

Abstract
The mixed lineage leukemia (MLL) family of proteins became known initially for the leukemia link of its founding member. Over the decades, the MLL family has been recognized as an important class of histone H3 lysine 4 (H3K4) methyltransferases that control key aspects of normal cell physiology and development. Here, we provide a brief history of the discovery and study of this family of proteins. We address two main questions: why are there so many H3K4 methyltransferases in mammals; and is H3K4 methylation their key function?
Keywords: MLL, Methylation, Histone, H3K4, Transcription, Epigenetics, Development
Background
The mixed lineage leukemia (MLL) gene was originally identified in humans due to its association with a common breakpoint found in a subset of incurable acute leukemias [1–4]. Most MLL gene mutations in leukemia are chromosome translocations that truncate the MLL gene and fuse it in frame to an ever-increasing number of different partner genes [5]. The role of MLL mutations in leukemia has been extensively explored in multiple reviews over the years [6–9]. Here, we focus on what is known about the function of the wild-type MLL protein and its related family members, a perhaps less studied but in some ways even more complicated topic.
History
When the human MLL cDNA was first sequenced [2–4], it was found to have a striking homology to the Drosophila gene trithorax [10]. Trithorax (or trx) is a founding member of the trithorax group (trxG) of proteins [11, 12] which were originally identified as regulators of Homeotic (or Homeobox, Hox) genes in Drosophila, a set of genes that are essential for body patterning in multi-cellular organisms [11–15]. TrxG proteins maintain the expression of Hox genes and this is antagonized by the repressive activity of the Polycomb group (PcG) of proteins [13, 15, 16]. Importantly, neither group of regulators is required for initiating Hox gene expression, but instead they are required to maintain expression patterns once they have been established by early acting transcription factors [15, 17]. Interestingly, both PcG and trxG genes were discovered in parallel with the Hox genes, due to the overall importance of this entire system for controlling development of the anterior–posterior axis [11–15]. Seminal work in mice showed that mammalian Mll behaves like a member of the trxG, in that Mll knockout (KO) mice display embryonic lethality and body plan defects (Embryonic day 9; E9) caused by altered Hox gene expression patterns [18, 19]. A key aspect of this work is that, similar to observations with trx mutants, Hox gene expression patterns initiate normally in Mll mutants but only break down at later stages of development [18, 19].
The mutually antagonistic nature of PcG and trxG function was highlighted by the observation that PcG/trxG double mutants in both Drosophila [20–22] and mice [23] produced embryos that were phenotypically closer to wild type than either individual mutation. Further support of this model came from work that revealed another class of genes, the so-called “enhancers of trithorax and Polycomb” (ETPs, [24]). For example, the Drosophila gene Additional sex combs (Asx) is a key member of this group [25] whose ETP function may be conserved in mammals [26]. Mutations in ETP genes enhance the activity of both PcG and trxG mutations, but alone they display phenotypes closer to wild type [25, 27].
Despite the early observations that PcG and trxG proteins balanced each other out and were required for maintenance but not initiation of gene expression, the genetics alone did not reveal any clues to the function of these proteins on a molecular level. A key aspect of this came from work on the SET domain.
The SET domain and methyltransferase activity
SET is an acronym [28] taken from the founder members of this family: Suppressor of variegation 3-9 (Su(var)3-9) [28]; Enhancer of Zeste (E(z)) [29]; and Trithorax [30]. All three are chromatin proteins, with Su(var)3-9 promoting repressive heterochromatin and E(z) and trx being members of the PcG and trxG, respectively. What was interesting to the field at the time was that these proteins, with apparently quite different functions, all contained a homologous protein domain indicating a possible similar activity. Using sequence homology, the SET domain was found in over 140 genes in multiple species including plants, bacteria and some viruses [31, 32]. The recognition that this domain was present in some plant N-methyltransferases led to the discovery that the SET domain in mammals was a lysine methyltransferase (KMT), capable of methylating lysine residues on histones [31].
Methylation had long been known to occur on histone proteins [33], including on specific lysine residues in a mono-, di- or tri-methyl form (reviewed in: [34–36]). A key discovery in understanding the distinct functions of different SET domain-containing proteins was that these domains had specificity for different lysine residues. For instance, EZH2, the mammalian homolog of the PcG protein E(z), specifically methylates lysine 27 on histone H3 (H3K27) [37] while SUV39H1, homolog of heterochromatic protein Su(var)3-9, methylates lysine 9 on histone H3 (H3K9) [31]. Interestingly, a viral SET domain-containing protein was identified with intrinsic H3K27 methyltransferase capabilities, raising the possibility that viral proteins may be able to alter the epigenome of the host [38].
Surprisingly, in contrast to SUV39H1 and EZH2, the MLL SET domain was initially thought to be functionally inactive [31]. However, subsequent work showed that it had methyltransferase activity specific for lysine 4 on histone H3 (H3K4) [39, 40], a modification known to be associated with active genes. H3K4 methyltransferase activity was found to be targeted directly to Hox genes by the full-length MLL protein and this resulted in the activation of their expression [39]. Previous work had identified Set1 in Saccharomyces cerevisiae as the major H3K4 methyltransferase in yeast [41–44], leading some to suggest that Set1 could be the yeast homolog of MLL/trx [45, 46]. However, subsequent work identified mammalian equivalents of Set1 [47–49], suggesting that although MLL and S. cerevisiae Set1 were related in their SET domains, the MLL protein had likely evolved to also take on other functions. In addition, the discovery of other MLL/trx-like SET domain-containing genes in Drosophila such as trithorax-related (trr) [50, 51] and Drosophila-Set1 (dSet1) [52, 53] indicated that MLL-like genes may represent a specific group in higher organisms that had diverged from the original Set1 protein in yeast (Fig. 1). This point is discussed in more detail below.
Fig. 1.
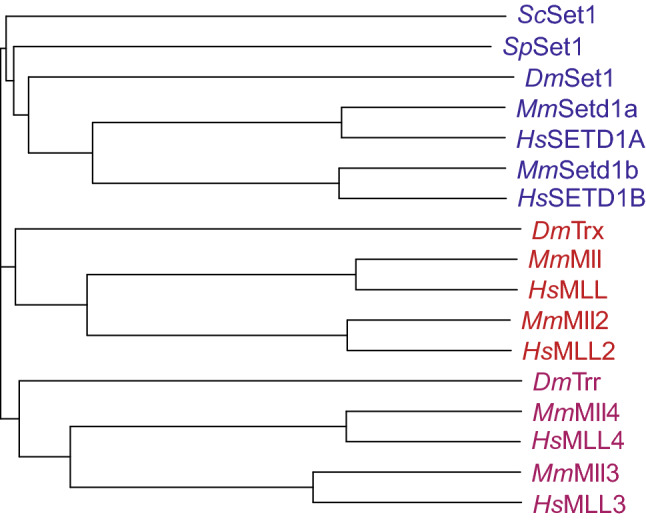
Sequence conservation of MLL family proteins. Evolutionary distances between MLL family protein sequences in human (Hs), mouse (Mm), Drosophila (Dm), Saccharomyces cerevisiae (Sc) and Schizosaccharomyces pombe (Sp), calculated using ClustalW. Horizontal lengths are proportional to sequence similarity distance
In the original studies that demonstrated MLL H3K4 methyltransferase activity, neither the purified SET domain [39] nor a purified MLL complex [40] was able to deposit trimethylation (H3K4me3). However, later work showed that a different MLL complex preparation actually had a preference for trimethylating H3K4, and this was associated with elevated H3K4me3 at Hox genes and stimulation of transcription from an in vitro chromatin template [54]. The apparently contradictory results were clarified by the observation that the MLL SET domain requires three cooperating proteins for full methyltransferase activity: WDR5, RbBP5 and ASH2L [55]. These components were not identified in the original MLL complex purification [40], but it is now known that WDR5, RbBP5 and ASH2L are common components of all H3K4me3 SET domain complexes, something that is also shared with Set1 in yeast [56].
Overall, the major importance of the SET domain work was that it provided a possible functional explanation for the antagonism of PcG vs trxG proteins. The ability to methylate specific lysine residues could help promote either gene activation (H3K4me3) or repression (H3K27me3), thus it was initially thought that the main function of these proteins was mostly explained by SET domain activity. Differential SET domain function also provides a possible functional explanation for the observation that the ETP Asx/ASXL1 is required for both trxG and PcG function [25, 26]. Asx has been found to interact with both the E(z) and Trx protein SET domains, controlling both activation and repression by modulating H3K4me3 and H3K27me3 levels at target loci [57]. However, the Asx family co-purifies in a complex containing the deubiquitinase BAP1 rather than EZH2 or MLL in both Drosophila and mammals [58], suggesting that further characterization of these interactions is required.
Despite these exciting observations, the fact that deletion of the MLL SET domain resulted in viable and fertile mice, albeit with developmental skeletal defects and disturbed Hox gene expression [59, 60], argued that the SET domain alone could not account for the main function of the MLL protein.
The MLL family
There are six members of the “MLL family” in mammals [56, 61]. The family is made up of three pairs of highly structurally-related proteins, with each pair related to a single Drosophila protein (Fig. 1) [56, 62]. There has been some confusion about the naming of the individual members, especially MLL2/4 [7], but we adopt the convention here of referring to the human gene at 19q13 as MLL2/KMT2B, which fits best with the structural relatedness of the proteins (see Figs. 1 and 2) as further described below [61]. MLL (MLL1 or KMT2A [1], human chromosome 11q23) pairs with MLL2 (KMT2B [63, 64], human chromosome 19q13) and these are both most closely related to trx itself [2–4, 10, 50, 62]. MLL3 (KMT2C, human chromosome 7q36 [65, 66]) pairs with MLL4 (KMT2D, human chromosome 12q13 [67, 68]) and these are highly related to the trithorax-related (trr) protein [50, 62]. Finally, SETD1A (KMT2F human chromosome 16p11 [47]) pairs with SETD1B (KMT2G human chromosome 12q24 [49]) and these are both closest to the Drosophila Set1 protein (dSet1) which in turn is the closest homolog to S. cerevisiae Set1 (Fig. 1) [52, 53, 62]. Although MLL5 (KMT2E [69]) was originally thought to be a member of the MLL family, the lack of intrinsic KMT activity and the observation that the MLL5 SET domain is divergent from the rest of the family has led to it being reclassified as representing a different subgroup of SET domain proteins [70].
Fig. 2.

Domain structure of mammalian MLL family proteins. The six human MLL family proteins are shown, with the approximate positions and sizes of identified domains. Numbers indicate the length of each protein. All contain a C-terminal SET domain which catalyses histone H3K4 methylation, as well as a variable number of DNA-binding and protein–protein interaction domains. RRM: RNA-recognition motif; PHD: plant homeodomain; FYRN/FYRC: FY-rich domain, N-/C-terminal; SET: Su(var)3-9/E(z)/Trithorax domain; HMG box: high mobility group box; Taspase cleavage site: recognition sequence cleaved by the threonine protease Taspase 1
Adapted from [56]
The main commonality between the MLL family members is that they share a highly related SET domain that is capable of methylating H3K4 [39, 47, 71–73]. MLL family members also all interact with the complex components WDR5, RbBP5, ASH2L and DPY30, which represent a core set of proteins required for full KMT activity of the SET domain [56, 62]. However, beyond the SET domain, different MLL family members display a different protein domain architecture ([56, 62] and see Fig. 2). Furthermore, despite residing in overlapping protein complexes, in some cases individual members can interact with unique sets of proteins, although interacting factors and unique domains are often shared between each of the partner pairs [7, 56]. Thus, although the MLL proteins are a “family” from the perspective that they represent an evolutionary and functional expansion of the SET1 system, it is worth considering that there are multiple other mammalian H3K4 KMTs (with less closely related KMT domains) such as SET7/9 [74] and the SMYD family of proteins [75, 76].
Since the cell has multiple ways of directing H3K4 KMT activity beyond the MLL family, it is probably more accurate to think of the SET domain as providing only a subset of each member’s function. This perspective fits with the observation that loss of the MLL SET domain does not phenocopy deletion of the entire gene [59, 77]. Following this logic, we would argue that these proteins, which happen to share H3K4 KMT activity and a similar evolutionary origin, are functionally distinct with unique biological roles in the cell, much in the same way an artist and a decorator may both use a paintbrush to different ends in their jobs. For this reason, the rest of this review will focus on the potentially unique roles of these proteins, how the individual proteins may be specifically directed towards key targets in the cell, and how their KMT activity may contribute to their overall function.
Function of H3K4 methylation
Before discussing MLL family member-specific activities, it is worth briefly touching on what the general role of H3K4 methylation is thought to be. Some of the earliest work showed that H3K4 methylation was highly associated with transcriptionally active Tetrahymena macronuclei [78]. This fit with later observations that indicated increased H3K4me2/3 was associated with elevated Hox gene expression as well as increased in vitro transcription on nucleosomal templates [39, 54, 79–81]. Yeast Set1 is recruited by the RNA polymerase II (RNA pol II) elongation machinery to target H3K4 methylation to actively transcribed genes [82]. Early experiments indicated that MLL could also associate with elongating RNA pol II [83], but more recent interactome studies with differentially phosphorylated versions of the RNA pol II C-terminal domain (CTD) failed to co-purify MLL family complex components [84]. Notably, the lack of correlation between MLL and RNA pol II binding within the gene body [85] also indicates that if this interaction ever occurs in vivo it is not sufficient for localisation.
More detailed analyses of the genome-wide distribution of H3K4 methylation supported these initial observations of a role in gene regulation but also suggested a slightly more nuanced pattern of activity based on the precise methylation status. H3K4me1 was found to be primarily associated with enhancers, and H3K4me2 was more generally distributed throughout active genes and enrichment for H3K4me3 marked active promoters [86–91]. The discovery of multiple protein domains possessing the ability to bind directly to methylated lysine residues provided a major functional insight into how H3K4me3 could impact promoter function. Examples of tandem chromodomains [92, 93], Plant HomeoDomain (PHD) zinc finger motifs [94] and Tudor domains [95–97], have been identified with specificity for H3K4me3, linking a number of protein activities with this modification. For example, the PHD motif is found in multiple proteins [56] thought to promote gene activation including nucleosome remodelers [56, 94, 98] and TAF3, a component of the RNA pol II pre-initiation complex [56, 79, 99]. Elegant in vitro studies indicated that TAF3 binding to H3K4me3 substrates could stimulate transcription by increasing the stability of the pre-initiation complex, allowing for rapid re-initiation through multiple rounds of transcription [79]. In contrast to the incredible range of proteins that bind to H3K4me3, proteins that specifically recognize H3K4me1 have been harder to find [56], although recent work suggests that the cohesin complex may be stabilized by binding to H3K4me1 [100]. However, the specifics of this interaction have yet to be established.
Despite the strong data linking H3K4 methylation to both enhancer and promoter function, SET1 KO or H3K4 point mutations that completely abolish H3K4me2/3 in yeast have a relatively mild phenotype [41]. In general, global loss of H3K4me3 induces very few transcriptional changes in most systems [101–103]. In addition, despite the fact that the breadth and height of H3K4me3 peaks correlate with higher levels of gene expression [104, 105], more recent work in mouse embryonic stem (ES) cells suggests that even where there are gene expression changes, there is very little correlation between the reduction in H3K4me3 levels and the extent of gene expression changes, at least globally [106]. Instead, the requirement for H3K4me3 appears to be gene specific, with the dependence on H3K4me3 appearing to be stronger at genes where there are potentially fewer “activating signals” maintaining expression. In addition, H3K4me1 appears to be dispensable for enhancer function in ES cells [107] and throughout Drosophila development [108]. These and other observations have led to the question of whether H3K4 methylation is actually required for promoting transcription or enhancer function, whether it has a much more subtle role in modulating gene expression, or even if it is simply a non-functional marker of past transcription [103]. Given the implied significance of the MLL family SET domains, this has important potential implications for their function.
Distinct biological phenotypes of MLL family members
MLL/MLL2
As described above, knockouts of the Mll gene in mice produced antero-posterior skeletal defects highly reminiscent of the Hox gene-mediated body pattern defects observed in trx mutant embryos [18, 19, 23]. Since embryonic death occurred relatively early, a full analysis of hematopoietic defects was not possible, but initial analysis of KO models indicated that MLL likely has a key role in normal hematopoietic development, primarily through the regulation of Hox genes [109–111]. Inducible Mll KO models were produced to more directly address MLL function in fetal and adult hematopoiesis, and although there were some specific differences between the KO models, in general MLL was found to be required for normal hematopoietic development [112–114]. These MLL phenotypes are contrasted by the observation that deletion of the MLL SET domain has very little impact on embryogenesis [59] or hematopoiesis [60]. Since loss of MLL has profound effects on gene expression, especially of the Hox genes [39, 60, 115], gene activation as mediated by MLL may require other domains of the protein (discussed below), likely by recruiting components of the MLL complex, such as MOF-mediated H4K16 acetylation (H4K16ac) [54, 60]. Interestingly, however, this work contrasts with what is observed in Drosophila where a point mutation in the SET domain of trx is sufficient to produce trx-dependent developmental defects such as homeotic phenotypes [116]. This could be due to the fact that there is some redundancy in the mammalian system between the different MLL family members, at least in terms of H3K4me2/3 deposition.
Despite the fact that MLL and MLL2 are quite similar proteins, KOs of Mll2 display a much more severe phenotype that result in early embryonic lethality with widespread evidence of apoptosis [61]. No body patterning defects were observed, but this could be due to the severity of the early-stage phenotype [61]. Interestingly, loss of MLL2 did have an impact on Hox gene expression, but mostly members of the HoxB cluster, whereas MLL primarily affects HoxA and C cluster genes [39, 61, 83]. Another major difference is that an inducible Mll2 KO has no effect on adult tissues or on normal blood development [117], except perhaps for a highly specific phenotype in macrophages [118]. Knockdown of MLL2 in mouse ES cells results in a reduction in H3K4me3 levels primarily at bivalent genes (whose promoters are marked with both H3K4me3 and H3K27me3), suggesting that MLL2 has a specific function at these genes [119, 120], although it is not clear whether this function is exclusive to MLL2. Notably, knockdown of MLL2 does not appear to disrupt the transcriptional changes at bivalent genes induced by retinoic acid (RA) [119, 120], even though the MLL2-associated factor AKAP95 is required for RA-mediated gene induction in ES cells [81].
MLL3/MLL4
Mutation of trr, the Drosophila homolog of MLL3/4, displays a vastly different phenotype to trx. Unlike trx, trr mutants do not display Hox gene defects or interact with either PcG or trxG mutations [50]. Instead, trr acts in retinal development and promotes hormone receptor-mediated gene activation [51] as well as functioning in the suppression of tissue growth [121].
The extent to which MLL3 and MLL4 play different roles in the cell is unclear. MLL4 has been shown to be only partly independent of MLL3 in adipogenesis and myogenesis [122]. Mll3 KO mice die at birth with no obvious morphological abnormalities, whereas an Mll4 KO results in embryonic lethality around day E9.5 [122], comparable to the Mll KO mouse, although whether Mll4 KOs affect Hox gene expression patterns has not been studied. Unlike MLL2, MLL4 can impact RA-regulated genes [123]. There is also an association of MLL3/4 mutations with some leukemias [124], and consistent with this both MLL3 and MLL4 are required for normal blood stem and progenitor cell function [125, 126].
SETD1A/SETD1B
dSet1 is responsible for most of the H3K4me3 in Drosophila [52, 127] and as previously mentioned is the closest relative to yeast Set1 (Fig. 1). Despite their high level of similarity, both SETD1A and SETD1B are individually required for normal mouse embryogenesis and display quite different KO phenotypes [128]. Setd1a KO embryos display a severe phenotype and die before gastrulation, while Setd1b KO embryos survive until E11.5, albeit in a severely growth-retarded condition [128]. In line with the embryonic phenotypes, Setd1a KO ES cells stop proliferating and die, while ES cells tolerate the loss of Setd1b [128]. Interestingly, only Setd1a KO ES cells display a global loss of H3K4 methylation [128]. Inducible KO models indicate SETD1A has a role in B cell development [129] and in erythropoiesis [130], but otherwise a Setd1a deletion does not display hematopoietic defects.
Overall, there are profound phenotypic differences between MLL family members, even between the highly related gene pairs, which make it clear that each protein has a unique function in the organism. Since most MLL family members are ubiquitously expressed, the most common explanation for this is that their H3K4 methyltransferase activity is differentially targeted within the nucleus. There is some evidence for this (discussed below), but it cannot fully explain the vast differences in phenotype between the different family members. Instead, we argue the biological data support the idea that each protein has a distinct molecular function, and that KMT activity is not the sole or even the major molecular role of each protein. In particular, while KOs of the different MLL family members have profound impacts on normal development, this is not always associated with changes in H3K4 methylation. Additionally, even though it is the only member to have been directly tested, it is clear that loss of MLL SET domain activity does not accurately phenocopy loss of the entire protein during development.
Distinct functional activities of MLL family members
MLL3/4 and enhancer function
In common with MLL/MLL2, MLL3/4 were initially found to be important for regulating expression of a subset of target genes [123]. However, a broader role for these proteins has since been identified at gene enhancers, sequences distal to target promoters. Given the initial identification of Trx in Drosophila, it is perhaps fitting that its homolog Trr was first shown to deposit H3K4me1 at enhancers [131] before a similar role was identified for MLL3 and MLL4 [122, 132]. Consistent with this, in vitro MLL3/4 show a reduced ability to trimethylate H3K4, relative to mono- and dimethylation [80, 122, 133].
Deletion of MLL4 disrupts levels of many features associated with enhancers, including H3K4me1, H3K27ac, Mediator, RNA pol II and enhancer transcription (eRNAs) [107, 122, 134], and MLL3/4 KOs disrupt CBP/p300 binding at these loci [135, 136]. A key feature of active enhancers is their ability to interact with target gene promoters, and these contacts appear to be dependent on MLL3/4. Double KOs of Mll3/4 in ES cells result in a reduction in short-range (< 100 kb) interactions, as measured by Hi-C, correlating with MLL3/4-dependent H3K4me1 loci [100]. Higher resolution analysis revealed specific loss of promoter–enhancer contacts for Sox2 [100], Nanog and Lefty [135]. Loss of MLL3/4 also resulted in reduced Rad21 (a subunit of the cohesin complex) localisation to enhancers [100], suggesting a potential mechanism for disruption of promoter–enhancer interactions.
Taken together, these results argue for a key role for MLL3/4 in the establishment or maintenance of enhancers. Is MLL3/4 simply required for deposition of H3K4me1, or are the non-SET domains important for enhancer function? This issue has been addressed using an ES cell line in which SET domain point mutations were introduced in the endogenous copies of Mll3 and Mll4, depleting global H3K4me1 levels [100, 107]. Whilst a reduction in cohesin binding at enhancers was observed [100], there were minimal effects on transcription, either of eRNAs or at target genes [107] and similar results were seen for MLL3/4 SET domain deletions [108]. Notably, only small reductions in H3K27ac at enhancers were observed with the point mutants, compared to the strong reductions in the double KO line [107]. Thus, the primary function of MLL3/4 at enhancers is likely independent of histone methylation, although how this is achieved is, as yet, unclear. It is worth noting, however, that whilst H3K4me1 levels were depleted they were not completely eliminated, leaving the importance of this histone modification in enhancer function still an open question.
MLL family protein complexes
There is not sufficient space in this review to fully explore the detailed and diverse compositions of MLL family complexes. However, here we touch on some of the different complex components, with a particular focus on MLL, to illustrate how differences in complex composition may explain some of the phenotypic differences observed among family members.
The stable core complexes of MLL and MLL2 are highly similar with very little to differentiate them, although each protein displays additional protein interactions that may be suggestive of different functions of the two proteins. Both MLL and MLL2 are targeted for proteolytic cleavage by the threonine aspartase Taspase 1 [40, 137–139], generating 320-kDa N-terminal (MLL-N) and 180-kDa C-terminal (MLL-C) fragments, in the case of MLL (Fig. 2). These fragments do not dissociate after cleavage, however; the FYRN and FYRC domains interact to form a single domain, holding the complex together [140]. Interestingly, cleavage separates the C-terminal SET domain from the N-terminal portion, which contains all of the known binding domains of MLL.
As discussed above, the MLL SET domain is functional only when in complex with WDR5, RbBP5 and ASH2L [55]. In addition, MLL stably interacts with a number of other proteins to modulate its localisation and activity. The N-terminus of MLL is associated with two key factors, menin and LEDGF (lens epithelium-derived growth factor) [141–145]. Menin is required for the expression of MLL target genes, including Hoxa9, Meis1, CDKN1B (p27) and CDKN2C (p18) [72, 143, 146] and this interaction is necessary for MLL fusion protein-mediated leukemogenesis [143, 147]. The interaction with menin (although not LEDGF) is shared with the MLL2 protein [72], but not with any other members of the MLL family. The interaction between MLL and menin generates a binding pocket for LEDGF, thus generating a ternary complex [148]. LEDGF promotes transcriptional activation [149, 150] and is essential for leukemogenesis [144], suggesting that a key role of menin may be to bridge the interaction between the two proteins. This transcriptional coactivator function [149, 150] could be a unique aspect of MLL complex activity. At least in some experiments, menin and LEDGF do not appear to be required to recruit MLL to target genes, with the N-terminal, menin-interacting, region of MLL being dispensable for recruitment of MLL constructs to Hoxa9 [151].
MLL has also been observed to interact with transcription cofactors, suggesting that a co-localisation of activities may be important for its function. For example, interactions have been identified between MLL and the lysine acetyltransferases (KATs) CBP [152], MOZ [153] and MOF [54], the latter of which is known to be important for MLL target gene expression, likely via H4K16 acetylation [54, 60]. Alone among CXXC domains, the domain within MLL has also been found to interact with the PAF1 complex [151, 154], which may provide a bridge to RNA pol II itself [155]. This again could be a unique aspect of MLL function, since MLL2 does not bind to the PAF1 complex [151].
Another distinct characteristic of the founder MLL is its negative regulation via interaction with repressive factors. Several proteins have been observed to immunoprecipitate with the CXXC domain region of MLL, originally defined as a repression domain due to the effects of these interactions on transcription [156]. Binding partners include the PcG proteins BMI-1 and HPC2, and the corepressors CtBP and HDAC1 [157]. In addition, the third PHD of MLL is bound by the cyclophilin CyP33 [158–161], and the extended PHD3/bromodomain region interacts with ASB, a substrate recognition subunit for the Elongin B/C-Cullin-SOCS box protein (ECS) E3 ubiquitin-ligase, targeting the methyltransferase for proteolysis during hematopoiesis [162].
MLL2 has very few unique known interacting partners, other than an interaction with AKAP95 [81]. The MLL3 and MLL4 proteins have multiple distinct interacting partners including the H3K27me3 demethylase UTX, PA1, PTIP and p53 [73, 163, 164]. However, there is very little that differentiates MLL3/4. Similarly, the SETD1A/B complexes are highly similar [47–49, 80], although the two proteins show distinct subnuclear distributions [49], indicating they are non-redundant. In addition to its apparently major role in promoting H3K4me3 at promoters in mammals, SETD1A also has an important non-SET role in regulating the DNA damage response [165]. Although unique complex components can help explain functional divergence of MLL family members, it is still difficult to fully explain why all six MLL family members are individually required for organism survival, producing distinct KO phenotypes.
Recruitment of MLL family members
To some extent, the term recruitment can be misleading, as it would seem to imply a deterministic intent in directing proteins to where they need to go. Single-molecule tracking (SMT) experiments of transcription factor (TF) binding in E. coli show that free diffusion coupled with non-specific DNA-TF interactions directs TFs towards their high-affinity binding sites in a process termed facilitated diffusion [166]. Weak, non-specific interactions likely represent a general way in which DNA-binding factors are funnelled down an affinity gradient towards their high-affinity binding sites [167]. Chromatin proteins tend to lack high-affinity DNA-binding domains and are much more likely to contain a large number of low-affinity chromatin and DNA-binding domains [98, 168]. Interestingly, in mammalian SMT experiments, the behavior of chromatin-like proteins suggests that they never freely diffuse and are instead “trapped” by a large number of low-level weak interactions that cause them to slowly “creep” along the surface of chromatin [169]. This behavior is consistent with the notion of multivalency (discussed in [168]). Multivalent interactions appear to be relatively common for chromatin factors, where many of the binding domains have only a low affinity for their substrate. Combining the affinities of multiple interactions can not only increase the strength of binding, but can also create a higher stringency for localisation to sites at which multiple epitopes must be present. This is thought to be a major way in which chromatin proteins search chromatin and then create stable complexes at specific chromatin sites [98, 106, 151, 168]. This idea is also consistent with recent work on the involvement of chromatin proteins in the self-organizing formation of phase-separated condensates that generate distinct regulatory domains [170, 171].
A notable characteristic of the MLL family proteins is the presence of multiple chromatin-interaction domains (Fig. 2), suggesting that a multivalency effect may be responsible for their genomic association. All four MLL proteins contain multiple PHD fingers [56, 172], but they do not show the same interaction specificities. PHD3 of MLL preferentially binds H3K4me3 tails [151, 161, 173], allowing the KMT to recognize its own product, and this appears to be important for MLL localisation at individual target genes [151, 161, 173]. MLL also contains an atypical bromodomain adjacent to PHD3 that does not appear to interact with acetylated lysine residues, although it has low stability in vitro and a fully intact domain has been difficult to work with [161, 174].
MLL and MLL2 contain a CXXC domain [56, 175], a protein domain known to recognize non-methylated CG-rich DNA (CpG islands) [176–179]. Interestingly, although MLL and MLL2 overall share a highly similar domain architecture with Drosophila trx, trx itself does not include a CXXC domain [4, 10], likely because of the relative lack of CpG islands in Drosophila [180], suggesting some divergence in localisation mechanisms between the species. Recognition of unmethylated CpGs is thought to be an important localisation mechanism for MLL [151, 181], and is required for myeloid transformation by the MLL–ENL fusion protein [181]. Although SETD1A/B do not contain a CXXC domain, they reside in a complex with the CXXC domain-containing protein CFP1 [47–49] which stabilizes SETD1A/B activity at promoters [106]. MLL3 and MLL4, in contrast, do not possess CXXC domains, consistent with the fact that while most active gene promoters contain a CpG island [182], only a small proportion of enhancers are at CpG islands [183].
The MLL CXXC domain binds CpG DNA with a Kd of 4.3 µM [178], and the PHD3 affinity for H3K4me3 tails is similar (Kd 19 µM [173] or 4.3 µM [161]). Individually, these low µM interactions are unlikely to provide sufficient binding affinity to stabilize MLL binding at target loci, suggesting that the combination of the two interactions is necessary for an association with chromatin. In support of this, in an experiment involving recruitment of MLL-N fragments to the HoxA9 locus in Mll KO MEFs, both the CXXC domain- and PHD finger-containing regions of the protein were required [151].
These domains cannot be sufficient for precise targeting of MLL, however; the same bivalent interaction (binding to unmethylated CpG and H3K4me3) is also used by the SETD1A/B complexes [106]. These are localized to active gene promoters via CFP1, which contains both a PHD and CXXC domain [48, 184, 185]. No single ChIP-seq dataset exists to compare the binding profiles of these different KMTs; however, by inference and in some cases by direct comparison, MLL, MLL2 and SETD1A/B all show partially distinct genomic distributions [85, 106, 120]. This strongly argues that despite the common binding motifs, additional activities are required to discriminate the binding profiles of these different proteins. For example, MLL and MLL2 contain additional DNA-binding motifs in the form of multiple HMG-like N-terminal AT hooks [156], which promote binding to AT-rich DNA.
The AT hook and menin/LEDGF interaction domains of MLL are dispensable for recruitment of MLL-N to HoxA9 [151], although without a genome-wide analysis it is difficult to know whether this applies to all MLL-bound loci. LEDGF itself contains a PWWP domain with specificity for H3K36me2 [186–190]; whilst MLL and LEDGF binding are not observed at all sites of H3K36me2, this may provide an additional stabilizing interaction for the complex at chromatin. Indeed, MLL binding at several target genes is reduced upon knockdown of the H3K36 methyltransferase ASH1L, although this has only been specifically observed in MLL rearranged leukemias [190].
One additional possibility is that binding of MLL to other chromatin proteins, for example, the KATs p300/CBP, MOF and MOZ, may provide further stabilizing interactions at specific loci. Whilst it is possible to argue that MLL “recruits” these KATs to target genes, the reverse is also plausible; p300/CBP, for example, interact with a large number of transcription factors [191]. Indeed, MLL has been shown to bind to CBP cooperatively with CREB or MYB, suggesting a synergism with additional factors, which may enhance specificity [152, 192]. Further, given that the KATs themselves contain chromatin-interaction domains (for example, bromodomains [191]) a more nuanced model would be that together these proteins generate a network of relatively low-affinity interactions, stabilizing the complex as a whole at chromatin.
A more sequence-specific mechanism for MLL3/4 interaction with chromatin has been proposed. Ectopic expression of CEBPβ or HOXA9 is sufficient to generate novel enhancers, including binding of MLL3/4 and deposition of H3K4me1 [122, 193], suggesting the potential for transcription factor-mediated localisation. However, the mechanism behind this process has not yet been elucidated. In addition, MLL3/4 also contain multiple chromatin-binding domains, so additional stabilizing interactions may be involved. In contrast to the H3K4me3-binding MLL PHD3 domain, the tandem PHD4-6 region of MLL4 (see Fig. 2) recognizes the histone H4 N-terminal tail, dependent on the absence of symmetric dimethylation of H4R3 [194]. A further interaction between ePHD6, a fragment containing PHD6 and the preceding zinc finger, and histidine 18 of a histone H4 tail peptide, has also been demonstrated [195], indicating multiple contacts between MLL4 and histone H4. These interactions are required for the in vitro methyltransferase activity of MLL4, and mutation of PHD6 disrupts expression of MLL4-dependent genes in vivo [195], although it is not clear what effect, if any, this has on enhancer localisation. Overall, there is no clear and obvious mechanistic explanation for how each MLL family protein could be uniquely and specifically localized to gene targets without the inclusion of additional recruitment factors.
Conclusions on the role of H3K4 KMT activity in MLL family activity
One of the questions we wanted to address is: why are there so many MLL KMTs in mammals? Since each protein is individually required for normal development, mammals have clearly evolved a need for all six MLL family members. However, the data are mixed on how important the individual KMT activities actually are. As discussed above, differential recruitment mechanisms of the MLL family do not seem to address this issue of specificity. Instead, differences in protein complex stoichiometry suggest that the requirement for each individual KMT could be determined to some extent by abundance [188]. This fits with the observation that SETD1A is the most abundant MLL family member [188] and it also appears to be responsible for most H3K4me3 in the cell [128]. If this forms part of the explanation, there must be some variation in stoichiometry across different tissues and/or stages of development; for example, MLL2 appears to be the major KMT responsible for H3K4me3 levels during oogenesis [196].
MLL KMT activity is not required for normal development or hematopoiesis [59, 60], but this contrasts with the observation that in Drosophila, point mutations in the trx SET domain are sufficient to cause Hox gene-mediated developmental defects [116]. Given the duplication of MLL family members in mammals, it is possible that some redundancy in H3K4 methyltransferase activity exists, despite their unique non-methyltransferase functions, so the effect of the MLL SET deletion is masked by the presence of MLL2 SET domain function [120]. This also argues that at least globally, the KMT activity is more crucial for some members than for others, and that many of the MLL proteins have evolved to have other functions. For example, an important role for MLL may be recruitment of MOF and the promotion of H4K16ac [54, 60]. Even SETD1A, which appears to be the major KMT in mammals, has additional activities not related to SET domain catalytic activity [165]. Alongside interactions with different protein complex members, it also remains possible that the MLL KMTs could have specificity for unique non-histone protein targets, such as methylation of p53 [197, 198].
One other key question is: how important is H3K4 methylation for the cell? In the case of MLL3/4, it seems clear that the proteins themselves are required for enhancer function, but MLL3/4-mediated H3K4me1 is not essential [107, 108]. The evidence is also fairly strong that H3K4me3 is not absolutely required for transcriptional activation or enhancer function [103], and in fact is not intrinsically required for cell survival [41]. The SET domain mutation in Drosophila trx suggests that although H3K4me3 may not be important for transcription or cell survival per se, it still could have a key role in normal development [116]. Similarly, some PcG mutations have modest effects on gene regulation in ES cells, but these same PcG mutant ES cells are unable to properly control gene expression transitions through differentiation [199]. This is consistent with the original observations of PcG and trxG mutations, where early development often proceeded normally, and only at later points did development and normal gene regulation begin to break down [15, 17]. It has been argued that the role of H3K4me3 is to reduce transcriptional noise and increase consistency between individual cells, rather than dictate the population mean gene expression level [103, 200]. As most analyses of H3K4me3 look at bulk population averages, these effects could have been missed in past experiments. Further, given the levels of cellular heterogeneity in a developing organism, this issue is likely of particular relevance compared to the steady state of cell lines.
One major function of histone marks, such as H3K4me3, then, may be to contribute to transcriptional memory, ensuring that active genes retain that state during successive cell divisions. Notably, Trx remains associated with DNA during S phase, suggesting that it may rapidly methylate nascent nucleosomes after deposition [201]. Interestingly, when unusual DNA structures disrupt DNA replication, histone marks are lost and gene regulation breaks down [202]. This fits with the potential role of H3K4 methylation in stabilizing promoter complex formation [79], especially through the cell cycle, thus increasing the probability that proper gene expression patterns are maintained. Indeed, histone marks are known to influence TF binding immediately following DNA replication [203]. In this way, H3K4me3 may provide less of an on–off switch than a way to decrease the stochastic nature of complex formation, thus ensuring the stability of gene expression patterns. This may explain why these marks and proteins are so often mutated in cancer, where increased stochasticity may make advantageous changes more likely in the expression of genes regulating proliferation and differentiation. Importantly, such a model would also explain why these systems are only absolutely crucial during the highly dynamic processes of multi-cellular organism development.
Acknowledgements
T. A. M. and N. T. C. are supported by the Medical Research Council (MRC, UK) Molecular Haematology Unit grant MC_UU_12009/6 and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) Programme. T. A. M. is a founding shareholder of OxStem Oncology (OSO), a subsidiary company of OxStem Ltd. The authors thank Rob Klose for his constructive comments on the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.der Ziemin-van der Poel S, et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA. 1991;88(23):10735–10739. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Djabali M, et al. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet. 1992;2(2):113–118. doi: 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- 3.Gu Y, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71(4):701–708. doi: 10.1016/0092-8674(92)90603-A. [DOI] [PubMed] [Google Scholar]
- 4.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71(4):691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 5.Meyer C, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 7.Ballabio E, Milne TA. Molecular and epigenetic mechanisms of MLL in human leukemogenesis. Cancers. 2012;4(3):904–944. doi: 10.3390/cancers4030904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne TA. Mouse models of MLL leukemia: recapitulating the human disease. Blood. 2017;129(16):2217–2223. doi: 10.1182/blood-2016-10-691428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazo AM, et al. The trithorax gene, a trans-acting regulator of the bithorax complex in Drosophila, encodes a protein with zinc-binding domains. Proc Natl Acad Sci USA. 1990;87(6):2112–2116. doi: 10.1073/pnas.87.6.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis EB. Genetic control of developmental pathways in Drosophila melanogaster. Proc XII Int Congress Genet. 1968;1:96–97. [Google Scholar]
- 12.Ingham P, Whittle R. Trithorax: a new homoeotic mutation of Drosophila melanogaster causing transformations of abdominal and thoracic imaginal segments. Mol Gen Genet. 1980;179:607–614. doi: 10.1007/BF00271751. [DOI] [Google Scholar]
- 13.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276(5688):565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 14.Duncan I, Montgomery G. E. B. Lewis and the bithorax complex: part II From cis-trans test to the genetic control of development. Genetics. 2002;161(1):1–10. doi: 10.1093/genetics/161.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassis JA, Kennison JA, Tamkun JW. Polycomb and trithorax group genes in Drosophila. Genetics. 2017;206(4):1699–1725. doi: 10.1534/genetics.115.185116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slifer EH. A mutant stock of Drosophila with extra sex-combs. J Exp Zool. 1942;90:31–40. doi: 10.1002/jez.1400900103. [DOI] [Google Scholar]
- 17.Struhl G, Akam M. Altered distributions of Ultrabithorax transcripts in extra sex combs mutant embryos of Drosophila. EMBO J. 1985;4(12):3259–3264. doi: 10.1002/j.1460-2075.1985.tb04075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu BD, et al. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378(6556):505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 19.Yu BD, et al. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci USA. 1998;95(18):10632–10636. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capdevila MP, Garcia-Bellido A. Genes involved in the activation of the bithorax complex of Drosophila. Wilehm Roux Arch Dev Biol. 1981;190(6):339–350. doi: 10.1007/BF00863271. [DOI] [PubMed] [Google Scholar]
- 21.Ingham PW. Differential expression of bithorax complex genes in the absence of the extra sex combs and trithorax genes. Nature. 1983;306(5943):591–593. doi: 10.1038/306591a0. [DOI] [PubMed] [Google Scholar]
- 22.Capdevila MP, Botas J, Garcia-Bellido A. Genetic interactions between the Polycomb locus and the Antennapedia and Bithorax complexes of Drosophila. Roux Arch Dev Biol. 1986;195(7):417–432. doi: 10.1007/BF00375746. [DOI] [PubMed] [Google Scholar]
- 23.Hanson RD, et al. Mammalian Trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc Natl Acad Sci USA. 1999;96(25):14372–14377. doi: 10.1073/pnas.96.25.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brock HW, van Lohuizen M. The Polycomb group—no longer an exclusive club? Curr Opin Genet Dev. 2001;11(2):175–181. doi: 10.1016/S0959-437X(00)00176-3. [DOI] [PubMed] [Google Scholar]
- 25.Milne TA, Sinclair DA, Brock HW. The Additional sex combs gene of Drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol Gen Genet. 1999;261(4–5):753–761. doi: 10.1007/s004380050018. [DOI] [PubMed] [Google Scholar]
- 26.Fisher CL, et al. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev Biol. 2010;337(1):9–15. doi: 10.1016/j.ydbio.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gildea JJ, Lopez R, Shearn A. A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics. 2000;156(2):645–663. doi: 10.1093/genetics/156.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tschiersch B, et al. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994;13(16):3822–3831. doi: 10.1002/j.1460-2075.1994.tb06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones RS, Gelbart WM. The Drosophila Polycomb-group gene Enhancer of zeste contains a region with sequence similarity to trithorax. Mol Cell Biol. 1993;13(10):6357–6366. doi: 10.1128/MCB.13.10.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stassen MJ, et al. The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech Dev. 1995;52(2–3):209–223. doi: 10.1016/0925-4773(95)00402-M. [DOI] [PubMed] [Google Scholar]
- 31.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 32.Schultz J, et al. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28(1):231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray K. The occurrence of epsilon-N-methyl lysine in histones. Biochemistry. 1964;3:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15(18):2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 35.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 36.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14(3):286–298. doi: 10.1016/S0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 37.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 38.Manzur KL, et al. A dimeric viral SET domain methyltransferase specific to Lys27 of histone H3. Nat Struct Biol. 2003;10(3):187–196. doi: 10.1038/nsb898. [DOI] [PubMed] [Google Scholar]
- 39.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10(5):1107–1117. doi: 10.1016/S1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10(5):1119–1128. doi: 10.1016/S1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 41.Briggs SD, et al. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15(24):3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roguev A, et al. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20(24):7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy PL, et al. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci USA. 2002;99(1):90–94. doi: 10.1073/pnas.221596698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santos-Rosa H, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 45.Nislow C, Ray E, Pillus L. SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol Biol Cell. 1997;8(12):2421–2436. doi: 10.1091/mbc.8.12.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller T, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA. 2001;98(23):12902–12907. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wysocka J, et al. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17(7):896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Skalnik DG. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280(50):41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 49.Lee JH, et al. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem. 2007;282(18):13419–13428. doi: 10.1074/jbc.M609809200. [DOI] [PubMed] [Google Scholar]
- 50.Sedkov Y, et al. Molecular genetic analysis of the Drosophila trithorax-related gene which encodes a novel SET domain protein. Mech Dev. 1999;82(1–2):171–179. doi: 10.1016/S0925-4773(98)00246-9. [DOI] [PubMed] [Google Scholar]
- 51.Sedkov Y, et al. Methylation at lysine 4 of histone H3 in ecdysone-dependent development of Drosophila. Nature. 2003;426(6962):78–83. doi: 10.1038/nature02080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ardehali MB, et al. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J. 2011;30(14):2817–2828. doi: 10.1038/emboj.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hallson G, et al. dSet1 is the main H3K4 di- and tri-methyltransferase throughout Drosophila development. Genetics. 2012;190(1):91–100. doi: 10.1534/genetics.111.135863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dou Y, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121(6):873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 55.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13(8):713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 56.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25(1):15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 57.Li T, et al. Additional sex combs interacts with enhancer of zeste and trithorax and modulates levels of trimethylation on histone H3K4 and H3K27 during transcription of hsp70. Epigenet Chromat. 2017;10(1):43. doi: 10.1186/s13072-017-0151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheuermann JC, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terranova R, et al. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci USA. 2006;103(17):6629–6634. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mishra BP, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7(4):1239–1247. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaser S, et al. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133(8):1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- 62.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.FitzGerald KT, Diaz MO. MLL2: a new mammalian member of the trx/MLL family of genes. Genomics. 1999;59(2):187–192. doi: 10.1006/geno.1999.5860. [DOI] [PubMed] [Google Scholar]
- 64.Huntsman DG, et al. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene. 1999;18(56):7975–7984. doi: 10.1038/sj.onc.1203291. [DOI] [PubMed] [Google Scholar]
- 65.Tan YC, Chow VT. Novel human HALR (MLL3) gene encodes a protein homologous to ALR and to ALL-1 involved in leukemia, and maps to chromosome 7q36 associated with leukemia and developmental defects. Cancer Detect Prev. 2001;25(5):454–469. [PubMed] [Google Scholar]
- 66.Ruault M, et al. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene. 2002;284(1–2):73–81. doi: 10.1016/S0378-1119(02)00392-X. [DOI] [PubMed] [Google Scholar]
- 67.Prasad R, et al. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15(5):549–560. doi: 10.1038/sj.onc.1201211. [DOI] [PubMed] [Google Scholar]
- 68.Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–342. doi: 10.1016/j.gene.2017.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emerling BM, et al. MLL5, a homolog of Drosophila trithorax located within a segment of chromosome band 7q22 implicated in myeloid leukemia. Oncogene. 2002;21(31):4849–4854. doi: 10.1038/sj.onc.1205615. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, et al. MLL5 (KMT2E): structure, function, and clinical relevance. CMLS. 2017;74(13):2333–2344. doi: 10.1007/s00018-017-2470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goo YH, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003;23(1):140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hughes CM, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13(4):587–597. doi: 10.1016/S1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 73.Cho YW, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282(28):20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang H, et al. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8(6):1207–1217. doi: 10.1016/S1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 75.Hamamoto R, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004;6(8):731–740. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 76.Ferguson AD, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19(9):1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 77.Mishra BP, Ansari KI, Mandal SS. Dynamic association of MLL1, H3K4 trimethylation with chromatin and Hox gene expression during the cell cycle. FEBS J. 2009;276(6):1629–1640. doi: 10.1111/j.1742-4658.2009.06895.x. [DOI] [PubMed] [Google Scholar]
- 78.Strahl BD, et al. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci USA. 1999;96(26):14967–14972. doi: 10.1073/pnas.96.26.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lauberth SM, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152(5):1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang Z, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154(2):297–310. doi: 10.1016/j.cell.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang H, et al. Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat Struct Mol Biol. 2013;20(10):1156–1163. doi: 10.1038/nsmb.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ng HH, et al. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11(3):709–719. doi: 10.1016/S1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 83.Milne TA, et al. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102(41):14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pineda G, et al. Proteomics studies of the interactome of RNA polymerase II C-terminal repeated domain. BMC Res Notes. 2015;8:616. doi: 10.1186/s13104-015-1569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kerry J, et al. MLL-AF4 spreading identifies binding sites that are distinct from super-enhancers and that govern sensitivity to DOT1L inhibition in leukemia. Cell Rep. 2017;18(2):482–495. doi: 10.1016/j.celrep.2016.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bernstein BE, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 87.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 88.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 89.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Flanagan JF, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438(7071):1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 93.Sims RJ, 3rd, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28(4):665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442(7098):86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 95.Bian C, et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011;30(14):2829–2842. doi: 10.1038/emboj.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee J, et al. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat Struct Mol Biol. 2008;15(1):109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu R, Wang GG. Tudor: a versatile family of histone methylation ‘readers’. Trends Biochem Sci. 2013;38(11):546–555. doi: 10.1016/j.tibs.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruthenburg AJ, et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145(5):692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131(1):58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 100.Yan J, et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018;28(2):204–220. doi: 10.1038/cr.2018.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Margaritis T, et al. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3′-end antisense transcription. PLoS Genet. 2012;8(9):e1002952. doi: 10.1371/journal.pgen.1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Clouaire T, et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26(15):1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Howe FS, et al. Is H3K4me3 instructive for transcription activation? BioEssays. 2017;39(1):1–12. doi: 10.1002/bies.201600095. [DOI] [PubMed] [Google Scholar]
- 104.Chen K, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47(10):1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benayoun BA, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2015;163(5):1281–1286. doi: 10.1016/j.cell.2015.10.051. [DOI] [PubMed] [Google Scholar]
- 106.Brown DA, et al. The SET1 complex selects actively transcribed target genes via multivalent interaction with CpG island chromatin. Cell reports. 2017;20(10):2313–2327. doi: 10.1016/j.celrep.2017.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dorighi KM, et al. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell. 2017;66(4):568–576. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rickels R, et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet. 2017;49(11):1647–1653. doi: 10.1038/ng.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hess JL, et al. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood. 1997;90(5):1799–1806. [PubMed] [Google Scholar]
- 110.Yagi H, et al. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92(1):108–117. [PubMed] [Google Scholar]
- 111.Ernst P, et al. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell. 2004;6(3):437–443. doi: 10.1016/S1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 112.McMahon KA, et al. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1(3):338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 113.Jude CD, et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1(3):324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gan T, et al. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24(10):1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang P, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Breen TR. Mutant alleles of the Drosophila trithorax gene produce common and unusual homeotic and other developmental phenotypes. Genetics. 1999;152(1):319–344. doi: 10.1093/genetics/152.1.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glaser S, et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenet Chromat. 2009;2(1):5. doi: 10.1186/1756-8935-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Austenaa L, et al. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012;36(4):572–585. doi: 10.1016/j.immuni.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 119.Hu D, et al. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol. 2013;20(9):1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Denissov S, et al. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141(3):526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 121.Kanda H, et al. The Drosophila ortholog of MLL3 and MLL4, trithorax related, functions as a negative regulator of tissue growth. Mol Cell Biol. 2013;33(9):1702–1710. doi: 10.1128/MCB.01585-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee J-E, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo C, et al. Global identification of MLL2-targeted loci reveals MLL2′s role in diverse signaling pathways. Proc Natl Acad Sci USA. 2012;109(43):17603–17608. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang W, Ernst P. SET/MLL family proteins in hematopoiesis and leukemia. Int J Hematol. 2017;105(1):7–16. doi: 10.1007/s12185-016-2118-8. [DOI] [PubMed] [Google Scholar]
- 125.Chen C, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25(5):652–665. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Santos MA, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514(7520):107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mohan M, et al. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol. 2011;31(21):4310–4318. doi: 10.1128/MCB.06092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bledau AS, et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development (Cambridge, England) 2014;141(5):1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- 129.Tusi BK, et al. Setd1a regulates progenitor B-cell-to-precursor B-cell development through histone H3 lysine 4 trimethylation and Ig heavy-chain rearrangement. FASEB J. 2015;29(4):1505–1515. doi: 10.1096/fj.14-263061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li Y, et al. Setd1a and NURF mediate chromatin dynamics and gene regulation during erythroid lineage commitment and differentiation. Nucleic Acids Res. 2016;44(15):7173–7188. doi: 10.1093/nar/gkw634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Herz HM, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26(23):2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hu D, et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33(23):4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wu L, et al. ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol Cell. 2013;49(6):1108–1120. doi: 10.1016/j.molcel.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lin-Shiao E, et al. KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev. 2018;32(2):181–193. doi: 10.1101/gad.306241.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang C, et al. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci USA. 2016;113(42):11871–11876. doi: 10.1073/pnas.1606857113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lai B, et al. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45(11):6388–6403. doi: 10.1093/nar/gkx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yokoyama A, et al. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100(10):3710–3718. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- 138.Hsieh JJ, Cheng EH, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115(3):293–303. doi: 10.1016/S0092-8674(03)00816-X. [DOI] [PubMed] [Google Scholar]
- 139.Hsieh JJ, et al. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23(1):186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Garcia-Alai MM, et al. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 2010;19(7):1432–1438. doi: 10.1002/pro.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yokoyama A, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24(13):5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Milne TA, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci USA. 2005;102(3):749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 144.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14(1):36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murai MJ, et al. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286(36):31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen YX, et al. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci USA. 2006;103(4):1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Caslini C, et al. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67(15):7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang J, et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482(7386):542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ge H, Si Y, Roeder RG. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998;17(22):6723–6729. doi: 10.1093/emboj/17.22.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brown-Bryan TA, et al. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol Cancer Res. 2008;6(8):1293–1307. doi: 10.1158/1541-7786.MCR-08-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Milne TA, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38(6):853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ernst P, et al. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol. 2001;21(7):2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paggetti J, et al. Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34 + cells. Oncogene. 2010;29(36):5019–5031. doi: 10.1038/onc.2010.254. [DOI] [PubMed] [Google Scholar]
- 154.Muntean AG, et al. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17(6):609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhu B, et al. The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev. 2005;19(14):1668–1673. doi: 10.1101/gad.1292105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci USA. 1994;91(22):10610–10614. doi: 10.1073/pnas.91.22.10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xia ZB, et al. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci USA. 2003;100(14):8342–8347. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fair K, et al. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol. 2001;21(10):3589–3597. doi: 10.1128/MCB.21.10.3589-3597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hom RA, et al. Molecular mechanism of MLL PHD3 and RNA recognition by the Cyp33 RRM domain. J Mol Biol. 2010;400(2):145–154. doi: 10.1016/j.jmb.2010.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Park S, et al. The PHD3 domain of MLL acts as a CYP33-regulated switch between MLL-mediated activation and repression. Biochemistry. 2010;49(31):6576–6586. doi: 10.1021/bi1009387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang Z, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell. 2010;141(7):1183–1194. doi: 10.1016/j.cell.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang J, Muntean AG, Hess JL. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood. 2012;119(5):1151–1161. doi: 10.1182/blood-2011-06-362079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lee S, et al. Crucial roles for interactions between MLL3/4 and INI1 in nuclear receptor transactivation. Mol Endocrinol. 2009;23(5):610–619. doi: 10.1210/me.2008-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kim J-H, et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Can Res. 2014;74(6):1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hoshii T, et al. A non-catalytic function of SETD1A regulates cyclin K and the DNA damage response. Cell. 2018;172(5):1007–1021. doi: 10.1016/j.cell.2018.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hammar P, et al. The lac repressor displays facilitated diffusion in living cells. Science (New York, N Y) 2012;336(6088):1595–1598. doi: 10.1126/science.1221648. [DOI] [PubMed] [Google Scholar]
- 167.Jen-Jacobson L, Engler LE, Jacobson LA. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure (London, England: 1993) 2000;8(10):1015–1023. doi: 10.1016/S0969-2126(00)00501-3. [DOI] [PubMed] [Google Scholar]
- 168.Ruthenburg AJ, et al. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8(12):983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Izeddin I, et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife. 2014;3:e02230. doi: 10.7554/eLife.02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cho WK, et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361(6400):412–415. doi: 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sabari BR, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361(6400):eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39(21):9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chang PY, et al. Binding of the MLL PHD3 finger to histone H3K4me3 is required for MLL-dependent gene transcription. J Mol Biol. 2010;400(2):137–144. doi: 10.1016/j.jmb.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Filippakopoulos P, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem Soc Trans. 2013;41(3):727–740. doi: 10.1042/BST20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Voo KS, et al. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol Cell Biol. 2000;20(6):2108–2121. doi: 10.1128/MCB.20.6.2108-2121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Birke M, et al. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002;30(4):958–965. doi: 10.1093/nar/30.4.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Allen MD, et al. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 2006;25(19):4503–4512. doi: 10.1038/sj.emboj.7601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bach C, et al. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 2009;28(6):815–823. doi: 10.1038/onc.2008.443. [DOI] [PubMed] [Google Scholar]
- 180.Aerts S, et al. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genom. 2004;5(1):34. doi: 10.1186/1471-2164-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ayton PM, Chen EH, Cleary ML. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol. 2004;24(23):10470–10478. doi: 10.1128/MCB.24.23.10470-10478.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bell JSK, Vertino PM. Orphan CpG islands define a novel class of highly active enhancers. Epigenetics. 2017;12(6):449–464. doi: 10.1080/15592294.2017.1297910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tate CM, Lee JH, Skalnik DG. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J. 2009;277:210–223. doi: 10.1111/j.1742-4658.2009.07475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thomson JP, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464(7291):1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pradeepa MM, et al. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8(5):e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Eidahl JO, et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013;41(6):3924–3936. doi: 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van Nuland R, et al. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33(10):2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Okuda H, et al. MLL fusion proteins link transcriptional coactivators to previously active CpG-rich promoters. Nucleic Acids Res. 2014;42(7):4241–4256. doi: 10.1093/nar/gkt1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhu L, et al. ASH1L links histone h3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016;6(7):770–783. doi: 10.1158/2159-8290.CD-16-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Voss AK, Thomas T. Histone lysine and genomic targets of histone acetyltransferases in mammals. BioEssays. 2018;40(10):e1800078. doi: 10.1002/bies.201800078. [DOI] [PubMed] [Google Scholar]
- 192.Goto NK, et al. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277(45):43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 193.Sun Y, et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell. 2018;34(4):643–658. doi: 10.1016/j.ccell.2018.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dhar SS, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26(24):2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu Y, et al. Structural insights into trans-histone regulation of H3K4 methylation by unique histone H4 binding of MLL3/4. Nat Commun. 2019;10(1):36. doi: 10.1038/s41467-018-07906-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Andreu-Vieyra CV, et al. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8(8):e1000453. doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huang J, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444(7119):629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 198.West LE, Gozani O. Regulation of p53 function by lysine methylation. Epigenomics. 2011;3(3):361–369. doi: 10.2217/epi.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Riising EM, et al. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell. 2014;55(3):347–360. doi: 10.1016/j.molcel.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 200.Benayoun BA, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158(3):673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Petruk S, et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell. 2012;150(5):922–933. doi: 10.1016/j.cell.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Guilbaud G, et al. Local epigenetic reprogramming induced by G-quadruplex ligands. Nat Chem. 2017;9(11):1110–1117. doi: 10.1038/nchem.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Petruk S, et al. Delayed accumulation of H3K27me3 on nascent DNA is essential for recruitment of transcription factors at early stages of stem cell differentiation. Mol Cell. 2017;66(2):247–257. doi: 10.1016/j.molcel.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]