

Eukaryotic ribosome biogenesis requires the action of approximately 200 trans-acting factors and the incorporation of 79 ribosomal proteins (RPs). The delivery of RPs to preribosomes is a major challenge for the cell because RPs are often highly basic and contain intrinsically disordered regions prone to nonspecific interactions and aggregation.
KEYWORDS: chaperone, PDCD2, ribosome biogenesis, Rps2, SSU, Tsr4, uS5
ABSTRACT
Eukaryotic ribosome biogenesis requires the action of approximately 200 trans-acting factors and the incorporation of 79 ribosomal proteins (RPs). The delivery of RPs to preribosomes is a major challenge for the cell because RPs are often highly basic and contain intrinsically disordered regions prone to nonspecific interactions and aggregation. To counteract this, eukaryotes developed dedicated chaperones for certain RPs that promote their solubility and expression, often by binding eukaryote-specific extensions of the RPs. Rps2 (uS5) is a universally conserved RP that assembles into nuclear pre-40S subunits. However, a chaperone for Rps2 had not been identified. Our laboratory previously characterized Tsr4 as a 40S biogenesis factor of unknown function. Here, we report that Tsr4 cotranslationally associates with Rps2. Rps2 harbors a eukaryote-specific N-terminal extension that is critical for its interaction with Tsr4. Moreover, Tsr4 perturbation resulted in decreased Rps2 levels and phenocopied Rps2 depletion. Despite Rps2 joining nuclear pre-40S particles, Tsr4 appears to be restricted to the cytoplasm. Thus, we conclude that Tsr4 is a cytoplasmic chaperone dedicated to Rps2.
INTRODUCTION
Ribosomes are the complex molecular machines responsible for translation. In eukaryotes, ribosome assembly begins in the nucleolus with the cotranscriptional binding of factors to the pre-rRNA. In Saccharomyces cerevisiae, more than 200 trans-acting factors are needed to properly fold, modify, and process the rRNAs and to assemble and export the preribosomal subunits to the cytoplasm, where they undergo final maturation (reviewed in references 1, to ,4). Ribosome assembly involves the incorporation of 79 ribosomal proteins (RPs), most of which join the assembling preribosomal particles in the nucleus (5–7). Many ribosomal proteins are poorly soluble as free proteins, as they contain disordered regions that adopt their final structure only after delivery to the ribosome. In addition, most ribosomal proteins are RNA binding proteins that are highly positively charged and prone to nonspecific interaction with RNA (8). Considering that eukaryotic RPs are synthesized in the cytoplasm and must be transported to the nucleus, their transport to the site of ribosome assembly is particularly challenging given the issues of solubility and nonspecific RNA interaction. To deal with these issues, cells have evolved dedicated RP chaperones and importins (reviewed in references 3 and 9).
Only a limited number of RP chaperones have been identified in yeast (3, 9, 10). Some chaperones are essential (10–14), while the deletion of others is not lethal but gives rise to significant growth defects (15–18). These chaperones appear dedicated to their client RPs, and owing to their unique interactions, there is no unified mechanism of binding to their client proteins. For example, Sqt1 uses its WD repeat β-propeller domain to bind Rpl10 (uL16) (14), Yar1 uses its ankyrin repeats to bind Rps3 (uS3) (19), and Acl4 uses its TPR repeats to bind Rpl4 (uL4) (20). In addition, RP chaperones bind to different regions of their client RPs: Sqt1 and Yar1 recognize the N termini of their clients (14, 19, 21), while Acl4 binds an internal loop of Rpl4 (15, 16, 20). Despite the diversity of these interactions, all chaperones have a shared role in facilitating RP expression. This is generally achieved via cotranslational recognition of the RP by the chaperone (14, 15) to promote RP solubilization by preventing aggregation (10, 14, 15, 17) or to protect RPs from proteasomal degradation (20). As most RPs are incorporated into preribosomal particles in the nucleus, they require import by karyopherins (Kaps) (22), and their chaperones typically remain associated with their client RP during nuclear import (reviewed in references 3 and 9). While RPs are often bound directly by the importin (10, 16, 20, 22, 23), some chaperones act as adaptors by facilitating the interaction with the importin (18, 24). Following nuclear import, the RP-chaperone complex is typically released from the importin in a RanGTP-dependent manner (reviewed in reference 25), and the RPs are subsequently loaded onto preribosomes.
Rps2 (uS5) binds to the central pseudoknot (CPK) of the small subunit (SSU [or 40S]), a critical rRNA feature that establishes the environment for the decoding center (26). Rps2 is needed for the nuclear export of pre-40S particles and their subsequent cytoplasmic maturation involving cleavage of the 20S rRNA intermediate in both budding and fission yeast (6, 27). Despite its role in SSU biogenesis, little is known regarding how Rps2 is chaperoned, imported, and delivered to the pre-40S subunit. In humans, an extraribosomal heterotrimeric subcomplex composed of Rps2, the arginine methyltransferase PRMT3, and PDCD2L is involved in pre-40S biogenesis, while a paralogous complex of Rps2, PRMT3, and PDCD2 may serve a redundant function (28). Additionally, an orthologous Rps2-PDCD2/Zfrp8 complex in Drosophila has been proposed (29). While a chaperone-like function for PDCD2L and PDCD2/Zfrp8 has been suggested (30), it has not been experimentally explored.
We previously identified TSR4 as an essential 46-kDa protein needed for SSU production (31). Tsr4 shows high sequence similarity to the metazoan proteins PDCD2L and PDCD2/Zfrp8 (30), suggesting that it may serve a similar function. Transcriptional repression of TSR4 results in accumulation of the 20S rRNA processing intermediate, but despite its impact on SSU biogenesis, Tsr4 does not cosediment with preribosomal complexes (31), hinting that it has an indirect role in 40S maturation. Here, we report that Tsr4 is a dedicated chaperone which cotranslationally associates with Rps2 to facilitate its expression. However, unlike its metazoan counterparts, Tsr4 does not appear to enter the nucleus, suggesting that its interaction with Rps2 is restricted to the cytoplasm.
RESULTS
N-terminal fragments of Rps2 sequester Tsr4.
Structural alignment of Rps2 (uS5) from S. cerevisiae 80S (PDB code 4V88) to S5 from Escherichia coli 70S (PDB code 4YBB) identified residues 74 to 231 of yeast Rps2 as constituting the evolutionarily conserved core of the protein, whereas residues 1 to 73 and 232 to 254 of Rps2 comprise N- and C-terminal extensions that are not present in the bacterial protein (Fig. 1A). Multiple-sequence alignment of Rps2 from various eukaryotic and prokaryotic species revealed that the N-terminal extension is conserved throughout eukaryotic Rps2 proteins (data not shown). Although significantly shorter, the C-terminal extension adopts a kinked alpha helix (Fig. 1A) that is conserved among eukaryotic Rps2 proteins (data now shown). To explore the functions of the eukaryotic extensions of Rps2, we generated a panel of seven green fluorescent protein (GFP)-tagged Rps2 truncations and deletions expressed under the control of the native RPS2 promoter on centromeric vectors (Fig. 1A). Expression of Rps21–73 or Rps21–223, containing residues 1 to 73 and 1 to 223, respectively, was strongly dominant negative (Fig. 1B) despite their expression from low-copy-number vectors using the native RPS2 promoter. Expression of the slightly longer Rps21–235 fragment was only slightly dominant negative, and this was the only truncation mutant that was able to complement the loss of RPS2, to any degree (Fig. 1C). Interestingly, the dominant negativity of Rps21–73 was partially alleviated by the addition of the C-terminal extension (amino acids [aa] 224 to 254) (Rps2Δcore) (Fig. 1B). Expression of the other fragments of Rps2 did not cause any obvious growth defect, and Western blotting showed that the dominant negative effect of expressing the N-terminal fragments did not correlate with higher protein expression levels (Fig. 1D). Thus, the dominant negative effect of the N-terminal fragments is most easily explained by sequestering a factor or set of factors.
FIG 1.

C-terminal truncations of Rps2-GFP are strongly dominant negative. (A, top) Structural alignment of E. coli S5 (purple) (from PDB code 4YBB) with yeast Rps2 (uS5) (multicolored) (from PDB code 4V88) generated with PyMOL. (Bottom) Cartoon depiction of the alignment of full-length Rps2 and deletion mutants with E. coli S5, colored as described above for the structural alignment. Amino acid positions are relative to yeast Rps2. (B) C-terminal truncations of Rps2 are dominant negative. Wild-type RPS2 and the indicated deletion mutants were ectopically expressed as GFP fusions in wild-type (BY4741) cells under the control of the RPS2 promoter. Tenfold serial dilutions were plated on glucose-containing medium lacking histidine and imaged after 2 days at 30°C. (C) Complementation assay of PGAL1-RPS2 cells (AJY3398) harboring the vectors described above for panel B, as shown by 10-fold serial dilutions plated on glucose-containing medium lacking histidine and grown for 2 days. (D) The expression of the Rps2-GFP fragments from the vectors in panel B was assayed in wild-type cells (BY4741) grown to exponential phase in glucose-containing medium lacking histidine. Rpl10 was used as a loading control. Proteins were extracted as described previously (65), separated by SDS-PAGE, and analyzed by Western blotting.
To identify potential binding partners of the fragments of Rps2-GFP, we expressed each fragment under the control of the galactose-inducible GAL1 promoter in a reg1-501 mutant strain that allows galactose induction in the presence of glucose (32). After brief expression of the Rps2-GFP fragments, extracts were prepared, and ribosomes were removed by ultracentrifugation prior to immunoprecipitation (IP) of the Rps2-GFP fragments to identify extraribosomal binding partners. Each fragment showed a slightly different set of copurifying binding partners (Fig. 2A). However, some copurifying species were common to the N-terminal fragments that were not present in the C-terminal fragments and vice versa. The N-terminal fragments all coimmunoprecipitated a species that migrated slightly above 50 kDa on SDS-PAGE gels (Fig. 2A, lanes 2 to 4) and was identified as Tsr4 by mass spectrometry. Although full-length Rps2-GFP comigrated with Tsr4 and masked its signal in SDS-PAGE gels (Fig. 2A, lane1), Tsr4 was detected by mass spectrometry in the wild-type (WT) sample as well. We also found unique spectra for Kap123 and Kap104 in the coimmunoprecipitation experiments with core-containing Rps2-GFP fragments (Fig. 2A, lanes 1, 3, 5, and 6). Kap123 is considered the primary importin for RPs (22), while Kap104 has been shown to interact with specific RPs (16, 18, 20, 23). This result suggests that the Rps2 core is needed for the recruitment of an importin.
FIG 2.

C-terminal truncations of Rps2-GFP sequester Tsr4. (A) The indicated Rps2-GFP proteins and GFP alone were expressed in strain AJY1134. Extracts were cleared of ribosomes by ultracentrifugation. GFP fusion proteins were immunoprecipitated and analyzed by SDS-PAGE and staining with Coomassie blue. The indicated bands (brackets) were excised for mass spectrometry. Proteins identified with >50 total peptides are listed below each lane, ordered by abundance. Proteins of particular interest are indicated by bold text. Asterisks, bait proteins; triangles, Tsr4. (B) The indicated Rps2-GFP fragments and GFP alone were expressed in AJY4363. Extracts were generated, and Tsr4-TEV-13myc was immunoprecipitated. The coprecipitating proteins (top) and inputs (bottom) were separated by SDS-PAGE and analyzed by Western blotting probing with anti-c-Myc and anti-GFP. (C) The toxic C-terminal truncations of Rps2-GFP, Rps21–73 and Rps21–223, were coexpressed in wild-type (BY4741) cells with a high-copy-number TSR4 or empty vector. Tenfold serial dilutions were plated on glucose-containing medium lacking histidine and uracil and grown for 3 days at 30°C. (D) Tsr4 association with endogenous Rps2 was determined by immunoprecipitation of Tsr4-TEV-13myc or Tsr4-GFP from cells expressing RPS2 (+) or repressed for RPS2 (−) using anti-myc beads. Rps2 was detected using an antibody against Rpl30 that cross-reacts with Rps2 (61). The signal for Tsr4-TEV-13myc in the IPs is denoted by asterisks, and a contaminating IgG species is denoted by triangles.
To independently verify that the N terminus of Rps2 facilitates its interaction with Tsr4, we performed the converse IP experiment. To this end, we genomically tagged TSR4 with TEV-13myc in the reg1-501 strain and briefly expressed the various Rps2-GFP fragments. We also expressed GFP alone as a negative control. Extracts were prepared, immunoprecipitations were done using Tsr4-TEV-13myc as bait, and Western blotting was used to detect coprecipitation of the GFP-tagged Rps2 fragments or GFP alone. The Rps2-GFP fragments harboring the N terminus coprecipitated with Tsr4 more abundantly than a fragment lacking the N terminus, while GFP alone did not coprecipitate, indicating specificity for the Rps2 fragments (Fig. 2B). This result suggests that while Tsr4 has some affinity for the core of Rps2, the N terminus is its primary binding site.
TSR4 is an essential gene that was first identified in a bioinformatic analysis of novel ribosome biogenesis factors and is required for the processing of the 20S rRNA intermediate (31). However, its specific function was not further explored. Because the Rps2 fragments harboring the N terminus all associated with Tsr4, while the fragments lacking it did not, we speculated that the N-terminal fragments were dominant negative due to sequestration of Tsr4. Consistent with this notion, the dominant negative defect of Rps21–73 was fully alleviated by expression of TSR4 from a high-copy-number vector (Fig. 2C). Surprisingly, the dominant negative effect of the longer Rps21–223 fragment was only partially suppressed by overexpression of TSR4, suggesting that it has a negative effect in addition to Tsr4 sequestration. Quantitative Western blot analyses using a GFP-tagged version of the 60S biogenesis factor Nmd3 for a standard curve indicated that the amount of Tsr4-GFP is roughly one-fifth the amount of Nmd3-GFP (data not shown). On average, there are approximately 7,500 Nmd3 molecules per cell (33). Thus, we estimate that each cell has about 1,500 Tsr4 molecules. This number is low compared to those of other RP chaperones like Yar1 and Sqt1, which are estimated to be present at about 11,000 and 7,600 molecules per cell, respectively (33). Consequently, low expression levels of Rps21–73 or Rps21–223 could effectively sequester the limited amount of Tsr4 available in a cell and have a profound effect on cell growth.
To ask if Tsr4 forms a complex with endogenous Rps2, we performed IP experiments using genomically expressed Tsr4-TEV-13myc as bait and used Western blotting to probe for the co-IP of native Rps2. Cells expressing genomic Tsr4-GFP were used for a negative control. Indeed, Rps2 was present in the 13myc-tagged Tsr4 IP but not the negative control (Fig. 2D, cf. lanes 4 and 6). Moreover, Rpl30 did not coimmunoprecipitate with Tsr4, indicating a specific association of Tsr4 with Rps2. To test if Tsr4 specifically associates with newly synthesized Rps2 and not preexisting Rps2 already incorporated into ribosomes, we transiently repressed the GAL1 promoter-driven expression of Rps2 for 2 h with the addition of 2% glucose to the medium, a sufficient time to disrupt Rps2 production and 40S ribosome biogenesis and export (6). We then assayed for copurification of Rps2 with Tsr4 from these cells. Indeed, no Rps2 was detected in the Tsr4 IP from these cells despite being readily detectable in the input (Fig. 2D, cf. lanes 5 and 2). These results suggest that Tsr4 transiently interacts with extraribosomal Rps2, prior to its assembly into ribosomes.
Because the Rps2-GFP immunoprecipitations (Fig. 2A) were done on samples precleared of ribosomes, we next asked if the most toxic N-terminal fragments of Rps2 could associate with ribosomes. Full-length and N-terminal fragments of Rps2 were expressed from centromeric vectors under the control of their cognate promoter in a PGAL1-RPS2 strain in which endogenous RPS2 expression was repressed with glucose for 5 h to eliminate competition between endogenous RPS2 and the plasmid-borne constructs. Extracts were subjected to ultracentrifugation through sucrose density gradients to separate extraribosomal proteins from ribosome-bound proteins. Full-length Rps2-GFP cosedimented with 40S, 80S, and polysomes, with very little present as free protein (Fig. 3A), consistent with it being a functional protein (Fig. 1C) that is incorporated into ribosomes. In contrast, Rps21–73 was present exclusively at the top of the gradient, indicating that it does not stably associate with ribosomes (Fig. 3B). A portion of the Rps21–223 pool was also present at the top of the gradient, but unlike Rps21–73, the larger Rps21–223 fragment also cosedimented with 40S subunits and 80S ribosomes in sucrose density gradients, with very small quantities detected in polysomes (Fig. 3C). It seems likely that the incorporation of the truncated Rps21–223 fragment into ribosomes disrupts either their maturation or function, accounting for the observation that the dominant negative effect of this fragment cannot be fully suppressed by overexpression of TSR4 (Fig. 2B). Notably, the UV trace from the cells expressing Rps21–223-GFP showed a deficit of 40S relative to 60S subunits compared to WT Rps2-GFP, indicating a defect in 40S biogenesis (compare Fig. 3C to Fig. 3A, black traces). The ability of N-terminal fragments of Rps2 to titrate out Tsr4, blocking its function in ribosome assembly and leading to a dominant negative phenotype, is reminiscent of genetic interactions between some RPs and their chaperones (14, 15, 21) and led us to hypothesize that Tsr4 is a dedicated chaperone for Rps2.
FIG 3.
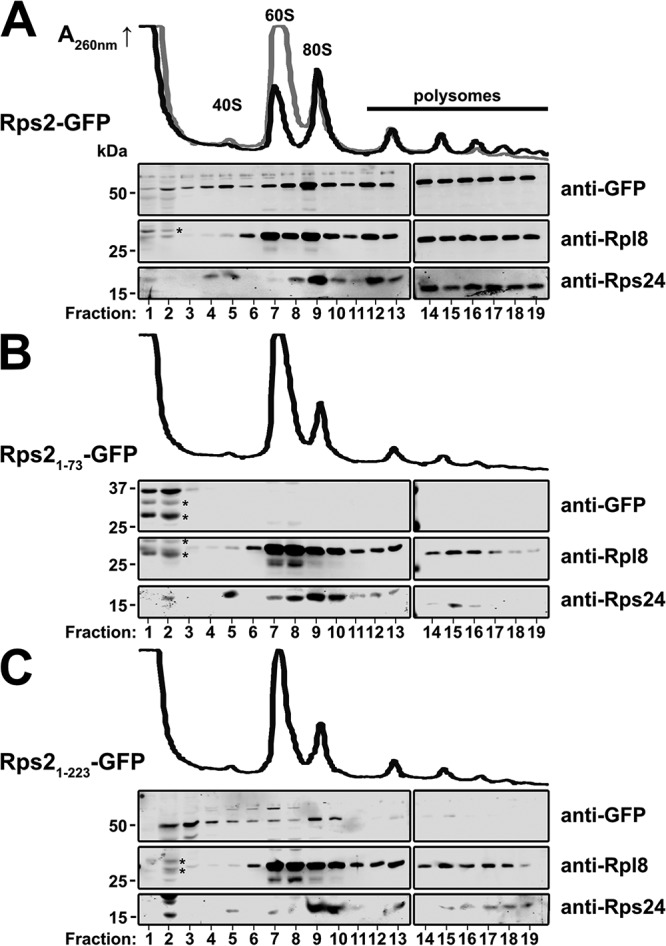
Sucrose density gradient sedimentation of Rps2-GFP fragments. The PGAL1-RPS2 strain AJY3398 containing an empty vector or vectors expressing GFP fusions of the indicated C-terminal Rps2 truncations under the control of the cognate promoter was cultured in glucose-containing medium for 5 h to repress genomic RPS2 expression prior to treatment with cycloheximide for 10 min. Extracts were generated and fractionated on sucrose density gradients, and proteins were precipitated. The sedimentation of relevant proteins was monitored by Western blotting following SDS-PAGE. Traces of continuous UV absorbance at 260 nm are shown. (A) Cells expressing wild-type Rps2-GFP (black trace) or no Rps2 (gray); (B and C) cells expressing Rps21–73-GFP and Rps21–223-GFP, respectively. Degradation products of the Rps2-GFP fragments are denoted by asterisks.
Residues within the extreme N terminus and a helical bundle of Rps2 facilitate the interaction with Tsr4.
Because the genetic interaction between RPS2 and TSR4 likely reflects a physical interaction, we sought to more finely map this interaction by identifying residues of Rps2 that are necessary for its interaction with Tsr4. We reasoned that we could use the dominant negative growth phenotype of Rps21–73 as a proxy for its interaction with Tsr4. Thus, Rps21–73 mutants that alleviate its toxicity would likely disrupt its interaction with Tsr4. To this end, we used error-prone PCR to generate a pool of amplicons containing random mutations in RPS21–73-GFP. We recombined the amplicons into a GAL1-inducible expression vector in vivo by homologous recombination. We plated the library on galactose-containing medium and screened for relief of the dominant negative growth phenotype. We isolated 110 colonies from approximately 3,000 transformants with various degrees of improved growth, of which 73 showed GFP fluorescence indicating expression of the fusion protein. Sanger sequencing identified 48 single point mutations within the RPS21–73 coding region, revealing 35 unique mutations that alleviated the dominant negativity of RPS21–73 (Fig. 4A and B and data not shown).
FIG 4.

Tsr4 recognizes two regions of the N terminus of Rps2. (A) Primary sequence of the N-terminal 73 residues of yeast Rps2, with charged residues indicated (+ and −) and secondary structure elements from PDB code 4V88 indicated at the top. The dashed line indicates a region not resolved in the structure under PDB code 4V88. Mutations that alleviated the dominant negative growth phenotype of Rps21–73-GFP are listed below the primary sequence. Strong mutants (white text), moderate mutants (bold black text), and weak mutants (black text) are indicated at the bottom. (B) Growth of a subset of mutants that relieved the dominant negative phenotype of Rps21–73-GFP. Tenfold serial dilutions of wild-type (BY4741) cells expressing the indicated mutants under the control of the galactose-inducible GAL1 promoter were plated on glucose- or galactose-containing medium lacking uracil and grown for 2 days at 30°C. (C) The helical bundle of Rps2 residues 34 to 73, shown as a cartoon depiction with a transparent electrostatic surface (from PDB code 4V88). Positive, negative, and neutral surfaces are indicated by blue, red, and white, respectively. Amino acids that can be mutated to alleviate the growth inhibition of Rps21–73 are shown as sticks, with residues included in panel B shown in yellow. (D) Strength of the interaction between Tsr4 and wild-type (WT) Rps21–73-GFP or the indicated Rps21–73-GFP mutants, as determined by their ability to coimmunoprecipitate with Tsr4-TEV-13myc.
Residues 1 to 33 of the N-terminal extension of Rps2 contain an RG-rich motif (Fig. 4A) commonly found in intrinsically disordered regions of RNA binding proteins (34). Consistent with its predicted disordered nature, this region of Rps2 has not been resolved in crystal or cryo-electron microscopy (EM) structures of 40S subunits. Five of the unique mutations identified in our screen resulted in substitutions within the RG-rich motif (Fig. 4A). Residues 34 to 73 fold into three short alpha helices that form a small helical bundle (Fig. 4A and C). The remaining 30 mutated residues were within this helical bundle (Fig. 4A and C), and the majority of these mapped to a positively charged surface (Fig. 4C). These results suggest that Tsr4, which has an overall net negative charge (pI = 4.5), binds the N-terminal extension of Rps2, in part, through electrostatic interactions.
To determine if these mutants lose interaction with Tsr4, we chose 10 representative mutants to investigate further (Fig. 4B). Of these, seven mapped to the N-terminal helical bundle of Rps2 (Fig. 4C, yellow residues). We expressed wild-type and mutant RPS21–73-GFP under the control of the galactose-inducible GAL1 promoter in the reg1-501 strain containing genomic TSR4-TEV-13myc. As a negative control, we expressed GFP alone. After a brief galactose induction, extracts were prepared, Tsr4-TEV-13myc was immunoprecipitated, and Western blotting was used to detect coprecipitation of the Rps21–73-GFP proteins or GFP. Wild-type Rps21–73-GFP showed a specific association with Tsr4, as GFP alone was not detected in the Tsr4 immunoprecipitation (Fig. 4D, left). All of the mutants showed a reduced association with Tsr4 compared to wild-type Rps21–73. The apparent differences in binding were not due to variable expression of the mutant proteins, as all showed similar levels of expression (Fig. 4D, right). Interestingly, the N-terminus-proximal mutations within Rps21–73-GFP appeared to have a stronger impact on its interaction with Tsr4 than did the mutations farther from the N terminus. Thus, the first 37 amino acids of Rps2 may be more critical for the interaction. We attempted to define the region(s) of Tsr4 needed for its interaction with Rps2 using the yeast two-hybrid assay. However, truncations from either end of the protein resulted in a loss of interaction and were uninformative (data not shown). Similar results were recently reported by a study that was published while this article was in review (35). Considering that the related YwqG protein folds into a single globular domain (30), Tsr4 does not seem amenable to domain mapping via serial truncations.
Tsr4 cotranslationally associates with Rps2 and is needed for its efficient expression.
Dedicated RP chaperones often bind their client proteins cotranslationally, soon after their interaction domain is presented from the exit tunnel of the ribosome (14, 15). For example, Sqt1 is a chaperone for Rpl10 (uL16) that cotranslationally associates with the N terminus of Rpl10 to allow copurification of RPL10 mRNA (14). Based on our observations that Tsr4 interacts with the N terminus of Rps2 (Fig. 2 and 4), we speculated that Tsr4 could similarly bind Rps2 cotranslationally. In order to test this idea, we affinity purified Tsr4-TAP and Sqt1-TAP and probed for specific mRNAs using reverse transcription-quantitative PCR (RT-qPCR). Cells were treated with cycloheximide (CHX) to arrest translation elongation. Extracts were made, and affinity purifications were done in the presence of CHX. We isolated the copurifying RNA and used it as the template in RT-qPCRs, amplifying either RPS2 or RPL10 mRNA for both the Tsr4 and Sqt1 purifications. We expected that Tsr4 would efficiently pull down RPS2 mRNA but not RPL10 mRNA, while Sqt1 would specifically purify RPL10 mRNA but not RPS2 mRNA. To display enrichment relative to the expected target mRNA of each bait, we first normalized the number of mRNA molecules isolated from immunoprecipitations to the number of mRNA molecules in the input and then set this value for the expected target mRNA for each bait to 1.0 (Fig. 5A). This analysis revealed that Tsr4-tandem affinity purification tag (TAP) enriched specifically for RPS2 mRNA but not RPL10 mRNA. Conversely, Sqt1-TAP enriched for RPL10 mRNA but not RPS2 mRNA. We also performed this experiment using Tsr4-GFP as bait and again saw an enrichment of the RPS2 mRNA over the RPL10 mRNA. This experiment, as performed, shows that Tsr4 can copurify RPS2 mRNA. However, it cannot inform us as to whether or not the copurification is due to a cotranslational association with nascent Rps2 or due to a direct binding of Tsr4 to the mRNA. To distinguish between these two possibilities, we repeated the Tsr4-TAP pulldowns and RT-qPCR for cells cultured and treated with or without CHX. Extracts were then prepared in the presence of CHX, for the CHX-treated cells, or in the presence of puromycin, for CHX-untreated cells. Puromycin induces the premature release of nascent peptides from the ribosome (36). Thus, if the association of Tsr4 with the RPS2 mRNA was due to cotranslational binding of nascent Rps2, then one would expect a loss of mRNA in the IP from the puromycin-containing extracts. Indeed, we saw an approximately 50% decrease of RPS2 mRNA in the puromycin-treated samples relative to the CHX-treated samples (Fig. 5B). This result suggests that the copurification of the RPS2 mRNA with Tsr4 is due, at least in part, to cotranslational binding of Tsr4 to nascent Rps2. However, whether or not Tsr4 binds directly to the RPS2 mRNA cannot be ruled out.
FIG 5.
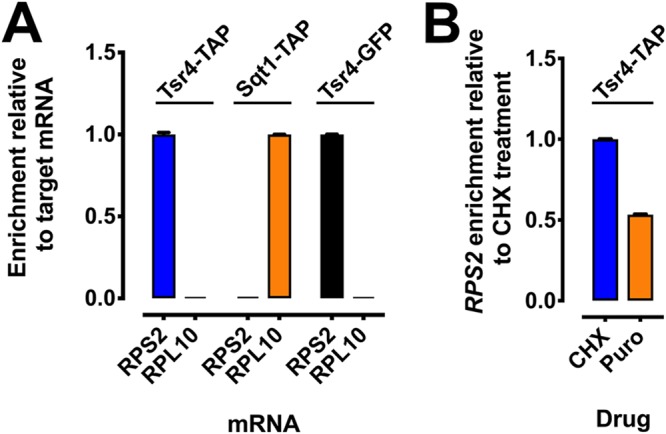
Tsr4 associates cotranslationally with nascent Rps2. (A) The association of Tsr4 and Sqt1 with the mRNAs of their client proteins, Rps2 and Rpl10, respectively, was determined by their ability to coimmunoprecipitate the mRNA of their client protein, as measured by RT-qPCR. To prevent ribosome runoff of mRNAs, cells were treated with CHX prior to harvest, and extracts and immunoprecipitations were done in the presence of CHX. Abundance is shown as mRNA enrichment relative to the expected target mRNA of each bait. The qPCRs were done in technical triplicates; error bars indicate the standard deviations for the ratios of mRNA molecules in the IP to the input for each replicate. (B) Cotranslational binding of Tsr4 with the RPS2 mRNA was determined using an approach similar to that described above for panel A, except that cell cultures were either treated or untreated with CHX prior to harvest, and extracts were prepared in the presence of CHX or puromycin (Puro), respectively, to impede or induce ribosome disassociation from nascent peptides, respectively. Abundance is shown as RPS2 mRNA enrichment relative to the CHX-treated sample.
Chaperones often facilitate the expression of their target RPs (10, 13, 14, 17). To test if Tsr4 similarly enhances the expression of Rps2, we transformed a vector encoding Rps2-GFP under the control of a galactose-inducible promoter into strains harboring either a wild-type or a temperature-sensitive allele of Tsr4 (tsr4-ts). The tsr4-ts mutant was unable to grow at the nonpermissive temperature of 37°C but was viable at 30°C and at room temperature (∼25°C). Cells were cultured in raffinose-containing medium at room temperature and then shifted to 37°C for 2 h to inactivate the temperature-sensitive Tsr4 protein. We then transiently expressed Rps2-GFP by the addition of galactose for 90 min. This experimental design allowed us to specifically probe for any Rps2 that was made only after the shift to 37°C. Extracts were prepared, and free proteins were separated from ribosomes by ultracentrifugation. Western blotting was performed on the input, the supernatant, and the pellet. This analysis showed that the levels of Rps2-GFP in the input, supernatant, and pellets were reduced in the strain background harboring tsr4-ts relative to the strain harboring the wild-type TSR4 allele (Fig. 6A). This decrease of Rps2-GFP was also apparent when cells were imaged by fluorescence microscopy after 60 min of induction (Fig. 6B). Together, these results suggest that functional Tsr4 is needed for the efficient expression of Rps2.
FIG 6.

Tsr4 is needed for efficient expression of Rps2. (A) The expression of Rps2-GFP was analyzed in tsr4-ts (AJY2873) cells compared to wild-type (WT) TSR4 cells (BY4741). Cells were shifted to a nonpermissive temperature for 2 h prior to the expression of Rps2-GFP for 90 min by the addition of galactose. Comparable amounts of extract were overlaid onto sucrose cushions prior to ultracentrifugation to separate free proteins (supernatant [Super]) from ribosomes (pellet). Rpl8 and Xrn1 were used as loading controls. The relative levels of Rps2 are quantified at the bottom as the ratio of Rps2 to Xrn1 and the ratio of Rps2 to Rpl8, where the ratio for the wild-type sample was set to a value of 1. (B) GFP fluorescence microscopy of TSR4 or tsr4-ts cells expressing Rps2-GFP. Cells were cultured as described above for panel A, except that Rps2-GFP was induced for 60 min after the 2-h shift to 37°C. (C) Tenfold serial dilutions of TSR4 (BY4741) and tsr4-ts (AJY2873) cells transformed with either an empty vector (pRS416) or a vector containing RPS2 (pAJ4244) under the transcriptional control of its cognate promoter after 3 days at the indicated temperatures on SD-ura medium containing glucose. (D) tsr4Δ cells (AJY4359) containing a URA3-marked TSR4 vector were transformed with HIS3-marked vectors containing TSR4 (pAJ4232) or RPS2 (pAJ4207) or an empty vector (pRS416), and complementation was assessed by plasmid shuffle on 5FOA-containing medium after 6 days of growth. (E) Tenfold serial dilutions of tsr4Δ cells harboring the TSR4 and RPS2 vectors from panel D were spotted onto SD-his medium containing glucose and grown for 3 days at 30°C. (F) Tenfold serial dilutions of PGAL1-3×HA-TSR4 (AJY4298) cells harboring an empty vector (pRS413) or a vector containing either TSR4 (pAJ4232) or RPS2 (pAJ4207) were grown on SD-his medium containing glucose for 3 days at 30°C. (G) Polysome profile traces of extracts generated from the cells described above for panel F cultured in SD-his medium containing glucose for 8 h to deplete endogenous Tsr4.
The need for a dedicated chaperone may be bypassed by increased production of its client protein (10, 14–17). We asked if the temperature sensitivity of a tsr4-ts mutant could be suppressed by an increased gene dosage of RPS2. Indeed, excess RPS2 expressed from a centromeric vector fully suppressed the slow-growth defect of the tsr4-ts mutant at 30°C and partially suppressed its lethality at 37°C (Fig. 6C). This suggests that increased expression of Rps2 stabilizes the product of tsr4-ts and/or bypasses the need for Tsr4 altogether. To determine if overexpression of RPS2 could bypass the lethality of a tsr4Δ mutant, we used a plasmid shuffle technique. We introduced HIS3-marked plasmids containing TSR4, RPS2, or nothing (empty vector) into a tsr4Δ strain harboring a URA3-marked vector containing TSR4. We then assayed for the ability of these HIS3-marked vectors to shuffle out the URA3-marked vector by their growth on medium containing the 5-fluoroorotic acid (5FOA). As expected, the HIS3-marked TSR4 vector complemented the tsr4Δ strain (Fig. 6D). The RPS2 vector suppressed the lethality of the tsr4Δ strain, while the empty vector did not support growth. We confirmed the absence of TSR4 in the RPS2-carrying cells by PCR (data not shown). To compare the growth phenotypes of the viable tsr4Δ strains, we spotted equivalent numbers of TSR4-complemented and RPS2-suppressed tsr4Δ cells onto glucose-containing medium lacking His. This revealed a slow-growth phenotype in the RPS2-suppressed tsr4Δ cells relative to the TSR4-complemeted tsr4Δ cells (Fig. 6E), suggesting only a partial bypass of tsr4Δ, akin to the suppression phenotype in the tsr4-ts cells (Fig. 6C). Partial suppression was also seen in cells in which TSR4 expression was repressed by a glucose-repressible promoter (PGAL1-3×HA-TSR4) (Fig. 6F). To ascertain whether or not the rescue of growth correlated with improved 40S biogenesis, we cultured PGAL1-3×HA-TSR4 cells harboring either TSR4, RPS2, or an empty vector in glucose-containing medium for 8 h. Extracts were generated and subjected to ultracentrifugation through sucrose density gradients. Polysome profiles from these samples revealed a strong defect in 40S biogenesis in cells containing the empty vector compared to TSR4-expressing cells, indicated by the large imbalance of free 40S relative to 60S subunits (Fig. 6G). These cells also displayed severely depressed levels of polysomes. Ectopic expression of RPS2 led to partial restoration of subunit imbalance and a slight restoration of polysomes compared to cells carrying the empty vector. Thus, increased gene dosage of RPS2, and a presumably concomitant increase in the production of Rps2 protein, can partially bypass the essential function of Tsr4, consistent with the notion that Tsr4 is a chaperone for Rps2.
Repression of TSR4 or RPS2 expression blocks pre-40S export.
Pre-40S particles exported from the nucleus contain 20S pre-RNA. In the cytoplasm, the endonuclease Nob1 cleaves 20S rRNA into the mature 18S (37, 38), liberating a small 5′ fragment of internal transcribed spacer 1 (ITS1) that is rapidly degraded by the exoribonuclease Xrn1 (39). Previous studies in S. cerevisiae and Schizosaccharomyces pombe, using fluorescent in situ hybridization (FISH) with an oligonucleotide probe specific to the D-A2 region of ITS1 of rRNA intermediates or using fluorescence microscopy using Rps7-GFP as a marker for 40S subunits, showed that depletion of Rps2 prevents pre-40S export (6, 27). If Tsr4 is a chaperone for Rps2, then one would expect that the depletion of Tsr4 should phenotypically mimic the depletion of Rps2. However, our laboratory previously reported that depletion of Tsr4 does not block the export of pre-40S particles from the nucleus (31), but that study used Rps2-GFP as a marker for pre-40S. Because Tsr4 is needed for the expression of Rps2 (Fig. 6A and B), the depletion of Tsr4 leads to the loss of newly synthesized Rps2-GFP, rendering Rps2-GFP ineffective as a reporter for the role of Tsr4 in pre-40S export.
To revisit the question of whether or not Tsr4 is important for pre-40S export, we generated strains in which RPS2 or TSR4 was placed under the transcriptional control of the glucose-repressible GAL1 promoter in an xrn1Δ background. Deletion of XRN1 allows the liberated ITS1 fragment to accumulate in the cytoplasm, providing a clearer change in the FISH signal when export is blocked (40). The transcription of RPS2 or TSR4 was repressed for 2 h by the addition of glucose, after which cells were prepared for FISH using an oligonucleotide probe that hybridizes specifically to the ITS1 region. As expected, the signal for ITS1 was predominantly cytoplasmic in the xrn1Δ strain that expresses both RPS2 and TSR4 (Fig. 7). Upon repression of RPS2 or TSR4 transcription, the signal for ITS1 accumulated in the nucleoplasm, indicating a defect in pre-40S export. Thus, like Rps2, Tsr4 is needed for pre-40S export.
FIG 7.
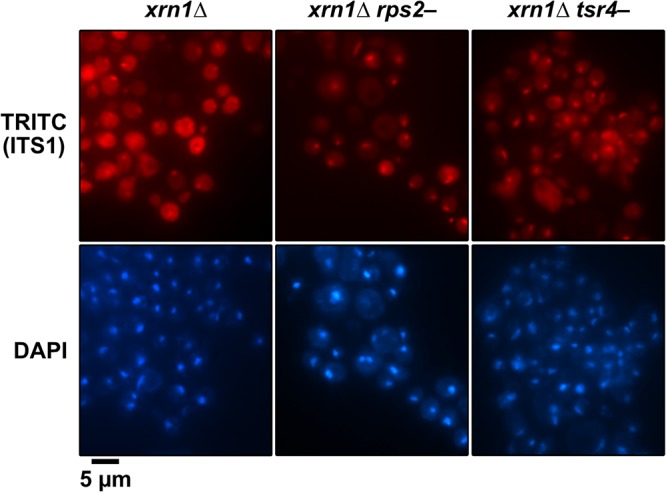
Repression of TSR4 blocks the nuclear export of pre-40S. The subcellular distribution of ITS1 was analyzed by FISH with a Cy3-labeled oligonucleotide complementary to the D-A2 region of ITS1 (tetramethyl rhodamine isothiocyanate [TRITC]) in xrn1Δ (RKY1977), PGAL1-RPS2 xrn1Δ (AJY4300), and PGAL1-3×HA-TSR4 xrn1Δ (AJY4297) cells after a switch to glucose-containing medium for 2 h. The position of the nucleus was determined by DAPI staining and the overall cell outline by differential interference contrast (DIC) microscopy.
Tsr4 appears restricted to the cytoplasm.
Dedicated RP chaperones typically remain bound to their client RPs during nuclear import (13, 15, 17, 18) to prevent their interaction with RNA cargo being exported from the nucleus and to facilitate loading of the RP onto preribosomes once inside the nucleus. Moreover, metazoan Tsr4 orthologs appear to shuttle between the nucleus and cytoplasm (28, 41). Tsr4 contains a weak putative, canonical bipartite nuclear localization signal (NLS) sequence (Fig. 8A), suggesting that Tsr4 may usher Rps2 into the nucleus. However, mutants containing alanine substitutions of lysine residues that disrupt the putative NLS (Tsr4ΔNLS-GFP) fully complemented the loss of Tsr4 (Fig. 8A), suggesting that this NLS is either nonfunctional or redundant with other import mechanisms. To further test the possibility that Tsr4 shuttles between the nucleus and the cytoplasm, we blocked Crm1 (Xpo1)-dependent nuclear export. Crm1 is a nuclear export receptor for proteins containing leucine-rich nuclear export sequences and is required for nuclear export of ribosomal subunits (reviewed in reference 42). In yeast, the Crm1T539C mutation sensitizes cells to inhibition by leptomycin B (LMB) (43). To test whether Crm1 inhibition has an effect on Tsr4 localization, we treated Crm1T539C cells expressing Tsr4-mCherry with LMB for 45 min. As a control for LMB efficacy, this strain also contains genomically encoded Nmd3-GFP, a Crm1 ligand required for 60S export whose steady-state distribution is cytoplasmic but rapidly accumulates in the nucleus upon Crm1 inhibition (43). Tsr4-mCherry did not accumulate in the nucleus following treatment with LMB, while Nmd3-GFP relocalized to the nucleus, as expected (Fig. 8B). This result suggests that Tsr4 does not shuttle between the nucleus and cytoplasm in a Crm1-dependent manner. Because Tsr4-mCherry fully complements the loss of TSR4, the absence of an apparent NLS and the inability to trap Tsr4 in the nucleus by inhibition of Crm1 lead us to conclude that it is unlikely that Tsr4 shuttles. We suggest that, unlike its metazoan counterparts, yeast Tsr4 hands Rps2 off to other factors prior to its entry into the nucleus. Our observation that the core of Rps2 coimmunoprecipitates karyopherins (Fig. 2A) raises the possibility that Tsr4 hands Rps2 off directly to import factors.
FIG 8.

Tsr4 does not shuttle between the nucleus and cytoplasm. (A) Growth of PGAL1-3×HA-TSR4 (AJY4298) cells harboring an empty vector (pRS416) or vectors containing TSR4-GFP (pAJ4186) or TSR4ΔNLS-GFP (pAJ4267), as shown by 10-fold serial dilutions plated on glucose-containing medium lacking uracil after 2 days. The primary sequence for residues 121 to 140 of Tsr4 is shown at the top. The putative bipartite NLS predicted by PSORT II (66) is underlined; alanine substitutions of Tsr4ΔNLS are indicated below the sequence. (B) Subcellular localization of ectopically expressed Tsr4-mCherry (pAJ4185) and genomically expressed Nmd3-GFP after 45 min of inhibition of Crm1T539C with 0.1 μM LMB in exponential-phase AJY1705 cells. Treatment with 1% ethanol (EtOH) was used in parallel as a vehicle control.
DISCUSSION
Rps2 is a universally conserved RP, but eukaryotic Rps2 contains unique N- and C-terminal extensions. Here, we provide evidence that these eukaryotic extensions of Rps2 drive its interaction with Tsr4, which we identify as a dedicated cytoplasmic chaperone for Rps2. A complementary study reporting that Tsr4 is a chaperone of Rps2 was published while this article was in review (35). Like other chaperones of RPs, Tsr4 finds its client protein cotranslationally, recognizing the N-terminal extension of Rps2 as it emerges from the ribosome. After translation, Tsr4 is released from Rps2 in the cytoplasm by an unknown mechanism (see below). We speculate that either concurrently with or soon after Tsr4 release, an importin binds to the conserved core of Rps2 and brings it to the nucleus, where it is delivered to pre-40S particles.
How does Tsr4 facilitate Rps2 expression?
Tsr4 is a modestly expressed protein which is easily titrated by even low levels of the N-terminal fragment of Rps2. This raises the question of how the small pool of Tsr4 finds nascent Rps2 in the large pool of translating ribosomes at a rate to sustain the active ribosome assembly pipeline. One possibility is that Tsr4 also has some affinity for the RPS2 mRNA, which helps localize it to the site of Rps2 translation. It is also possible that Tsr4 assembly with the N terminus of Rps2 itself is promoted by additional factors. The Ccr4-Not complex has recently been implicated in cotranslational assembly of the regulatory cap of the proteasome (44). Intriguingly, TSR4 was identified in a screen that also identified multiple components of the NOT complex, suggesting a functional link between Tsr4 and the Ccr4-Not complex (45). Alternatively, the limiting levels of Tsr4 in cells and the rate at which it engages with nascent Rps2 may set the level of Rps2. Excess RPs, including Rps2, are rapidly targeted for proteasomal degradation by the ubiquitin ligase Tom1 and become insoluble in the absence of Tom1 (46, 47). Thus, Rps2 may be constitutively expressed and degraded, and only the population that is captured cotranslationally by Tsr4 is competent for assembly into pre-40S particles in the nucleus. A similar model for the cotranslational assembly of multimeric protein complexes has recently been proposed as a means to protect highly basic or hydrophobic regions of proteins prior to assembly (48).
How is Rps2 handled after Tsr4 is released?
Our data suggest that Tsr4 does not enter the nucleus (Fig. 8), indicating that Rps2 is released from Tsr4 and handed off to an import factor. This raises the question of how Tsr4 is released from Rps2. When bound to the ribosome, the N terminus of Rps2 makes an intramolecular interaction with its C terminus (Fig. 1A) (26). Because Tsr4 binds to the N terminus of Rps2, a tempting model is that the C terminus of Rps2 facilitates Tsr4 release. Supporting this notion, the fusion of the C terminus to the N-terminal fragment of Rps2 partially alleviated the toxicity of the N-terminal fragment (Fig. 1B, cf. Rps2Δcore and Rps21–73). However, we have not been able to demonstrate that the C terminus of Rps2 affects the interaction with Tsr4 in vitro (Fig. 2B and data not shown). Subsequent to or concomitant with the release from Tsr4, Rps2 must bind an importin so that it can be ferried into the nucleus. Based on our mass spectrometry results of Rps2 IPs (Fig. 2A), its nuclear import is likely facilitated by the nuclear importins Kap123 and/or Kap104. Kap123 is one of the primary importins of the cell (22), while Kap104 has been implicated in the transport of certain RPs (16, 18, 20). Typically, when an RP is imported into the nucleus, its chaperone remains associated until the RP is loaded onto a preribosome (reviewed in references 3 and 9). The observation that Tsr4 appears to be a strictly cytoplasmic chaperone for Rps2 makes it unique, as it would be the first example of a chaperone that is released prior to its client’s nuclear import.
Differences between S. cerevisiae Tsr4/Rps2 and homologous systems.
Tsr4 is evolutionarily conserved among eukaryotes (30). PDCD2/Zfrp8 in Drosophila directly interacts with Rps2, is both cytoplasmic and nuclear, and appears to serve a function in 40S production, but the significance of its interaction with Rps2 has not been extensively characterized (29, 41, 49). Humans have two Tsr4 homologs, PDCD2 and PDCD2L (30), and expression of human PDCD2 can fully complement the loss of Drosophila PDCD2/Zfrp8, indicating functional conservation (49). The Bachand group showed that the two human proteins also bind to Rps2 and have partially redundant roles in 40S production (28); however, PDCD2L was characterized more extensively than PDCD2. Unlike yeast Tsr4, PDCD2L shuttles into the nucleus where it binds to pre-40S particles (28, 31) and harbors a leucine-rich nuclear export signal, suggesting that it may also function as an export adaptor (28). Interestingly, while PDCD2L binds pre-40S subunits, PDCD2 does not. Moreover, knockdown of PDCD2 is lethal and causes a greater 40S defect than that caused by deletion of PDCD2L. Because yeast Tsr4 does not bind nascent 40S and is essential for 40S production (31), yeast Tsr4 may serve a role more similar to that of PDCD2 than to that of PDCD2L, despite the greater similarity of yeast Tsr4 to PDCD2L than to PDCD2.
Both PDCD2L and PDCD2 also form extraribosomal and heterotrimeric complexes with Rps2 and the methyltransferase PRMT3 (28). The formation of a homologous complex in yeast is unlikely because yeast Tsr4 and Hmt1 (Rmt1), the yeast homolog of PRMT3, appear to be spatially separated, as Hmt1 is found in the nucleus (50); however, we have not tested their interaction. PRMT3 and Hmt1 methylate two arginine residues in the N terminus of Rps2 (51–54), but their modification is substoichiometric (52, 55), and mutation of these residues to lysine or alanine in yeast Rps2 had no obvious effect on growth (data not shown), suggesting that their methylation is unimportant for basic cellular function and viability. In a more recent study, the Bachand group showed that PRMT3 does not cotranslationally associate with Rps2 (56), but whether or not PDCD2L or PDCD2 cotranslationally associates with Rps2 was not explored. Notably, Drosophila PDCD2/Zfrp8 copurifies with translation machinery, suggesting that cotranslational capture of Rps2 may be a conserved function of Tsr4 orthologs (41). Interestingly, the human zinc finger protein ZNF277 was identified as a cotranslational interactor of Rps2; however, knockdown of ZNF277 does not impair 40S production, suggesting that the ZNF277-Rps2 complex has a role outside ribosome biogenesis (56). Higher eukaryotic organisms appear to have evolved a more elaborate mechanism for handling Rps2 than that of S. cerevisiae and have even possibly adapted some extraribosomal functions for Rps2-containing complexes.
MATERIALS AND METHODS
Strains, growth media, and genetic methods.
All S. cerevisiae strains and sources are listed in Table 1. AJY2873 was generated by integration of a fragment of pAJ1915 to delete nucleotides 190 to 266 of URA3 in the tsr4-ts strain obtained as described previously (57). AJY3398 and AJY4357 were generated by genomic integration of KanMX6-PGAL1 into the genomic locus of RPS2 in BY4741 and AJY4352, respectively. AJY4297 and AJY4300 were generated by integration of KanMX6-PGAL1-3×HA into the genomic locus of TSR4 (58) and KanMX6-PGAL1 (58) into the genomic locus of RPS2 in CH1305 (59), respectively, and the XRN1 locus in the resultant strains was disrupted with URA3. AJY4298 was generated by integration of KanMX6-PGAL1-3×HA (58) into the genomic locus of TSR4 in BY4741. AJY4353 was generated by integration of TAP-HIS3MX6 into the genomic locus of TSR4 in BY4741. AJY4359 was generated by disruption of the genomic locus of TSR4 with integration of KanMX6 (58) in BY4743 (Open Biosystems). The resultant strain was transformed with pAJ4183 and was subsequently sporulated and dissected. AJY4352 was generated by integration of TEV-13myc::Natr into the genomic locus of TSR4 in BY4741. AJY4363 was generated by genomic integration of TSR4-TEV-13myc::Natr amplified from AJY4352 into AJY1134. All yeast strains were cultured at 30°C in either YPD (2% peptone, 1% yeast extract, 2% dextrose), YPgal (2% peptone, 1% yeast extract, 1% galactose), or synthetic dropout (SD) medium containing 2% dextrose unless otherwise noted. When appropriate, media were supplemented with 150 to 250 μg/ml G418 or 100 μg/ml nourseothricin. All plasmids and sources are listed in Table 2.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| AJY1134 | MATa pep4-3 prb1-1122 ura3-52 leu2-3,112 reg1-501 gal1 | 32 |
| AJY1705 | MATα leu2Δ0 ura3Δ0 met15Δ0 CRM1T539C-HA NMD3-GFP::KanMX6 | 43 |
| AJY1951 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 SQT1-TAP::HIS3MX6 | 67 |
| AJY2873 | MATa ura3Δ0 leu2Δ0 his3Δ1 can1Δ::LEU2-MFA1pr::His3 tsr4-ts::ura3Δ | This study |
| AJY3398 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 KanMX6::PGAL1-RPS2 | This study |
| AJY4251 | MATa TSR4-GFP::HIS3MX6 leu2Δ0 ura3Δ0 | 68 |
| AJY4297 | MATa ade2 ade3 lys2-801 ura3-52 KanMX6::PGAL1-3×HA-TSR4 xrn1Δ::URA3 | This study |
| AJY4298 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 KanMX6::PGAL1-3×HA-TSR4 | This study |
| AJY4300 | MATa ade2 ade3 lys2-801 ura3-52 KanMX6::PGAL1-RPS2 xrn1Δ::URA3 | This study |
| AJY4352 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TSR4-TEV-13myc::Natr | This study |
| AJY4353 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TSR4-TAP::HIS3MX6 | This study |
| AJY4357 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 TSR4-TEV-13myc::Natr KanMX6::PGAL1-RPS2 | This study |
| AJY4359 | MATa ura3Δ0 leu2Δ0 his3Δ1 tsr4Δ::KanMX/pAJ4183 (TSR4 URA3 CEN ARS) | This study |
| AJY4363 | MATa pep4-3 prb1-1122 ura3-52 leu2-3,112 reg1-501 gal1 TSR4-TEV-13myc::Natr | This study |
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Open Biosystems |
| RKY1977 | MATa ade2 ade3 leu2 lys2-801 ura3-52 xrn1Δ | 69 |
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Reference |
|---|---|---|
| pAJ1399 | RPS2-GFP HIS3 CEN ARS | 70 |
| pAJ3576 | RPS21–73-GFP HIS3 CEN ARS | This study |
| pAJ3577 | RPS274–254-GFP HIS3 CEN ARS | This study |
| pAJ3578 | RPS21–223-GFP HIS3 CEN ARS | This study |
| pAJ4081 | PGAL1-RPS21–73-GFP URA3 CEN ARS | This study |
| pAJ4082 | PGAL1-RPS21–223-GFP URA3 CEN ARS | This study |
| pAJ4183 | TSR4 URA3 CEN ARS | This study |
| pAJ4184 | TSR4 URA3 2μ ARS | This study |
| pAJ4185 | TSR4-mCherry URA3 CEN ARS | This study |
| pAJ4186 | TSR4-GFP URA3 CEN ARS | This study |
| pAJ4207 | RPS2 HIS3 CEN ARS | This study |
| pAJ4219 | PGAL1-RPS21–73-GFPΔBsrGI URA3 CEN ARS | This study |
| pAJ4220 | RPS21–235-GFP HIS3 CEN ARS | This study |
| pAJ4231 | RPS2Δcore-GFP HIS3 CEN ARS | This study |
| pAJ4232 | TSR4 HIS3 CEN ARS | This study |
| pAJ4233 | RPS274–223-GFP HIS3 CEN ARS | This study |
| pAJ4234 | RPS2224–254-GFP HIS3 CEN ARS | This study |
| pAJ4241 | PGAL1-RPS2Δcore-GFP URA3 CEN ARS | This study |
| pAJ4242 | PGAL1-RPS274–223-GFP URA3 CEN ARS | This study |
| pAJ4243 | PGAL1-RPS2224–254-GFP URA3 CEN ARS | This study |
| pAJ4244 | RPS2 URA3 CEN ARS | This study |
| pAJ4261 | PGAL1-RPS2-GFP URA3 CEN ARS | This study |
| pAJ4267 | TSR4ΔNLS-GFP URA3 CEN ARS | This study |
| pAJ4270 | PGAL1-GFP URA3 CEN ARS | This study |
| pAJ4271 | PGAL1-RPS274–254-GFP URA3 CEN ARS | This study |
| pAJ4451 | PGAL1-RPS21–73_F14S-GFP URA3 CEN ARS | This study |
| pAJ4452 | PGAL1-RPS21–73_G15D-GFP URA3 CEN ARS | This study |
| pAJ4453 | PGAL1-RPS21–73_N18D-GFP URA3 CEN ARS | This study |
| pAJ4454 | PGAL1-RPS21–73_W35R-GFP URA3 CEN ARS | This study |
| pAJ4455 | PGAL1-RPS21–73_P37L-GFP URA3 CEN ARS | This study |
| pAJ4456 | PGAL1-RPS21–73_V38D-GFP URA3 CEN ARS | This study |
| pAJ4457 | PGAL1-RPS21–73_R43G-GFP URA3 CEN ARS | This study |
| pAJ4458 | PGAL1-RPS21–73_H59R-GFP URA3 CEN ARS | This study |
| pAJ4459 | PGAL1-RPS21–73_L61S-GFP URA3 CEN ARS | This study |
| pAJ4460 | PGAL1-RPS21–73_E65K-GFP URA3 CEN ARS | This study |
| pRS413 | HIS3 CEN ARS | 71 |
| pRS416 | URA3 CEN ARS | 71 |
| pRS426 | URA3 2μ ARS | 71 |
Random PCR mutagenesis of Rps21–73-GFP.
Random mutations in PGAL1-RPS21–73-GFP were generated by error-prone PCR using Taq polymerase and pAJ4081 as the template with oligonucleotides that hybridize to the sequence upstream of the GAL1 promoter and to the GFP coding sequence. The vector pAJ4219 was linearized with the restriction enzymes BsrGI and BamHI, cleaned up with a Zymoclean gel DNA recovery kit (Zymo Research), cotransformed with the mutant amplicon library into BY4741, and plated onto synthetic dropout medium without uracil (SD-ura medium) containing glucose to allow gap rescue of the linearized vector via the promoter and GFP sequences. Approximately 3,000 colonies were screened on SD-ura medium containing galactose for their ability to alleviate the toxicity caused by the expression of the Rps21–73-GFP fragment. Vectors expressing mutants with reduced toxicity and that showed GFP expression were rescued from yeast, transformed back into BY4741 to verify their alleviation of the toxicity, and sequenced.
Immunoprecipitation.
For purifying the panel of Rps2-GFP fragments, strain AJY1134 containing the appropriate galactose-inducible vectors was cultured in glucose-containing medium until exponential phase. Expression was then induced for 90 min by the addition of galactose to a 1% final concentration. Subsequent steps were performed on ice or at 4°C unless otherwise noted. Cells were washed once in buffer A (20 mM HEPES-KOH [pH 8.0], 110 mM potassium acetate [KOAc], 40 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF] and benzamidine, and 1 μM leupeptin and pepstatin) and resuspended in buffer A, and extracts were prepared by vortexing with glass beads. Extracts were clarified by centrifugation at 18,213 × g for 15 min and normalized to A260 units, and Triton X-100 was added to a final concentration of 0.1% (vol/vol). The extracts were subjected to ultracentrifugation at 70 krpm for 15 min on a TLA100.3 rotor (Beckman-Coulter) to remove ribosomes. Supernatants were incubated with GFP-trap (Chromotek) for 90 min. The beads were washed three times with buffer A-w (buffer A supplemented with 0.1% [vol/vol] Triton X-100), resuspended in 1× Laemmli buffer, and heated at 99°C for 3 min. Samples were separated on 6 to 18% SDS-PAGE gradient gels, and bands of interest were excised. Protein-containing gel slices were prepared for in-gel digestion, and peptides were recovered for identification by mass spectrometry as described previously (57). The resultant peptides were run for 30 min on a Dionex LC and Orbitrap Fusion 1 system (Thermo Scientific) for liquid chromatography-tandem mass spectrometry (LC-MS/MS). Data were processed in Scaffold v4.8.3 (Proteome Software, Inc.), and a protein threshold of a 99% minimum and a 2-peptide minimum and a peptide threshold of a 0.5% false discovery rate (FDR) were applied.
To assess the interactions between Tsr4-TEV-13myc and the Rps2-GFP fragments or between Tsr4-TEV-13myc and the Rps21–73-GFP mutants, we transformed the appropriate galactose-inducible vectors into AJY4363. Cells were cultured; extracts were prepared as described above, without ribosome removal; and immunoprecipitations were done with anti-c-Myc magnetic beads (catalog no. 88842; Pierce). IPs and inputs were separated on 6 to 18% SDS-PAGE gradient gels and subjected to Western blotting (see below). To assess the interaction of Tsr4-TEV-13myc and endogenous Rps2, we cultured AJY4352, AJY4357, and AJY4251 in galactose-containing medium for 3 h. A final concentration of 2% glucose was added to each culture, and cells were cultured an additional 2 h prior to harvesting. Extracts were made, and IPs and Western blotting were done as described above for the AJY4363 samples, except that the IPs were resuspended in Laemmli buffer lacking beta-mercaptoethanol to avoid the light chain of IgG from masking the native Rps2 Western blot signal.
Sucrose density gradient analysis.
AJY3398 was transformed with pAJ1399, pAJ3576, pAJ3578, and pRS413. Cells were cultured in glucose-containing medium lacking histidine for 5 h at 30°C to deplete endogenous Rps2 and then treated with 100 μg/ml CHX for 10 min at 30°C. For polysome profile analysis of Tsr4-depleted cells, expression of TSR4 was repressed for 8 h in AJY4298 cells transformed with pAJ4232, pAJ4207, and pRS413, and cells were harvested at comparable densities. Extracts and sucrose density gradient fractionation were done as previously described (60), except that the lysis buffer contained 100 μg/ml of CHX and 7 mM beta-mercaptoethanol.
Western blotting.
Primary antibodies used in this study were anti-c-Myc monoclonal 9e10 (BioLegend), anti-GFP (M. Rout), anti-Rpl8 (K.-Y. Lo), anti-Rps24 (our laboratory), anti-Rpl10 (our laboratory), and anti-Xrn1 (our laboratory), and anti-Rpl30 (61) was used to detect both Rpl30 and Rps2, which were previously referred to as L32 and S4, respectively (62). Secondary antibodies used were goat anti-mouse antibody–IRDye 800CW (Li-Cor Biosciences) and goat anti-rabbit antibody–IRDye 680RD (Li-Cor Biosciences). Blots were imaged with an Odyssey CLx infrared imaging system (Li-Cor Biosciences) using Image Studio (Li-Cor Biosciences).
Assay for cotranslational association by immunoprecipitation and RT-qPCR.
For strains expressing the TAP-tagged bait proteins, cultures of AJY1951 and AJY4353 were grown to exponential phase and then treated with 100 μg/ml of CHX for 10 min at 30°C prior to harvesting. After harvesting, cells were washed with buffer B (50 mM Tris [pH 7.5], 100 mM NaCl, 1.5 mM MgCl2, 100 μg/ml CHX, 1 mM PMSF and benzamidine, and 1 μM leupeptin and pepstatin) and resuspended in the same buffer supplemented with RiboLock RNase inhibitor (catalog no. EO0384; Thermo Scientific), and extracts were prepared by vortexing with glass beads. Extracts were clarified by centrifugation at 18,213 × g for 10 min and normalized by A260 units, and Triton X-100 was added to a final concentration of 0.1% (vol/vol). A total of 3% of each normalized extract was taken as the input. The normalized extracts were incubated with Dynabeads (Invitrogen) coupled to rabbit IgG (Sigma), prepared as described previously (63), for 90 min. The beads were washed three times with buffer B-w (buffer B supplemented with 0.1% Triton X-100). RNAs from the IPs and inputs were isolated using acid-phenol-chloroform as described previously (64) and resuspended in nuclease-free water. DNA was removed from the input samples using a Turbo DNA-free kit (catalog no. AM1907; Invitrogen). To compare the effects of CHX and puromycin treatments, AJY4353 cells were cultured to exponential phase and then split in half and treated with 100 μg/ml CHX or left untreated for 10 min at 30°C. Extracts and IPs were done as described above, except that the lysis and wash buffers were supplemented with either 100 μg/ml CHX or 100 μM puromycin for the CHX-treated cells and untreated cells, respectively. For GFP-tagged Tsr4, AJY4251 was cultured, and affinity purification was done similarly except that extracts were generated in buffer A (see “Immunoprecipitation,” above) supplemented with RiboLock RNase inhibitor and washes were done using buffer A-w, both supplemented with 100 μg/ml CHX, and protein G-Dynabeads (Invitrogen) precoated with rabbit anti-GFP (M. Rout) were used for immunoprecipitation.
Fifty nanograms of RNA from each IP and input was used as the template for cDNA synthesis using a qScript cDNA SuperMix kit (catalog no. 95048-025; Quantabio). For qPCR, 10% (for the TAP-tagged samples) and 13% (for the GFP-tagged samples) of each reverse transcription (RT) reaction mixture was used as the template using a PerfeCTa SYBR green FastMix low ROX kit (catalog no. 95074-250; Quantabio). qPCR for each oligonucleotide pair was done in technical triplicate. The following program on an Applied Biosystems ViiA 7 real-time PCR instrument was used: an initial hold step at 50°C for 2 min followed by an initial denaturation and polymerase activation step at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and elongation and data collection at 60°C for 1 min. A ramp rate of 1°C/s between thermocycling was used. The following oligonucleotide pairs were used: RPS2-Forward (5′-GAAGATGTCTACACCCAATCTAACG-3′) and RPS2-Reverse (5′-GAGTCAAGAAACCGTATGTGTTACC-3′) (amplicon size of 100 bp), and RPL10-Forward (5′-AGATACCAAAAGAACAAGCCTTACC-3′) and RPL10-Reverse (5′-GTAGATTCTGATCTTGGAGTCTGGA-3′) (amplicon size of 75 bp). Oligonucleotide pairs were designed using Primer3web software version 4.1.0 (http://primer3.ut.ee).
The threshold cycle (CT) for each reaction was incorporated into the standard curve to derive the number of mRNA molecules present using ViiA 7 software (Applied Biosystems). The standard curve was generated using 13, 1.3, and 0.13 ng of genomic DNA from BY4741, corresponding to about 1,000,000, 100,000, and 10,000 genomic loci for each target gene. The number of RNA molecules isolated in the whole IP (IPRNA) and the number of molecules present in the entire input (InputRNA) were extrapolated. The ratio of IPRNA to InputRNA was calculated for each reaction. The average ratio and standard deviation for each triplicate set were determined. The ratios were then normalized to the average ratio of the target mRNA for each bait to display enrichment relative to the target mRNA. For comparing CHX and puromycin treatments, the ratios were normalized to the value for the CHX-treated sample. The normalized values were plotted in GraphPad Prism version 7.0c.169 for Mac iOS (GraphPad Software).
Rps2-GFP expression in the tsr4-ts strain.
Strains AJY2873 and BY4741 containing pAJ4261 were grown to mid-log phase in 100 ml of SD-ura medium containing raffinose and then shifted to 37°C for 2 h. Rps2-GFP expression was induced for 90 min with 1% galactose prior to harvesting the cells. All subsequent steps were carried out on ice or at 4°C. Cells were washed and resuspended in buffer A, and extracts were made by glass bead lysis, clarified for 10 min at 18,213 × g, and normalized to 1 A260 unit in a final volume of 100 μl. Half of each normalized extract was overlaid onto a 50-μl 15% sucrose cushion in the same buffer and subjected to ultracentrifugation at 70 krpm for 15 min on a TLA100 rotor. Fractions were taken by manual pipetting, and Laemmli sample buffer was added to a 1× concentration. The samples were heated at 99°C for 3 min. Equal relative amounts of each sample were separated on a 6 to 18% SDS-PAGE gradient gel and subjected to analysis by Western blotting.
Fluorescent in situ hybridization.
RKY1977, AJY4297, and AJY4300 cells were grown to saturation in galactose-containing medium and diluted 5-fold in fresh glucose-containing medium and continued to grow for 2 h. Formaldehyde was added to a 4.5% final concentration, and cultures were gently agitated at 30°C for 30 min. Cells were washed twice with KSorb buffer (1.2 M sorbitol, 0.1 M potassium phosphate buffer [pH 7.0]) and then incubated in KSorb buffer with 50 μg/ml Zymolyase T20 for 15 min at 37°C in the presence of 20 mM vanadyl ribonucleoside complex (VRC), 28 mM β-mercaptoethanol, and 1 mM PMSF. Cells were gently pelleted, washed three times with ice-cold KSorb buffer, and resuspended in KSorb buffer before applying them to Teflon-coated immunofluorescence slides (catalog no. 18357; Polysciences, Inc.) precoated with polylysine. Slides were incubated in a moist chamber at room temperature for 10 min, excess cells were gently aspirated, and the slides were stored in 70% ethanol at −20°C. Cells were rehydrated by washing twice with 2× SSC (300 mM NaCl, 30 mM sodium citrate [pH 7.0]) and then incubated in a prehybridization solution (10% dextran sulfate, 50% deionized formamide, 1× Denhardt’s solution, 2 mM VRC, 4× SSC, 0.2% purified bovine serum albumin [BSA], 25 μg yeast tRNA, and 500 μg/ml single-stranded DNA [ssDNA]) for 1 h at 72°C in a moist chamber. The solution was replaced with a prehybridization solution containing 1 μM Cy3-labeled oligonucleotide D-A2 probe (5′-ATGCTCTTGCCAAAACAAAAAAATCCATTTTCAAAATTATTAAATTTCTT-3′). Slides were incubated in a moist chamber at 72°C for 1 h, followed by overnight incubation at 37°C. Wells were then washed with 2× SSC at 37°C and then with 1× SSC at room temperature containing 0.1% NP-40 for 30 min each. Cells were incubated for 2 min with 4′,6′-diamidino-2-phenylindole (DAPI) at 1 μg/ml in phosphate-buffered saline (PBS), washed twice with PBS, and mounted in Aqua-Poly/Mount (Polysciences, Inc.). Fluorescence was visualized on a Nikon E800 microscope fitted with a Plan Apo 100×/1.4-numerical-aperture objective and a Photometrics CoolSNAP ES camera controlled by NIS-Elements AR2.10 software, and photos were processed with Affinity Designer.
Fluorescence microscopy of GFP-tagged proteins.
Cells were cultured as described in the figure legends. The fluorescence signal was captured using a 500-ms exposure time on a Nikon E800 microscope fitted with a Plan Apo 100×/1.4-numerical-aperture objective and a Photometrics CoolSNAP ES or PCO sCMOS pco.edge camera controlled by NIS-Elements AR2.10 software, and photos were processed with Affinity Designer.
ACKNOWLEDGMENTS
This work was supported by NIH grants GM108823 and GM127127 to A.W.J. and by a fellowship from The University of Texas at Austin Graduate School to J.J.B. The Proteomics Facility in the Center for Biomedical Research Support at The University of Texas at Austin is supported in part by CPRIT grant RP110782.
We thank M. Rout, K.-Y. Lo, and J. Warner for antibodies. We also thank K. Robbins, J. Recchia-Rife, and J. Ream for their help with experiments and L. Leblanc for advice regarding RT-qPCR. We thank P. Sutjita for helpful comments regarding the manuscript.
J.J.B. and A.W.J. conceptualized and performed experiments, analyzed and interpreted data, and composed and edited the manuscript and figures. S.M. performed the FISH analysis and interpreted the data.
REFERENCES
- 1.Woolford JL, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195:643–681. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kressler D, Hurt E, Baßler J. 2017. A puzzle of life: crafting ribosomal subunits. Trends Biochem Sci 42:640–654. doi: 10.1016/j.tibs.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Peña C, Hurt E, Panse VG. 2017. Eukaryotic ribosome assembly, transport and quality control. Nat Struct Mol Biol 24:689–699. doi: 10.1038/nsmb.3454. [DOI] [PubMed] [Google Scholar]
- 4.Baßler J, Hurt E. 2019. Eukaryotic ribosome assembly. Annu Rev Biochem 88:281–306. doi: 10.1146/annurev-biochem-013118-110817. [DOI] [PubMed] [Google Scholar]
- 5.Jakob S, Ohmayer U, Neueder A, Hierlmeier T, Perez-Fernandez J, Hochmuth E, Deutzmann R, Griesenbeck J, Tschochner H, Milkereit P. 2012. Interrelationships between yeast ribosomal protein assembly events and transient ribosome biogenesis factors interactions in early pre-ribosomes. PLoS One 7:e32552. doi: 10.1371/journal.pone.0032552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira-Cerca S, Pöll G, Gleizes P-E, Tschochner H, Milkereit P. 2005. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol Cell 20:263–275. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Pöll G, Braun T, Jakovljevic J, Neueder A, Jakob S, Woolford JL, Tschochner H, Milkereit P. 2009. rRNA maturation in yeast cells depleted of large ribosomal subunit proteins. PLoS One 4:e8249. doi: 10.1371/journal.pone.0008249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jäkel S, Mingot J-M, Schwarzmaier P, Hartmann E, Görlich D. 2002. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J 21:377–386. doi: 10.1093/emboj/21.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillet B, Mitterer V, Kressler D, Pertschy B. 2017. Hold on to your friends: dedicated chaperones of ribosomal proteins. Dedicated chaperones mediate the safe transfer of ribosomal proteins to their site of pre-ribosome incorporation. Bioessays 39:1–12. doi: 10.1002/bies.201600153. [DOI] [PubMed] [Google Scholar]
- 10.Ting Y-H, Lu T-J, Johnson AW, Shie J-T, Chen B-R, Kumar S S, Lo K-Y. 2017. Bcp1 is the nuclear chaperone of Rpl23 in Saccharomyces cerevisiae. J Biol Chem 292:585–596. doi: 10.1074/jbc.M116.747634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisinger DP, Dick FA, Denke E, Trumpower BL. 1997. SQT1, which encodes an essential WD domain protein of Saccharomyces cerevisiae, suppresses dominant-negative mutations of the ribosomal protein gene QSR1. Mol Cell Biol 17:5146–5155. doi: 10.1128/mcb.17.9.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaper S, Fromont-Racine M, Linder P, de la Cruz J, Namane A, Yaniv M. 2001. A yeast homolog of chromatin assembly factor 1 is involved in early ribosome assembly. Curr Biol 11:1885–1890. doi: 10.1016/S0960-9822(01)00584-X. [DOI] [PubMed] [Google Scholar]
- 13.Iouk TL, Aitchison JD, Maguire S, Wozniak RW. 2001. Rrb1p, a yeast nuclear WD-repeat protein involved in the regulation of ribosome biosynthesis. Mol Cell Biol 21:1260–1271. doi: 10.1128/MCB.21.4.1260-1271.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pausch P, Singh U, Ahmed YL, Pillet B, Murat G, Altegoer F, Stier G, Thoms M, Hurt E, Sinning I, Bange G, Kressler D. 2015. Co-translational capturing of nascent ribosomal proteins by their dedicated chaperones. Nat Commun 6:7494. doi: 10.1038/ncomms8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pillet B, García-Gómez JJ, Pausch P, Falquet L, Bange G, de la Cruz J, Kressler D. 2015. The dedicated chaperone Acl4 escorts ribosomal protein Rpl4 to its nuclear pre-60S assembly site. PLoS Genet 11:e1005565. doi: 10.1371/journal.pgen.1005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelter P, Huber FM, Kunze R, Flemming D, Hoelz A, Hurt E. 2015. Coordinated ribosomal L4 protein assembly into the pre-ribosome is regulated by its eukaryote-specific extension. Mol Cell 58:854–862. doi: 10.1016/j.molcel.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch B, Mitterer V, Niederhauser J, Stanborough T, Murat G, Rechberger G, Bergler H, Kressler D, Pertschy B. 2012. Yar1 protects the ribosomal protein Rps3 from aggregation. J Biol Chem 287:21806–21815. doi: 10.1074/jbc.M112.365791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kressler D, Bange G, Ogawa Y, Stjepanovic G, Bradatsch B, Pratte D, Amlacher S, Strauß D, Yoneda Y, Katahira J, Sinning I, Hurt E. 2012. Synchronizing nuclear import of ribosomal proteins with ribosome assembly. Science 338:666–671. doi: 10.1126/science.1226960. [DOI] [PubMed] [Google Scholar]
- 19.Holzer S, Ban N, Klinge S. 2013. Crystal structure of the yeast ribosomal protein rpS3 in complex with its chaperone Yar1. J Mol Biol 425:4154–4160. doi: 10.1016/j.jmb.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 20.Huber FM, Hoelz A. 2017. Molecular basis for protection of ribosomal protein L4 from cellular degradation. Nat Commun 8:14354. doi: 10.1038/ncomms14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West M, Hedges JB, Chen A, Johnson AW. 2005. Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol Cell Biol 25:3802–3813. doi: 10.1128/MCB.25.9.3802-3813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rout MP, Blobel G, Aitchison JD. 1997. A distinct nuclear import pathway used by ribosomal proteins. Cell 89:715–725. doi: 10.1016/S0092-8674(00)80254-8. [DOI] [PubMed] [Google Scholar]
- 23.Mitterer V, Gantenbein N, Birner-Gruenberger R, Murat G, Bergler H, Kressler D, Pertschy B. 2016. Nuclear import of dimerized ribosomal protein Rps3 in complex with its chaperone Yar1. Sci Rep 6:36714. doi: 10.1038/srep36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calviño FR, Kharde S, Ori A, Hendricks A, Wild K, Kressler D, Bange G, Hurt E, Beck M, Sinning I. 2015. Symportin 1 chaperones 5S RNP assembly during ribosome biogenesis by occupying an essential rRNA-binding site. Nat Commun 6:6510. doi: 10.1038/ncomms7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bange G, Murat G, Sinning I, Hurt E, Kressler D. 2013. New twist to nuclear import: when two travel together. Commun Integr Biol 6:e24792. doi: 10.4161/cib.24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 27.Perreault A, Bellemer C, Bachand F. 2008. Nuclear export competence of pre-40S subunits in fission yeast requires the ribosomal protein Rps2. Nucleic Acids Res 36:6132–6142. doi: 10.1093/nar/gkn625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landry-Voyer A-M, Bilodeau S, Bergeron D, Dionne KL, Port SA, Rouleau C, Boisvert F-M, Kehlenbach RH, Bachand F. 2016. Human PDCD2L is an export substrate of CRM1 that associates with 40S ribosomal subunit precursors. Mol Cell Biol 36:3019–3032. doi: 10.1128/MCB.00303-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minakhina S, Naryshkina T, Changela N, Tan W, Steward R. 2016. Zfrp8/PDCD2 interacts with RpS2 connecting ribosome maturation and gene-specific translation. PLoS One 11:e0147631. doi: 10.1371/journal.pone.0147631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burroughs AM, Aravind L. 2014. Analysis of two domains with novel RNA-processing activities throws light on the complex evolution of ribosomal RNA biogenesis. Front Genet 5:424. doi: 10.3389/fgene.2014.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Lee I, Moradi E, Hung N-J, Johnson AW, Marcotte EM. 2009. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol 7:e1000213. doi: 10.1371/journal.pbio.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hovland P, Flick J, Johnston M, Sclafani RA. 1989. Galactose as a gratuitous inducer of GAL gene expression in yeasts growing on glucose. Gene 83:57–64. doi: 10.1016/0378-1119(89)90403-4. [DOI] [PubMed] [Google Scholar]
- 33.Ho B, Baryshnikova A, Brown GW. 2018. Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst 6:192.e3–205.e3. doi: 10.1016/j.cels.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 35.Rössler I, Embacher J, Pillet B, Murat G, Liesinger L, Hafner J, Unterluggauer JJ, Birner-Gruenberger R, Kressler D, Pertschy B. 7 May 2019. Tsr4 and Nap1, two novel members of the ribosomal protein chaperOME. Nucleic Acids Res doi: 10.1093/nar/gkz317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pestka S. 1971. Inhibitors of ribosome functions. Annu Rev Microbiol 25:487–562. doi: 10.1146/annurev.mi.25.100171.002415. [DOI] [PubMed] [Google Scholar]
- 37.Fatica A, Oeffinger M, Dlakić M, Tollervey D. 2003. Nob1p is required for cleavage of the 3′ end of 18S rRNA. Mol Cell Biol 23:1798–1807. doi: 10.1128/mcb.23.5.1798-1807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fatica A, Tollervey D, Dlakić M. 2004. PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA 10:1698–1701. doi: 10.1261/rna.7123504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stevens A, Hsu CL, Isham KR, Larimer FW. 1991. Fragments of the internal transcribed spacer 1 of pre-rRNA accumulate in Saccharomyces cerevisiae lacking 5′→3′ exoribonuclease 1. J Bacteriol 173:7024–7028. doi: 10.1128/jb.173.21.7024-7028.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moy TI, Silver PA. 1999. Nuclear export of the small ribosomal subunit requires the ran-GTPase cycle and certain nucleoporins. Genes Dev 13:2118–2133. doi: 10.1101/gad.13.16.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan W, Schauder C, Naryshkina T, Minakhina S, Steward R. 2016. Zfrp8 forms a complex with fragile-X mental retardation protein and regulates its localization and function. Dev Biol 410:202–212. doi: 10.1016/j.ydbio.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutten S, Kehlenbach RH. 2007. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 17:193–201. doi: 10.1016/j.tcb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Hedges J, West M, Johnson AW. 2005. Release of the export adapter, Nmd3p, from the 60S ribosomal subunit requires Rpl10p and the cytoplasmic GTPase Lsg1p. EMBO J 24:567–579. doi: 10.1038/sj.emboj.7600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panasenko OO, Somasekharan SP, Villanyi Z, Zagatti M, Bezrukov F, Rashpa R, Cornut J, Iqbal J, Longis M, Carl SH, Peña C, Panse VG, Collart MA. 2019. Co-translational assembly of proteasome subunits in NOT1-containing assemblysomes. Nat Struct Mol Biol 26:110–120. doi: 10.1038/s41594-018-0179-5. [DOI] [PubMed] [Google Scholar]
- 45.Collart MA, Kassem S, Villanyi Z. 2017. Mutations in the NOT genes or in the translation machinery similarly display increased resistance to histidine starvation. Front Genet 8:61. doi: 10.3389/fgene.2017.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sung M-K, Porras-Yakushi TR, Reitsma JM, Huber FM, Sweredoski MJ, Hoelz A, Hess S, Deshaies RJ. 2016. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife 5:e19105. doi: 10.7554/eLife.19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sung M-K, Reitsma JM, Sweredoski MJ, Hess S, Deshaies RJ. 2016. Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol Biol Cell 27:2642–2652. doi: 10.1091/mbc.E16-05-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiber A, Döring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G, Bukau B. 2018. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature 561:268–272. doi: 10.1038/s41586-018-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minakhina S, Changela N, Steward R. 2014. Zfrp8/PDCD2 is required in ovarian stem cells and interacts with the piRNA pathway machinery. Development 141:259–268. doi: 10.1242/dev.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henry MF, Silver PA. 1996. A novel methyltransferase (Hmt1p) modifies poly(A)+-RNA-binding proteins. Mol Cell Biol 16:3668–3678. doi: 10.1128/mcb.16.7.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yagoub D, Hart-Smith G, Moecking J, Erce MA, Wilkins MR. 2015. Yeast proteins Gar1p, Nop1p, Npl3p, Nsr1p, and Rps2p are natively methylated and are substrates of the arginine methyltransferase Hmt1p. Proteomics 15:3209–3218. doi: 10.1002/pmic.201500075. [DOI] [PubMed] [Google Scholar]
- 52.Lipson RS, Webb KJ, Clarke SG. 2010. Rmt1 catalyzes zinc-finger independent arginine methylation of ribosomal protein Rps2 in Saccharomyces cerevisiae. Biochem Biophys Res Commun 391:1658–1662. doi: 10.1016/j.bbrc.2009.12.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swiercz R, Cheng D, Kim D, Bedford MT. 2007. Ribosomal protein rpS2 is hypomethylated in PRMT3-deficient mice. J Biol Chem 282:16917–16923. doi: 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- 54.Swiercz R, Person MD, Bedford MT. 2005. Ribosomal protein S2 is a substrate for mammalian PRMT3 (protein arginine methyltransferase 3). Biochem J 386:85–91. doi: 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young BD, Weiss DI, Zurita-Lopez CI, Webb KJ, Clarke SG, McBride AE. 2012. Identification of methylated proteins in the yeast small ribosomal subunit: a role for SPOUT methyltransferases in protein arginine methylation. Biochemistry 51:5091–5104. doi: 10.1021/bi300186g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dionne KL, Bergeron D, Landry-Voyer A-M, Bachand F. 2019. The 40S ribosomal protein uS5 (RPS2) assembles into an extra-ribosomal complex with human ZNF277 that competes with the PRMT3-uS5 interaction. J Biol Chem 294:1944–1955. doi: 10.1074/jbc.RA118.004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Aroya S, Coombes C, Kwok T, O’Donnell KA, Boeke JD, Hieter P. 2008. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell 30:248–258. doi: 10.1016/j.molcel.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. doi:. [DOI] [PubMed] [Google Scholar]
- 59.Kranz JE, Holm C. 1990. Cloning by function: an alternative approach for identifying yeast homologs of genes from other organisms. Proc Natl Acad Sci U S A 87:6629–6633. doi: 10.1073/pnas.87.17.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Black JJ, Wang Z, Goering LM, Johnson AW. 2018. Utp14 interaction with the small subunit processome. RNA 24:1214–1228. doi: 10.1261/rna.066373.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vilardell J, Warner JR. 1997. Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol Cell Biol 17:1959–1965. doi: 10.1128/mcb.17.4.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mager WH, Planta RJ, Ballesta JG, Lee JC, Mizuta K, Suzuki K, Warner JR, Woolford J. 1997. A new nomenclature for the cytoplasmic ribosomal proteins of Saccharomyces cerevisiae. Nucleic Acids Res 25:4872–4875. doi: 10.1093/nar/25.24.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP. 2007. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods 4:951–956. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- 64.Zhu J, Liu X, Anjos M, Correll CC, Johnson AW. 2016. Utp14 recruits and activates the RNA helicase Dhr1 to undock U3 snoRNA from the preribosome. Mol Cell Biol 36:965–978. doi: 10.1128/MCB.00773-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kushnirov VV. 2000. Rapid and reliable protein extraction from yeast. Yeast 16:857–860. doi:. [DOI] [PubMed] [Google Scholar]
- 66.Nakai K, Horton P. 1999. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 24:34–36. doi: 10.1016/S0968-0004(98)01336-X. [DOI] [PubMed] [Google Scholar]
- 67.Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 68.Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. 2003. Global analysis of protein localization in budding yeast. Nature 425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 69.Johnson AW, Kolodner RD. 1995. Synthetic lethality of sep1 (xrn1) ski2 and sep1 (xrn1) ski3 mutants of Saccharomyces cerevisiae is independent of killer virus and suggests a general role for these genes in translation control. Mol Cell Biol 15:2719–2727. doi: 10.1128/mcb.15.5.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW. 2008. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol Cell Biol 28:3151–3161. doi: 10.1128/MCB.01674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]