanism of ETH resistance in M. tuberculosis.
Mutations in ethR, the regulator of ethA
ethA is negatively regulated by EthR (Rv3855), a transcriptional repressor belonging to the TetR family. The two genes are oriented opposite to each other, separated by a 73-bp intergenic region that contains the ethA promoter and to which EthR binds. A strain with a transposon insertion in ethR was highly sensitive to ETH, while ethR overexpression increased the resistance to ETH (45). Two mutations in ethR have been identified (Table 2) in highly ETH-resistant clinical strains, which represent 4% of ETH-resistant clinical strains screened; however, these two strains also contained the c-15t mutation in the promoter region of inhA, and one of the two had a mutation in ethA as well (62). Since the majority of ETH-resistant clinical isolates have mutations in ethA and/or inhA, ethR might only play a minor role in ETH resistance in M. tuberculosis clinical isolates.
CONCLUSIONS AND THE FUTURE
The mechanisms of resistance to INH and ETH in M. tuberculosis are both simple and complex (Fig. 3). The main mechanisms of resistance are mutations in katG and ethA, the activators of INH and ETH, respectively, preventing the formation of the INH-NAD or ETH-NAD adduct, and mutations in inhA, the target of INH and ETH, leading to titration of the drug or reduced binding of the INH-NAD or ETH-NAD adduct to InhA. Numerous mutations in multiple genes have been identified in INH- or ETH-resistant M. tuberculosis clinical isolates (Tables 1 to 3). Ultimately, the validation of a mutation responsible for INH or ETH resistance requires transferring the point mutation into wild-type M. tuberculosis and measuring the level of resistance. This is far beyond the scope of most studies that describe mutations associated with drug resistance in M. tuberculosis.
FIGURE 3.

Relationship among the genes and proteins involved in the resistance to INH and ETH in M. tuberculosis. Connections in red indicate a negative relationship (degradation of an active molecule, negative regulator of an enzyme) that would lead to resistance to INH and/or ETH; in green are positive actions that would increase a strain fitness or susceptibility to the drugs. The dashed line points to an interaction that does not result directly in INH resistance or susceptibility. doi:10.1128/microbiolspec.MGM2-0014-2013.f3
It is often reported that a certain percentage (up to 30%) of INH-resistant M. tuberculosis clinical isolates have no mutation in any of the genes studied, leading some to conclude that there is still much more to discover about INH and ETH mode of action and resistance. Yet most studies sequenced only a small fraction of the genes known to confer INH resistance such as katG and inhA, looking only at the region around codon 315 of katG or the regulatory region of inhA. However, more than 300 mutations have been identified in katG from amino acids 1 to 735 (Table 3), and mutations outside of the S315 region have also been shown to be highly defective in INH activation. Sequencing only a fraction of the katG gene around the S315 region might leave mutations responsible for the drug resistance phenotype undiscovered. Nevertheless, in studies where the katG and inhA genes and their regulatory regions are entirely sequenced, the mechanism of resistance is not identified in up to 5% of the INH-resistant clinical isolates.
Additional mechanisms of resistance likely remain to be identified, and a worthwhile endeavor may be to revisit the early studies on INH mechanisms of action to determine other factors of INH and ETH resistance in M. tuberculosis. Knowledge of resistance genes and mutations has provided the means to develop GenoType MTBDRplus, a rapid nucleic acid–based test for assessing INH and RIF resistance (128). Moreover, it may provide strategies to develop new drugs that bypass the known mechanisms of drug resistance. For example, the natural product pyridomycin inhibits InhA without requiring KatG activation and is therefore active against highly INH-resistant M. tuberculosis clinical isolates carrying a katG mutation (129). The identification of additional genes contributing to INH and ETH resistance will also expand our understanding of the mechanisms of drug action. Finally, in addition to specific mutations that confer resistance to every cell in a population, new studies that reveal the way in which a cell can become transiently phenotypically resistant to INH or ETH will be important in developing better ways to kill M. tuberculosis with INH and ETH and shorten chemotherapy.
REFERENCES
- 1.Behnisch R, Mietzsch F, Schmidt H. 1950. Chemical studies on thiosemicarbazones with particular reference to antituberculous activity. Am Rev Tuberc 61:1–7. [DOI] [PubMed] [Google Scholar]
- 2.Domagk G 1950. Investigations on the antituberculous activity of the thiosemicarbazones in vitro and in vivo. Am Rev Tuberc 61:8–19. [DOI] [PubMed] [Google Scholar]
- 3.Schatz A, Waksman SA. 1944. Effect of streptomycin upon Mycobacterium tuberculosis and related organisms. Proc Soc Exp Biol Med 57:244–248. [Google Scholar]
- 4.Lehmann J 1946. para-Aminosalicylic acid in the treatment of tuber-culosis. Lancet 247:15. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein JW, Lott A, Steinberg BA, Yale HL. 1952. Chemotherapy of experimental tuberculosis. Am Rev Tuberc 65:357–374. [DOI] [PubMed] [Google Scholar]
- 6.Fox HH. 1952. The chemical approach to the control of tuberculosis. Science 116:129–134. [DOI] [PubMed] [Google Scholar]
- 7.Medical Research Council Investigation. 1950. Treatment of pulmonary tuberculosis with streptomycin and para-amino-salicylic acid. Br MedJ 2:1073–1085. [PMC free article] [PubMed] [Google Scholar]
- 8.Medical Research Council Investigation. 1952. The treatment of pulmonary tuberculosis with isoniazid. Br Med J 2:735–746. [PMC free article] [PubMed] [Google Scholar]
- 9.Crofton J, Mitchison DA. 1948. Streptomycin resistance in pulmonary tuberculosis. Br Med J 2:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crofton J 1959. Chemotherapy of pulmonary tuberculosis. Br Med J 1:1610–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.WHO. 2012. Global Tuberculosis Report 2012. Geneva, Switzerland.
- 12.Cegielski P, Nunn P, Kurbatova EV, Weyer K, Dalton TL, Wares DF, Iademarco MF, Castro KG, Raviglione M. 2012. Challenges and controversies in defining totally drug-resistant tuberculosis. Emerg Infect Dis 18:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domagk G, Offe HA, Siefken W. 1952. [Additional investigations in experimental chemotherapy of tuberculosis (neoteban)]. Dtsch Med Wochenschr 77:573–578. [DOI] [PubMed] [Google Scholar]
- 14.Chorine V 1945. Action of nicotinamide on bacilli of the species Mycobacterium. Compt Ren 220:150–151. [Google Scholar]
- 15.Barclay WR, Ebert RH, Kochweser D. 1953. Mode of action of isoniazid. Am Rev Tuberc 67:490–496. [DOI] [PubMed] [Google Scholar]
- 16.Pope H 1953. Antagonism of isoniazid by certain metabolites. Am Rev Tuberc 68:938–939. [DOI] [PubMed] [Google Scholar]
- 17.Russe HP, Barclay WR. 1955. The effect of isoniazid on lipids of the tubercle bacillus. Am Rev Tuberc 72:713–717. [DOI] [PubMed] [Google Scholar]
- 18.Ebina T, Motomiya M, Munakata K, Kobuya G. 1961. Effect of isoniazid on fatty acids in Mycobacterium. CR Seances Soc Biol Fil 155:1176–1178. [PubMed] [Google Scholar]
- 19.Gangadharam PRJ, Harold FM, Schaefer W. 1963. Selective inhibition of nucleic acid synthesis in Mycobacterium tuberculosis by isoniazid. Nature 198:712–714. [DOI] [PubMed] [Google Scholar]
- 20.Winder FG, Brennan P. 1965. Effect of isoniazid on lipid metabolism in Mycobacterium tuberculosis. Biochem J 96:77P. [Google Scholar]
- 21.Bekierkunst A 1966. Nicotinamide-adenine dinucleotide in tubercle bacilli exposed to isoniazid. Science 152:525–526. [DOI] [PubMed] [Google Scholar]
- 22.Davis WE, Weber MM. 1977. Specificity of isoniazid on growth inhibition and competition for an oxidized nicotinamide adenine dinucleotide regulatory site on the electron transport pathway in Mycobacterium phlei. Antimicrob Agents Chemother 12:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herman RP, Weber MM. 1980. Site of action of isoniazid on the electron transport chain and its relationship to nicotinamide adenine dinucleotide regulation in Mycobacterium phlei. Antimicrob Agents Chemother 17:450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takayama K, Wang L, David HL. 1972. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takayama K, Schnoes HK, Armstrong EL, Boyle RW. 1975. Site of inhibitory action of isoniazid in the synthesis of mycolic acids in Mycobacterium tuberculosis. J Lipid Res 16:308–317. [PubMed] [Google Scholar]
- 26.Davidson LA, Takayama K. 1979. Isoniazid inhibition of the synthesis of monounsaturated long-chain fatty acids in Mycobacterium tuberculosis H37Ra. Antimicrob Agents Chemother 16:104–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnsson K, Schultz PG. 1994. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J Am Chem Soc 116:7425–7426. [Google Scholar]
- 28.Lei B, Wei CJ, Tu SC. 2000. Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of inhA inhibitor. J Biol Chem 275:2520–2526. [DOI] [PubMed] [Google Scholar]
- 29.Wilming M, Johnsson K. 1999. Spontaneous formation of the bio-active form of the tuberculosis drug isoniazid. Angew Chem Int Ed Engl 38:2588–2590. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Heym B, Allen B, Young D, Cole S. 1992. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358:591–593. [DOI] [PubMed] [Google Scholar]
- 31.Rozwarski DA, Grant GA, Barton DH, Jacobs WR Jr, Sacchettini JC. 1998. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279:98–102. [DOI] [PubMed] [Google Scholar]
- 32.Dessen A, Quemard A, Blanchard JS, Jacobs WR Jr, Sacchettini JC. 1995. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267:1638–1641. [DOI] [PubMed] [Google Scholar]
- 33.Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR Jr Blanchard JS. 1995. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 34:8235–8241. [DOI] [PubMed] [Google Scholar]
- 34.Marrakchi H, Laneelle G, Quemard A. 2000. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 146(Pt 2):289–296. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen M, Quemard A, Broussy S, Bernadou J, Meunier B. 2002. Mn(III) pyrophosphate as an efficient tool for studying the mode of action of isoniazid on the InhA protein of Mycobacterium tuberculosis. Antimicrob Agents Chemother 46:2137–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rawat R, Whitty A, Tonge PJ. 2003. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA 100:13881–13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilcheze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, Jacobs WR Jr. 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J Bacteriol 182:4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR Jr. 2006. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med 12:1027–1029. [DOI] [PubMed] [Google Scholar]
- 39.Grumbach F, Rist N, Libermann D, Moyeux M, Cals S, Clavel S. 1956. Experimental antituberculous activity of certain isonicotinic thioamides substituted on the nucleus. CR Hebd Seances Acad Sci 242:2187–2189. (In French.) [PubMed] [Google Scholar]
- 40.Brouet G, Marche J, Rist N, Chevallier J, Lemeur G. 1959. Observations on the antituberculous effectiveness of alpha-ethyl-thioisonicotinamide in tuberculosis in humans. Am Rev Tuberc 79:6–18. [DOI] [PubMed] [Google Scholar]
- 41.Petty TL, Mitchell RS. 1962. Successful treatment of advanced isonizid-and streptomycin-resistant pulmonary tuberculosis with ethionamide, pyrazinamide, and isoniazid. Am Rev Respir Dis 86:503–512. [DOI] [PubMed] [Google Scholar]
- 42.Middlebrook G 1952. Sterilization of tubercle bacilli by isonicotinic acid hydrazide and the incidence of variants resistant to the drug in vitro. Am Rev Tuberc 65:765–767. [PubMed] [Google Scholar]
- 43.Schaefer WB. 1954. The effect of isoniazid on growing and resting tubercle bacilli. Am Rev Tuberc 69:125–127. [DOI] [PubMed] [Google Scholar]
- 44.Winder FG, Collins PB, Whelan D. 1971. Effects of ethionamide and isoxyl on mycolic acid synthesis in Mycobacterium tuberculosis BCG. J Gen Microbiol 66:379–380. [DOI] [PubMed] [Google Scholar]
- 45.Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, Locht C, Besra GS. 2000. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem 275:28326–28331. [DOI] [PubMed] [Google Scholar]
- 46.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE 3rd. 2000. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA 97:9677–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fraaije MW, Kamerbeek NM, Heidekamp AJ, Fortin R, Janssen DB.2004. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J Biol Chem 279:3354–3360. [DOI] [PubMed] [Google Scholar]
- 48.Vannelli TA, Dykman A, Ortiz de Montellano PR. 2002. The anti-tuberculosis drug ethionamide is activated by a flavoprotein mono-oxygenase. J Biol Chem 277:12824–12829. [DOI] [PubMed] [Google Scholar]
- 49.Wang F, Langley R, Gulten G, Dover LG, Besra GS, Jacobs WR Jr, Sacchettini JC. 2007. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med 204:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dover LG, Alahari A, Gratraud P, Gomes JM, Bhowruth V, Reynolds RC, Besra GS, Kremer L. 2007. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrob Agents Chemother 51:1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hok TT. 1964. A comparative study of the susceptibility to ethionamide, thiosemicarbazone, and isoniazid of tubercle bacilli from patients never treated with ethionamide or thiosemicarbazone. Am Rev Respir Dis 90:468–469. [DOI] [PubMed] [Google Scholar]
- 52.Stewart SM, Hall E, Riddell RW, Somner AR. 1962. Bacteriological aspects of the use of ethionamide, pyrazinamide and cycloserine in the treatment of chronic pulmonary tuberculosis. Tubercle 43:417–431. [DOI] [PubMed] [Google Scholar]
- 53.Lefford MJ. 1966. The ethionamide sensitivity of British pre-treatment strains of Mycobacterium tuberculosis. Tubercle 47:198–206. [DOI] [PubMed] [Google Scholar]
- 54.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR Jr. 1988. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc Natl Acad Sci USA 85:6987–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4:1911–1919. [DOI] [PubMed] [Google Scholar]
- 56.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR Jr. 1994. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263:227–230. [DOI] [PubMed] [Google Scholar]
- 57.Takayama K, Wang L, Merkal RS. 1973. Scanning electron microscopy of the H37Ra strain of Mycobacterium tuberculosis exposed to isoniazid. Antimicrob Agents Chemother 4:62–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dias MV, Vasconcelos IB, Prado AM, Fadel V, Basso LA, de Azevedo WF Jr Santos DS. 2007. Crystallographic studies on the binding of isonicotinyl-NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. J Struct Biol 159:369–380. [DOI] [PubMed] [Google Scholar]
- 59.Brossier F, Veziris N, Truffot-Pernot C, Jarlier V, Sougakoff W. 2006. Performance of the genotype MTBDR line probe assay for detection of resistance to rifampin and isoniazid in strains of Mycobacterium tuberculosis with low- and high-level resistance. J Clin Microbiol 44:3659–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller B, Streicher EM, Hoek KG, Tait M, Trollip A, Bosman ME, Coetzee GJ, Chabula-Nxiweni EM, Hoosain E, Gey van Pittius NC, Victor TC, van Helden PD, Warren RM. 2011. inhA promoter mutations: a gateway to extensively drug-resistant tuberculosis in South Africa? Int J Tuberc Lung Dis 15:344–351. [PubMed] [Google Scholar]
- 61.Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. 2003. ethA, inhA, and katG loci of ethionamide-resistant clinical Myco-bacterium tuberculosis isolates. Antimicrob Agents Chemother 47:3799–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brossier F, Veziris N, Truffot-Pernot C, Jarlier V, Sougakoff W. 2011. Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 55:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vadwai V, Ajbani K, Jose M, Vineeth VP, Nikam C, Deshmukh M, Shetty A, Soman R, Rodrigues C. 2012. Can inhA mutation predict ethionamide resistance? Int J Tuberc Lung Dis 17:129–130. [DOI] [PubMed] [Google Scholar]
- 64.Miesel L, Weisbrod TR, Marcinkeviciene JA, Bittman R, Jacobs WR Jr. 1998. NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J Bacteriol 180:2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vilcheze C, Weisbrod TR, Chen B, Kremer L, Hazbon MH, Wang F, Alland D, Sacchettini JC, Jacobs WR Jr. 2005. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother 49:708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, Garcia-Garcia L, Leon CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de Leon A, Alland D. 2006. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 50:2640–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cardoso RF, Cardoso MA, Leite CQ, Sato DN, Mamizuka EM, Hirata RD, de Mello FF, Hirata MH. 2007. Characterization of ndh gene of isoniazid resistant and susceptible Mycobacterium tuberculosis isolates from Brazil. Mem Inst Oswaldo Cruz 102:59–61. [DOI] [PubMed] [Google Scholar]
- 68.Lee AS, Teo AS, Wong SY. 2001. Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 45:2157–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee AS, Lim IH, Tang LL, Telenti A, Wong SY. 1999. Contribution of kasA analysis to detection of isoniazid-resistant Mycobacterium tuberculosis in Singapore. Antimicrob Agents Chemother 43:2087–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vilcheze C, Av-Gay Y, Attarian R, Liu Z, Hazbon MH, Colangeli R, Chen B, Liu W, Alland D, Sacchettini JC, Jacobs WR Jr. 2008. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol 69:1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vilcheze C, Av-Gay Y, Barnes SW, Larsen MH, Walker JR, Glynne RJ, Jacobs WR Jr. 2011. Coresistance to isoniazid and ethionamide maps to mycothiol biosynthetic genes in Mycobacterium bovis. Antimicrob Agents Chemother 55:4422–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Newton GL, Buchmeier N, Fahey RC. 2008. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol Mol Biol Rev 72:471–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu X, Vilcheze C, Av-Gay Y, Gomez-Velasco A, Jacobs WR Jr. 2011. Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 55:3133–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rawat M, Johnson C, Cadiz V, Av-Gay Y. 2007. Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem Biophys Res Commun 363:71–76. [DOI] [PubMed] [Google Scholar]
- 75.Projahn M, Koser CU, Homolka S, Summers DK, Archer JA, Niemann S. 2011. Polymorphisms in isoniazid and prothionamide resistance genes of the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother 55:4408–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang XD, Gu J, Wang T, Bi LJ, Zhang ZP, Cui ZQ, Wei HP, Deng JY, Zhang XE. 2011. Comparative analysis of mycobacterial NADH pyrophosphatase isoforms reveals a novel mechanism for isoniazid and ethionamide inactivation. Mol Microbiol 82:1375–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Middlebrook G 1954. Isoniazid resistance and catalase activity of tubercle bacilli. Am Rev Tuberc 69:471–472. [DOI] [PubMed] [Google Scholar]
- 78.Middlebrook G, Cohn ML. 1953. Some observations on the pathogenicity of isoniazid-resistant variants of tubercle bacilli. Science 118:297–299. [DOI] [PubMed] [Google Scholar]
- 79.Winder F 1960. Catalase and peroxidase in mycobacteria. Possible relationship to the mode of action of isoniazid. Am Rev Respir Dis 81: 68–78. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Garbe T, Young D. 1993. Transformation with katG restores isoniazid-sensitivity in Mycobacterium tuberculosis isolates resistant to a range of drug concentrations. Mol Microbiol 8:521–524. [DOI] [PubMed] [Google Scholar]
- 81.Mokrousov I, Narvskaya O, Otten T, Limeschenko E, Steklova L, Vyshnevskiy B. 2002. High prevalence of KatG Ser315Thr substitution among isoniazid-resistant Mycobacterium tuberculosis clinical isolates from northwestern Russia, 1996 to 2001. Antimicrob Agents Chemother 46:1417–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wengenack NL, Uhl JR, St Amand AL, Tomlinson AJ, Benson LM, Naylor S, Kline BC, Cockerill FR 3rd, Rusnak F. 1997. Recombinant Mycobacterium tuberculosis KatG(S315T) is a competent catalase-peroxidase with reduced activity toward isoniazid. J Infect Dis 176:722–727. [DOI] [PubMed] [Google Scholar]
- 83.Saint-Joanis B, Souchon H, Wilming M, Johnsson K, Alzari PM, Cole ST. 1999. Use of site-directed mutagenesis to probe the structure, function and isoniazid activation of the catalase/peroxidase, KatG, from Myco-bacterium tuberculosis. Biochem J 338(Pt 3):753–760. [PMC free article] [PubMed] [Google Scholar]
- 84.Ramaswamy S, Musser JM. 1998. Molecular genetic basis of anti-microbial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis 79:3–29. [DOI] [PubMed] [Google Scholar]
- 85.van Doorn HR, Kuijper EJ, van der Ende A, Welten AG, van Soolingen D, de Haas PE, Dankert J. 2001. The susceptibility of Mycobacterium tuberculosis to isoniazid and the Arg→Leu mutation at codon 463 of katG are not associated. J Clin Microbiol 39:1591–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Soolingen D, de Haas PE, van Doorn HR, Kuijper E, Rinder H, Borgdorff MW. 2000. Mutations at amino acid position 315 of the katG gene are associated with high-level resistance to isoniazid, other drug resistance, and successful transmission of Mycobacterium tuberculosis in the Netherlands. J Infect Dis 182:1788–1790. [DOI] [PubMed] [Google Scholar]
- 87.Wei CJ, Lei B, Musser JM, Tu SC. 2003. Isoniazid activation defects in recombinant Mycobacterium tuberculosis catalase-peroxidase (KatG) mutants evident in InhA inhibitor production. Antimicrob Agents Chemother 47:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramaswamy SV, Reich R, Dou SJ, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA. 2003. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 47:1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cohn ML, Oda U, Kovitz C, Middlebrook G. 1954. Studies on isoniazid and tubercle bacilli. I. The isolation of isoniazid-resistant mutants in vitro. Am Rev Tuberc 70:465–475. [DOI] [PubMed] [Google Scholar]
- 90.Cohn ML, Kovitz C, Oda U, Middlebrook G. 1954. Studies on isoniazid and tubercle bacilli. II. The growth requirements, catalase activities, and pathogenic properties of isoniazid-resistant mutants. Am Rev Tuberc 70:641–664. [DOI] [PubMed] [Google Scholar]
- 91.Pym AS, Saint-Joanis B, Cole ST. 2002. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect Immun 70:4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gagneux S, Burgos MV, DeRiemer K, Encisco A, Munoz S, Hopewell PC, Small PM, Pym AS. 2006. Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathog 2:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Escalante P, McKean-Cowdin R, Ramaswamy SV, Williams-Bouyer N, Teeter LD, Jones B, Graviss EA. 2013. Can mycobacterial katG genetic changes in isoniazid-resistant tuberculosis influence human disease features? Int J Tuberc Lung Dis 17:641–51. [DOI] [PubMed] [Google Scholar]
- 94.Pym AS, Domenech P, Honore N, Song J, Deretic V, Cole ST. 2001. Regulation of catalase-peroxidase (KatG) expression, isoniazid sensitivity and virulence by furA of Mycobacterium tuberculosis. Mol Microbiol 40:879–889. [DOI] [PubMed] [Google Scholar]
- 95.Zahrt TC, Song J, Siple J, Deretic V. 2001. Mycobacterial FurA is a negative regulator of catalase-peroxidase gene katG. Mol Microbiol 39:1174–1185. [DOI] [PubMed] [Google Scholar]
- 96.Ando H, Kitao T, Miyoshi-Akiyama T, Kato S, Mori T, Kirikae T. 2011. Downregulation of katG expression is associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 79:1615–1628. [DOI] [PubMed] [Google Scholar]
- 97.Lee JH, Ammerman NC, Nolan S, Geiman DE, Lun S, Guo H, Bishai WR. 2012. Isoniazid resistance without a loss of fitness in Mycobacterium tuberculosis. Nat Commun 3:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rosner JL. 1993. Susceptibilities of oxyR regulon mutants of Escherichia coli and Salmonella typhimurium to isoniazid. Antimicrob Agents Chemother 37:2251–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sreevatsan S, Pan X, Zhang Y, Deretic V, Musser JM. 1997. Analysis of the oxyR-ahpC region in isoniazid-resistant and -susceptible Myco-bacterium tuberculosis complex organisms recovered from diseased humans and animals in diverse localities. Antimicrob Agents Chemother 41:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE 3rd, Stover CK. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641–1643. [DOI] [PubMed] [Google Scholar]
- 101.Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. 2004. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol 52:1291–1302. [DOI] [PubMed] [Google Scholar]
- 102.Baker LV, Brown TJ, Maxwell O, Gibson AL, Fang Z, Yates MD, Drobniewski FA. 2005. Molecular analysis of isoniazid-resistant Myco-bacterium tuberculosis isolates from England and Wales reveals the phylogenetic significance of the ahpC −46A polymorphism. Antimicrob Agents Chemother 49:1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Upton AM, Mushtaq A, Victor TC, Sampson SL, Sandy J, Smith DM, van Helden PV, Sim E. 2001. Arylamine N-acetyltransferase of Mycobacterium tuberculosis is a polymorphic enzyme and a site of isoniazid metabolism. Mol Microbiol 42:309–317. [DOI] [PubMed] [Google Scholar]
- 104.Payton M, Auty R, Delgoda R, Everett M, Sim E. 1999. Cloning and characterization of arylamine N-acetyltransferase genes from Mycobacterium smegmatis and Mycobacterium tuberculosis: increased expression results in isoniazid resistance. J Bacteriol 181:1343–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhakta S, Besra GS, Upton AM, Parish T, Sholto-Douglas-Vernon C, Gibson KJ, Knutton S, Gordon S, DaSilva RP, Anderton MC, Sim E. 2004. Arylamine N-acetyltransferase is required for synthesis of mycolic acids and complex lipids in Mycobacterium bovis BCG and represents a novel drug target. J Exp Med 199:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alland D, Kramnik I, Weisbrod TR, Otsubo L, Cerny R, Miller LP, Jacobs WR Jr, Bloom BR. 1998. Identification of differentially expressed mRNA in prokaryotic organisms by customized amplification libraries (DECAL): the effect of isoniazid on gene expression in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 95:13227–13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. [DOI] [PubMed] [Google Scholar]
- 108.Wilson M, DeRisi J, Kristensen HH, Imboden P, Rane S, Brown PO, Schoolnik GK. 1999. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci USA 96:12833–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mdluli K, Slayden RA, Zhu Y, Ramaswamy S, Pan X, Mead D, Crane DD, Musser JM, Barry CE 3rd. 1998. Inhibition of a Mycobacterium tuberculosis beta-ketoacyl ACP synthase by isoniazid. Science 280:1607–1610. [DOI] [PubMed] [Google Scholar]
- 110.Kremer L, Dover LG, Morbidoni HR, Vilcheze C, Maughan WN, Baulard A, Tu S, Honore N, Deretic V, Sacchettini JC, Locht C, Jacobs WRJ, Besra G. 2003. Inhibition of InhA activity, but not KasA activity, induces formation of a KasA-containing complex in mycobacteria. J Biol Chem 278:20547–20554. [DOI] [PubMed] [Google Scholar]
- 111.Colangeli R, Helb D, Sridharan S, Sun J, Varma-Basil M, Hazbon MH, Harbacheuski R, Megjugorac NJ, Jacobs WR Jr, Holzenburg A, Sacchettini JC, Alland D. 2005. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol Microbiol 55:1829–1840. [DOI] [PubMed] [Google Scholar]
- 112.Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR Jr. 2000. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J Bacteriol 182:1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen P, Bishai WR. 1998. Novel selection for isoniazid (INH) resistance genes supports a role for NAD+-binding proteins in mycobacterial INH resistance. Infect Immun 66:5099–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ducasse-Cabanot S, Cohen-Gonsaud M, Marrakchi H, Nguyen M, Zerbib D, Bernadou J, Daffe M, Labesse G, Quemard A. 2004. In vitro inhibition of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA by isoniazid. Antimicrob Agents Chemother 48:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Argyrou A, Vetting MW, Aladegbami B, Blanchard JS. 2006. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat Struct Mol Biol 13:408–413. [DOI] [PubMed] [Google Scholar]
- 116.Wang F, Jain P, Gulten G, Liu Z, Feng Y, Ganesula K, Motiwala AS, Ioerger TR, Alland D, Vilcheze C, Jacobs WR Jr, Sacchettini JC. 2010. Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob Agents Chemother 54:3776–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ho YM, Sun YJ, Wong SY, Lee AS. 2009. Contribution of dfrA and inhA mutations to the detection of isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 53:4010–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Argyrou A, Jin L, Siconilfi-Baez L, Angeletti RH, Blanchard JS. 2006. Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry 45:13947–13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McMurry L, Petrucci RE Jr, Levy SB. 1980. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc Natl Acad Sci USA 77:3974–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ainsa JA, Blokpoel MC, Otal I, Young DB, De Smet KA, Martin C. 1998. Molecular cloning and characterization of Tap, a putative multi-drug efflux pump present in Mycobacterium fortuitum and Mycobacterium tuberculosis. J Bacteriol 180:5836–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Silva PE, Bigi F, Santangelo MP, Romano MI, Martin C, Cataldi A, Ainsa JA. 2001. Characterization of P55, a multidrug efflux pump in Mycobacterium bovis and Mycobacterium tuberculosis. Antimicrob Agents Chemother 45:800–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gupta AK, Katoch VM, Chauhan DS, Sharma R, Singh M, Venkatesan K, Sharma VD. 2010. Microarray analysis of efflux pump genes in multidrug-resistant Mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb Drug Resist 16:21–28. [DOI] [PubMed] [Google Scholar]
- 123.Wang K, Pei H, Huang B, Zhu X, Zhang J, Zhou B, Zhu L, Zhang Y, Zhou FF. 2013. The expression of ABC efflux pump, Rv1217c-Rv1218c, and its association with multidrug resistance of Mycobacterium tuberculosis in China. Curr Microbiol 66:222–226. [DOI] [PubMed] [Google Scholar]
- 124.Balganesh M, Kuruppath S, Marcel N, Sharma S, Nair A, Sharma U. 2010. Rv1218c, an ABC transporter of Mycobacterium tuberculosis with implications in drug discovery. Antimicrob Agents Chemother 54:5167–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Domenech P, Reed MB, Barry CE 3rd. 2005. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun 73:3492–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li XZ, Zhang L, Nikaido H. 2004. Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 48:2415–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pasca MR, Guglierame P, De Rossi E, Zara F, Riccardi G. 2005. mmpL7 gene of Mycobacterium tuberculosis is responsible for isoniazid efflux in Mycobacterium smegmatis. Antimicrob Agents Chemother 49: 4775–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hillemann D, Rusch-Gerdes S, Richter E. 2007. Evaluation of the GenoType MTBDRplus assay for rifampin and isoniazid susceptibility testing of Mycobacterium tuberculosis strains and clinical specimens. J Clin Microbiol 45:2635–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hartkoorn RC, Sala C, Neres J, Pojer F, Magnet S, Mukherjee R, Uplekar S, Boy-Rottger S, Altmann KH, Cole ST. 2012. Towards a new tuberculosis drug: pyridomycin: nature’s isoniazid. EMBO Mol Med 4:1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Valvatne H, Syre H, Kross M, Stavrum R, Ti T, Phyu S, Grewal HM. 2009. Isoniazid and rifampicin resistance-associated mutations in Mycobacterium tuberculosis isolates from Yangon, Myanmar: implications for rapid molecular testing. J Antimicrob Chemother 64:694–701. [DOI] [PubMed] [Google Scholar]
- 131.Caws M, Duy PM, Tho DQ, Lan NT, Hoa DV, Farrar J. 2006. Mutations prevalent among rifampin- and isoniazid-resistant Mycobacterium tuberculosis isolates from a hospital in Vietnam. J Clin Microbiol 44:2333–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rindi L, Bianchi L, Tortoli E, Lari N, Bonanni D, Garzelli C. 2005. Mutations responsible for Mycobacterium tuberculosis isoniazid resistance in Italy. Int J Tuberc Lung Dis 9:94–97. [PubMed] [Google Scholar]
- 133.Huang WL, Chen HY, Kuo YM, Jou R. 2009. Performance assessment of the GenoType MTBDRplus test and DNA sequencing in detection of multidrug-resistant Mycobacterium tuberculosis. J Clin Microbiol 47:2520–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fenner L, Egger M, Bodmer T, Altpeter E, Zwahlen M, Jaton K, Pfyffer GE, Borrell S, Dubuis O, Bruderer T, Siegrist HH, Furrer H, Calmy A, Fehr J, Stalder JM, Ninet B, Bottger EC, Gagneux S. 2012. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:3047–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang M, Yue J, Yang YP, Zhang HM, Lei JQ, Jin RL, Zhang XL, Wang HH. 2005. Detection of mutations associated with isoniazid resistance in Mycobacterium tuberculosis isolates from China. J Clin Microbiol 43:5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luo T, Zhao M, Li X, Xu P, Gui X, Pickerill S, DeRiemer K, Mei J, Gao Q. 2010. Selection of mutations to detect multidrug-resistant Mycobacterium tuberculosis strains in Shanghai, China. Antimicrob Agents Chemother 54:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Doustdar F, Khosravi AD, Farnia P, Masjedi MR, Velayati AA. 2008. Molecular analysis of isoniazid resistance in different genotypes of Mycobacterium tuberculosis isolates from Iran. Microb Drug Resist 14: 273–279. [DOI] [PubMed] [Google Scholar]
- 138.Rahim Z, Nakajima C, Raqib R, Zaman K, Endtz HP, van der Zanden AG, Suzuki Y. 2012. Molecular mechanism of rifampicin and isoniazid resistance in Mycobacterium tuberculosis from Bangladesh. Tuberculosis (Edinb) 92:529–534. [DOI] [PubMed] [Google Scholar]
- 139.Cardoso RF, Cooksey RC, Morlock GP, Barco P, Cecon L, Forestiero F, Leite CQ, Sato DN, Shikama Mde L, Mamizuka EM, Hirata RD, Hirata MH. 2004. Screening and characterization of mutations in isoniazid-resistant Mycobacterium tuberculosis isolates obtained in Brazil. Antimicrob Agents Chemother 48:3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kiepiela P, Bishop KS, Smith AN, Roux L, York DF. 2000. Genomic mutations in the katG, inhA and aphC genes are useful for the prediction of isoniazid resistance in Mycobacterium tuberculosis isolates from Kwazulu Natal, South Africa. Tuber Lung Dis 80:47–56. [DOI] [PubMed] [Google Scholar]
- 141.Rouse DA, Li Z, Bai GH, Morris SL. 1995. Characterization of the katG and inhA genes of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 39:2472–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo H, Seet Q, Denkin S, Parsons L, Zhang Y. 2006. Molecular characterization of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis from the USA. J Med Microbiol 55:1527–1531. [DOI] [PubMed] [Google Scholar]
- 143.Lavender C, Globan M, Sievers A, Billman-Jacobe H, Fyfe J. 2005. Molecular characterization of isoniazid-resistant Mycobacterium tuberculosis isolates collected in Australia. Antimicrob Agents Chemother 49:4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Silva MS, Senna SG, Ribeiro MO, Valim AR, Telles MA, Kritski A, Morlock GP, Cooksey RC, Zaha A, Rossetti ML. 2003. Mutations in katG, inhA, and ahpC genes of Brazilian isoniazid-resistant isolates of Mycobacterium tuberculosis. J Clin Microbiol 41:4471–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Poudel A, Nakajima C, Fukushima Y, Suzuki H, Pandey BD, Maharjan B, Suzuki Y. 2012. Molecular characterization of multidrug-resistant Mycobacterium tuberculosis isolated in Nepal. Antimicrob Agents Chemother 56:2831–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sekiguchi J, Miyoshi-Akiyama T, Augustynowicz-Kopec E, Zwolska Z, Kirikae F, Toyota E, Kobayashi I, Morita K, Kudo K, Kato S, Kuratsuji T, Mori T, Kirikae T. 2007. Detection of multidrug resistance in Mycobacterium tuberculosis. J Clin Microbiol 45:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.N’Guessan KR, Dosso M, Ekaza E, Kouakou J, Jarlier V. 2008. Molecular characterisation of isoniazid-resistant Mycobacterium tuberculosis isolated from new cases in Lagunes region (Cote d’Ivoire). Int J Antimicrob Agents 31:498–500. [DOI] [PubMed] [Google Scholar]
- 148.Lin HL, Kim H, Yun Y, Park CG, Kim B, Park Y, Kook Y. 2007. Mutations of katG and inhA in MDR M. tuberculosis.Tuberc Respir Dis 63:128–138. [Google Scholar]
- 149.Basso LA, Zheng R, Musser JM, Jacobs WR Jr, Blanchard JS. 1998. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis: enzymatic characterization of enoyl reductase mutants identified in isoniazid-resistant clinical isolates. J Infect Dis 178:769–775. [DOI] [PubMed] [Google Scholar]
- 150.Sholto-Douglas-Vernon C, Sandy J, Victor TC, Sim E, Helden PD. 2005. Mutational and expression analysis of tbnat and its response to isoniazid. J Med Microbiol 54:1189–1197. [DOI] [PubMed] [Google Scholar]
- 151.Ristow M, Mohlig M, Rifai M, Schatz H, Feldmann K, Pfeiffer A. 1995. New isoniazid/ethionamide resistance gene mutation and screening for multidrug-resistant Mycobacterium tuberculosis strains. Lancet 346:502–503. [DOI] [PubMed] [Google Scholar]
- 152.Yoon JH, Nam JS, Kim KJ, Choi Y, Lee H, Cho SN, Ro YT. 2012. Molecular characterization of drug-resistant and -susceptible Mycobacterium tuberculosis isolated from patients with tuberculosis in Korea. Diagn Microbiol Infect Dis 72:52–61. [DOI] [PubMed] [Google Scholar]
- 153.Chan RC, Hui M, Chan EW, Au TK, Chin ML, Yip CK, AuYeang CK, Yeung CY, Kam KM, Yip PC, Cheng AF. 2007. Genetic and phenotypic characterization of drug-resistant Mycobacterium tuberculosis isolates in Hong Kong. J Antimicrob Chemother 59:866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leung KL, Yip CW, Yeung YL, Wong KL, Chan WY, Chan MY, Kam KM. 2010. Usefulness of resistant gene markers for predicting treatment outcome on second-line anti-tuberculosis drugs. J Appl Microbiol 109:2087–2094. [DOI] [PubMed] [Google Scholar]
- 155.Boonaiam S, Chaiprasert A, Prammananan T, Leechawengwongs M. 2010. Genotypic analysis of genes associated with isoniazid and ethionamide resistance in MDR-TB isolates from Thailand. Clin Microbiol Infect 16:396–399. [DOI] [PubMed] [Google Scholar]
- 156.Lee H, Cho SN, Bang HE, Lee JH, Bai GH, Kim SJ, Kim JD. 2000. Exclusive mutations related to isoniazid and ethionamide resistance among Mycobacterium tuberculosis isolates from Korea. Int J Tuberc Lung Dis 4:441–447. [PubMed] [Google Scholar]
- 157.Marttila HJ, Soini H, Huovinen P, Viljanen MK. 1996. katG mutations in isoniazid-resistant Mycobacterium tuberculosis isolates recovered from Finnish patients. Antimicrob Agents Chemother 40:2187–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Khanna A, Raj VS, Tarai B, Sood R, Pareek PK, Upadhyay DJ, Sharma P, Rattan A, Saini KS, Singh H. 2010. Emergence and molecular characterization of extensively drug-resistant Mycobacterium tuberculosis clinical isolates from the Delhi Region in India. Antimicrob Agents Chemother 54:4789–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ramaswamy SV, Dou SJ, Rendon A, Yang Z, Cave MD, Graviss EA. 2004. Genotypic analysis of multidrug-resistant Mycobacterium tuberculosis isolates from Monterrey, Mexico. J Med Microbiol 53:107–113. [DOI] [PubMed] [Google Scholar]
- 160.Siddiqi N, Shamim M, Hussain S, Choudhary RK, Ahmed N, Prachee, Banerjee S, Savithri GR, Alam M, Pathak N, Amin A, Hanief M, Katoch VM, Sharma SK, Hasnain SE. 2002. Molecular characterization of multidrug-resistant isolates of Mycobacterium tuberculosis from patients in North India. Antimicrob Agents Chemother 46:443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Torres MJ, Criado A, Gonzalez N, Palomares JC, Aznar J. 2002. Rifampin and isoniazid resistance associated mutations in Mycobacterium tuberculosis clinical isolates in Seville, Spain. Int J Tuberc Lung Dis 6:160–163. [PubMed] [Google Scholar]
- 162.Pretorius GS, van Helden PD, Sirgel F, Eisenach KD, Victor TC. 1995. Mutations in katG gene sequences in isoniazid-resistant clinical isolates of Mycobacterium tuberculosis are rare. Antimicrob Agents Chemother 39:2276–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cockerill FR 3rd, Uhl JR, Temesgen Z, Zhang Y, Stockman L, Roberts GD, Williams DL, Kline BC. 1995. Rapid identification of a point mutation of the Mycobacterium tuberculosis catalase-peroxidase (katG) gene associated with isoniazid resistance. J Infect Dis 171:240–245. [DOI] [PubMed] [Google Scholar]
- 164.Ghiladi RA, Medzihradszky KF, Rusnak FM, Ortiz de Montellano PR. 2005. Correlation between isoniazid resistance and superoxide reactivity in Mycobacterium tuberculosis KatG. J Am Chem Soc 127:13428–13442. [DOI] [PubMed] [Google Scholar]
- 165.Bolotin S, Alexander DC, Chedore P, Drews SJ, Jamieson F. 2009. Molecular characterization of drug-resistant Mycobacterium tuberculosis isolates from Ontario, Canada. J Antimicrob Chemother 64:263–266. [DOI] [PubMed] [Google Scholar]
- 166.Mo L, Zhang W, Wang J, Weng XH, Chen S, Shao LY, Pang MY, Chen ZW. 2004. Three-dimensional model and molecular mechanism of Mycobacterium tuberculosis catalase-peroxidase (KatG) and isoniazid-resistant KatG mutants. Microb Drug Resist 10:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ando H, Kondo Y, Suetake T, Toyota E, Kato S, Mori T, Kirikae T. 2010. Identification of katG mutations associated with high-level isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jnawali HN, Hwang SC, Park YK, Kim H, Lee YS, Chung GT, Choe KH, Ryoo S. 2013. Characterization of mutations in multi-and extensive drug resistance among strains of Mycobacterium tuberculosis clinical isolates in Republic of Korea. Diagn Microbiol Infect Dis 76:187–196. [DOI] [PubMed] [Google Scholar]
- 169.Doustdar F, Khosravi AD, Farnia P, Masjedi MR, Velayati AA. 2008. Molecular analysis of isoniazid resistance in different genotypes of Mycobacterium tuberculosis isolates from Iran. Microb Drug Resist 14:273–279. [DOI] [PubMed] [Google Scholar]
- 170.Rouse DA, DeVito JA, Li Z, Byer H, Morris SL. 1996. Site-directed mutagenesis of the katG gene of Mycobacterium tuberculosis: effects on catalase-peroxidase activities and isoniazid resistance. Mol Microbiol 22:583–592. [DOI] [PubMed] [Google Scholar]
- 171.Heym B, Alzari PM, Honore N, Cole ST. 1995. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 15:235–245. [DOI] [PubMed] [Google Scholar]
- 172.Yuan X, Zhang T, Kawakami K, Zhu J, Li H, Lei J, Tu S. 2012. Molecular characterization of multidrug- and extensively drug-resistant Mycobacterium tuberculosis strains in Jiangxi, China. J Clin Microbiol 50:2404–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ramirez MV, Cowart KC, Campbell PJ, Morlock GP, Sikes D, Winchell JM, Posey JE. 2010. Rapid detection of multidrug-resistant Mycobacterium tuberculosis by use of real-time PCR and high-resolution melt analysis. J Clin Microbiol 48:4003–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fang Z, Doig C, Rayner A, Kenna DT, Watt B, Forbes KJ. 1999. Molecular evidence for heterogeneity of the multiple-drug-resistant Mycobacterium tuberculosis population in Scotland (1990 to 1997). J Clin Microbiol 37:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Abe C, Kobayashi I, Mitarai S, Wada M, Kawabe Y, Takashima T, Suzuki K, Sng LH, Wang S, Htay HH, Ogata H. 2008. Biological and molecular characteristics of Mycobacterium tuberculosis clinical isolates with low-level resistance to isoniazid in Japan. J Clin Microbiol 46:2263–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Khadka DK, Eampokalap B, Panitchakorn J, Ramasoota P, Khusmith S. 2007. Multiple mutations in katG and inhA identified in Thai isoniazid-resistant Mycobacterium tuberculosis isolates. Southeast Asian J Trop Med Public Health 38:376–382. [PubMed] [Google Scholar]
- 177.Kim SY, Park YJ, Kim WI, Lee SH, Ludgerus Chang C, Kang SJ, Kang CS. 2003. Molecular analysis of isoniazid resistance in Mycobacterium tuberculosis isolates recovered from South Korea. Diagn Microbiol Infect Dis 47:497–502. [DOI] [PubMed] [Google Scholar]
- 178.Minh NN, Van Bac N, Son NT, Lien VT, Ha CH, Cuong NH, Mai CT, Le TH. 2012. Molecular characteristics of rifampin- and isoniazid- resistant Mycobacterium tuberculosis strains isolated in Vietnam. J Clin Microbiol 50:598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zakerbostanabad S, Titov LP, Bahrmand AR. 2008. Frequency and molecular characterization of isoniazid resistance in katG region of MDR isolates from tuberculosis patients in southern endemic border of Iran. Infect Genet Evol 8:15–19. [DOI] [PubMed] [Google Scholar]
- 180.Zenteno-Cuevas R, Zenteno JC, Cuellar A, Cuevas B, Sampieri CL, Riviera JE, Parissi A. 2009. Mutations in rpoB and katG genes in Mycobacterium isolates from the Southeast of Mexico. Mem Inst Oswaldo Cruz 104:468–472. [DOI] [PubMed] [Google Scholar]
- 181.Soudani A, Hadjfredj S, Zribi M, Messaadi F, Messaoud T, Masmoudi A, Fendri C. 2011. Genotypic and phenotypic characteristics of Tunisian isoniazid-resistant Mycobacterium tuberculosis strains. J Microbiol 49:413–417. [DOI] [PubMed] [Google Scholar]
- 182.Haas WH, Schilke K, Brand J, Amthor B, Weyer K, Fourie PB, Bretzel G, Sticht-Groh V, Bremer HJ. 1997. Molecular analysis of katG gene mutations in strains of Mycobacterium tuberculosis complex from Africa. Antimicrob Agents Chemother 41:1601–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Soolingen D, van Embden JD. 1996. Characterization of the catalase-peroxidase gene (katG) and inhA locus in isoniazid-resistant and -susceptible strains of Mycobacterium tuberculosis by automated DNA sequencing: restricted array of mutations associated with drug resistance. J Infect Dis 173:196–202. [DOI] [PubMed] [Google Scholar]
- 184.Lipin MY, Stepanshina VN, Shemyakin IG, Shinnick TM. 2007. Association of specific mutations in katG, rpoB, rpsL and rrs genes with spoligotypes of multidrug-resistant Mycobacterium tuberculosis isolates in Russia. Clin Microbiol Infect 13:620–626. [DOI] [PubMed] [Google Scholar]
- 185.Waksman SA. 1964. The Conquest of Tuberculosis. University of California Press, Berkeley. [Google Scholar]
- 186.Winder FG, Collins PB. 1970. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J Gen Microbiol 63: 41–48. [DOI] [PubMed] [Google Scholar]