

Abstract
Regulation of Ca2+ homeostasis in the extracellular space plays an important role in neuronal function. Several modeling studies and recent measurements have demonstrated that modest action potential or synaptic activity can result in a significant reduction in extracellular calcium ([Ca]o2+). Changes in [Ca]o2+ can regulate intracellular signaling enzymes, such as Ca2+/calmodulin–dependent protein kinase II, and influence neuronal function at synaptic and nonsynaptic sites. The change in [Ca]o2+ can affect several types of ion channels and neurotransmitter receptors and activate a Ca2+-sensitive receptor in neuronal membranes. Depletion of [Ca]o2+ may function as an activity-dependent extracellular messenger that regulates nervous system function during development, learning, and disease.
Keywords: Cadherin, Calcium-sensitive receptor, CaMKII, Extracellular calcium, Long-term potentiation, Synaptic plasticity
On a molecular level, the function of the nervous system is to regulate the transmembrane flux of calcium ions. (This was paraphrased from a provocative remark by Dr. Bertil Hille in a lecture on ion channels at the Society for Neurosciences meeting.) Whether it is to control muscle contraction, secretion, or synaptic transmission, the nervous system acts ultimately by regulating the level of intracellular Ca2+ levels. For some time, there has been speculation that changes in the concentration of Ca2+ outside neurons may provide an important activity-dependent signal that could regulate neuronal excitability and synaptic transmission. However, whether the availability of extracellular calcium ions ([Ca]o2+) could be depleted by the activity-dependent influx of Ca2+ and whether this would be sufficient to have a functional effect remain open questions. Because of the extremely low resting concentration of this cation inside cells, neural activity can increase intracellular Ca2+ concentration easily 10- to 100-fold by activating Ca2+-permeable channels in the neuronal membrane or intracellular organelles storing Ca2+. But with 2 mM Ca2+ in the extracellular medium, the Ca2+ drawn into neurons by impulse activity would only lower the concentration outside the cell a relatively small amount and for only a short period of time.
In considering this question, three major issues must be addressed.
Is there significant depletion of [Ca]o2+ accompanying neural activity? If so, what is the magnitude and temporal dynamics of the depletion? How does this vary with patterns of neural activity and with different subcellular regions of the neuron?
How could the reduction in [Ca]o2+ be detected by the neuron?
What are the functional consequences of this activity-dependent depletion of [Ca]o2+ in normal and pathological situations?
There is now substantial evidence bearing on all of these questions, indicating that activity-dependent regulation of [Ca]o2+ can have effects on synaptic transmission, axonal excitability, intracellular signaling enzymes, synaptic plasticity, and disease.
Can Neural Activity Reduce the Concentration of [Ca]o2+?
Early studies using Ca2+-sensitive electrodes (Nicholson and others 1977; Benninger and others 1980; Heinemann and others 1990) demonstrated that free Ca2+ in the extrasynaptic space of stratum pyramidale is significantly reduced following 10-Hz stimulation for several seconds to the stratum radiatum (Fig. 1). With long trains of action potentials, free Ca2+ can decrease from 2 mM to less than 1.5 mM. The reduction in [Ca]o2+ is mimicked by exogenous application of glutamate and persists for several seconds following the end of either electrical stimulation or glutamate exposure. The paradigms used in these studies resemble conditions that are more likely observed during epileptic seizure activity rather than during physiologically relevant synaptic activity. However, physiologically relevant changes in [Ca]o2+ might be produced by less intense stimulation in close proximity to the cell membrane or in other microdomains beyond the detection limits of ion-sensitive electrodes.
Fig. 1.

[Ca2+]o can drop following synaptic activity. A, Recordings with Ca2+-selective microelectrodes from guinea pig stratum pyramidale (SP) are shown during a 5-sec 10-Hz stimulus train in stratum radiatum (SR) (black bar). Adapted from Benninger and others (1980), Fig. 2, with permission from Elsevier Science. B, Recordings with Ca2+-selective microelectrodes from rat SR show a drop in [Ca]2+o following a 15-sec exposure to N-methyl-D-aspartate (NMDA) (black bar). Adapted from Heinemann and others (1990), Fig. 3, with permission from Elsevier Science. C, Evoked postsynaptic currents (EPSCs) and fluorescence measurements in SP following five 100-Hz stimuli applied to SR imaged using a cell-impermeant Ca2+-sensitive dye (Rhod-5N) and intracellular recordings show a decrease in Rhod-5N fluorescence following theta stimulation to SR. Note that the drop in fluorescence begins at the onset of the EPSC in SP (red line) and continues for several hundred milliseconds. Adapted from Rusakov and Fine (2003), Fig. 2, with permission from Cell Press. In both A and B, [Ca]2+o drops from baseline levels (dotted line) to 1.45 mM and less than 1 mM following either synaptic stimulation (A) or NMDA exposure (B), respectively.
[Ca]o2+ Depletion Occurs Following Modest Synaptic Activity
Estimates of the degree of Ca2+ depletion during modest activity have been made using mathematical modeling. These calculations support the findings that regulation of [Ca]o2+ is highly dependent on firing frequency (Vassilev and others 1997) and determined by such factors as channel density, glia, and synapse size (Egelman and Montague 1999; Rusakov 2001). One particular model assumes that N-methyl-D-aspartate (NMDA) receptors act as the primary Ca2+ sink (Vassilev and others 1997); α-amino-3-hydroxy-5-methylisoxazole 4-propionic acid (AMPA) receptors may function as Ca2+ sinks as well. Increasing the stimulation frequency from 3 to 100 Hz resulted in a prolonged reduction in [Ca]o2+, which recovered over several hundred milliseconds following termination of stimulation. Mathematical modeling suggests that extracellular Ca2+ depletion increases with increasing spine length and glial ensheathment.
In a recent paper, Rusakov and Fine (2003) have used a novel Ca2+ imaging approach to measure changes in [Ca]o2+ following synaptic activity. In this study, [Ca]o2+ was measured using a membrane-impermeant Ca2+-sensitive dye. A dye with a low Ca2+-binding affinity was used so that it would be sensitive to the high concentration of Ca2+ in the extracellular space. The authors observed a modest decrease (2%) in [Ca]o2+ at stratum pyramidale CA1 neurons following short trains of activity (5 bursts at 100 Hz) (Fig. 1C). The authors proposed that NMDA receptors in the active state function as large Ca2+ sinks and that even modest activity produces a drop in [Ca]o2+. Rusakov and Fine argue for the contribution of modest activity in modulating [Ca]o2+ over tens of milliseconds rather than several seconds as in pathological states such as epilepsy and hypoxia.
Factors Affecting Activity-Dependent Depletion of [Ca]o2+
The density and distribution of Ca2+-permeable channels (e.g., voltage-activated Ca2+ channels, NMDA and AMPA receptors) and pumps (e.g., Ca2+-ATPase and Na+/Ca2+–exchanger) determine the degree of depletion following neural activity. Large transient decreases in Ca2+ (dropping to < 0.7 mM for tens of milliseconds) are predicted to occur at the nerve terminal and spines due to the high density of NMDA receptors present as well as slow activation kinetics of these receptors (Egelman and Montague 1998, 1999). The Ca2+-ATPase and Na+/Ca2+–exchanger are differentially located at the synapse as well. The plasma membrane Ca2+-ATPase is located primarily on presynaptic terminals and may function to replenish [Ca]o2+ following synaptic activity (Juhaszova and others 2000), whereas the Na+/Ca2+–exchanger is located at extrasynaptic sites and may function to regulate intracellular Ca2+ homeostasis (Jeon and others 2003). However, during pathological states of activity (e.g., under hypoxia), the Na+/Ca2+–exchanger may in fact become a Ca2+ sink as it can operate in “reverse mode” to decrease [Ca]o2+. An additional factor that can modulate [Ca]o2+ is the presence of glia. Most central excitatory synapses are ensheathed in glia. The presence of glia can restrict repletion of extracellular Ca2+ as well as slow neurotransmitter clearance due to a decrease in the permeability of the extracellular space to small molecules (Vassilev and others 1997; Rusakov 2001).
How Is a Reduction in [Ca]o2+ Detected by Neurons?
Neurons and excitable cells can respond to changes in [Ca]o2+ through a number of mechanisms that either directly or indirectly sense the local environment. The resting membrane potential is established by an electrochemical gradient through selective permeability of ionic channels to Na+, K+, Cl−, and Ca2+; transiently decreasing [Ca]o2+ will decrease the driving force and subsequently decrease the magnitude of Ca2+ currents. Many other cation-selective channels are modulated by alterations in [Ca]o2+. Gating kinetics of the Na+ channel are directly modified by the level of [Ca]o2+ (Armstrong and Cota 1999), and some nonselective cation channels are activated by decreases in [Ca]o2+ (Xiong and others 1997). Fluctuations in [Ca]o2+ may also directly affect neurotransmitter release as it displays a steep fourthorder dependence on [Ca]o2+ (Dodge and Rahamimoff 1967); therefore, modest changes in [Ca]o2+ of several hundred micromolar can result in significant effects on synaptic transmission.
Glutamate and γ-aminobutyric acid (GABA) are the main excitatory and inhibitory neurotransmitter systems in the brain and function predominately through fast synaptic neurotransmission via ionotropic receptors. Glutamate and GABA also function through G protein-coupled metabotropic receptors (mGluR and GABABR). In addition to being activated by agonist, they are directly modulated by changes in [Ca]o2+ (Kubokawa and others 1996; Kubo and others 1998; Galvez and others 2000; Tabata and others 2002). Both mGluR and GABABR modulate synaptic efficacy through interactions with channels, intracellular Ca2+ stores, and adenylyl cyclase. The presence of elevated [Ca2+]o can enhance receptor-G protein coupling and downstream signaling, but regulation of these receptors by [Ca2+]o is complicated and may be indirect (Galvez and others 2000; Nash and others 2001).
A Ca2+-sensitive receptor (CaSR) has been identified, which specifically detects changes in [Ca2+]o. The CaSR was initially cloned and characterized in bovine and human parathyroid gland (Brown and others 1993; Garrett and others 1995) and brain (Ruat and others 1995, reviewed in Brown and MacLeod 2001) (see Box 1). The CaSR has been identified in many tissues and may serve as a general homeostatic mechanism for regulating extracellular Ca2+ levels. Though there is debate as to the endogenous ligand for the CaSR, it is activated by cations (La3+ > Gd3+ > Ca2+ > Mg2+), polyamines (spermine), and antibiotics (neomycin). The CaSR couples to intracellular Ca2+ accumulation through both nonselective cation channels and activation of phospholipase C (Fig. 2). Selective allosteric modulators and antagonists have been developed that will aid in elucidating its function in the CNS (Nemeth 2002).
Box 1: Ca2+ Sensing Can Be Mediated by Family C, G-Protein-Coupled Receptors.
The Ca2+-sensitive receptor (CaSR) is part of the family C, G-protein-coupled receptors. A noticeable feature of the receptor is that the ligand or Ca2+-binding domain is formed by a large extracellular domain followed by a seven-transmembrane domain and a short cytoplasmic tail. This contrasts with prototypic Gprotein-coupled receptors (e.g., β-adrenergic receptor), which lack a large extracellular domain where ligand binding occurs within the transmembrane domains. This family of receptors includes mGluR, GABABR, and several putative pheromone and taste receptors. The extracellular domain incorporates the agonist-binding domain and is a structural homolog of the leucine/isoleucine/valine–binding protein (Hoshino and Kose 1989) and bacterial periplasmic–binding proteins (O’Hara and others 1993). The cysteine-rich domain is involved in signaling ligand binding from the agonist domain to the transmembrane helices. The activation mechanism has been termed the “Venuslytrap” model of receptor activation due to the resemblance to the Venus-flytrap, which possesses a hinged jaw that closes on ingesting food (Bockaert and Pin 1999). The CaSR can couple through Gi/o to the inhibition of adenylyl cyclase and through Gq to activate phospholipase C (and phospholipase D); in addition, the CaSR can directly modulate nonselective cation channels through Gβγ subunits (Brown and MacLeod 2001). Several isoforms of the mGluR are modulated by Ca2+ as well as the GB1 subunit of the GABABR (Galvez and others 2000; Nash and others 2001).
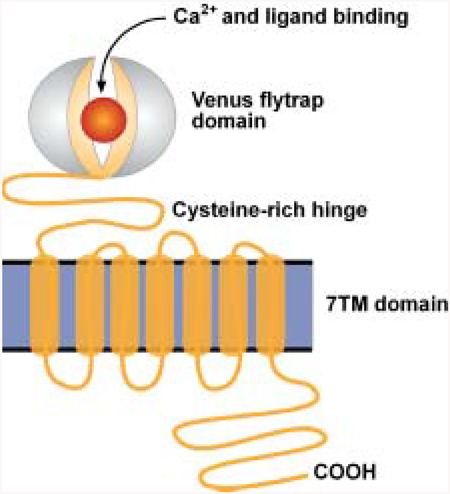
Fig. 2.

Activity-mediated changes in [Ca]o2+ regulate presynaptic and postsynaptic function. A number of proteins can function to sense changes in [Ca]o2+ following synaptic activity. The Ca2+-sensitive receptor (CaSR), n-cadherin, and mGluR located at the synapse modify presynaptic and postsynaptic neuronal activity following changes in [Ca]o2+. Several factors can alter the degree of extracellular Ca2+ depletion following activity: glia that can restrict Ca2+ diffusion into the cleft, presence of the CaSR on presynaptic versus postsynaptic neurons, and transporters that can terminate glutamate signaling. Transient drops in [Ca]o2+ act through cadherins, the CaSR, and channels to modulate several pathways through decreased activation of phospholipase C (or phospholipase D), CaMKII, and β-catenin/Wnt–mediated signaling with effects on neurotransmitter release as well as long-term synaptic plasticity; perturbations can result in pathological states.
Second Messenger Signaling Mediated by [Ca2+]o
Several lines of evidence demonstrate that fluctuations in [Ca2+]o can modulate second messenger signaling. Activation of the CaSR receptor by increased [Ca2+]o results in activation of a number of signaling pathways, for example, β-catenin/Wnt (Chakrabarty and others 2003) and MAPK (Yamaguchi and others 2000). The cell-adhesion molecule n-cadherin requires Ca2+ to undergo dimerization; cell-cell cadherin signaling results in activation of the β-catenin/Wnt signaling complex, with downstream effects on synaptic growth and enhancement (Huntley and others 2002). Either removal of [Ca2+]o or blocking cadherin dimerization interferes with the induction of long-term plasticity in hippocampal slice (Tang and others 1998). This mechanism would allow fluctuations in [Ca2+]o produced by neuronal activity to signal recent neuronal activity.
Increases in intracellular Ca2+ via NMDA receptors and Ca2+ channels result in activation (through autophosphorylation and subsequent Ca2+-independent activity) of Ca2+/CaM–dependent protein kinase (CaMKII). A novel regulation of this enzyme is that the autophosphorylation state is highly dependent on [Ca2+]o (Scholz and Palfrey 1998; Eshete and Fields 2001). Both dorsal root ganglia and hippocampal neurons respond to increases in [Ca2+]o by increasing the proportion of CaM kinase in the autophosphorylated state. This occurs without any detectable changes in intracellular Ca2+ concentration (Eshete and Fields 2001). Bursts of synaptic activity that can transiently decrease [Ca2+]o can modulate the state of CaMKII, a kinase strongly implicated in learning and memory. It is not understood how CaMKII is regulated by alterations in [Ca2+]o, but this may involve interactions with other Ca2+-sensing proteins. Interestingly, the studies of extracellular Ca2+ depletion in dorsal root ganglia neurons were performed in cultures without synapses, suggesting a role for extracellular Ca2+ depletion in modulating axonal properties (e.g., axonal excitability and conduction) partly by CaMKII (Eshete and Fields 2001).
Activity-Dependent Depletion of Ca2+ in Normal and Disease States
Activity-dependent depletion of Ca2+ may have consequences on a number of cellular processes such as shortterm plasticity, cell adhesion, and cell function in disease states.
It is long accepted that short-term depression (STD) results from a reduction in exocytosis of the readily releasable pool of synaptic vesicles in the presynaptic neuron following a stimulus. Rusakov and Fine (2003) argue that STD may result in part from a reduction in [Ca2+]o. To test this hypothesis, they measured STD in slice cultures. Blockade of NMDA receptors with the NMDA receptor antagonist D-2-amino-5-phosphonopentanoic acid reduced the degree of STD and increased the fluorescence signal with the Ca2+-sensitive impermeant dye. They also demonstrated that the addition of dextran to the superfusate to impair transmitter diffusion resulted in an increase in STD.
Studies in hippocampal slice suggest that depletion of synaptic Ca2+ produced by high-frequency stimulation is necessary for induction of long-term potentiation (LTP). Superfusion with either peptide competitors or antibodies to n-cadherin during the LTP-induction protocol interferes with the induction of early LTP (Tang and others 1998). However, if [Ca2+]o is elevated during the LTP inducing stimulus, block by either peptide or anticadherin antibodies is no longer effective. This suggests that during synaptic activity, there is a functional change in cadherin structure that is dependent on an activity-dependent reduction in [Ca2+]o in the synaptic cleft. The protocadherin arcadlin (activity-regulated cadherin-like protein) also plays a role in the induction of LTP; reducing [Ca2+]o during superfusion with antiarcadlin antibodies interferes with the induction of early LTP (Yamagata and others 1999).
The CaSR is a critical component in modulating parathyroid hormone secretion involved in Ca2+ homeostasis and bone metabolism. Several disease states result from mutations in the CaSR, including familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism (reviewed in Hendy and others 2000). Neurological deficits are also observed in neonatal severe hyperparathyroidism such as impairments in memory and cognition as well as symptoms resembling amyotrophic lateral sclerosis (Numann and others 1984; Cole and others 1990; Jackson and others 1998). These neurological deficits suggest that the CaSR is important in synaptic function. Hippocampal CaSR expression in rodents begins to appear at postnatal day 5 and steadily increases until 30 days when it begins to stabilize. This increase in CaSR expression coincides with the ability to produce LTP, a developmental period when a high level of synaptic plasticity and refinement is occurring (Chattopadhyay and others 1997).
Conclusion
Depletion of Ca2+ functions as an activity-dependent extracellular messenger to regulate several forms of synaptic plasticity during both development and learning and potential disease states. The extent of activitydriven changes in [Ca]o2+ is highly dependent on such factors as the types of synapses, presence of glia, and receptors present. Equally important are various mechanisms maintaining Ca2+ homeostasis in the extracellular space. The CaSR couples through a number of signaling pathways with the ability to directly modulate neuronal activity; impairments in this receptor have effects on cognition, suggesting a possible role in synaptic function. Ca2+-mediated cell adhesion through cadherins has an important role in directing plastic changes that occur during development and learning. It is therefore critical that [Ca]o2+ be tightly maintained for normal synaptic function to occur, in part through Ca2+ pumps and exchangers. Deregulation in Ca2+ sensing through impairments in any of these proteins could have profound effects on functioning of the brain.
References
- Armstrong CM, Cota G. 1999. Calcium block of Na+ channels and its effect on closing rate. Proc Natl Acad Sci U S A 96(7):4154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger C, Kadis J, Prince DA. 1980. Extracellular calcium and potassium changes in hippocampal slices. Brain Res 187(1): 165–82. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Pin JP. 1999. Molecular tinkering of G protein-coupled receptors: an evolutionary success. Embo J 18(7):1723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, and others. 1993. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366(6455):575–80. [DOI] [PubMed] [Google Scholar]
- Brown EM, MacLeod RJ. 2001. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81(1):239–97. [DOI] [PubMed] [Google Scholar]
- Chakrabarty S, Radjendirane V, Appelman H, Varani J. 2003. Extracellular calcium and calcium sensing receptor function in human colon carcinomas: promotion of E-cadherin expression and suppression of beta-catenin/TCF activation. Cancer Res 63(1):67–71. [PubMed] [Google Scholar]
- Chattopadhyay N, Legradi G, Bai M, Kifor O, Ye C, Vassilev PM, and others. 1997. Calcium-sensing receptor in the rat hippocampus: a developmental study. Brain Res Dev Brain Res 100(1):13–21. [DOI] [PubMed] [Google Scholar]
- Cole D, Forsythe CR, Dooley JM, Grantmyre EB, Salisbury SR. 1990. Primary neonatal hyperparathyroidism: a devastating neurodevelopmental disorder if left untreated. J Craniofac Genet Dev Biol 10(2):205–14. [PubMed] [Google Scholar]
- Dodge FA Jr., Rahamimoff R. 1967. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol 193(2):419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. 1998. Computational properties of peridendritic calcium fluctuations. J Neurosci 18(21):8580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. 1999. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys J 76(4):1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshete F, Fields RD. 2001. Spike frequency decoding and autonomous activation of Ca2+-calmodulin-dependent protein kinase II in dorsal root ganglion neurons. J Neurosci 21(17):6694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, Malitschek B, and others. 2000. Ca(2+) requirement for high-affinity gammaaminobutyric acid (GABA) binding at GABA(B) receptors: involvement of serine 269 of the GABA(B)R1 subunit. Mol Pharmacol 57(3):419–26. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC, and others. 1995. Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J Biol Chem 270(21):12919–25. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Stabel J, Rausche G. 1990. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res 83:197–214. [DOI] [PubMed] [Google Scholar]
- Hendy GN, D’Souza-Li L, Yang B, Canaff L, Cole DE. 2000. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat 16(4):281–96. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Kose K. 1989. Cloning and nucleotide sequence of braC, the structural gene for the leucine-, isoleucine-, and valine-binding protein of Pseudomonas aeruginosa PAO. J Bacteriol 171(11):6300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley GW, Gil O, Bozdagi O. 2002. The cadherin family of cell adhesion molecules: multiple roles in synaptic plasticity. Neuroscientist 8(3):221–33. [DOI] [PubMed] [Google Scholar]
- Jackson CE, Amato AA, Bryan WW, Wolfe GI, Sakhaee K, Barohn RJ. 1998. Primary hyperparathyroidism and ALS: is there a relation? Neurology 50(6):1795–9. [DOI] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS. 2003. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron 38(6):965–76. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Church P, Blaustein MP, Stanley EF. 2000. Location of calcium transporters at presynaptic terminals. Eur J Neurosci 12(3):839–46. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Miyashita T, Murata Y. 1998. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 279(5357):1722–5. [DOI] [PubMed] [Google Scholar]
- Kubokawa K, Miyashita T, Nagasawa H, Kubo Y. 1996. Cloning and characterization of a bifunctional metabotropic receptor activated by both extracellular calcium and glutamate. FEBS Lett 392(1):71–6. [DOI] [PubMed] [Google Scholar]
- Nash MS, Saunders R, Young KW, Challiss RA, Nahorski SR. 2001. Reassessment of the Ca2+ sensing property of a type I metabotropic glutamate receptor by simultaneous measurement of inositol 1,4,5-trisphosphate and Ca2+ in single cells. J Biol Chem 276(22):19286–93. [DOI] [PubMed] [Google Scholar]
- Nemeth EF. 2002. The search for calcium receptor antagonists (calcilytics). J Mol Endocrinol 29(1):15–21. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Bruggencate GT, Steinberg R, Stockle H. 1977. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci U S A 74(3): 1287–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numann PJ, Torppa AJ, Blumetti AE. 1984. Neuropsychologic deficits associated with primary hyperparathyroidism. Surgery 96(6): 1119–23. [PubMed] [Google Scholar]
- O’Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, and others. 1993. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 11(1):41–52. [DOI] [PubMed] [Google Scholar]
- Ruat M, Molliver ME, Snowman AM, Snyder SH. 1995. Calcium sensing receptor: molecular cloning in rat and localization to nerve terminals. Proc Natl Acad Sci U S A 92(8):3161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA. 2001. The role of perisynaptic glial sheaths in glutamate spillover and extracellular Ca(2+) depletion. Biophys J 81(4):1947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Fine A. 2003. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron 37(2):287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz WK, Palfrey HC. 1998. Activation of Ca2+/calmodulindependent protein kinase II by extracellular calcium in cultured hippocampal pyramidal neurons. J Neurochem 71(2):580–91. [DOI] [PubMed] [Google Scholar]
- Tabata T, Aiba A, Kano M. 2002. Extracellular calcium controls the dynamic range of neuronal metabotropic glutamate receptor responses. Mol Cell Neurosci 20(1):56–68. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. 1998. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron 20(6):1165–75. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. 1997. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J 72(5):2103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Lu W, MacDonald JF. 1997. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc Natl Acad Sci U S A 94(13):7012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Sugiura H, Maru E, Dominique M, Irie Y, and others. 1999. Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. J Biol Chem 274(27):19473–9. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Chattopadhyay N, Kifor O, Sanders JL, Brown EM. 2000. Activation of p42/44 and p38 mitogen-activated protein kinases by extracellular calcium-sensing receptor agonists induces mitogenic responses in the mouse osteoblastic MC3T3-E1 cell line. Biochem Biophys Res Commun 279(2):363–8. [DOI] [PubMed] [Google Scholar]