

Abstract
Background
Patients with microsatellite instability-high (MSI-H) colorectal cancer (CRC) generally have a better prognosis than patients with microsatellite stable (MSS) CRC. However, some MSI-H CRC patients do not gain overall survival benefits from immune checkpoint-blockade treatment. In other words, heterogeneity within the subgroup of MSI-H tumors remains poorly understood. Thus, an in-depth molecular characterization of MSI-H tumors is urgently required.
Methods
Here, we use nonnegative matrix factorization (NMF)-based consensus clustering to define CRC MSI-H subtypes in The Cancer Genome Atlas and a French multicenter cohort GSE39582. CIBERSORT was used to calculate the proportions of 22 lymphocytes in tumor tissue in MSI-H subtypes.
Results
MSI-H CRC samples basically clustered into two subgroups (MSI-H1 and MSI-H2). MSI-H1 was characterized by a lower BRAF mutational status, higher frequency of chromosomal instability, global hypomethylation, and worse survival than MSI-H2. Further examination of the immune landscape showed that macrophages of the M2 phenotype were enriched in MSI-H1, which may be associated with poor prognosis in this subgroup.
Conclusions
Our results illustrate the genetic heterogeneity in MSI-H CRCs and macrophages may serve as good targets for anticancer therapy in MSI-H1.
Graphical abstract

Electronic supplementary material
The online version of this article (10.1186/s12964-019-0397-4) contains supplementary material, which is available to authorized users.
Keywords: Colorectal cancer, Microsatellite instability-high, Subtyping, Tumor-associated macrophages, Tumor-infiltrating lymphocytes
Background
Much effort has been devoted to the molecular subtyping of colorectal cancer (CRC) based on gene expression profiles [1–3]. According to the current widely accepted consensus molecular subtype (CMS) classification system, microsatellite instability-high (MSI-H) samples belong to the CMS1 subtype and are characterized by hypermutation and CpG island methylator phenotype (CIMP). The MSI-H status (commonly linked to a high mutational burden) may be associated with a better prognosis due to the accumulation of somatic mutations [4, 5]. However, we previously found that even in a hypermutated subpopulation can be further divided into two groups with either a high or low prognostic index [6]. In addition, MSI-H tumors generally respond well to immunotherapy by anti-PD-1 immune checkpoint inhibition [7]. Nevertheless, JAK1 loss-of-function mutations, with a prevalence of 20% in MSI-positive CRC are associated with the upregulation of genes associated with resistance to anti-PD-1 treatment [8]. Thus, the MSI-H population may also display different expression patterns that are masked by higher variations relative to other subtypes, such as CMS2–4.
In this study, we used nonnegative matrix factorization (NMF)-based consensus clustering to define MSI-H CRC subtypes. Intriguingly, we found that MSI-H CRC can be separated into two different subtypes with distinct molecular profiles, which help us better understand the heterogeneity within MSI-H.
Materials and methods
Somatic mutation data retrieval and processing
CRC somatic mutation data and clinical information were downloaded from The Cancer Genome Atlas (TCGA) data portal (05/03/2015). Silent mutations, RNA mutations, and any mutation in the intron, 5′ untranslated region (UTR), and 3’UTR were discarded. Retained mutational profiles of each case were used to refine the list of mutated genes in a total of 59 MSI-H tumors [9]. Lastly, clinical information of each patient (Additional file 1: Table S1) was added to mutational information via unique patient ID.
Gene expression data processing and normalization
Level 3 tumor mRNA expression data sets (RNASeqV2) were obtained from TCGA (October 2015). The GSE39582 (Affymetrix HG U133 Plus 2.0 arrays) dataset was downloaded from the Gene Expression Omnibus (GEO). Raw CEL files were processed using the affy package of BioConductor [10]. Then, MAS5 algorithm was used for background correction, normalization and summarization of single probes for all probe sets. Analysis of differentially expressed mRNA was performed using the DEGSeq package for R/Bioconductor [11]. Genes with expression levels < 1 (RNA-Seq by Expectation Maximization (RSEM)-normalized counts) in more than 50% of samples were removed. Significant differentially expressed mRNAs were selected according to a false discovery rate (FDR)-adjusted P value < 0.05 and fold change > 2 conditions. NMF was performed using the NMF package for R [12].
HM450k data retrieval and processing
DNA methylation data (HumanMethylation450) were downloaded from TCGA data portal (11/13/2016). The methylation level of each probe was measured with a beta value ranging from 0 to 1; this value is calculated as the ratio of the methylated signal to the sum of the methylated and unmethylated signals. Probes with an “NA” value in more than 10% of the CRC samples were discarded. Next, the Bioconductor package limma was used to identify differentially methylated sites (DMSs) in the remaining probes [13]. Significant DMSs were selected according to FDR-adjusted P value < 0.01 and beta value change > 0.2. All heatmaps were generated using the pheatmap package in R (64-bit, version 3.0.2).
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses
GO and KEGG enrichment analyses were performed using the clusterProfiler package from BioConductor [14]. Significantly enriched GO terms and pathways were selected according to an FDR-adjusted P value < 0.01.
Network construction and hub gene definition
Coexpression network construction was performed as described in our previous work [15]. Hub genes were those with an extremely high level of connectivity in a given network. Connectivity reflects how frequently a node interacts with other nodes and the sum of the weights across all edges of a node. Here, the top 50 genes with the highest connectivity in each module that were reasonable to display were defined as hub genes as previously described [16].
Survival analysis
Survival differences between MSI-H1 and MSI-H2 were tested by the Kaplan-Meier method and analyzed with the log-rank test with functions survfit and survdiff in the survival package for R [17]. Cox univariate model was carried out with function coxph in the R package survival. A P value < 0.05 was considered significant.
Deciphering lymphocytes in tumor tissue in MSI-H populations
To accurately quantify the relative amount of distinct lymphocytes in tumor tissue, CIBERSORT was used to calculate the proportions of 22 lymphocytes in tumor tissue [18]. The permutations were set to > = 100, and quantile normalization (QN) of the input expression mixture was set to FALSE for TCGA RNAseq data.
Immunohistochemistry (IHC) of Zhejiang University cancer institute (ZUCI) dMMR samples
Subtyping of 28 ZUCI dMMR frozen tissue samples was based on RNA-seq dataset. Matched formalin fixed paraffin-embedded (FFPE) samples were collected from pathology department. IHC staining and semi-quantitative analysis were performed as our previous work [19]. The four μm sections were incubated with the anti-CD68 (1:500 dilution, Cell Signaling Technology, 76437) and CD163 (1:500 dilution, Cell Signaling Technology, 93498) antibody, respectively.
Results
MSI-H CRC clusters into two distinct subtypes
A stochastic NMF algorithm was used to assess whether any clusters were present in the MSI-H CRC cases. To determine the best factorization rank r, a critical parameter in NMF, different values two to six was calculated. Then, the best r value was chosen according to some quality measures, such as the first value of r for which the cophenetic coefficient starts decreasing, the first value where the residual sum of squares (RSS) curve presents an inflection point [20], and direct visual inspection of the consensus matrix. Notably, r = 2 met all of these quality criteria, in other words; the MSI-H tumors fell into two separate subgroups (Fig. 1a). Of the 59 MSI-H samples annotated in TCGA, 11 were classified into MSI-H group 1 (MSI-H1) and 48 were classified into MSI-H group 2 (MSI-H2). The ratio of MSI-H1 to MSI-H2 was approximately 1:4 (Fig. 1b).
Fig. 1.

MSI-H CRC cases clustered into two gene expression-based subtypes. a, Clustering of 59 MSI-H CRC cases in TCGA by NMF. Correlation matrix heatmaps correspond to ranks 2 to 6. b, The association of clinicopathologic factors and MSI-H subtypes is shown on the top. Heatmap displaying the expression pattern of MSI-H subtypes
To validate the MSI-H subtypes found in TCGA dataset, we used the GSE39582 dataset [21] with 77 dMMR (deficient in DNA mismatch repair system (MMR)) samples to explore clustering (Additional file 2: Figure S1). Consistent with the two subtypes of MSI-H CRC in TCGA, two clusters were also observed in the French multicenter cohort (MSI-H1: MSI-H2 was approximately 1:3, Additional file 3: Figure S2). MSI-H CRC has a relatively good prognosis when diagnosed as stage II and a poor prognosis when diagnosed as advanced stages, suggesting that stage IV MSI-H CRC may differ considerably at the molecular level from stage II MSI-H CRC. As stage II CRC has the highest prevalence of MSI (81% in TCGA and 69% in GSE39582), a separate MSI subtyping was performed for stage II CRC only. Results showed that MSI subtyping remained basically unchanged (MSI-H1 and MSI-H2), suggesting that MSI-H CRC molecular subtyping was independent of tumor stages (Additional file 4: Figure S3 and Additional file 5: Figure S4).
MSI-H1 harbors a lower BRAF mutational frequency and a higher frequency of chromosomal instability
The ratio of MSI-H1: MSI-H2 closely corresponds to Lynch syndrome-related tumors and sporadic MSI tumors, which are two major classes of CRC. To test this hypothesis, we extracted the methylation probes specific to MLH1 and BRAF (V600E) status and found that MLH1 hypermethylation was always accompanied by BRAF mutations in both MSI-H1 and MSI-H2, indicating that MSI-H1 did not correspond to Lynch syndrome, because MLH1 deficiency and BRAF mutations rarely occur simultaneously in this disorder. Additionally, consistent with previous findings, a mutual exclusivity was observed between BRAF and KRAS mutations (Fig. 1b).
In addition, there was no significant association between MSI-H subtypes and tumor locations, sex or American Joint Committee on Cancer (AJCC) stages according to Fisher’s exact test (Fig. 1b). Both subtypes were enriched in stage II/III (75%) and right-sided colon (80%). However, we found that MSI-H1 was enriched in POLE mutations (55% in MSI-H1 vs. 30% in MSI-H2). Interestingly, a lower BRAF mutational frequency was observed in MSI-H1 (36%) than in MSI-H2 (56%), while no difference was found in the KRAS status. This was also the case in GSE39582, with a BRAF mutational frequency of 35% and 43% for MSI-H1 and MSI-H2, respectively. However, approximately two-fold higher chromosomal instability was observed in MSI-H1 than in MSI-H2 (50% vs. 29%, GSE39582).
The tumor immune regulatory network was disrupted in MSI-H1 subtype
To better understand the molecular difference between MSI-H1 and MSI-H2, it is necessary to determine specific genes that are enriched in each subtype. Differential expression analysis between MSI-H1 and MSI-H2 identified 1,669 and 944 differentially expressed genes (DEGs) in TCGA and GSE39582, respectively, with 298 shared between the two datasets (Fig. 2a, Additional file 6: Table S2). Intriguingly, only 130 genes were down-regulated in MSI-H1. A remarkable 11-fold increase in up-regulated genes found in MSI-H1 inspired us to examine their functional enrichment. GO revealed that these genes were mainly enriched in the immune response, such as regulation of cell adhesion, T cell activation, and lymphocyte differentiation (Fig. 2b). For example, a much higher expression level of CCL2/5, CXCL12, CD86, and CD163 was observed in MSI-H1. KEGG pathway enrichment analysis revealed that MSI-H1 was enriched in genes corresponding to the PI3K-Akt (P = 1.7E-7, FDR adjusted), Ras (P = 7.8E-4), Rap1 (P = 6.2E-7), and Chemokine (P = 1.1E-3) signaling pathways (Fig. 2c). We also performed gene set enrichment analyses (GSEA) to decipher the molecular signatures underlying MSI-H1 and MSI-H2 by using the much broader landscape of signatures of hallmark processes collected in the MSigDB database [22]. Notably, epithelial to mesenchymal transition, apical junction, myogenesis, inflammatory response, and KRAS signaling up were enriched in MSI-H1 (Additional file 7: Figure S5 a~d). While E2F targets, MYC targets, G2M checkpoint, glycolysis, mtorc1 signaling, and oxidative phosphorylation had high scores in MSI-H2 (Additional file 7: Figure S5 e~j).
Fig. 2.

Characterization of functional enrichment of DEGs reside in the MSI-H subtypes. a, Venn diagram displaying the number of genes that overlapped within DEGs and DMSs identified in TCGA and GSE39582 datasets. b, GO enrichment of up-regulated DEGs found in MSI-H1. c, KEGG pathway enrichment of up-regulated DEGs found in MSI-H1. d, Coexpression regulatory network identified by WGCNA. Genes that were not coexpressed were assigned to the gray group. Each vertical line corresponds to a gene, and branches are expression modules of highly interconnected groups of genes. In total, seven modules ranging from 146 to 2,939 genes in size were identified in MSI-H2. The yellow module associated with the immune response was selected for visualization
Considering the substantial difference in expression profiles between MSI-H1 and MSI-H2, we speculated that the intrinsic regulatory network also differs to a great extent. To this end, we used weighted correlation network analysis (WGCNA) to determine core gene regulatory modules. Interestingly, seven modules consisting of at least 100 genes were discovered in MSI-H2. Notably, the yellow module represented by CCR1, CD163, CD2, CD52, CD53, and CD86 specific to MSI-H2 was linked to the immune response (Fig. 2d). This observation indicated that the subtle immune regulatory network was lost in MSI-H1, although thousands of immune-related genes were switched on.
MSI-H subtypes have distinct methylation patterns
The transcriptome may be too volatile to be affected by some driver mutations. Thus, global DNA methylation pattern was also interrogated between MSI-H subtypes to determine whether the discrepancy between subtypes extended beyond driver mutations. Consistent with the expression pattern, the methylation pattern fell into two main clusters (Fig. 3). A slight global hypomethylated status was observed in MSI-H1. Further in-depth analysis revealed that 3,101 CpGs that covered 1,353 genes were differentially methylated between MSI-H1 and MSI-H2 (Additional file 8: Table S3). Among them, 310 genes overlapped with DEGs identified in TCGA (Fig. 2a). Interestingly, the abovementioned immune-related DEGs were rarely associated with epigenetic regulation (P > 0.05, χ2 test).
Fig. 3.

Clustering of 59 MSI-H CRC samples based on DNA methylation profiles. Although the methylation clusters were not identical to the MSI-H subtypes based on expression data, the main trend was preserved
In CRC, the dMMR status is strongly associated with the CIMP [21]. Therefore, we wanted to determine whether the CIMP is associated with MSI-H subtypes. Notably, in contrast to chromosomal instability, a much higher frequency of samples with the CIMP was observed in MSI-H2 (67%) than in MSI-H1 (42%, GSE39582), which, to an extent, was consistent the global hypomethylated status of MSI-H1 samples assembled in TCGA. Additionally, no MLH1-associated CpGs were differentially methylated; in other words, MSI-H subtypes had little association with MLH1 methylation status. In addition, the extent of MLH1 hypermethylation was even higher in MSI-H2 (71%) than in MSI-H1 (55%).
MSI-H1 has a much poorer prognosis than MSI-H2
Previous studies commonly propose that BRAF mutations are associated with a poor prognosis [23, 24]. We thus compared overall survival (OS) between MSI-H subtypes using the Cox proportional hazards model. Interestingly, patient survival in MSI-H1 was significantly poorer than that in MSI-H2 (Additional file 9: Figure S6, hazard ratio (HR) = 2.464, 95% confidence interval (CI), 1.13–5.37, P = 0.019). The 5-year OS rates were 61% (95% CI, 45 to 84%) and 82% (95% CI, 73 to 93%), respectively. As stage II CRC would be clinically most relevant to MSI-H subtypes, we also compared OS between stage II MSI-H subtypes. The findings held true even after adjusting for stages likely to influence the results (Fig. 4). Nonetheless, it is not known why MSI-H2 CRCs with a higher BRAF mutational frequency have a better outcome.
Fig. 4.
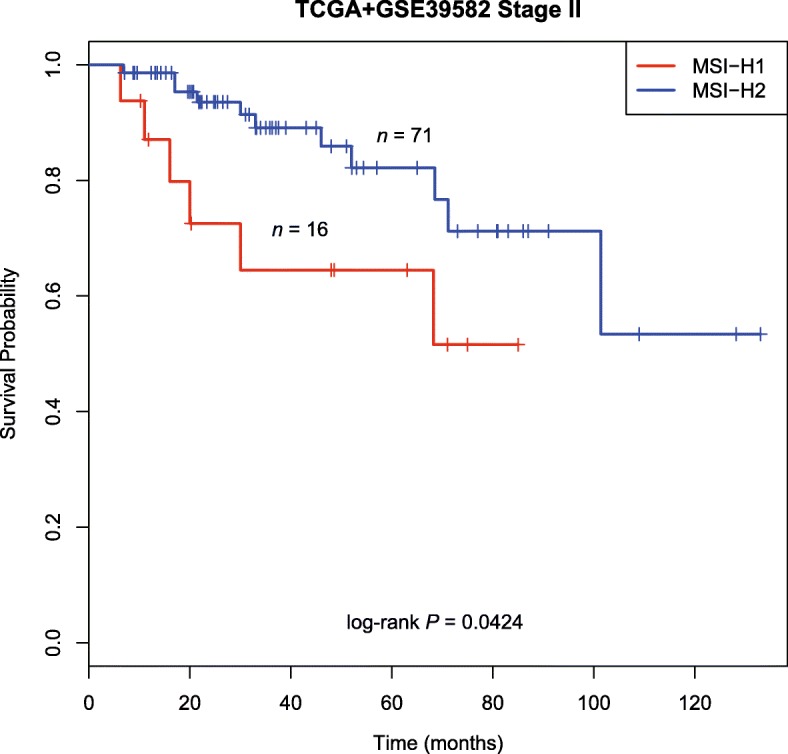
Survival status of stage II MSI-H CRC subtypes. Kaplan-Meier curves showing OS according to MSI-H subtypes. Clearly, a better prognosis was observed for MSI-H2 than for MSI-H1
Worse prognosis of MSI-H1 may be associated with the enrichment of M2 macrophages and PD-L2 expression
The lower frequency of BRAF mutations in MSI-H1 accompanied by a relatively worse prognosis inspired us to identify the potential tumor microenvironment component underlying this contradictory association. First, we compared the tumor mutational burden (TMB) between MSI-H1 and MSI-H2 because it has a direct impact on the abundance of neoantigens. However, MSI-H1 harbored a similar TMB to MSI-H2 (median mutational frequency ~ 31/million bases, P = 0.28, Wilcoxon’s test). Thus, the difference may be related to the participating cell population in the tumor immune microenvironment (TIME), which plays an important role in the poor outcomes in MSI-H1. Deconvolution of 22 lymphocytes in tumor tissue using CIBERSORT revealed a significant enrichment of M2 macrophages in MSI-H1 (Fig. 5a and b, Additional file 10: Table S4, P < 0.001). Tumor-associated macrophages (TAMs) of the M2 phenotype are well-known to promote tumor proliferation and are associated with a poor prognosis in different cancer types [25, 26]. CD163 and CD206, two canonical markers of M2 TAMs, had a much higher expression level in MSI-H1 compared to MSI-H2 both in TCGA and GSE39582 (Fig. 5c-f, P < 0.001). We then examined the expression of TAM markers (CD68 and CD163) in serial FFPE sections from selected dMMR CRC cases. Notably, we found that CD68 and CD163 had a much higher expression level in MSI-H1 than in MSI-H2 (Fig. 5g-j). This trend also held true for CCL2 and CCL5 (Additional file 11: Figure S7, P < 0.01). Tumor-derived CCL2/5 are significant indicators of early relapse and infiltration of TAMs, which contribute to cancer cell proliferation, inflammatory microenvironment of tumors, immune response evasion and angiogenesis [27, 28].
Fig. 5.

Identification of underlying molecular profiles associated with worse outcomes in MSI-H1. The infiltration of M2 macrophages was more pronounced in MSI-H1 than in MSI-H2 using TCGA (a) and GSE39582 (b) data. For c~f, boxplot distribution of CD163 and CD206 expression levels using TCGA and GSE39582 data. As for g~j, representative IHC staining for M2 TAM markers (CD68 and CD163) of serial sections from MSI-H1 and MSI-H2. For k and l, boxplot distribution of PD-L2 expression levels using TCGA and GSE39582 data
Furthermore, we found that the expression of the immune checkpoint molecule PD-L2 (Programmed cell death 1 ligand 2, also known as PDCD1LG2 or CD273) was significantly higher (> 3-fold) in MSI-H1 than inMSI-H2 (Fig. 5k and l, P < 0.001). However, no significant difference was observed in other immune checkpoint molecules such as CTLA-4, PD-1/PD-L1, and LAG-3.
Discussion
To the best of our knowledge, this is the first systematic subtyping of MSI-H CRC. Both transcriptome and DNA methylome uncovered that two major subtypes were presented in MSI-H CRC, suggesting that MSI-H subtypes was not associated with specific driver mutations. The MSI-H status is well known to confer good prognosis in CRC [2, 29, 30]. Nevertheless, in this study, we found that one-quarter of the MSI-H CRC cases displayed distinct molecular features and poor prognoses. As patients with MSI-H2 CRC had a higher frequency of BRAF alterations but a better outcome, we believe that the BRAF status is nonsignificant in MSI samples. By evaluating 477 MSI CRC cases, Taieb et al. [31] found that no prognostic role of BRAF mutations in patients with MSI CRC. Barras et al. [32] found that CRC cases with BRAF mutations can be classified into two subtypes and should not be regarded as having a unique biology that may be exploited for drug targeting. Furthermore, Shimada et al. [33] demonstrated that patients with non-V600E mutant-type cancer had a better OS than patients with V600E mutant-type cancer, which is in line with previous observations [34, 35]. In addition, Laura et al. [36] found that height is a stronger risk factor for CRCs with BRAF mutations or MSI, suggesting that molecular pathological epidemiology (MPE) demonstrates the strengths of interdisciplinary integration to study CRC because more and more evidence indicates that diet, smoking, lifestyle, and the microbiome may influence the genome, epigenome, transcriptome, proteome, and metabolome of tumor [37]. Thus, more careful attention should be paid in the treatment of heterogeneous MSI-H population.
Due to defects in DNA MMR, MSI-H tumors have a much higher rate of nonsynonymous mutations, leading to an increased number of neoepitopes and cytotoxic tumor-infiltrating lymphocytes (CTLs). This is the reason why patients with MSI-H CRC generally have more favorable outcomes with immune checkpoint-blockade treatment than patients with microsatellite instability low (MSI-L) or microsatellite stable (MSS) CRC [38]. However, this phenomenon alone can hardly explain the worse survival in MSI-H1 because these tumors also possess huge somatic mutations. In fact, unlike in early-stage disease, MSI is even linked to poorer survival in metastatic CRC [39]. It is tempting to believe that some underlying differences exist in the TIME. Bernhard et al. [40] found that immunoscore was superior to microsatellite instability in predicting patients’ disease-specific recurrence and survival. Coexpression subnetwork construction suggested that the immune regulatory equilibrium in MSI-H1 is off kilter. Further deconvolution of immune infiltrates revealed the enrichment of immunosuppressive M2 TAMs in MSI-H1. M2 TAMs are good targets for anticancer therapy through either ablation or redifferentiation away from protumoral towards antitumoral states because they express canonical markers [41, 42].
PD-L2, which resembles PD-L1, the other ligand of PD-1, is an inhibitor of effector T cells, had a much higher expression in MSI-H1 than in MSI-H2. Overexpression of PD-L2 is associated with poor survival in CRC [43]. Although PD-L2 expression is generally correlated with the expression of PD-L1, PD-L2 is also present in the absence of PD-L1 in subsets of tumor samples [44]. The response rates of a clinical trial including 172 pembrolizumab-treated head and neck squamous cell carcinoma (HNSCC) patients with recurrent or metastatic disease were 23.0% (95% CI, 16.0–31.4) and 26.6% (95% CI, 18.0–36.7) in PD-L1- and PD-L2- positive patients, respectively. These were both much higher than the response rates in the PD-L1- and PD-L2-negative patients (5.9%; 95% CI, 0.1–28.7). Additionally, the overall response rate (ORR) was greatest in patients who were positive for both PD-L1 and PD-L2 and was 2-fold higher (27.5%; 95% CI, 18.6–37.8) than that in patients whose tumors were positive only for PD-L1 (11.4%; 95% CI, 3.2–26.7) [44]. Thus, nominate PD-L2 as a potential novel therapeutic target may be more effective in MSI-H1 CRC.
Conclusions
In summary, our study showed that not all MSI-H CRC cases share the same molecular characteristics and clinical outcomes. M2 macrophages and PD-L2 reside in the TIME may counteract the prognostic benefit offered by the large amount of neoantigens produced by dMMR tumors. Finally, the heterogeneity in MSI-H tumors indicates that PD-1/PD-L1 does not fit all and that more clinical measures should be addressed for selected patients to improve survival.
Additional files
Table S1. Clinical information of TCGA and GSE39582 samples used in this study. (XLSX 43 kb)
Figure S1. Clustering of 77 MSI-H CRCs in GSE39582 by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 830 kb)
Figure S2. MSI-H CRC patients (GSE39582) fell into two gene expression-based subtypes. (PDF 2907 kb)
Figure S3. Clustering of 48 stage II MSI-H CRCs in TCGA by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 488 kb)
Figure S4. Clustering of 53 stage II MSI-H CRCs in GSE39582 by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 557 kb)
Table S2. DEGs identified between MSI-H1 and H2 for TCGA and GSE39582. (XLSX 359 kb)
Figure S5. GSEA analysis revealed functional enrichment differences between MSI-H1 and MSI-H2. GSEA was performed for MSI-H CRCs using the hallmark gene signatures collected from MSigDB. (PPTX 276 kb)
Table S3. Differentially methylated sites between MSI-H1 and MSI-H2 identified in this study. (XLSX 251 kb)
Figure S6. Survival status of MSI-H CRC subtypes. (PDF 175 kb)
Table S4. Deconvolution of TCGA and GSE39582 TIL populations by CIBERSORT. (XLSX 52 kb)
Figure S7. Boxplot distribution of CCL2 and CCL5 expression level between MSI-H1 and MSI-H2 by using TCGA and GSE39582 data. (PDF 412 kb)
Acknowledgements
Not applicable.
Abbreviations
- CIMP
CpG island methylator phenotype
- CMS
Consensus molecular subtype
- CRC
Colorectal cancer
- CTLs
Cytotoxic tumor-infiltrating lymphocytes
- DEGs
Differentially expressed genes
- DFS
Disease-free survival
- DMSs
Differentially methylated sites
- FDR
False discovery rate
- GEO
Gene Expression Omnibus
- GO
Gene Ontology
- GSEA
Gene set enrichment analyses
- HNSCC
Head and neck squamous cell carcinoma
- HR
Hazard ratio
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- Mb
Million bases
- MMR
Mismatch repair system
- MSI-H
Microsatellite instability-high
- MSI-H1
MSI-H group 1
- MSI-H2
MSI-H group 2
- MSI-L
Microsatellite instability low
- MSS
Microsatellite stable
- NMF
Nonnegative matrix factorization
- OS
Overall survival
- QN
Quantile normalization
- TAMs
Tumor-associated macrophages
- TCGA
The Cancer Genome Atlas
- TIME
Tumor immune microenvironment
- TMB
Tumor mutational burden
Authors’ contributions
Conception and design: WH, SZ, Provision of study materials or patients: SZ, Collection and assembly of data: WH, YY, LQ, JC, Data analysis and interpretation: WH, YY, WG, Manuscript writing: All authors. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81802883) and Fundamental Research Funds for the Central Universities (grant number 2018FZA7012) to Wangxiong Hu.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the TCGA repository, https://cancergenome.nih.gov/.
Ethics approval and consent to participate
The Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine approved this study.
Consent for publication
All authors have agreed to publish this manuscript.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wangxiong Hu, Phone: +86-571-87784606, Email: wxhu@zju.edu.cn.
Yanmei Yang, Email: yangyanmei@zju.edu.cn.
Lina Qi, Email: linaqi@zju.edu.cn.
Jiani Chen, Email: chen_jiani@zju.edu.cn.
Weiting Ge, Email: geweiting@zju.edu.cn.
Shu Zheng, Phone: +86-571-87784501, Email: zhengshu@zju.edu.cn.
References
- 1.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17:79–92. doi: 10.1038/nrc.2016.126. [DOI] [PubMed] [Google Scholar]
- 3.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, Kim C, Taniyama Y, Kim SI, Choi HJ, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18:6531–6541. doi: 10.1158/1078-0432.CCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 6.Hu W, Yang Y, Ge W, Zheng S. Deciphering molecular properties of hypermutated gastrointestinal cancer. J Cell Mol Med. 2018. [DOI] [PMC free article] [PubMed]
- 7.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sveen A, Johannessen B, Tengs T, Danielsen SA, Eilertsen IA, Lind GE, Berg KCG, Leithe E, Meza-Zepeda LA, Domingo E, et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017;9:46. doi: 10.1186/s13073-017-0434-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–1350. doi: 10.1038/nm.4191. [DOI] [PubMed] [Google Scholar]
- 10.Gautier L, Cope L, Bolstad BM. Irizarry RA: affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Feng Z, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 12.Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu G, Wang LG, Han Y, He QY. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu W, Yang Y, Li X, Huang M, Xu F, Ge W, Zhang S, Zheng S. Multi-omics approach reveals distinct differences in left- and right-sided Colon Cancer. Mol Cancer Res. 2017. [DOI] [PubMed]
- 16.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therneau TM, Grambsch PM. Modeling survival data: extending the cox model. New York: Springer; 2000. [Google Scholar]
- 18.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Hu W, Zhou J, Huang Y, Peng J, Yuan Y, Yu J, Zheng S. CLCA1 suppresses colorectal cancer aggressiveness via inhibition of the Wnt/beta-catenin signaling pathway. Cell Commun Signal. 2017;15:38. doi: 10.1186/s12964-017-0192-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchins LN, Murphy SM, Singh P, Graber JH. Position-dependent motif characterization using non-negative matrix factorization. Bioinformatics. 2008;24:2684–2690. doi: 10.1093/bioinformatics/btn526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yaeger R, Cercek A, Chou JF, Sylvester BE, Kemeny NE, Hechtman JF, Ladanyi M, Rosen N, Weiser MR, Capanu M, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer. 2014;120:2316–2324. doi: 10.1002/cncr.28729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, Van Cutsem E, Xie T, Bosman FT, Roth AD, Delorenzi M. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012;30:1288–1295. doi: 10.1200/JCO.2011.39.5814. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, Miyashita T, Tajima H, Ninomiya I, Munesue S, et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer. 2016;19:1052–1065. doi: 10.1007/s10120-015-0579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR, van der Burg SH. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73:2480–2492. doi: 10.1158/0008-5472.CAN-12-3542. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, He W, Huang J, Wang B, Li H, Cai Q, Su F, Bi J, Liu H, Zhang B, et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat Commun. 2018;9:3826. doi: 10.1038/s41467-018-06152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svensson S, Abrahamsson A, Rodriguez GV, Olsson AK, Jensen L, Cao Y, Dabrosin C. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin Cancer Res. 2015;21:3794–3805. doi: 10.1158/1078-0432.CCR-15-0204. [DOI] [PubMed] [Google Scholar]
- 29.Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautes-Fridman C, Laurent-Puig P, Fridman WH. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22:4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 30.Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, Newcomb PA. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taieb Julien, Le Malicot Karine, Shi Qian, Penault Lorca Frédérique, Bouché Olivier, Tabernero Josep, Mini Enrico, Goldberg Richard M., Folprecht Gunnar, Luc Van Laethem Jean, Sargent Daniel J., Alberts Steven R., Francois Emile Jean, Laurent Puig Pierre, Sinicrope Frank A. Prognostic Value ofBRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. Journal of the National Cancer Institute. 2016;109(5):djw272. doi: 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barras D, Missiaglia E, Wirapati P, Sieber OM, Jorissen RN, Love C, Molloy PL, Jones IT, McLaughlin S, Gibbs P, et al. BRAF V600E mutant colorectal Cancer subtypes based on gene expression. Clin Cancer Res. 2017;23:104–115. doi: 10.1158/1078-0432.CCR-16-0140. [DOI] [PubMed] [Google Scholar]
- 33.Shimada Y, Tajima Y, Nagahashi M, Ichikawa H, Oyanagi H, Okuda S, Takabe K, Wakai T. Clinical significance of BRAF non-V600E mutations in colorectal Cancer: a retrospective study of two institutions. J Surg Res. 2018;232:72–81. doi: 10.1016/j.jss.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 34.Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015;26:2092–2097. doi: 10.1093/annonc/mdv290. [DOI] [PubMed] [Google Scholar]
- 35.Amaki-Takao M, Yamaguchi T, Natsume S, Iijima T, Wakaume R, Takahashi K, Matsumoto H, Miyaki M. Colorectal Cancer with BRAF D594G mutation is not associated with microsatellite instability or poor prognosis. Oncology. 2016;91:162–170. doi: 10.1159/000447402. [DOI] [PubMed] [Google Scholar]
- 36.Hughes LA, Williamson EJ, van Engeland M, Jenkins MA, Giles GG, Hopper JL, Southey MC, Young JP, Buchanan DD, Walsh MD, et al. Body size and risk for colorectal cancers showing BRAF mutations or microsatellite instability: a pooled analysis. Int J Epidemiol. 2012;41:1060–1072. doi: 10.1093/ije/dys055. [DOI] [PubMed] [Google Scholar]
- 37.Ogino S, Nowak JA, Hamada T, Milner DA, Jr, Nishihara R. Insights into pathogenic interactions among environment, host, and tumor at the crossroads of molecular pathology and epidemiology. Annu Rev Pathol. 2019;14:83–103. doi: 10.1146/annurev-pathmechdis-012418-012818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O, Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. Integrative analyses of colorectal cancer show Immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 41.Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018. [DOI] [PubMed]
- 42.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, Song H, Xie D, Jiang X, Wang H. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157–167. doi: 10.1136/gutjnl-2015-310514. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Yao H, Li C, Liang L, Zhang Y, Shi H, Zhou C, Chen Y, Fang JY, Xu J. PD-L2 expression in colorectal cancer: independent prognostic effect and targetability by deglycosylation. Oncoimmunology. 2017;6:e1327494. doi: 10.1080/2162402X.2017.1327494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, Lunceford J, Cheng J, Chow LQM, Seiwert TY, et al. PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clin Cancer Res. 2017;23:3158–3167. doi: 10.1158/1078-0432.CCR-16-1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical information of TCGA and GSE39582 samples used in this study. (XLSX 43 kb)
Figure S1. Clustering of 77 MSI-H CRCs in GSE39582 by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 830 kb)
Figure S2. MSI-H CRC patients (GSE39582) fell into two gene expression-based subtypes. (PDF 2907 kb)
Figure S3. Clustering of 48 stage II MSI-H CRCs in TCGA by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 488 kb)
Figure S4. Clustering of 53 stage II MSI-H CRCs in GSE39582 by NMF. Correlation matrix heatmaps correspond to rank 2 to 6. (PDF 557 kb)
Table S2. DEGs identified between MSI-H1 and H2 for TCGA and GSE39582. (XLSX 359 kb)
Figure S5. GSEA analysis revealed functional enrichment differences between MSI-H1 and MSI-H2. GSEA was performed for MSI-H CRCs using the hallmark gene signatures collected from MSigDB. (PPTX 276 kb)
Table S3. Differentially methylated sites between MSI-H1 and MSI-H2 identified in this study. (XLSX 251 kb)
Figure S6. Survival status of MSI-H CRC subtypes. (PDF 175 kb)
Table S4. Deconvolution of TCGA and GSE39582 TIL populations by CIBERSORT. (XLSX 52 kb)
Figure S7. Boxplot distribution of CCL2 and CCL5 expression level between MSI-H1 and MSI-H2 by using TCGA and GSE39582 data. (PDF 412 kb)
Data Availability Statement
The datasets generated and/or analysed during the current study are available in the TCGA repository, https://cancergenome.nih.gov/.