

Abstract
Objective
To develop imaging biomarkers of diseases in the Lewy body spectrum and to validate these markers against postmortem neuropathologic findings.
Methods
Four cognitively normal participants with Parkinson disease (PD), 4 with PD with cognitive impairments, and 10 with dementia with Lewy bodies underwent amyloid imaging with [11C]Pittsburgh compound B (PiB) and dopamine transporter (DAT) imaging with [11C]Altropane. All 18 had annual neurologic examinations. All cognitively normal participants with PD developed cognitive impairment before death. Neuropathologic examinations assessed and scored Braak Lewy bodies, Thal distribution of amyloid, Consortium to Establish a Registry for Alzheimer's Disease neuritic amyloid plaques, Braak neurofibrillary tangles, and cerebral amyloid angiopathy, as well as total amyloid plaque burden in the superior frontal, superior parietal, occipital, and inferior temporal cortical regions. PET data were expressed as the standardized uptake value ratio with cerebellar reference. Analyses accounted for the interval between imaging and autopsy.
Results
All 18 patients met neuropathologic criteria for Lewy body disease; the DAT concentration was low in each case. All patients with elevated [11C]PiB retention measured in a neocortical aggregate had β-amyloid deposits at autopsy. [11C]PiB retention significantly correlated with neuritic plaque burden and with total plaque burden. [11C]PiB retention also significantly correlated with the severity of both Braak stages of neurofibrillary tangle and Lewy body scores. Neuritic plaque burden was significantly associated with neurofibrillary tangle pathology.
Conclusion
Antemortem [11C]Altropane PET is a sensitive measure of substantia nigra degeneration. [11C]PiB scans accurately reflect cortical amyloid deposits seen at autopsy. These findings support the use of molecular imaging in the evaluation of patients with Lewy body diseases.
The Lewy body spectrum of diseases include Parkinson disease (PD), PD with dementia (PDD), and dementia with Lewy bodies (DLB). They all share underlying pathologic changes, including aggregates of α-synuclein that accumulate in substantia nigra neurons; in PDD and DLB, synuclein containing neurons and neurites spread into cortical regions.1–3 In addition, neuropathologic changes characteristic of Alzheimer disease (AD), including β-amyloid (Aβ) plaques and neurofibrillary tangles (NFT), are commonly observed at autopsy in both PDD and DLB.4–6 Because of these overlapping pathologic findings, the differential diagnosis between PDD/DLB and AD is often uncertain. In 2016, the Alzheimer's Disease-Related Dementias summit report identified several key challenges for Lewy body disease (LBD) research, including developing neuroimaging biomarkers to aid in the diagnosis and validating such markers against postmortem neuropathology.7 In initial steps to address these goals, we and others have previously shown that amyloid burden measured in life with the PET radioligand [11C]Pittsburgh compound B (PiB) was increased in DLB and to a lesser extent in PDD compared to normal healthy elderly patients and patients with PD with normal cognition.8–11 Furthermore, high levels of amyloid in PD, PDD, and DLB were also associated with faster rates of cognitive decline.12,13 We and others have also demonstrated that dopamine transporter (DAT) imaging aids in discriminating PDD and DLB from AD.14–17 Validation of these biomarkers, however, requires postmortem verification. With this goal in mind, we describe the concordance between molecular imaging and neuropathologic findings in a cohort of parkinsonian patients with cognitive impairments and dementia.
Methods
Eighteen participants in the Lewy body spectrum of disease were recruited from Massachusetts General Hospital's Movement and Memory Disorder Units into a longitudinal study from 2006 to 2011 (table 1). All 18 underwent [11C]PiB imaging, neurologic examination, and detailed neuropsychological evaluation at their first visit, followed by annual clinical and cognitive testing.10 Ten of these participants also underwent DAT imaging with [11C]Altropane.18 At baseline examination and imaging, the clinical diagnoses were DLB in 10, PD with normal cognition in 4, PD with mild cognitive impairment (MCI) in 3, and PDD in 1. At the final study visit, the diagnoses were DLB in 10, PD-MCI in 4, and PDD in 4. Participants with DLB met clinical consensus criteria for probable DLB, including the presence of at least 2 of the following: parkinsonism, visual hallucinations, fluctuations of cognition, and REM sleep behavioral disorder.2 Participants with PD met the diagnostic criteria for idiopathic PD of the United Kingdom Parkinson's Disease Society Brain Bank Diagnostic Criteria.19 Participants with PD-MCI and PDD met respective level II PD-MCI20 and PDD21 diagnostic criteria of the Movement Disorders Society, with objective impairment in at least 2 cognitive domains. In contrast to subjects with PD-MCI, those with PDD demonstrated impairment in daily function on the basis of their cognitive impairment. Interviews with caregivers were acquired in all cases.
Table 1.
Demographic summary

In all participants but 1, [11C]PiB PET was acquired on a Siemens/CTI ECAT HR+ scanner (63 parallel planes, axial field of view 15.2 cm, in-plane resolution 4.1-mm full width at half-maximum, slice width 2.4 mm; Siemens, Munich, Germany). In 1 participant, [11C]PiB was acquired on a GE PC4096 scanner (2D mode, 15 image planes, 10.0-cm axial field of view, 7.0-mm transaxial resolution, 6.0-mm slice interval, 39 frames, 8 × 15 seconds, 4 × 60 seconds, 27 × 120 seconds; GE, Milwaukee, WI). [11C]PiB data were acquired with a 39-frame dynamic protocol (8 × 15 seconds, 4 × 60 seconds, and 27 × 120 seconds), reconstructed, and corrected for scatter, attenuation, and randoms with vendor-supplied software. [11C]PiB PET data were spatially transformed into the PET native space with Statistical Parametric Mapping (SPM12, Wellcome Trust Centre for Neuroimaging, London, UK) using early frame data (0–8 minutes after injection). [11C]PiB data were recorded as the standardized uptake value ratio (SUVR) with whole cerebellar reference that included gray and white matter, as reported22 using data from 40 to 60 minutes after injection. A probabilistic template space atlas based on the FreeSurfer GTM segmentation23 was used to define regions of interest. For comparison with global neuropathologic measures of amyloid deposition, we evaluated an aggregate region of interest comprising the frontal, lateral temporal, and retrosplenial regions (FLR).24 FLR binding for the 1 dataset acquired on the GE PC4096 scanner was interpolated from precuneus binding, a site of early and robust amyloid deposition that correlates strongly (>0.9) with FLR cortical [11C]PiB retention (r = 0.92 in this dataset). Regional PiB binding was unavailable for this participant. High [11C]PiB binding was taken as FLR SUVR ≥1.2 on the basis of a gaussian mixture model on a reference dataset of clinically normal elderly.25
We assessed striatal DAT concentration with Altropane [2β-carbomethoxy-3β(4-fluorophenyl)-n-(1-iodoprop-1-en-3-yl) nortropane], a cocaine analog DAT ligand with fast kinetics26 and high DAT selectivity.27 [11C]Altropane PET was acquired on a Siemens/CTI ECAT HR+ scanner, as described previously,18 and compared to 20 age-matched healthy controls (age 72.7 ± 8.4 years; 7 male, 13 female; Mini-Mental State Examination score 29.1 ± 1.4). Briefly, [11C]Altropane was prepared onsite, and 15 mCi [11C]Altropane was injected as a bolus, followed by a 60-minute dynamic acquisition. PET data were reconstructed and corrected for attenuation with vendor-provided software. The DAT concentration was estimated with specific binding of [11C]Altropane, which was computed in regions of interest, including the putamen, using the SUVR measured between 40 and 60 minutes after injection, with whole cerebellar reference.
By the time of death, all participants with DLB and PD were cognitively impaired. Postmortem brain tissue was examined to determine the accuracy of the clinical diagnosis and to rate the severity of LBD and AD pathologic changes. At the time of autopsy, brains were divided at the midline, with one-half then cut into coronal slabs for freezing at −80°C and the other half fixed in 10% buffered formalin. Tissue blocks were prepared from the formalin-fixed side after 10 to 14 days of fixation, processed on a Thermo Scientific Excelsior ES tissue processor (Thermo Fisher Scientific, Waltham, MA), and embedded in paraffin. All sections were cut on a microtome at 7 µmol/L and stained with Luxol fast blue/hematoxylin & eosin for routine assessment. Bielschowsky silver stain was performed on sections from select blocks. Immunohistochemistry for hyperphosphorylated tau (polyclonal rabbit anti-human tau, Dako, Glostrup, Denmark) at a titration of 1:6,000, Aβ (monoclonal mouse anti-human Aβ clone 6F/3D, Dako, Glostrup, Denmark) at a titration of 1:600, and α-synuclein (mouse anti-α-synuclein LB509, Life Technologies Corp, Frederick, MD) at a titration of 1:200 was also performed on sections from select blocks and processed on a Leica Bond RX automated stainer (Leica Biosystems, Wetzlar, Germany) according to the manufacturer's instructions.
AD neuropathologic changes were scored according to the current National Institute on Aging–Alzheimer's Association's neuropathologic assessment guidelines28 with the ABC rating scale, where A refers to Thal amyloid phase,29 a measure of the hierarchical involvement of brain regions of any plaque (Thal range 0–5, A range 0–3); B is a measure of NFT pathology based on Braak staging (Braak NFT stage range 0–6, B range 0–3)30; and C is a numeric equivalent of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuritic plaque score (range 0–3).31 Cerebral amyloid angiopathy severity was rated with the Vonsattel score (range 0–3).32 Lewy body pathology was scored with the Braak staging system on a scale from 0 to 6 (table 2).1,33
Table 2.
PET and neuropathologic findings

In addition, because the currently used CERAD scoring system for regional plaque burden is restricted to the evaluation of neuritic and cored plaques (using Bielschowsky silver stain and immunohistochemistry for hyperphosphorylated tau in our institution),31 we measured total regional amyloid plaque burden (using immunohistochemistry for Aβ), accounting for both neuritic/cored and nonneuritic/noncored plaques containing fibrillar amyloid. We evaluated regional amyloid severity on a 4-point scale (0–3), where 0 = absent, 1 = sparse, 2 = moderate, and 3 = severe plaque pathology within a tissue section. To enable regional cortical correlations of total amyloid plaque burden with [11C]PiB retention, 2 neuropathologists (N.C., M.F.) independently assigned a regional amyloid severity score in the superior frontal, superior parietal, pericalcarine, and inferior temporal regions while blinded to each other's score. Scores were then reviewed for consensus. Of the 72 blind assessments, there was complete agreement in 60 (83%). The remaining 12 (17%) showed a difference in score in each instance of only 1 degree, and these were modified to reflect consensus among the 2 pathologists. Regional amyloid severity measurements were not available for the 1 participant acquired on the GE PC4096 scanner. To facilitate comparison of PiB correlations with CERAD and with total amyloid plaque burden, we computed average CERAD scores and average regional amyloid severity scores across the same regions. All neuropathologic evaluations were blinded to clinical diagnosis, DAT concentration, and [11C]PiB retention.
Neuropathology correlates of [11C]PiB retention were investigated with partial Spearman correlations that controlled for the interval between imaging and autopsy. These adjusted correlations are reported in the text. Significant correlations were further explored by incorporating Vonsattel scores as covariates. For standard neuropathologic measures, [11C]PiB retention in the FLR aggregate region was used. For regional amyloid severity measurement, the regional imaging correlate was matched to hemisphere and to the region of interest. Correlations between neuropathology measures were assessed via Spearman coefficients. Differences between neuropathology measures based on disease status and differences in DAT concentration between imaged cases and healthy participants were evaluated with Student 2-tailed t tests and Mann-Whitney U tests as appropriate. All analyses were conducted with R Software.
Standard protocol approvals, registrations, and patient consents
This study was approved by the Institutional Review Board of Partners Healthcare, Inc. All participants gave written informed consent and received a small stipend for participation.
Data availability
Data are available on request.
Results
The clinical diagnosis of PD or DLB was confirmed neuropathologically in all 18 cases on the basis of extensive neuronal loss in the substantia nigra with Lewy bodies (table 2). All cases had Braak Lewy body scores ≥3, but the Braak stage was significantly higher (p = 0.002) in the DLB group than in the PD-MCI and PDD groups. The [11C]Altropane DAT PET reflected the resultant dopamine deficiency; putamen uptake was reduced in all 10 cases acquired (mean ± SD 1.67 ± 0.24 SUVR) compared with healthy controls (mean ± SD 3.03 ± 0.39 SUVR, p < 0.0001). Other neuropathologic findings were similar across all 18 participants. Specifically, no diagnostic group differences were noted for ABC scores and estimates of cerebral amyloid angiopathy.
Most participants (17 of 18) had evidence for Aβ deposits, as measured by CERAD neuritic plaque scores ≥1 and Thal amyloid distribution scores ≥1. In most cases, [11C]PiB retention accurately reflected the extent of amyloid deposits. One participant had no neuritic plaques and Thal phase 0; in this case, [11C]PiB retention was minimal (SUVR <1.00, patient 1 [DLB]. Of note, comparably low [11C]PiB retention was also observed in 4 participants with sparse or moderate neuritic plaque burden (CERAD scores of 1–2) and Thal phase A scores ranging from 1 to 3 (participants 11 and 14–16). At PET, 2 of these mismatched cases were cognitively normal with PD and 2 had PD-MCI; by the time of death, all had developed cognitive impairment. In these instances, the median interval between [11C]PiB scan and death was 6.0 (range 4.7–7.5) years compared to the overall median of 4.4 (range 0.2–7.6) years.
[11C]PiB retention correlated with CERAD neuritic plaques score (r = 0.62, p = 0.0075) (figure 1A). [11C]PiB retention lacked a significant correlation with Thal amyloid phase (r = 0.45, p = 0.07; figure 1B). Because [11C]PiB labels nonneuritic and neuritic plaques,11 the CERAD score is likely an underestimate of total amyloid burden. We therefore measured the total amyloid plaque burden—neuritic and nonneuritic Aβ—in each cortical lobe on a scale of 0 to 3+. [11C]PiB retention correlated with this measure of regional amyloid severity averaged over the 4 regions sampled (r = 0.49, p = 0.046). By region, local [11C]PiB retention correlated with regional total amyloid burden in occipital (r = 0.68, p = 0.0035) and superior parietal (r = 0.56, p = 0.025) but not in inferior temporal or superior frontal regions (figure 2). The correlation in the occipital region remained significant after controlling for multiple comparisons (Bonferroni, p = 0.013). To directly compare the [11C]PiB correlation with CERAD to the [11C]PiB correlation with total amyloid plaque burden, we computed average CERAD scores across the same regions. [11C]PiB retention correlated with average CERAD scores as well (r = 0.68, p = 0.0028). Thus, [11C]PiB retention significantly correlated with measures of amyloid plaque burden.
Figure 1. Boxplots of [11C]PiB retention as a function of neuritic plaque score and Thal amyloid phase in Lewy body disease.

Partial correlations adjusting for the interval between [11C]Pittsburgh compound B (PiB) PET and death: (A) neuritic plaque score (r = 0.62, p = 0.0075) and (B) Thal amyloid phase (r = 0.45, p = 0.07). A typical cut point for [11C]PiB positivity is 1.2. Filled circles indicate dementia with Lewy bodies; open diamonds, Parkinson disease with mild cognitive impairment and Parkinson disease with dementia. SUVR = standardized uptake value ratio.
Figure 2. [11C]PiB retention was significantly correlated with total amyloid deposits in occipital and superior parietal regions.

Partial correlations controlling for the interval between PET and autopsy: (A) occipital (r = 0.68, p = 0.0035), (B) superior parietal (r = 0.56, p = 0.025), (C) inferior temporal (r = 0.31, p = 0.3), and (D) superior frontal (r = 0.15, p = 0.6). Filled circles indicate dementia with Lewy bodies; open diamonds, Parkinson disease with mild cognitive impairment and Parkinson disease with dementia. PiB = [11C]Pittsburgh compound B; SUVR = standardized uptake value ratio.
An intriguing finding was the relationship between [11C]PiB uptake and non-Aβ pathologic findings. Braak neurofibrillary stage (and B score) correlated strongly with [11C]PiB retention (r = 0.73, p = 0.001) and with Braak Lewy body stage (r = 0.54, p = 0.025), with the latter result largely driven by the DLB group of participants (figure 3). In contrast, [11C]Altropane retention was not significantly correlated with Braak Lewy body stage in this cohort (p = 0.21) (figure 3). Across the established neuropathology metrics of ABC and Braak Lewy body stages, only the CERAD neuritic score and Braak NFT stages were significantly correlated (r = 0.50, p = 0.04).
Figure 3. β-Amyloid as detected by [11C]PiB was positively correlated with both neurofibrillary tangles and Lewy bodies.
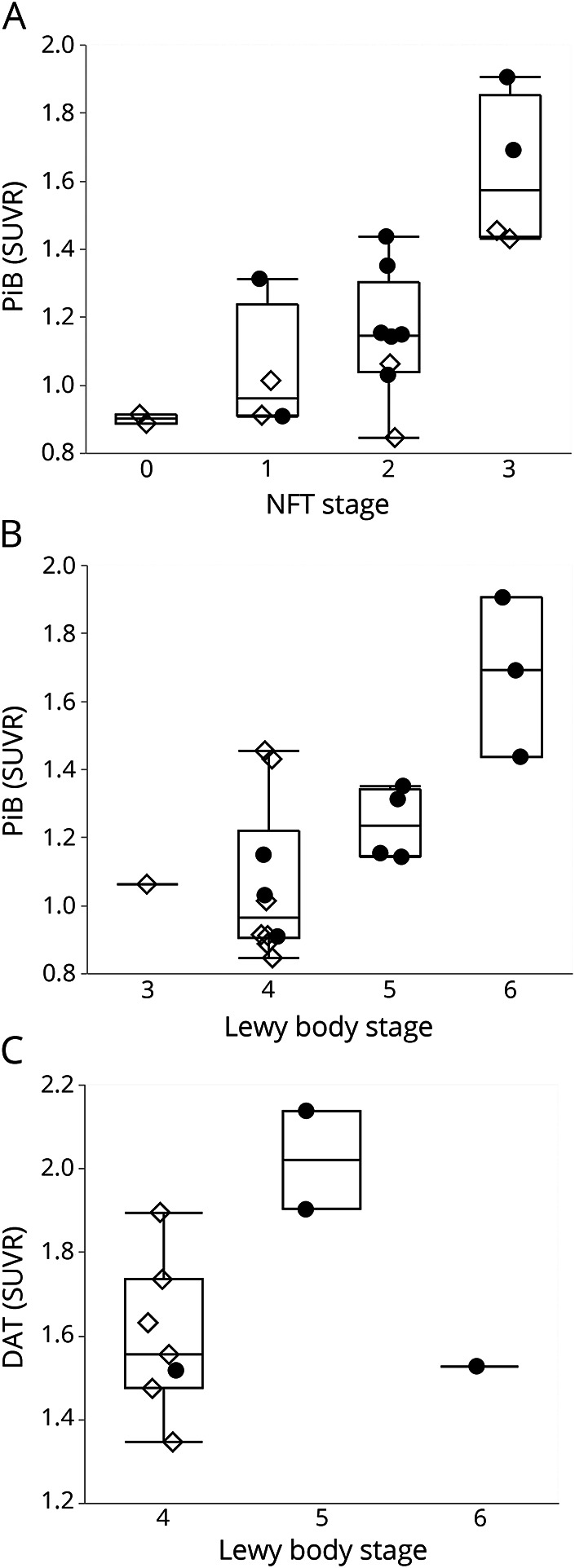
Partial correlations controlling for the interval between PET and autopsy: (A) neurofibrillary tangles (NFT) stage with [11C]Pittsburgh compound B (PiB) (r = 0.73, p = 0.001), (B) Lewy body stage with PiB (r = 0.54, p = 0.025), and (C) Lewy body stage with [11C]Altropane (p = 0.21). Filled circles indicate dementia with Lewy bodies. Open diamonds, Parkinson disease with mild cognitive impairment and Parkinson disease with dementia. DAT = dopamine transporter; SUVR = standard uptake value ratio.
Discussion
The results of this study indicate that DAT and amyloid molecular neuroimaging reflects the underlying pathologic changes that characterize the Lewy body spectrum of diseases. Using DAT imaging to support the diagnosis of PD is well documented17,34 and approaches 90% in sensitivity and specificity. In the present study, [11C]Altropane uptake was decreased in all 10 cases in whom it was performed, with no differences across the diagnostic groups.
The place of amyloid scans in the Lewy body set of diseases is less well documented and accepted. One of the first autopsies to examine the accuracy of [11C]PiB for detecting Aβ amyloid was in fact a case with DLB.35 This article was a single case report, and 2 small studies in PDD have since been completed.36,37 In contrast to LBD, however, there have been ample reports affirming [11C]PiB accuracy in detecting antemortem the amyloid deposits seen at autopsy in elderly participants with MCI and AD.22,38–40 Overall, the sensitivity and specificity have been very high, with demonstrated binding to both neuritic and nonneuritic yet fibrillar forms of Aβ.41–43 Our findings in the Lewy body spectrum of diseases largely confirm these reports and extend them to specify the nature and distribution of the [11C]PiB-neuropathology relationship.
PiB binds all fibrillar forms of amyloid, avidly staining both cored neuritic plaques and nonneuritic fibrillar diffuse plaques that are frequently present in both PDD and DLB. Because the CERAD plaque score measures only neuritic plaques, it is likely an underestimate of the total amyloid burden. Even so, [11C]PiB retention was significantly correlated with the postmortem CERAD score. To gain a more comprehensive understanding of [11C]PiB-amyloid correlations, we generated a total plaque score in 4 cortical regions that combined neuritic and nonneuritic fibrillar Aβ. [11C]PiB retention correlated with this total plaque score. When we analyzed each region individually, [11C]PiB retention and the total plaque score correlated significantly in the occipital region after multiple comparison testing and nominally in the superior parietal cortical region. Regional atrophy, which would reduce [11C]PiB retention by partial volume averaging with CSF, may have degraded regional correlations.
In this LBD cohort, [11C]PiB uptake did not increase monotonically with increasing Thal amyloid phase, a measure developed to capture the sequential spread of amyloid deposits across brain regions over the course of AD.29 This unexpected finding contrasts with prior reports in AD,22,38,44 raising the possibility that the hierarchical involvement of amyloid plaque topography in LBD may be distinct from AD. Even so, there was a moderately strong correlation of marginal significance between [11C]PiB uptake and Thal amyloid phase that would require a larger sample to evaluate at full power. These results suggest that the conventional neuropathologic measure of Thal amyloid phase may not readily capture cortical [11C]PiB retention in LBD, reinforcing the value of the total plaque score, a metric that accounts for combined neuritic and nonneuritic fibrillar plaque counts.
In this study, [11C]PiB uptake was also related to non-Aβ pathologic changes. There was a strong correlation between [11C]PiB retention and NFT severity. Because neuritic plaques and NFT are significantly correlated in postmortem studies,4 this finding further validates [11C]PiB imaging for detecting this relationship in life. We also found a significant correlation between [11C]PiB retention and Lewy body stages: the higher the Lewy body score, the higher the [11C]PiB uptake. This observation also is consistent with underlying pathologic changes known from PD and DLB postmortem studies4,45 and contrasted with the lack of significant correlation between [11C]Altropane uptake and Lewy body stages. These correlations support the view that Aβ, tau, and α-synuclein have the capacity to promote each other's aggregation.46–49 It is in this sense that [11C]PiB as a biomarker of Aβ shows additional promise as an index of NFT and Lewy bodies in PDD and DLB.
The strengths of this study include a well-characterized clinical population who underwent state-of-the-art neuroimaging and then, at death, careful neuropathologic examinations. Most of the patients were men, and all were white, so how well these findings relate to a more diverse population remains unknown. Lack of autopsy data on the healthy controls who underwent [11C]Altropane imaging is another theoretical limitation, although the absence of parkinsonism in otherwise healthy controls is likely to be a reliable predictor of an intact substantia nigra. Another possible weakness is the relatively small sample size, but there were enough patients to render unequivocal conclusions about the fidelity of amyloid brain scans to detect pathologic changes in the Lewy body spectrum of diseases.
Acknowledgment
The authors thank Joseph J. Locascio (Department of Neurology, Massachusetts General Hospital) for statistical counsel and Parisa Oviedo (Department of Neurology, Massachusetts General Hospital) for research assistance.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- DAT
dopamine transporter
- DLB
dementia with Lewy bodies
- FLR
frontal, lateral temporal, and retrosplenial regions
- LBD
Lewy body disease
- MCI
mild cognitive impairment
- NFT
neurofibrillary tangles
- PD
Parkinson disease
- PDD
Parkinson disease with dementia
- PiB
Pittsburgh compound B
- SUVR
standardized uptake value ratio
Appendix. Authors
Study funding
Supported by the National Alzheimer's Coordinating Center Collaborative Project 5 U01 AG016976-11 (to S.N.G., K.A.J., and J.H.G.), the National Institute of Neurologic Disorders and Stroke (to S.N.G., K.A.J., and J.H.G.; to K.A.J. alone), the National Institute on Aging (to K.A.J.), the Alzheimer's Disease Association (to K.A.J.), the National Parkinson Foundation (to S.N.G., K.A.J., and J.H.G.), and the Michael J. Fox Foundation (to S.N.G. and J.H.G.).
Disclosure
J. Shirvan reports fellowship funding by Biogen and Idec. N. Clement, S. Katz, R. Ye, and A. Schultz report no disclosures relevant to the manuscript. K. Johnson has served as a paid consultant for Bayer, GE Healthcare, Janssen Alzheimer's Immunotherapy, Siemens Medical Solutions, Genzyme, Novartis, Biogen, Roche, ISIS Pharma, AZTherapy, GEHC, Lundberg, and Abbvie. He is a site coinvestigator for Lilly/Avid, Pfizer, Janssen Immunotherapy, and Navidea. He has spoken at symposia sponsored by Janssen Alzheimer's Immunotherapy and Pfizer. He receives funding from NIH grants R01EB014894, R21 AG038994, R01 AG026484, R01 AG034556, P50 AG00513421, U19 AG10483, P01 AG036694, R13 AG042201174210, R01 AG027435, and R01 AG037497 and Alzheimer's Association grant ZEN-10-174210. T. Gomez-Isla participated as speaker at an Eli Lilly and Co–sponsored educational symposium and serves on an Eli Lilly Data Monitoring Committee. She receives research funding from NIH grants AG005134, AG036694, and AG043511. M. Frosch reports a sponsored research agreement with Biogen. He has received honoraria from NIH, Northwestern University, and the University of Michigan. J. Growdon reports honoraria from the National Institute on Aging and JAMA and serves on the Advisory Board for Neuroimmune Holding. S. Gomperts has served on the Advisory Board for Acadia Pharmaceuticals and Sanofi. He receives funding from NIH grants R01 AG054551, R01 AG062208, P50 AG005134, R21 NS109833, and DOD CDMRP/W81XW1810516 and the Lewy Body Dementia Association. Go to Neurology.org/N for full disclosures.
References
- 1.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 2.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 4.Colom-Cadena M, Gelpi E, Charif S, et al. Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 2013;72:1203–1212. [DOI] [PubMed] [Google Scholar]
- 5.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuboi Y, Dickson DW. Dementia with Lewy bodies and Parkinson's disease with dementia: are they different? Parkinsonism Relat Disord 2005;11(suppl 1):S47–S51. [DOI] [PubMed] [Google Scholar]
- 7.Holtzman D. ADRD Summit 2016 Report to the National Advisory Neurological Disorders and Stroke Council. US Department of Health and Human Services [online]. Available at: aspehhsgov/adrd-summit-2016-report-national-advisory-neurological-disorders-and-stroke-council. Accessed September 15, 2016. [Google Scholar]
- 8.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging 2012;33:2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord 2012;27:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomperts SN. Imaging the role of amyloid in PD dementia and dementia with Lewy bodies. Curr Neurol Neurosci Rep 2014;14:472. [DOI] [PubMed] [Google Scholar]
- 12.Gomperts SN, Locascio JJ, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 2013;80:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomperts SN, Marquie M, Locascio JJ, Bayer S, Johnson KA, Growdon JH. PET radioligands reveal the basis of dementia in Parkinson's disease and dementia with Lewy bodies. Neurodegener Dis 2016;16:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKeith I, O'Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol 2007;6:305–313. [DOI] [PubMed] [Google Scholar]
- 15.Walker RW, Walker Z. Dopamine transporter single photon emission computerized tomography in the diagnosis of dementia with Lewy bodies. Mov Disord 2009;24(suppl 2):S754–S759. [DOI] [PubMed] [Google Scholar]
- 16.Gomperts SN. Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Continuum 2016;22:435–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaasinen V, Vahlberg T. Striatal dopamine in Parkinson disease: a meta-analysis of imaging studies. Ann Neurol 2017;82:873–882. [DOI] [PubMed] [Google Scholar]
- 18.Marquie M, Locascio JJ, Rentz DM, et al. Striatal and extrastriatal dopamine transporter levels relate to cognition in Lewy body diseases: an (11)C altropane positron emission tomography study. Alzheimers Res Ther 2014;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson's disease. Arch Neurol 1993;50:140–148. [DOI] [PubMed] [Google Scholar]
- 20.Litvan I, Goldman JG, Tröster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord 2012;27:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007;22:1689–1707; quiz 1837. [DOI] [PubMed] [Google Scholar]
- 22.Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 2015;138:1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greve DN, Salat DH, Bowen SL, et al. Different partial volume correction methods lead to different conclusions: an (18)F-FDG-PET study of aging. Neuroimage 2016;132:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedden T, Van Dijk KR, Becker JA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci 2009;29:12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mormino EC, Betensky RA, Hedden T, et al. Amyloid and APOE epsilon4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 2014;82:1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischman AJ, Bonab AA, Babich JW, et al. [(11)C, (127)I] Altropane: a highly selective ligand for PET imaging of dopamine transporter sites. Synapse 2001;39:332–342. [DOI] [PubMed] [Google Scholar]
- 27.Elmaleh DR, Fischman AJ, Shoup TM, et al. Preparation and biological evaluation of iodine-125-IACFT: a selective SPECT agent for imaging dopamine transporter sites. J Nucl Med 1996;37:1197–1202. [PubMed] [Google Scholar]
- 28.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 30.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 32.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991;30:637–649. [DOI] [PubMed] [Google Scholar]
- 33.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 34.Bajaj N, Hauser RA, Grachev ID. Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2013;84:1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh compound B confirmed at autopsy: a case report. Arch Neurol 2007;64:431–434. [DOI] [PubMed] [Google Scholar]
- 36.Akhtar RS, Xie SX, Brennan L, et al. Amyloid-beta positron emission tomography imaging of Alzheimer's pathology in Parkinson's disease dementia. Mov Disord Clin Pract 2016;3:367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burack MA, Hartlein J, Flores HP, Taylor-Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology 2010;74:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikonomovic MD, Buckley CJ, Heurling K, et al. Post-mortem histopathology underlying beta-amyloid PET imaging following flutemetamol F 18 injection. Acta Neuropathol Commun 2016;4:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol 2012;11:669–678. [DOI] [PubMed] [Google Scholar]
- 40.Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement 2015;11:964–974. [DOI] [PubMed] [Google Scholar]
- 41.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain 2007;130:2607–2615. [DOI] [PubMed] [Google Scholar]
- 42.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 2008;131:1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niedowicz DM, Beckett TL, Matveev S, et al. Pittsburgh compound B and the postmortem diagnosis of Alzheimer disease. Ann Neurol 2012;72:564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thal DR, Beach TG, Zanette M, et al. [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of amyloid-beta pathology. Alzheimers Dement 2015;11:975–985. [DOI] [PubMed] [Google Scholar]
- 45.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol 2002;59:102–112. [DOI] [PubMed] [Google Scholar]
- 46.Masliah E, Rockenstein E, Veinbergs I, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA 2001;98:12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Gerson JE, Kayed R. Alpha-synuclein oligomers induce a unique toxic tau strain. Biol Psychiatry 2018;84:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sengupta U, Guerrero-Muñoz MJ, Castillo-Carranza DL, et al. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol Psychiatry 2015;78:672–683. [DOI] [PubMed] [Google Scholar]
- 49.Bennett RE, DeVos SL, Dujardin S, et al. Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol 2017;187:1601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available on request.