

Abstract
Highly cytotoxic agents have found an important niche in targeted anticancer therapy. Here we develop a new light release strategy for the targeting of one of these agents, 2-pyrrolinodoxorubicin, showing dramatic enhancements in toxicity with light and single digit nM potency.
Grphical Abstract
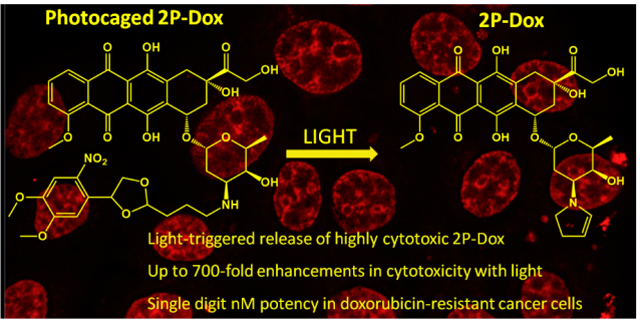
A novel light-based strategy is used to deliver 2-pyrrolinodoxorubicin showing excellent cytotoxicity in doxorubicin-resistant cancer cell lines.
One of the major contributing factors for the failure of promising anticancer drugs in the clinic is off target toxicity. 1,2 This problem is particularly acute when one considers highly-potent cytotoxins as potential anticancer therapeutics.2 While they are potent anticancer agents, the high toxicity typically leads to a very narrow therapeutic window that prevents clinical use. Instead, research has focused on developing novel methods using antibodies to target these molecules directly to the tumor itself.3–6
A promising alternative strategy for directing the release of highly-potent cytotoxins involves the use of light. To date, photodynamic therapy (PDT) has proven a popular and effective strategy for treatment of cancer with light.7–9 PDT relies on an administered photosensitizer that is activated using wavelengths of light between 650–800 nm. Once activated, the photosensitizer (PS) creates singlet oxygen, which is cytotoxic to cancerous cells. However, the deeper regions of tumors are typically hypoxic, making photodynamic therapy ineffective for larger tumors. Moreover, the short-lived nature of singlet oxygen prevents its diffusion into deeper regions of the tumor where light cannot penetrate.10
A more recent alternative to PDT is photoactivated chemotherapy where a standard cancer chemotherapeutic is converted into a light-activatable form.11,12 A common strategy is to attach the molecule to a photocage,13 a re-leasable group that blocks the anticancer agent’s activity until illumination. We and others have pursued this strategy by caging conventional anticancer agents such as doxorubicin, cisplatin, etc.12,14–21 Each of these advances has focused on delivery of anticancer agents with moderate cytotoxicity. Release of a more potent drug should enable deeper tissue activation as less efficient release should still lead to the cytotoxic effect.
Here we describe the photoactivation of one of these highly potent agents, 2-pyrrolino doxorubicin (2P-Dox)22,23 (Figure 1a). 2P-Dox is a derivative of the standard cancer chemotherapeutic doxorubicin (Dox) which is prescribed for the treatment of a variety of cancers including ovarian, breast, and lung cancer, as well as leukemia.24 2P-Dox is 100–1000 fold more potent than Dox in vitro against a number of Dox resistant cell lines, and has IC50 values in the low nM to high pM range.25–28 The extreme potency of 2P-Dox is a result of its ability to form an aminal adduct with an amino group of a guanine base in close vicinity to its binding site in DNA.27 2P-Dox is also not a substrate of the P-gp pump which is the key factor for resistance of Dox.29 Research efforts have focused on targeting 2P-Dox through antibody or peptide conjugation,22,30,31 but here we describe the synthesis and analysis of a photoactivatable version of 2P-Dox.
Fig. 1. 2-pyrrolinodoxorubicin (2P-Dox) and its prodrugs.
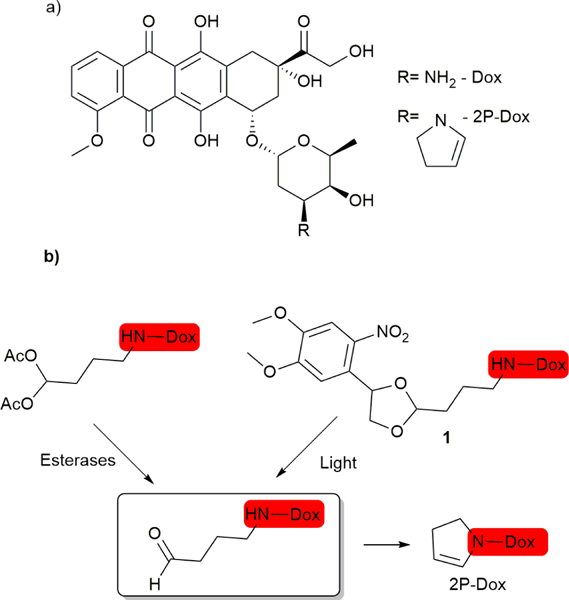
(a). Structure of Dox and 2P-Dox. (b) A previously described esterase activated prodrug of 2P-Dox and the light-activated prodrug of 2P-Dox described here.
We have based on our strategy on the di-acetoxy prodrug of 2P-Dox25,27. The acetates on this produg are cleaved intracellularly by esterases releasing the latent aldehyde which cyclizes to form 2P-Dox (Figure 1b). We reasoned that a suitable photo-caging group for the latent aldehyde13 would lead to a light-activatable 2P-Dox (1, Figure 1b).
Kantevari et al. previously utilized a bis(4,5-dimethoxy-2-nitrophenyl)ethylene glycol for the photorelease of various aldehydes and ketones through acetal protection. 32 We have chosen to use a modified version of this strategy for our drug delivery. To prepare compound 1 (Scheme 1), we started by a chlorination of commercially available nitro-veratryl alcohol (2), which was then reacted with triphenyl phosphine to generate phosphonium salt 3. The salt was then reacted with formaldehyde under Wittig reaction conditions to give a styrene which was dihydroxylated to give diol 4. The diol was acetal protected with commercially available 3-cyanopropionaldehyde dimethyl acetal (5) to give diastereomeric acetals, and the nitrile group was reduced with DIBAL-H33 to give aldehyde 6 which was coupled to doxorubicin under reductive amination conditions to yield (1). The overall yield was 41%. We also prepared a control compound, 7, formed by the reductive amination of aldehyde 6 and cyclohexylamine.
Scheme 1.
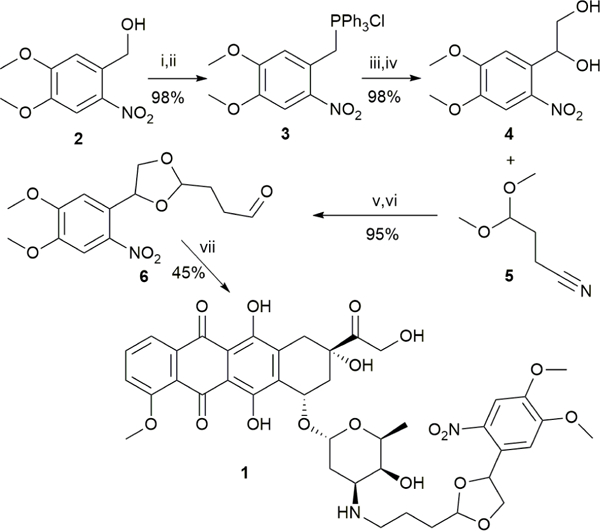
(i) SOCl2; (ii) PPh3/solvent ; (iii) formaldehyde/water; (iv) OsO4/NMO/CH2Cl2/H2O; (v) PPTS/benzene; (vi) DIBAL-H/CH2Cl2; (vii) NaCNBH3/H2O
With (1) in hand, we investigated the rates of release of 2P-Dox under UV illumination via thin layer chromatography (Figure 2). Cleavage begins to take place after 2 min with 88% cleavage after 60 min irradiation (Figure 2a,b). A clean MS showing 2P-Dox was observed after 60 min (Figure 2c). No cleavage was observed in the dark (not shown). HPLC traces of the reaction before and after illumination were consistent with the TLC results (Fig. S1, ESI†)
Fig. 2. Thin layer chromatography release assay.
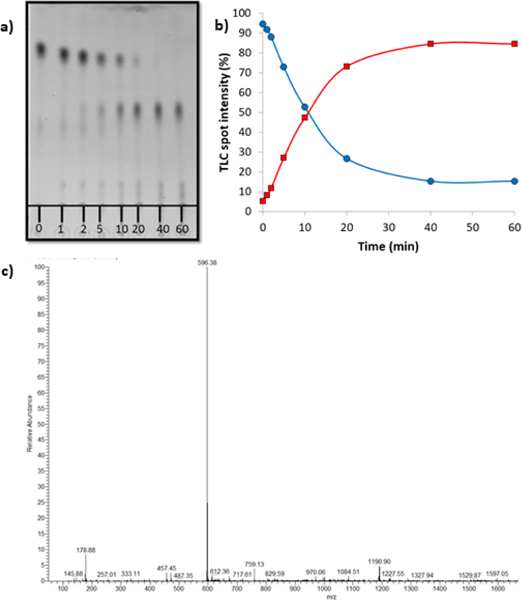
(a) A 20 μM solution of 1 in PBS buffer was irradiated under 380 nm light (9.0 mW/cm2) and the release of 2P-Dox was monitored via TLC. (b) Graph representing the increase in intensity of the lower Rf TLC spot corresponding to 2P-Dox over time. (c) ESI-MS of the solution after 60 min reaction time showing the clean formation of 2P-Dox. Calculated (M+ H)+ = 596.21; Observed = 596.38.
Once we determined that light had no effect on cytotoxicity (Fig. S2a, ESI†) we proceeded with cellular studies to demonstrate that the 2P-Dox released led to the expected enhancement in cytotoxicity. We performed the cellular viability assay in three human cancer cell lines: MCF-7 (breast), A2780 (ovarian) and A2780ADR (doxorubicin resistant ovarian) using the CellTiter-Blue method to evaluate the light-induced effects of compound 1 as well control compound 7 (Table 1, Figure 3, Fig. S2-S5, ESI†). We observed excellent photo-induced toxicity of compound 1 with impressive 327–750 fold increase over dark toxicity. 1 exhibited remarkably low IC50 values ranging from 0.1–50 nM in presence of light (380 nm, 30 min); these values were comparable to that obtained for 2P-Dox alone. The control compound 7 showed no toxicity; demonstrating that the cytotoxic effects are completely due to released drug (Fig. S5, ESI†). In accord to previously reported observations25–27 , we found that 2P-Dox displays higher potency than doxorubicin and also retains its activity in the doxorubicin-resistant cell line A2780ADR, where it is 330-fold more active than doxorubicin alone in A2780ADR cells.
Table 1.
IC50 values (μM) as obtained from cell titer blue cell survival assays in human cancer cell lines.
| MCF-7 | A2780 | A2780ADR | ||||
| (+) Light | (-) Light | (+) Light | (-) Light | (+) Light | (-) Light | |
| 1 | 0.05 ± 0.01 | 18.7 ± 2.4 | 0.002 ± 0.001 | 1.5 ± 0.6 | 0.003 ± 0.001 | 0.98 ± 0.26 |
| Dox | 1.3 ± 0.2 | 1.4 ± 0.1 | 0.076 ± 0.016 | 0.095 ± 0.021 | 0.99 ± 0.25 | 1.3 ± 0.3 |
| 2P-Dox | 0.010 ± 0.007 | 0.013 ± 0.005 | n.d. | 0.0017 ± 0.0004 | n.d. | 0.002 ± 0.001 |
| 7 | >50 | >50 | >50 | >50 | >50 | >50 |
Fig. 3. Cell culture assays with compound 1.
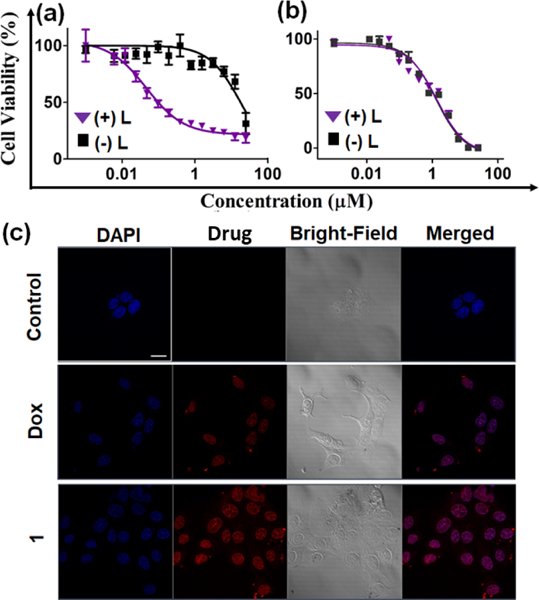
Cell viability as determined by CellTiter-Blue assay in MCF-7 cells treated with (a) compound 1 and (b) doxorubicin in absence (black squares) or presence of light (380 nm, 9.0 mW/cm2, 30 min, purple triangles). (c) Confocal microscopic images of MCF-7 cells alone (first row) or treated with 10 μM of doxorubicin (Dox, second row) and compound 1 (third row) for 2 h. The columns from left to right show: DAPI nuclear staining (blue channel), Dox or compound 1 (red channel), the brightfield image, and a merge of the red and blue channels. Scale bar = 10 μm.
We next exploited the fluorescent properties of doxorubicin and compound 1 to quantify the cellular uptake in MCF-7 cell lines using flow cytometry (Fig. S6, ESI†). The obtained data clearly indicates that compound 1 showed higher cellular uptake than doxorubicin. Several studies have shown that N-alkylation or O-alkylation of DOX with lipophilic moieties can improve both the amount and rate of cell uptake,34–36 and we expect a similar phenomenon is operating here. The enhanced uptake also likely contributes to the improved cytotoxicity of compound 1 relative to Dox. To understand the sub-cellular localization, we carried out confocal microscoopic studies. The images revealed the nuclear co-localization of compound 1 and doxorubicin (Figure 3c). Taken together, our light-releasable compound 1 mirrors the activity of the parent drug 2P-Dox, yet has significantly reduced activity in the dark.
Here we have shown for the first time that we can generate 2P-Dox in a light dependent manner. The large enhancements in activity suggest that this approach will have an effective therapeutic window. Although UV light is poorly tissue penetrating, one could envision the use of alternate protecting groups that can be released with longer wavelengths of light that are more tissue penetrating.37,38,39,40 The high potency of 2P-Dox will further improve tissue penetration as even poorly illuminated deep tissues will contain enough released 2P-Dox for the cytotoxic effect. More broadly, this work opens up new opportunities for repurposing of highly potent cytotoxins for effective cancer chemotherapy.
Supplementary Material
Acknowledgments
M.C.T.H. acknowledges support of this work by the NIH (CA167582). P.D. acknowledges the Altria Corporation for a graduate fellowship. K.M.’s postdoctoral fellowship was supported by the Virginia Commonwealth Health Review Board (236–03-16).
Footnotes
Conflicts of Interest
M.C.T.H. is a part of a company, LightSwitch Bio, that is developing related technologies for commercial application.
References
- 1.Hopkins AL, Nat. Chem. Biol, 2008, 4, 682–690. [DOI] [PubMed] [Google Scholar]
- 2.Wollowitz S, Drug Dev. Res, 2010, 71, 420–428. [Google Scholar]
- 3.Sassoon I and Blanc V, Antibody-drug conjugate (ADC) clinical pipeline: a review, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Lambert JM and Berkenblit A, Ann. Rev. Med, 2018, 69,191–207. [DOI] [PubMed] [Google Scholar]
- 5.Beck A, Goetsch L, Dumontet C and Corvà’ N, Nat. Rev. Drug. Disc, 2017, 16, 315. [DOI] [PubMed] [Google Scholar]
- 6.Nani RR, Gorka AP, Nagaya T, Yamamoto T, Ivanic J, Kobayashi H and Schnermann MJ, ACS Cent. Sci, 2017, 3, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolmans DE, Fukumura D and Jain RK, Nat. Rev. Cancer, 2003, 3, 380. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Jiang C, Longo JPF, Azevedo RB, Zhang H and Muehlmann LA, Acta Pharm. Sin. B, 2018, 8, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnett R, Chem. Soc. Rev, 1995, 24, 19–33. [Google Scholar]
- 10.Maisch T, Baier J, Franz B, Maier M, Landthaler M, Szeimies R-M and Bäumler W, Proc. Natl. Acad. Sci. USA, 2007, 104, 7223–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrer NJ, Salassa L and Sadler PJ, Dalton Trans, 2009, 10690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reeβing F and Szymanski W, Curr. Med. Chem, 2018, 24, 4905–4950. [DOI] [PubMed] [Google Scholar]
- 13.Klán P, Solomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A and Wirz J, Chem. Rev, 2012, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibsen S, Zahavy E, Wrasdilo W, Berns M, Chan M and Esener S, Pharm. Res, 2010, 27, 1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi SK, Thomas T, Li MH, Kotlyar A, Desai A and Baker JR, Chem. Commun, 2010, 46, 2632–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong PT, Tang S, Cannon J, Chen D, Sun R, Lee J, Phan J, Tao K, Sun K, Chen B, Baker JR and Choi SK, Bioconj. Chem, 2017, 28, 3016–3028. [DOI] [PubMed] [Google Scholar]
- 17.Dcona MM, Sheldon JE, Mitra D and Hartman MC, Bioorg. Med. Chem. Lett, 2017, 27, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dcona MM, Mitra D, Goehe RW, Gewirtz DA, Lebman DA and Hartman MC, Chem. Commun, 2012, 48, 4755–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi M, Skwarczynski M, Prakash H, Hirota S, Kimura T, Hayashi Y and Kiso Y, Bioorg. Med. Chem, 2008, 16, 5389–5397. [DOI] [PubMed] [Google Scholar]
- 20.Mitra K, Gautam S, Kondaiah P and Chakravarty AR, Angew. Chem. Int. Ed, 2015, 54, 13989–13993. [DOI] [PubMed] [Google Scholar]
- 21.Mitra K, Lyons CE and Hartman MC, Angew. Chem. Int. Ed, 2018, 57, 10263–10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy A, Schally AV, Halmos G, Armatis P, Cai RZ, Csernus V, Kovacs M, Koppan M, Szepeshazi K and Kahan Z, Proc. Natl. Acad. Sci. USA, 1998, 95, 1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy A, Armatis P, Cai RZ, Szepeshazi K, Halmos G and Schally AV, Proc. Natl. Acad. Sci. USA, 1997, 94, 652–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young RC, Ozols RF and Myers CE, N. Engl. J. Med, 1981, 305, 139–153. [DOI] [PubMed] [Google Scholar]
- 25.Cherif A and Farquhar D, J. Med. Chem, 1992, 35, 3208–3214. [DOI] [PubMed] [Google Scholar]
- 26.Farquhar D, Cherif A, Bakina E and Nelson JA, J. Med. Chem, 1998, 41, 965–972. [DOI] [PubMed] [Google Scholar]
- 27.Zwelling LA, Altschuler E, Cherif A and Farquhar D, Cancer Res, 1991, 51, 6704–6707. [PubMed] [Google Scholar]
- 28.Nagy A, Armatis P and Schally AV, Proc. Natl. Acad. Sci. USA, 1996, 93, 2464–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castex C, Merida P, Blanc E, Clair P, Rees AR and Temsamani J, Anti-Cancer Drug, 2004, 15, 609–617. [DOI] [PubMed] [Google Scholar]
- 30.Nagy A, Schally AV, Armatis P, Szepeshazi K, Halmos G, Kovacs M, Zarandi M, Groot K, Miyazaki M, Jungwirth A and null others, Proc. Natl. Acad. Sci. USA, 1996, 93,7269–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeffrey SC, Nguyen MT, Andreyka JB, Meyer DL, Doronina SO and Senter PD, Bioorg. Med. Chem. Lett, 2006, 16, 358–362. [DOI] [PubMed] [Google Scholar]
- 32.Kantevari S, Narasimhaji CV and Mereyala HB, Tetrahedron, 2005, 61, 5849–5854. [Google Scholar]
- 33.Paquette LA, Backhaus D, Braun R, Underiner TL and Fuchs K, J. Am. Chem. Soc, 1997, 119, 9662–9671. [Google Scholar]
- 34.Lothstein L, Rodrigues PJ, Sweatman TW and Is-rael M, Anticancer Drugs, 1998, 9, 58–66. [DOI] [PubMed] [Google Scholar]
- 35.Israel M, Sweatman TW, Seshadri R and Koseki Y, Cancer Chemother. Pharmacol, 1989, 25, 177–183. [DOI] [PubMed] [Google Scholar]
- 36.Israel M, Seshadri R, Koseki Y, Sweatman TW and Idriss JM, Cancer Treat. Rev, 1987, 14, 163–167. [DOI] [PubMed] [Google Scholar]
- 37.Jagdeo J, Austin E, Mamalis A, Wong C, Ho D and Siegel DM, Lasers Surg. Med, 2018, 50, 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barolet D, Semin. Cutan. Med. Surg, 2008, 27, 227–238. [DOI] [PubMed] [Google Scholar]
- 39.Shell TA and Lawrence DS, Acc. Chem. Res, 2015, 48, 2866–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorka AP, Nani RR, Zhu J, Mackem S and Schnermann MJ, J. Am. Chem. Soc, 2014, 136, 14153–14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.