

Abstract
Background aims.
Chimeric antigen receptor (CAR) T-cell therapy targeting CD19 has demonstrated remarkable success in targeting B-cell malignancies but is often complicated by serious systemic toxicity in the form of cytokine release syndrome (CRS). CRS symptoms are primarily mediated by interleukin 6 (IL-6), and clinical management has focused on inhibition of IL-6 signaling. The cellular source and function of IL-6 in CRS remain unknown.
Methods.
Using co-culture assays and data from patients on our clinical CAR T-cell trials, we investigated the cellular source of IL-6, as well as other CRS-associated cytokines, during CAR T-cell activation.We also explored the effect that IL-6 has on T-cell function.
Results.
We demonstrated that IL-6 is secreted by monocyte-lineage cells in response to CAR T-cell activation in a contact-independent mechanism upon T-cell engagement of target leukemia. We observed that the presence of antigen-presenting cell-derived IL-6 has no impact on CAR T-cell transcriptional profiles or cytotoxicity. Finally, we confirm that CART cells do not secrete IL-6 in vivo during clinical CRS.
Discussion.
These findings suggest that IL-6 blockade will not affect CD19 CAR T-cell-driven anti-leukemic cytotoxicity, permitting enhanced control of CRS while maintaining CAR T-cell efficacy.
Keywords: chimeric antigen receptor, cytokine release syndrome, interleukin 6
Introduction
The primary toxicity associated with highly active cellular therapy using CD19 chimeric antigen receptor (CD19 CAR) T cells is the hyper-inflammatory state known as cytokine release syndrome (CRS) [1].This toxicity is characterized by clinical symptoms ranging from a mild influenza-like syndrome to extreme elevations in core body temperature and life-threatening multi-organ failure. In our original report of CD19 CART cells for acute lymphoblastic leukemia (ALL), we described significant elevations in several serum cytokines, most notably interleukin (IL)-2, IL-6 and interferon-gamma (INF-γ) in patients with CRS [1]. The first patient treated in our phase 1 study experienced life-threatening toxicity in the form of distributive shock requiring multiple vasopressors and respiratory failure requiring prolonged mechanical ventilation. Administration of the anti-IL-6 receptor agent tocilizumab several days into this critical illness resulted in prompt hemodynamic stabilization, pointing to the central role of IL-6 in causing these symptoms. Other groups have observed a similar IL-6-driven syndrome with CD19 CAR T cells, and treatment with anti–IL-6 therapy has become standard practice for the management of severe CRS [2,3]. Use of this agent has been cautious, however, as we have a limited understanding of the cellular source of IL-6 during CRS, whether IL-6 is secreted by CAR T cells themselves as a means of homeostatic support in a rapidly dividing cell population, or if IL-6 promotes CART-cell efficacy. Several cellular sources of IL-6 have been identified, including macrophages, dendritic cells and B and T lymphocytes [4–7].T cells have been identified as the primary source of pathologic IL-6 in models of multiple sclerosis [7], and T-cell-derived IL-6 has been implicated in mediating a positive feedback loop driving TH17 cell differentiation [8]. Innate cells are often the primary sources of IL-6; however, these studies permit that T cells themselves may be the source of IL-6 observed in CRS. Although classical T-cell activation in response to infection and auto-antigens has been studied for decades, the effect of CAR-mediated T-cell activation is poorly understood, and CAR-driven activation may generate distinct cytokine support needs and have a distinct effect on T-cell-mediated IL-6 production. In the absence of a deeper understanding of the role of IL-6 in CAR T-cell function and in CRS, balancing the management of severe toxicity with optimization of anti-tumor activity has been driven by empirical trial and error.
A recent report from our group has provided initial insight into the physiology of CRS. In the first prospective examination of a large panel of serum cytokines in pediatric and adult patients receiving CD19 CAR T-cell therapy for ALL, we observed that elevations in IFN-γ, IL-6, IL-8, soluble IL-2 receptor-α (sIL-2Rα), soluble IL-6 receptor (sIL-6R), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1α (MIP-1α), macrophage inflammatory protein 1β (MIP-1β) and granulocyte-macrophage colony stimulating factor (GM-CSF) were associated with the development of severe CRS [9]. Early elevations in IFN-γ, serum glycoprotein 130 (sgpl30), a component of sIL-6R, and sIL-1RA were predictive markers of development of severe CRS. Both T-cell expansion and baseline disease burden have been speculated to be primary determinants of severity of CRS; however, expansion itself was not associated with development of CRS. Similarly, disease burden alone did not provide any further predictive modeling over serum cytokine levels. Examination of all patients who received tocilizumab therapy upon manifestation of severe CRS demonstrated a consistent and rapid resolution of toxicity after administration, with discontinuation of vasopressors within 24–36 h, confirming the clinical significance of IL-6 in mediating toxicity.
The immunologic cascade that results from CAR-mediated T-cell activation, as opposed to native T-cell receptor (TCR)-mediated activation, and the resulting cellular events that lead to the biochemical derangements of CRS have clear clinical relevance; two adult patients treated at the University of Pennsylvania have died while experiencing CRS, and many more have experienced significant morbidity. Other institutions have experienced similar morbidity and mortality as a result of CRS [3, 10].The study discussed above [9] provided a detailed cytokine profile of patients who experienced CRS, and we observed that cytokine dynamics in CRS are almost identical to those in hemotophagocytic lymphohistiocytosis (HLH).This inflammatory syndrome is driven by macrophage activation, suggesting that CAR T cells are unlikely to be lone actors in CRS, and other immune cells may be key players. Our clinical understanding thus far has hinged on elevations in IL-6, and although IL-6 seems to drive clinical symptoms, it is clear that it is one of several cytokines that becomes up-regulated during CART-cell activation in vivo, suggesting that a network of cytokine signaling contributes to CRS.
To further elucidate the cellular drivers of CRS, we isolated antigen-presenting cells (APCs) derived from the monocyte lineage and performed in vivo and in vitro co-culture experiments to identify the cellular events leading to the cytokine elevations associated with this clinical syndrome. Here, we observe that although T cells alone are sufficient for the production of some CRS-associated cytokines, both activated T cells and APCs are necessary for production of IL-6 and this dependence is not reliant on cell-to-cell contact.We further identify that monocyte-derived cells are responsible for IL-6 secretion that results in response to CAR-mediated T-cell activation. Finally, we demonstrate that CAR-activated T-cell function is not affected by the presence of IL-6-secreting APCs and confirm that CAR T cells from patients experiencing CRS do not produce IL-6. These details of the CRS cascade provide not only a deeper immunologic understanding of this syndrome but also further opportunity for safe management of CRS without compromising CART-cell efficacy.
Methods
Xenograft studies and patient samples
Six to 10-week old NOD-SCID-γc–/– (NSG) mice were obtained from the Jackson Laboratory or bred in-house under an approved Institutional Animal Care and Use Committee protocol and maintained in pathogen-free conditions. Patient leukemia and T cells were obtained under a Children’s Hospital of Philadelphia Institutional Review Board-approved protocol (CHP959 and CHP784, respectively). T-cell engineering for this study has been described previously [1]. Animals were given 106 primary human ALL cells via tail vein, followed by 5 × 106 CART cells (11% CAR positive) 7 days later. Peripheral blood was collected via retro-orbital sinus and submitted to the University of Pennsylvania Human Immunology Core for cytokine quantification.
Isolation of normal donor monocytes and T cells and T-cell engineering
Primary human T cells and monocytes from normal donors were procured through the University of Pennsylvania Human Immunology Core. For all co-culture experiments, T cells and monocytes were obtained from the same donor. T cells were combined at a ratio of 1:1 CD4:CD8 cells at a concentration of 106 cells/mLT-cell culture media with stimulatory microbeads coated with antibodies directed against CD3 and CD28 (Life Technologies, catalog #111.32D) at a concentration of three beads/cell, as had been reported previously [11].Twenty-four hours after initial stimulation, T cells were exposed to lentiviral vector encoding the CD19 CAR construct at a multiplicity of infection (MOI) of 5–10 particles/cell. Stimulatory beads were removed on day 7, and cells were counted and volumes measured serially until growth and size trends indicated cells were rested down, at which time they were frozen. Cells were then thawed 12–18 h before in vivo injection or in vitro co-culture. Untargeted T cells were cultured in the same manner but were not treated with lentiviral vector.
Lentiviral vector preparation
High-titer, replication-defective lentiviral vectors were produced using 293T human embryonic kidney cell. HEK293T cells were seeded at 107 cells perT150 tissue culture flask 24 h before transfection, as previously described [12]. On the day of transfection, cells were treated with 7 μg of pMDG.1,18 μg of pRSV.rev, 18 μg of pMDLg/p.RRE packaging plasmids and 15 μg of transfer plasmid in the presence of either Express-In Transfection Reagent (Open Biosystems) or Lipofectamine 2000 transfection reagent (Life Technologies, catalog #11668019). Transfer plasmids containing CAR constructs were modified so that expression of the CAR was under control of the EF-1α promoter.Viral supernatants were harvested 24 h and 48 h after transfection and concentrated by ultracentrifugation overnight at 10 500g. Twenty-four hours after initial stimulation, T cells were exposed to lentiviral vector at a concentration of 5–10 infectious particles per T cell and then cultured as described earlier.
Production of monocyte lineage cells
Monocytes were collected as described earlier and differentiated in vitro [13]. Monocytes (2 × 106) were plated in 1 mL of RPMI 1640 supplemented with supplemented with 0.1 mmol/L MEM Non-Essential Amino Acids, 2 mmol/L L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin (Life Technologies) and 10% fetal calf serum and cultured for 4 days. Cells were then harvested using 2 mmol/L ethylenediaminetetraacetic acid (EDTA) and stained with CD14, CD45, CD68 and CD163 to confirm macrophage differentiation. To produce dendritic cell lineages, monocytes were plated at 6 × 106 in 1 mL of cR10 and treated with 0.2 μg/mL human IL-4 (R&D Systems, #204-IL-050) and 0.2 μg/mL GM-CSF (R&D Systems, #215-GM-050). On day 4 cells were either harvested using 2 mmol/L EDTA (immature dendritic cells), or cultures were treated with 100 ng/mLLPS (Sigma-Aldrich, #L2630) and 0.05 μg/ mL IFN-γ (R&D Systems, #285-IF-100; mature dendritic cells). Twenty-four hours later, cells were harvested with 2 mmol/L EDTA and stained with CD45, CD80 and CD86 to confirm immature DC and mature DC differentiation.
Co-culture assay
T cells were engineered as described earlier, and monocyte lineages were differentiated as described. Nalm-6 ALL cell lines were used as targets. Cells were combined at a ratio of 50 T cells, 10 targets and 1 APC in 150 μL of cR10. Twenty microliters of supernatant was aspirated after 18 h and replaced with 20 μL cR10. Twenty microliters was then aspirated again at 48 hours. For trans-well co-culture assays, T cells and targets were cultured as described for our standard co-culture assay. Pooled monocytes were seeded in ThinCert cell culture inserts (Greiner Bio-One, product #662610) placed in each well of the 24-well plate. Co-cultures were incubated at 37°C, and cells from both the inserts and the wells were collected at 18 and 48 h for RNA isolation as described later in the article.
Measurement of cytokine levels
Cytokine concentration determination from animal serum and from culture supernatants was performed by the University of Pennsylvania Human Immunology Core using the Millipore Luminex 200 system and Milliplex Human Cytokine/Chemokine 21 Plex Assay (EMD Millipore; products #40–012 and #HCY4MG-64K-PX21). Measurements were performed using standard product protocols.
RNA extraction
Total RNA was prepared from cell pellets lysed in Qiagen Buffer RLT (Qiagen, product #79216). Lysates were processed and RNA extracted using RNA Clean & Concentrator-5 columns (Zymo Research, product #D4003) according to the manufacturer’s protocol. Total RNA quality and yield were assessed using either an Agilent 2100 Bioanalyzer with Eukaryotic Total RNA Pico chips (Agilent Technologies, product #5067) or a Biophotometer (Eppendorf, product D30) equipped with a Hellma TrayCell microvolume ultramicro cell (Hellma Analytics).
nanoString nCounter assay
Gene expression was measured on the nanoString nCounter SPRINT Profiler (NanoString Technologies) using the nCounter Human Immunology v2 gene expression Code Set (NanoString Technologies). Samples were prepared and processed according to the manufacturer’s recommendations. Briefly, 50 ng total RNA was hybridized in solution to the nCounter Human Immunology v2 gene expression Code Set for 18 h at 65°C. Hybridized samples were then loaded into the nCounter SPRINT cartridge (NanoString Technologies), which was then sealed and placed in the instrument for processing and analysis.
CD107a degranulation assay
Co-culture experiments were setup as described earlier. After 18 h, this culture was combined with an antibody cocktail consisting of anti-CD107a-e660 (eBiosciences, catalog #50–1079), and stimulatory antibodies directed against CD28 (clone 9.3) and CD49d (BD Biosciences, catalog #555051) for 1 h, as previously described [14]. Intracellular protein transport was halted by addition of GolgiStop (BD Biosciences, catalog #554724) and cells were incubated for an additional 3 h. Cells were then harvested and stained for CD8 and CD107a (BD Biosciences) and analyzed on an Accuri C6 Flow Cytometer.
Statistical analysis
All statistical analysis was performed using Prism 6 (GraphPad Software) using analysis of variance testing for group comparisons. Fisher’s exact test was used for comparison of percentages. All comparisons that are reported as significant reached P < 0.05, or as calculated after Bonferroni corrections for multiple comparisons. Unsupervised clustering and Nanostring analysis was performed with nSolver version 2.5 (Nanostring Technologies).
Results
Combining CD19 CAR T cells and targets fails to mimic clinically observed CRS in xenograft mice
To evaluate the role of CAR-activated T cells in CRS, we created a patient-derived xenograft model of an aggressive and multiply refractory pediatric ALL. The malignant cells used to establish this xenograft were derived from the first patient with ALL treated at our institution (patient 1 or CHP959–100) [1]. As reported, this patient experienced grade 4 toxicity, including need for prolonged vasopressor support and mechanical ventilation. Clinical CRS was accompanied by a significant elevation in serum IL-6 (approximately a 1000-fold increase on day 6 after CD19 CAR T-cell infusion compared with pre-infusion baseline) and rapid resolution of symptoms after administration of anti-IL-6R antibody therapy. To evaluate the role of this patient’s CAR T cells in producing CRS-associated cytokines in vivo, NSG mice were engrafted with 106 primary ALL cells from patient CHP959–100, followed by injection of 5 × 106 CD19 CAR T cells from the same patient 7 days later. A subgroup of animals was also given tocilizumab (100 μg via intraperitoneal injection) every other day after CAR T-cell infusion. Measurement of serum cytokine levels on day 3 after CAR T-cell infusion demonstrated measurable levels of IFN-γ, IL-2 and GM-CSF but no detectable IL-6, independent of tocilizumab administration (Figure 1). A similar pattern was observed when animals were engrafted an ALL cell line (Nalm-6) and treated with CD19 CART cells from a normal donor (Supplementary Figure S1), supporting that this lack of IL-6 production was not a patient-specific phenomenon, and suggesting that the cellular component responsible for the significant IL-6 production observed clinically was absent in these immunodeficient xenografts.
Figure 1.
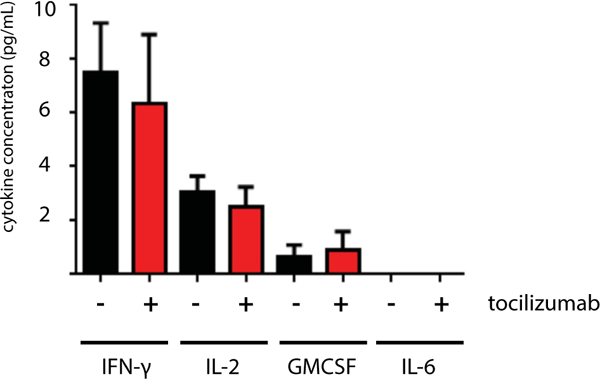
Serum cytokine concentrations in xenograft mice bearing primary pediatric ALL treated with CD19 CART cells. NSG mice were given 106 primary ALL cells and 5 × 106 autologous CD19 CART cells 7 days later. Serum was collected 3 days after T-cell delivery, and a subgroup of animals was given tocilizumab on days 1 and 3 after T cells. Human cytokine concentrations are measured in picograms per milliliter.
Antigen-stimulated CAR T-cell activation in the presence of APCs results in elevated levels of CRS-associated cytokines
On the basis of the similarities in serum cytokine profiles between CRS and HLH, we next evaluated the role that APCs of the monocyte lineage may play in cytokine production.We combined either CD19 CAR T cells or untargeted T cells with a CD19+ ALL cell line (Nalm-6) in the presence of APCs in vitro at cell ratios of 10 T cells: 50 targets: 1 APC, and collected culture supernatants after 18 h of co-culture. As demonstrated in our prospective clinical study [9], early elevations are observed in serum IFN-γ in patients with CRS. We found that IFN-γ was detected whenever T cells were activated by targets, with no significant difference dependent on the presence of APCs (Figure 2A). Moderate levels of GM-CSF were secreted when T cells were combined with targets; however, significant elevations were noted when APCs were included in co-culture (Figure 2B), suggesting either enhancement of T-cell-based secretion by APCs, or two cellular sources of GM-CSF. IL-2, classically considered a CD4 T-cell factor, demonstrated a similar pattern, with some secretion when T cells and targets were combined and significant enhancement with the inclusion of APCs in co-culture (Figure 2C).This pattern differed when examining IL-8 and IL-6. Similar levels of IL-8 were observed in cultures of APCs alone, APCs combined with targets and APCs combined with targets and untargeted T cells (Figure 2D). Levels significantly increased when APCs were combined with targets and targeted T cells (Figure 2D).Together these data suggest that APCs secrete low levels of IL-8 independent of T cells or targets, but the combination of targets, targeted T cells and APCs resulted in high levels of IL-8. IL-6 levels followed a similar pattern (Figure 2E); low levels were observed when APCs were alone, combined with targets and combined with untargeted T cells and targets. Levels significantly rose when APCs were combined with activated T cells and targets.
Figure 2.
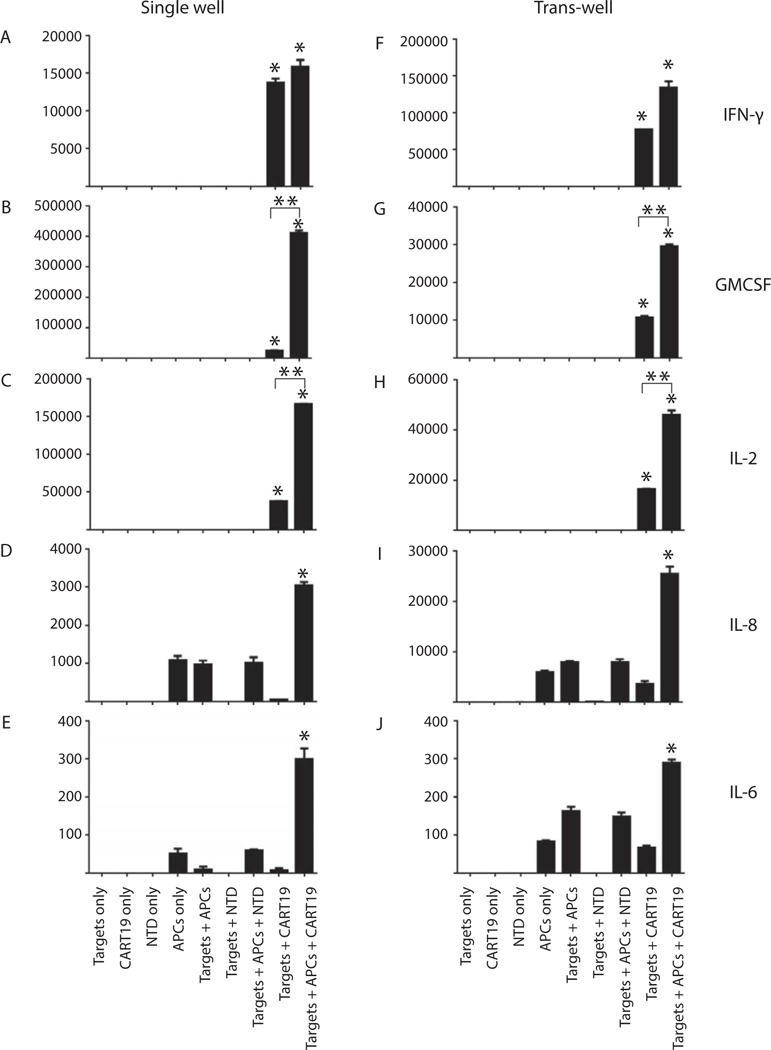
Cytokine expression after cellular co-culture. T cells, targets and APCs were combined at a ratio of 10:50:1, respectively. Supernatants were collected after 18 h of co-culture. Cytokine levels are measured in picograms per milliliter. *Statistical difference across the group; P < 0.05. **Statistical difference between the two groups; P < 0.05.
To clarify whether the APC–T-cell interaction was mediated by cell-to-cell contact or by soluble factors, we set up the same co-cultures in parallel with co-cultures which separated T cells and targets from APCs using trans-well inserts. The same numbers of T cells and targets were placed in the plate wells, and the same number of APCs were placed in trans-well inserts. As shown in Figure 2F–J, relative quantities of cytokine secretion were unchanged for IFN-γ, GM-CSF, IL-2 and IL-8. Interestingly, the trans-well separation resulted in a relative increase in IL-6 when APCs were combined with targets in the presence or absence of untargeted T cells. However, the highest IL-6 levels were still observed when CD19 CART cells were combined with targets and APCs, demonstrating a statistically significant elevation compared with all other co-cultures (P = 0.001).These studies demonstrate that the cytokine network induced upon CAR-mediated T-cell activation in the presence of APCs is not dependent on cell-to-cell contact between T cells and APCs, and the combination of targets, CAR T cells and APCs is necessary for high levels of IL-6 production.
Monocyte-lineage cells differentially secrete CRS-associated cytokines
We next sought to identify which APC lineages were responsible for IL-6 production. We isolated monocytes and cultured them in vitro to produce differentiated progeny of the monocyte lineage, including immature dendritic cells, mature dendritic cells and macrophages [13] (we did not include osteoclasts because these cells do not play a role in antigen presentation). These undifferentiated monocytes and differentiated lineages were combined with CD19 CAR T cells and targets in co-culture as described. Culture supernatants were collected at 18 and 48 h. Consistent with findings from Figure 2, IFN-γ levels were elevated in the presence of each APC lineage, as well as in the absence of APCs, suggesting that IFN-γ is produced by activated CAR T cells independent of APCs (Figure 3A). Levels increased marginally after 48 h of culture, suggesting that the majority of IFN-γ secretion occurs early. GM-CSF levels demonstrated a significant increase between the 18 and 48-h time points, with cultures containing immature dendritic cells producing the most GM-CSF, followed by mature DCs and macrophages (Figure 3B). Activated T cells alone produced very little GM-CSF, as did cultures of activated T cells with monocytes, suggesting that GM-CSF secretion is driven by differentiated monocyte-lineage cell populations. Interestingly, IL-2 levels demonstrated a large peak at the 48-h time point when activated T cells were combined with mature dendritic cells and a smaller but significant peak when T cells were combined with macrophages (Figure 3C). Low levels of IL-2 were detected after 48 h when T cells were combined with targets alone or in the presence of immature dendritic cells, but the presence of monocytes did not appear to result in significant IL-2 production. Although some low level IL-8 was detected in nearly all cultures containing APCs, a 3log increase was detected when activated T cells were combined with macrophages compared with T cells and targets alone at the 18-h time point (Figure 3D).The presence of immature dendritic cells also yielded high IL-8 levels at 18 h, with more modest elevations in the monocyte cultures. The presence of mature dendritic cells did not significantly alter IL-8 concentration. Finally, IL-6 levels were observed to be highest when activated T cells were combined with immature dendritic cells after 48 h of culture, with a greater than 100 × increase in cytokine concentration compared with CART cells and targets alone (Figure 3E). Modest elevations were detected in cultures containing activated T cells and mature DCs and macrophages. Of note, nearly no IL-6 was detected in the absence of APCs, but a low level was produced in the absence of targets (T cells and immature DCs alone); these levels were several logs below those observed when CAR T cells were combined with targets and APCs.
Figure 3.
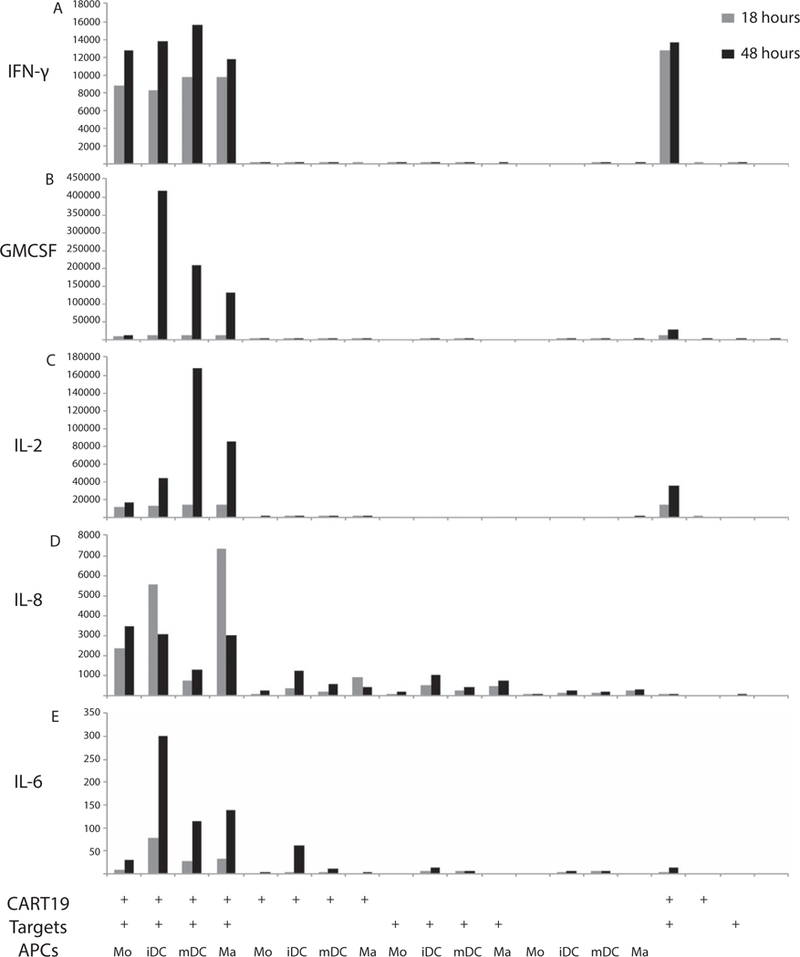
Cytokine secretion from co-culture experiments combining monocyte-lineage cells with T cells and targets. Monocyte-lineage cells were differentiated in vitro, and T cells, targets and APCs were combined at a ratio of 10:50:1, respectively. Supernatants were collected at 18 and 48 h and analyzed for cytokine concentrations, measured in picograms per milliliter.
APC-derived IL-6 does not affect CAR T-cell transcription or cytotoxicity
The experiments shown here demonstrate which APC and activated T-cell combinations resulted in production of CRS-associated cytokines but did not definitively demonstrate which cell type (APC or CAR T cell) was responsible for cytokine secretion in these co-cultures.To clarify which cells were producing which cytokines, we conducted trans-well co-culture experiments and performed transcriptional analysis on discrete cell populations. As depicted by regression analysis of 697 genes related to immune activation, there was no detectible difference in transcriptional profile of activated CAR T cells in the presence or absence of APCs (Figure 4A, R2 = 0.951, P> 0.5).We next examined the profiles of APCs alone compared with APCs combined with untargeted T cells and Nalm-6 leukemia and found no change in APC transcriptional profile (Figure 4B, R2 = 0.934, P > 0.5). Finally, we compared APC transcription profiles when combined with untargeted T cells and Nalm-6 or CD19 CART cells and Nalm-6 and found significant variability in APC transcriptional profiles (Figure 4C, R2 = 0.830, P = 0.0017).These data demonstrate that APCs have no effect on CAR T-cell transcriptional programs but that CAR-activated T cells significantly alter APC transcriptional phenotypes. From the same trans-well studies, we mapped RNA constructs to their cell of origin. We found that IFN-γ is made exclusively by T cells, IL-2 and GM-CSF are predominantly made by T cells, IL-8 is largely made by APCs and IL-6 is exclusively made by APCs (Figure 5), delineating the cellular origins of these CRS-associated cytokines.
Figure 4.
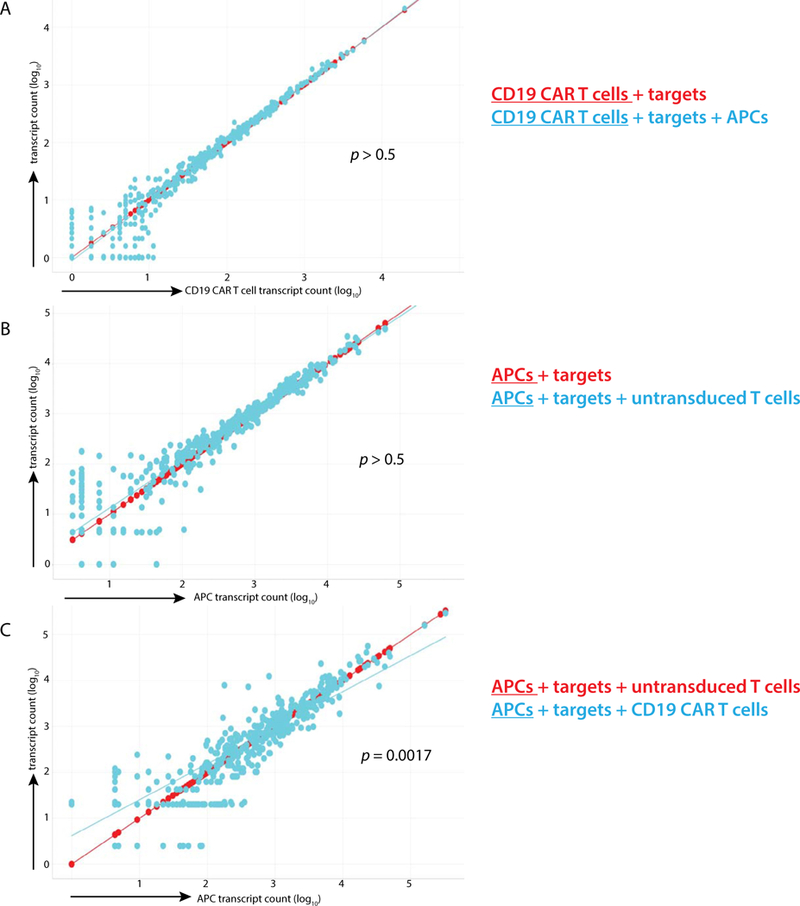
Transcriptional analysis of isolated cell populations. T cells and targets were isolated from APCs using trans-well inserts and co-cultured for 18 h. Six hundred and ninety-seven RNA transcripts were quantified from each cell population, and log counts of each are displayed. Transcriptional profiles of (A) CD19 CART cells when combined with targets and when combined with targets and pooled monocytes, (B) APCs when combined with targets and with targets and untargeted T cells and (C) APCs when combined with targets and untargeted T cells, and when combined with targets and targeted T cells are presented as linear regression plots.
Figure 5.
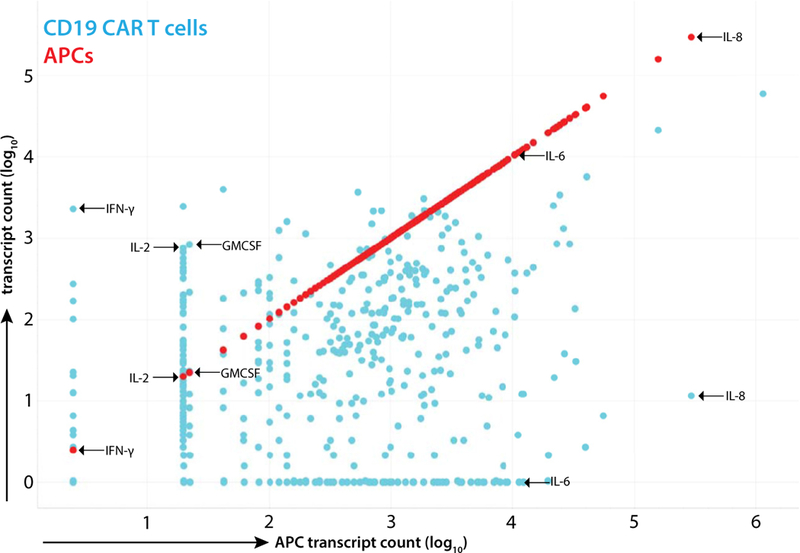
Transcript profile of activated CD19 CART cells and monocyte-lineage APCs. Cells were harvested from trans-well co-culture of CD19 CART cells, Nalm-6 leukemia and pooled monocytes after 18 h. Transcript counts from T cells are displayed in blue and counts from APCs in red. Full quantification of each transcript is presented in Supplementary Table S1.
To evaluate whether CD19 CAR T-cell activity is altered by IL-6 in a transcription-independent manner, we performed co-culture experiments and evaluated T-cell cytotoxic activity. CD19+ ALL cells, APCs and T cells were combined as described, and T cells were harvested after of co-culture. To control for the effects that nonspecific CAR signaling may have on cytotoxicity assessment, we engineered T cells to express either no CAR (Figure 6A), a CAR directed at the irrelevant antigen GD2 (Figure 6B) or the CD19 CAR (Figure 6C). Cytotoxicity was measured by upregulation of CD107a, a measure of T-cell degranulation. No degranulation was detected when untargeted T cells or GD2 CART cells are combined with ALL cells, and similar levels of degranulation were detected when CD19 CART cells encounter CD19+ leukemia in the presence or absence of APCs.
Figure 6.
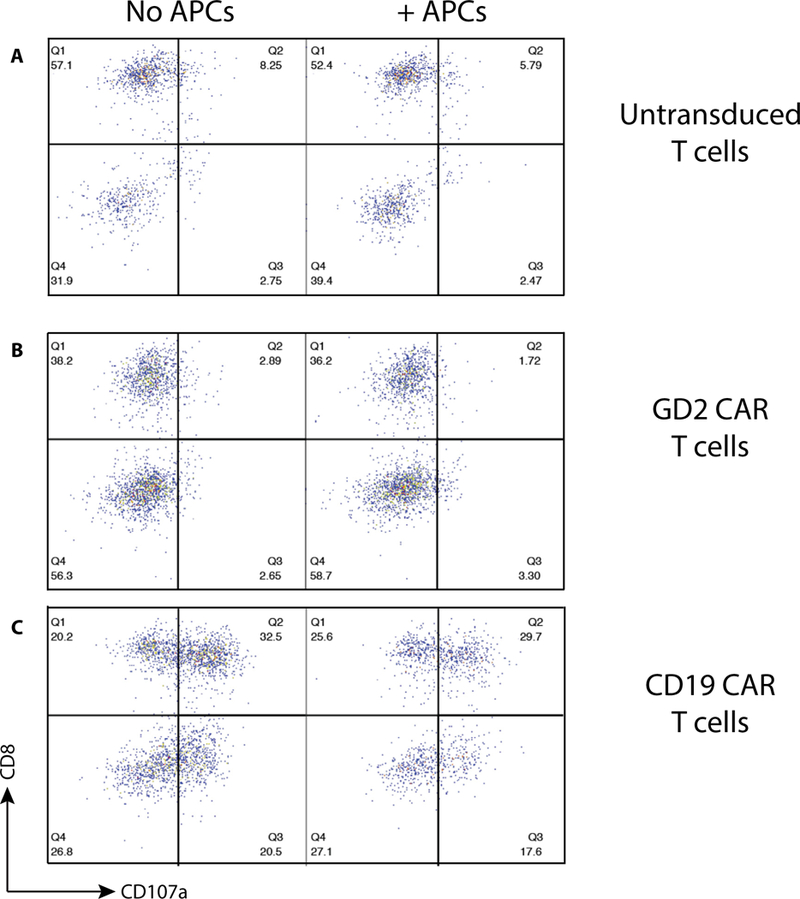
T-cell degranulation in the presence of APCs. T cells expressing (A) no CAR molecule, (B) GD2-targeted CAR or (C) CD19-targeted CAR were combined with CD19+ target ALL cell line Nalm-6. Degranulation was measured by quantification of CD107a surface expression.
CD19 CAR T cells from patients with CRS do not produce IL-6
Cytokine analysis of patients who have received CD19 CAR T-cell therapy for leukemia demonstrated that while many cytokines are elevated during CRS, only a few contribute to a predictive model of which patients will go on to develop grade 4 CRS after T-cell infusion [9]. We collected peripheral blood and isolated mononuclear cells from patients who had received CD19 CAR T-cell therapy for treatment of ALL as part of our phase I clinical trial on their first day of fever after T-cell infusion. Seven of 10 patient samples had detectable peripheral CART cells, and of these patients, 3 experienced grade 2 CRS, 1 grade 3, and 3 grade 4 (severe CRS).The remaining three samples had no detectable peripheral T cells but only circulating ALL cells; of these patients, one was classified as grade 3 and two grade 4 CRS. We performed unsupervised clustering analysis on these samples and found distinct transcriptional profiles for grade 2–3 compared with grade 4 CRS (Figure 7).T cells from patients that developed severe CRS had elevations in granzyme B, perforin, IFN-γ, Zap70, EOMES and Lag-3 transcripts, and suppressed levels of tumor necrosis factor-α, IL-1β and CCR7 compared with those with grade 2–3 CRS. B-cell transcripts, such as CD79, Pax5 and CD19, were only elevated in the three samples with circulating leukemia. (For complete transcript level quantification see Supplementary Table S1.)
Figure 7.
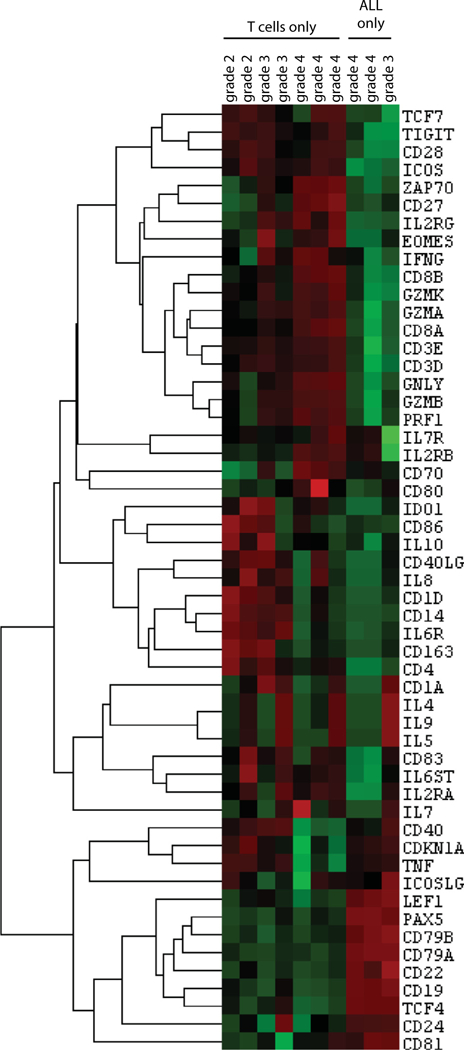
Transcriptional profile of peripheral blood mononuclear cells collected from patients with ALL treated with CD19 CART cells. Peripheral blood was collected on first day of fever after engineered T-cell infusion. Seven patients had T cells detectable in peripheral blood with no detectable ALL, and three patients had only ALL cells and no detectable T cells.
Having demonstrated that CAR T cells do not produce IL-6 in vitro, we sought to confirm this finding in a relevant clinical context. Transcriptional analysis of patients experiencing fever who went on to develop CRS revealed that none of the samples containing CART cells demonstrated detectable levels of IL-6 transcript, with all IL-6 transcript levels measuring below the lower limit of detection (<1 copy of RNA transcript per cell; Figure 7). Similarly, no samples containing only leukemia cells had detectable levels of IL-6, confirming that in these patients, neither the T cells nor leukemia were responsible for IL-6 production. Examination of the T cells from this collection using light microscopy demonstrated a highly activated phenotype, with large, irregular nuclei, open chromatin and irregular plasma membranes (Supplementary Figure S3), confirming that activated CART cells do not produce the IL-6 that drives CRS.
Discussion
Although engineered T-cell therapies have been tested in clinical trials for 2 decades, only the past 5 years have witnessed significant efficacy of this therapy [15–18]. In a classic example of an unexpected toxicity of a first-in-human therapy, highly active CD19 CAR T-cell therapy has been complicated by the development of CRS in a significant percentage of patients treated across several institutions, with 25–30% requiring intensive care unit support. The clinical description of this syndrome has been robust [19], however we have lacked biological insight into which patients will develop CRS, the cellular source of IL-6, and the role IL-6 has in CAR T-cell activity needed to fully inform clinical management. Here we demonstrate that monocyte-lineage APCs produce IL-6 in response to CAR-mediated T-cell recognition of leukemia and that IL-6 itself has no impact on T-cell transcription or cytotoxicity. This represents a paradigm shift in our understanding of cytokine release syndrome. At our institution, the vast majority of patients with ALL have developed CRS, with 27% experiencing severe CRS requiring tocilizumab [20]. Anti-IL-6R therapy has proven effective in managing this toxicity and in the majority of cases has led to a rapid clinical improvement. The patient whose cells were used in this current study experienced an abrupt improvement of an ongoing respiratory and hemodynamic insufficiency, and this has been true for several other patients at our center. Our previous report defined a biochemical method to predict development of severe CRS, and this report suggests that inhibition of IL-6 clinically will not affect anti-leukemic activity. Our findings demonstrate that monocyte-lineage-derived IL-6 does not alter CAR T-cell transcriptional signatures and that this transcriptional stability corresponds to stability of cytotoxic activity. Our clinical observations have hinted toward this: relapse rates appear to be similar among patients who receive tocilizumab and those who do not [20]. Additionally, our previous in vivo studies have demonstrated durable disease eradication in xenograft models of ALL (which lack monocyte-lineage APCs) when animals are treated with CD19 CART cells [21]. Combined, these observations suggest that APCs and APC-produced IL-6 are not necessary for CAR T-cell activity in vivo and are likely simply bystanders to target killing. On the basis of the data presented here, we have designed a clinical trial in which tocilizumab administration can occur at the first sign of CRS after CAR T-cell infusion. We anticipate that this will greatly reduce the morbidity associated with CRS while preserving the efficacy of this highly active cell therapy.
CRS is not unique to CD19 CART cells, but appears to be a physiology shared by other potent T-cell activating therapies. The bi-specific antibody blinatumomab, which engages CD3 and CD19 resulting in co-localization and T-cell activation, has also been shown to cause an IL-6–driven CRS that can be similarly mitigated with tocilizumab [22]. T cells expressing transgenic TCRs directed against the cancer-testis antigen NY-ESO-1 have caused significant IL-6 elevations and CRS in patients with myeloma and even solid malignancies [23]. As more cellular and antibody-based T-cell therapies are developed for malignant, and nonmalignant conditions [24], many more patients will be at risk for development of CRS, and early IL-6 blockade has the potential to greatly reduce the burden of this toxicity.
Our findings demonstrated that monocyte-derived immature dendritic cells yielded the greatest IL-6 signal in response to CAR-mediated T-cell activation. Although our in vitro system lacks endothelial cells and other stromal components that may have additional clinical significance in the manifestation and consequences of CRS, the identification of the cellular source of IL-6, along with the confirmation that CD19 CAR T cells from patients experiencing CRS do not produce IL-6, highlights the central physiology of this syndrome. The next step in the continued understanding of CRS is identification of the soluble factor released upon CAR-mediated T-cell activation that drives APC-derived IL-6 secretion.
Interestingly, combination of APCs with co-cultures lacking activated T cells also yielded some IL-6 production. In vitro monocytes culture inherently results in cellular activation [13], and we speculate that this explains the background level of APC-derived cytokine secretion. Combination with activated T cells resulted in a significant increase in IL-6 levels, suggesting again that an unmeasured factor promotes IL-6 secretion when T cells become activated.
Natural T cells do not stimulate this degree of systemic IL-6 secretion in response to infection, and thus we hypothesize that this activity is driven by an element of CAR signaling that differs from TCR signaling. To identify the downstream CAR T-cell factor that triggers APC IL-6 secretion, we have performed preliminary studies inhibiting IFN-γ and GM-CSF in our in vitro co-cultures. These inhibitions did not appear to suppress IL-6 secretion, suggesting that other T-cell-derived elements are responsible APC activation. As presented here, IL-2 may be another candidate, and we have performed similar co-culture experiments and measured levels of IL-1β, IL-4, IL-5, IL-10 and TNF-α (Supplementary Figure S2). Cytokine secretion patterns suggested that IL-5, IL-10 and TNF-a are produced, at least in part, by activated CAR T cells, and one or several of these molecules may be the signal driving IL-6 secretion from monocyte-lineage cells. Interestingly, in no co-culture study did we identify detectable levels of IL-1β, making this an unlikely candidate. It is also possible that the driver of IL-6 secretion is not derived directly from CART cells, but is instead comes from dying leukemic cells.
Interestingly, we observed higher levels of IL-6 when APCs were combined with targets and T cells and APCs were separated in the trans-well setting. One hypothesis is that direct cell-to-cell contact between targets and APCs inhibits secretion of IL-6, via inhibitory signaling from cell-surface molecules. Alternatively, the micropore material of the trans-well insert may provide nonspecific stimulation to the APCs that is not delivered by the inert plastic of the traditional plate well and that this stimulation results in enhanced IL-6 secretion. The overall pattern, however, remained the same, with the only statistically relevant increase in IL-6 secretion resulting from the combination of CD19 CAR T cells, targets and APCs, suggesting that IL-6 secretion by APCs is stimulated not by cell-to-cell contact but instead by a soluble factor present when CART cells engage targets.
Transcriptional mapping using our trans-well system allowed us to identify the cellular sources of all cytokines evaluated. Not surprisingly, IFN-γ was produced by T cells only, consistent with our findings from cytokine quantitation presented in Figure 3. Perhaps a bit more surprising was that IL-2, GM-CSF and IL-8 were produced by both cell populations, albeit with clear predominance coming from either APCs or T cells. Low levels of IL-2 were derived from APCs, while the majority came from T cells. Examination of secretion patterns from Figure 3 demonstrated that when CAR T cells were combined with targets, IL-2 levels in supernatants were ~40 000 pg/mL at 48 h, similar to that seen when CAR T cells and targets were combined with monocytes and immature dendritic cells. The presence of mature dendritic cells and macrophages, however, resulted in significantly higher IL-2 levels, nearing 160 000 pg/ mL (a fourfold increase). Although these differences in concentration are not entirely correlated to the differences in transcription, it is likely that the monocyte lineages responsible for IL-2 production are mature dendritic cells and macrophages. Several possibilities may explain the significant differences in cytokine quantity not explained by changes in transcription. Altered receptor expression or recycling on part of the APCs may cause a fluctuation in the soluble IL-2 present at time of collection. Alternatively, some APCs may secrete other soluble factors that enhance stability or lower consumption of IL-2, whereas others lead to IL-2 degradation. GM-CSF demonstrated a nearly identical pattern, with increased secretion when activated T cells were combined with APCs, and transcriptional evidence of two cellular sources. In this case, immature dendritic cells appear to be the source of APC-derived GM-CSF, with mature DCs and macrophages also contributing, albeit to a lesser degree. IL-8, however, presents a different story. Detection at the protein level only occurred when activated T cells were combined with APCs, with very low levels detected when T cells were alone or with targets, while transcriptional analysis again demonstrated two cellular sources. APC IL-8 transcript levels were ~4log greater than T-cell transcript levels, which may explain these dynamics. IL-2 and GM-CSF transcripts both demonstrated a ~ 2log discrepancy. Finally, IL-8 was the only cytokine that demonstrated significantly greater levels at the 18-hour time point, with all others peaking at 48 h, suggesting that this molecule may be an early component of the CRS cascade.
Improved toxicity management will accelerate the broad application of this highly effective therapy, making it available to more patients at more centers. Improved understanding of the underlying biology of this toxicity is a seminal step in our progress toward the broad use of cellular therapies for cancer.
Supplementary Material
Acknowledgments
We thank A. Seif and H. Bassiri for review of this manuscript, and J. Perazelli, J. Storm, L. Winestone and B. Moghimi for their technical assistance.
This work was supported in part by grants from Stand Up To Cancer–St. Baldrick’s Pediatric Dream Team Translational Research grant (SU2C-AACR-DT1113) and Pennsylvania Department of Health (to SAG), as well as the St. Baldrick’s Foundation and Gabrielle’s Angel Foundation for Cancer Research (to DMB). Additional funds provided by the First Annual Immuno-oncology Forum (to DMB). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Footnotes
Disclosure of interest: BLL has patents and intellectual property related to the work described in this article that have been licensed to Novartis. SAG has a consulting agreements with and receives research funding from Novartis. The other authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.jcyt.2017.04.001.
References
- [1].Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013;368(16):1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CART cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014;6(224):224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine-directed therapies. Annu Rev Med 2015;66:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Leech MD, Barr TA, Turner DG, Brown S, O’Connor RA, Gray D, et al. Cutting edge: IL-6-dependent autoimmune disease: dendritic cells as a sufficient, but transient, source. J Immunol 2013;190(3):881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med 2012;209(5):1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Trinschek B, Luessi F, Haas J, Wildemann B, Zipp F, Wiendl H, et al. Kinetics of IL-6 production defines T effector cell responsiveness to regulatoryT cells in multiple sclerosis. PLoS ONE 2013;8(10):e77634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008;29(4):628–36. [DOI] [PubMed] [Google Scholar]
- [9].Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov 2016;6(6):664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol 2016;34(10):1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood 2003;102(6):2004–13. [DOI] [PubMed] [Google Scholar]
- [12].Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, et al. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res 2014;2(11):1059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Han TH, Jin P, Ren J, Slezak S, Marincola FM, Stroncek DF. Evaluation of 3 clinical dendritic cell maturation protocols containing lipopolysaccharide and interferon-gamma. J Immunother 2009;32(4):399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Singh N, Kulikovskaya I, Barrett DM, Binder-Scholl G, Jakobsen B, Martinez D, et al. T cells targeting NY-ESO-1 demonstrate efficacy against disseminated neuroblastoma. Oncoimmunology 2016;5(1):e1040216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000;96(3):785–93. [PubMed] [Google Scholar]
- [16].Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther 2002;5(6):788–97. [DOI] [PubMed] [Google Scholar]
- [17].Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006;12(20 Pt 1):6106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lamers CH, Langeveld SC, Groot-van Ruijven CM, Debets R, Sleijfer S, Gratama JW. Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunol Immunother 2007;56(12):1875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124(2):188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013;24(8):717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013;121(26):5154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015;21(8):914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016;353(6295):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.