

Abstract
Protein disorder plays a crucial role in signal transduction and is key for many cellular processes including transcription, translation, and cell cycle. Within the intrinsically disordered protein interactome, the α-helix is commonly used for binding, which is induced via a disorder-to-order transition. Because the targeting of protein—protein interactions (PPIs) remains an important challenge in medicinal chemistry, efforts have been made to mimic this secondary structure for rational inhibitor design through the use of stapled peptides. Cap-dependent mRNA translation is regulated by two disordered proteins, 4E-BP1 and eIF4G, that inhibit or stimulate the activity of the m7G cap-binding translation initiation factor, eIF4E, respectively. Both use an α-helical motif for eIF4E binding, warranting the investigation of stapled peptide mimics for manipulating eIF4E PPIs. Herein, we describe our efforts toward this goal, resulting in the synthesis of a cell-active stapled peptide for further development in manipulating aberrant cap-dependent translation in human diseases.
Graphcal Abstract
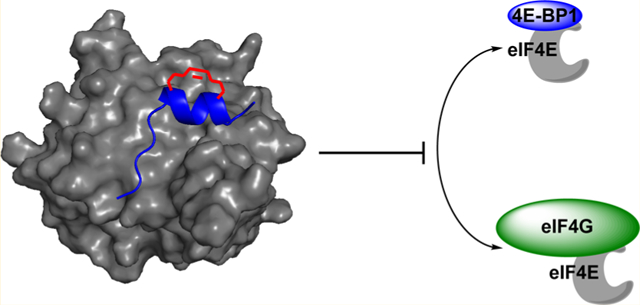
INTRODUCTION
Intrinsically disordered proteins (IDPs) and proteins containing intrinsically disordered regions (IDRs) are a growing family of key regulatory and signaling proteins that either entirely or partially lack tertiary structure;1–3 however, upon binding to a protein partner, many undergo a disorder-to-order transition.4,5 A common secondary structure used by IDPs and IDRs is the α-helix,6,7 which in the field of peptide inhibitor design has been mimicked through hydrocarbon peptide stapling.8,9 While many successful examples of stapled peptides have been reported, including those against IDRs (e.g. p53, NOTCH and FIP),8–18 the approach is not universal. In these cases, the peptides, although more helical, display similar to weaker binding affinities for their target proteins.19–21 Recent kinetic studies have indicated that this may be because of slower association rates (ka) in comparison to the linear peptides,22 as most IDPs and IDRs often exhibit fast ka’s.23 Even with full-length proteins, the impact of helix stabilization on enhancing IDP/IDR binding affinity has been shown to be system-dependent.24–29 Because potent lead molecules are often preorganized into a bioactive conformation, particularly inhibitors of protein—protein interactions (PPIs),30 further analysis is warranted to investigate the structural and kinetic properties of a peptide that may predict successful stapled peptide development.
One crucial cellular process that is regulated by IDPs and IDRs is the initiation of cap-dependent mRNA translation.31 This type of protein translation is aberrantly activated in many human diseases, including cancer32,33 and neurodegeneration,34,35 providing rationale for the development of peptidomimetics to inhibit the PPIs involved in its initiation. Eukaryotic translation initiation factor 4E (eIF4E), the m7GpppX-cap-binding protein, is the rate-limiting translation initiation factor whose activity is regulated by two IDPs, 4E-BP1 and eIF4G (Figure 1A).31 4E-BP1 functions as a gatekeeper of cap-dependent mRNA translation via its ability to bind and inhibit eIF4E.36 Unlike most IDPs, 4E-BP1 has been shown to be almost entirely disordered in the free state;37 however, its eIF4E-binding domain (Asp55—Arg63) folds into a short, yet high affinity α-helical structure upon association.31,38 The PPI is mediated by a conserved binding motif, YXXXXLϕ (X: variable; ϕ: hydrophobic), that is found in all eIF4E binding proteins including eIF4G (Figure 1B).39 Upon phosphorylation,40 the canonical helix of 4E-BP1 becomes destabilized41 allowing eIF4G, a scaffolding translation initiation factor,42 to compete for binding to eIF4E.43 Similar to 4E-BP1, eIF4G uses an IDR within the protein (Asp613–Gln62l) for binding, which undergoes a disorder-to-order transition to form a short α-helix nearly identical to that of 4E-BP1 (Figure 1B).31,44 eIF4G binding to eIF4E stimulates formation of the eIF4F translation initiation factor complex that drives cap-dependent translation.45 Both 4E-BP1 and eIF4G also contain second binding sites outside of the canonical helical domain; however, these sites are of low affinity.38,44,46 As eIF4E overexpression and 4E-BP1 hyper-phosphorylation are common mechanisms by which cap-dependent translation becomes dysregulated in disease,32,33 we became interested in developing stapled peptide mimics of these disordered proteins as inhibitors of eIF4E PPIs. However, because 4E-BP1 and eIF4G form similar bound structures, we were uncertain which would provide an optimal scaffold for stapled peptide development. Herein, we detail our efforts toward this goal and describe our use of linear peptide-binding kinetics in assessing the 4E-BP1 and eIF4G sequences for stapled peptide design.
Figure 1.
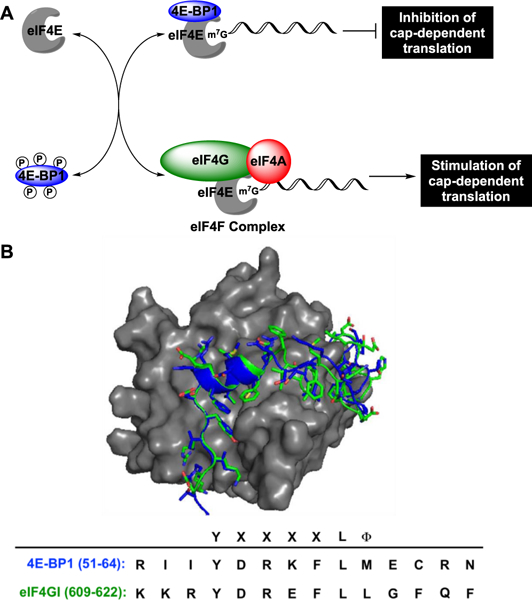
Regulation of cap-dependent translation initiation by 4E-BP1 and eIF4G. (A) Biological scheme. (B) Overlay and amino acid sequences of the eIF4E-binding regions for 4E-BP1 (blue; PDB: 4UED) and eIF4GI (green; PDB: 5T46) bound to eIF4E (gray).
RESULTS AND DISCUSSION
Spectroscopic Analysis of Linear 4E-BP1 and eIF4G Peptides.
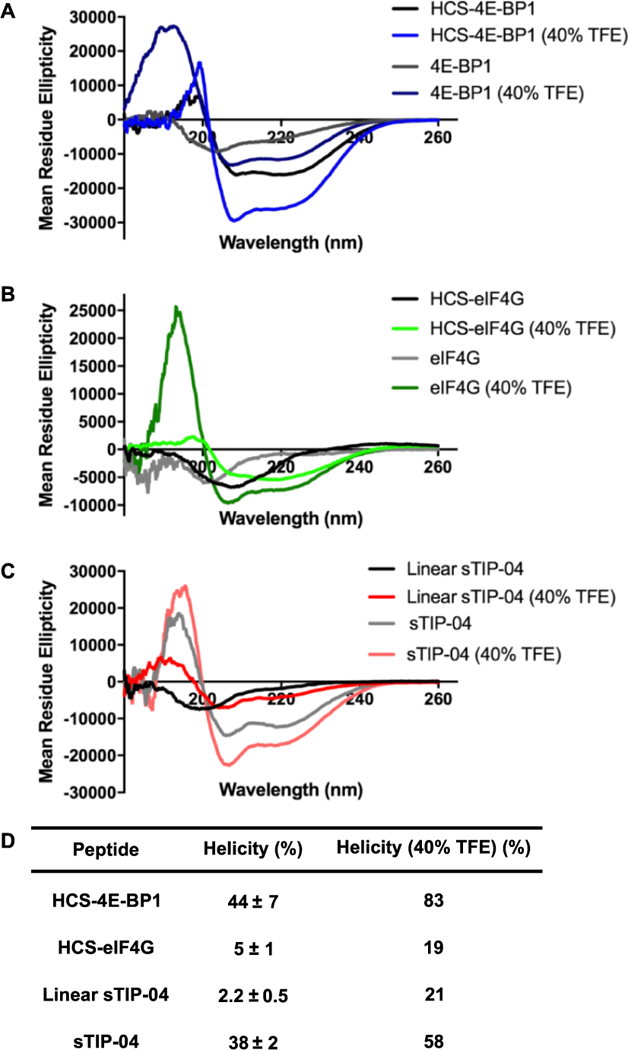
To begin our investigations, we first measured the helical propensities of the linear 4E-BP1 and eIF4G peptides. For each, we used the sequences resolved by X-ray crystallography: GTRIIYDRKFLMECRN for 4E-BP1 and KKRYDREFLLGFQF for eIF4GI.31,38,44 Although the peptides are of differing lengths (16 amino acids for 4E-BP1 and 14 amino acids for eIF4G), we wanted to ensure that both were similarly charged (+2). The peptides were synthesized chemically and contained a N-terminal acetyl cap and C-peptide was found to be 6-fold more helical than the eIF4G peptide (16 and 2.5%, respectively). Both peptides became more helical in the presence of helix-inducing solvent 2,2,2-trifluoroethanol (TFE) (Figure 2); however, its effect on eIF4G was much more pronounced and a 10-fold enhancement was observed (only 2.3-fold for 4E-BP1). These results demonstrate that the 4E-BP1 peptide contains some helical preorganization, even in the absence of eIF4E, while eIF4G does not. This has also been observed via NMR studies, which showed that residues Arg56—Arg63 of 4E-BP1 adopt a ~20% helical structure in the free state.47 Additionally, based on the TFE experiments, the overall helical propensity of 4E-BP1 appears to be greater than that of eIF4G, which is likely attributed to the disorder-promoting N-terminus of eIF4G (KKR).48 This was confirmed experimentally, and a linear 4E-BP1 peptide containing KKR in place of RII was found to be only 8 and 28% helical in the absence and presence of TFE, respectively (Figure 2).
Figure 2.
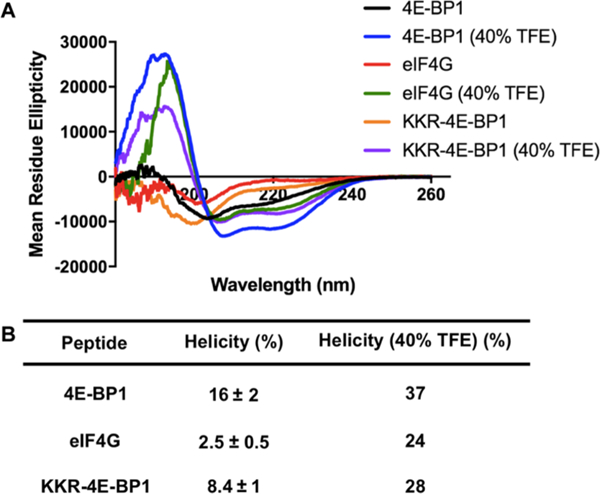
Linear peptide helicities as determined via CD spectroscopy in 5 mM sodium phosphate buffer, pH 7.4, 25 °C. (A) Spectra. (B) Table of average helicity values measured at 25–100 μM ± standard deviation across the concentrations tested. For the TFE experiments, only 100 μM peptide was used.
Kinetic Analysis of Linear 4E-BP1 and eIF4G Peptides.
To determine if any kinetic differences exist between the 4E-BP1 and eIF4G peptides, we next assessed binding to eIF4E via a surface plasmon resonance (SPR) assay. Of note, measurement of binding via fluorescence anisotropy with fluorescein isothiocyanate (FITC)-labeled peptides was also attempted; however, a dose-dependent increase in fluorescence intensity was observed upon eIF4E titration. We attributed this to structural reorganization of the peptide (i.e. helix formation) upon binding, which may relieve some fluorescence quenching of the fluorophore.49,50 For SPR, m7GDP-bound eIF4E was immobilized on a HisCap sensor chip (ForteBio) via its N-terminal His10 tag. 4E-BP1 or eIF4G peptide (12.5–200 nM) was then flowed over the immobilized protein. The association (ka) and dissociation (kd) rates and binding constant (Kd) were obtained by fitting to a 1:1 interaction model based on at least two independent data sets (sensorgrams can be found in Figures S4 and S5; tabulated results are shown in Table S1). Because IDP/IDR kinetics and structure have been shown to be affected by temperature,26,48,51–53 we performed these experiments from 10 to 30 °C. Higher temperature SPR analyses were also attempted; however, eIF4E instability was observed prohibiting testing.
As shown in Figure 3A, while the ka’s of the eIF4G peptide were temperature insensitive (1.7–2.0 × 106 M−1 s−1), the 4E-BP1 peptide showed a 3-fold increase in association rate (0.4–1.3 × 106 M−1 s−1). This was not due to temperature-induced structural differences, as determined via CD studies, although a small increase in 4E-BP1 helicity was observed (15–19% helical) (Figure S6A). To determine if these differences were due to peptide length, we performed a similar analysis on a shorter 4E-BP1 peptide (51RIIYDRKFLMECRN64). As can be seen in Figure 3A, this peptide exhibited consistent ka’s across the temperatures tested like eIF4G, but with a maximum association rate similar to that of the longer 4E-BP1 sequence (Tables S1 and S2). Accordingly, no differences in secondary structure were found between the two 4E-BP1 peptides at 25 °C (Figure S7). At most temperatures examined, particularly the higher temperatures tested, the eIF4G peptide exhibited a kinetic advantage over both 4E-BP1 peptides (~2-fold; Tables S2 and S3). We speculated that this may be because of its lack of secondary structure as disorder has been hypothesized to be a kinetic advantage of IDPs/IDRs.23,54 Indeed, this was corroborated with the KKR-4E-BP1 peptide, and similar ka’s of 2.0 × 106 M−1 s−1 (KKR-4E-BP1) and 1.7 × 106 M−1 s−1 (eIF4G) (p = 0.1169) were observed at 25 °C (0.8 × 106 M−1 s−1 for 4E-BP1; p = 0.0004). Thus, these results indicate that the 4E-BP1 and eIF4G peptides may utilize distinct mechanisms of association for binding to eIF4E, potentially reflective of their differences in the secondary structure.
Figure 3.
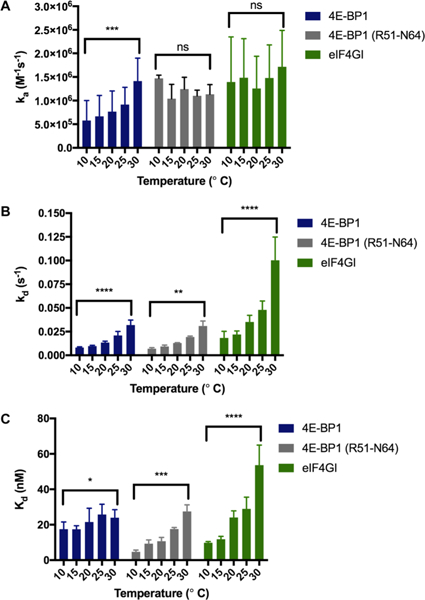
Kinetic analyses of 4E-BP1, 4E-BP1 (R51–N64), and eIF4G peptides via SPR. The effect of temperature on (A) ka (B) kd and (C) kd. Statistical significance was determined using unpaired, two-tailed Student’s t tests. ns = not significant (p > 0.05). (A) ***: p = 0.0004. (B) ****: p = <0.0001; **: p = 0.0015. (C) *: p = 0.0162; ***: p = 0.0005; ****: p = <0.0001. Please see Tables S1–S3 for tabulated results, p values and statistical analyses.
With respect to the dissociation kinetics, all peptides exhibited increases in kd over the temperatures tested (3.9–5.5-fold) (Figure 3B and Table S1). Because gross structural changes in the peptides were not observed over this temperature range (Figure S6A,B), we attribute these differences to weaker stability of the peptide—protein complexes at higher temperatures,55 perhaps because of eIF4E surface restructuring based on its known plasticity.56 Overall, kd’s for the 4E-BP1 peptide were slower than that of eIF4G (Tables S1 and S3). This was irrespective of the 4E-BP1 sequence and 4E-BP1 (R51–N64) exhibited identical dissociation kinetics to that of the 4E-BP1 peptide (Figure 3; Tables S1 and S2). We hypothesize that these differences may be due to an enhanced complementarity of the eIF4E-4E-BP1 peptide interaction, particularly at the C-terminus of the peptide. Based on the X-ray crystal structures of 4E-BP1 and eIF4G peptides bound to eIF4E31 and MD simulations of eIF4G peptide binding,56 the C-terminus of the eIF4G peptide helix is distorted because of steric effects and subsequent reorganization of residues Phe616 and Phe620. Thus, our findings indicate that with increased temperature, the eIF4G peptide may sample more non-productive binding conformations.
Finally, we analyzed the binding constants. For 4E-BP1, little change in Kd was observed over the temperatures tested (17–24 nM); however, for eIF4G, a larger increase in Kd was measured (9.8–54 nM; 5.5-fold). Interestingly, a similar increase in affinity was also found with 4E-BP1 (R51–N64) (4.7–27 nM; 5.8-fold; p = 0.0005); however, like its association kinetics, the highest Kd measured was similar to that of 4E-BP1 (Figure 3; Tables S1 and S2). Because the ka’s of eIF4G were unaffected by temperature, this change in affinity appears to be due to kd. While binding affinities of IDPs/IDRs have been typically shown to correlate with ka,57 a recent report found that this is not always the case.58 Using stopped-flow fluorescence spectroscopy, the binding affinities of disordered peptide mutants for PDZ domains were found to correlate with changes in kd rather than ka because of side-chain interactions that occurred late and cooperatively after the rate-limiting barrier.58 Deviations between the 4E-BP1 and eIF4G peptides were observed primarily at 30 °C (Tables S2 and S3). Thus, at higher and more physiologically relevant temperatures, 4E-BP1 binding seems to be preferred possibly because of its greater complementarity to eIF4E and its increased population of bioactive conformation (vide supra). These findings are of potential relevance to the mechanism of eIF4E activity, as 4E-BP1-mediated inhibition of eIF4E is disrupted only upon phosphorylation of 4E-BP1, which disrupts the helix dipole favoring 4E-BP1 release.41
To further investigate the binding mechanisms of the 4E-BP1 and eIF4G peptides, we also probed the influence of long-range electrostatic interactions by performing our kinetics experiments over a range of salt concentrations (sensorgrams can be found in Figure S8; tabulated results are shown in Table S4).52 As shown in Figure 4A, the association of both peptides was enhanced with increasing salt concentration, indicating that the net interactions are attractive even in the absence of charge-charge interactions (2.2-fold, 0.9–2.0 × 106 M−1 s−1 for 4E-BP1; and 3.1-fold, 1.7–5.3 × 106 M−1 s−1 for eIF4G).59 This was not due to a change in structure, as no differences were observed via CD (Figure S6C,D). Although 4E-BP1 (R51–N64) showed no increase in association, at all salt concentrations tested, the ka’s were statistically insignificant from those of 4E-BP1, yet statistically different from those of eIF4G (Tables S4 and S6). With respect to dissociation, while the kd of the 4E-BP1 peptide did not change (0.023–0.016 s−1), that of eIF4G showed a 5-fold increase (0.046–0.27 s−1) (Figure 4B). These changes in association and dissociation resulted in a 2-fold increase in Kd for the eIF4G peptide (27–56 nM), yet a 3-fold decrease in Kd for 4E-BP1 (25–7.9 nM) (Figure 4C), demonstrating that electrostatic interactions have differential effects on eIF4G and 4E-BP1 binding to eIF4E (Tables S4 and S5). Interestingly, although 4E-BP1 (R51–N64) exhibited similar ka and kd measurements to the 4E-BP1 peptide, a 3-fold increase in Kd was observed at 1.0 M salt concentration similar to eIF4G (Figure 4; Tables S4 and S6). These results are surprising considering the loss of only two amino acids predicted to be unstructured, and point to a potential importance of these residues in stabilizing a salt bridge between Arg51 of 4E-BP1 and Glu140/Asp147 of eIF4E to anchor the N-terminus of the peptide to eIF4E.31 In eIF4G, the corresponding Lys609 has been shown to be disordered and not able to engage in a similar interaction with these acidic residues.31 Thus, the negative impact of salt on these two peptides is likely because of different effects. Together with the temperature analyses, these studies indicate that although 4E-BP1 and eIF4G form similar bound structures, differences exist in the stability of the complexes and their binding mechanisms and kinetics.
Figure 4.
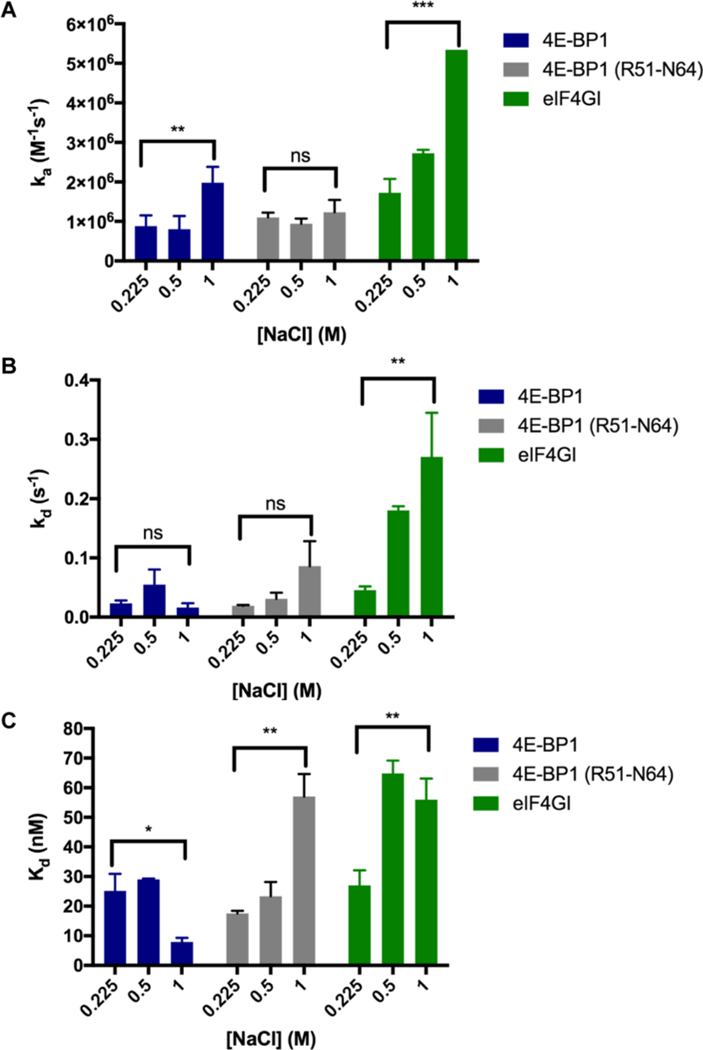
Kinetic analyses of 4E-BP1, 4E-BP1 (R51–N64), and eIF4G peptides via SPR. The effect of ionic strength at 25 °C on (A) ka, (B) kd, and (C) Kd. Statistical significance was determined using unpaired, two-tailed Student’s t tests. ns = not significant (p > 0.05). (A) **: p = 0.0025; ***: p = 0.0002. (B) **: p = 0.0023. (C) *: p = 0.0161; **: p = 0.0024 and 0.0041, respectively. Please see Tables S4–S6 for tabulated results, p values, and statistical analyses. In (A), the error bar for eIF4G is too small to see (Table S4).
Design, Synthesis, and Spectroscopic Analysis of 4E-BP1 and eIF4G Stapled Peptides.
Based on our spectroscopic and kinetic data, we predicted that 4E-BP1 would provide an optimal sequence for stapled peptide development. Thus, to determine how the observed differences between the linear 4E-BP1 and eIF4G peptides may affect the capacity to be constrained, we designed 4E-BP1 and eIF4G hydrocarbon-stapled (HCS) peptides. Based on knowledge of the key residues of 4E-BP1 and eIF4G for binding to eIF4E,31 solvent-exposed Lys57/Glu61 of 4E-BP1 and Glu615/Gly619 of eIF4G were chosen as staple sites. Computational analyses were then performed to examine the effects of staple length and stereochemistry. From these analyses, 8-atom linkers containing L-configuration alkenyl amino acids were predicted to be optimal for stabilization of the i, i + 4 helices (Figures S1–S3). Importantly, these results were in-line with those previously identified in the field for stabilizing similar helices.10 4E-BP1 and eIF4G stapled peptides (HCS-4E-BP1 and HCS-eIF4G; Table 1) were synthesized via Fmoc solid-phase peptide synthesis. Fmoc-(S)-2-(4′-pentenyl)alanine was incorporated at the stapling sites and macrocyclization was subsequently performed using Grubbs I-catalyzed ring-closing metathesis. Similar to the linear peptides, each contained a N-terminal acetyl cap and a C-terminal amide.
Table 1.
SPR Analysis of Linear and HCS-4E-BP1 and -eIF4G and sTIP-04 Peptides at 25 °C
| Peptide | Sequence | Ka×106 (M−1s−1) | kd×10(s−1) | Kd (nM) |
|---|---|---|---|---|
| 4E-BP1 | 0.82 ± 0.3 | 21 ±4 | 26 ±6 | |
| HCS-4E-BP1 | 1.5 ± 0.2 | 5.3 ±4 | 4.1 ±3 | |
| eIF4G | 1.7 ± 0.3 | 48 ± 9 | 29 ±6 | |
| HCS-eIF4G | 0.33 ± 0.01 | 30 ±4 | 90 ±0.4 | |
| Linear sTIP-04 | 2.0 ± 0.1 | 123 ± 14 | 62 ±6 | |
| sTIP-04 | 0.83 ± 0.05 | 5.2 ± 0.1 | 6.3 ± 0.4 | |
We first measured the CD spectra of the HCS peptides. As shown in Figure 5A,B, while HCS-4E-BP1 was found to exhibit enhanced helicity in comparison to the linear peptide (16–44%), the helicity of HCS-eIF4G was nearly the same as that of the linear sequence (2.5–5%; Figure 5D). In the presence of TFE, the helicity of HCS-4E-BP1 was improved to 83%, a marked increase over that of the linear peptide in the same solvent (37%). Because peptide stock concentration was determined via amino acid analysis, this observation is not because of differences in concentration and may indicate the formation of a unique helical structure in this solvent that cannot be obtained in the absence of the chemical constraint. On the other hand, for HCS-eIF4G, based on the CD spectrum, no enhancement in structural ordering was obtained in TFE, indicating that stapling resulted in the formation of a nonhelical macrocyclic peptide. This may be because of steric clash between the amino acid side chains of Phe616 and Phe620 and the alkenyl staple which prevents formation of the bioactive helical conformation.31 Thus, the effect of stapling was IDR-dependent and secondary structure content was enhanced only for the already preorganized 4E-BP1 peptide.
Figure 5.
Stapled peptide helicities as determined via CD spectroscopy in 5 mM sodium phosphate buffer, pH 7.4, 25 °C. (A) 4E-BP1 and HCS-4E-BP1 spectra. (B) eIF4G and HCS-eIF4G spectra. (C) Linear and sTIP-04 spectra. (D) Table of average helicity values measured at 25–100 μM ± standard deviation across the concentrations tested. For the TFE experiments, only 100 μM peptide was used.
Prior to our studies, an eIF4E-binding peptide based on the eIF4G sequence and optimized via phage display was reported.56 This peptide contained key mutations predicted to strengthen the intramolecular hydrogen-bonding within the N-terminus of the helix (Asp613 → Ser and Phe616 → Gln) and side-chain packing against eIF4E within the C-terminus (Phe620 → Leu) (linear sTIP-04 in Table 1).56 Subsequently, a stapled peptide analogue was designed, which exhibited enhanced helicity and binding affinity (sTIP-04 in Table 1).60 To compare our results with eIF4G/HCS-eIF4G to this mutant peptide, the linear and stapled sTIP-04 peptides were chemically synthesized and analyzed for helicity. In contrast to our results with HCS-eIF4G, sTIP-04 was confirmed to be as helical as the HCS-4E-BP1 peptide (38 and 44%, respectively; Figure 5C,D). This is in spite of the fact that linear sTIP-04 exhibited spectra and helical propensity similar to that of eIF4G (2.2 and 2.5%, respectively). Thus, the mutations do not promote structural ordering of the linear peptide in solution, but do enable the sequence to achieve a helical structure upon chemical constraint. Based on MD simulation and crystallography studies, this effect was found to be primarily due to the Gln616 and Leu620 residues, as a corresponding peptide containing only a Ser613 mutation-retained helical deviation at the C-terminus like eIF4G.60 From these examples, it is evident that secondary structure content of a linear IDR peptide does not predict a priori helical stabilization through stapling.
Kinetic Analysis of 4E-BP1, eIF4G, and sTIP-04 Stapled Peptides.
We then assayed kinetics of the stapled peptides using SPR, and the results at 25 °C are summarized in Table 1 (sensorgrams can be found in Figure S9). For the HCS-4E-BP1 peptide, improvements in both ka and kd were observed over the linear peptide, yielding a 6-fold decrease in Kd from 25 to 4.1 nM. Thus, stapling of 4E-BP1 yielded a peptidomimetic with overall greater complementarity to eIF4E because of stabilization of its bioactive conformation. With HCS-eIF4G, however, the peptide was found to exhibit a reduced binding affinity of 90 nM compared to the linear sequence (29 nM) primarily due to a 5-fold reduction in ka, as has been observed with other IDR stapled peptides.22 This finding was not surprising considering our helicity studies, which indicated the formation of a nonhelical macrocyclic peptide which likely has to undergo significant reorganization for binding to eIF4E. For sTIP-04, a decrease in ka was also observed (2.4-fold); but in this case, it was accompanied by a >20-fold enhancement in kd showcasing the ability of its mutations to enhance complementarity and stabilize an eIF4E-bioactive structure. Like its helicity, the binding affinity of sTIP-04 was similar to that of HCS-4E-BP1 (6.3 nM).
To probe this in more detail and further investigate the impact of linear peptide sequence on stapled peptide design, temperature, and salt studies were performed on linear sTIP-04. As shown in Figure S10, this peptide exhibited similar trends with respect to ka, kd, and Kd to the eIF4G sequence; however, linear sTIP-04 exhibited faster dissociation kinetics leading to less favorable binding affinities (results at 25 °C can be found in Table 1; tabulated data at other temperatures in Table S1; p values in Table S7). As residues Tyr612, Phe616, and Phe620 are known to participate in π—π stacking interactions to promote optimal anchoring to the eIF4E surface,60 loss of this intramolecular networking is clearly detrimental to binding. On the other hand, the conformational restriction of sTIP-04, as evidenced by its enhanced helicity, overcame this loss to drive tight binding to eIF4E by providing an ordered helical scaffold for displaying the residues key for the interaction (Tyr612, Arg614, and Leu617).31
With increasing salt concentration, unexpected results were obtained. As shown in Figure S11, a 4-fold decrease in Kd was observed with linear sTIP-04 at 1.0 M salt concentration (62–14 nM; p = 0.0197), which is similar to what was observed with the 4E-BP1 peptide (Figure 4C; p values in Table S8). Interestingly, this was the only correlative feature identified between these linear peptides that yielded stapled peptides with enhanced activity. This was not due to an increase in ka, as found with 4E-BP1, but resultant of a 48-fold decrease in kd (tabulated data in Table S4). In addition to shielding long-range electrostatic interactions, increased ionic strength can also alter the solvation of a peptide or protein. In the case of 4E-BP1 and linear sTIP-04, this effect stabilizes the interaction with eIF4E, albeit through different kinetic mechanisms. Recently, the incorporation of hydrocarbon staples has been shown to provide an analogous influence, altering the architecture of water molecules surrounding a peptide—protein interface.61 Although more examples would be required, our results with eIF4E-binding peptides indicate that this type of kinetic analysis may play an important predictive role in assessing peptide sequences for solvent-exposed hydrocarbon stapling.
In Vitro Inhibitory Activity of 4E-BP1 and eIF4G-Stapled Peptides.
As a follow-up to our kinetics experiments, we probed the ability of the peptides to inhibit eIF4E PPIs. To do so, we utilized our recently reported catalytic enzyme-linked click chemistry assay (cat-ELCCA) for the eIF4E—4E-BP1 PPI, which is a chemiluminescence-based assay that detects the interaction of full-length proteins and is applicable for compound screening.62 As shown in Figure 6, 4E-BP1 peptides were more potent antagonists of the interaction in comparison to the eIF4G peptides with HCS-4E-BP1 showing the greatest inhibitory activity (IC50 value of 3.1–6.7 nM). Importantly, this measurement was similar to that of full-length 4E-BP1, which was previously found to exhibit an IC50 value of 11 nM in PPI cat-ELCCA.62 Binding affinities measured via SPR were found to correlate with biochemical potencies, and the linear 4E-BP1 peptide showed only a slightly weakened IC50 value of 20–48 nM. For the eIF4G series, sTIP-04 was most potent as expected (IC50 value of 17–31 nM), followed by the linear eIF4G (137–195 nM), linear sTIP-04 (384–633 nM), and HCS-eIF4G (4500–7300 nM). We hypothesize that the significantly weaker activity of HCS-eIF4G may be due to its likely structure as a macrocycle rather than a helix, which would not be able to readily compete with the tight binding 4E-BP1 protein. To demonstrate that the activity of these peptides is on-mechanism, we also prepared a HCS-4E-BP1 analogue containing alanine mutations at Leu59 and M60 (HCS-4E-BP1 (LMAA)), which have been shown to be critical for binding to eIF4E.62,63 Although almost as helical as HCS-4E-BP1 (35%, Figure S12), this peptide was found to exhibit significantly diminished inhibition in cat-ELCCA (IC50 value of 3500–18 000 nM).
Figure 6.
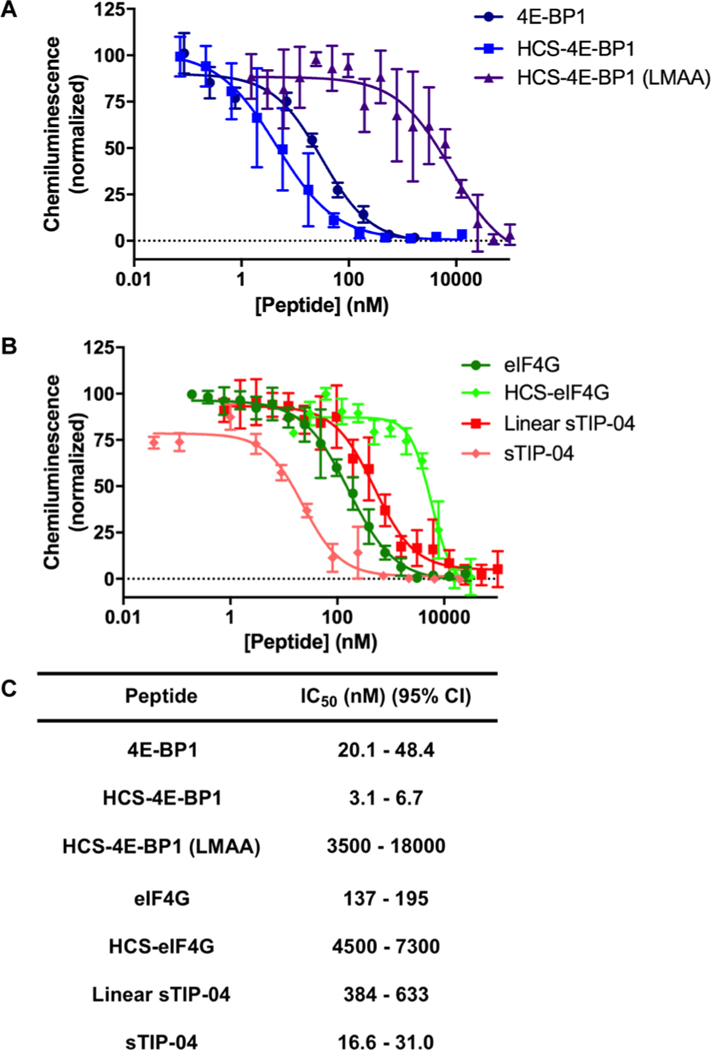
In vitro inhibitory activities of HCS-4E-BP1 and -eIF4G peptides as determined via PPI cat-ELCCA. (A) Inhibition of the eIF4E—4E-BP1 PPI by 4E-BP1, HCS-4E-BP1, and HCS-4E-BP1 (LMAA) peptides; and (B) eIF4G, HCS-eIF4G, linear sTIP-04, and sTIP-04 peptides. (C) Table of IC50 values presented as 95% confidence intervals.
Cellular Activity of HCS-4E-BP1 and sTIP-04.
Encouraged by our in vitro results, we next examined the cellular activity of HCS-4E-BP1 and sTIP-04 in comparison to the inactive HCS-4E-BP1 (LMAA) peptide. As a model, we utilized MDA-MB-231 triple-negative breast cancer cells which exhibit activated eIF4F complex driving cap-dependent translation.64 Prior to testing, we determined each peptide’s cellular permeability via confocal microscopy using FITC-labeled analogues. As shown in Figures S13 and S14 and Movies S1–S3, even in the presence of 10% fetal bovine serum (FBS), all peptides were found to be taken up into cells after 5 h incubation.
To test the inhibitory effect on eIF4E PPIs, we utilized a m7GDP cap pull-down assay, which is an affinity purification method for eIF4E and its protein-binding partners (Figure 7A).65 MDA-MB-231 cells cultured with 10% FBS were treated with varying concentrations of peptide (0–5 μM) and lysates were analyzed via m7GDP affinity chromatography. As can be seen in Figure 7B–D, HCS-4E-BP1 and sTIP-04 exhibited dose-responsive inhibitory activity of eIF4E binding to both eIF4G and 4E-BP1 as visualized by western blot, whereas HCS-4E-BP1 (LMAA) was inactive as expected. In line with our SPR and cat-ELCCA results, the activities of HCS-4E-BP1 and sTIP-04 were nearly identical in this cell-based assay. Of note, no changes in total eIF4E, eIF4G, or 4E-BP1 protein levels were observed following peptide treatment (Figure S15).
Figure 7.
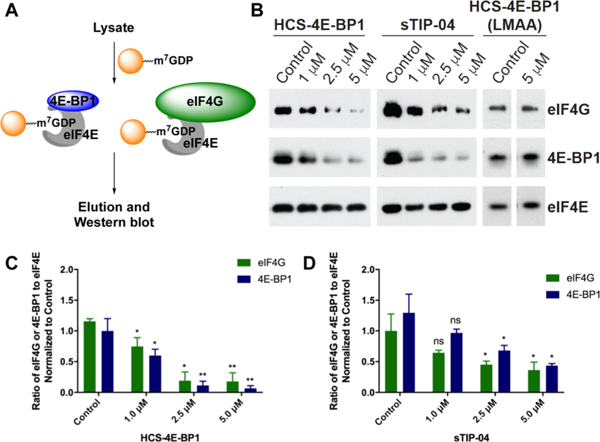
Cellular inhibitory activities of the sTIP-04, HCS-4E-BP1, and HCS-4E-BP1 (LMAA) peptides as determined via m7GDP cap affinity chromatography. (A) Assay scheme. (B) Inhibition of the eIF4E–4E-BP1 and eIF4E–eIF4G PPIs in MDA-MB-231 cells. In all cases, cells were treated with peptides dissolved in water for 6 h and eIF4E was used as a normalization control. (C) Quantitation of the HCS-4E-BP1 data in (B) from triplicate biological replicates. (D) Quantitation of the sTIP-04 data in (B) from triplicate biological replicates. Please see Tables S9 and S10 for p values.
We then measured the effect of the peptides on cellular growth. Surprisingly, despite exhibiting comparable activities in the cell-based m7GDP cap pull-down assay, sTIP-04 exhibited a much more dramatic antiproliferative effect in comparison to HCS-4E-BP1 (Figure 8A,B). Importantly, HCS-4E-BP1 (LMAA) showed no activity (Figure 8C). As the weaker activity of HCS-4E-BP1 more closely resembles what has been observed in MDA-MB-231 cells overexpressing a non-phosphorylatable 4E-BP1 protein64 or those treated with rapamycin to inhibit 4E-BP1 phosphorylation,64,66 which have been shown to be cytostatic, this sTIP-04 activity was unexpected. Because stapled peptides have been demonstrated to disrupt the plasma membrane, particularly those with a high isoelectric point (pI) like sTIP-04 (pI ≈ 11),67 we tested whether this activity was due to nonspecific cellular lysis using the lactate dehydrogenase (LDH) cytotoxicity assay, which measures the release of LDH following loss of membrane integrity.68 As shown in Figure 8D, no LDH leakage was observed. Thus, sTIP-04 may exhibit off-target effects in cells, perhaps because of the fact that it is based on a mutant sequence. Future proteomics studies will be performed to address this question.
Figure 8.
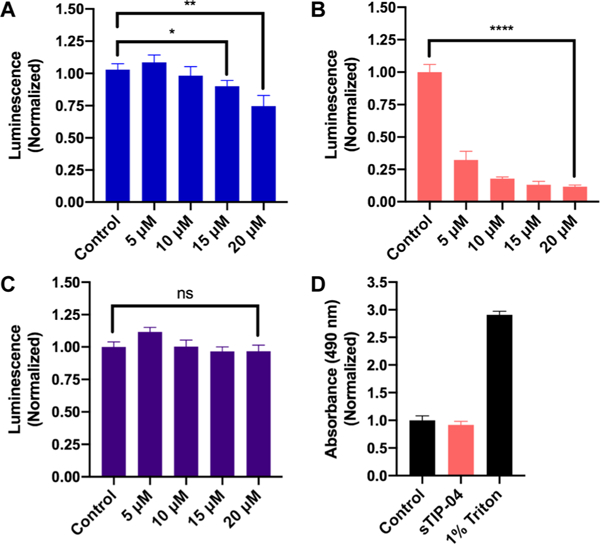
Antiproliferative effects of (A) HCS-4E-BP1, (B) sTIP-04, and (C) HCS-4E-BP1 (LMAA) as determined via the CellTiter-Glo assay. Statistical significance was determined using unpaired, two-tailed Student’s t tests. ns = not significant (p > 0.05). (A) *: p = 0.0259; **: p = 0.0066; (B) ****: p = <0.0001. (D) Measurement of cell membrane damage by sTIP-04 (15 μM) using the LDH cytotoxicity assay using water and 1% Triton as negative and positive controls, respectively.
CONCLUSIONS
In conclusion, we have performed spectroscopic and kinetic studies to evaluate the IDR sequences of 4E-BP1, eIF4G, and an eIF4G mutant, linear sTIP-04, for the development of stapled peptides targeting eIF4E, the m7GpppX-cap-binding translation initiation factor. Through these efforts, we have demonstrated that while intrinsic helicity cannot be used to predict peptide sequences that will benefit from this type of conformational constraint, analysis of binding kinetics can play an important role in selecting an IDR peptide for stapled peptide design. By challenging the peptide—protein interactions with thermal and electrostatic perturbations, we revealed the significance of assessing peptide complementarity for its protein-binding partner and desolvation effects. This was evidenced through correlation of the linear 4E-BP1 and eIF4G kinetics and corresponding stapled peptide properties, where the more complementary 4E-BP1 sequence led to HCS-4E-BP1 with improved affinity and bioactivity, whereas eIF4G stapling resulted in the synthesis of a nonhelical macrocycle because of structural deviations at its C-terminus. While the linear sTIP-04 peptide did not exhibit high complementarity for eIF4E like 4E-BP1, its binding affinity was similarly improved in high salt concentrations, which may mimic the local environmental changes induced by the hydrocarbon staple. As this was the only correlative feature between these sequences, future efforts will be devoted toward addressing this hypothesis using additional PPI models. As IDR-mediated PPIs are widespread in biology and involved in many important biological processes, we are excited by the possibility that a similar type of analysis could be performed on other systems to enable the development of new stapled peptide tools for use in biological interrogation and target validation for drug discovery.
The development of HCS-4E-BP1, which exhibited promising in vitro and cellular activity, is particularly important, as the existing small-molecule inhibitors of the eIF4E—eIF4G PPI, 4EGI-1,69 and 4E1RCat,70 contain structural features that classify them as pan-assay interference compounds or PAINS.71–74 Moreover, these molecules have been shown to induce several phenotypes not observed upon eIF4E knockdown or knockout, indicative of off-target effects.69,75–85 Thus, there remains a great need for fully profiled chemical probes targeting eIF4E and its PPIs so that rigorous validation of this target can be performed in disease models. It is our hope that further optimization of HCS-4E-BP1 and related constrained peptides will enable the development of such a probe. Future work will be focused on improving the cellular activity of HCS-4E-BP1 peptides, as there remains a large gap between in vitro and cellular potencies, and assessing in vivo pharmacokinetic properties and efficacy. Additional efforts will be made toward solving the structure of HCS-4E-BP1 bound to eIF4E to elucidate its binding interactions with the protein and how they compare with the linear peptide, as has recently been done with sTIP-04 analogues.86
EXPERIMENTAL SECTION
Materials.
Linear 4E-BP1, 4E-BP1 (R51—N64), eIF4G, KKR-4E-BP1, and sTIP-04 peptides were purchased from New England Peptides and dissolved in H2O. Fmoc-protected amino acids and Rink amide MBHA resin were purchased from P3 Biosystems and used as received. Fmoc-(S)-2-(4-pentenyl)alanine-OH was purchased from Aldrich and used as received. eIF4E (9742), 4E-BP1 (9644), and eIF4G (2858) antibodies were purchased from Cell Signaling Technology.
Data and Statistical Analysis.
All data were analyzed using GraphPad Prism version 6.0c for Mac OS X (GraphPad Software, www.graphpad.com). Two-sided t-tests were performed using Prism; equal variance between samples being compared was established. Graphs show mean ± standard deviation as described in the figure legends.
CD Spectroscopy.
Peptides were dissolved in buffer (5 mM sodium phosphate buffer, pH 7.4) at concentrations between 25 and 100 μM. CD spectra were recorded on a Jasco I-1500 CD-Spectropolarimeter. Data was collected between 180 and 260 nm with a step resolution of 0.1 nm and a speed of 50 nm/s. Five accumulations were taken with the response time set to 1 s, bandwidth to 5 nm, and pathlength of 0.1 cm. The α-helical content of each peptide was calculated using previously reported equations.87 For determination of temperature dependence, 100 μM samples were equilibrated to 10, 15, 20, 25, and 30 °C, and measurements were taken as mentioned above at each temperature. For each peptide, triplicate analyses were performed.
Protein Expression and Purification.
Plasmid pET19bpp-eIF4E-contained human eIF4E subcloned into a pET19b vector modified to include a 10× His tag and a PreScission protease cut-site between the tag and the beginning of the eIF4E gene (a kind gift from Dr. George Garcia). eIF4E DNA was amplified from the PFN29K vector and subcloned into the modified pET19b vector using NdeI and BamH1 restriction sites introduced with primers eIF4E-FOR-Nde1 (5′-GGTACATATGGCGACTGTCGAACCGGA-3′) and eIF4E-REV-BamH 1 (5′ -CATCGGATCCTTAAACAACAAACC-TAT-3′). The pET19bpp-eIF4E plasmid was transformed into BL21(DE3) cells. Cells were grown at 37 °C to an OD600 of 0.6, induced with 1 mM IPTG, and grown for 16 h at 16 °C. The cells were pelleted and lysed through sonication in lysis buffer [50 mM sodium phosphate, pH 7.4, 100 mM NaCl, 0.5 mM phenyl-methylsulfonyl fluoride, 0.5 mM ethylenediaminetetraacetic acid (EDTA), 5 mM β-mercaptoethanol, 0.5% Tween-20, and 1 mM dithiothreitol (DTT)]. The cell lysate was centrifuged at 38 000g for 2 h and then incubated with m7GDP cap-affinity resin for 45 min. The resin was washed with lysis buffer and phosphate buffered saline (PBS; pH 7.4). Pure eIF4E was eluted with 100 μM m7GTP in 50 mM sodium phosphate, pH 7.4, 100 mM NaCl, and 5% glycerol. The protein was aliquoted and stored without concentrating (~0.3 mg/ mL) at −20 °C. The yield was 1.5 mg from 2 L of cell culture.
SPR Spectroscopy.
SPR was performed using a SensiQ Pioneer instrument and a HisCap chip (three-dimensional hydrogel surface) with 3 channels in series. Channel 1 was activated with NiCl2 and 10× His-eIF4E was immobilized by injection into the running buffer (10 mM HEPES—NaOH, pH 7.5, 225 mM NaCl, 10 mM imidazole, 0.1% Tween-20, and 5% glycerol). The concentration was adjusted to about 600 RU. Peptide (50 μL) was injected at 50 μL/min over a range of 10 concentrations in duplicate and random order (188 nM to 4.0 μM) at 25 °C. The chip was regenerated in between each cycle with an injection 500 mM EDTA followed by reactivation with NiCl2 and reimmobilization of 10× His-eIF4E. For temperature studies, the chip was equilibrated to 10, 15, 20, 25, or 30 °C. Salt studies were conducted at 25 °C with running buffer adjusted to 225, 500, or 1000 mM. The association rate (ka), dissociation rate (kd), and binding constant (Kd) were obtained by fitting to a 1:1 interaction model using global data analysis with the Qdat software, which employs the Levenberg—Marquardt nonlinear regression algorithm. Triplicate experiments were performed for each analysis.
Peptide Synthesis and Purification.
HCS-4E-BP1, HCS-eIF4G, sTIP-04, and HCS-4E-BP1 (LMAA) were synthesized on a 0.2 mmol scale in a 20 mL fritted syringe using MBHA Rink amide resin (0.2–0.4 mmol/g loading). In brief, the resin was swelled for 20 min at 25 °C in 1:1 solution of dimethylformamide (DMF) and dichloromethane. Fmoc groups were removed following the addition of a 20% piperidine/DMF solution (10 mL) and gentle agitation for a total of 20 min at 25 °C. After each Fmoc deprotection and amino acid couplings, the resin was thoroughly washed with N-methyl-2-pyrrolidone (NMP), CH2Cl2, and DMF. Amino acid couplings were performed by addition of amino acid (1 mmol) pre-activated with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (0.9 mmol), N,N-diisopropylethylamine (2 mmol) in NMP (5 mL), and agitated for 2–3 h at 25 °C. The peptide was stapled on resin by bubbling nitrogen gas in a dichloroethylene solution of Grubbs I catalyst at 6 mM for at least 2 h.88 The reaction was monitored through 10–20 mg resin test cleavages via liquid chromatography—mass spectrometry (LC—MS). The procedure was repeated until the reaction went to complete conversion, usually after three replicates. After stapling, the N-terminus was acetylated or modified with FITC (separated by 2 β-alanine residues),88 and the peptide was cleaved from the resin using Trifluoroacetic acid/ thioanisole/water/triisopropylsilane (90:4:4:2) for 4 h at 25 °C. The resulting solution was added to glacial ether (~200 mL) for peptide precipitation. The precipitates were then collected, dissolved, and purified via reversed-phase-high performance liquid chromatography (RP-HPLC). RP-HPLC was performed using binary gradients of solvents A and B, where A is 0.1% HCO2H in water and B is 0.1% HCO2H in acetonitrile or 0.1% HCO2H in methanol. Analytical RP-HPLC was performed using an Agilent 1260 Infinity HPLC equipped with a ZORBAX Eclipse SB-C18 column (4.6 × 150 mm; 5 μm) at a flow rate of 1 mL/min, with detection at 214 and 254 nm. Preparative RP-HPLC was performed using an Agilent 1260 Infinity HPLC equipped with a PrepHT SB-C18 column (21.2 × 150 mm; 5 μm) at a flow rate of 18.6 mL/min, with detection at 214 and 254 nm. Fractions containing the desired peptide were confirmed by LC—MS using an Agilent Q-TOF HPLC—MS. Only fractions containing peptide at ≥95% analytical purity were lyophilized, redissolved in 1:1 acetic acid/water, and lyophilized again for use. Stock solutions of HCS-4E-BP1, sTIP-04, and HCS-4E-BP1 (LMAA) were dissolved in H2O. Stock solutions of HCS-eIF4G, FITC-HCS-4E-BP1, FITC-HCS-sTIP-04, and FITC-HCS-4E-BP1 (LMAA) were dissolved in DMSO. Peptide stock concentrations were determined via amino acid analysis.
PPI cat-ELCCA.
IC50 values were determined using PPI cat-ELCCA as reported.62 In brief, biotinylated HaloTag-eIF4E62 [10 μL of 50 nM in buffer A (50 mM phosphate buffer (pH 7.4), 100 mM NaCl, 0.01% Tween-20, 2 mM DTT)] was immobilized in the wells of a white, high-binding capacity streptavidin-coated 384-well plate (cat #15505). Following overnight incubation at 4 °C, the wells were washed 3× with buffer A with a 5 min incubation in between each wash. Varying concentrations of peptide were then added (5 μL total volume) followed by methyltetrazine-labeled 4E-BP162 (5 μL of 8 nM in buffer A). The plate was then incubated for 1 h at 25 °C. The wells were again washed 3× with buffer A as described above. trans-Cyclooctene-labeled horseradish peroxidase62 (10 μL of 1 μM in buffer A) was then added and the plate was agitated for 1 h at 25 °C. After final washing with buffer A (3 × 50 μL) and buffer B (50 mM phosphate buffer (pH 7.4); (3 × 50 μL)), 30 μL of the SuperSignal West Pico Chemiluminescent Substrate (prepared following the kit instructions; Pierce) was added and chemiluminescence data was collected on a BioTek Cytation3. At least triplicate analyses of technical quadruplicates were measured for each peptide.
General Cell Culture.
MDA-MB-231 cells, a kind gift from Dr. Nouri Neamati and authenticated by STR profiling, were grown in RPMI-1640 media supplemented with 10% FBS (Atlanta Biologicals), 2 mM glutamine and 1% penicillin—streptomycin.
Confocal Microscopy.
MDA-MB-231 cells were grown in 12-well plates on poly-L-lysine-coated coverslips. Cells were treated with FITC peptides at 1 μM for 5 h. Cells were washed twice with ice-cold 1×PBS, and then fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were washed twice with 1 × PBS and then treated with 5 μg/μL of Hoechst 33342 to stain the nucleus. Cells were again washed thrice with 1×PBS and mounted on a glass slide with Prolong Antifade Media. Images at 20× were taken in BioTek Cytation 3 Cell Imaging Multi-Mode reader. Images at 60× (with oil) were taken using a Nikon A1 Spectral confocal microscope. Z stack images were processed in ImageJ and Adobe Photoshop. Figures were formatted in Adobe Illustrator. 3D movies were prepared in Imaris Image Analysis software. Triplicate analyses were performed.
m7GDP Cap Affinity Assay and Western Blot.
The cap pull-down assay was carried out as previously described.89,90 Briefly, MDA-MB-231 were grown in 6 cm dishes and treated with peptides for 6 h. Cells were then lysed in cap pull-down buffer [50 mM HEPES–KOH (pH 7.5), 150 mM KCl, 1 mM EDTA, 2 mM DTT and 0.1% Tween 20] containing protease inhibitors. Cell lysate was centrifuged at 15 000 rpm for 25 min. The supernatant was subsequently incubated for 2 h at 4 °C with m7GDP-agarose resin.90 Beads were washed 3X with the cap pull-down buffer, 1× with TBS and 1× with water. Proteins were eluted by boiling in 2×LDS sample buffer for 10 min at 70 °C, resolved on a 4–12% bis–Tris gel, and transferred to polyvinylidene fluoride membrane in Towbin’s buffer. The membrane was blocked in 5% milk for 1 h at 25 °C, and then incubated with a primary antibody (overnight at 4 °C) and secondary antibody (1 h at 25 °C). Proteins were visualized by autoradiography. In all cases, the eIF4E level was used for normalization. For total cell lysate assays, cells were lysed using RIPA buffer (10 mM Tris-HCl, 150 mM NaCl, 1% Triton, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, pH 7.2) supplemented with 10 μg/mL aprotinin, 5 μg/mL leupeptin, 7 μg/ mL pepstatin, 10 mM NaF, 2 mM sodium orthovanadate, 10 mM β-glycerophosphate, and 2 mM sodium pyrophosphate. Lysates were then sonicated thoroughly on ice. Protein concentrations were normalized by the BCA assay (Pierce). Figures were formatted in Adobe Illustrator. Triplicate biological replicates were performed.
Densitometry.
Scanned western blot images were processed using ImageJ software. The pixels in each band gave a raw reading. The baseline was selected as a background and was subtracted from each raw reading. Ratios of 4E-BP1/eIF4G to eIF4E were calculated and normalized to control. Graphs were plotted in GraphPad Prism.
Cell Viability Assay.
The CellTiter-Glo assay kit was purchased from Promega and was performed according to the manufacturer’s instructions. Briefly, 2000 cells were plated in a white, 96-well tissue culture-treated plate. Cells were treated with the peptides at different concentrations in triplicate and incubated for 6 days. On the sixth day, the cell culture media was replaced with 100 μL of OptiMEM and then lysed with 100 μL of CellTiter-Glo reagent. Total luminescence was read within 1 h using a BioTek Cytation 3 reader. Data was normalized and processed in a GraphPad Prism. Triplicate biological replicates were performed.
LDH Assay.
The LDH Cytotoxicity Assay kit was purchased from Cayman Chemical and was performed according to the manufacturer’s instructions. Briefly, 5000 MDA-MB-231 cells were plated in a clear-bottomed, 96-well plate. After 16 h, the media was changed to OptiMEM and cells were treated with the respective amount of peptide. 1% Triton was used as a positive control for maximum release of LDH. After 24 h, 100 μL of supernatant was removed and centrifuged at 2000 rpm for 5 min. This supernatant was then mixed with 100 μL of LDH reaction solution and incubated at 37 °C. Absorbance at 490 nm was read within 2 h. Triplicate biological replicates were performed.
Supplementary Material
ACKNOWLEDGMENTS
We thank Linda Barthel of the Microscopy and Image Analysis Laboratory at the University of Michigan NCRC for assistance with confocal microscopy. This work was supported through a generous start-up package from the University of Michigan College of Pharmacy, a discovery grant from the American Brain Tumor Association (A.L.G.), and the NIH (R01 CA202018 to A.L.G., T32 GM007767 to E.E.G., and T32 GM008353 to J.M.S.).
ABBREVIATIONS
- CD
circular dichroism
- eIF4E
eukaryotic translation initiation factor 4E
- eIF4G
eukaryotic translation initiation factor 4G
- HCS
hydrocarbon staple
- IDP
intrinsically disordered protein
- PPI
protein—protein interaction
- SPR
surface plasmon resonance
- TFE
2,2,2-trifluoroethanol
- 4E-BP1
eIF4E binding protein
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.9b00068.
Molecular formula strings (CSV)
Structures of eIF4E; virtual sequential alanine mutagenesis; determination of staple length and stereochemistry; linear 4E-BP1 peptide sensorgrams; linear eIF4G peptide sensorgrams; CD spectra; CD spectra of 4E-BP1 (R51-N64); representative linear 4E-BP1 and eIF4G peptide sensorgrams; representative sensorgrams for stapled peptides; kinetic analyses; CD spectra of HCS-4E-BP1 (LMAA); Cellular permeability of FITC-labeled sTIP-04, HCS-4E-BP1 and HCS-4E-BP1 (LMAA); Cellular permeability of FITC-labeled sTIP-04, HCS-4E-BP1 and HCS-4E-BP1 (LMAA); western blot of total protein levels; SPR results; statistical significance between 4E-BP1 (R51-N64) and the 4E-BP1/eIF4G peptides at each temperature; statistical significance between 4E-BP1 and eIF4G peptides at each temperature; and tabulated SPR results (PDF)
Cell permeability movie of HCS-4E-BP1 (AVI)
Cell permeability movie of sTIP-04 (AVI)
Cell permeability movie of HCS-4E-BP1 (LMAA) (AVI)
The authors declare no competing financial interest.
REFERENCES
- (1).van der Lee R; Buljan M; Lang B; Weatheritt RJ; Daughdrill GW; Dunker AK; Fuxreiter M; Gough J; Gsponer J; Jones DT; Kim PM; Kriwacki RW; Oldfield CJ; Pappu RV; Tompa P; Uversky VN; Wright PE; Babu MM Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Oldfield CJ; Dunker AK Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- (3).Wright PE; Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wright PE; Dyson HJ Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Csizmok V; Follis AV; Kriwacki RW; Forman-Kay JD Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chem. Rev. 2016, 116, 6424–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Oldfield CJ; Cheng Y; Cortese MS; Romero P; Uversky VN; Dunker AK Coupled Folding and Binding with α-Helix-Forming Molecular Recognition Elements. Biochemistry 2005, 44, 12454–12470. [DOI] [PubMed] [Google Scholar]
- (7).Fuxreiter M; Simon I; Friedrich P; Tompa P Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [DOI] [PubMed] [Google Scholar]
- (8).Walensky LD; Bird GH Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cromm PM; Spiegel J; Grossmann TN Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10, 1362–1375. [DOI] [PubMed] [Google Scholar]
- (10).Schafmeister CE; Po J; Verdine GL An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar]
- (11).Walensky LD; Kung AL; Escher I; Malia TJ; Barbuto S; Wright RD; Wagner G; Verdine GL; Korsmeyer SJ Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bernal F; Tyler AF; Korsmeyer SJ; Walensky LD; Verdine GL Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Moellering RE; Cornejo M; Davis TN; Bianco CD; Aster JC; Blacklow SC; Kung AL; Gilliland DG; Verdine GL; Bradner JE Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grossmann TN; Yeh JTH; Bowman BR; Chu Q; Moellering RE; Verdine GL Inhibition of oncogenic Wnt signaling through direct targeting of -catenin. Proc. Natl. Acad. Sci., U.S.A. 2012, 109, 17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bernal F; Wade M; Godes M; Davis TN; Whitehead DG; Kung AL; Wahl GM; Walensky LD A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).LaBelle JL; Katz SG; Bird GH; Gavathiotis E; Stewart ML; Lawrence C; Fisher JK; Godes M; Pitter K; Kung AL; Walensky LD A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 2012, 122, 2018–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To K-H; Olson KA; Kesavan K; Gangurde P; Mukherjee A; Baker T; Darlak K; Elkin C; Filipovic Z; Qureshi FZ; Cai H; Berry P; Feyfant E; Shi XE; Horstick J; Annis DA; Manning AM; Fotouhi N; Nash H; Vassilev LT; Sawyer TK Stapled α—helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci., U.S.A. 2013, 110, E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mitra S; Montgomery JE; Kolar MJ; Li G; Jeong KJ; Peng B; Verdine GL; Mills GB; Moellering RE Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer. Nat. Commun. 2017, 8, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Okamoto T; Zobel K; Fedorova A; Quan C; Yang H; Fairbrother WJ; Huang DCS; Smith BJ; Deshayes K; Czabotar PE Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem. Biol. 2013, 8, 297–302. [DOI] [PubMed] [Google Scholar]
- (20).Okamoto T; Segal D; Zobel K; Fedorova A; Yang H; Fairbrother WJ; Huang DCS; Smith BJ; Deshayes K; Czabotar PE Further insights into the effects of pre-organizing the BimBH3 helix. ACS Chem. Biol. 2014, 9, 838–839. [DOI] [PubMed] [Google Scholar]
- (21).Bird GH; Gavathiotis E; LaBelle JL; Katz SG; Walensky LD Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem. Biol. 2014, 9, 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Miles JA; Yeo DJ; Rowell P; Rodriguez-Marin S; Pask CM; Warriner SL; Edwards TA; Wilson AJ Hydrocarbon constrained peptides - understanding preorganisation and binding affinity. Chem. Sci. 2016, 7, 3694–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dogan J; Gianni S; Jemth P The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [DOI] [PubMed] [Google Scholar]
- (24).Bienkiewicz EA; Adkins JN; Lumb KJ Functional Consequences of Preorganized Helical Structure in the Intrinsically Disordered Cell-Cycle Inhibitor p27Kip1f. Biochemistry 2002, 41, 752–759. [DOI] [PubMed] [Google Scholar]
- (25).Gianni S; Morrone A; Giri R; Brunori M A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem. Biophys. Res. Commun. 2012, 428, 205–209. [DOI] [PubMed] [Google Scholar]
- (26).Rogers JM; Steward A; Clarke J Folding and Binding of an Intrinsically Disordered Protein: Fast, but Not ‘Diffusion-Limited’. J. Am. Chem. Soc. 2013, 135, 1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Iesmantavicius V; Dogan J; Jemth P; Teilum K; Kjaergaard M Helical propensity in an intrinsically disordered protein accelerates ligand binding. Angew. Chem. Int. Ed. 2014, 53, 1548–1551. [DOI] [PubMed] [Google Scholar]
- (28).Jhong S-R; Li C-Y; Sung T-C; Lan Y-J; Chang K-J; Chiang Y-W Evidence for an induced-fit process underlying the activation of apoptotic BAX by an intrinsically disordered BimBH3 peptide. J. Phys. Chem. B 2016, 120, 2751–2760. [DOI] [PubMed] [Google Scholar]
- (29).Schmidtgall B; Chaloin O; Bauer V; Sumyk M; Birck C; Torbeev V Dissecting mechanism of coupled folding and binding of an intrinsically disordered protein by chemical synthesis of conformationally constrained analogues. Chem. Commun. 2017, 53, 7369–7372. [DOI] [PubMed] [Google Scholar]
- (30).Pelay-Gimeno M; Glas A; Koch O; Grossmann TN Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Marcotrigiano J; Gingras A-C; Sonenberg N; Burley SK Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 1999, 3, 707–716. [DOI] [PubMed] [Google Scholar]
- (32).Pelletier J; Graff J; Ruggero D; Sonenberg N Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bhat M; Robichaud N; Hulea L; Sonenberg N; Pelletier J; Topisirovic I Targeting the translation machinery in cancer. Nat. Rev. Drug Discovery 2015, 14, 261–278. [DOI] [PubMed] [Google Scholar]
- (34).Costa-Mattioli M; Sossin WS; Klann E; Sonenberg N Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gkogkas CG; Khoutorsky A; Ran I; Rampakakis E; Nevarko T; Weatherill DB; Vasuta C; Yee S; Truitt M; Dallaire P; Major F; Lasko P; Ruggero D; Nader K; Lacaille J-C; Sonenberg N Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2012, 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Richter JD; Sonenberg N Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [DOI] [PubMed] [Google Scholar]
- (37).Fletcher CM; McGuire AM; Gingras A-C; Li H; Matsuo H; Sonenberg N; Wagner G 4E Binding Proteins Inhibit the Translation Factor eIF4E without Folded Structure. Biochemistry 1998, 37, 9–15. [DOI] [PubMed] [Google Scholar]
- (38).Peter D; Igreja C; Weber R; Wohlbold L; Weiler C; Ebertsch L; Weichenrieder O; Izaurralde E Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol. Cell 2015, 57, 1074–1087. [DOI] [PubMed] [Google Scholar]
- (39).Mader S; Lee H; Pause A; Sonenberg N The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol. Cell. Biol. 1995, 15, 4990–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Martineau Y; Azar R; Bousquet C; Pyronnet S Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene 2013, 32, 671–677. [DOI] [PubMed] [Google Scholar]
- (41).Tait S; Dutta K; Cowburn D; Warwicker J; Doig AJ; McCarthy JEG Local control of a disorder-order transition in 4E-BP1 underpins regulation of translation via eIF4E. Proc. Natl. Acad. Sci., U.S.A. 2010, 107, 17627–17632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Marcotrigiano J; Lomakin IB; Sonenberg N; Pestova TV; Hellen CUT; Burley SK A Conserved HEAT Domain within eIF4G Directs Assembly of the Translation Initiation Machinery. Mol. Cell 2001, 7, 193–203. [DOI] [PubMed] [Google Scholar]
- (43).Haghighat A; Mader S; Pause A; Sonenberg N Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995, 14, 5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gmner S; Peter D; Weber R; Wohlbold L; Chung M-Y; Weichenrieder O; Valkov E; Igreja C; Izaurralde E The structures of eIF4E-eIF4G complexes reveal an extended interface to regulate translation initiation. Mol Cell 2016, 64, 467–479. [DOI] [PubMed] [Google Scholar]
- (45).Merrick WC eIF4F: a retrospective. J. Biol. Chem. 2015, 290, 24091–24099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lukhele S; Bah A; Lin H; Sonenberg N; Forman-Kay JD Interaction of the eukaryotic initiation factor 4E with 4E-BP2 at a dynamic bipartite interface. Structure 2013, 21, 2186–2196. [DOI] [PubMed] [Google Scholar]
- (47).Kim D-H; Lee C; Cho Y-J; Lee S-H; Cha E-J; Lim JE; Sabo TM; Griesinger C; Lee D; Han K-H A pre-structured helix in the intrinsically disordered 4EBP1. Mol. BioSyst. 2015, 11, 366–369. [DOI] [PubMed] [Google Scholar]
- (48).Uversky VN Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [DOI] [PubMed] [Google Scholar]
- (49).Johnson OT; Kaur T; Garner AL A conditionally fluorescent peptide reporter of secondary structure modulation. ChemBioChem 2019, 20, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Goldberg JM; Batjargal S; Chen BS; Petersson EJ Thioamide quenching of fluorescent probes through photoinduced electron transfer: mechanistic studies and applications. J. Am. Chem. Soc. 2013, 135, 18651–18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kjaergaard M; Nørholm A-B; Hendus-Altenburger R; Pedersen SF; Poulsen FM; Kragelund BB Temperature-dependent structural changes in intrinsically disordered proteins: Formation of α-helices or loss of polyproline II? Protein Sci. 2010, 19, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Shammas SL; Travis AJ; Clarke J Remarkably fast coupled folding and binding of the intrinsically disordered transactivation domain of cMyb to CBP KIX. J. Phys. Chem. B 2013, 117, 13346–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hackl EV Effect of temperature on the conformation of natively unfolded protein 4E-BP1 in aqueous and mixed solutions containing trifluoroethanol and hexafluoroisopropanol. Protein J. 2015, 34, 18–28. [DOI] [PubMed] [Google Scholar]
- (54).Shoemaker BA; Portman JJ; Wolynes PG Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Schraml M; von Proff L Temperature-dependent antibody kinetics as a tool in antibody lead selection. Methods Mol. Biol. 2012, 901, 183–194. [DOI] [PubMed] [Google Scholar]
- (56).Zhou W; Quah ST; Verma CS; Liu Y; Lane DP; Brown CJ Improved eIF4E binding peptides by phage display guided design: plasticity of interacting surfaces yield collective effects. PLoS ONE 2012, 7, e47235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Prakash MK Insights on the role of (dis)order from protein-protein interaction linear free-energy relationships. J. Am. Chem. Soc. 2011, 133, 9976–9979. [DOI] [PubMed] [Google Scholar]
- (58).Haq SR; Chi CN; Bach A; Dogan J; Engstrom A; Hultqvist G; Karlsson OA; Lundstrom P; Montemiglio LC; Strømgaard K; Gianni S; Jemth P Side-chain interactions form late and cooperatively in the binding reaction between disordered peptides and PDZ domains. J. Am. Chem. Soc. 2012, 134, 599–605. [DOI] [PubMed] [Google Scholar]
- (59).Marqusee S; Baldwin RL Helix stabilization by Glu-...Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci., U.S.A. 1987, 84, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Lama D; Quah ST; Verma CS; Lakshminarayanan R; Beuerman RW; Lane DP; Brown CJ Rational optimization of conformational effects induced by hydrocarbon staples in peptides and their binding interfaces. Sci. Rep. 2013, 3, 3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sim AYL; Verma C How does a hydrocarbon staple affect peptide hydrophobicity? J. Comp. Chem. 2015, 36, 773–784. [DOI] [PubMed] [Google Scholar]
- (62).Song JM; Menon A; Mitchell DC; Johnson OT; Garner AL High-throughput chemical probing of full-length protein-protein interactions. ACS Comb. Sci. 2017, 19, 763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Grosso S; Pesce E; Brina D; Beugnet A; Loreni F; Biffo S Sensitivity of global translation to mTOR inhibition in REN cells depends on the equilibrium between eIF4E and 4E-BP1. PLoS ONE 2011, 6, e29136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Avdulov S; Li S; Van Michalek V; Burrichter D; Peterson M; Perlman DM; Manivel JC; Sonenberg N; Yee D; Bitterman PB; Polunovsky VA Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004, 5, 553–563. [DOI] [PubMed] [Google Scholar]
- (65).Sonenberg N; Rupprecht KM; Hecht SM; Shatkin AJ Eukaryotic mRNA cap binding protein: purification by affinity chromatography on sepharose-coupled m7GDP. Proc. Natl. Acad. Sci., U.S.A. 1979, 76, 4345–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Noh W-C; Mondesire WH; Peng J; Jian W; Zhang H; Dong J; Mills GB; Hung M-C; Meric-Bernstam F Determinants of rapamycin sensitivity in breast cancer cells. Clin. Cancer Res. 2004, 10, 1013–1023. [DOI] [PubMed] [Google Scholar]
- (67).Bird GH; Mazzola E; Opoku-Nsiah K; Lammert MA; Godes M; Neuberg DS; Walensky LD Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Li Y-C; Rodewald LW; Hoppmann C; Wong ET; Lebreton S; Safar P; Patek M; Wang L; Wertman KF; Wahl GM A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep. 2014, 9, 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Moerke NJ; Aktas H; Chen H; Cantel S; Reibarkh MY; Fahmy A; Gross JD; Degterev A; Yuan J; Chorev M; Halperin JA; Wagner G Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [DOI] [PubMed] [Google Scholar]
- (70).Cencic R; Hall DR; Robert F; Du Y; Min J; Li L; Qui M; Lewis I; Kurtkaya S; Dingledine R; Fu H; Kozakov D; Vajda S; Pelletier J Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc. Natl. Acad. Sci., U.S.A. 2011, 108, 1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (72).Baell J; Walters MA Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [DOI] [PubMed] [Google Scholar]
- (73).Devine SM; Mulcair MD; Debono CO; Leung EWW; Nissink JWM; Lim SS; Chandrashekaran IR; Vazirani M; Mohanty B; Simpson JS; Baell JB; Scammells PJ; Norton RS; Scanlon MJ Promiscuous 2-aminothiazoles (PrATs): a frequent hitting scaffold. J. Med. Chem. 2015, 58, 1205–1214. [DOI] [PubMed] [Google Scholar]
- (74).Baell JB; Ferrins L; Falk H; Nikolakopoulos G PAINS: relevance to tool compound discovery and fragment-based screening. Aust. J. Chem. 2013, 66, 1483–1494. [Google Scholar]
- (75).Mokas S; Mills JR; Garreau C; Fournier M-J; Robert F; Arya P; Kaufman RJ; Pelletier J; Mazroui R Uncoupling stress granule assembly and translation initiation inhibition. Mol. Biol. Cell 2009, 20, 2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Fan S; Li Y; Yue P; Khuri FR; Sun S-Y The eIF4E/ eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia 2010, 12, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).McMahon R; Zaborowska I; Walsh D Noncytotoxic inhibition of viral infection through eIF4F-independent suppression of translation by 4EGi-1. J. Virol. 2011, 85, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Tamburini J; Green AS; Bardet V; Chapuis N; Park S; Willems L; Uzunov M; Ifrah N; Dreyfus F; Lacombe C; Mayeux P; Bouscary D Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [DOI] [PubMed] [Google Scholar]
- (79).Redondo N; García-Moreno M; Sanz MA; Carrasco L Translation of viral mRNAs that do not require eIF4E is blocked by the inhibitor 4EGI-1. Virology 2013, 444, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Descamps G; Gomez-Bougie P; Tamburini J; Green A; Bouscary D; Maïga S; Moreau P; Le Gouill S; Pellat-Deceunynck C; Amiot M The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br. J. Cancer 2012, 106, 1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Willimott S; Beck D; Ahearne MJ; Adams VC; Wagner SD Cap-translation inhibitor, 4EGI-1, restores sensitivity to ABT-737 apoptosis through cap-dependent and -independent mechanisms in chronic lymphocytic leukemia. Clin. Cancer Res. 2013, 19, 3212–3223. [DOI] [PubMed] [Google Scholar]
- (82).Schwarzer A; Holtmann H; Brugman M; Meyer J; Schauerte C; Zuber J; Steinemann D; Schlegelberger B; Li Z; Baum C Hyperactivation of mTORCl and mTORC2 by multiple oncogenic events causes addiction to eIF4E-dependent mRNA translation in T-cell leukemia. Oncogene 2015, 34, 3593. [DOI] [PubMed] [Google Scholar]
- (83).Attar-Schneider O; Drucker L; Zismanov V; Tartakover-Matalon S; Lishner M Targeting eIF4GI translation initiation factor affords an attractive therapeutic strategy in multiple myeloma. Cell. Signal. 2014, 26, 1878–1887. [DOI] [PubMed] [Google Scholar]
- (84).Chen L; Aktas BH; Wang Y; He X; Sahoo R; Zhang N; Denoyelle S; Kabha E; Yang H; Yefidoff-Freedman R; Supko JG; Chorev M; Wagner G; Halperin JA Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Yefidoff-Freedman R; Chen T; Sahoo R; Chen L; Wagner G; Halperin JA; Aktas BH; Chorev M 3-Substituted indazoles as configurationally locked 4EGI-1 mimetics and inhibitors of the eIF4E/eIF4G interaction. ChemBioChem 2014, 15, 595–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Lama D; Liberatore A-M; Frosi Y; Nakhle J; Tsomaia N; Bashir T; Lane DP; Brown CJ; Verma CS; Auvin S Structural insights reveal a recognition feature for tailoring hydrocarbon stapled-peptides against the eukaryotic translation initiation factor 4E protein. Chem. Sci. 2019, 10, 2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Shepherd NE; Hoang HN; Abbenante G; Fairlie DP Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127, 2974–2983. [DOI] [PubMed] [Google Scholar]
- (88).Kim Y-W; Grossmann TN; Verdine GL Synthesis of all-hydrocarbon stapled a-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [DOI] [PubMed] [Google Scholar]
- (89).Edery I; Altmann M; Sonenberg N High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIF-4E and affinity purification using a simplified cap-analog resin. Gene 1988, 74, 517–525. [DOI] [PubMed] [Google Scholar]
- (90).Kaur T; Menon A; Garner AL Synthesis of 7-benzylguanosine cap-analogue conjugates for eIF4E targeted degradation. Eur. J. Med. Chem. 2019, 166, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.