

Abstract
Artificial macrocycles recently became popular as a novel research field in drug discovery. As opposed to their natural twins, artificial macrocycles promise to have better control on synthesizability and control over their physicochemical properties resulting in druglike properties. Very few synthetic methods allow for the convergent, fast but diverse access to large macrocycles chemical space. One synthetic technology to access artificial macrocycles with potential biological activity, multicomponent reactions, is reviewed here, with a focus on our own work. We believe that synthetic chemists have to acquaint themselves more with structure and activity to leverage the design aspect of their daily work.
Keywords: macrocycle, multicomponent reaction, bioavailability, convergence, bioactivity, druglike properties, synthetic pathway, property space
Graphical abstract
1. Introduction
Macrocycles are magic. Many natural products are macrocyclic with complex stereochemistries and structures. They are often highly dynamic cyclic molecules with complex conformational hypersurfaces which allows them to undergo a variety of unusual receptor interactions. The conformational dynamic also determines transport properties to facilitate crossing cellular membranes. However, relatively few macrocycles are approved drugs despite often high biological activities seen in natural product macrocycles. Issues are the lacking transport properties of macrocycles through biological membranes, the pathway to go for most intracellular targets. The passive cellular permeability of macrocycles with a size over 1000 Da sharply drops. However, the chemical space from 500–1000 Da seems to be a sweat spot for passive permeability and remains a largely unexplored chemical space. Another issue is the complex synthetic access towards natural macrocycles often in a very sequential multistep fashion. In contrast, artificial macrocycles can be synthesized sometimes in a straightforward short synthetic route. Macrocycles are considered to cover the space in between small molecules and biologics. Small molecules require defined, narrow and deep receptor pockets whereas monoclonal antibodies (mAbs) as classical representatives of biologics can efficiently address large and flat receptor surfaces. Therefore, a pressing question is ‘can macrocycles be elaborated as a useful class of druglike compounds with a complementary space in between small molecule and biologics drugs?’ The field of macrocycles in synthesis and for medicinal chemistry applications is exploding and is well beyond the current essay.1 Thus, we will give a personal overview on artificial macrocycles, their binding mode to receptors, rules for library design, properties, and their synthesis with a focus on work from the authors laboratory.
2. Macrocycle Properties and Receptor Binding
Natural macrocycles have been recently extensively classified according to receptor-binding modes and different regions within the macrocycle structure.1i According to this definitions, macrocycles can be subdivided into three different regions further discussed in chapter 5. To further understand the receptor–macrocycle interactions and how knowledge for library design can be extracted, we build an extensive database of synthetic and natural macrocycles and structural information from the protein data bank (Figure 1)1n.
Figure 1.
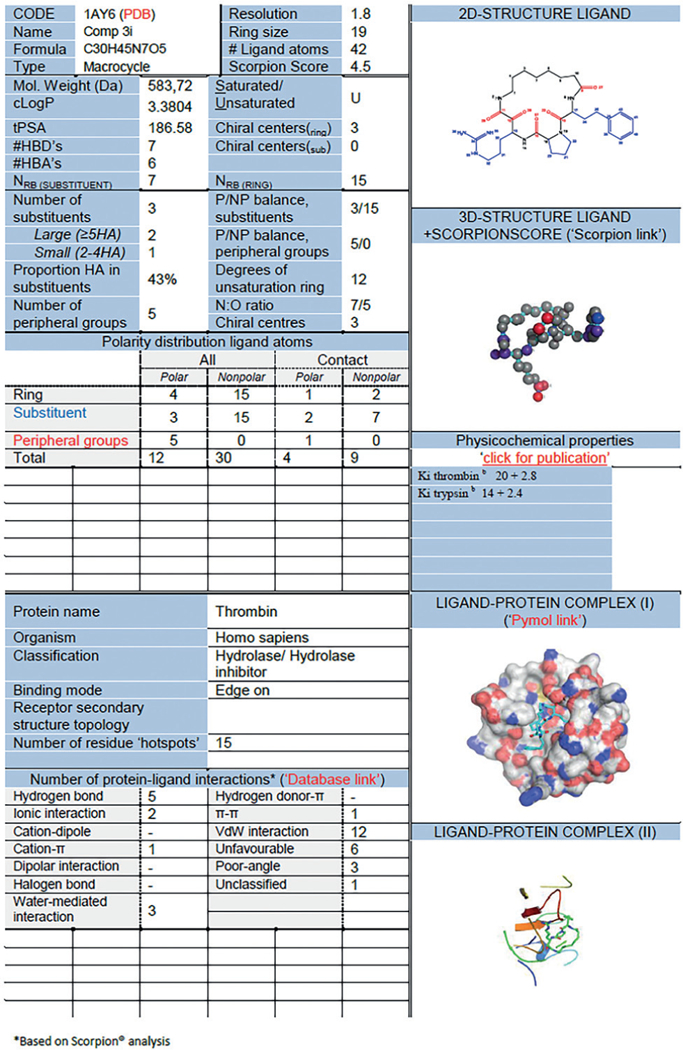
Example of an entry card of our macrocycle database for a α-ketoamide thrombin inhibitor (PDB ID 1AY6)1o
Strikingly, the majority of current target proteins investigated in industry are old targets already known before the deciphering of the human genome, so-called pregenomic targets. These include GPCRs and kinases, for example, where small molecules based on the receptor characteristics can be readily developed. Truly genomic targets, how-ever, are currently a minor focus in pharmaceutical industry. It is argued that current libraries used in high-through-put screening of genomic targets do often not provide good starting points for follow-up medicinal chemistry projects. Often such targets are protein–protein interactions (PPIs) with rather poorly defined, flexible pockets, large and flat surface areas, and are unsuitable for current chemical space of pharma libraries. If successful PPI (ant-)agonists approach the market they are mostly from the domain of bio-logics, for example, monoclonal antibodies (mAbs).
A typical example of a PPI is the programmed death-1 (PD1) and its ligand PDL1, which are expressed on T-cells and cancer cells, respectively. The approved mAbs pembrolizumab and nivolumab both target the cell surface receptor PD1, while atezolizumab, durvalumab, and avelumab target PDL1 and are celebrated new anticancer agents with a never seen before long-term remission, and even cure in hard-to-treat cancers.2 An analysis of the PPI is shown in Figure 2 based on our recently solved co-crystal structure of human PD1-PDL1.3 Several typical features of PPIs can be observed: 1) a large buried surface area of 1970 Å2; 2) a rather flat surface formed by the extended β-sheet network of immunoglobulin-type fold in both proteins; 3) the affinity of both protein is synergistically induced by numerous small contributions of hydrophobic and polar interactions in which each of them are not very strong. Thus, typical computational methods of small-molecule draggability score the PPI PD1-PDL1 very low.4 While optimized small molecules bind to their receptor with a rather high ligand efficacy, natural or artificial macrocycles often show a close similarity of binding to the natural protein interacting partners. The binding energy is scattered around the full macrocyclic scaffold: Few key interactions of anchor points are interconnected by larger strings of connecting units like anchoring amino acids in the interface of interacting proteins.
Figure 2.
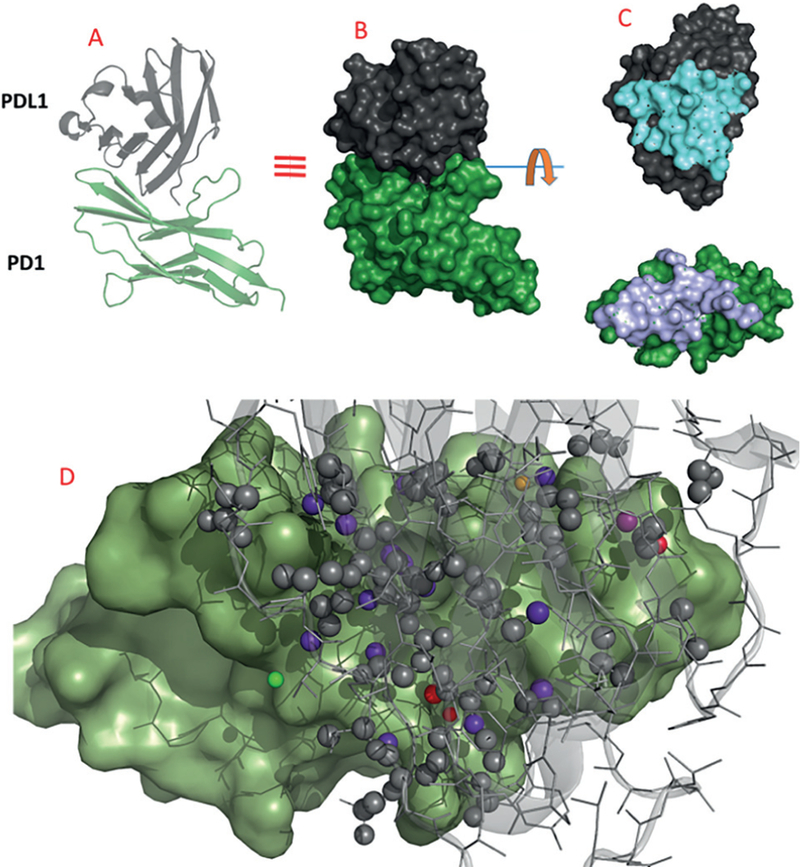
The archetypical difficult PPI PD1-PDL1 (PDB ID 4ZQK). Cartoon presentation with grey PDL1 on top and green PD1 below. Noteworthy the all β-sheet fold of both interacting proteins leading to a large and flat buried surface area. B: Surface presentation. C: The separated proteins by a 90° clockwise and anticlockwise rotation through the orthogonal axis, and showing the footprints (cyan and blue surfaces) of the interacting proteins. D: The heavy atoms of PDL1 making direct contact to PD1 are shown as balls. Their color coding is according to their contribution (scorpion score) to the energy with interaction ranging from grey (minor contribution) to red (major contribution).
An example of an artificial macrocycle binding to the interface of the IL17 dimer is shown in Figure 3.5
Figure 3.
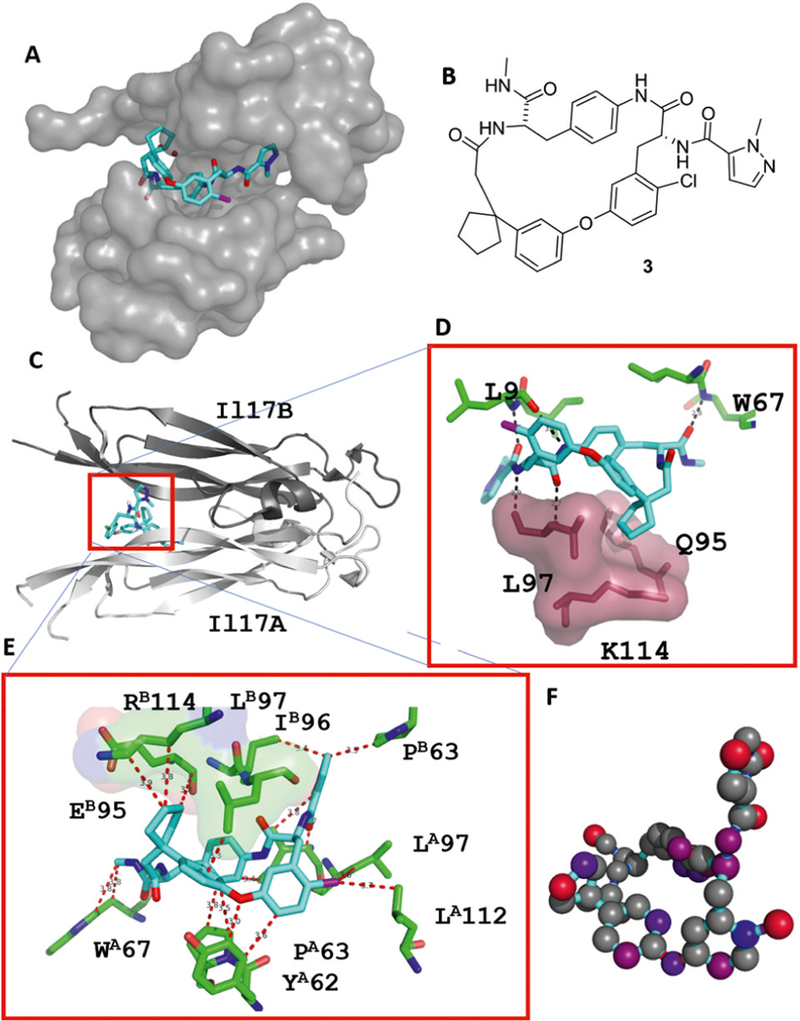
IL17-macrocycle interaction. A: An artificial macrocycle binding into a grove in the IL17A dimer interface (PDB ID 5HI4). B: 2D structure of macrocycle. C–E: Close-up view of two different macrocycle– receptor interacting regions. D: A spirocyclopentyl group hot spot undergoing multiple hydrophobic interactions with a receptor lysine, leucine, and glutamine. E: Macrocycle–receptor interaction overview, including hydrogen bindings, π, and hydrophobic interactions. F: Atomic hot spots of the macrocycle according to their contribution to the binding to the IL17 receptor. It was calculated using scorpion software and the color code is rendered according to their contribution to the energy, the interaction ranging from grey (minor contribution) to red (major contribution).6
3. Synthetic Approaches toward Artificial Macrocycles Using MCR
A well-known challenge in macrocycle synthesis is the cycle formation over oligo- or polymerization. Paul Ruggli and Karl Ziegler have introduced the high-dilution principle, according to which low concentrations of the starting acyclic precursor favor cyclization over chain formation.7 Another challenge relates to the exploration of the natural macrocycles for drug discovery since synthesizing such compounds in a timely and divers fashion is difficult, especially when a series of molecules for structure–activity relationship (SAR) elucidation or screening libraries is needed.
Moreover, cyclization methods are required that are working in a general fashion with a wide variety of substrates and functional groups. Therefore, development of short and efficient synthetic approaches with only few steps is necessary. A number of highly interesting synthetic routes have been developed including rapid and efficient methodologies such as DNA-encoded chemistry, enzyme-catalyzed ring closures, special classes of structurally ordered macrocycles such as stapled peptides, or accessing peptide macrocycles from genetically encoded polypeptides, which, however, are beyond the current assay and have been extensively reviewed elsewhere.8 Noteworthy, the majority of methods focuses on peptide macrocycles. In contrast, multicomponent reaction (MCR) chemistry is very well used in synthesis of a diverse range of macrocycles and is also able to generate great levels of molecular diversity and complexity at low synthetic costs.9 MCRs such as Ugi and Passerini reactions have been used to develop many strategies towards macrocycle libraries. These reactions are used for macrocyclization directly or to synthesize linear precursors which can be cyclized whether by MCRs or other procedures. Already in 1979 Failli and Immer10 described for the first time the use of Ugi MCR for the one-pot macrocyclization of N,C-terminal unprotected linear hexapeptides 1 to head-to-tail cyclic peptide 4. Surprisingly, the product which was isolated from the reaction of triglycine (5) with isobutyraldehyde (2) and cyclohexyl isocyanide (3) was the substituted cyclic hexaglycine 6 derived from a double MCR of the starting materials (Scheme 1). Obviously, the formation of the cyclic hexapeptide of the tripeptide is favored.
Scheme 1.
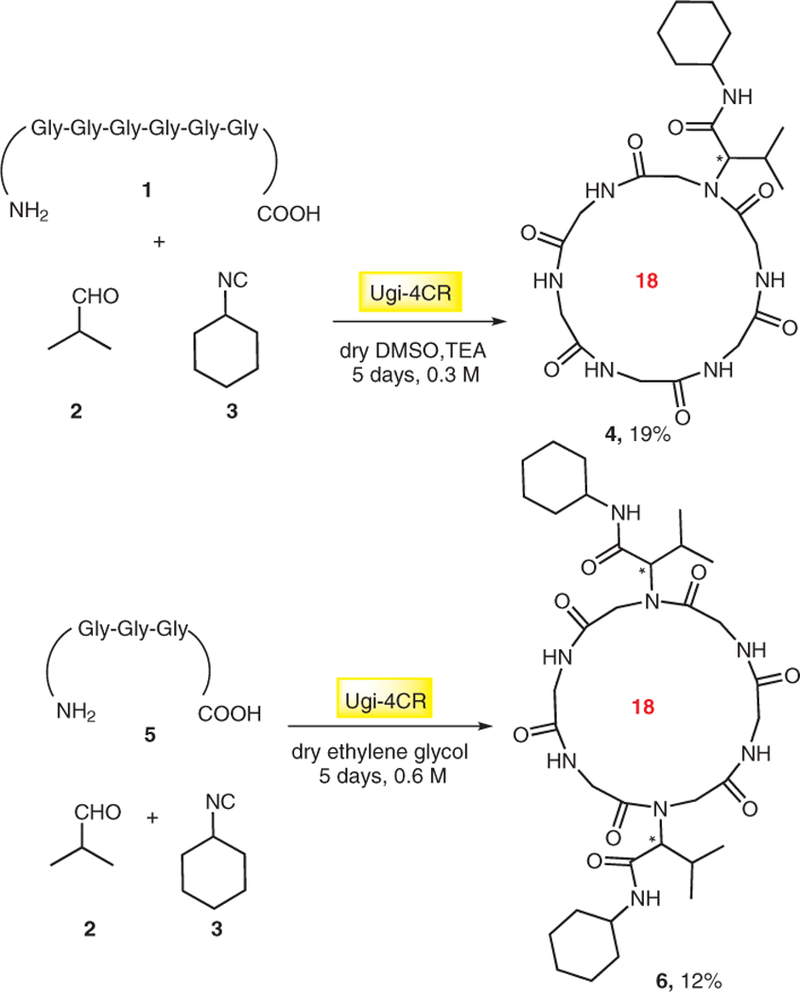
First in time peptide macrocyclization strategy using the Ugi reaction
An alternative synthetic approach to macrocycles is the synthesis of the functionalized linkers via MCR followed by different ring-closure methods. For example, Zhu et al. used the Ugi four component reaction (Ugi-4CR) followed by an intramolecular SNAr-based cycloetherification to synthesize macrocycles with an endo aryl-aryl ether bond found in vancomycin.11 The Ugi reaction of an aldehyde 7, an amine 8, an ω-(3-hydroxyphenyl) alkanecarboxylic acid 9, and an isocyanide 10 afforded the desired dipeptide amides 11 as a mixture of two diastereomers in a ratio of 1:1, separable by preparative TLC. Then, cycloetherification has been done smoothly in DMF using potassium carbonate as a base to form 16-membered macrocycle 12 in very good yield (Scheme 2). Interestingly, an unusual aprotic solvent toluene plus the additive ammonium chloride was used in the Ugi reaction.
Scheme 2.
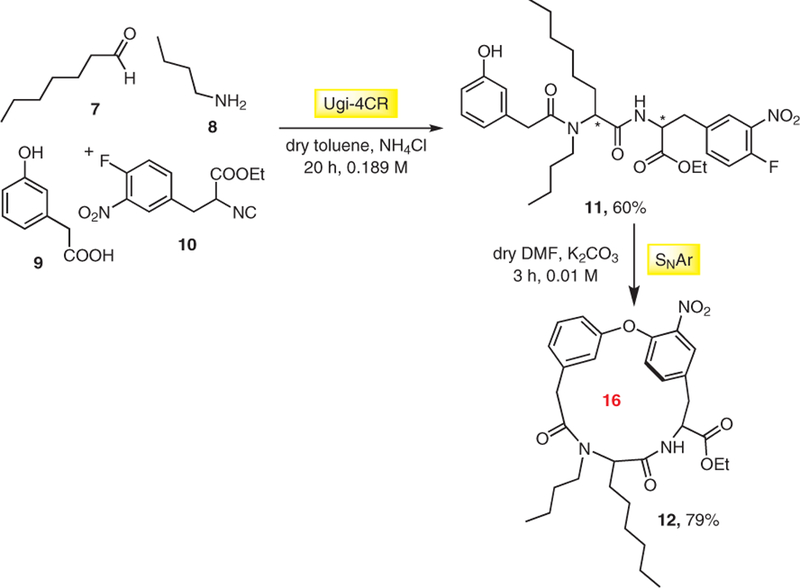
Synthesis of biaryl ether containing macrocycle
Zhu et al. also introduced a tandem Ugi-3CR and intra-molecular ‘click’ ring closure as a straightforward route to macrocycles of type 17.12 The reaction between ω-azidoam-ine 14, aldehyde 13, and alkynyl isocyanide 15 gives 5-ami-nooxazole 16 as an intermediate which undergoes intramolecular [3+2] cycloaddition between alkyne and azide to afford macrocycle 17. By this method different 14-, 15-, and 16-membered ring macrocycles were synthesized in 24– 76% yields (Scheme 3).
Scheme 3.
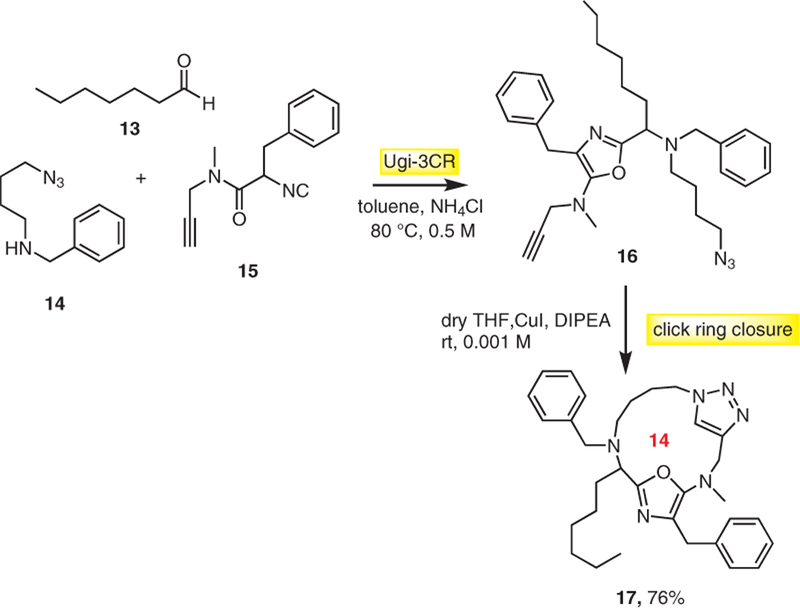
One-pot synthesis of macrocycles by tandem Ugi-3CR/click ring closure
A new protocol for the synthesis of macrocyclodepsipeptide 22 (Scheme 4) is via three-component reaction of α,α-disubstituted α-isocyanoacetamide 19, an aldehyde 18, and an amino alcohol 20 followed by saponification and cyclization under acidic conditions.13 5-Iminooxazoline 21 acts as an internal activator of the vicinal carboxylic acid under mild acidic conditions to afford macrocycle 22 in 48% overall yield.
Scheme 4.
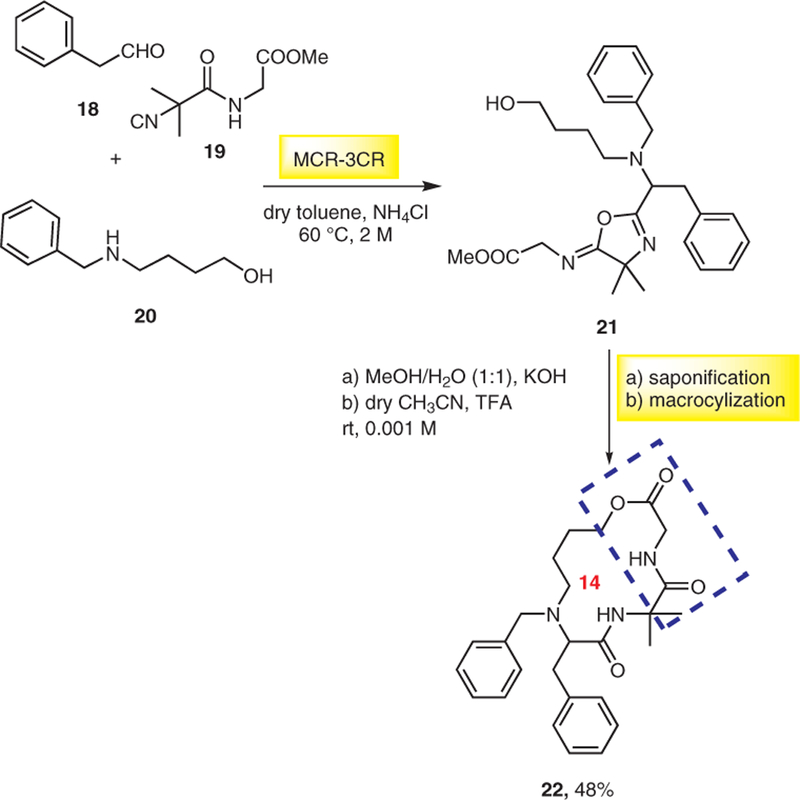
Synthesis of macrocyclodepsipeptide; the depsipeptide unit is highlighted by the blue-dotted box
Wessjohann and co-workers, other pioneers in MCR macrocycle chemistry developed a new strategy for the synthesis of cyclic RGD pentapeptoids via consecutive Ugi reactions.14 The targeted compound 29 was synthesized by two consecutive U-4CRs in which acyclic amino acid precursors 27 and 28 were synthesized in 68% and 85% yields, respectively. Acyclic amino acid 28, after ester hydrolysis and Cbz deprotection, underwent the third Ugi reaction in the presence of tert-butyl isocyanide and paraformaldehyde to yield cyclopeptoid 29 in overall 33% yield under pseudo-high-dilution conditions (Scheme 5).
Scheme 5.
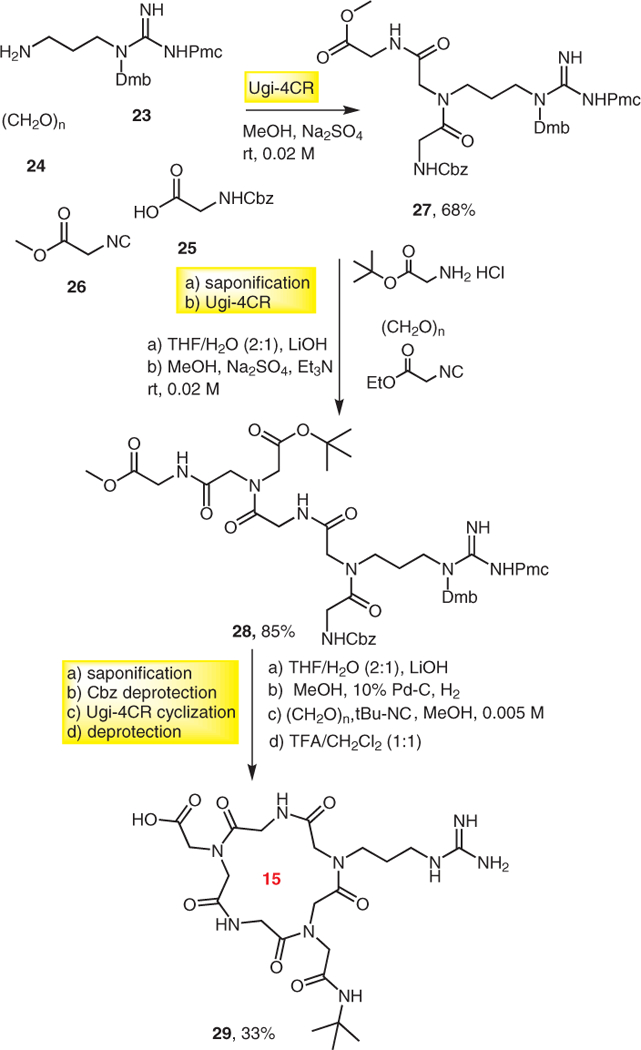
Synthesis of cyclopeptoid by triple U-4CRs
A new synthetic strategy relying on multiple multicomponent macrocyclizations including bifunctional building blocks (MiBs) was developed by Wessjohann.15 This approach has proven to be suitable for the rapid construction of challenging macrobicycles such as cryptands, crypto-phanes, and steroid-based cages. As an example, synthesis of cryptand 35 using diacids and diisocyanides as bifunctional building blocks is outlined in Scheme 6. The first Ugi-MiB was carried out by using diacid 30 and diisocyanide 33 to give macrocycle 34, which upon cleavage of the ester groups to two carboxylic functionalities, reacts as diacid in the second Ugi-MiB to afford macrobicycle 35 in 36% yield.
Scheme 6.
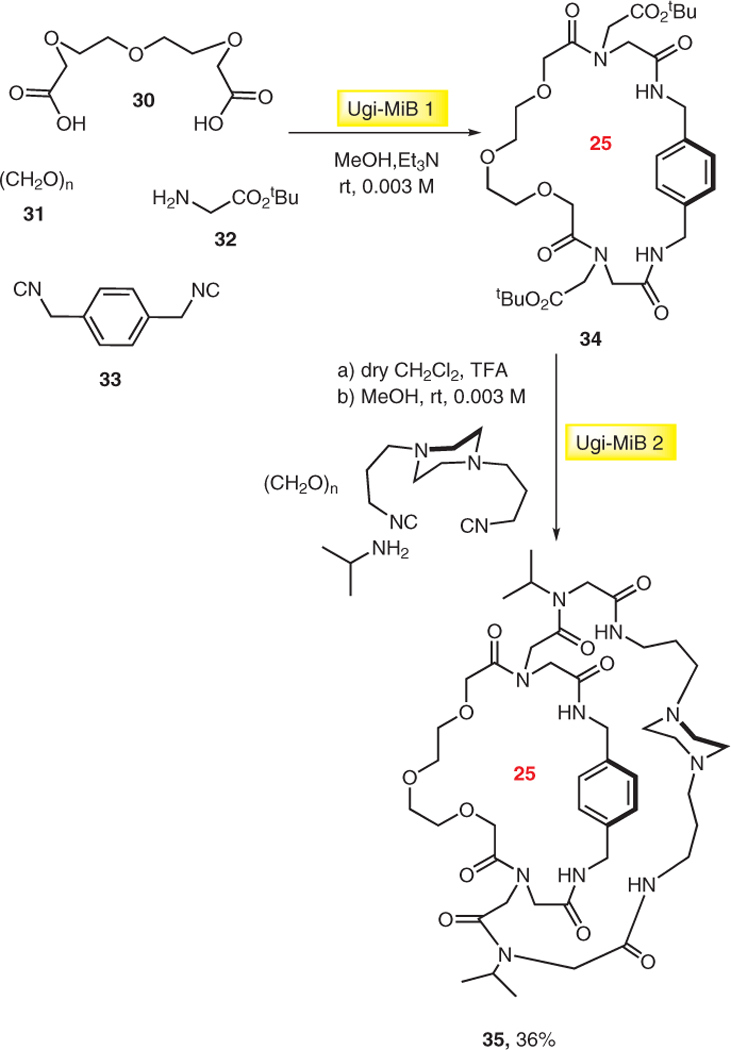
Synthesis of cryptand 35 by two sequential double Ugi-4CR-based macrocyclizations
Yudin introduced a general technology platform for the synthesis of cyclic peptides and derivatives by amphoteric azridine aldehyde dimers.16 This versatile synthetic approach leads to a multitude of cyclic peptide derivatives of different ring size with unusual side-chain modifications. This strategy has also been used for the synchronized synthesis of peptide-based macrocycles by digital microfluidics which is of potential interest for the fast and automated synthesis of libraries of compounds for applications in drug discovery and high-throughput screening.16a Cyclic peptides of type 37 and 38 were synthesized based on the Ugi-4CR by using amphiphilic aziridino aldehydes 36 (Scheme 7). Firstly, amino aldehyde and a linear peptide form an imine, which undergoes cyclization in the presence of isocyanide to give peptidic macrocycles with various ring sizes (9–18 atoms) depending on the used linear peptide.
Scheme 7.
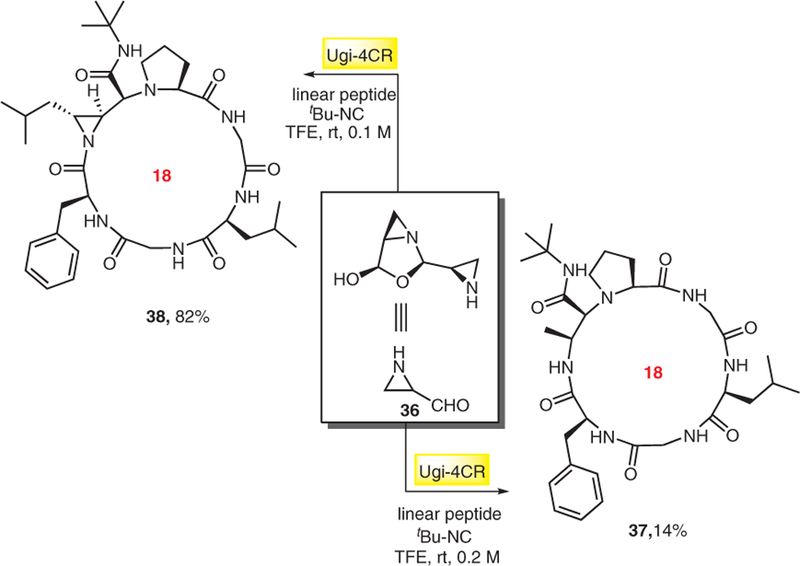
Aziridine carbaldehyde as versatile building block for macrocycle syntheses
Yudin also used the convertible isocyanide (N-isocya-nimino)triphenylphosphorane which was first introduced by Ramazani17 in the Ugi oxadiazole formation for the head-to-tail synthesis of 15-, 18-, 21-, and 24-membered rings 40 from linear peptide precursors 39 and aldehydes (Scheme 8).18 Interestingly all of the oxadiazole-containing macrocycles tested in the PAMPA assay displayed higher membrane permeability than cyclosporin A, the prototype of a macrocyclic bioavailable drug.
Scheme 8.
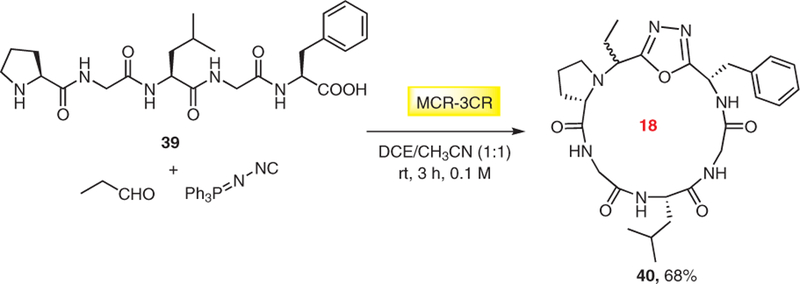
MCR synthesis of oxadiazole containing macrocyclic peptides
Recently, in order to improve pharmacological properties through the N-alkylation of the macrocycles and to access specific secondary structures of biological relevance, Rivera’s group introduced the use of Ugi reaction in side-chain to side-chain and side-chain to terminus macrocyclization of peptides.19 Linear peptide building blocks 41 and 43 were first synthesized either by a standard Fmoc solid-phase procedure or by a stepwise solution-phase synthesis, and then Ugi strategy was applied in the cyclization step by using commercially available isocyanides. Also, a smaller pentapeptide have been successfully cyclized by the Ugi reaction to 44 to show the flexibility in the tethering peptide side chains to the termini (Scheme 9).
Scheme 9.
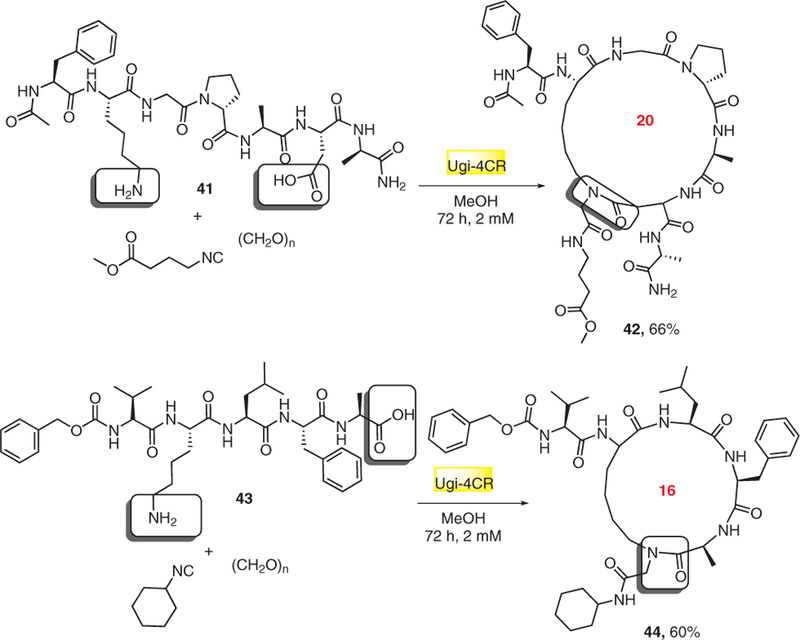
Macrocycles 42 and 44 represent side-chain to side-chain and side-chain to terminus peptide macrocyclization, respectively
A new multicomponent methodology by using the Ugi– Smiles reaction for the cyclization of 3-nitrotyrosine-containing peptides was reported by Rivera.20 Different derivatives of oligopeptides bearing the 3-nitrotyrosine residue at the C-terminus 45 and 47 were subjected to Ugi–Smiles macrocyclizations in the presence of paraformaldehyde and n-dodecyl isocyanide to give a variety of structurally novel N-aryl-bridged cyclic lipopeptides 46 and 48, respectively, in good yields (Scheme 10).
Scheme 10.
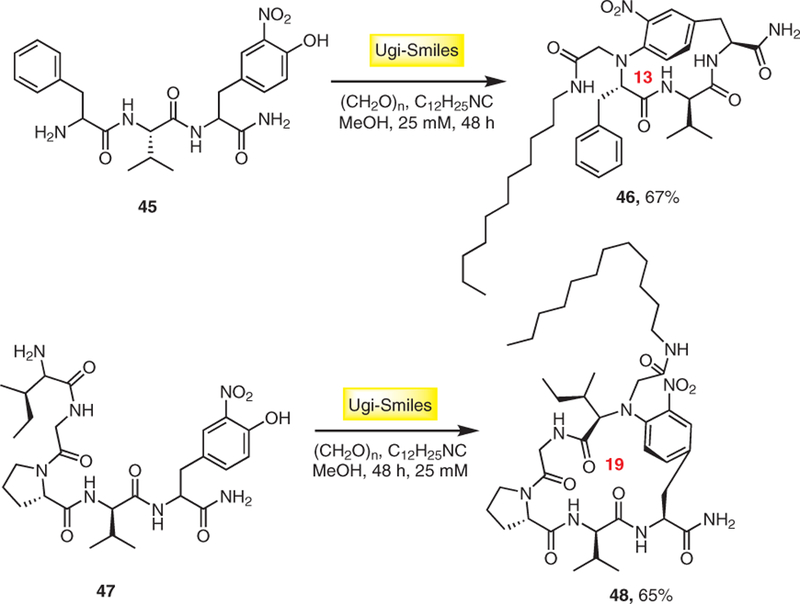
Novel N-aryl-bridged cyclic lipopeptides
In an early attempt to synthesize libraries of artificial macrocycles we investigated ideas for their rapid synthe-sis.21 We devised the concept to build up the linear precursors by using convergent MCR chemistry and including orthogonal functional groups for the end-game macrocyclization. For example, we synthesized linear precursors using different Passerini MCRs with terminal alkenes in two side chains to macrocyclize the ring by ring-closing metathesis (RCM) (Scheme 11). However, during the process of library expansion, we realized that while the initial Passerini MCRs 51 were working quite well over a wide range of substrates, the subsequent metathesis reaction was generally low yielding and had a very limited substrate scope in terms of ring size, side-chain diversity, and positioning of the orthogonal functional groups.
Scheme 11.
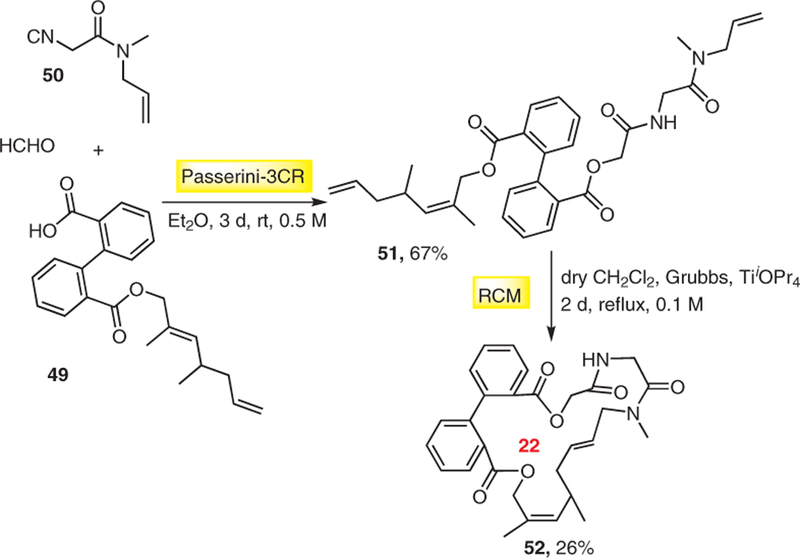
Fast macrocycle synthesis using MCR followed by RCM
Topologically, there are five different pathways to form macrocycles based on bifunctional starting materials using the Ugi-4CR reaction (Scheme 12). We will focus here on all of the possibilities for the synthesis of bifunctional building blocks in order to accomplish cyclization by MCR. Applying this concept, we have developed methods that can quickly and accurately convert small molecules into macrocycles via Ugi reaction. This approach provides a very short and versatile pathway to synthesize macrocycle libraries through isocyanide-based multicomponent reactions (IM-CRs).
Scheme 12.
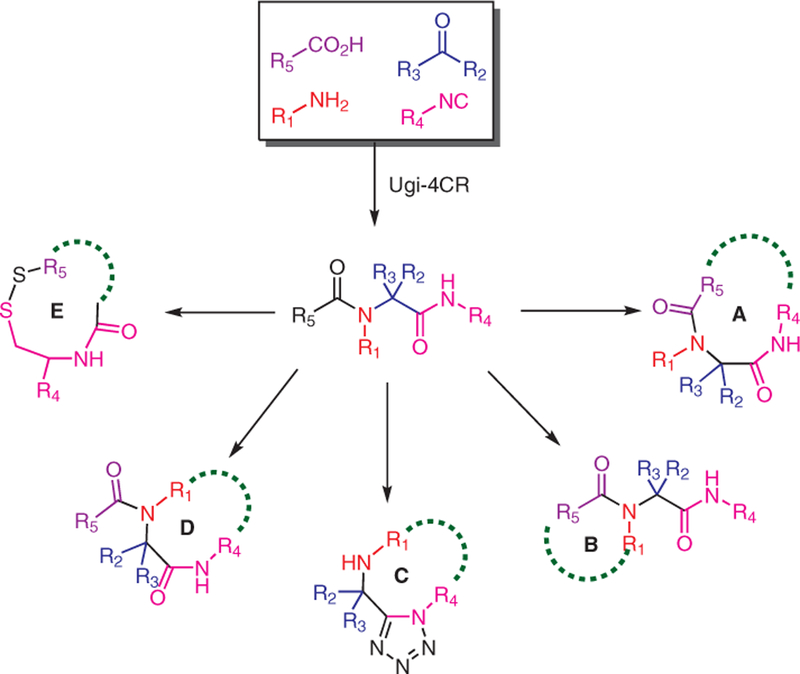
Topologically possible pathways for direct macrocyclizations using Ugi-4CR in the Dömling lab
Thus, we reacted α-isocyano-ω-carboxylic acids 53 of different lengths (Scheme 12 and Scheme 13, method A), which can be accessed in three steps from the commercially available amino acids, with the oxo and amine components to yield various 12- to 16-membered macrocycles 54 through an Ugi ring closure.22 Surprisingly, in this approach free isocyano carboxylic acid does not work, but the corresponding potassium salt with NH4Cl additive works nicely, and under the optimized conditions macrocycles with various size and different substituted α-isocyano-ω-carboxylic acids with additional amide and urea motifs can be synthesized (Scheme 13).
Scheme 13.

Macrocycles derived from α-isocyano-ω-carboxylic acids using Ugi-4CR
Another strategy developed by us, involves the ring opening of cyclic carboxylic acid anhydrides with diamines which was then applied to the head-to-tail cyclization of artificial medium- and macrocycles (Schemes 12 and 14, method B). A large library of terminal amino acids of different chain lengths was synthesized in good to excellent yields from α,ω-amino carboxylic acids 55 (Scheme 14). The ring closure was accomplished through an exponential diversification step using Ugi MCR resulting in complex macrocycles 56.23 The two-step reaction sequence can also be performed without isolation of the intermediate α,ω-amino acids thus providing a one-pot one-step version of the macrocycle synthesis. To the best of our knowledge this is the shortest de novo macrocycle synthesis described ever. Moreover, the reaction is very general. Assuming 100 derivatives of each starting material class, a macrocycle space of 1004 = 100 million can be accessed.
Scheme 14.
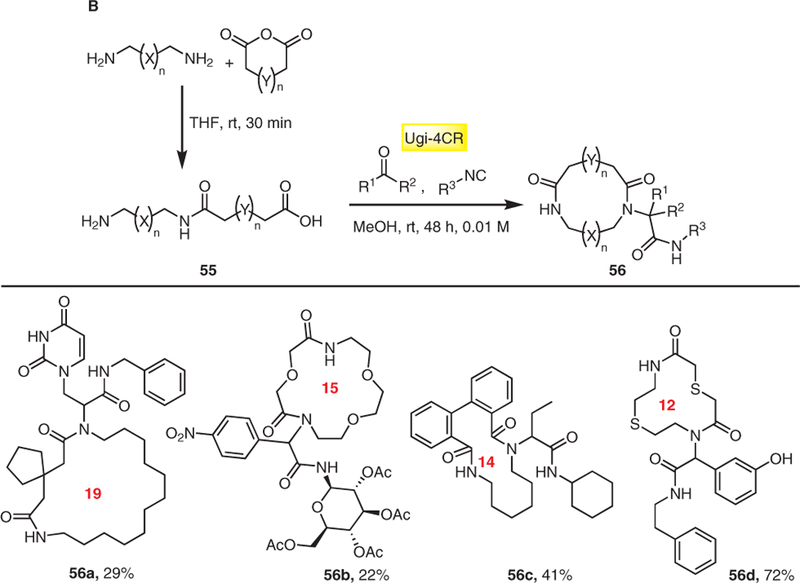
Macrocycles derived from α,ω-amino carboxylic acids using Ugi-4CR
Another IMCR macrocyclization strategy developed by us involves simple starting materials such as diamines, isocyanide esters, and aldehydes which leads to macrocylces with three points of diversity. In this strategy α-iso-cyano-ω-amines 57 and aldehydes in the presence of the azide source like TMSN3 leads to tetrazole macrocycles 58 (Schemes 12 and 15, method C)24a. Different α-isocyano-ω-amines of variable length were obtained in excellent purity and good yields (42–60%) by direct coupling of diamines with isocyanide esters. The macrocyclic ring closure was carried out through Ugi tetrazole reaction (UT-MCR) to afford macrocycles of size 11–19 in moderate yields of 21– 66% after purification by column chromatography (Scheme 15). To introduce more diversity, the same linker α-isocya-no-ω-amine 57 was also reacted with aldehyde and carboxylic acid to give macrocycles 59 by the Ugi MCR (Schemes 12 and 15, method D)24b. These are other examples of two-step de novo and very general macrocycles syntheses.
Scheme 15.
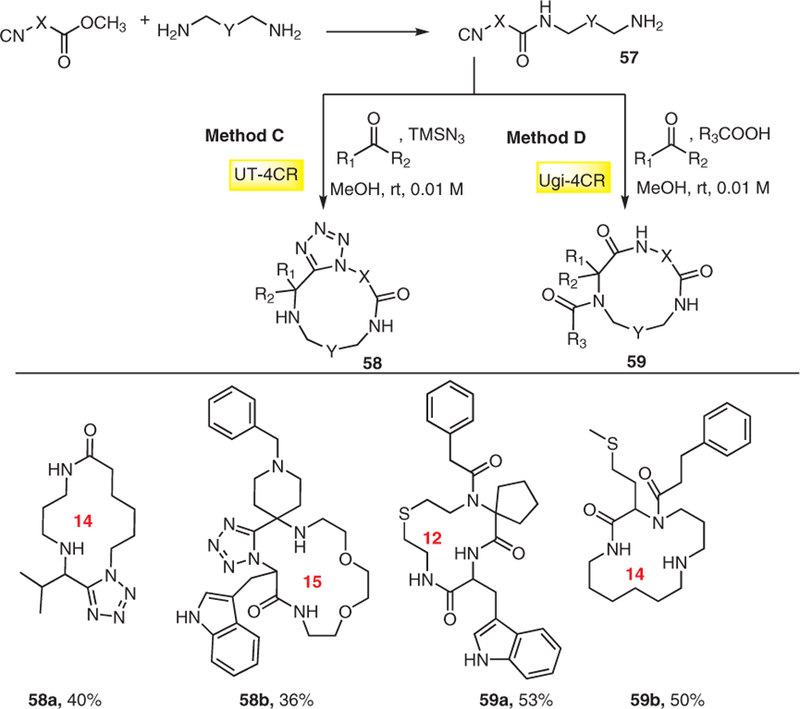
Macrocycles derived from α-isocyano-ω-amines using UT-MCR and classical Ugi MCR
We introduced the concept of ‘sulfur switch’ in the Ugi reaction, which leads to a diverse array of artificial disulfide-bridged macrocycles. In this strategy, the solid, odorless and configurationally stable cysteine-derived isocyanide was for the first time introduced in the Ugi-4CR, which fits well for head-to-tail disulfide formation.25 Reaction of Fmoc-Cys(Trt)-OH, amine, cysteine isocyanide, and aldehyde in a MeOH/THF/DMF solvent mixture afforded Ugi adduct 60, followed by iodine-mediated oxidative cyclization to give disulfide-bridged peptidomimetics 61 in good to excellent yields (Scheme 16).
Scheme 16.
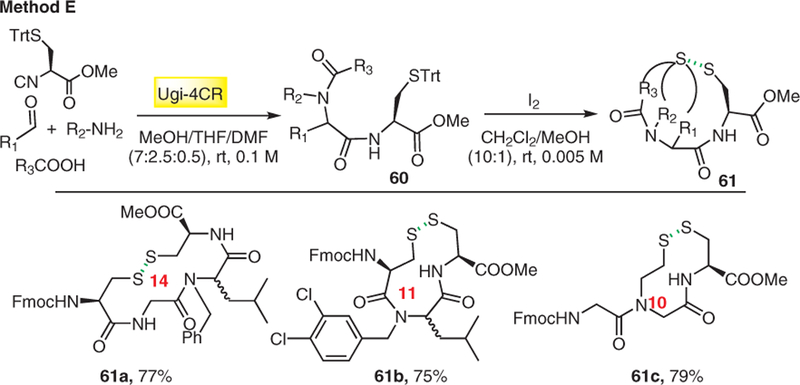
Disulfide bridged peptidomimetics 61
Next, we introduced a general strategy to macrocycles via union of two orthogonal MCRs, by using UT-MCR, an MCR of great interest due to the formation of α-amino tetrazoles, bioisosteres to cis-amides. The linker α-isocyano-ω-carboxylic acids were then macrocyclized by an U-4CR in the presence of primary amine and oxo component (Scheme 17).22 The first UT-MCR was performed by the reaction of an aldehyde, tritylamine, TMSN3, and a bifunctional ester protected amino acid derived isocyanide to give α-amino tetrazole in excellent yields, followed by deprotection and coupling reaction with an isocyano carboxylic acid to yield the α-isocyano-ω-carboxylic acid linker 62. Next, Ugi reaction for the macrocyclic ring closure was carried out in the presence of a primary amine and an oxo component in methanol as solvent to afford highly decorated macrocycles 63 of size 12–21 in moderate yields. In another approach, the Passerini MCR was used for the macrocyclic ring-closure step by using aliphatic, aromatic, and heterocyclic oxo components as aldehydes and ketones to yield macrocyclic depsipeptides 64 as shown in Scheme 17.26
Scheme 17.
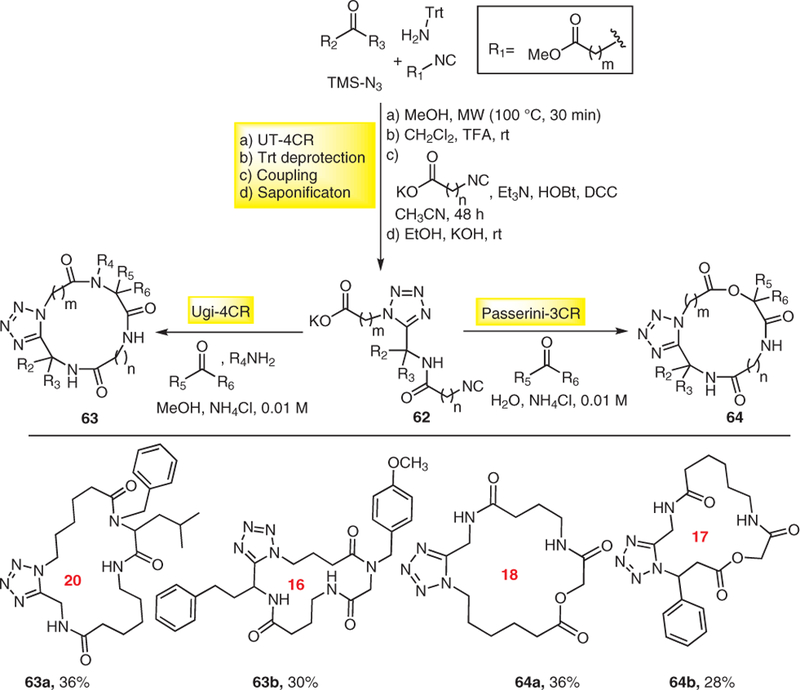
UT-MCR/Ugi-4CR, and UT-MCR/Passerini MCR derived macrocycle synthesis
To introduce even more diversity into the macrocycle linker portion, another well-established Ugi MCR, the U-5C-4CR, was also used to synthesize 21-membered macrocycle 66 from an unprotected α-amino acid (S)-proline, benzaldehyde, and diamine-derived monoisocyanide. The use of (S)-proline resulted in good diastereoselectivity in compound 65. After diastereomer separation by chromatography, the major diastereomer was reacted further in a sequence involving N-deprotection, coupling, saponification, and macrocycle formation by Ugi-4CR to afford 66 (Scheme 18).22 A key feature of macrocycle 66 is its tertiary amine as part of the macrocycles skeleton which potentially improves water solubility and blood-brain barrier penetration.
Scheme 18.
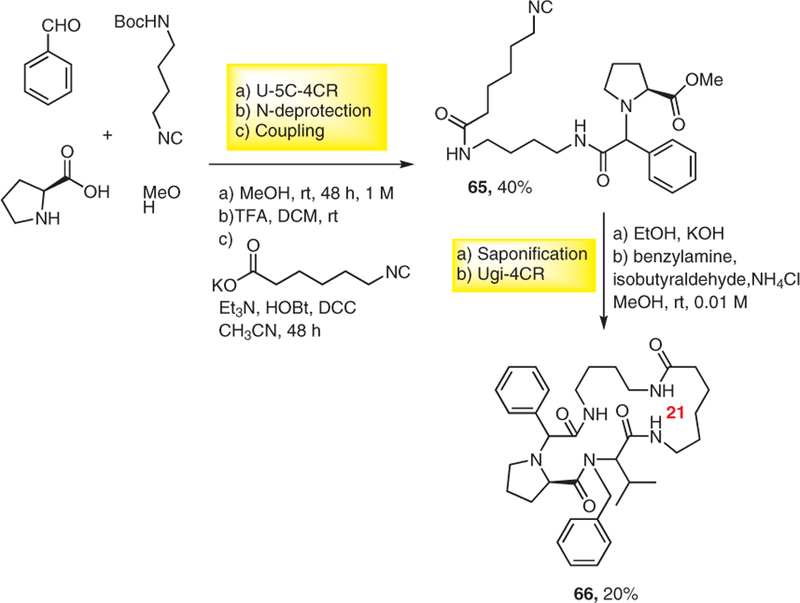
Mixed U-5C-4CR/Ugi-4CR strategy derived macrocycle synthesis pathway.
Overall, we have established up to now more than ten different synthetic routes towards variable artificial macrocycle scaffolds in one to maximum five sequential steps. This gives us a representative coverage of an interesting and large chemical space of macrocycles with affordable chemical accessibility.
4. Design Rules for Membrane Crossing Macrocycles
Membrane crossing of drugs is crucial for their biological activity because the great majority of molecular targets of drugs are intracellular. Moreover, the most preferred application form of drugs is oral. The role model of an orally bioavailable natural product, FDA-approved macrocyclic drug, is cyclosporine A with F = 30% in humans (Figure 4). Interestingly, cyclosporin A shows quite some conformational flexibility depending if it is receptor bound, or crystallized from an aprotic or protic solvent. While structural and conformational determinants of macrocycle cell permeability have been investigated, much more experimental and theoretical analysis work has to be performed to find the sweet spots of macrocyclic cell permeability.27
Figure 4.
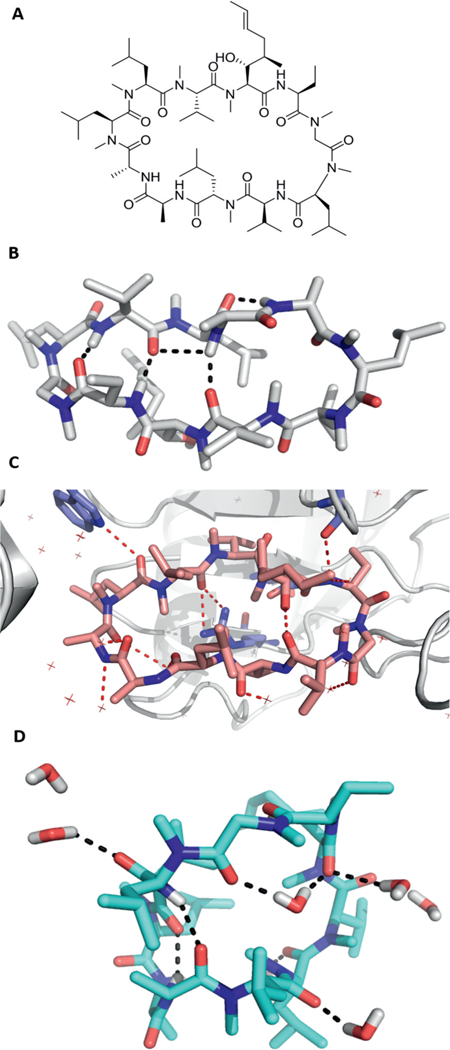
Different conformations of cyclosporin A in different environments. A: 2D structure of cyclosporin A; B: cyclosporin A crystallized from CCl4 (CCDC ID P212121). Four intramolecular hydrogen bondings are shown as black-dotted lines; C: cyclosporin A bound to the cistrans prolyl isomerase cyclophilin A (PDB ID 2X2C). All polar atoms are involved in hydrogen bondings to either the receptor or involved in a water network (red-dotted lines). One intramolecular hydrogen bond exists between the secondary hydroxy group and an amide carbonyl oxygen; D: cyclosporin A crystallized from H2O (CCDC ID P212121) forming a different hydrogen-bonding network then crystallized from CCl4 (B).
A simplified model of macrocycle passive membrane diffusion is shown in Figure 5.28 In the aqueous intra- and extracellular phase the macrocycle exists in a conformational ensemble to maximize the hydrogen-bonding contacts of the polar atoms with the water molecules, thus increasing the water solubility. Whereas in the lipid phase the macrocycle undergoes a conformational change to hide the polar atoms through intramolecular hydrogen bonding and exposing the hydrophobic moieties, thus increasing lipid solubility. According to this model the conformational dynamic of macrocycles allows for different physicochemical properties dependent on the solvent environment, such as polar surface area, lipophilicity, volume, and shape. Pictorially, macrocycles have been described to behave like chameleons.28 A consequence of this model is that highly dynamic macrocycles which can undergo a hydrophilic to hydrophobic side-chain exposure should facilitate passive membrane diffusion. Thus, macrocycles with intramolecular hydrogen-bonding opportunities could be candidates with improved passive membrane permeation, a topic of high interest in our laboratory.
Figure 5.
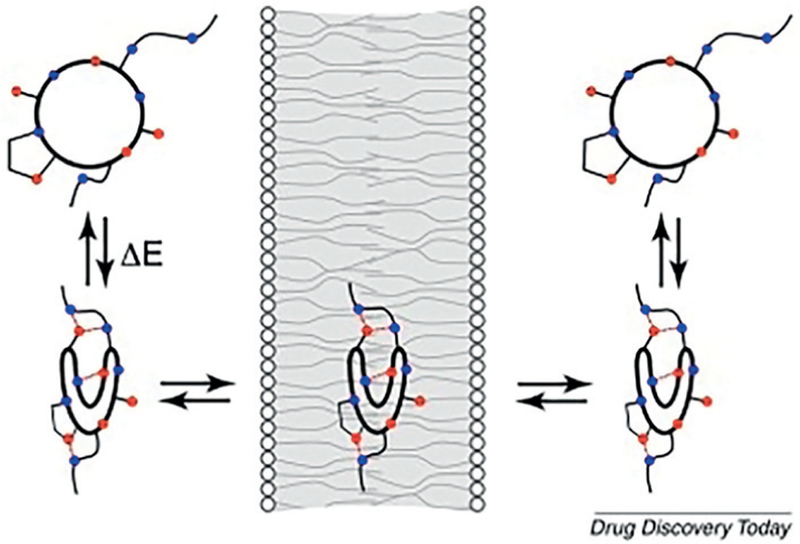
Simplified pictorial model for macrocycle passive membrane permeation (Reprinted with permission from ref. 28. Copyright 2018 Elsevier).
Thus, a key question in the field of macrocycles in drug discovery is can we propose guidelines for the design of macrocycles with a higher propensity for passive membrane permeation?
Key to passive membrane transport is the balance lipophilicity to polarity and the size and shape of molecules. Amongst polarity, hydrogen bondings play an outstanding role. Too many hydrogen-bonding motifs in molecules can have a deleterious effect on membrane penetration as a high water desolvation penalty has to be payed and unfavorable interactions of the polarized hydrogen with the aliphatic fatty acid side chains in the interior of the mem-brane. However, hydrogen bond is not hydrogen bond. The position is the overall molecule and their involvement in possible intramolecular contacts are important factors which can be used to design molecule penetrable yet polar. Size and molecular weight are part of predictive rules of oral bioavailability of small molecules and such molecules should have a MW < 500 Da according to Lipinsky rules. On the other hand, libraries of cyclic peptides show a much reduced membrane permeability at MWs above 1000 Da.29 Thus, it is speculated that 1000 Da constitutes a cut-off up-per size limit for druglike compounds.30
While torsion-angle preference is not systematically applied in small-molecule drug discovery, its investigation and influence on conformational dynamic, target occupancy, and transport properties is even less investigated in macrocycles.31 Sporadic investigations on some systems provide rather narrow rules which can be applied only to particular macrocycle systems. Recent examples include, for example, the conformational analysis of 14-membered macrocyclic ethers.32 Clearly much more in-depth research is needed to provide a general framework of macrocyclic SAR.
N-Methylation (alkylation) chemistry and biology of N-methylated proteins and peptides has been reviewed exten-sively.33 Peptide to peptoid substitutions has been shown to increase cell permeability in cyclic hexapeptides.34 It was found that N-substitutions maintained permeability but also increased conformational heterogeneity. Promisingly, diversification with nonproteinogenic side chains increased permeability up to threefold. Strikingly, in orally bioavailable cyclosporin A 7 out of 11 amide groups are N-methylated (Figure 6). On top of multiple other results, recent evidence suggested that N-methylated cis-peptide bonds at certain locations may promote the intestinal permeability of peptides through a suitable conformational preorganiza-tion.35 Site-specific chemical N-methylation of peptides can be challenging and is mostly performed through coupling of N-methyl amino acid building blocks. Using MCR technologies also allows for the site-specific N-substitution of secondary amide-Ns. Interestingly, using Ugi-type reactions allows not only for the site-specific N-methylation but many more substituted alkyl and aryl groups can be smoothly introduced (Figure 6). For example, by using the morpholino ethylamine building block an N-morpholino ethyl side chain can be introduced, which in addition to hiding the secondary amide hydrogen-bond donor also brings considerable water-solubility improvements of the overall structure.
Figure 6.
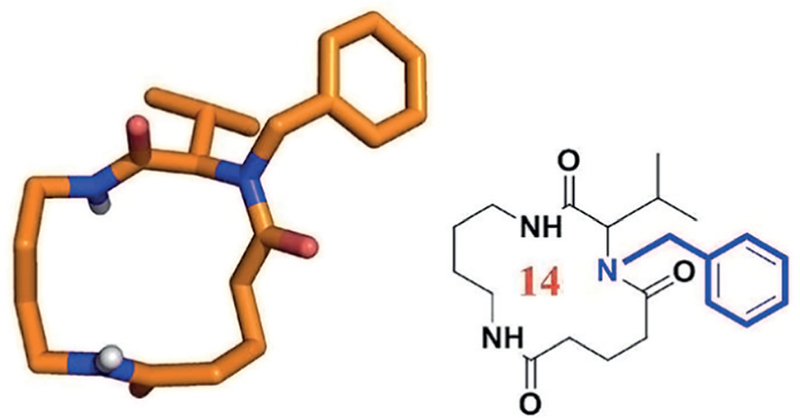
Example of an N-benzylated macrocyclic amide introduced by an Ugi macro ring closure (CCDC ID 1408656).
Overall, incorporation of peptoid residues into cyclic peptides and other macrocycles can maintain or improve cell permeability, while increasing access to diverse side-chain functionality thus allowing for straightforward property tuning.
Lipophilicity is a key property of druglike compounds, often highly important for efficient target binding and determines membrane penetration, water solubility, and metabolic stability and thus toxicology. Exaggerated lipophilicity can also lead to erroneous screening results.
For macrocyclic structures, it seems to be important that a balanced lipophilicity is maintained which can be adapted to the dielectricity constant of the solvents water and membrane lipids. Dynamic macromolecules which can exist in different conformations and expose or hide their polar surface area according to the nature of the solvent seems to be an important factor of passive membrane permeation. The lipophilicity of dynamic macrocycles is not static and calculation of cLogP based on 2D topology provides only a very limited picture. The cLogP and MW play an overarching role to obtain druglike properties. The libraries based on different MCR scaffolds designed in our laboratory show such a balanced property profile (Figure 7).
Figure 7.
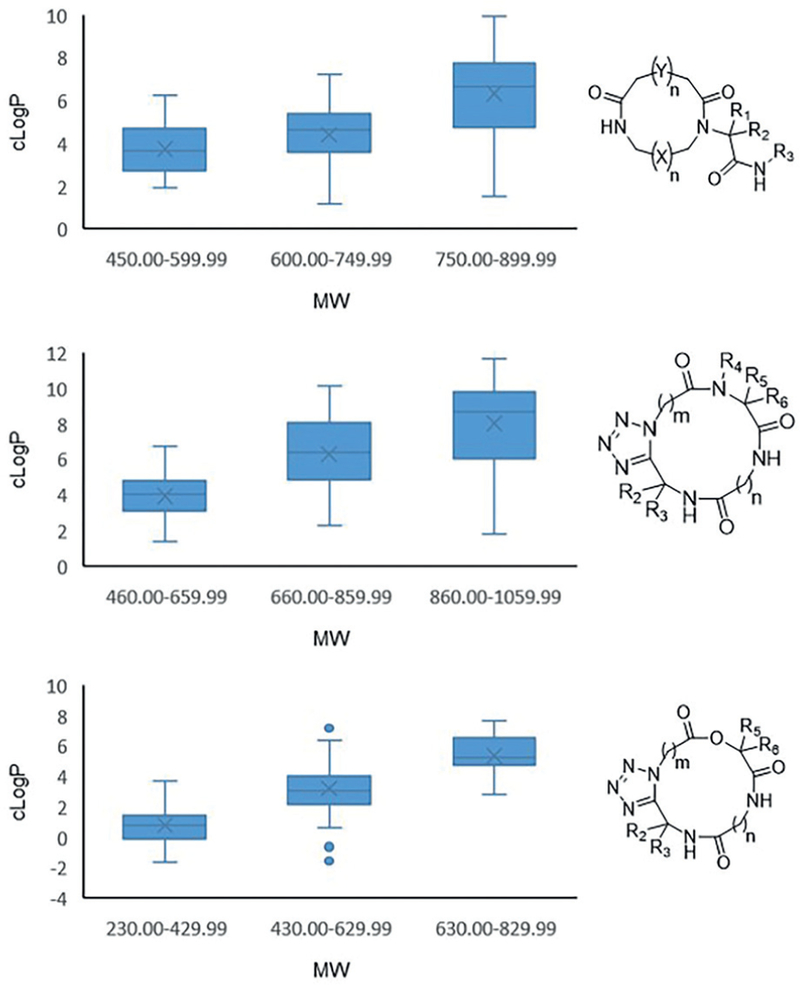
cLogP vs MW box plot of three different scaffolds shown in Scheme 13 and 16. Randomly 100 macrocycles were generated, and the properties calculated.
Thus, in the lipophilicity design of macrocyclic compounds not only the cLogP based on 2D structures but also the conformational dynamic must play a role and lipophilicities calculated based on their 3D structures are required.
Secondary amide to bioisostere transformations can help to improve druglike properties and passive permeation by reducing hydrogen-bond donor count, avoiding unfavorable polar lipid interactions and by influencing the dynamic behavior. Nature uses multiple molecular replacements in macrocycles, e.g. double bonds, small heterocycles – such as oxazoles and thiazoles –benzene rings, or S–S bonds. An example of a heterocycle in cyclic peptides was already mentioned before in the MCR synthesis of oxadiazole containing macrocyclic peptides (40), in which secondary amide-oxa-diazole replacement led to a great improvement on PAMPA permeability. The 1,5-disubstituted tetrazole is another example and consists of a well-known cis-amide bioisostere.36 Multiple powerful synthetic methods for the synthesis of 1,5-disubstituted tetrazole are known, and novel methods have been described including MCRs.37 We have developed several synthetic methods to incorporate the 1,5-disubsti-tuted tetrazole moiety into macrocycles, either into the macro ring or as a side chain (Figure 8).22 Central nervous system multiparameter optimization (CNS MPO) analysis of matched molecular pairs containing a secondary amide or a 1,5-disubstituted tetrazole reveals a large boost of predicted membrane penetration.38
Figure 8.
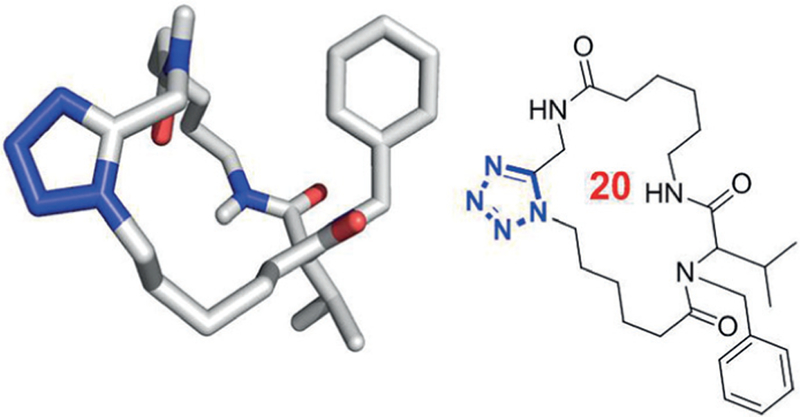
Example of incorporation of a cisamide isosteric 1,5-di substituted tetrazole moiety into a macrocycle (CCDC ID 1408649)
Other bioisosteres worthwhile to mention here is the triazole moiety, which has been utilized in β-turn mimetic peptides39 and as an isostere of the amide bond.40
Depsipetides are natural structures where in a peptide sequence one or several amide groups are replaced by an ester group thus leading to adjacent or alternating amide and ester groups. An archetypical example is the ionophore valinomycin. Replacement of a secondary amide group by an ester is leading to the reduction of hydrogen-bond donors and can thus lead to an increase of membrane permeation while leaving the overall polarity of the molecule similar. On the other hand, it has to be kept in mind that ester groups are rather easy to cleave spontaneously or enzymatically. The depsipetide moiety can be naturally introduced using the Passerini MCR which can lead to α-hydroxycarbonyl amides. We have developed several methods for the macro ring closure via the Passerini reaction which can be used to synthesize artificial macrocycles with depsipetide motifs (Figure 9 and Figure 10).26
Figure 9.
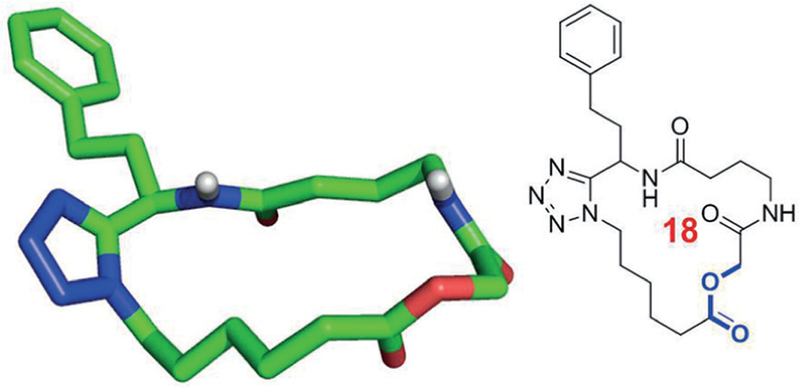
An artificial macrocycle with a depsipeptide motive incorporated into the ring structure (CCDC ID 1442896)
Figure 10.
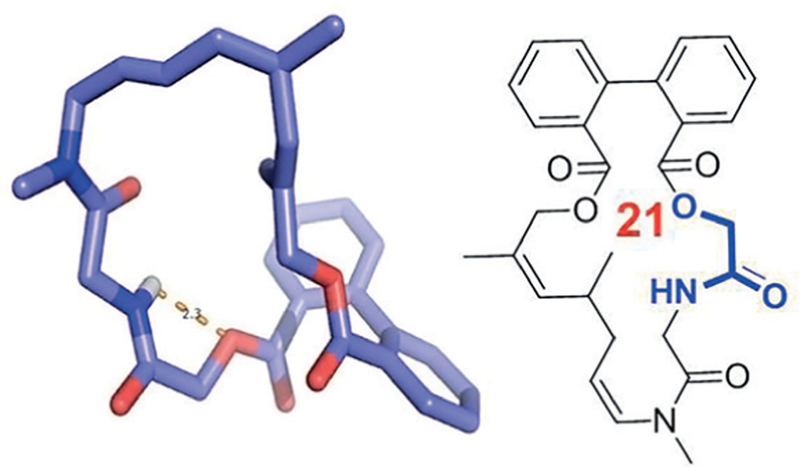
Example of a macrocycle featuring atropisomeric biphenyl moiety, a depsipeptide motif, two stiffening double bonds, and a weak intramolecular hydrogen bond (CCDC ID 200226)
Intramolecular hydrogen bonding can help to stabilize certain conformations which can expose or hide hydropho-bic substituents and thus help membrane crossing.41 This is also true for small cycles and the subtle balance between the strength of the hydrogen-bond interaction, geometry of the newly formed ring system, and the relative energies of the open and closed conformations in polar and unpolar environments has been analyzed in detail.42
For example, the structure–permeability relationships of cyclic hexapeptide diastereomers containing γ-amino acid compounds clearly showed much more water solubility (containing statine elements), better membrane permeability, and higher stability to liver microsomes than similar non-γ-amino acid containing derivatives.43 The permeability of the γ-amino acid containing macrocycles (Figure 11) correlated well with the extent of intramolecular hydrogen bonding observed in the solution structures determined in the low-dielectric solvent CDCl3, and the best compounds showed an oral bioavailability up to 21% in rat. Thus, the incorporation of γ-amino acids offers a route to increase backbone diversity and improve ADME properties in cyclic peptide scaffolds. γ-Amino acid moieties can be beneficially incorporated into artificial macrocycles using MCR routes.22
Figure 11.
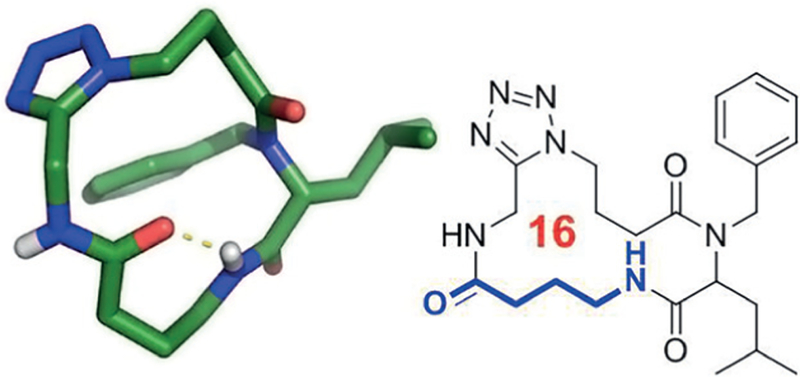
Example of an intramolecular hydrogen bond formed through a γ-amino acid linker unit in the macrocycles (CCDC ID 1408653)
Beyond the incorporation of γ-amino acids into macrocycles, the modulation of apparent lipophilicity through intramolecular hydrogen bonding in macrocyclic molecules in general is supported by intrinsic cell permeability and intestinal absorption data in rat and human.41 An additional possibility for designing intramolecular hydrogen bonding comprise the exo- to endocyclic amide hydrogen-bonding formation.
Recent evidence suggested that an exocyclic secondary amide group can play a key role in the assembly of the secondary structure of macrocyclic peptides.16b
This is in accordance with our findings in the solid-phase structures of several Ugi macrocycles derived from α-amino-ω-carboxylic acids, oxo components, and isocyanides. In all investigated structures a hydrogen bonding was found between the exocyclic secondary amide group towards an amide group in the macrocyclic ring (Figure 12).
Figure 12.
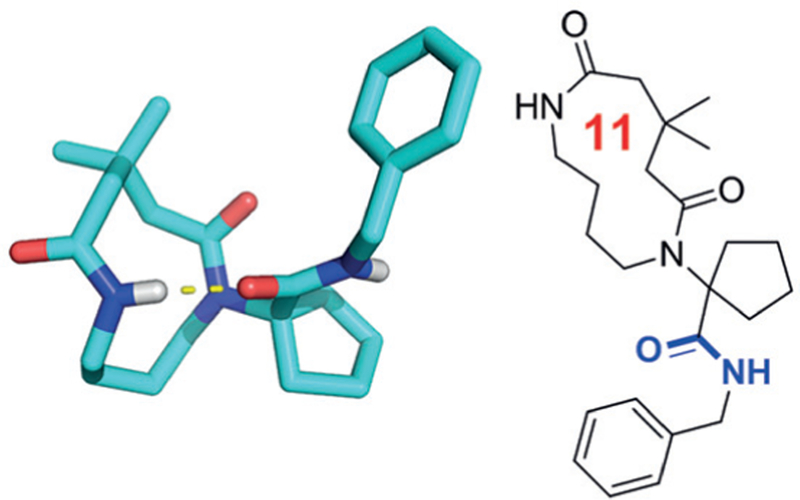
Example of a exo- to endocyclic hydrogen bonding in solid phase (CCDC ID 1508125)
5. Design Rules for Screening Libraries of Macrocycles
Screening of compound libraries is currently the most successful approach in early drug discovery. Therefore, much effort is placed into optimization of the screening libraries for high-throughput screening (HTS). Factors improving drug likeliness include water solubility, lipophilicity, number of rotatable bonds, MW, polar surface area, sp3 character (escape from flatland), exclusion of toxicophores, in- or exclusion of electrophiles (to yield covalent receptor adducts), etc. and have been discussed extensively in medicinal chemistry literature. On the other hand, modern post genomic targets need a different chemical space which is not adequately represented in current screening decks. Such chemical space is populated, for example, with compounds beyond current drug likeliness rules, for example, r-o-5. Recently, Whitty et al. investigated the question ‘How proteins bind macrocycles’ and thus analyzed 22 cocrystal structures of natural macrocycles binding to different pro-teins.1i To better describe the observed interactions the authors distinguished distinct macrocycle regions (Figure 1): 1) the ring atoms (black) which define the macrocyclic scaffold; 2) the peripheral atoms (red), comprising small groups such as methyl, carbonyl, hydroxyl, and halogens that consist of a single heavy atom directly bound to the macro ring; and 3) substituent atoms (blue), comprising larger structures connected to the ring. Different binding modes of macrocycles to their receptors could be observed including edge-, face-on, and compact binding mode. Whitty et al. proposed a set of rules for the design of libraries of pharmacologically active macrocycles by analyzing a number of macrocycle structural features:1i, 28
-
1)
Structural diversity in the substituent atoms, ring at-om, and peripheral atom region is an important consideration when designing MC libraries for drug discovery, as they bind equally likely to a receptor hot spot.
-
2)
Structural and polar diversity in peripheral groups is particularly important for good protein binding and cellular activity also to ensure adequate polar surface (PSA) area, which is critical for good aqueous solubility.
-
3)
The physicochemical balance of one polar (O or N) atom per two or three nonpolar (C, S, Cl) atoms should be targeted to yield a clogP similar to oral conventional drugs, while the PSA scales with MW is typically much higher.
-
4)
A diverse, general-purpose macrocycle library with large and small substituents distributed around the ring will have utility across a wide range of different protein binding-site topologies including edge-on, face-on, and compact binding modes. Conformational flexibility for a given macrocycle or class can help to switch between compact and elongated shapes.
-
5)
Substantial degree of unsaturation in the macrocycle skeleton by the introduction of alkenes, amide groups, or smaller (hetero)cycles is important likely improving rigidity and thus providing compact shapes for increased passive membrane permeation.
In our analysis of a more extensive set of ca. 100 protein macrocycle structures, including artificial macrocycles on top of natural products, we came to similar conclusions.1n During the design of artificial macrocycles we consider these design rules and analyses the features of our libraries and compare them with the Whitty rules. Clearly, macrocyclic scaffolds can be designed in a way to obey certain physicochemical rules.
6. Computational Macrocyclic Methods
We have recently introduced AnchorQuery, an interactive, web-based, and specialized pharmacophore search technology that brings interactive virtual screening of novel protein–protein inhibitors to the desktop.44 AnchorQuery leverages the concept of anchors, amino acid residues that bury a large amount of solvent accessible surface area at the protein–protein interface. Every compound in our >31 million MCR accessible virtual library contains an anchor analogue and a functional group that is a chemical mimic of a specific amino acid.45 AnchorQuery pharmacophore queries always include an anchor feature in addition to the standard hydrophobic, ionic, and hydrogen bond donors. Anchor-Query can be used together with the companion technology PocketQuery to provide an extract PPI inhibitor starting point pharmacophore from PPI structure.46 The virtual screening technology has been successfully used to discover multiple MCR scaffolds against the p53-MDM2/MDMX and PDK2 PPIs.47 The virtual screening technology is very powerful to discover novel ligands especially if a deep concave and hydrophobic pocket is present in the protein target. We are currently implementing a similar VS technology based on the wealth of macrocyclic MCR chemistry (Figure 13).48a However, populating a meaningful representation of a 3D conformational space is a highly demanding, not completely solved problem and has been reviewed recently.48b Many different software technologies have been proposed for efficient macrocycle conformational sampling and compared against each other.49 We are using the free macrocycle conformer generator of MOLOC which gives a useful 3D space presentation and gives comparable good results to commercial software.50 The efficient macrocycle conformational sampling together with the fast synthetic access to the MCR macrocycle space and a powerful pharmacophore VS platform will be established to facilitate macrocycle drug discovery for difficult biological targets. Using this pharmacophore approach, we were already able to discover a potent 15-membered macrocyclic inhibitor of the p53-MDM2 PPI (Figure 14). 23
Figure 13.

Above: ANCHOR.QUERY platform work flow. Below: different conformers of a 13-membered macrocycle with a biphenyl unit and generated with MOLOC.
Figure 14.
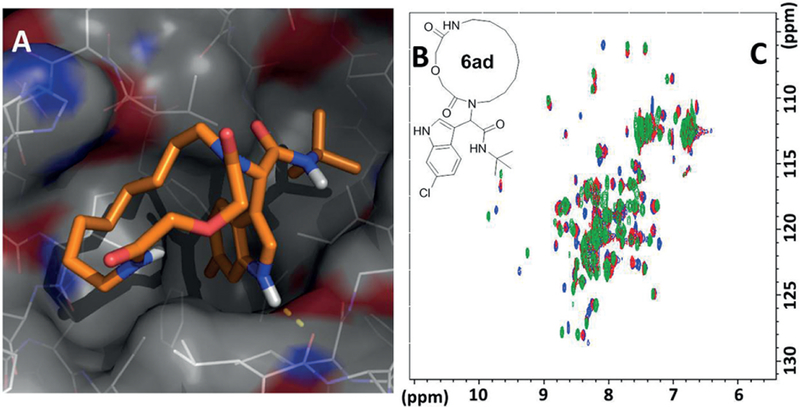
Articial macrocyclic p53-MDM2 protein–protein interaction antagonists.Docking picture (A) of compound 6ad in MDM2 receptor (PDB ID 1YCR) based on HSQC binding studies (B)
7. Future View
Artificial macrocycles are an emerging and largely underexploited part of chemical space where potentially drugs for difficult genomic targets can be discovered. Current pharmaceutical libraries are largely unsuitable to target the large and interesting class of post genomic targets, and artificial macrocycles promise to fill the gap between small molecules and the large molecular weight biologics. While artificial macrocycles can have advantages over their natural twins such as better control over synthesis, ADMET properties and target binding, fast and convergent synthesis pathways are underdeveloped. We foresee a couple of topical areas for future research in the macrocyclic chemical and biological space (Figure 15). While there seems to be a MW cut-off of 1000 Da for passive membrane transportation, there is also indication that specifically the macrocyclic space between 500–1000 Da is virtually unexplored but holds promise to harbor a large number of macrocycles with druglike ADMET properties and therefore represents a vast opportunity for those prepared to venture into new territories of drug discovery. In our laboratory novel strategies are elaborated to synthetically access specifically the space of 500–1000 Da using convergent chemistries including MCR. To fully leverage the potential of artificial macrocycles the structural factors enabling membrane penetration and the dependency on the cycle dynamic has to be thoroughly investigated. The poorly understood dynamic of the macrocycles also plays an important role in receptor binding, and better understanding could be very helpful in the screening of virtual libraries of artificial macrocycles. Large-scale investigations of PPI structures, for example, loops reveal another promising area for macrocycle drug design. The successful application of virtual screening against protein receptors or pharmacophore-based, however, needs considerable refinements of the computational sampling and representation of the 3D conformer space of macrocycles. The good news for synthetic organic chemistry is that the current state-of-the-art thus provides a considerable chance to develop novel, general, fast, stereoselective, and diverse routes and methodologies to this intriguing scaffold class. Multicomponent reaction technology can play a considerable role in this efforts by building on its strengths such as convergence, diversity, synthetic simplicity, and ability to cover a considerable chemical space. Macrocycles are magic.
Figure 15.
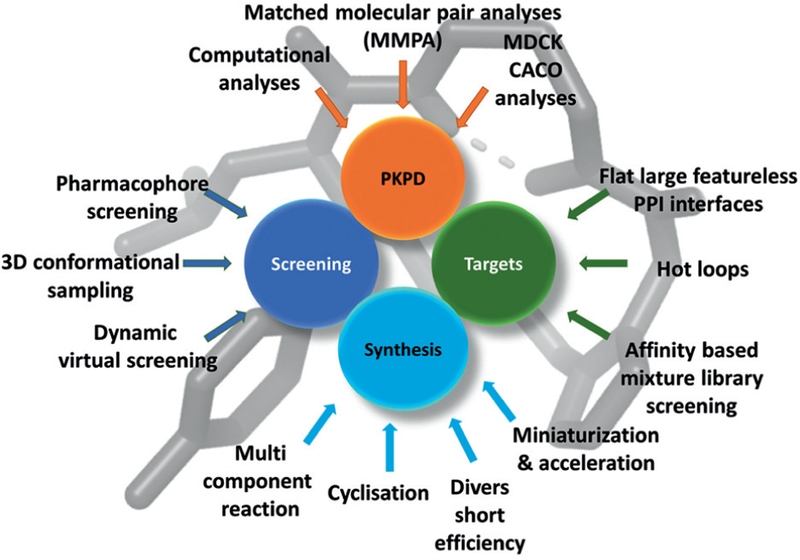
Some contemporary and future topics of research in the macrocycle area
Acknowledgment
The work in the PIs laboratory was financially supported from the NIH (NIH 2R01GM097082-05) and by the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115489, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ inkind contribution. Moreover, funding has also been received from the European Union’s Horizon 2020 research and innovation programme under MSC ITN ‘Accelerated Early stage drug dIScovery’ (AEGIS), grant agreement No 675555. EMMA is grateful for a PhD stipendship from the Egypt government. SS is grateful for a postdoctoral fellowship from KWF (Grant Agreement No. 10504).
Biographical Sketches

Prof. Alexander Dömling has held the chair for Drug Design at the University of Groningen since 2011. He studied chemistry and biology at the Technische Universität München and obtained his PhD under the guidance of Ivar Ugi. After a postdoc under a Humboldt Fellowship in the group of the Nobel Laureate Barry Sharpless, he founded the biotech company Morphochem, later Carmolex Inc. and most recently Telesis and SMIO BV. After his habilitation,he worked as full professor at the University of Pittsburgh in the School of Pharmacy. His interests are centered around MCR chemistry and its application to problems in drug discovery. His special focus is centered around the question how to leverage the huge MCR space. Thus, he is working on MCRcentered pharmacophore methods structure-based drug design,and MCR-centered fragment-based drug design methods and extreme miniaturization to library synthesis. He is the author of more than 200 scientific articles, reviews, and book contributions. He has applied for more than 30 patents. His long-term vision is to bring a novel drug to patients in an indicated area of unmet medical needs.

Eman Abdelraheem was born in Sohag, Egypt. She obtained her BSc degree in Chemistry and did her MSc in Organic Chemistry under the guidance of Prof. Ahmed K. El-Shafei at Sohag University, Egypt. During her master’s degree, she worked on the project ‘Synthesis of some New Spiro and Fused Heterocyclic Compounds’. Currently, she is pursuing her PhD under the supervision of Prof. Alexander Dömling at Drug Design Department, University of Groningen, Netherlands since 2013. She is working on multicomponent reaction (MCR) chemistry for the synthesis of ‘Artificial Macrocycles and its Application as Potent p53–MDM2 Inhibitors’. She is the author of more than 20 scientific articles and reviews, and she joined for more than 10 international conferences. For her PhD studies, she was awarded an Egyptian PhD Scholarship.

Dr. Shabnam Shaabani studied chemistry at the Shahid Beheshti University (SBU), Tehran, Iran. She did her MSc and PhD under the supervision of Prof. Ahmad Shaabani. She was member of the exceptional talent center at SBU. Dr. Shabnam Shaabani is a visionary young scientist and her dream is to design, synthesize, and develop molecules for unmet medical needs such as orphan diseases. Currently, she is doing her postdoc at the Drug Design Group under Prof. Dömling’s supervision at the University of Groningen, Groningen, The Netherlands, working on PETlabeled PD1 small molecule ligands to develop companion diagnostics for immune oncology therapy. Her research interests include nanocatalysis, multicomponent reactions (MCR), application of MCR towards chemical biology, drug discovery in immune oncology and orphan diseases, and miniaturization and automation in chemistry (‘Drug Discovery at the Speed of Sound’). She is the author of more than 20 scientific articles and book contributions. Dr. Shabnam Shaabani is aiming topursue an academic career.
References
- (1) (a).Driggers EM; Hale SP; Lee J; Terrett NK Nat. Rev. Drug Discov 2008, 7, 608. [DOI] [PubMed] [Google Scholar]; (b) Frost JR; Smith JM; Fasan R Curr. Opin. Struct. Biol 2013, 23, 571. [DOI] [PubMed] [Google Scholar]; (c) Gavenonis J; Sheneman BA; Siegert TR; Eshelman MR; Kritzer JA Nat. Chem. Biol 2014, 10, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Heinis C Nat. Chem. Biol 2014, 10, 696. [DOI] [PubMed] [Google Scholar]; (e) Hunsdiecker H; Erlbach H Chem. Ber 1947, 80, 129. [Google Scholar]; (f) Mäde V; Els-Heindl S; Beck-Sickinger AG Beilstein J. Org. Chem 2014, 10, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Marsault E; Peterson ML J. Med. Chem 2011, 54, 1961. [DOI] [PubMed] [Google Scholar]; (h) Masson G; Neuville L; Bughin C; Fayol A; Zhu J Multicomponent Syntheses of Macrocycles, In Synthesis of Heterocycles via Multicomponent Reactions II; Orru RVA; Ruijter E, Eds.; Springer: Berlin, Heidelberg, 2010, 1. [Google Scholar]; (i) Villar EA; Beglov D; Chennamadhavuni S; Porco JA Jr.; Kozakov D; Vajda S; Whitty A Nat. Chem. Biol 2014, 10, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wessjohann L; Ruijter E; Garcia-Rivera D; Brandt W Mol. Diversity 2005, 9, 171. [DOI] [PubMed] [Google Scholar]; (k) White CJ; Yudin AK Nat. Chem 2011, 3, 509. [DOI] [PubMed] [Google Scholar]; (l) Yudin AK Chem. Sci 2015, 6, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Gartner ZJ; Tse BN; Grubina R; Doyon JB; Snyder TM; Liu DR Science 2004, 305, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) http://www.drugdesign.nl/publications/macrocycle-database/; (o) http://www.drugdesign.nl/wpcontent/uploads/2018/04/1AY6.pdf.
- (2).Dömling A; Holak TA Angew. Chem. Int. Ed 2014, 53, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zak KM; Kitel R; Przetocka S; Golik P; Guzik K; Musielak B; Dömling A; Dubin G; Holak TA Structure 2015, 23, 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kozakov D; Hall DR; Napoleon RL; Yueh C; Whitty A; Vajda SJ Med. Chem 2015, 58, 9063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang W; Groves MR; Dömling A MedChemComm 2018, 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kuhn B; Fuchs JE; Reutlinger M; Stahl M; Taylor NR J. Chem. Inf. Model 2011, 51, 3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Ziegler K; Eberle H; Ohlinger H Eur. J. Org. Chem 1933, 504, 94. [Google Scholar]; (b) Ruggli P Eur. J. Org. Chem 1912, 392, 92. [Google Scholar]
- (8) (a).Verdine GL; Hilinski GJ Drug Discovery Today: Technol 2012, 9, e41. [Google Scholar]; (b) Connors WH; Hale SP; Terrett NK Curr. Opin. Chem. Biol 2015, 26, 42. [DOI] [PubMed] [Google Scholar]; (c) Smith JM; Frost JR; Fasan, J. Org. Chem 2013, 78, 3525. [DOI] [PubMed] [Google Scholar]
- (9).Koopmanschap G; Ruijter E; Orru RV A. Beilstein J. Org. Chem 2014, 10, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Failli A; Immer H; Götz M Can. J. Chem 1979, 57, 3257. [Google Scholar]
- (11).Cristau P; Vors JP; Zhu JP Org. Lett 2001, 3, 4079. [DOI] [PubMed] [Google Scholar]
- (12).Pirali T; Tron GC; Zhu JP Org. Lett 2006, 8, 4145. [DOI] [PubMed] [Google Scholar]
- (13).Pirali T; Tron GC; Masson G; Zhu JP Org. Lett 2007, 9, 5275. [DOI] [PubMed] [Google Scholar]
- (14).Vercillo OE; Andrade CKZ; Wessjohann LA Org. Lett 2008, 10, 205. [DOI] [PubMed] [Google Scholar]
- (15).Rivera DG; Wessjohann LA J. Am. Chem. Soc 2009, 131, 3721. [DOI] [PubMed] [Google Scholar]
- (16) (a).Hili R; Rai V; Yudin AK J. Am. Chem. Soc 2010, 132, 2889. [DOI] [PubMed] [Google Scholar]; (b) Zaretsky S; Scully CCG; Lough AJ; Yudin AK Chem. Eur. J 2013, 19, 17668. [DOI] [PubMed] [Google Scholar]
- (17).Ramazani A; Rezaei A Org. Lett 2010, 12, 2852. [DOI] [PubMed] [Google Scholar]
- (18).Frost JR; Scully CCG; Yudin AK Nat. Chem 2016, 8, 1105. [DOI] [PubMed] [Google Scholar]
- (19).Vasco AV; Perez CS; Morales FE; Garay HE; Vasilev D; Gavin JA; Wessjohann LA; Rivera DG J. Org. Chem 2015, 80, 6697. [DOI] [PubMed] [Google Scholar]
- (20).Morejon MC; Laub A; Westermann B; Rivera DG; Wessjohann LA Org. Lett 2016, 18, 4096. [DOI] [PubMed] [Google Scholar]
- (21).Beck B; Larbig G; Mejat B; Magnin-Lachaux M; Picard A; Herdtweck E; Dömling A Org. Lett 2003, 5, 1047. [DOI] [PubMed] [Google Scholar]
- (22).Liao GP; Abdelraheem EMM; Neochoritis CG; Kurpiewska K; Kalinowska-Tluscik J; McGowan DC; Dömling A Org. Lett 2015, 17, 4980. [DOI] [PubMed] [Google Scholar]
- (23).Madhavachary R; Abdelraheem EMM; Rossetti A; Twarda-Clapa A; Musielak B; Kurpiewska K; Kalinowska-Tłuścik J; Holak TA; Dömling A Angew. Chem. Int. Ed 2017, 56, 10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24) (a).Abdelraheem EMM; de Haan MP; Patil P; Kurpiewska K; Kalinowska-Tłuścik J; Shaabani S; Dömling A Org. Lett 2017, 19, 5078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Abdelraheem EMM; Khaksar S; Kalinowska-Tłuścik J; Shaabani S; Dömling AJ Org. Chem 2018, 83, 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Vishwanatha TM; Bergamaschi E; Dömling A Org. Lett 2017, 19, 3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Abdelraheem EMM; Kurpiewska K; Kalinowska-Tłuścik J; Dömling AJ Org. Chem 2016, 81, 8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Over B; Matsson P; Tyrchan C; Artursson P; Doak BC; Foley MA; Hilgendorf C; Johnston SE; Lee MD IV.; Lewis J Nat. Chem. Biol 2016, 12, 1065. [DOI] [PubMed] [Google Scholar]
- (28).Whitty A; Zhong M; Viarengo L; Beglov D; Hall DR; Vajda, Drug Discovery Today 2016, 21, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pye CR; Hewitt WM; Schwochert J; Haddad TD; Townsend CE; Etienne L; Lao Y; Limberakis C; Furukawa A; Mathiowetz AM; Price DA; Liras S; Lokey RS J. Med. Chem 2017, 60, 1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Matsson P; Kihlberg JJ Med. Chem 2017, 60, 1662. [DOI] [PubMed] [Google Scholar]
- (31).Schärfer C; Schulz-Gasch T; Ehrlich H-C; Guba W; Rarey M; Stahl MJ Med. Chem 2013, 56, 2016. [DOI] [PubMed] [Google Scholar]
- (32).Clyne DS; Weiler L Tetrahedron 2000, 56, 1281. [Google Scholar]
- (33).Chatterjee J; Rechenmacher F; Kessler H Angew. Chem. Int. Ed 2013, 52, 254. [DOI] [PubMed] [Google Scholar]
- (34).Schwochert J; Turner R; Thang M; Berkeley RF; Ponkey AR; Rodriguez KM; Leung SS; Khunte B; Goetz G; Limberakis C Org. Lett 2015, 17, 2928. [DOI] [PubMed] [Google Scholar]
- (35).Marelli UK; Ovadia O; Frank AO; Chatterjee J; Gilon C; Hoffman A; Kessler H Chem. Eur. J 2015, 21, 15148. [DOI] [PubMed] [Google Scholar]
- (36) (a).Herr RJ Bioorg. Med. Chem 2002, 10, 3379. [DOI] [PubMed] [Google Scholar]; (b) Zabrocki J;Dunbar JB Jr.; Marshall KW; Toth MV; Marshall GR J. Org. Chem 1992, 57, 202. [Google Scholar]; (c) Zabrocki J; Smith GD; Dunbar JB; Iijima H; Marshall GR J. Am. Chem. Soc 1988, 110, 5875. [Google Scholar]
- (37).Chandgude AL; Dömling A Eur. J. Org. Chem 2016, 2383.
- (38).Wager TT; Hou X; Verhoest PR; Villalobos A ACS Chem. Neurosci 2010, 1, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Oh K; Guan ZB Chem. Commun 2006, 3069. [DOI] [PubMed]
- (40) (a).Brik A; Alexandratos J; Lin YC; Elder JH; Olson AJ; Wlodawer A; Goodsell DS; Wong CH ChemBioChem 2005, 6, 1167. [DOI] [PubMed] [Google Scholar]; (b) Angell Y; Burgess KJ Org. Chem 2005, 70, 9595. [DOI] [PubMed] [Google Scholar]; (c) Bock VD; Speijer D; Hiemstra H; van Maarseveen JH Org. Biomol. Chem 2007, 5, 971. [DOI] [PubMed] [Google Scholar]; (d) Hitotsuyanagi Y; Motegi S; Hasuda T; Takeya K Org. Lett 2004, 6, 1111. [DOI] [PubMed] [Google Scholar]; (e) Horne WS; Stout CD; Ghadiri MR J. Am. Chem. Soc 2003, 125, 9372. [DOI] [PubMed] [Google Scholar]; (f) Horne WS; Yadav MK; Stout CD; Ghadiri MRJ Am.Chem. Soc 2004, 126, 15366. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Goncalves V; Gautier B;Regazzetti A; Coric P; Bouaziz S; Garbay C; Vidal M;Inguimbert N Bioorg. Med. Chem. Lett 2007, 17, 5590. [DOI] [PubMed] [Google Scholar]
- (41).Alex A; Millan DS; Perez M; Wakenhut F; Whitlock GA MedChemComm 2011, 2, 669. [Google Scholar]
- (42).Kuhn B; Mohr P; Stahl M J. Med. Chem 2010, 53, 2601. [DOI] [PubMed] [Google Scholar]
- (43).Bockus AT; Lexa KW; Pye CR; Kalgutkar AS; Gardner JW; Hund KC; Hewitt WM; Schwochert JA; Glassey E; Price DA J. Med. Chem 2015, 58, 4581. [DOI] [PubMed] [Google Scholar]
- (44).Koes D; Khoury K; Huang Y; Wang W; Bista M; Popowicz GM; Wolf S; Holak TA; Dömling A; Camacho CJ PLoS One 2012, 7, e32839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45) (a).Dömling A; Wang W; Wang K Chem. Rev 2012, 112, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dömling A Chem. Rev 2006, 106, 17. [DOI] [PubMed] [Google Scholar]
- (46) (a).Koes DR; Camacho CJ Nucleic Acids Res 2012, 40, W387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koes DR; Camacho CJ Bioinformatics 2011, 28, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47) (a).Popowicz GM; Czarna A; Wolf S; Wang K; Wang W; Dömling A; Holak TA Cell Cycle 2010, 9, 1104. [DOI] [PubMed] [Google Scholar]; (b) Khoury K; Popowicz GM; Holak TA; Dömling A MedChemComm 2011, 2, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang YJ; Wolf S; Koes D; Popowicz GM; Camacho CJ; Holak TA; Dömling A ChemMedChem 2012, 7, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shaabani S; Neochoritis CG; Twarda-Clapa A; Musielak B; Holak TA; Dömling A MedChemComm 2017, 8, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Srivastava S; Beck B; Wang W; Czarna A; Holak TA; Dömling AJ Comb. Chem 2009, 11, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang K; Doemling A Cancer Res 2009, 69.; (g) Czarna A; Beck B; Srivastava S; Popowicz GM; Wolf S; Huang Y; Bista M; Holak TA; Dömling A Angew. Chem. Int. Ed 2010, 49, 5352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Huang Y; Wolf S; Bista M; Meireles L; Camacho C; Holak TA; Dömling A Chem. Biol. Drug Des 2010, 76, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Popowicz GM; Dömling A; Holak TA Angew. Chem. Int. Ed 2011, 50, 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Christner SM; Clausen DM; Beumer JH; Parise RA; Huang Y; Dömling AS; Eiseman JL Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research,; 2012, 72 (8 Suppl.), Abstract Nr 4726; Mar 31–Apr 4. [Google Scholar]; (k) Wang W; Cao H; Wolf S; Camacho-Horvitz MS; Holak TA; Dömling A Bioorg. Med. Chem 2013, 21, 3982. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Huang Y; Wolf S; Beck B; Köhler L-M; Khoury K; Popowicz GM; Goda SK; Subklewe M; Twarda A; Holak TA ACS Chem. Biol 2014, 9, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Neochoritis C; Estrada-Ortiz N; Khoury K; Dömling A Annu. Rep. Med. Chem 2014, 49, 167. [Google Scholar]; (n) Neochoritis CG; Wang K; Estrada-Ortiz N; Herdtweck E; Kubica K; Twarda A; Zak KM; Holak TA; Dömling A Bioorg. Med. Chem. Lett 2015, 25, 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Surmiak E; Twarda-Clapa A; Zak KM; Musielak B; Tomala MD; Kubica K; Grudnik P; Madej M; Jablonski M; Potempa J ACS Chem. Biol 2016, 11, 3310. [DOI] [PubMed] [Google Scholar]; (p) Twarda-Clapa A; Krzanik S; Kubica K; Guzik K; Labuzek B; Neochoritis CG; Khoury K; Kowalska K; Czub M; Dubin G; Dömling A; Skalniak L; Holak TA J. Med. Chem 2017, 60, 4234. [DOI] [PubMed] [Google Scholar]; (q) Kroon E; Schulze JO; Süß E; Camacho CJ; Biondi RM; Dömling A Angew. Chem. Int. Ed 2015, 54, 13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48) (a).Koes DR; Dömling A; Camacho CJ Protein Science 2018, 27, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hawkins PCD J. Chem. Inf. Model 2017, 57, 1747. [DOI] [PubMed] [Google Scholar]
- (49) (a).Chen I-J; Foloppe N Bioorg. Med. Chem 2013, 21, 7898. [DOI] [PubMed] [Google Scholar]; (b) Watts KS; Dalal P; Tebben AJ; Cheney DL; Shelley JC J. Chem. Inf. Model 2014, 54, 2680. [DOI] [PubMed] [Google Scholar]; (c) Sindhikara D; Spronk SA; Day T; Borrelli K; Cheney DL; Posy SL J. Chem. Inf.Model 2017, 57, 1881. [DOI] [PubMed] [Google Scholar]; (d) Coutsias EA; Lexa KW; Wester MJ; Pollock SN; Jacobson MP J. Chem. Theory Comput 2016, 12, 4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gerber PR; Gubernator K; Müller K Helv. Chim. Acta 1988, 71, 1429. [Google Scholar]