

Abstract
The biological potency of natural products has been exploited for decades. Their inherent structural complexity and natural diversity might hold the key to efficiently address the urgent need for the development of novel pharmaceuticals. At the same time, it is that very complexity, which impedes necessary chemical modifications such as structural diversification, to improve the effectiveness of the drug. For this purpose, Cytochrome P450 enzymes, which possess unique abilities to activate inert sp3‐hybridised C−H bonds in a late‐stage fashion, offer an attractive synthetic tool. In this review the potential of cytochrome P450 enzymes in chemoenzymatic lead diversification is illustrated discussing studies reporting late‐stage functionalisations of natural products and other high‐value compounds. These enzymes were proven to extend the synthetic toolbox significantly by adding to the flexibility and efficacy of synthetic strategies of natural product chemists, and scientists of other related disciplines.
Keywords: chemoenzymatic synthesis, cytochrome P450 enzymes, drug discovery, late-stage diversification, natural products
1. Introduction
Natural products such as taxol, artemisinin, monensin, amphotericin – to name but a few – belong to the group of molecules that changed the world as we know it, and will continue to do so.1 Their structural complexity and diversity are unmatched, and arise from a constant evolutionary refinement by host organisms.2,3 Similarly, efficient natural product diversification is essential in the field of drug discovery, in order to meet the demand for novel drugs and drug analogues with improved specificity, or to test their metabolism in the body.4 This occurs preferably using chemical late‐stage modifications.5, 6, 7 However, due to natural products’ inherent complexity, such task is difficult to accomplish in a selective fashion. Often, chemically highly similar derivatives require independent synthetic access,8 violating the desired time and material efficiency.9
Bond polarity dictates the type of reactivity; the polarity profile inherent in a molecule regulates how to install functional groups (FGs). The insertion of FGs at a late stage of the synthesis, however, is difficult due to the inertness of sp3‐hybridised C−H bonds within the carbon backbone. In an attempt to avoid problems and foresee solutions to the installation of FGs, the retrosynthetic approach consolidated in the chemical community.10, 11, 12, 13 In contrast, nature developed its enzymatic machinery providing elegant tools to somewhat bend these rules. Among other enzymes, particularly cytochrome P450 enzymes (P450s) stand out with their ability to activate sp3‐hybridised C−H bonds for oxygen functionalisation. Moreover, this even happens with outstanding chemo‐, regio‐ and stereo‐selectivities, thereby oxidising substrates more inert than their own surrounding protein framework.14, 15, 16
The increasing need for sustainability is changing the rules of synthetic chemistry considerably, driving the application of Green Chemistry principles,17, 18, 19 the current trend toward protecting‐group‐free syntheses20 and the continuous interest in the total synthesis of natural products in more elegant ways.21 This calls for the integration of enzymes into the synthetic toolbox, supported by the discovery of a broad diversity of novel enzymes (e. g. by metagenomics) and the recent success in tuning enzymes in vitro to process requirements by rational engineering and directed enzyme evolution.22, 23, 24, 25, 26, 27, 28 As a consequence, the term “Biocatalytic retrosynthesis” was introduced in 2013, which revitalised the attention given to the use of enzymes for upgrading synthetic routes to complex targets, implementing an effective tool to achieve FG installation in a highly selective fashion under mild reaction conditions.29, 30, 31, 32, 33
Natural products form the basis of drugs targeting a range of severe diseases, like cancer or malaria, and can serve as an inspiration in drug discovery. Between 1940s to the end of 2014, every second small molecule drug approved was either a natural product or a direct derivative of it34,35 despite the fundamental shift in the drug industry towards synthetic small molecule libraries driven by combinatorial chemistry.36 Generally, the generation of a diverse compounds library to cover a diverse chemical space increases the chances to identify or improve a promising lead compound, while the occupation of a focused chemical space is sufficient for fine‐tuning.37,38 When dealing with complex molecules like natural products, late‐stage functionalisation offers an effective starting point for further diversification (Figure 1). This strategy has become popular only very recently and is about to enter the toolbox of medicinal chemistry.5,7 Enzymes such as P450s are well known for their ability to install oxygenated FGs into primary and secondary metabolites, often operating at a later stage of the biosynthesis.15 Thus, in combination with the broad range of available P450s and their diversified reaction scope, functionalisation via C−H activation embodies a method of immense potential for easy and rapid diversification of complex natural products.
Figure 1.
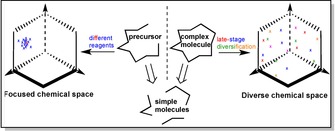
Focused and diverse chemical space covered by compound libraries of complex molecules. The two different libraries are generated either by using different reagents to synthesise similar final products from precursor, or by applying late‐stage diversification to an already synthesised complex molecule, respectively.
This review will highlight the power of using cytochrome P450 enzymes for the concept of biocatalytic late‐stage diversification by demonstrating the current progress in this field with recent examples. The importance of natural products will be introduced, and the elegancy of late‐stage Csp3‐H bond activation to achieve efficient natural product diversification outlined. First reports proved the efficient use of cytochrome P450 enzymes in total synthesis of natural products.39, 40, 41 Complementary to a recent summary focusing on the applications of oxygenases for total syntheses,42 this review will prioritise P450‐catalysed late‐stage diversification of natural products for drug discovery.
1.1. Cytochrome P450 Enzymes
The more “noble and late” transition metal catalysts (Pd, Rh, Pt, Au, etc.) have proven very effective in activating unreactive chemical bonds over other more reactive ones. They dominated the field of homogeneous catalysis (e. g. Pd‐catalysed cross‐coupling reactions) up to now.43 However, their toxicity, low abundance and high price have often outweighed their advantages, and are no more in agreement with the need for a sustainable catalyst.44 The focus of recent research has thus increasingly shifted towards coinage metals such as Co‐, Ni‐ and particularly Fe‐based catalysts, albeit their benignity should be used as a “selling point” with care.45 Iron is the most abundant transition metal on earth and plays an important role in nature. Providing some interesting all‐round characteristics for catalysis, a lot of progress has been made on its use for the activation of C−H bonds.44 Nonetheless, this stands in no comparison whatsoever to the capabilities that biological “Fe‐catalysts” exhibit in nature in the form of cytochrome P450 enzymes.
The energy required for breaking the inert C−H bond is partially compensated with forming a strong O−H bond (Scheme 1).46 The binding of the substrate results in a spin shift (low to high spin state) of the Fe‐complex, allowing Fe(III) to Fe(II) reduction by a corresponding reductase as the redox partner. Subsequently, a ferryl oxo porphyrin radical cation species (compound I) is formed, which abstracts the hydrogen atom of the substrate radically.47,48 The strength of the FeO−H bond is the driving force controlling the hydrogen transfer to yield compound II.14 Product formation then occurs via a radical rebound mechanism.48
Scheme 1.
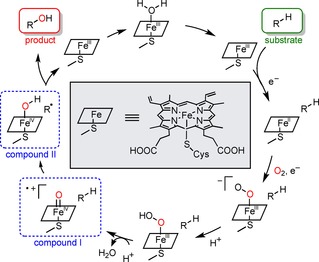
The P450 reaction mechanism catalysing the hydroxylation of a C−H bond. Marked in blue are the hydrogen abstraction step by compound I and the subsequent radical rebound with compound II to form the hydroxylated product.47,48
Of course, the large protein framework, the exact geometry of the active‐site and the heme‐ligand surrounding the iron ion are of high importance.14,49
However, essential for the P450’s ability to activate the inert C−H bonds is the directly coordinated cysteine thiolate.46,48,50,51 Chemically, a strong electron donor like thiolate seems counterintuitive for the design of a strong oxidant. In actual fact, the electron “push” to the iron centre generates the necessary “pull” for the C−H abstraction (Scheme 2).14 This happens by increasing the basicity sufficiently, while sacrificing some of its redox potential.50 This helps to effectively balance side reactions like uncoupling,51 which is the undesired production of H2O2 as a by‐product releasing reactive oxygen species that can deactivate the P450 enzyme itself.52
Scheme 2.
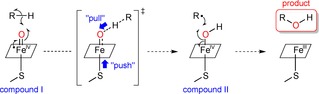
The “push” effect of the electron‐rich thiolate in compound I to enable a stronger “pull” and abstract the hydrogen from the C−H bond.14 The formed compound II leads to product formation via radical rebound.
The majority of P450s exist as a pair of individual heme and reductase proteins since the terminal monooxygenase is no electron‐transfer domain itself. The electrons required to reduce the P450 iron centre are provided by NAD(P)H and transferred via a FAD‐containing reductase and a ferredoxin unit. Only a few P450s have their corresponding redox partners integrated within the same polypeptide and are therefore self‐sufficient.53 The dependence on an efficient electron‐transfer by different cofactors often makes the reduction of P450s the rate‐limiting factor. As eukaryotic P450s are additionally membrane‐bound while their bacterial equivalents are soluble, self‐sufficient fusion systems like the bacterial BM3 (CYP102 A1) are usually more inviting for application.54
1.2. Natural Diversity of Natural Products
Natural selection is the key mechanism of evolution that enables ‘nature to live on the edge‘ meaning the constant development and improvement towards the status quo in order to replicate, adapt and survive.55 One way to get a potential advantage over competitors is the generation of unique natural products. However, useful, potent biological activity was a rare feature among them. Therefore a larger number of natural products synthesised increased the probability of obtaining a “lucky hit”56, 57, 58 that would enhance the fitness of the organism.
In this fashion, natural product diversification served as a natural screening platform for further structural exploration (cf. diversity‐oriented synthesis).
Firn and Jones56 nicely illustrated the origin for chemical diversity of natural products with the help of a matrix grid (Figure 2) stating that it is mainly the result of the broad substrate tolerance of some enzymes involved in the biosynthesis. Thus, a combination of only three enzymes can generate eleven different product molecules if they also tolerate derivatives of the starting material as substrates.
Figure 2.
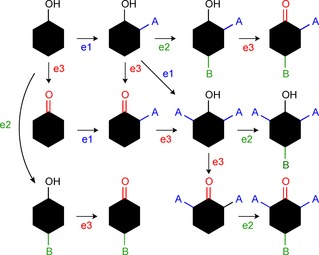
Matrix grid to illustrate chemical diversity from broad substrate tolerance of enzymes e1, e2 and e3.56
Cytochrome P450 enzymes are well known for catalysing a wide range of distinct reaction types counting 20 already in 1996 and the number is still growing.59 When possessing a relaxed specificity, these enzymes can drive diversification as shown in the example of the Gibberellin biosynthetic pathway.60 The refinement of the ent‐kaurene intermediate results in more than 136 distinct products using practically only four different P450 enzymes (Scheme 3). The oxidations happen in a combinatorial pattern similar to the matrix grid shown in Figure 2, thus increasing the diversity polynomially. Remarkably, in this pathway the alkene – the only chemically reactive functional group present – is left intact, although two of the P450s involved in oxidative modifications are multifunctional.61 Every manipulation in this diversification process involves a C−H activation, yet many complex natural products like the one shown in the box with variable functionality are formed seemingly easily using only four enzymes.
Scheme 3.
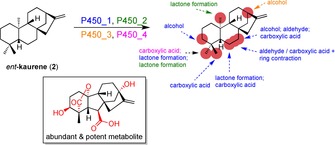
The diversification of ent‐kaurene (2) by four P450 enzymes occurring in the Gibberellin biosynthetic pathway.60,61 The positions accessible by different P450s are marked in red and the enzymes with their corresponding reactions are colour‐coded. One of the most abundant metabolites with known potency is shown in the box with the introduced functional groups coloured in red.
At the same time the lack of selectivity hampers the probability for synthesising a particularly attractive product in useful quantities. This may represent a major challenge in the preparative applications of P450s in vitro.
1.3. Drug Discovery – Need for Diversification Tools
Some natural products are considered active compounds due to their excellent pharmacological profiles, thus lending themselves as attractive starting points in drug discovery. However, their diversity and complexity are a gift and a curse at the same time. Their quality as lead molecules is likely to be suitable, but effective diversification is essential in order to improve their drug quality.4 The latter often proves to be challenging. Natural products are inadequate for modern high‐throughput screening (HTS) with biochemical assays in the pharmaceutical industry,62 they often fail to comply with the desired physiochemical properties (e. g. Lipinski's Rule of Five)34,63,64 and difficulties are faced in accessing sufficient amounts of the target natural product.34,65,66 Supply by extraction from natural resources by fermentation using metabolically engineered organisms or total synthesis can be cost intensive,67 incompatible68 or tedious,8 respectively. Nonetheless, in recent years the focus in drug discovery is increasingly shifting back towards natural product scaffolds.34,36,69 They received more attention in the light of (i) the misjudgement regarding the success of the discovery of small, synthetic lead compounds by HTS methods,36,64,69 (ii) the growing antibiotic resistance70 and need for new antibiotics,71,72 (iii) the change of strategy from a conventional target‐oriented to a modern diversity‐oriented synthesis (DOS)73, 74, 75 and (iv) the shortcomings in the productivity in combination with expenses for clinical trials.76
Traditionally, a target‐oriented strategy was used in drug discovery to find the best‐fitting drug molecule for a predefined protein target in need of modulation. However, the success rate of the molecule's actual target‐modulating abilities was unpredictable due to the biological complexity of living systems.73,77 By leaving unknown both the target within a certain pathway as well as the compound expected to modulate it, modern DOS allows a certain degree of freedom for the final lead structure. This strategy aims to divergently synthesise a collection of compounds to cover a large chemical space around a specific template (Figure 3).73,77
Figure 3.
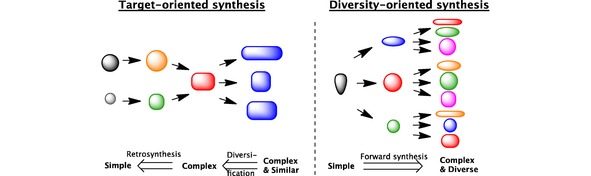
Concept of target‐ and diversity‐oriented synthesis illustrated with colour and shape to code for diversity and complexity, respectively.77
Synthetic strategies are planned using the retrosynthetic approach,78,79 which is based upon the step‐wise detachment of the product molecule with maximum efficiency until its origin is found in commercially available starting materials. Often, for highly similar derivatives of the desired product completely new pathways must be developed because the synthesis of complex molecules requires reactive sites of suitable polarity placed at the correct positions.8 The deliberate choice of methods for carbon backbone construction with the help of appropriate polar functional groups is therefore an important factor in the retrosynthetic strategy.80 Common synthetic strategies include the umpolung process, FG interconversion or use of protecting groups.12
Similarly, in a manner resembling the do‐it‐yourself furniture assembly, where the item is built up from small pieces following step‐by‐step assembly instructions, this synthetic approach also had to be devised in the reverse direction beforehand (Figure 4).
Figure 4.
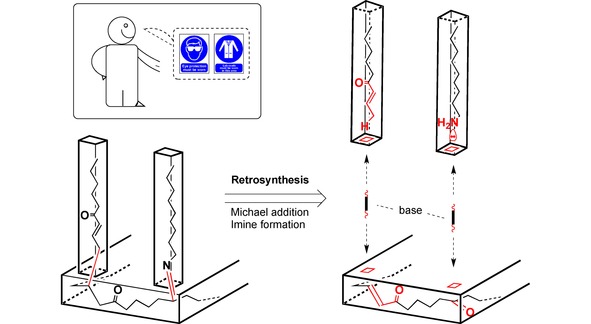
Symbolic illustration of the retrosynthetic approach applied to both molecules and do‐it‐yourself furniture assembly.
Analogously to the demand for specific local functionalisation in the construction of complex molecular scaffolds, if assembly holes are missing on the furniture material or wrong screws have been used, it will be impossible to construct the desired product as planned. Sheer force or the help of tools such as a drilling machine would be required. This is very similar in synthetic chemistry; to modify or functionalise the target molecule by activating an sp3‐hybridised C−H bond (i. e. “drill a hole”) extreme conditions like high temperatures or toxic metal catalysts are required.81, 82, 83, 84, 85
This comparison nicely illustrates the obstacles that need to be overcome when attempting a late‐stage modification of a complex molecular scaffold possessing various other reactive groups. The number of publications on C−H activation and their application in the synthesis of pharmaceuticals are steadily increasing, reflecting the ongoing chemical research on this topic.86
Such late‐stage refinement adds a great deal of flexibility to synthetic strategies and diversification purposes in the drug discovery industry making it such an attractive and powerful tool.
Excellent reviews have summarised approaches like C−H activation used in the synthesis86, 87, 88, 89 or diversification of natural products.4,90, 91, 92, 93, 94 In the following chapter, the application of P450 enzymes in diversity‐oriented syntheses is illustrated with some recent examples.
2. Examples
2.1. Macrocycle Synthesis and Diversification
Macrocycles hold great therapeutic capacities because their ring structure is conformationally defined, often resulting in high affinity and selectivity for protein targets.95 Several natural products that have been exploited successfully as powerful therapeutics possess a macrocyclic skeleton.96
The recent example of Gilbert et al. therefore represents an appealing template for macrocycle functionalisation using P450s.97 The P450 enzyme of choice was the bacterial PikC from the pikromycin pathway, which had been characterised by the Sherman group,98 then engineered to make it self‐sufficient by fusion to the known RhFRED reductase domain.99 The enzyme's natural anchoring mechanism involves a salt bridge and hydrogen bonding network to the desosamine sugar functionality of glycosylated macrolactone substrates.100,101 This mechanism enables substrate specificity for the macrolide antibiotics in the pikromycin and methymycin pathways.99 In a recent study, the Sherman group took advantage of this mechanism by replacing the desosamine sugar with a synthetic anchoring group.102 The possibility to add or remove the anchoring group, and the relationship between its size and rigidity and the regioselectivity of the enzymatic hydroxylation makes this a highly versatile strategy in synthetic chemistry (Scheme 4).
Scheme 4.
The structure of the anchoring group for PikC substrate recognition can be changed for controlling the regioselectivity of the enzymatic hydroxylation.102
A nice demonstration was thus given at the end of last year with the diversification of macrocycles 4–6.97 From the common intermediate 3, 12‐ and 11‐membered products 4 and S,S‐5, as well as the latter's diastereomeric equivalent R,R‐6, were formed by endo‐ and exocyclisation, respectively. Diversification could then be further driven by the attachment of different linker groups a–e (Scheme 5). These were synthesised via azide‐alkyne click chemistry, thus improving once more the substrate engineering method by allowing access to high‐throughput linker synthesis. The linkers differed in length and shape i. e. ortho‐, meta‐ and para‐substituted benzene or absence of phenyl group.
Scheme 5.
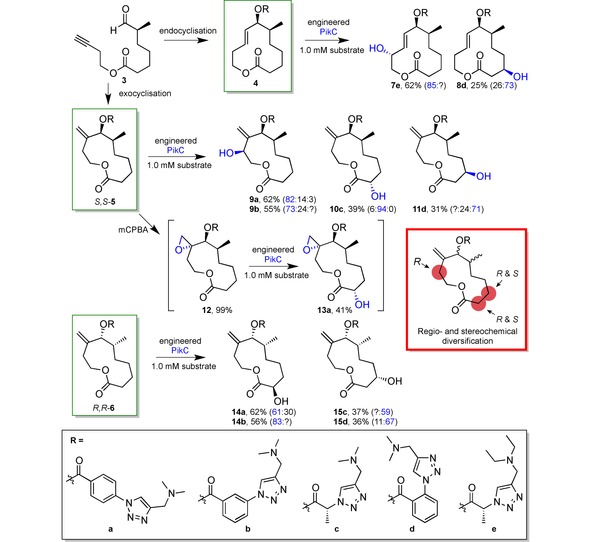
Late‐stage diversification of macrocycles 4, S,S‐5 and R,R‐6 (green boxes) with only a single P450 (PikC) directed by the different linkers a – e (black box).97 The product ratio is given in brackets highlighting the product with blue colour. Question marks indicate unknown fraction of corresponding regioisomers. Yields of preparative reactions ranged between 25 % and 62 %. In the red box the regio‐ and stereoselectivity profile of the exocyclisation products of 12 is summarised.
In substrate 4 longer linkers, i. e. a and b provided regiochemical access to C−H bonds distal of the linker, while shorter ones (c and d) oxidised C−H bonds proximal to the directing group. In contrast, for diastereomer R,R‐6 linkers a and b led to distal oxidation like c and d, similar to 4 where the shortest linker e oxidised the proximal allylic proton.
Neither epoxidation, nor amine linker oxidation was observed, although linker e had intentionally been designed as the diethylamine to avoid N‐demethylation observed in some cases. To test orthogonal chemoselectivity for C−H oxidation, 5 was oxidised to its epoxide 12 and PikC then oxidised it to 13 a. Noteworthy, albeit left unmentioned by the authors, the presence of the epoxide changed the selectivity towards a distal position compared to product 9 a, due to the missing allylic activation. Proximal allylic oxidation also occurred in substrate 6, albeit with very low selectivity, and some unidentified minor products were formed from 4 and 5, which was not discussed in the paper. More diverse linker modifications are yet to be explored.
No total turnover numbers (TTN) were provided – the maximum number of chemical conversions of substrate molecules per enzyme molecule over the catalyst's lifetime. However, preparative experiments were conducted on a 30–60 mg scale.
The combination of esterification and click reactions enables access to a range of different products with regio‐ and stereoselectivity, where the linker length and geometry is the decisive diversification factor. The full extent of this method's potential has certainly not materialised so far, and more is yet to come in the use of directing groups in combination with enzyme catalysis. However, the above mentioned study has demonstrated that a single enzyme can selectively functionalise macrocycles bearing so many C−H bonds in a controllable manner with the help of linkers.
2.2. Simulation‐Guided Mutagenesis of P450 for Selectivity Prediction
Using directed evolution of an enzyme for tuning it towards desired selectivities requires extensive time and effort due to the need for screening all of the generated variants.16,23 This can be minimised by deriving the most beneficial mutations, generating smaller libraries with site‐specific mutagenesis and verifying mutations experimentally.103 The semi‐rational approach is made increasingly efficient combining structural and biochemical data with computational data. Rather than generating a maximum diversity of active site geometries,22,104,105 more focused libraries can thereby be generated like this.
Cytochrome P450 enzymes,50,106, 107, 108 particularly BM3,109,110 have been the subject of intense research computationally.. With macrocycles as the substrate, the study of Urlacher et al. in 2018 demonstrated the power of in silico engineering to impose control on the selectivity of P450 enzyme BM3 on the diterpenoid β‐cembrenediol.111 In a previous study the Urlacher group had already investigated the BM3 on the β‐cembrenediol.112,113 The carbon backbone of this macrocycle of neuroprotective function has 16 possible oxidation sites, and three of them are accessible with the wild‐type enzyme (Figure 5). Using first‐sphere active site mutagenesis, a small library of mutants was generated, of which two showed excellent regioselectivity either for positions C9 (product 18) or C10 (product 19), and quite good stereoselectivity in each case. Another mutant, BM3V78A/F87A, was unselective, though in addition to 18 and 19 the epoxide product 17 was yielded as well.
Figure 5.
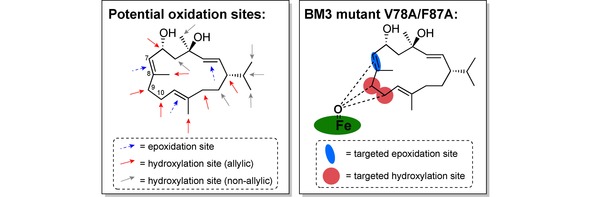
The potential oxidation sites of β‐cembrenediol on the left, and the three positions unselectively targeted by BM3 mutant V78A/F87A on the right.111
Using the mutant V78A/F87A as the starting template, a subsequent study computationally investigated what active site residue would significantly influence the enzyme's selectivity for 16 upon mutation.111 The strategy was based on the premise that the substrate's binding mode within the active site determines the selectivity of reaction. Given multiple conformational modes possible for a large molecule like a macrocycle, careful conformational analysis and substrate docking simulations were necessary to obtain possible enzyme‐substrate complexes. However, with those in hand the binding density surface (BDS, i. e. the 2D surface of two probability density functions) could be mapped for different positions of 16 keeping in mind the reaction mechanisms and optimal distance for the reaction to occur. The BDS of the chemo‐ and regioselectivity of BM3V78A/F87A catalysing the oxidation of 16 is shown in Scheme 6. The major hotspot shown in red indicates that the hydroxylation reactions are favoured over the epoxidation towards product 17, supporting the experimental data. Further BDS graphs were modelled giving valuable information about the regioselectivity between the hydroxylation products 18 and 19 and their stereoselectivities, or that of the epoxidation reaction.
Scheme 6.
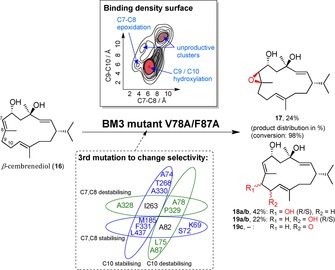
β‐cembrenediol (16) is converted to epoxide 17 and hydroxylation products 18 and 19 by the BM3 mutant V78A/F87A.111 The percentage refers to the product distribution. The mapped binding density surface indicates a preference for products 18 and 19. Binding free energy of different active site residues suggested additional mutations and predicted a potential selectivity outcome by stabilising the corresponding binding mode.
In combination with modelled protein‐substrate orientations, the binding free energies of individual active site residues could be calculated, indicating whether this amino acid would stabilise or destabilise the substrate position and thus the corresponding product formation.
An in silico mutation simulation then identified the most favourable triple mutants. Experiments were performed to test the suggested mutations and it was found that six out of ten caused an increase in desired product by at least 2‐fold. In one case both products 17 and 18 were eliminated completely, and production of 19 was increased more than 5‐fold.
The objective to construct a computational methodology of enzyme engineering to predict a shift in enzyme selectivity was well accomplished, albeit enzyme activity itself was not included in the calculations as noted by the authors: It was observed that four of ten triple mutants showed significantly lower conversion than the imperfect template. Computational predictions can provide essential information for application in enzyme engineering for diversification This presents a significant paradigm on how to generate and control the distribution of products.
2.3. Facile Transformation of an Antibiotic Macrolide to other Members of its Class via Late‐Stage Diversification using Natural P450 Enzymes
The availability of novel antimicrobial drugs is severely limited urgently demanding solutions when considering the mounting crisis of antibiotic resistance and the simultaneous lack of economic incentive for antibiotic drug discovery.70 Natural products present a renewed hope especially for the resolution of this crisis because they had once paved the way for the Golden age of antibiotic discovery.71,72 Novel chemical structures may prove to be the key against evolving pathogens. Macrolides are an important class of antibiotics, but do not always comply with the desired characteristics for pharmaceutical use.114 Chemical modifications of these naturally occurring therapeutics is thus essential and tools for their derivatisation desirable. In fact, derivatives of the same bioactive chemical scaffold represent the main source of “novel” antibiotics.115
The Sherman group had already studied several biocatalytic syntheses of macrolactone glycosides in the past.116 Recently, they examined the interconnection of pharmaceutically valued macrolides within this compound family via P450 diversification, thus giving an example of the application of this enzyme class in actual drug discovery.117 Following up on its first biocatalytic total synthesis, antibiotic M‐4365 G1 (20) was successively functionalised to a set of juvenimicin and rosamicin macrolides possessing the same core structure. The executing tools were three P450s TyII, JuvD and MycCl, all part of the biosynthesis of closely related compounds: macrolactone glycosides tylosin, juvenimicin and mycinamicin, respectively (Scheme 7). Using self‐sufficient P450‐RhFRED fusion proteins, several mg of product per 1 L overexpression culture could be obtained in preparative‐scale reactions.
Scheme 7.
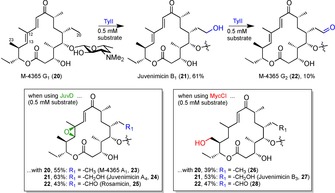
Late‐stage diversification of natural product M‐4365 G1 (20) with three natural P450s to obtain the improved antibiotic forms (21–28).117 The functional groups introduced bare the same colour to the respectively catalysing P450 enzyme.
The concept of interconnecting different antibiotic families from a common scaffold via late‐stage adjustments with the best‐fitting P450s of biosynthesis of this compound family worked out well. Barely functionalised compound 20 offered itself as a good starting point to be oxidised twice up to the aldehyde 22 by P450 TyII. From each of the compounds 20–22 either an epoxide at positions C12 and C13 or a hydroxyl group at position C23 could be introduced with P450s JuvD or MycCI, respectively.
The authors reasoned their P450 enzyme selection as follows: (i) Epoxidation of 22 leads to rosamicin 25. Logically, first choice were thus P450s JuvC and JuvD due to their high sequence similarity to the established P450 enzymes RosC (87 %) and RosD (84 %) of the rosamicin biosynthesis.118 (ii) However, since JuvC's activity on 20 was disappointing, the job was allocated to TyII. (iii) MycCI was employed due to its characterised task to hydroxylate this position in the Mycinamicin biosynthetic pathway, and its unusual substrate flexibility.119
Unfortunately, the sequential application of P450s JuvD and MycCI on either one of compounds 20–22 was not reported, and neither was that of TyII on any of compounds 23, 24, 26 and 27. Therefore, a complete assessment of the ease of transformation from one antibiotic to another by P450 enzymes can therefore not be made. Furthermore, while the performance of TyII in first oxidation to 21 was satisfactory (61 % isolated yield), the low yield obtained in the second oxidation step (∼10 %) forced the authors to prepare 22 with the help of chemical methods by using a copper(I)/TEMPO catalyst system.
Nevertheless, given the number of studies that have explored the derivatisation of known antibiotics, the biocatalytic component in this study makes it stand out. It highlights the capacity of P450s in the application to the drug discovery process, allowing facile access of a variety of chemical derivatives. Two different functional groups at three individual positions could be selectively introduced to pharmaceutically active natural products. The fact that this can be achieved post‐synthetically at a late‐stage fashion to a complex macrolide heralds both the establishment of P450 enzymes into drug discovery, as well as the benefit of recourse to natural products.
2.4. Artemisinin Functionalisation with Absolute Regio‐ and Stereoselectivities using Active‐Site Fingerprinted and Engineered BM3 Variants
As an alternative to performing exhaustive computational simulations on the active site of BM3 towards the designed generation of substrate diversity, the Fasan group developed a ‘fingerprinting method’ to map the binding pocket of cytochrome P450 enzyme BM3 variants in order to predict the regioselectivity exhibited towards terpenes.120
The workflow is illustrated in Scheme 8A. Five different molecules were selected as substrate probes covering a range of different molecular scaffolds present in many terpenes of pharmaceutical value. The template BM3 mutant FL#62 exhibited high activity on all of these probes and was thus chosen as the template for mutagenesis. A mutant library was generated by using site‐saturation mutagenesis on six positions within the FL#62’s active site that were known to have an impact directly on the enzyme's binding pocket. With the help of high‐throughput screening the activity of the variants towards the five molecular skeletons gave an indication on the size and geometry of the active site. This in turn was used to predict the activity of a given variant for substrates of similar molecular structure, thus verifying the fingerprint. In 87–97 % of times the predictions were found to be correct. Interestingly, most enzymes (5/6) having different fingerprints also displayed diverse regioselectivity, which again was verified experimentally.
Scheme 8.
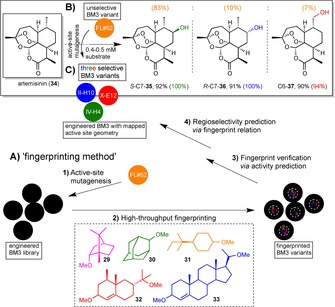
Late‐stage functionalisation of artemisinin (34) with BM3 variants after active‐site geometry fingerprinting and active site mutagenesis.121 The percentage yield is indicated, and in brackets the product distribution is shown in the same colour as the corresponding BM3 mutant. A) The application of a developed high‐throughput fingerprinting method120 on FL#62, a known variant of BM3 is illustrated. Active‐site mutagenesis on FL#62 afforded a library of mutants, which active‐site geometries were mapped to identify three BM3 variants appropriate for the substrate of choice. B) FL#62 itself catalysed the transformation of artemisinin (30) yielding an improvable product distribution of 83 : 10 : 7 for 35, 36, 37, respectively, with a TTN of 340. C) The three identified BM3 variants of FL#62 hydroxylated 30 at two different positions, IV‐H4 (362 TTN) and II‐H10 (270 TTN) with absolute (100 %) regio‐ and R‐ and S‐stereoselectivity, and the X‐E12 (113 TTN) with high regioselectivity (94 %).121
Conclusively, this ‘fingerprinting method’ of mapping the active site geometry yielded BM3 variants of diversified regioselectivity that are able to target positions across the whole molecular scaffold of terpenes.
Yet, would this method also be transferable to other substrates? This question was answered with a second paper from the group, applying the method to the popular example of the anti‐malarial drug artemisinin (43).121 P450 enzymes are involved in artemisinin's biosynthesis and thus semi‐synthetic supply is applicable,122,123 which is managed through methods of metabolic engineering and synthetic biology.124,125 Late‐stage diversification of the drug can be achieved through synthetic chemistry tools used to synthesise new analogues or metabolic products,93,126 often with aid of organometallic catalysis.127,128
The authors aimed at obtaining P450 enzymes that would catalyse the late‐stage functionalisation of artemisinin with high regio‐ and stereoselectivity.121 Their strategy was the application of the described ‘fingerprinting method’. They thus started off with the same BM3 mutant FL#62, which had proven to possess a broad substrate scope including multiple ring scaffolds. FL#62 was indeed accepting 34 as a substrate with 340 total turnovers to yield 35–37 (83 : 10 : 7) with an unselective product distribution (Scheme 8B). A small library of mutants was yielded by rational driven site‐saturation mutagenesis of the active site, regiochemically diverse variants predicted by P450 fingerprinting, verified experimentally and the three most promising mutants (Il‐H10, X‐E12 and IV‐H4) identified (Scheme 8B). These catalysed the formation of S−C7‐35, R−C7‐36 and C6‐37 with 100, 100 and 94 % selectivity (Scheme 8C).
The success of the developed ‘fingerprinting method’ on a complex, yet fragile compound of high pharmaceutical interest like artemisinin speaks for itself. Vital for the positive result was the large and unselective binding pocket of the starting template BM3 FL#62 that could be fine‐tuned towards the target substrate. Given an effective high‐throughput screening method, the method's ease of execution makes the extremely high selectivities obtained even more remarkable.
2.5. Late‐Stage Functionalisation of Steroidal Scaffolds with Engineered P450 Variants
One major challenge of mutagenesis is to find an adequate balance between the enzyme's selectivity and specific activity,129 since improving one usually diminishes the other.120 Therefore alternatives to mutagenesis have been explored130,131 including the use of directing groups such as those in section 2.1. to determine the selectivity and substrate tolerance102 or in section 2.3. for substrate recognition by the enzymes.101,119 While clearly very compelling, such alternatives to improve the selectivity are likely to suffer from a lack of wide‐ranging applicability.
Example A)
Already back in 2011, the Reetz group published an example of P450 mutagenesis for hydroxylating the complex steroidal scaffold with near complete regioselectivity.132 Their approach followed a typical ‘evolve‐and‐screen’ strategy of a BM3 variant, however, combinatorial active site saturation test (CAST) coupled with rational design reduced the amount of screening – bottleneck of directed evolution133 – immensely. CAST efficiently generates focused libraries by mutating all relevant residues in proximity to the active site134 and had originally been developed within the Reetz group, to broaden the substrate tolerance.135 The BM3(F87A) variant chosen for their study already unselectively catalysed the transformation of testosterone (37) to a mixture of the 2β‐ and 15β‐alcohols. The F87A mutation of the template BM3 has long been known to change the substrate orientation and thus the regioselectivity,136 and has been widely used.137
Favourable residues were selected with the help of mechanistic and structural data, which were grouped and filtered to render cooperative effects more likely, and the amino‐acid alphabet was partly reduced: Having focused at first on only three sites, the same number of combinatorial libraries was generated and the screening thus reduced to 8,700 transformants.132 These included two mutants, KSA‐1 and KSA‐14, which catalysed the hydroxylation of testosterone at positions C2, 41, and C15, 42, with 97 % and 96 % regioselectivity, respectively, and complete stereoselectivity for the β‐H (Scheme 9A). As another example, progesterone was tested as a substrate. Products 2β‐OH, 43, and 16β‐OH, 44, were formed with 100 % and 91 % selectivity using mutants KSA‐9 and again KSA‐1, respectively. According to computational calculations, the extremely high regioselectivity is due to the restriction of 37 and 38 to two defined orientations within the enzyme's binding pocket.
Scheme 9.
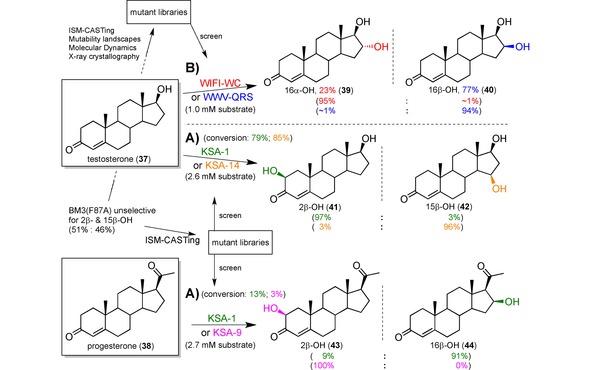
Three different studies showed a proof‐of‐concept late‐stage functionalisation of testosterone (37) and/or progesterone (38) with excellent regio‐ and/or stereoselectivities using engineered P450 mutants. The product distributions are given in brackets and coloured according to the respective enzyme variant that catalysed the reaction. A) Using iterative saturation mutagenesis (ISM) the lack of selectivity of P450BM3 variant F87A for positions 2β and 15β (1 : 1 mixture) was shifted almost completely towards either one to afford 41 and 42 with 97 % and 96 % selectively over the other.132 B) With the help of molecular dynamic simulations, X‐ray data and an explored mutability landscape, ISM‐CASTing was performed on the residues lining the binding pocket of the same BM3 variant F87 A to change the regioselectivity to position C16 with stereoselectivities of 95 % and 94 % ee for both enantiomers 39 and 40, respectively.129 Percentage yield is given for an up‐scaling transformation carried out with the respective best mutant.
Example B)
In a follow‐up study, the Reetz group extended the scope of their BM3 variants to the C16 position of testosterone, setting target to both 16α and 16β stereoisomers.129 Synthetically, C−H activation at this position in steroids is of high interest because it allows easier access to biologically active glucocorticoids. Rather than using a mutant that had already shown some C16 hydroxylation, here the authors started with the same BM3(F87 A) mutant as before. To guide mutagenesis identifying the appropriate amino acid residues, even more trust was put into rational design and molecular dynamics computations were carried out supportively. The main rational element was a protein mutability landscape (Figure 6), which is the systematic analysis of the effect any amino acid at a particular position has on the enzyme's performance.138,139
Figure 6.
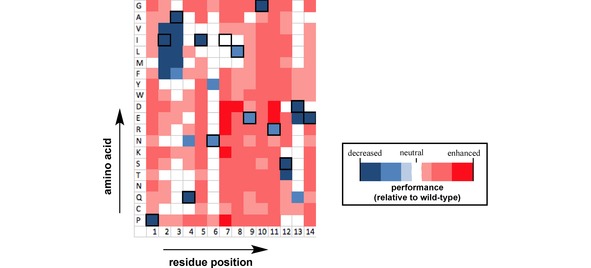
Example of a mutability landscape in the form of a heat map for different amino acids at 14 residue positions within an enzyme.138
Such a map of the sequence‐function relationship was generated for the F87A template enzyme by analysing previous data from Commandeur et al.,140, 141, 142 Payne et al.,143 and their own lab,144 and by screening mutants available from a previous study.145 For example the group of Commandeur had identified residue 72 to be important for substrate orientation, and showed that it was completely inverting the stereoselectivity of C16 hydroxylation.141 The analysis was directed towards four aims: (i) conversion of testosterone, a C16 hydroxylation considering both (ii) α‐ and (iii) β‐protons, in contrast to the previously observed (iv) 15β‐product.
With the mutability landscape at hand, the residues were filtered, grouped and five of them selected each for libraries A and B. Three cycles of iterative saturation mutagenesis (ISM) was performed: The method CAST was first applied to library A (950 mutants screened). Then the mutant with the best selectivity for each 16α‐ and 16β‐OH products was used as the template to generate one library B in each case (2×950 mutants screened). Again, the best mutant of every library B was optimised into four further enzyme variants with the help of MD simulations (2×5 mutants screened). Even after the third ISM cycle and the computationally guided third step, the 16β‐OH selective mutant had to be fine‐tuned further because a strong trade‐off between activity and selectivity was observed. This strategy resulted in mutant WIFI‐WC catalysing the hydroxylation of 37 to afford 16α‐39 with 96 % selectivity, 95 % conversion and 8660 TTN, and mutant WWV‐QRS with 92 % selectivity for 16β‐40, 92 % conversion and 9044 TTN (Scheme 9B). Furthermore, the best mutants found also catalysed the C16 hydroxylations of four other steroids, namely androstenedione, nandrolone, boldenone and norethindrone, with exquisite selectivity and activity.
While Reetz et al. had already achieved significant improvements in selectivity and relative rate profiles with ITM and CAST approaches in 2011 (Scheme 9A),132 they criticised the trade‐off in activity and the amount of screening (2×9000 mutants screened) themselves later. In comparison, in the study from 2018129 activities close to the one exhibited by the BM3 wild type towards fatty acids with far less screening effort (3×950+X mutants screened) could be obtained. Of course, other factors like enzyme stability have not been considered here, but still this proof‐of‐principle study awakens hope for a greater involvement of such enzymes in industry, and elegantly demonstrates the potential of a well thought‐through semi‐rational directed evolution approach.
2.6. Late‐Stage Diversification of Lead Compounds with Liver Microsomes
The synthesis of drug metabolites represents an integral part of every drug discovery programme in order to predict the half‐time of a drug or to test the toxicity of its metabolic products. Obviously, human liver P450s are in the focus of research in drug discovery because they are responsible for the degradation of drugs and xenobiotics in the body.15 With their broad substrate tolerance these liver P450s are attractive tools for predicting in vivo drug interactions via in vitro reactions146, 147, 148 or drug metabolite synthesis via late‐stage modifications.149,150
Often, preclinical drug screening involves the use of liver models to understand the downstream process of the drug in a natural environment simulation.
Since the human liver cannot be used for this purpose, animal livers and liver microsomes – vesicles of fragmented endoplasmic reticulum containing P450s – are employed as models151 and even BM3 variants instead.152,153 Many metabolites can be formed from the corresponding drug precursor as shown in Scheme 10 with the example of the lorcaserin metabolism by human liver microsomes.153 Structural derivatisation in a late‐stage fashion is therefore preferred for the chemical metabolite synthesis, and liver microsomes are a practical tool to use.154
Scheme 10.
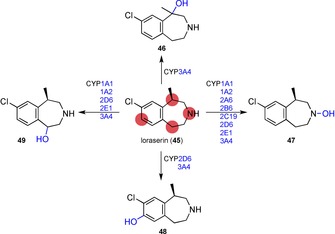
Metabolism of the 5‐hydroxytryptamine 2 C agonist lorcaserin by P450s of human liver microsomes.153
In two very recent studies, Obach et al. searched for new inhibitors targeting the human phosphodiesterase‐2 (PDE2),155,156 which is one of a group of enzymes that hydrolyse the phosphodiester bond within second messenger molecules cyclic adenosine monophosphate and cyclic guanosine monophosphate. PDE2s are thus important for the human physiology, for example cardiac and vascular processes. Consequently, PDE2 inhibitors are of high clinical relevance for treatment of stroke or heart rate regulation.157 In the first study, Obach and co‐workers improved a lead candidate for PDE2 inhibition by hydroxylation using monkey liver microsomes. The newly generated candidate had more favourable physiochemical properties while retaining the parent potency.155
The research group then extended their work to the transformation of nine lead compounds applying liver microsomes of eight different species each case.156 Their aim was to obtain a better structure‐activity understanding for PDE2s in order to enable improvement of target potency, metabolic lability and membrane permeability. The nine leads were incubated with the liver microsomes and the metabolic product profile analysed by HPLC to identify the most promising of the applied species. The product of scale‐up experiments was fractionated by HPLC, isolated and checked for purity by UHPLC‐UV‐MS and its structure determined by quantitative NMR. The latter technique had been established in a previous study as a handy tool to allow for structure and concentration determination of the metabolites.158 This way 36 new analogues were generated, which were tested for PDE2 inhibition, drug clearance and membrane permeability. Up to seven derivatives of the parent lead compound were generated.
A better potency was observed for some of the metabolites. In the case of compound 51 c the demethylation of parent lead 50 carried out by hamster microsomes increased the potency by 10‐times (Scheme 11). Analogue 51 c also had a greater metabolic stability, but at the cost of a slightly diminished membrane permeability. Following an N‐demethylation to compound 51 a decreased the potency slightly, albeit still better than lead 50. However, the 10‐fold decrease in permeability makes 51 a unsuitable. In total, most derivatives showed a mild to significant decrease in potency and often lost out in at least one more of the other two features in comparison to its parent lead compound. At most, the same quality could be maintained.
Scheme 11.
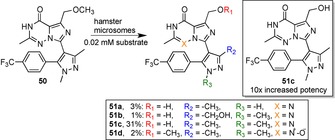
Late‐stage diversification of human phosphodiesterase‐2 (PDE2) inhibitor 50 using liver microsomes of different animals. The analogue 51 c was observed to have a 10‐fold increased potency, an increased metabolic stability and a slightly decreased membrane permeability relative to the parent lead compound 50.156
The analytical analysis came with the handy feature that only less than a micromole of the lead compound is required for this process. Furthermore, reactions are carried out under mild conditions in aqueous solutions of known concentrations. This clearly represents a great time‐saver compared to chemical reactions of milligram scale at harsher conditions. Notwithstanding, the authors admitted that some screening effort is required to identify the liver microsomes of the best‐fitting species and good fortune to be able to produce a favourable structure‐activity relationship.
This study illustrates nicely the applicability of liver microsomes for late‐stage diversification purposes of lead drug compounds. In hindsight, the authors did not discover a novel improved drug because compound 51 c was already the figurehead of the previous publications described before,155 despite their extension of diversity via more substrates and other species’ liver microsomes. This indicates the limitation and requirement of the aforementioned luck factor. However, in view of late‐stage diversification this work is a convincing example of achieving this within a few simple working steps. Process development for the synthesis of larger quantities will certainly remain a significant challenge.
2.7. Late‐Stage Functionalisation of the Antileukemic Agent Parthenolide with Engineered BM3 Variants to Improve its Potency
Another example of the Fasan group, who again applied their active‐site geometry ‘fingerprinting’ method in combination with active site mutagenesis,120 gives an idea of how newly generated functionalisation can be used for further diversification. Kolev et al. studied the emerging natural product parthenolide as substrate due to its extraordinary properties against various diseases,159 particularly its anticancer activity.160,161 However, its low solubility in water and stability at various conditions necessitates the synthesis of derivatives, and thus attracted a lot of attention from research. The reactive position C13 and the C1−C10 double bond have been accessed chemically, however, only the C‐13 analogue DMAPT was convincing enough to enter clinical trials.162
In 2014, Fasan et al. therefore devised cytochrome P450 enzymes to access positions C9 and C14.159 Indeed, their previously engineered P450BM3 variant FL#62 catalysed the hydroxylation of parthenolide (52) to afford the epoxide 53, C9‐54 and C14‐55 in a product ratio of 77 : 13 : 10 and >1000 total turnovers. Their ‘fingerprinting’ method resulted in mutant enzymes III‐D4 (4980 TTN), XII‐F12 (1310 TTN) and XII−D8 (60 TTN) having 90, 80 and 95 % selectivity for the products, respectively (Scheme 12). These alcohols were then further diversified by chemical benzoylation using acid chloride reactants carrying aromatic substituents to furnish derivatives that had a greater potency against primary acute myelogenous leukaemia (AML) cells (e. g. 56 and 57) than 52. In addition, Fasan showed a greater selectivity against malignant over healthy Bone Marrow (BM) cells motivating the authors to classify positions C9 and C14 as ‘hot‐spots’.
Scheme 12.
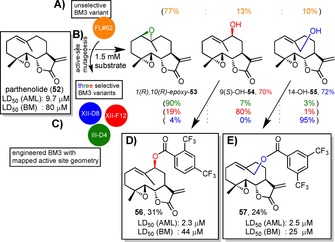
Late‐stage functionalisation of parthenolide (52) with P450BM3 variants after active‐site geometry fingerprinting120 and active site mutagenesis.159 The percentage yield of a preparative scale synthesis is indicated in the colour of the respective best mutant that catalysed it. Product distributions are shown in brackets and coloured according to the respective enzyme variant that catalysed the reaction. A) FL#62 catalysed the transformation of parthenolide yielding an improvable product distribution of 77 : 13 : 10 for 53, 54 and 55, respectively. B) Active‐site mutagenesis on FL#62 was used just like in Scheme 9 to identify three selective BM3 mutants: III‐D4 (4980 TTN), XII‐F12 (1310 TTN) and XII‐D8 (60 TTN). C) Parthenolide was hydroxylated at three different positions to form an epoxide 1(R),10(R)‐epoxy‐53, 9(S)‐OH‐54 and 14‐OH‐55 with absolute stereo‐ and excellent regioselectivities (90 %, 80 % and 95 %, respectively). D) Further derivatisation was carried out with the hydroxylated products and compounds 56 and 57 were found to have better LD50 values for primary acute myelogenous leukaemia and normal bone marrow cells compared to 52.
The Fasan group were able to further improve their enzymatic synthesis in 2016 and designed new analogues by extending the diversification scope.163 In addition to acylation and alkylation reactions, carbene insertion were applied as alternative O−H functionalisations. At the same time, the new compounds avoided the potentially labile ester linkage to hydrolysis, and few showed even better potency. Carbamate analogues were also added to the diversification spectrum.164 A diverse library of parthenolide derivatives was generated, tested against a range of different malignant cell lines and compared via a heat map. This provided the authors with a tumour‐cell specific anticancer activity profile of various molecular scaffolds. In the end, compounds bearing high cytotoxicity against every malignant cell type with up to 14‐fold increased activity compared to 52 were identified.
These studies nicely demonstrate the power of P450 enzymes for late‐stage functionalisation as an entry to the diversification of parthenolide alcohols by different chemical reactions.159,163,164 The latter has set the fundament for tailoring a library of different analogues to successfully improve the potency and selectivity to malignant cells in comparison to parthenolide itself. Noteworthy is that the authors included healthy cell lines as controls in order to analyse the selectivity of the compounds towards malignant cells over healthy cells. This was a point of appropriate criticism for many studies that did not assess the cytotoxicity on healthy cells, thus creating the illusion that parthenolide was only toxic to tumour cells. However, similar to many other studies, Fasan et al. did not reveal the water solubility and thus bioavailability of their lead compounds, which remains the greatest obstacle for entering clinical application.162
3. Conclusion
Natural products will exert a substantial influence on drug discovery in the future. This is not only reflected by their increasing re‐emphasis, but also by the immense existing chemical space of natural products that has yet to be fully explored. Clearly, the vast diversity and complexity of natural products is currently unmatched by man‐made synthetic compound libraries. Statistics show that the discovery rate of structurally unique scaffolds of natural products remains stable despite the ever‐increasing number of known structures.75
The use of sp3‐hybridised C−H bond activation has advanced considerably in the last decade and has proven very useful in many natural product total syntheses.165 The convincing atom and step economies of C−H functionalisation as well as the possibility of powerful manipulations in a late‐stage fashion add significant benefits and flexibility to the synthetic strategies. However, the activation of unreactive carbon centres usually requires metal catalysts, which are unsustainable and in many cases lack selectivity.5,7,165, 166, 167 Cytochrome P450 enzymes thus offer an attractive, albeit complex alternative that is in line with environmentally friendly research. The promise to exploit their unique benefits for synthetic application led to extensive effort in protein engineering and many promising studies, which demonstrate its potential for expansion of the chemical toolbox (Figure 7).168,169
Figure 7.
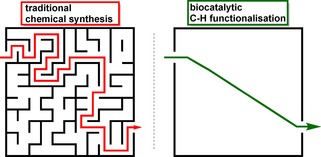
Comparison of functional group installation en route via traditional chemical synthesis versus late‐stage biocatalytic C−H functionalization.
Therefore, this review attempted to illustrate the potential of P450s in lead diversification from the perspective of drug discovery. This was done by presenting novel studies reporting late‐stage functionalisation of natural products and other high‐value compounds, and by complementing the recent review of Renata et al.42 who looked at the first endeavours to introduce P450 enzymes in total syntheses of natural products.
As seen in the examples presented above, P450s work beautifully for the diversification of complex scaffolds post‐synthetically by providing nucleophilic or electrophilic handles that allow further attachment of different building blocks. In this late‐stage manner, novel therapeutic analogues could therefore be generated with synthetic ease resulting in derivatives of improved potency and physiochemical properties.
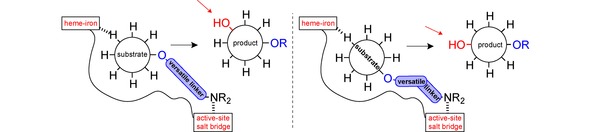
It is worth noting that self‐sufficient P450 enzymes work best because their natural fusion to redox partners permits easier handling and accounts for the observed highest turnover numbers and coupling efficiencies.170 It is thus not astonishing that BM3 is such a prominent P450 candidate and used in the majority of the examples shown here. However, artificial fusion to redox partners may be used to increase efficiency of other P450s and interestingly, a variation in redox partners also offers an opportunity to change the enzyme's selectivity.171
Although substrates of P450s often include high value molecules like fine chemicals and pharmaceuticals, the application of P450s in industrial processes is still limited due to several issues such as instability, poor solvent tolerance and low substrate solubility, or cofactor dependence. In general, these issues can be successfully addressed by immobilisation and use of whole‐cell catalysts, bi‐phasic reaction systems and a cofactor regeneration system, respectively.25,31,54,172,173 Moreover, P450s sometimes also possess an unfavourable (regio‐)selectivity or are too specific. The range of creative strategies to tackle this problem as seen above via anchoring groups, computational modelling, a ‘fingerprinting method’ or simple directed evolution not only paints a promising picture, but also gives an idea of methods yet to be invented to address other P450 drawbacks. While use of cytochrome P450 enzymes in synthesis and in industry seems still in its infancy, it clearly holds an immense potential,168,171 as demonstrated by studies to evolve P450s even into catalysing reactions totally unknown to nature.22,94,174, 175, 176 Eventually, the elegancy and success in protein engineering will be the determining factor, as underlined by the recent Nobel prize in chemistry, awarded to Frances H. Arnold for developing and furnishing the technique of directed evolution of enzymes to a wider audience.177
Biocatalysis on a whole is on the rise. Its ‘fourth wave’178 or ‘golden age’179 of biocatalysis is approaching rapidly, as indicated by an increasing frequency of innovative discoveries, the sprouting influential ideas and the joining of forces among chemists, biologists and engineers. It can be anticipated that advanced computational approaches and laboratory automation will further accelerate the transition of conventional organic synthesis180,181 and scaffold diversification,182,183 towards a modern productive science that uses all kinds of sustainable chemical and enzymatic technologies on an equal footing.184,185
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Nico D. Fessner obtained his MSci in Chemistry and was the recipient of the Joachim Steinke Prize for Excellence in Organic Chemistry at Imperial College London, where he graduated in the group of Prof. Alan Spivey. After working on a project in neuroscience in the group of Dr. Matthieu Louis in Barcelona, he started his PhD under the supervision of Prof. Anton Glieder at the Graz University of Technology as part of the European H2020 MSCA‐ITN project OXYtrain (No.722390).

Acknowledgements
This project received funding from the European Union's Horizon 2020 research and innovation programme, OXYtrain MSCA‐ITN, under grant agreement No. 722390. I would like to thank my academic mentor Prof. A. Glieder for suggesting to write this review. I am grateful to Dr. S. Schmidt for critical reading the manuscript and for fruitful discussions. I would also like to thank Anna Hatzl for proofreading and helpful comments.
N. D. Fessner, ChemCatChem 2019, 11, 2226.
References
- 1. Nicolau K. C., Montagnon T., Molecules That Changed the World, Wiley-VCH, 2008. [Google Scholar]
- 2. Osbourn A., Ross R., Carter G., No Natural Products: Discourse, Diversity, and Design, Wiley & Sons Inc., 2014. [Google Scholar]
- 3. Rahman A. -u., Studies in Natural Products Chemistry Vol. 59, Elsevier Inc., 2018. [Google Scholar]
- 4. Li G., Lou H.-X., Med. Res. Rev. 2018, 38, 1255–1294. [DOI] [PubMed] [Google Scholar]
- 5. Cernak T., Dykstra K. D., Tyagarajan S., Vachal P., Krska S. W., Chem. Soc. Rev. 2016, 45, 453–754. [DOI] [PubMed] [Google Scholar]
- 6. Wencel-Delord J., Glorius F., Nat. Chem. 2013, 5, 369–375. [DOI] [PubMed] [Google Scholar]
- 7. Blakemore D. C., Castro L., Churcher I., Rees D. C., Thomas A. W., Wilson D. M., Wood A., Nat. Chem. 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- 8. Piel J., Natural Products via Enzymatic Reactions, Springer-Verlag Heidelberg, 2010. [Google Scholar]
- 9. Trost B., J. Org. Chem. 2014, 79, 9913–9913. [DOI] [PubMed] [Google Scholar]
- 10. Harmata M., Stategies and Tactics in Organic Synthesis Vol. 8, Elsevier Ltd., Amsterdam, 2012. [Google Scholar]
- 11. Warren S., Wyatt P., Organic Synthesis The Disconnection Approach 2nd Ed, Wiley & Sons Inc., Chichester, 2008. [Google Scholar]
- 12. Clayden J., Greeves N., Warren S., Organic Chemistry 2nd Ed, Oxford University Press, 2013. [Google Scholar]
- 13. Šunjić V., Peroković V. Petrović, Organic Chemistry from Retrosynthesis to Asymmetric Synthesis, Springer International Publishing, New York, 2016. [Google Scholar]
- 14. Groves J. T., Nat. Chem. 2014, 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Montellano P. Ortiz, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer Science & Business Media, 2015. [Google Scholar]
- 16. Urlacher V. B., Girhard M., Trends Biotechnol. 2012, 30, 26–36. [DOI] [PubMed] [Google Scholar]
- 17. Sheldon R. A., Green Chem. 2017, 19, 1–320. [Google Scholar]
- 18. Hollmann F., Arends I. W. C. E., Buehler K., Schallmey A., Bühler B., Green Chem. 2011, 13, 226–265. [Google Scholar]
- 19. Woodley J. M., Trends Biotechnol. 2008, 26, 321–327. [DOI] [PubMed] [Google Scholar]
- 20. Young I. S., Baran P. S., Nat. Chem. 2009, 1, 193–205. [DOI] [PubMed] [Google Scholar]
- 21. Baran P. S., J. Am. Chem. Soc. 2018, 140, 4751–4755. [DOI] [PubMed] [Google Scholar]
- 22. Arnold F. H., Angew. Chem. Int. Ed. 2018, 57, 4143–4148; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4212–4218. [Google Scholar]
- 23. Wang J. bo, Li G., Reetz M. T., Chem. Commun. 2017, 53, 3916–3928. [DOI] [PubMed] [Google Scholar]
- 24. Cheng F., Zhu L., Schwaneberg U., Chem. Commun. 2015, 51, 9760–9772. [DOI] [PubMed] [Google Scholar]
- 25. Behrendorff J. B. Y. H., Huang W., Gillam E. M. J., Biochem. J. 2015, 467, 1–15. [DOI] [PubMed] [Google Scholar]
- 26. Roiban G. D., Reetz M. T., Chem. Commun. 2015, 51, 2208–2224. [DOI] [PubMed] [Google Scholar]
- 27. McIntosh J. A., Farwell C. C., Arnold F. H., Curr. Opin. Chem. Biol. 2014, 19, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reetz M. T., Tetrahedron 2012, 68, 7530–7548. [Google Scholar]
- 29. Turner N., Humphreys L., Biocatalysis in Organic Synthesis: The Retrosynthesis Approach, Royal Society Of Chemistry, 2018. [Google Scholar]
- 30. Hönig M., Sondermann P., Turner N. J., Carreira E. M., Angew. Chem. Int. Ed. 2017, 56, 8942–8973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9068–9100. [Google Scholar]
- 31. de Souza R. O. M. A., Miranda L. S. M., Bornscheuer U. T., Chem. Eur. J. 2017, 23, 12040–12063. [DOI] [PubMed] [Google Scholar]
- 32. Green A. P., Turner N. J., Perspect. Sci. 2016, 9, 42–48. [Google Scholar]
- 33. Turner N. J., O'reilly E., Nat. Chem. Biol. 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]
- 34. Newman D. J., Cragg G. M., J. Nat. Prod. 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 35. Patridge E., Gareiss P., Kinch M. S., Hoyer D., Drug Discovery Today 2016, 21, 204–207. [DOI] [PubMed] [Google Scholar]
- 36. Harvey A. L., Edrada-Ebel R., Quinn R. J., Nat. Rev. Drug Discovery 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]
- 37. Welsch M. E., Snyder S. A., Stockwell B. R., Curr. Opin. Chem. Biol. 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barnes E. C., Kumar R., Davis R. A., Nat. Prod. Rep. 2016, 33, 372–381. [DOI] [PubMed] [Google Scholar]
- 39. Loskot S. A., Romney D. K., Arnold F. H., Stoltz B. M., J. Am. Chem. Soc. 2017, 139, 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Werneburg M., Hertweck C., ChemBioChem 2008, 9, 2064–2066. [DOI] [PubMed] [Google Scholar]
- 41. Henrot M., Richter M. E. A., Maddaluno J., Hertweck C., De Paolis M., Angew. Chem. Int. Ed. 2012, 51, 9587–9591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9725–9729. [Google Scholar]
- 42. King-Smith E., Zwick C. R., Renata H., Biochemistry 2017, 57, 403–412. [DOI] [PubMed] [Google Scholar]
- 43. Su B., Cao Z. C., Shi Z. J., Acc. Chem. Res. 2015, 48, 886–896. [DOI] [PubMed] [Google Scholar]
- 44. Fürstner A., ACS Cent. Sci. 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Egorova K. S., Ananikov V. P., Angew. Chem. Int. Ed. 2016, 55, 12150–12162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12334–12347. [Google Scholar]
- 46. Benkovic S. J., Hammes-Schiffer S., Science (80-. ). 2003, 301, 1196–1202. [DOI] [PubMed] [Google Scholar]
- 47. Guengerich F. P., ACS Catal. 2018, 8, 10964–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Munro A. W., Girvan H. M., Mason A. E., Dunford A. J., McLean K. J., Trends Biochem. Sci. 2013, 38, 140–150. [DOI] [PubMed] [Google Scholar]
- 49. Schyman P., Lai W., Chen H., Wang Y., Shaik S., J. Am. Chem. Soc. 2011, 133, 7977–7984. [DOI] [PubMed] [Google Scholar]
- 50. Shaik S., Cohen S., Wang Y., Chen H., Kumar D., Thiel W., Chem. Rev. 2010, 110, 949–1017. [DOI] [PubMed] [Google Scholar]
- 51. Yosca T. H., Yosca T. H., Rittle J., Krest C. M., Onderko E. L., Silakov A., Calixto J. C., Behan R. K., Green M. T., Science (80-. ). 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Albertolle M. E., Guengerich F. Peter, J. Inorg. Biochem. 2018, 186, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hannemann F., Bichet A., Ewen K. M., Bernhardt R., Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 330–344. [DOI] [PubMed] [Google Scholar]
- 54. Lundemo M. T., Woodley J. M., Appl. Microbiol. Biotechnol. 2015, 99, 2465–2483. [DOI] [PubMed] [Google Scholar]
- 55. Long A., Liti G., Luptak A., Tenaillon O., Nat. Rev. Genet. 2015, 16, 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Firn R. D., Jones C. G., Nat. Prod. Rep. 2003, 20, 382–391. [DOI] [PubMed] [Google Scholar]
- 57. Firn R. D., Jones C. G., Mol. Microbiol. 2000, 37, 989–994. [DOI] [PubMed] [Google Scholar]
- 58. Radman M., Nature 1999, 401, 866–867. [DOI] [PubMed] [Google Scholar]
- 59. Bernhardt R., J. Biotechnol. 2006, 124, 128–145. [DOI] [PubMed] [Google Scholar]
- 60. Fischbach M. A., Clardy J., Nat. Chem. Biol. 2007, 3, 353–355. [DOI] [PubMed] [Google Scholar]
- 61. Tudzynski B., Appl. Microbiol. Biotechnol. 2005, 66, 597–611. [DOI] [PubMed] [Google Scholar]
- 62. Rishton G. M., Am. J. Cardiol. 2008, 101, 43–49.18243858 [Google Scholar]
- 63. Koehn F. E., MedChemComm 2012, 3, 854–865. [Google Scholar]
- 64. Campbell I. B., Macdonald S. J. F., Procopiou P. A., Drug Discovery Today 2018, 23, 219–234. [DOI] [PubMed] [Google Scholar]
- 65. David B., Wolfender J.-L., Dias D. A., Phytochem. Rev. 2015, 14, 299–315. [Google Scholar]
- 66. Newman D. J., Pharmacol. Ther. 2016, 162, 1–9. [DOI] [PubMed] [Google Scholar]
- 67. Chemat F., Vian M. A., Cravotto G., Int. J. Mol. Sci. 2012, 13, 8615–8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Krivoruchko A., Nielsen J., Curr. Opin. Biotechnol. 2015, 35, 7–15. [DOI] [PubMed] [Google Scholar]
- 69. Rodrigues T., Reker D., Schneider P., Schneider G., Nat. Chem. 2016, 8, 531–541. [DOI] [PubMed] [Google Scholar]
- 70. Ventola C. L., P T 2015, 40, 277–83. [PMC free article] [PubMed] [Google Scholar]
- 71. Wright G. D., Nat. Prod. Rep. 2017, 34, 694–701. [DOI] [PubMed] [Google Scholar]
- 72. Moloney M. G., Trends Pharmacol. Sci. 2016, 37, 689–701. [DOI] [PubMed] [Google Scholar]
- 73. Schreiber S. L., Science (80-. ). 2000, 287, 1964–1969. [Google Scholar]
- 74. Bebbington M. W. P., Chem. Soc. Rev. 2017, 46, 5059–5109. [DOI] [PubMed] [Google Scholar]
- 75. Pye C. R., Bertin M. J., Lokey R. S., Gerwick W. H., Linington R. G., Proc. Mont. Acad. Sci. 2017, 114, 5601–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hay M., Thomas D. W., Craighead J. L., Economides C., Rosenthal J., Nat. Biotechnol. 2014, 32, 40–51. [DOI] [PubMed] [Google Scholar]
- 77. Burke M. D., Schreiber S. L., Angew. Chem. Int. Ed. 2004, 43, 46–58; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 48–60. [Google Scholar]
- 78. Corey E. J., Cheng X.-M., Logic of Chemical Synthesis, Wiley-VCH Verlag GmBH & Co. KGaA, New York, 1995. [Google Scholar]
- 79. Corey E. J., Angew. Chem. Int. Ed. 1991, 30, 455–465; [Google Scholar]; Angew. Chem. 1991, 103, 469–479. [Google Scholar]
- 80. Newhouse T., Baran P. S., Hoffmann R. W., Chem. Soc. Rev. 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yu J.-Q., Catalytic Transformations via C−H Activation Vol. 1 & 2, Georg Thieme Verlag KG, 2016. [Google Scholar]
- 82. Pérez P., Alkane C−H Activation by Single-Site Metal Catalysis, Springer Science & Business Media, 2016. [Google Scholar]
- 83. Dyker G., Handbook of C−H Transformations: Applications in Organic Synthesis, Wiley-VCH Verlag GmBH & Co. KGaA, 2008. [Google Scholar]
- 84. Mas-Ballesté R., Que L., Science (80-. ). 2006, 312, 1885–1886. [DOI] [PubMed] [Google Scholar]
- 85. Labinger J. A., Bercaw J. E., Nature 2002, 417, 507–514. [DOI] [PubMed] [Google Scholar]
- 86. Yamaguchi J., Yamaguchi A. D., Itami K., Angew. Chem. Int. Ed. 2012, 51, 8960–9009; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9092–9142. [Google Scholar]
- 87. McMurray L., O'Hara F., Gaunt M. J., Chem. Soc. Rev. 2011, 40, 1885–1898. [DOI] [PubMed] [Google Scholar]
- 88. Chen D. Y. K., Youn S. W., Chem. Eur. J. 2012, 18, 9452–9474. [DOI] [PubMed] [Google Scholar]
- 89. Qiu Y., Gao S., Nat. Prod. Rep. 2016, 33, 562–581. [DOI] [PubMed] [Google Scholar]
- 90. Shugrue C. R., Miller S. J., Chem. Rev. 2017, 117, 11894–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Robles O., Romo D., Nat. Prod. Rep. 2014, 31, 318–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Morrison K. C., Hergenrother P. J., Nat. Prod. Rep. 2014, 31, 6–14. [DOI] [PubMed] [Google Scholar]
- 93. Karimov R. R., Hartwig J. F., Angew. Chem. Int. Ed. 2018, 57, 4234–4241; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4309–4317. [Google Scholar]
- 94. Zhang X., Li S., Nat. Prod. Rep. 2017, 34, 1061–1089. [DOI] [PubMed] [Google Scholar]
- 95. Driggers E. M., Hale S. P., Lee J., Terrett N. K., Nat. Rev. Drug Discovery 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- 96. Yu X., Sun D., Molecules 2013, 18, 6230–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gilbert M. M., DeMars M. D., Yang S., Grandner J. M., Wang S., Wang H., Narayan A. R. H., Sherman D. H., Houk K. N., Montgomery J., ACS Cent. Sci. 2017, 3, 1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xue Y., Wilson D., Zhao L., Liu H. W., Sherman D. H., Chem. Biol. 1998, 5, 661–667. [DOI] [PubMed] [Google Scholar]
- 99. Li S., Podust L. M., Sherman D. H., J. Am. Chem. Soc. 2007, 129, 12940–12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sherman D. H., Li S., Yermalitskaya L. V., Kim Y., Smith J. A., Waterman M. R., Podust L. M., J. Biol. Chem. 2006, 281, 26289–26297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li S., Chaulagain M. R., Knauff A. R., Podust L. M., Montgomery J., Sherman D. H., Proc. Natl. Acad. Sci. USA 2009, 106, 18463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Negretti S., Narayan A. R. H., Chiou K. C., Kells P. M., Stachowski J. L., Hansen D. A., Podust L. M., Montgomery J., Sherman D. H., J. Am. Chem. Soc. 2014, 136, 4901–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Steiner K., Schwab H., Comput. Struct. Biotechnol. J. 2012, 2, e201209010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Toscano M. D., Woycechowsky K. J., Hilvert D., Angew. Chem. Int. Ed. 2007, 46, 3212–3236; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3274–3300. [Google Scholar]
- 105. Ruff A. J., Dennig A., Schwaneberg U., FEBS J. 2013, 280, 2961–2978. [DOI] [PubMed] [Google Scholar]
- 106. Nair P. C., McKinnon R. A., Miners J. O., Drug Metab. Rev. 2016, 48, 434–452. [DOI] [PubMed] [Google Scholar]
- 107. Stjernschantz E., Vermeulen N. P. E., Oostenbrink C., Expert Opin. Drug Metab. Toxicol. 2008, 4, 513–527. [DOI] [PubMed] [Google Scholar]
- 108. Shaik S., De Visser S., in Cytochrome P450 (Ed.: P. Ortiz de Montellano), Springer Science & Business Media, 2005. [Google Scholar]
- 109. Roccatano D., J. Phys. Condens. Matter 2015, 27, 273102. [DOI] [PubMed] [Google Scholar]
- 110. Tian L., Friesner R. A., J. Chem. Theory Comput. 2009, 5, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Petrović D., Bokel A., Allan M., Urlacher V. B., Strodel B., J. Chem. Inf. Model. 2018, 58, 848–858. [DOI] [PubMed] [Google Scholar]
- 112. Le-Huu P., Heidt T., Claasen B., Laschat S., Urlacher V. B., ACS Catal. 2015, 5, 1772–1780. [Google Scholar]
- 113. Le-Huu P., Petrović D., Strodel B., Urlacher V. B., ChemCatChem 2016, 8, 3755–3761. [Google Scholar]
- 114. Omura S., Macrolide Antibiotics: Chemistry, Biology, and Practice, Academic Press, 2002. [Google Scholar]
- 115. Fischbach M. A., Walsh C. T., Science (80-. ). 2009, 325, 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hansen D. A., Rath C. M., Eisman E. B., Narayan A. R. H., Kittendorf J. D., Mortison J. D., Yoon Y. J., Sherman D. H., J. Am. Chem. Soc. 2013, 135, 11232–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lowell A. N., DeMars M. D., Slocum S. T., Yu F., Anand K., Chemler J. A., Korakavi N., Priessnitz J. K., Park S. R., Koch A. A., J. Am. Chem. Soc. 2017, 139, 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Iizaka Y., Higashi N., Ishida M., Oiwa R., Ichikawa Y., Takeda M., Anzai Y., Kato F., Antimicrob. Agents Chemother. 2013, 57, 1529–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. DeMars M. D., Sheng F., Park S. R., Lowell A. N., Podust L. M., Sherman D. H., ACS Chem. Biol. 2016, 11, 2642–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhang K., El Damaty S., Fasan R., J. Am. Chem. Soc. 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]
- 121. Zhang K., Shafer B. M., DeMars M. D., Stern H. A., Fasan R., J. Am. Chem. Soc. 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Renault H., Bassard J. E., Hamberger B., Werck-Reichhart D., Curr. Opin. Plant Biol. 2014, 19, 27–34. [DOI] [PubMed] [Google Scholar]
- 123. Xie D. Y., Ma D.-M., Judd R., Jones A. L., Phytochem. Rev. 2016, 15, 1093–1114. [Google Scholar]
- 124. Kung S. H., Lund S., Murarka A., McPhee D., Paddon C. J., Front. Plant Sci. 2018, 87, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Paddon C. J., Keasling J. D., Nat. Rev. Microbiol. 2014, 12, 355–367. [DOI] [PubMed] [Google Scholar]
- 126. Chaturvedi D., Goswami A., Saikia P. P., Barua N. C., Rao P. G., Chem. Soc. Rev. 2010, 39, 435–454. [DOI] [PubMed] [Google Scholar]
- 127. Huang X., Bergsten T. M., Groves J. T., J. Am. Chem. Soc. 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]
- 128. Gormisky P. E., White M. C., J. Am. Chem. Soc. 2013, 135, 14052–14055. [DOI] [PubMed] [Google Scholar]
- 129. Acevedo-Rocha C. G., Gamble C. G., Lonsdale R., Li A., Nett N., Hoebenreich S., Lingnau J. B., Wirtz C., Fares C., Hinrichs H., ACS Catal. 2018, 8, 3395–3410. [Google Scholar]
- 130. Polic V., Auclair K., Bioorg. Med. Chem. 2014, 22, 5547–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Auclair K., Polic V., in Monooxygenase, Peroxidase Peroxygenase Prop. Mech. Cytochrome P450. Adv. Exp. Med. Biol. (Eds.: E. Hrycay, S. Badiera), Springer Science & Business Media, 2015. [Google Scholar]
- 132. Kille S., Zilly F. E., Acevedo J. P., Reetz M. T., Nat. Chem. 2011, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- 133. Lutz S., Bornscheuer U., Protein Engineering Handbook Vol. 1–2, Wiley-VCH, 2009. [Google Scholar]
- 134. Reetz M. T., Angew. Chem. Int. Ed. 2011, 50, 138–174; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 144–182. [Google Scholar]
- 135. Reetz M. T., Bocola M., Carballeira J. D., Zha D., Vogel A., Angew. Chem. Int. Ed. 2005, 117, 4264–4268. [Google Scholar]
- 136. Oliver C. F., Modi S., Sutcliffe M. J., Primrose W. U., Lian L. Y., Roberts G. C. K., Biochemistry 1997, 36, 1567–1572. [DOI] [PubMed] [Google Scholar]
- 137. Whitehouse C. J. C., Bell S. G., Wong L.-L., Chem. Soc. Rev. 2012, 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- 138. van der Meer J. Y., Biewenga L., Poelarends G. J., ChemBioChem 2016, 17, 1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Acevedo-Rocha C., Ferla M., Reetz M., in Protein Eng. Methods Mol. Biol. (Eds.: U. Bornscheuer, M. Höhne), Humana Press, 2018. [Google Scholar]
- 140. Rea V., Kolkman A. J., Vottero E., Stronks E. J., Ampt K. A. M., Honing M., Vermeulen N. P. E., Wijmenga S. S., Commandeur J. N. M., Biochemistry 2012, 51, 750–760. [DOI] [PubMed] [Google Scholar]
- 141. Venkataraman H., de Beer S. B. A., van Bergen L. A. H., van Essen N., Geerke D. P., Vermeulen N. P. E., Commandeur J. N. M., ChemBioChem 2012, 13, 520–523. [DOI] [PubMed] [Google Scholar]
- 142. Vottero E., Rea V., Lastdrager J., Honing M., Vermeulen N. P. E., Commandeur J. N. M., J. Biol. Inorg. Chem. 2011, 16, 899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Geronimo I., Denning C. A., Rogers W. E., Othman T., Huxford T., Heidary D. K., Glazer E. C., Payne C. M., Biochemistry 2016, 55, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hoebenreich S., Zilly F. E., Acevedo-Rocha C. G., Zilly M., Reetz M. T., ACS Synth. Biol. 2015, 4, 317–331. [DOI] [PubMed] [Google Scholar]
- 145. Khatri Y., Jóźwik I. K., Ringle M., Ionescu I. A., Litzenburger M., Hutter M. C., Thunnissen A. M. W. H., Bernhardt R., ACS Chem. Biol. 2018, 13, 1021–1028. [DOI] [PubMed] [Google Scholar]
- 146. Wienkers L. C., Heath T. G., Nat. Rev. Drug Discovery 2005, 4, 825–833. [DOI] [PubMed] [Google Scholar]
- 147. Kirchmair J., Göller A. H., Lang D., Kunze J., Testa B., Wilson I. D., Glen R. C., Schneider G., Nat. Rev. Drug Discovery 2015, 14, 387–404. [DOI] [PubMed] [Google Scholar]
- 148. Tyzack J. D., Hunt P. A., Segall M. D., J. Chem. Inf. Model. 2016, 56, 2180–2193. [DOI] [PubMed] [Google Scholar]
- 149. Guengerich F. P., Nat. Rev. Drug Discovery 2002, 1, 359–366. [DOI] [PubMed] [Google Scholar]
- 150. Winkler M., Geier M., Hanlon S. P., Nidetzky B., Glieder A., Angew. Chem. Int. Ed. 2018, 10.1002/anie.201800678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Asha S., Vidyavathi M., Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [DOI] [PubMed] [Google Scholar]
- 152. Sawayama A. M., Chen M. M. Y., Kulanthaivel P., Kuo M. S., Hemmerle H., Arnold F. H., Chem. Eur. J. 2009, 15, 11723–11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Usmani K. A., Chen W. G., Sadeque A. J. M., Drug Metab. Dispos. 2012, 40, 761–771. [DOI] [PubMed] [Google Scholar]
- 154. Cusack K. P., Koolman H. F., Lange U. E. W., Peltier H. M., Piel I., Vasudevan A., Bioorg. Med. Chem. Lett. 2013, 23, 5471–5483. [DOI] [PubMed] [Google Scholar]
- 155. Stepan A. F., Tran T. P., Helal C. J., Brown M. S., Chang C., O'Connor R. E., De Vivo M., Doran S. D., Fisher E. L., Jenkinson S., ACS Med. Chem. Lett. 2018, 9, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Obach R. S., Walker G. S., Sharma R., Jenkinson S., Tran T. P., Stepan A. F., J. Med. Chem. 2018, 61, 3626–3640. [DOI] [PubMed] [Google Scholar]
- 157. Shafiee-Nick R., Afshari A. R., Mousavi S. H., Rafighdoust A., Askari V. R., Mollazadeh H., Fanoudi S., Mohtashami E., Rahimi V. B., Mohebbi M., Biomed. Pharmacother. 2017, 94, 541–556. [DOI] [PubMed] [Google Scholar]
- 158. Walker G. S., Bauman J. N., Ryder T. F., Smith E. B., Spracklin D. K., Obach R. S., Drug Metab. Dispos. 2014, 42, 1627–1639. [DOI] [PubMed] [Google Scholar]
- 159. Kolev J. N., O'Dwyer K. M., Jordan C. T., Fasan R., ACS Chem. Biol. 2014, 9, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Babaei G., Aliarab A., Abroon S., Rasmi Y., Aziz S. G. G., Biomed. Pharmacother. 2018, 106, 239–246. [DOI] [PubMed] [Google Scholar]
- 161. Gach K., Długosz A., Janecka A., Naunyn-Schmiedeberg′s Arch. Pharmacol. 2015, 388, 477–486. [DOI] [PubMed] [Google Scholar]
- 162. Seca A., Silva A., Pinto D., in Stud. Nat. Prod. Chem. (Ed.: A. Rahman), Elsevier, 2017. [Google Scholar]
- 163. Tyagi V., Alwaseem H., O'Dwyer K. M., Ponder J., Li Q. Y., Jordan C. T., Fasan R., Bioorg. Med. Chem. 2016, 24, 3876–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Alwaseem H., Frisch B. J., Fasan R., Bioorg. Med. Chem. 2018, 26, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Chen K., Lei X., Curr. Opin. Green Sustain. Chem. 2018, 11, 9–14. [Google Scholar]
- 166. White M. C., Zhao J., J. Am. Chem. Soc. 2018, jacs.8b05195. [Google Scholar]
- 167. Davies H. M. L., Morton D., ACS Cent. Sci. 2017, 3, 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li F., Zhang X., Renata H., Curr. Opin. Chem. Biol. 2019, 49, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Boström J., Brown D. G., Young R. J., Keserü G. M., Nat. Rev. Drug Discovery 2018, DOI 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- 170. Ciaramella A., Minerdi D., Gilardi G., Rend. Lincei 2017, 28, 169–181. [Google Scholar]
- 171. Wei Y., Ang E. L., Zhao H., Curr. Opin. Chem. Biol. 2018, 43, 1–7. [DOI] [PubMed] [Google Scholar]
- 172. Hammerer L., Winkler C. K., Kroutil W., Catal. Lett. 2018, 148, 787–812. [Google Scholar]
- 173. Sheldon R. A., Woodley J. M., Chem. Rev. 2018, 118, 801–838. [DOI] [PubMed] [Google Scholar]
- 174. Brandenberg O. F., Fasan R., Arnold F. H., Curr. Opin. Biotechnol. 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hammer S. C., Knight A. M., Arnold F. H., Curr. Opin. Green Sustain. Chem. 2017, 7, 23–30. [Google Scholar]
- 176. Davis H., Plowright A., Valeur E., Nat. Rev. Drug Discovery 2017, 16, 681–698. [DOI] [PubMed] [Google Scholar]
- 177.“The Nobel Prize in Chemistry 2018. NobelPrize.org. Nobel Media AB 2018. Sat. 10 Nov 2018,” can be found under https://www.nobelprize.org/prizes/chemistry/2018/summary/, 2018.
- 178. Bornscheuer U. T., Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376, 1–7. [DOI] [PubMed] [Google Scholar]
- 179. Turner N. J., Kumar R., Curr. Opin. Chem. Biol. 2018, 43, A1–A3. [DOI] [PubMed] [Google Scholar]
- 180. Coley C. W., Rogers L., Green W. H., Jensen K. F., ACS Cent. Sci. 2017, 3, 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ley S. V., Fitzpatrick D. E., Ingham R. J., Myers R. M., Angew. Chem. Int. Ed. 2015, 54, 3449–3464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3514–3530. [Google Scholar]
- 182. Li J., Ballmer S. G., Gillis E. P., Fujii S., Schmidt M. J., Palazzolo A. M. E., Lehmann J. W., Morehouse G. F., Burke M. D., Science (80-. ). 2015, 347, 1221–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Klucznik T., Mikulak-Klucznik B., McCormack M. P., Lima H., Szymkuć S., Bhowmick M., Molga K., Zhou Y., Rickershauser L., Gajewska E. P., Chem 2018, 4, 522–532. [Google Scholar]
- 184. Lipshutz B. H., Curr. Opin. Green Sustain. Chem. 2018, 11, 1–8. [Google Scholar]
- 185. Romney D. K., Arnold F. H., Lipshutz B. H., Li C. J., J. Org. Chem. 2018, 83, 7319–7322. [DOI] [PubMed] [Google Scholar]