

Abstract
Background
Kinetoplastids are a flagellated group of protists, including some parasites, such as Trypanosoma and Leishmania species, that can cause diseases in humans and other animals. The genomes of these species enclose a fraction of retrotransposons including VIPER and TATE, two poorly studied transposable elements that encode a tyrosine recombinase (YR) and were previously classified as DIRS elements. This study investigated the distribution and evolution of VIPER and TATE in kinetoplastids to understand the relationships of these elements with other retrotransposons.
Results
We observed that VIPER and TATE have a discontinuous distribution among Trypanosomatidae, with several events of loss and degeneration occurring during a vertical transfer evolution. We were able to identify the terminal repeats of these elements for the first time, and we showed that these elements are potentially active in some species, including T. cruzi copies of VIPER. We found that VIPER and TATE are strictly related elements, which were named in this study as VIPER-like. The reverse transcriptase (RT) tree presented a low resolution, and the origin and relationships among YR groups remain uncertain. Conversely, for RH, VIPER-like grouped with Hepadnavirus, whereas for YR, VIPER-like sequences constituted two different clades that are closely allied to Crypton. Distinct topologies among RT, RH and YR trees suggest ancient rearrangements/exchanges in domains and a modular pattern of evolution with putative independent origins for each ORF.
Conclusions
Due to the presence of both elements in Bodo saltans, a nontrypanosomatid species, we suggested that VIPER and TATE have survived and remained active for more than 400 million years or were reactivated during the evolution of the host species. We did not find clear evidence of independent origins of VIPER-like from the other YR retroelements, supporting the maintenance of the DIRS group of retrotransposons. Nevertheless, according to phylogenetic findings and sequence structure obtained by this study and other works, we proposed separating DIRS elements into four subgroups: DIRS-like, PAT-like, Ngaro-like, and VIPER-like.
Electronic supplementary material
The online version of this article (10.1186/s13100-019-0175-2) contains supplementary material, which is available to authorized users.
Keywords: Tyrosine recombinase, DIRS, Transposable elements, Phage integrase, Hepadnavirus, Retroelements
Background
Trypanosomatid species are among the most ancient eukaryotes, comprising more than ten genera that include both monoxenous insect parasites and dixenous species, which alternate between insects and vertebrates (or plants) [1–3]. Species of Trypanosoma and Leishmania cause some human pathologies, such as Chagas disease (T. cruzi), sleeping sickness (T. brucei) and leishmaniasis (genus Leishmania) and are transmitted by invertebrate vectors [4]. In addition to the importance of these species as pathogens, they have several unique features. The protein-coding genes of trypanosomatids are transcribed polycistronically, and the mature monocistronic mRNAs are generated by a trans-splicing mechanism and polyadenylation [5]. Moreover, gene expression in these species is mostly regulated posttranscriptionally, since all polycistronic precursor RNAs seem to be transcribed at approximately the same rate [6].
More than 10 years ago, the reference genome sequences of three trypanosomatids, T. brucei, T. cruzi and L. major, were published [7–9]. These genomes are highly repetitive (over 50% in T. cruzi), mainly due to large gene families of surface molecules, subtelomeric repeats and transposable elements (TEs) (exclusively retrotransposons) [8]. Recently, Pita et al. (2019) compared the repetitive DNA portions among these three genomes using genome-wide, low-coverage Illumina sequencing. These authors estimated that the genome fraction corresponding to retrotransposons ranges from 12.6% in T. cruzi to 5.7% in T. brucei and only 1.6% in L. major [10].
Retrotransposons are eukaryotic mobile elements that move through an RNA intermediate. They constitute an important source of genetic variation and have actively shaped the structure, function, and evolution of genomes [11–14]. Although considerable attention has been devoted to the two major groups of retrotransposons (LTR and non-LTR elements [15, 16]), recent results have confirmed the presence of another ubiquitous group of retroelements [17–21] that encode a tyrosine recombinase (YR) gene instead of integrase (IN) or endonuclease (EN). Wicker et al. (2007) classified the YR-containing retroelements in the order DIRS, named based on the first described YR retrotransposon, the DIRS1 from the slime mold Dictyostelium discoideum [22].
YR elements, in general, contain three long ORFs (open reading frames). ORF1 was considered a putative gag-like gene, given the similarities in size and position with the upstream region of LTR retrotransposons. These elements also have ORFs encoding a YR and a reverse transcriptase/RNase H (RT/RH) that are frequently extensively overlapped [23]. In some DIRS elements, it is also possible to find a coding region for a methyltransferase (MT) or a hydrolase whose functions are still unknown. These elements present a diversified structure of repeats that can be primarily of two types: (1) terminal inverted repeats (ITRs) and an internal complementary region (ICR) that is complementary to the beginning of the left ITR and to the end of the right ITR or (2) “split” direct repeats [24].
Wicker et al. (2007) divided the DIRS order into three subfamilies, DIRS, Ngaro, and VIPER. Recently, Poulter and Butler (2015) divided the YR retrotransposons into three major subgroups: DIRS, PAT-like, and Ngaro elements. This subdivision was based on a phylogenetic tree of the RT/RH domains and on structural differences, which led to the separation of PAT-like from the DIRS subgroup. VIPER (vestigial interposed retroelement) sequences were not included in the latter analyses. This retroelement was encountered in T. cruzi [25], T. brucei and T. vivax [26] and was considered an extinct retrotransposon due to the absence of potentially functional copies and to the apparent lack of conserved terminal repeat regions [26, 27]. Moreover, there is another element, TATE (telomere associated transposable element), that could be classified as DIRS and was found in the genome of L. braziliensis as in tandem clusters inserted at the same relative position within the telomeric hexamer repeats (GGG↑TTA) [28].
Little is known about VIPER and TATE in trypanosomatids, and several questions about their structure, functioning and origin remain unanswered. In this study, we investigated the distribution of these elements in the available trypanosomatid genomes and provided evidence that some of their copies could still be active in some species. We also performed an evolutionary analysis of retrotransposon RT, RH and YR proteins. Although we failed to fully elucidate the relationships among YR elements, some differences in topologies were recovered for distinct domains, suggesting the occurrence of a modular pattern of evolution with ancient changes in domains. Moreover, our results suggested a close relationship between VIPER-like sequences and the hepadnavirus RH region that may reflect a shared origin of this domain. This paper represents a first step in characterizing the functioning and impact of YR-containing TEs in kinetoplastids.
We used the retrotransposon group nomenclature according to the classification system of Wicker et al. (2007) [29]: order LINE; superfamilies Copia, Gypsy, Bel-Pao, and Retrovirus from order LTR; superfamilies within order DIRS were updated to DIRS-like, PAT-like, Ngaro-like, and VIPER-like. The non-italicized term “DIRS” was used to refer to all the YR-containing retrotransposons (order DIRS).
Results
VIPER and TATE present a discontinuous distribution in trypanosomatid species
A total of 44 trypanosomatid genomes were investigated in this study for the presence of VIPER and TATE together with the genome of Bodo saltans, a free-living kinetoplastid species from the Bodonidae family. Almost all analyzed species presented significant tblastn hits for at least one protein of VIPER or TATE (Additional file 1). Only Leishmania tarentolae, Leptomonas seymouri, Lotmaria passim, Phytomonas sp. isolates EM1 and HART1 and Trypanosoma rangeli did not present any significant hit, suggesting the complete loss of these retrotransposons.
For most species, VIPER and TATE seem to have degenerated, as seen in Additional file 2; nevertheless, we were not able to evaluate the conservation of several copies located in the extremities of contigs/scaffolds or missing data regions. Moreover, it is important to consider that the definition of the gag-like ORF was mostly based on positional information, since this analysis revealed a highly divergent ORF that did not present significant signs for specific domains in most of the evaluated species.
When the status of the copies (potentially encoding, likely degenerate, absent or inconclusive) was summarized on the topology of the trypanosomatid species tree [based on previous studies [1, 30–39]) (Fig. 1), we could observe a patchy distribution of elements among species. The presence of VIPER and TATE in B. saltans, representing the outgroup of the tree, suggests that both TEs were already present in the last common ancestor of this species and trypanosomatids, which diverged more than 460 million years ago (mya) [1]. This ancient existence makes the study of the conservation of these elements throughout trypanosomatid evolution even more exciting.
Fig. 1.

Distribution and conservation of VIPER and TATE among trypanosomatids. The tree was drawn based on several studies (see text). Branches are not drawn to scale. The presence of at least one potentially encoding copy of the element in the species is indicated as a dark green rectangle. The presence of only degenerate copies is represented by a light green rectangle. Species, where it was not possible to conclude on the conservation of the element, have a question mark, and XXXX represents the probable complete absence of the element
VIPER is a potentially encoding retrotransposon in some Trypanosomatidae species
VIPER seems to have been completely lost in the majority of Leishmania species, Endotrypanum monterogeii, Lo. passim, Lep. seymouri, Phytomonas sp. isolate EM1 and HART1, and P. francai. Several other species presented degenerate copies of VIPER. In L. braziliensis, L. panamensis and L. peruviana, which are three closely related species [40, 41], one of the significant hits corresponds to a VIPER-derived gene that was already reported [42].
We found at least one potentially encoding copy of VIPER in B. saltans, T. theileri, Angomonas deanei, Crithidia fasciculata, C. mellificae, and Lep. pyrrhocoris. Table 1 and Additional file 3 show a summary of these results. In T. cruzi, from four different sequenced genome assemblies, only the Dm28c strain presented putative complete encoding copies.
Table 1.
Summary of VIPER copy analyzes from the distinct genomes presenting at least one potentially encoding copy
| Species | No of analyzed copies | No of copies with at least one potentially encoding ORF | No of copies with the three potentially encoding ORFs | No of potential encoding copies with YR and RT/RT related domains | No of copies with SDRs | Structure of SDRs size in base pairs, percentage of identity | No of putative autonomous copiesa | Mean identity among copies in nu (total) and aa levels (for each ORF)b |
|---|---|---|---|---|---|---|---|---|
| B. saltans | 10 | 7 | 4 | 1 | 1c |
5′ → A1 3′ → B1 → B2 → A2 A: 83 bp, 100%; B: 57 bp, 100% |
0 |
nu: na Gag-like: na YR (5, 150 aa): 32.7% RT/RH (7, 294 aa): 33% |
| T. cruzi Dm28c | 15 | 15 | 12 | 12 | 7 |
5′ → A1 3′ → B1 → A2 → B2 A: 173 bp, 100%; B: 219 bp, 100% |
7 |
nu (13): 95% Gag-like (13, 492 aa): 97.9% YR (15, 338 aa): 98.2% RT/RH (14, 948 aa): 96.8% |
| T. theileri | 15 | 5 | 3 | 2 | 1 (incomplete) |
5′ absent 3′ → A1 → A2 A: 95 bp, 100%; |
0 |
nu: na Gag-like (3, 240 aa): 31.1% YR (2, 272 aa): 47.1% RT/RH (5, 701 aa): 57.6% |
| C. fasciculata | 15 | 7 | 4 | 1 | 3 |
5′ → A1 3′ → B1 → A2 → B2 1: (A: 136 bp, 99%; B: 288 bp, 100%) 2: (A: 144 bp, 100%; B: 308 bp, 100%) 3: (A: 414 bp, 93%; B: 294 bp, 99%) |
1 |
nu: na Gag -like (3, 240 aa): 31.1% YR (5, 278 aa): 41.6% RT/RH (5, 701 aa): 40.8% |
| C. mellificae | 15 | 4 | 1 | 0 |
? all copies in short or end of contigs |
na | 0 |
nu: na Gag-like: na YR (3, 507 aa): 100% RT/RH (2, 393 aa): 43% |
| A. deanei | 4 | 4 | 1 | 0 | 0 | na | 0 |
nu: na Gag-like (3, 240 aa): 31.1% YR (2, 272 aa): 48.4% RT/RH (6, 611 aa): 26.7% |
| Lep. pyrrhocoris | 15 | 14 | 2 | 0 | 0 | na | 0 |
nu: na Gag-like (2, 396 aa): 99.7% YR (13, 430 aa): 61.6% RT/RH (11, 1091 aa): 72.1% |
aputative autonomous copies are those containing the tree expected ORFs plus direct repeats
bidentity was estimated only for regions with confident alignment. The number of sequences and the size of alignment is shown in parenthesis
cthe pattern of repeats is distinct than expected and the distance from the end of the ORF and the next repeat is quite large and unexpected. Thus, it is not possible to state whether this repetition is indeed part of the element
For most species, the nucleotide alignment of VIPER copies was impaired due to the high divergence, indicating that the events of mobilization separating these copies occurred a long time ago. The difference is also high at the amino acid level. In contrast, in T. cruzi Dm28c, the nucleotide alignment from conserved copies enabled the identification of the boundaries of the element and the isolation of complete copies that show only 5% mean divergence.
We observed that the VIPER copies of T. cruzi and C. fasciculata presented split direct repeats (SDRs) with a pattern similar to that found for Ngaro and PAT-like elements [18] ordered as A1 at the 5′ end and B1, A2 and B2 at the 3′ end (Fig. 2). For some copies, the related repeats present 100% identity. The 3′ repeats were not present in the Repbase T. cruzi VIPER consensus sequence, whereas the 5′ repeat was absent in T. theileri. One copy of B. saltans presented putative SDRs with a distinct structure of repeats ordered as A1 at the 5′ end and B1, B2, A2 at the 3′ end. For A. deanei, it was not possible to evaluate the presence of repeats due to the small size of the contig.
Fig. 2.

Schematic structure of the potentially complete VIPER and TATE retroelements found in trypanosomatids. Repbase consensus sequences and the SIRE element were also represented. Rectangles of different colors represent the ORFs encoding Gag-like (blue), YR (yellow), and RT / RH (pink), whose position was drawn according to the reading phase (+ 1, + 2 or + 3). The arrows indicate the repeats. The scaffold and position of copies are displayed next to the species name. The figure is drawn near to scale (except for some repeats), which is distinct for VIPER and TATE. The represented L. braziliensis TATE copy possesses a frameshift in the third ORF represented as two dashes. NNNN represents missing data regions
Vasquez et al. (1999, 2000) described the existence of a truncated non-autonomous version of VIPER, called SIRE (short interspersed repetitive element), which was defined by the first 185 bp of VIPER 5′ portion, an exclusive internal region of approximately 30 bp, and 220 bp of the VIPER 3′ end [25, 43]. With the identification of T. cruzi VIPER SDRs, it is possible to observe that the SIRE element corresponds to the region encompassing A2 and B2 repeats (Fig. 2). This finding can open discussions about the SIRE origin (central or 5′ degeneration) and mobility capacity (mobile elements with the help of machinery provided by autonomous VIPER copies or just remnants of VIPER insertions).
In some VIPER copies, we observed the expected domains for YR (DNA_BRE_C super family) and RT/RH (RT_like super family and RNase_H_like super family) proteins. For the Gag-like protein, it was not expected to find a conserved domain. In this study, we found several different predicted domains (Additional file 3): 1) the SMC_N super family in copies from A. deanei, C. fasciculata, C. mellificae, and T. cruzi CL Brener Non-Esmeraldo-like; 2) the PHA03247 super family, downstream of the SMC_N domain in C. mellificae; and 3) the CwlO1 super family or AcrA in almost all T. cruzi Dm28c copies. Most of these domains are classified as a model that may span more than one domain and seems to be found in a vast number of nonrelated proteins [as checked in the Conserved Domain Database (CDD)]. Moreover, all these domains were predicted with high e-values (although below the threshold), indicating very low sequence similarity. Thus, it is difficult to determine whether the domains are indeed present in the proteins and the function they could perform (see Additional file 4 for more information).
In T. cruzi Dm28c, T. theileri and Lep. pyrrhocoris copies, we also found a CH-box (CX2CX4HX4C) motif that is often present in Gag proteins from retroviruses and LTR-retrotransposons [44, 45] and in some YR-retrotransposons [18]. This region is known to form a zinc finger with nucleic-acid-binding properties that is involved in packaging the RNA genome [46]. This motif was previously found in the Gag-like protein for the T. cruzi CL Brener VIPER consensus obtained by Lorenzi et al. (2006). In contrast, we were not able to find the major homology region (MHR - a 22-aa sequence located in the capsid domain of several Gag proteins) that was also predicted by these authors. We found no conserved CH-box in the copies from B. saltans, A. deanei, C. fasciculata, and C. mellificae.
TATE, another YR retrotransposon in kinetoplastids
As shown in Fig. 1, TATE seems to have been completely lost in Phytomonas sp. isolates EM1 and Hart1, Lo. passim, Lep. seymouri, L. tarentolae and in most Trypanosoma species (except T. theileri). In several others, including most Leishmania species, TATE copies are possibly degenerate. We found potentially encoding copies of TATE in B. saltans, C. fasciculata, C. mellificae, S. culicis, L. braziliensis MHOM/BR/75/M2904, Lep. pyrrhocoris and T. theileri. Due to the phylogenetic proximity of L. panamensis and L. peruviana to L. braziliensis [40, 41], we expected to find similar patterns in the three species. However, most copies in L. peruviana and L. panamensis are in missing data regions. The same is found for the L. braziliensis MHOM/BR/75/M2903 strain. Table 2 and Additional file 5 summarize these results.
Table 2.
Summary of TATE copy analyzes from the distinct genomes presenting at least one potentially encoding copy
| Species | No of analyzed copies | No of copies with at least one potentially encoding ORF | No of copies with the three potentially encoding ORFs | No of potential encoding copies with YR and RT/RT related domains | No of copies with SDRs | Structure of SDRs size in base pairs, percentage of identity | No of putative autonomous copies* | Mean identity among copies in nu (total) and aa levels (for each ORF)** |
|---|---|---|---|---|---|---|---|---|
| B. saltans | 5 | 2 |
2 *one ORF with the three genes |
0 | 1 |
5′ → A1, A2 3′ → B1 → B2 A: 73 bp, 99%; B: 161 bp, 81% |
0 |
nu: na Gag-like, YR, RT/RH (2, 1464 aa): 49% |
| T. theileri | 15 | 7 | 4 | 0 | 1 (incomplete/missing data) |
5′ → A1 3′ → A2 A: 91 bp, 100%;; B: missing data |
0 |
nu: na Gag-like: na YR (3, 584 aa): 45.3% RT/RH (4, 698 aa): 86.9% |
| C. fasciculata | 15 | 7 | 4 | 0 | 0 | na | 0 |
nu: na Gag -like (3, 240 aa): 31.1% YR (7, 727 aa): 44.8% RT/RH (3, 473 aa): 53.6% |
| C. mellificae | 11 | 3 | 1 | 1 | 1 (incomplete/missing data) |
5′ → A1 3′ → A2 A: 357 bp, 100%; B: missing data |
0 |
nu: na Gag-like (2, 240 aa): 25.6% YR (3, 434 aa): 36.3% RT/RH: 1 sequence |
| S. culicis | 15 | 1 | 1 | 0 | 0 | na | 0 | 1 sequence |
| Lep. pyrrhocoris | 15 | 11 | 4 | 0 | 0 | na | 0 |
nu: na Gag-like (5, 194 aa): 29.7% YR (10, 468 aa): 48.9% RT/RH (8, 882 aa): 55.6% |
| L. braziliensis | 15 | 15 | 7 | 6 | 1 |
5′ → A1 3′ → B1 → A2 → B2 A: 381 bp, 96%; B: 294 bp, 99% |
? |
nu: 90% Gag-like (8, 450 aa): 73.3% YR (15, 982 aa): 95.6% RT/RH (14, 954 aa): 96% |
*putative autonomous copies are those containing the tree expected ORFs plus direct repeats
**identity was estimated only for regions with confident alignment. The number of sequences and the size of alignment is shown in parenthesis
? the copy where the SDRs were identified presents a frameshift mutation in the third ORF, however, we do not rule out the possibility that the genome could have additional copies that were not detected due to assembling issues
Two potentially encoding copies from B. saltans presented a single ORF structure, while copies from other species exhibited the expected three ORFs. The divergence of copies from the same species is high, except for L. braziliensis, which showed a mean of 90% nucleotide identity.
The TATE element seems to have a pattern of terminal repeats similar to that found for VIPER, although their identification was compromised in most species due to the proximity of copies to the end of contigs and due to the presence of more than one copy in tandem in the same scaffold. In L. braziliensis, at least one copy presented a pattern of SDRs, although the 3′ repeats were not shown in the Repbase TATE consensus sequence (Fig. 2). Moreover, we noticed more complex patterns of repetitions (Additional file 5) in some copies of B. saltans and L. braziliensis, but they could be a result of wrong genome assembly or chimeric natural sequences due to insertion of copies inside existent ones.
In L. braziliensis, we observed the expected domains for YR (DNA_BRE_C super family), and in the N-terminus region of several copies, this domain was preceded by a SPOR super family domain (predicted with low significance). This domain is involved in binding peptidoglycan and is found in proteins involved in sporulation and cell division, such as FtsN, DedD, and CwlM. Functional studies may help to address the role (if any) of this region. For ORF3, in addition to the RT_like superfamily, a Peptidase_A17 domain was found. Although this last domain is described as a homolog of aspartic proteinases at PFAM, it is, in fact, an RH domain and was inaccurately named [47]. The Gag-like protein of all TATE copies has no predicted domain and no clear CH-box.
Evolution of VIPER and TATE in the trypanosomatid genomes
We performed phylogenetic reconstructions of VIPER and TATE elements based on the RT/RH and YR proteins. In both cases, both TEs revealed reciprocally monophyletic, although the YR tree presented a low phylogenetic resolution and few strongly established relationships (data not shown). Figure 3 shows the RT/RH majority-rule consensus tree obtained through BA, presenting the expected separation into two large clades corresponding to the different elements.
Fig. 3.
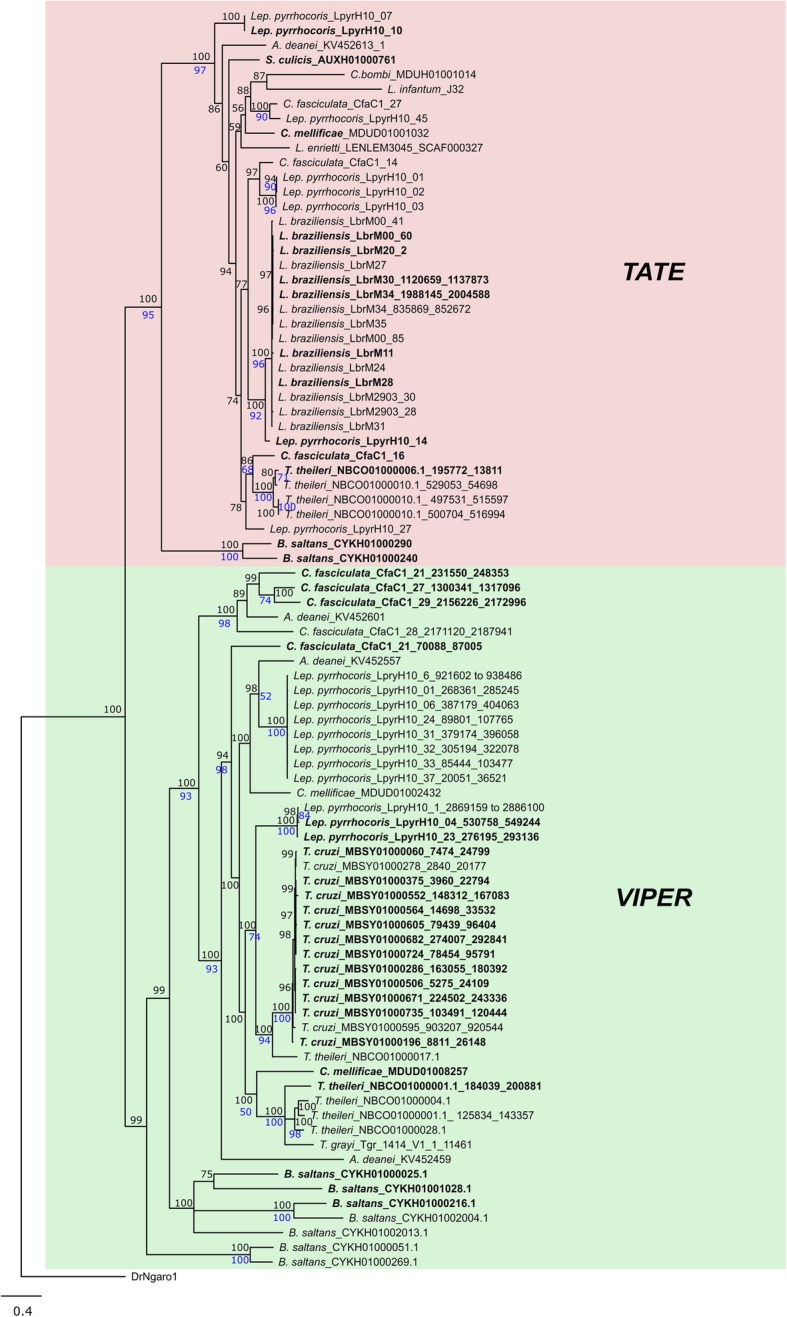
VIPER and TATE phylogenetic relationships among kinetoplastids. BA tree (mixed model + G, 336 aa) of VIPER and TATE copies of kinetoplastid species based on the RT/RH amino acid sequence. The RT/RH sequence of DrNgaro1 was used as an outgroup. Posterior probability values obtained in the BA are indicated above the nodes, and clades with support less than 50% were collapsed; bootstrap values obtained in the ML analysis are indicated below the nodes in blue. Sequences that are part of putative encoding copies (with the three ORFs potentially encoding) are highlighted in bold
To understand the evolutionary history of these elements, we compared their phylogeny with the host species phylogenetic relationships, a standard procedure for TE evolutionary analysis [48–50]. For both TEs, B. saltans encompass the earlier offshoot, which is expected once this species is a nonparasitic close relative [51]. However, it is possible to find several incongruities in derived clades, such as the unexpected clustering of copies from distant species (for example, one VIPER copy of C. mellificae grouped with T. theileri and T. grayi) and scattered branching of copies of the same species (such as TATE copies from Lep. pyrrhocoris and C. fasciculata and VIPER copies from Lep. pyrrhocoris, T. theileri, C. fasciculata, C. mellificae and A. deanei). Those incongruities, however, cannot be explained by horizontal transfer, since the divergence among copies is not lower than expected (results not shown). This tree also shows cases of intragenomic mobilization and diversification (for example, TATE from L. braziliensis and VIPER from T. cruzi and Lep. Pyrrhocoris). Potentially encoding copies are present in several clades.
Relationship of VIPER and TATE with other retroelements and YR-proteins
We found few significant hits for VIPER and TATE outside Kinetoplastida in the NCBI blastp searches. Hits for RT/RH and YR were recovered from Oceanobacter sp and Candidatus Handelsmanbacteria. We initially included these sequences in the phylogenetic analysis. However, the distribution of those revealed interspersed in the TATE and VIPER clades, raising the hypothesis of genomic sequence contamination that was strongly suggested after more in-depth investigations. Thus, these sequences were not used in the final phylogenies. Another significant hit for VIPER YR encompassed the sequence WP_106215579.1 from the bacterium Kineococcus rhizosphaerae, annotated as an integrase. We included this and other homologous proteins (retrieved after further blastp searches) in the phylogeny. Additional file 6 outlines the discussion concerning this issue.
ML and BA trees recovered similar topologies, whose results are presented in the form of the Bayesian majority rule consensus tree recovered for each major domain (Figs. 4, 5 and 6). Each of these trees summarizes the support values recovered through both ML and BA as distinct node labels.
Fig. 4.

RT phylogenetic relationships. BA tree (mixed model + G, 197 aa) based on the RT amino acid sequences of different groups of retroelements. Possible encoding copies of VIPER and TATE are highlighted in bold. Posterior probability values obtained in the are indicated above the nodes, and clades with support less than 50% were collapsed; bootstrap values from ML analysis are presented in blue below the nodes (only values above 50). The tree was rooted with the LINE clade
Fig. 5.

RH phylogenetic relationships. BA tree (mixed model + G, 146 aa) based on the RH amino acid sequences of different groups of retroelements. Possible encoding copies of VIPER and TATE are highlighted in bold. Posterior probability values are indicated above the nodes, and clades with support less than 50% were collapsed; bootstrap values from ML analysis are presented in blue below the nodes (only values above 50). The tree was rooted with the Eukaryote RH clade
Fig. 6.

Phylogenetic relationships of YR family proteins. Unrooted BA tree (mixed model + G, 157 aa) based on the YR amino acid sequences from DIRS retrotransposons (DIRS-like, PAT-like, Ngaro-like, and VIPER-like) along with other YR family proteins: resolvase [site-specific recombinases involved in chromosome dimer resolution in archaea (XerA) and bacteria (XerC/D)], FLP (yeast Flp-like flippases), phage integrase, prokaryote transposons, Crypton, uncharacterized YR (proteins retrieve by blastp searches). YR sequences from the distinct groups shown by Wang et al. (2018) are in red: shufflon (shufflon-specific DNA recombinases); DAI (dusA-associated integrases); phage integrases; IntC (integrases from integrative and conjugative elements); IntI (integron integrases); IntG (genomic island integrases); XerA (site-specific recombinases involved in chromosome dimer resolution in archaea), Cre (phage P1-like recombinases); archaeal SSV-type, pNOB8-type and pTN3-type integrases; and integrases encoded by haloarchaeal SNJ2, BJ1-like and phiCh1-like viruses. Possible encoding copies of VIPER and TATE are highlighted in bold. Posterior probability values are indicated above the nodes, and clades with support less than 50% were collapsed; bootstrap values from ML analysis are presented in blue below the nodes (only values above 50)
Figure 4 shows the RT BA tree with a general topology less resolved than that found in other studies employing the same domain [26, 52], recovering only the well-supported clustering of Gypsy, Caulimovirus, and retroviruses. The lack of clustering of all YR elements could indicate an independent origin of these elements or merely the ancient separation of clades. As concerns the groups of YR elements, it was not possible to recover the monophyly either of DIRS-like (EvenDirs and DIRS-1_PH grouped in separate clades) or of PAT-like (three lineages), as also evidenced by Lorenzi et al. (2006) and Poulter and Butler (2015). In fact, the monophyly of these groups was only supported by Goodwin and Poulter (2004, 2005), who recovered a clade containing Kangaroo and Pat together with DIRS-like sequences in an RT/RH tree. Even so, Ngaro-like and VIPER-like sequences formed monophyletic groups (support of 100% BA, 99% ML and 100% BA, 81% ML, respectively). This finding confirms the close relationship of VIPER and TATE elements, both of which are encountered in trypanosomatids and share a similar structure despite sequence divergence.
The RH tree (Fig. 5) shows the positioning of retroviruses out of the Gypsy/caulimovirus clade [also seem by Malik and Eickbush (2001)]. Moreover, we observed an unexpected close relationship between VIPER and hepadnavirus, which are enveloped viruses with reverse-transcribed DNA genomes [53]. This possible relationship was never reported previously. In the RH tree from Malik and Eickbush (2001), VIPER sequences were not included, and in the Goodwin and Poulter (2004) RT/RH tree, both VIPER and Hepadnavirus sequences are located as polytomic branches together with Ngaro-like, caulimovirus/Gypsy, and DIRS-like/PAT-like sequences. It is possible that this relationship was masked in the last study due to the concatenation of RH with the RT domain. As occurred with RT, RH also shows unsatisfactory resolution to the clustering of elements belonging to different groups of YR elements, since DIRS-like and PAT-like sequences are scattered in the tree. Several of these results may be attributable to an outcome of the lower conservation of RH domains with the consequent loss of phylogenetic signal. Nevertheless, as the grouping of VIPER and hepadnavirus is strongly supported and was recovered under both ML and BA approaches, it is not possible to reject the possibility of changes/rearrangements in domains. Corroborating this similarity, when the RH portion of Hepatitis B virus (HBV) is isolated and used as a query on NCBI PSI-blastp searches [(excluding Hepadnaviridae (taxid:10404)], B. saltans VIPER sequences are retrieved in the third iteration.
For YR, in addition to VIPER and TATE sequences and the putative integrases mentioned above, we also included several sequences from different known groups of YR proteins. The phylogeny recovered was presented as unrooted, given the low resolution of the clades and the absence of a well-supported outgroup choice (Fig. 6). The relation of the Crypton group of DNA transposons with DIRS retroelements suggested by this tree had already been described [18], although we were not able to recover the monophyly of the different groups. This failure also occurred for VIPER and TATE sequences that were not recovered as reciprocally monophyletic groups. Interestingly, we can observe unpredicted grouping of YR elements with shufflon-specific DNA recombinase and some integrases, such as CRE (phage P1-like recombinases) and BJ1 (integrase from BJ1-like virus). Moreover, the YR tree suggests that those putative integrase sequences retrieved from NCBI after blastp searches likely correspond to some uncharacterized groups of YR proteins, since to the best of our knowledge, we have included representative sequences from the current characterized groups, and they did not cluster with any of them. In fact, these sequences constituted a single clade that seems to be closely related to some PAT-like and Ngaro-like elements.
Discussion
Influence of genome assemblies and TE detection methodology in the genomic analysis of VIPER and TATE
In this work, we used more than 40 Kinetoplastida genomes with different levels of assembly. Repetitive DNA sequences represent a challenge to the process of genome assembly, and information about these sequences is frequently incomplete. This problem can especially occur with next-generation sequencing approaches due to short reads [54, 55]. For Lep. pyrrhocoris, for example, despite the good quality of the assemblage at the chromosomal level, with 37X coverage [56], several TE copies were found near missing data regions. Another shortcoming of this kind of study is related to the incongruence between results performed with the same genome in different steps of data cleaning and assembly or with the same strain sequenced with distinct technologies. For example, the complete absence of VIPER evidenced in T. rangeli in this study contrasts with the remnants identified by other authors [38], which probably evaluated raw data. Moreover, in this study, we identified putative complete VIPER copies in the T. cruzi Dm28c strain that was sequenced using PacBio technology (GenBank assembly accession: GCA_002219105.2), which generates long reads and, consequently, better-quality assembly of the repetitive genome fraction [57]. When we previously analyzed the first available assembly for this strain (GenBank assembly accession: GCA_000496795.1 - sequenced by 454 technology), we were able to find encoding copies of VIPER but none with terminal repeat structure (data not shown). This finding suggests that we may be losing information about complete and putative encoding copies for other species. Nevertheless, despite these limitations, genomic data continue to be a valuable source for TE studies, being widely used to better understand the elements in the genomic contexts of different organisms [58–62], and in this report, we describe significant findings of VIPER and TATE elements.
Several programs and pipelines are now available to detect and annotate repetitive sequences, mainly using detection based on similarity, structure or repetitiveness [63]. These tools are crucial for large-scale approaches and for providing an overall view of the TE composition of genomes. Here, we study two TE families applying a less automated search based on similarity, though with a careful evaluation of copies nevertheless. The most challenging work was the definition of the boundaries of the elements. Thus, we also used a pipeline for the automatic identification of TEs in the two most relevant genomes (T. cruzi for VIPER and L. braziliensis for TATE) to confirm our manual evaluations. We did not observe advantages of using these pipelines to identify the boundaries of the elements, since even in these cases, we had to proceed with manual inspection of identified copies, and in most cases, they were predicted to be shorter than those we found.
VIPER and TATE: ancient residents and survivors of kinetoplastids genomes
We presented the first investigation of VIPER and TATE elements from a wide-scale phylogenetic perspective. Our in silico searches show a discontinuous distribution of both TEs in the host species, whose evolution was better understood with the phylogenetic analyses. In this study, we adopted the most parsimonious scenario according to the current data for all evolutionary inferences, implying that the future addition of new genome sequences or new species may lead to changes in assumptions.
Initially, Lorenzi et al. (2006) proposed that VIPER would have colonized the genomes of Trypanosoma species after the separation of this genus and Leishmania due to the absence of VIPER in the Leishmania species that were previously analyzed. Our data fully challenge this scenario because we found potentially encoding copies or remnants of VIPER not only in B. saltans [as already reported by Jackson et al. (2016)] but also in species of the Leishmaniinae subfamily. Although the presence of these elements in B. saltans could be indicative of horizontal transfer, sequence divergences support vertical transmission. In fact, this pattern of evolution seems to be shared for several gene families where B. saltans appears to have retained many genes that were present in its ancestor with the trypanosomatids and that ended up being lost discontinuously in some specific groups throughout the evolution of trypanosomatids [64, 65].
Due to the divergence found among VIPER and TATE copies within the same species and the pattern of separation of copies into distinct clades, we can suggest the existence of numerous cases of ancestral polymorphism. However, the topology of the tree indicates that most of the VIPER and TATE polymorphisms arose after the separation of trypanosomatids with the ancestor of B. saltans. Despite the inconclusions in some points of the phylogeny, we can suggest several independent loss events of the elements along with the trypanosomatid evolution. The absence of significant hits supports the complete degeneration of TATE in most Trypanosoma species and VIPER in most Leishmania species, which can be taken with some confidence once several genomes have an adequate assembly level.
In this study, for the first time, we described potentially complete and encoding copies of VIPER in the T. cruzi Dm28c strain, including terminal repeats. Only degenerate copies were found in the T. cruzi CL Brener genome in this and in other works [25–27]. We cannot conclude whether the differences between T. cruzi strains are due to genome assembly problems or due to genetic differences between strains. In the course of writing, another T. cruzi Dm28c PacBio assembly was published by Berná et al. (2018) [66]. These researchers found approximately 200 copies of VIPER that were considered defective. In a brief search of the potentially encoding VIPER identified in our work against this genome, we were able to find matches with 100% identity, indicating that the assignment of T. cruzi Dm28c VIPER copies as defective by these authors was probably due to differences in interpretation. Additionally, these authors did not identify the terminal repeat regions, and the copies were considered to have 3.423 bp, considerably smaller than the complete copies that we found. Although the DNA_BRE_C domain was predicted only with less significant e-values (CD-search e-value of approximately 0.1), the composition and conservation of ORFs and the presence of putative SDRs suggest that these T. cruzi Dm28c VIPER copies are potentially active. RNA-Seq reads mapping VIPER were found for T. cruzi Dm28c during epimastigote growth in vitro [67]. However, we need to be cautious concluding TE activity in trypanosomatids based only on this type of data, since those reads could be only a transitory molecule resulting from global RNA expression. On the other hand, peptides from Gag-like protein were detected during the differentiation process from T. cruzi Dm28c noninfective epimastigotes into infective metacyclic trypomastigotes [68], a stronger sign of possible activity. In addition, we found potential encoding VIPER copies in some other species.
We also obtained significant results for the TATE element, such as the terminal repeat pattern in L. braziliensis and the presence of potentially encoding copies in some other species. The L. braziliensis genome presents good coverage and assembly (Peacock et al. 2007), but the repetitive nature of TATE and its disposition in tandem made the analysis more difficult. The complete copy that we found in this genome contains a frameshift at the end of the third ORF; however, we believe that additional full and potentially active copies could be found in an improved genome assembly. Assembling issues may also have hindered better characterization in other species. Recent analysis of differential expression across the life cycle stages of L. braziliensis reveals that TATE RNAs are overrepresented in amastigote and metacyclic forms [69]. TATE sequences also constituted 38% of small interfering RNAs (siRNAs) associated with L. braziliensis Argonaute1 protein [70]. The presence of siRNA from TATE in this species suggests that an endogenous RNAi pathway could be responsible for controlling the transposition of this element [70, 71].
Considering the putative complete VIPER and TATE copies, we encountered some terminal repeats sharing 100% identity. For Ngaro-like and PAT-like elements, a model of reverse transcription was proposed including steps of annealing between portions of the repeats [23, 24]. This model implies that repeat sequences A1 and A2 and B1 and B2 should be at least nearly identical in the time of insertion. If VIPER and TATE possess a similar reverse transcription process, the conservation of repeats is also an indication of recent activity.
In conclusion, the presence of VIPER and TATE in B. saltans suggests that the two elements were already present in the last common ancestor of the studied species more than 400 mya and have remained active in some species. This “survival” for such an extended period raises questions about the coevolution of these sequences with their host genomes, such as the way that these sequences have escaped from inactivation and how the genomes control these sequences. Alternatively, these elements could have experienced reactivation events. Furthermore, with so many millions of years of coevolution, it is natural to think about the impact of these elements on the evolution of species and how TEs may have contributed to the organization and functionality of the kinetoplastid genomes.
Putative modular evolution of YR retroelements
Analysis of RT, RH and YR domains was performed separately, which is important because we already had suggestions of incongruent relationships among the proteins [26] and because it is known that retrotransposons and retroviruses have a modular evolution profile with recurrent events of domain rearrangements or even a reticulated origin of its components [72–74]. The ability to recover the evolutionary history of these sequences depends on the time in which these elements arose and the selective pressures to which they were exposed. These details have a direct influence on the quality and reliability of sequence alignments. According to our previous assumption, we observed no resolution for some clades, possibly due to the lack of phylogenetic signal that may reflect very ancient separation events, possibly before the divergence of the major eukaryotic groups. Thus, it is quite likely that most of the incongruent results found in this study in the comparisons between topologies obtained for different domains are an outcome of a weak phylogenetic signal. This effect seems to be the case in, for example, the recurrent polytomy detected for DIRS-like and PAT-like and by the absence of monophyly of VIPER-like sequences in the RH and YR phylogenies. Despite this finding, we showed here for the first time that VIPER and TATE elements form a monophyletic group in the RT phylogeny, suggesting a common origin of both TEs. This result questions the recent findings of Pita et al. (2019), which suggested TATE as a distinct group from other DIRS elements based on an RT tree [10]. Nevertheless, it is not clear if authors have used VIPER sequences in their phylogeny.
Although the monophyly of VIPER-like could be confidently assigned by the most conserved of the evaluated domains (RT), the origin of these domains and their relationship with other retrotransposons were not clearly established. In fact, considering the three evaluated domains, there are no significant results supporting either independent or common origins for different YR retroelements. In this sense, YR-element groups were all placed at the same level in a highly polytomous RT phylogeny. In the RH phylogeny, YR-elements grouped with Gypsy-caulimovirus and Hepadnavirus, and in the YR phylogeny, they grouped with Crypton, the uncharacterized YRs and some other integrase/recombinase sequences. In the last case, Crypton revealed polyphyletic, which was already expected, since previous phylogenetic evaluations recovered four groups with weakly supported relationships [75]. Poulter and Butler (2015) suggested that the origin of YR retrotransposons possibly occurred many times, resulting in three groups: DIRS-like, PAT-like and Ngaro. The independent origin hypothesis was also proposed by Goodwin and Poulter (2004) and Lorenzi et al. (2006). In this study, we were not able to find strong evidence of independent origins for the different YR elements; therefore, our results cannot rule out a scenario of common ancient origin.
The contrasting clustering of YR elements among RT, RH and YR suggests that they could share a modular evolutionary pattern with distinct sources for the different domains. In this sense, whereas the RT-domain may share common origins with LTR-retrotransposons Gypsy (as evidenced more clearly by other authors [18, 26]), the RH-domain evidenced a close relationship with Hepadnavirus. Moreover, the YR domain of YR elements may stem from Crypton or some integrase/recombinase sequences (or even may have given rise to them). Such a modular evolution pattern was already proposed for YR retroelements by Poulter and Butler (2015), which suggested that these elements had an origin from a combination of Crypton DNA elements and an RT/RH from a Gypsy LTR-retrotransposon. Although our data do not disagree with this proposal in its total, we can suggest alternative scenarios, such as the origin of their YR directly from some prokaryotic or viral integrases/recombinases. Similarly, the reverse relation, where viral and prokaryotic YR sequences originate from a YR element, could also have occurred. More in-depth studies of homologous sequences to YR retroelements and other retrotransposons will possibly help to refine this scenario.
Another intriguing relationship recovered by this study is that between the RH domain of VIPER-like TEs and hepadnaviruses. In fact, the phylogeny revealed that VIPER-like RH sequences are more closely related to hepadnavirus than to the other YR retroelements. This finding may suggest that RH sequences suffered additional rearrangements along the evolutionary history of YR retroelements. According to Lauber et al. (2017), hepadnavirus and nackedvirus (a nonenveloped HBV-related family of fish viruses [76]) separated more than 400 mya before the rise of tetrapods. This evolutionary period coincides with the minimum time of origin of VIPER and TATE elements, suggesting a possible shared source of the RH domain of VIPER-like and the ancestor of hepadnavirus and nackednavirus.
In general, as we did not find clear evidence of independent origins of VIPER-like from the other YR retroelements, we support the maintenance of the DIRS group of retrotransposons. Based on the scenario presented in this study and others, we propose dividing the YR-containing retroelements into four groups, which were named according to the first described element (these names were also the ones chosen by other authors) followed by the indication “like”: DIRS-like, PAT -like, Ngaro-like and VIPER-like. This proposition complements other works that mention the classification of these elements [18, 29, 77]. PAT-like, Ngaro-like, and VIPER-like elements show a similar terminal repeat pattern, whereas DIRS-like elements have important repeat structure differences [18] and consequently differ in the mechanism of reverse transcription [22, 23]. Since other studies revealed that PAT-like and DIRS-like are phylogenetically closely related [17, 20, 24], their separation is mainly due to the structural difference of the terminal repeats [18]. According to the classification of Wicker et al. (2007), the subgroups correspond to superfamilies from the order DIRS. Although the order DIRS is not demonstrated to be monophyletic, this classification helps to refer to YR-containing retrotransposons.
Conclusions
This report presents a comprehensive description of the structure, distribution, and evolution of VIPER and TATE retrotransposons in trypanosomatid species, suggesting that these elements are notably ancient components of those genomes and have survived and remained active in some species or were reactivated during host species evolution. We also investigated the relationship of these elements with other retrotransposons. Our data do not support independent origins for different YR retroelements but suggest the occurrence of ancient exchanges of domains. We also proposed to separate the elements of the order DIRS into four subgroups according to phylogenetic findings and sequence structure: DIRS-like, PAT-like, Ngaro-like, and VIPER-like.
Methods
Genomes and in silico searches
The genomes were retrieved from TriTrypDB [78], or NCBI [79], and their information is available in Additional file 7. Local tblastn searches were performed against each genome using the amino acid sequences of the three proteins of VIPER and TATE, whose canonical sequences were obtained from Repbase database [80]. Hits were considered significant when their respective e-values were lower than e− 10. To confirm the identity of sequences, we used CENSOR software [81], a tool that screens query sequences against the Repbase database.
Characterization of copies
All putative VIPER and TATE copies were retrieved together with 8 kb of flanking regions (whenever available) using an in-house Perl script. Redundancy was further eliminated using the CD-HIT-est tool [82] and through manual review. Up to fifteen of the most conserved copies of each species were carefully analyzed by individual manual inspection. The analyzed copies of VIPER and TATE are provided in Additional files 8 and 9, respectively. The presence of ORFs was analyzed using the NCBI ORFfinder tool. NCBI CD-search [83] was used to check for domains in the predicted protein sequences. RT/RH and YR ORFs were identified mainly by their similarity to VIPER and TATE canonical proteins or by the predicted domains. ORF 1 or gag-like was predicted primarily by its proximity or overlap with ORF 2 and by the lack of orthologues in other species. Copies were considered potentially encoding when they presented three ORFs encoding proteins with significant size (> 300 aa for gag-like, > 250 aa for YR and > 600 aa for RT/RH). Putative complete copies of VIPER and TATE possessing repeats are provided in Additional files 10 and 11, respectively.
VIPER and TATE DNA sequences were aligned with MUSCLE program [84], and the alignments were visually inspected to identify the limits of the retrotransposon copies. To verify the presence of terminal repeat structures, the potentially complete copies were analyzed by blastn with the parameters “align two or more sequences” and “somewhat similar sequences (blastn)” using the same sequence as query and subject.
To evaluate our analysis strategy, mainly avoiding erroneous determination of element limits, we also used an automated pipeline to search for VIPER in T. cruzi Dm28c and TATE in L. braziliensis MHOM/BR/75/M2904. We used the detection TE programs (based on assembled genomes) from PiRATE, as automated in Galaxy platform [63, 85].
To detect VIPER activity in T. cruzi Dm28c, we analyzed the presence of RNA-seq reads and peptides from VIPER in results of published transcriptomic [67] and proteomic studies of this strain [68]. As most VIPER copies are annotated as hypothetical proteins, we retrieved all TriTrypDB gene IDs and UniProt IDs from VIPER copies and searched for them in the tables containing all genes and proteins identified in the studies using in-house Python code.
Phylogenetic analyses of TATE and VIPER
To understand the evolutionary history of VIPER and TATE, we inferred the phylogeny of RT/RH and YR amino acid sequences separately. All ORFs presenting significant sizes were included, and the sequence DrNgaro (AAN71721.1) from Danio rerio was used as an outgroup. Amino-acid sequences were aligned with the MUSCLE program, as available in MEGA 7 [96]. For RT/RH, only the regions encompassing domains 1–7 [52] and domains D10 to D134 [74], respectively, were trimmed from the original sequences, whereas for YR the region encompassing boxes 1 and 2 [26], added to the intervening sequence was evaluated. The alignments are available in the Additional files 12 and 13.
Phylogenetic analyses were then performed for each of these datasets using Bayesian Analysis (BA) in MrBayes 3.2.6 [86] under a mixed amino acid model. The Markov Chain Monte Carlo (MCMC) of the BA was run for 10,000,000 generations, sampling trees every 1000 generations, and burning 25% of the initial results. Additionally, a maximum likelihood (ML) analysis was performed under the rapid bootstrap algorithm, which also searches for best-scoring ML trees. This bootstrap analysis was conducted for 1000 replicates using RAxML-HPC2 implemented on CIPRES [87] following the amino acid substitution model with the lowest Bayesian Information Criterion (BIC), as measured in ModelTest-NG (Darriba et al. 2019) (LG + G4 + F and WAG+G4 for RT/RH and YR, respectively). Phylogenetic trees resulting from BA and ML searches were visualized and edited in FigTree 1.4.2 [88] and Evolview tool, respectively.
Evolution of DIRS elements
Looking for VIPER- and TATE-related sequences, online blastp searches were performed on NCBI against the nonredundant protein sequences (nr) database using the canonical sequences of VIPER and TATE RT/RH and YR proteins as queries. Sequences with significant e-values (e < 10− 5) were included in the phylogenies. Additionally, for YR, other related sequences were retrieved by blastP searches and included in the phylogeny. Full information about these searches is available in Additional file 6.
To contemplate the diversity of YR-containing TEs in the phylogenies, we analyzed all TEs assigned as autonomous DIRS in the Repbase (note that it includes all YR-containing elements). A total of 413 sequences were retrieved and analyzed for the presence of ORFs and domains. Of those sequences, 128 presented the three expected conserved domains (RT-, RH-, and YR-related domains) and were selected. We then filtered this dataset, eliminating monophyletic sequences from the same species with less than 35% divergence. This preliminary analysis was performed using Neighbor-Joining under the Poisson correction method, as performed in MEGA 7 [89]. Approximately 30 sequences remained in the dataset after this initial filtering.
The RT and RH datasets were further complemented by the inclusion of representative sequences from each of the following related groups: LINE, Gypsy, Copia, Bel-Pao, hepadnavirus, caulimovirus, and retrovirus (see Additional file 14). For RH, it was also possible to include eukarya sequences. For YR, we added the sequences found in kinetoplastids, the related proteins found by blastp searches and several known related proteins representatives of distinct groups {Flippase, Phage integrase, Resolvase (XerC and XerD) and Prokaryotic Transposase [also used by Lorenzi et al. (2006)]}. We also included one representative sequence of each of the major clades recovered by Wang et al. (2018) [90] (see Additional file 15).
For the RT-related and RNA-directed RNA polymerase sequences, the region encompassing domains 1–7 [52] was first trimmed from the original sequences and then subjected to two multiple progressive alignments approaches: 1) the first performed with ClustalW under the BLOSUM protein weight matrix, individually for each major sampling group (DIRS/Pat/Ngaro, VIPER/TATE, LINE, Gypsy, Copia, Bel-Pao, hepadnavirus, caulimovirus, retrovirus); 2) the second performed with MAFFT [91] for the entire set of sequences, employing the E-INS-i iterative refinement method with the help of DASH (Database of Aligned Structural Homologs) homologous structures. In both steps, alignment was checked regarding homology assignments within each of the seven RT domains, and variable regions between domains were withdrawn, whereas gaps were filled with “X”.
For RH, two alignment approaches were also performed using ClustalW and Muscle after an initial trimming of the region encompassing domains D10 to D134 [74]. For YR, boxes 1 and 2 [26] were first located, and the surrounding N-terminus and C-terminus regions of both domains were deleted. The alignment was then performed with the same criteria described above using ClustalW and Muscle but with a distinct grouping strategy at the first step [DIRS/Pat/Ngaro, VIPER/TATE, flippase, phage integrase, resolvase (XerC and XerD) and prokaryotic transposase].
Phylogenetic analyses were performed under BA and ML, as described above. Whereas mixed models were employed for BA analyses, ML analyses were performed under individual amino acid substitution models selected in ModelTest-NG (LG + G4, RTREV+G4 + F and LG + G4, for RH, RT and YR, respectively). RT analyses were rooted using the non-LTR LINE clade (ingi, Jockey, bilbo, Lian, TRAS1 and SLACS) as an outgroup, as previously supported [52], whereas RH phylogenies were rooted by eukarya sequences, following Malik and Eickbush (2001). For YR, the phylogenies were presented as unrooted phylograms. All the alignments are provided in Additional files 16, 17 and 18.
Additional files
Table. The number of significant hits (e-value cut-off of 10− 10) found on tblastn searches for VIPER and TATE proteins against each genome. (PDF 20 kb)
Table. Summary of the analyzes of the 15 most conserved copies of VIPER and TATE elements in each species. (XLSX 98 kb)
Table. Summary of main features of potentially encoding VIPER copies. (PDF 72 kb)
Additional information. Domains predicted for VIPER and TATE proteins (PDF 87 kb)
Table. Summary of main features of potentially encoding TATE copies (PDF 71 kb)
Additional information. Evaluation of VIPER and TATE similar sequences outside Kinetoplastida. (PDF 147 kb)
Table. List of genomes used for in silico searches. (PDF 25 kb)
Sequences. VIPER copies analyzed in all species are provided together with the flanking region (TXT 2890 kb)
Sequences. TATE copies analyzed in all species are provided together with the flanking region (TXT 2700 kb)
Sequences. Putative complete VIPER copies (TXT 52 kb)
Sequences. Putative complete TATE copies (TXT 31 kb)
Alignments. Alignment of RT/RH sequences of VIPER and TATE (FAS 30 kb)
Alignments. Alignment of YR sequences of VIPER and TATE (FAS 15 kb)
Table. List of sequences used for RT phylogeny (PDF 29 kb)
Table. List of sequences used for YR phylogeny (PDF 29 kb)
Alignments. Alignment of RT sequences of different groups of retroelements (FAS 24 kb)
Alignments . Alignment of RH sequences of different groups of retroelements (FAS 17 kb)
Alignments . Alignment of YR sequences of DIRS retrotransposons (DIRS-like, PAT-like, Ngaro-like, and VIPER-like) along with other YR family proteins (FAS 23 kb)
Acknowledgments
To Dr. S Beverley and The Genome Institute, Washington University School of Medicine, and all the EuPathDB (Eukaryotic Pathogen Genomics Resource) research communities who have contributed for prepublication access of the trypanosomatid genome sequences. To Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the student’s fellowships.
Authors’ contributions
AL conceived the study; YCR and AL performed the BLAST and analysis of copies. DSV, CMBS, and ALKL helped in the analysis of TE copies and writing. LJR performed the phylogenetic analyses. YCR, LJR and AL analyzed the results and wrote the manuscript. MAK assisted in interpretation of the results and manuscript writing. All authors read and approved the final manuscript.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-453812/2014–7).
Availability of data and materials
Genomic coordinates of VIPER and TATE copies analyzed here are available in the Additional file 2, and the sequences are available in Additional files 8 and 9.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yasmin Carla Ribeiro and Lizandra Jaqueline Robe contributed equally to this work.
Contributor Information
Yasmin Carla Ribeiro, Email: yacrib@gmail.com.
Lizandra Jaqueline Robe, Email: lizbiogen@gmail.com.
Danila Syriani Veluza, Email: danilaveluza@gmail.com.
Cyndia Mara Bezerra dos Santos, Email: cyndiamara@gmail.com.
Ana Luisa Kalb Lopes, Email: anakalblopes@gmail.com.
Marco Aurélio Krieger, Email: mkrieger@fiocruz.br.
Adriana Ludwig, Email: adriana.ludwig@pq.cnpq.br, Email: adriludwig@gmail.com.
References
- 1.Lukes J, Skalický T, Týč J, Votýpka J, Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol. 2014;195:115–122. doi: 10.1016/j.molbiopara.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Simpson AGB, Stevens JR, Lukeš J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006;22:168–174. doi: 10.1016/j.pt.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Fernandes AP, Nelson K, Beverley SM. Evolution of nuclear ribosomal RNAs in kinetoplastid protozoa: perspectives on the age and origins of parasitism. Proc Natl Acad Sci U S A. 1993;90:11608–11612. doi: 10.1073/pnas.90.24.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussbaum K, Honek J, Cadmus CM, Efferth T. Trypanosomatid parasites causing neglected diseases. Curr Med Chem. 2010;17:1594–1617. doi: 10.2174/092986710790979953. [DOI] [PubMed] [Google Scholar]
- 5.Campbell DA, Thomas S, Sturm NR. Transcription in kinetoplastid protozoa: why be normal? Microbes Infect. 2003;5:1231–1240. doi: 10.1016/j.micinf.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Kramer S. Developmental regulation of gene expression in the absence of transcriptional control: the case of kinetoplastids. Mol Biochem Parasitol. 2012;181:61–72. doi: 10.1016/j.molbiopara.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 8.El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran A-N, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 9.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pita S, Díaz-Viraqué F, Iraola G, Robello C. The Tritryps comparative Repeatome: insights on repetitive element evolution in Trypanosomatid pathogens. Genome Biol Evol. 2019;11:546–551. doi: 10.1093/gbe/evz017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein SJ, O’Neill RJ. Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosom Res. 2018;26:5–23. doi: 10.1007/s10577-017-9569-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kazazian HH. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 13.Casola C, Betrán E. The genomic impact of gene Retrocopies: what have we learned from comparative genomics, population genomics, and transcriptomic analyses? Genome Biol Evol. 2017;9:1351–1373. doi: 10.1093/gbe/evx081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Waminal NE, Choi H-I, Perumal S, Lee S-C, Nguyen VB, et al. Rapid amplification of four retrotransposon families promoted speciation and genome size expansion in the genus Panax. Sci Rep. 2017;7:9045. doi: 10.1038/s41598-017-08194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Havecker ER, Gao X, Voytas DF. The diversity of LTR retrotransposons. Genome Biol. 2004;5:225. doi: 10.1186/gb-2004-5-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eickbush TH, Jamburuthugoda VK. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 2008;134:221–234. doi: 10.1016/j.virusres.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin TJ, Poulter RT. The DIRS1 group of retrotransposons. Mol Biol Evol. 2001;18:2067–2082. doi: 10.1093/oxfordjournals.molbev.a003748. [DOI] [PubMed] [Google Scholar]
- 18.Poulter RTM, Butler MI. Tyrosine Recombinase Retrotransposons and Transposons. Microbiol Spectr. 2015;3:MDNA3–0036-2014. doi: 10.1128/microbiolspec.MDNA3-0036-2014. [DOI] [PubMed] [Google Scholar]
- 19.Piednoël M, Gonçalves IR, Higuet D, Bonnivard E. Eukaryote DIRS1-like retrotransposons: an overview. BMC Genomics. BioMed Central Ltd. 2011;12:621. doi: 10.1186/1471-2164-12-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodwin TJD, Poulter RTM, Lorenzen MD, Beeman RW. DIRS retroelements in arthropods: identification of the recently active TcDirs1 element in the red flour beetle Tribolium castaneum. Mol Gen Genomics. 2004;272:47–56. doi: 10.1007/s00438-004-1028-2. [DOI] [PubMed] [Google Scholar]
- 21.Muszewska A, Steczkiewicz K, Ginalski K. DIRS and Ngaro retrotransposons in fungi. PLoS One. 2013;8:e76319. doi: 10.1371/journal.pone.0076319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cappello J, Handelsman K, Lodish HF. Sequence of Dictyostelium DIRS-1: an apparent retrotransposon with inverted terminal repeats and an internal circle junction sequence. Cell. 1985;43:105–115. doi: 10.1016/0092-8674(85)90016-9. [DOI] [PubMed] [Google Scholar]
- 23.Goodwin TJD, Poulter RTM. A new group of tyrosine recombinase-encoding retrotransposons. Mol Biol Evol. 2004;21:746–759. doi: 10.1093/molbev/msh072. [DOI] [PubMed] [Google Scholar]
- 24.Poulter RTM, Goodwin TJD. DIRS-1 and the other tyrosine recombinase retrotransposons. Cytogenet Genome Res. 2005;110:575–588. doi: 10.1159/000084991. [DOI] [PubMed] [Google Scholar]
- 25.Vazquez M, Ben-Dov C, Lorenzi H, Moore T, Schijman A, Levin MJ. The short interspersed repetitive element of Trypanosoma cruzi, SIRE, is part of VIPER, an unusual retroelement related to long terminal repeat retrotransposons. Proc Natl Acad Sci U S A. 2000;97:2128–2133. doi: 10.1073/pnas.050578397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorenzi HA, Robledo G, Levin MJ. The VIPER elements of trypanosomes constitute a novel group of tyrosine recombinase-enconding retrotransposons. Mol Biochem Parasitol. 2006;145:184–194. doi: 10.1016/j.molbiopara.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Bringaud F, Ghedin E, El-Sayed NM, Papadopoulou B. Role of transposable elements in trypanosomatids. Microbes Infect. 2008;10:575–581. doi: 10.1016/j.micinf.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 30.Cortez AP, Ventura RM, Rodrigues AC, Batista JS, Paiva F, Añez N, et al. The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology. 2006;133:159–169. doi: 10.1017/S0031182006000254. [DOI] [PubMed] [Google Scholar]
- 31.Du Y, Maslov DA, Chang KP. Monophyletic origin of beta-division proteobacterial endosymbionts and their coevolution with insect trypanosomatid protozoa Blastocrithidia culicis and Crithidia spp. Proc Natl Acad Sci U S A. 1994;91:8437–8441. doi: 10.1073/pnas.91.18.8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flegontov P, Votýpka J, Skalický T, Logacheva MD, Penin AA, Tanifuji G, et al. Paratrypanosoma is a novel early-branching trypanosomatid. Curr Biol. 2013;23:1787–1793. doi: 10.1016/j.cub.2013.07.045. [DOI] [PubMed] [Google Scholar]
- 33.Harkins KM, Schwartz RS, Cartwright RA, Stone AC. Phylogenomic reconstruction supports supercontinent origins for Leishmania. Infect Genet Evol. 2016;38:101–109. doi: 10.1016/j.meegid.2015.11.030. [DOI] [PubMed] [Google Scholar]
- 34.Kato H, Cáceres AG, Hashiguchi Y. First Evidence of a Hybrid of Leishmania (Viannia) braziliensis/L. (V.) peruviana DNA Detected from the Phlebotomine Sand Fly Lutzomyia tejadai in Peru. PLoS Negl Trop Dis. 2016;10:e0004336. doi: 10.1371/journal.pntd.0004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly S, Ivens A, Manna PT, Gibson W, Field MC. A draft genome for the African crocodilian trypanosome Trypanosoma grayi. Sci Data. 2014;1:140024. doi: 10.1038/sdata.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Runckel C, DeRisi J, Flenniken ML. A draft genome of the honey bee trypanosomatid parasite Crithidia mellificae. PLoS One. 2014;9:e95057. doi: 10.1371/journal.pone.0095057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarz RS, Bauchan GR, Murphy CA, Ravoet J, de Graaf DC, Evans JD. Characterization of two species of Trypanosomatidae from the honey bee Apis mellifera : Crithidia mellificae Langridge and McGhee, and Lotmaria passim n. gen., n. sp. J Eukaryot Microbiol. 2015;62:567–583. doi: 10.1111/jeu.12209. [DOI] [PubMed] [Google Scholar]
- 38.Stoco PH, Wagner G, Talavera-Lopez C, Gerber A, Zaha A, Thompson CE, et al. Genome of the Avirulent human-infective trypanosome—Trypanosoma rangeli. PLoS Negl Trop Dis. 2014;8:e3176. doi: 10.1371/journal.pntd.0003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teixeira MMG, Borghesan TC, Ferreira RC, Santos MA, Takata CSA, Campaner M, et al. Phylogenetic validation of the genera Angomonas and Strigomonas of Trypanosomatids harboring bacterial endosymbionts with the description of new species of Trypanosomatids and of Proteobacterial symbionts. Protist. 2011;162:503–524. doi: 10.1016/j.protis.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Valdivia HO, Reis-Cunha JL, Rodrigues-Luiz GF, Baptista RP, Baldeviano GC, Gerbasi RV, et al. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics. 2015;16:715. doi: 10.1186/s12864-015-1928-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 2015;5:8550. doi: 10.1038/srep08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ludwig A, Krieger MA. Genomic and phylogenetic evidence of VIPER retrotransposon domestication in trypanosomatids. Mem Inst Oswaldo Cruz Fundação Oswaldo Cruz. 2016;111:765–769. doi: 10.1590/0074-02760160224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vázquez M, Lorenzi H, Schijman AG, Ben-Dov C, Levin MJ. Analysis of the distribution of SIRE in the nuclear genome of Trypanosoma cruzi. Gene. 1999;239:207–216. doi: 10.1016/S0378-1119(99)00387-X. [DOI] [PubMed] [Google Scholar]
- 44.Katz RA, Skalka AM. The retroviral enzymes. Annu Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 45.Boeke JD, Corces VG. Transcription and reverse transcription of retrotransposons. Annu Rev Microbiol. 1989;43:403–434. doi: 10.1146/annurev.mi.43.100189.002155. [DOI] [PubMed] [Google Scholar]
- 46.Rein A, Harvin DP, Mirro J, Ernst SM, Gorelick RJ. Evidence that a central domain of nucleocapsid protein is required for RNA packaging in murine leukemia virus. J Virol. 1994;68:6124–6129. doi: 10.1128/jvi.68.9.6124-6129.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Majorek KA, Dunin-Horkawicz S, Steczkiewicz K, Muszewska A, Nowotny M, Ginalski K, et al. The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014;42:4160–4179. doi: 10.1093/nar/gkt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludwig A, Valente VLS, Loreto ELS. Multiple invasions of Errantivirus in the genus drosophila. Insect Mol Biol. 2008;17:113–124. doi: 10.1111/j.1365-2583.2007.00787.x. [DOI] [PubMed] [Google Scholar]
- 49.Wallau GL, Ortiz MF, Loreto ELS. Horizontal transposon transfer in eukarya: detection, bias, and perspectives. Genome Biol Evol. 2012;4:689–699. doi: 10.1093/gbe/evs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silva JC, Loreto EL, Clark JB. Factors that affect the horizontal transfer of transposable elements. Curr Issues Mol Biol. 2004;6:57–71. [PubMed] [Google Scholar]
- 51.Simpson AGB, Gill EE, Callahan HA, Litaker RW, Roger AJ. Early evolution within kinetoplastids (euglenozoa), and the late emergence of trypanosomatids. Protist. 2004;155:407–422. doi: 10.1078/1434461042650389. [DOI] [PubMed] [Google Scholar]
- 52.Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990;9:3353–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Bustos A, Cuadrado A, Jouve N. Sequencing of long stretches of repetitive DNA. Sci Rep. 2016;6:36665. doi: 10.1038/srep36665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flegontov P, Butenko A, Firsov S, Kraeva N, Eliáš M, Field MC, et al. Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania. Sci Rep. 2016;6:23704. doi: 10.1038/srep23704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakano K, Shiroma A, Shimoji M, Tamotsu H, Ashimine N, Ohki S, et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum Cell. 2017;30:149–161. doi: 10.1007/s13577-017-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dufresne M, Lespinet O, Daboussi M-J, Hua-Van A. Genome-wide comparative analysis of pogo-like transposable elements in different Fusarium species. J Mol Evol. 2011;73:230–243. doi: 10.1007/s00239-011-9472-1. [DOI] [PubMed] [Google Scholar]
- 59.de la Chaux N, Wagner A. BEL/Pao retrotransposons in metazoan genomes. BMC Evol Biol. 2011;11:154. doi: 10.1186/1471-2148-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keidar D, Doron C, Kashkush K. Genome-wide analysis of a recently active retrotransposon, au SINE, in wheat: content, distribution within subgenomes and chromosomes, and gene associations. Plant Cell Rep. 2018;37:193–208. doi: 10.1007/s00299-017-2213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guyot R, Darré T, Dupeyron M, de Kochko A, Hamon S, Couturon E, et al. Partial sequencing reveals the transposable element composition of Coffea genomes and provides evidence for distinct evolutionary stories. Mol Gen Genomics. 2016;291:1979–1990. doi: 10.1007/s00438-016-1235-7. [DOI] [PubMed] [Google Scholar]
- 62.Slotkin RK. The case for not masking away repetitive DNA. Mob DNA. 2018;9:15. doi: 10.1186/s13100-018-0120-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berthelier J, Casse N, Daccord N, Jamilloux V, Saint-Jean B, Carrier G. A transposable element annotation pipeline and expression analysis reveal potentially active elements in the microalga Tisochrysis lutea. BMC Genomics. 2018;19:378. doi: 10.1186/s12864-018-4763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson AP, Otto TD, Aslett M, Armstrong SD, Bringaud F, Schlacht A, et al. Kinetoplastid Phylogenomics reveals the evolutionary innovations associated with the origins of parasitism. Curr Biol. 2016;26:161–172. doi: 10.1016/j.cub.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Souza DAS, Pavoni DP, Krieger MA, Ludwig A. Evolutionary analyses of myosin genes in trypanosomatids show a history of expansion, secondary losses and neofunctionalization. Sci Rep. 2018;8:1376. doi: 10.1038/s41598-017-18865-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berná L, Rodriguez M, Chiribao ML, Parodi-Talice A, Pita S, Rijo G, et al. Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microb genomics. 2018;4. [DOI] [PMC free article] [PubMed]
- 67.Santos CMB, Ludwig A, Kessler RL, Rampazzo RCP, Inoue AH, Krieger MA, et al. Trypanosoma cruzi transcriptome during axenic epimastigote growth curve. Mem Inst Oswaldo Cruz. 2018;113:e170404. [DOI] [PMC free article] [PubMed]
- 68.Amorim JC, Batista M, da Cunha ES, Lucena ACR, de Lima CVP, Sousa K, et al. Quantitative proteome and phosphoproteome analyses highlight the adherent population during Trypanosoma cruzi metacyclogenesis. Sci Rep. 2017;7:9899. doi: 10.1038/s41598-017-10292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruy PDC, Monteiro-Teles NM, Miserani Magalhães RD, Freitas-Castro F, Dias L, Aquino Defina TP, et al. Comparative transcriptomics in Leishmania braziliensis: disclosing differential gene expression of coding and putative noncoding RNAs across developmental stages. RNA Biol. 2019:1–22. [DOI] [PMC free article] [PubMed]
- 70.Atayde VD, Shi H, Franklin JB, Carriero N, Notton T, Lye L-F, et al. The structure and repertoire of small interfering RNAs in Leishmania (Viannia) braziliensis reveal diversification in the trypanosomatid RNAi pathway. Mol Microbiol. 2013;87:580–593. doi: 10.1111/mmi.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lye L-F, Owens K, Shi H, Murta SMF, Vieira AC, Turco SJ, et al. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010;6:e1001161. doi: 10.1371/journal.ppat.1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malik HS, Henikoff S, Eickbush TH. Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res. 2000;10:1307–1318. doi: 10.1101/gr.145000. [DOI] [PubMed] [Google Scholar]
- 73.Terzian C, Pélisson A, Bucheton A. Evolution and phylogeny of insect endogenous retroviruses. BMC Evol Biol. 2001;1:3. doi: 10.1186/1471-2148-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malik HS, Eickbush TH. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001;11:1187–1197. doi: 10.1101/gr.185101. [DOI] [PubMed] [Google Scholar]
- 75.Kojima KK, Jurka J. Crypton transposons: identification of new diverse families and ancient domestication events. Mob DNA. 2011;2:12. doi: 10.1186/1759-8753-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lauber C, Seitz S, Mattei S, Suh A, Beck J, Herstein J, et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 2017;22:387–399.e6. doi: 10.1016/j.chom.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9:411–412. doi: 10.1038/nrg2165-c1. [DOI] [PubMed] [Google Scholar]
- 78.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2009;38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2012;40:D13–D25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jurka J, Kapitonov VVV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 81.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marchler-Bauer A, Bryant SH. CD-search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gatew Comput Environ Work GCE 2010. IEEE; 2010. p. 1–8.
- 88.Rambaut A. FigTree [Internet]. 2008. Available from: https://github.com/rambaut/figtree/releases
- 89.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang J, Liu Y, Liu Y, Du K, Xu S, Wang Y, et al. A novel family of tyrosine integrases encoded by the temperate pleolipovirus SNJ2. Nucleic Acids Res. 2018;46:2521–2536. doi: 10.1093/nar/gky005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47(W1):W5–W10. doi: 10.1093/nar/gkz342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table. The number of significant hits (e-value cut-off of 10− 10) found on tblastn searches for VIPER and TATE proteins against each genome. (PDF 20 kb)
Table. Summary of the analyzes of the 15 most conserved copies of VIPER and TATE elements in each species. (XLSX 98 kb)
Table. Summary of main features of potentially encoding VIPER copies. (PDF 72 kb)
Additional information. Domains predicted for VIPER and TATE proteins (PDF 87 kb)
Table. Summary of main features of potentially encoding TATE copies (PDF 71 kb)
Additional information. Evaluation of VIPER and TATE similar sequences outside Kinetoplastida. (PDF 147 kb)
Table. List of genomes used for in silico searches. (PDF 25 kb)
Sequences. VIPER copies analyzed in all species are provided together with the flanking region (TXT 2890 kb)
Sequences. TATE copies analyzed in all species are provided together with the flanking region (TXT 2700 kb)
Sequences. Putative complete VIPER copies (TXT 52 kb)
Sequences. Putative complete TATE copies (TXT 31 kb)
Alignments. Alignment of RT/RH sequences of VIPER and TATE (FAS 30 kb)
Alignments. Alignment of YR sequences of VIPER and TATE (FAS 15 kb)
Table. List of sequences used for RT phylogeny (PDF 29 kb)
Table. List of sequences used for YR phylogeny (PDF 29 kb)
Alignments. Alignment of RT sequences of different groups of retroelements (FAS 24 kb)
Alignments . Alignment of RH sequences of different groups of retroelements (FAS 17 kb)
Alignments . Alignment of YR sequences of DIRS retrotransposons (DIRS-like, PAT-like, Ngaro-like, and VIPER-like) along with other YR family proteins (FAS 23 kb)
Data Availability Statement
Genomic coordinates of VIPER and TATE copies analyzed here are available in the Additional file 2, and the sequences are available in Additional files 8 and 9.